

Abstract
The induction of acetylcholine receptor (AChR) clustering by neurally released agrin is a critical, early step in the formation of the neuromuscular junction. Laminin, a component of the muscle fiber basal lamina, also induces AChR clustering. We find that induction of AChR clustering in C2 myotubes is specific for laminin-1; neither laminin-2 (merosin) nor laminin-11 (a synapse-specific isoform) are active. Moreover, laminin-1 induces AChR clustering by a pathway that is independent of that used by neural agrin. The effects of laminin-1 and agrin are strictly additive and occur with different time courses. Most importantly, laminin- 1–induced clustering does not require MuSK, a receptor tyrosine kinase that is part of the receptor complex for agrin. Laminin-1 does not cause tyrosine phosphorylation of MuSK in C2 myotubes and induces AChR clustering in myotubes from MuSK−/− mice that do not respond to agrin. In contrast to agrin, laminin-1 also does not induce tyrosine phosphorylation of the AChR, demonstrating that AChR tyrosine phosphorylation is not required for clustering in myotubes. Laminin-1 thus acts by a mechanism that is independent of that used by agrin and may provide a supplemental pathway for AChR clustering during synaptogenesis.
The highly specialized postsynaptic structures at the adult neuromuscular junction arise and are maintained in part through signals conveyed by specific components of the basal lamina (Burden et al., 1979; Hall and Sanes, 1993). The most prominent feature of the postsynaptic muscle membrane is the high density (>10,000/μm2) of acetylcholine receptors (AChRs)1 on the myotube surface (Fertuck and Salpeter, 1976). During synaptogenesis, AChRs, which are diffusely distributed on myotubes before innervation, accumulate at the nascent synapse soon after the growth cone first contacts the myotube membrane. Shortly thereafter a defined basal lamina appears between the nerve terminal and the muscle cell. This local postsynaptic accumulation of AChRs is caused by two mechanisms: an increase in local receptor synthesis and the redistribution of preexisting surface AChRs (Hall and Sanes, 1993).
The clustering of preexisting AChRs beneath the nerve terminal is thought to occur through the action of agrin, a component of the adult synaptic basal lamina that is synthesized and released by motor neurons (McMahan, 1990). Agrin is a large protein with several sites of alternative splicing that affect its biological activity (Rupp et al., 1991; Ferns et al., 1992, 1993; Ruegg et al., 1992; Tsim et al., 1992). The isoforms most active in clustering AChRs are found in neurons, whilst muscle cells synthesize less active isoforms (Ferns et al., 1993; Hoch et al., 1993). Treatment of myotube cultures with agrin causes coaggregation of the AChR and other components of the adult synapse, such as acetylcholinesterase, laminin, and heparan sulfate proteoglycan (Wallace, 1989; Nitkin and Rothschild, 1990). Recently, experiments using knock-out mice have unequivocably established the importance of agrin in synaptogenesis. Muscles from agrin-deficient mice fail to make differentiated neuromuscular junctions; although muscles in these animals have AChR clusters, they are smaller, and few are associated with nerve fibers (Gautam et al., 1996).
Agrin mediates AChR clustering by binding and activating a signaling receptor on the muscle surface that is specific for the neural agrin isoform (Bowen et al., 1996). Although the muscle receptor that binds agrin and mediates its action on AChR clustering has not been completely identified (Nastuk et al., 1991), one of its components appears to be MuSK (muscle specific kinase), a receptor tyrosine kinase specifically expressed by skeletal muscle. MuSK is upregulated at the onset of muscle differentiation and is localized to the adult neuromuscular junction (Valenzuela et al., 1995). Agrin treatment of cultured myotubes induces tyrosine phosphorylation of MuSK, and in intact myotubes, agrin can be crosslinked to MuSK, presumably via an accessory protein (Glass et al., 1996). Experiments using mice homozygous for a MuSK gene disruption show that MuSK is necessary for formation of the neuromuscular junction. MuSK-null mice do not form AChR clusters, and myotubes made from these mutant mice fail to respond to neural agrin (DeChiara et al., 1996; Glass et al., 1996). Thus, MuSK is required for agrin-induced AChR clustering and appears to be part of the receptor complex that binds agrin and mediates its action on muscle cells. The precise molecular mechanism by which agrin-induced activation of MuSK leads to AChR clustering is not known but may involve phosphorylation of the AChR itself (Wallace et al., 1991; Qu and Huganir, 1994; Ferns et al., 1996). In C2 myotubes, agrin induces a transient phosphorylation of the β subunit of the AChR that precedes clustering; moreover, inhibitors that block the AChR phosphorylation also block AChR clustering (Wallace, 1994; Ferns et al., 1996).
Besides agrin, a number of other factors stimulate AChR clustering in cultured muscle cells. These include laminin (Vogel et al., 1983), VVA-B4, a lectin that selectively binds a synapse-specific carbohydrate (Martin and Sanes, 1995), and two proteins, basic fibroblast growth factor and heparin-binding growth-associated molecule, that are active when bound to polystyrene beads (Peng et al., 1991, 1995). Neither the mechanism by which these factors act, nor their physiological role, is known.
The possible role of laminin in synapse formation is of particular interest as it is present at the earliest synapses and is colocalized with AChR clusters formed both in vivo and in vitro (Bayne et al., 1984; Chiu and Sanes, 1984; Daniels et al., 1984; Gordon et al., 1993). In the adult, laminin is a major component of the muscle fiber basal lamina, with distinct synaptic and extrasynaptic isoforms. Laminin is a heterotrimer composed of three chains, α, β, and γ, and multiple forms of each chain are encoded by separate genes (e.g., α1-5, β1-3, and γ1-2). The type of laminin isoform produced is dependent upon its chain composition (Timpl et al., 1982; Burgeson et al., 1994). Several different laminin isoforms with distinct distribution patterns are found in skeletal muscle (Sanes et al., 1990; Lentz et al., 1997; Miner et al., 1997). The isoforms that have been identified in muscle are laminin-1 (α1β1γ1), laminin-2 (merosin; α2β1γ1), which is the predominant isoform found in adult muscle (Leivo and Engvall, 1988), and laminin-11 (a synapse-specific isoform; α5β2γ1), a recently characterized heterotrimer that contains two chains, α5 and β2 (s-laminin), that are only expressed at the adult endplate (Hunter et al., 1989; Martin et al., 1995; Patton, B.L., and J.R. Sanes, personal communication). The α1 chain of laminin-1 is expressed early in skeletal muscle development (Sewry et al., 1995), and its expression increases in dystrophic and inflammatory myopathies, indicating that it may play a role in muscle regeneration (Mundegar et al., 1995).
Because of its potential physiological importance, we have investigated the mechanism by which laminin-1 induces AChR clustering in C2 myotubes. We find that laminin-1, but not laminin-2 or -11, induce AChR clustering and that the effects of laminin-1 are additive to those of agrin. Laminin-1–induced AChR clustering does not require MuSK and is not accompanied by tyrosine phosphorylation of the AChR. The laminin-1 signaling pathway is thus different from that used by agrin and may serve as an additional or supplemental mechanism for AChR clustering during the formation and maintenance of the neuromuscular junction.
MATERIALS AND METHODS
Antibodies and Reagents
A peptide corresponding to the last 20 amino acid residues of rat MuSK (CSIHRILQRMCERAEGTVGV; Valenzuela et al., 1995) was synthesized and used to produce polyclonal antibodies in rabbits by Research Genetics (Huntsville, AL). Anti-phosphotyrosine antibodies were purchased from Upstate Biotechnology Inc. (mAb 4G10; Lake Placid, NY) and from Transduction Laboratories (mAb PY20; Lexington, KY). The AChR β subunit antibody, mAb 124, was a gift from John Lindstrom (University of Pennsylvania, Philadelphia, PA). Rhodamine-conjugated α-bungarotoxin was purchased from Molecular Probes, Inc. (Eugene, OR). Laminin-1 (EHS tumor-derived), α-bungarotoxin, and CNBr-activated Sepharose beads were purchased from Sigma Chemical Co. (St. Louis, MO). Toxin-conjugated Sepharose beads were prepared as described previously by Gu and Hall (1988). Laminin-2 (isolated from human placenta) was purchased from GIBCO BRL (Gaithersburg, MD). Laminin-11 (isolated from a Schwann cell sarcoma) was a gift from Arlene Chiu (Beckman Research Institute of the City of Hope, Duarte, CA). Soluble recombinant neural (C-Ag4,8) and muscle (C-Ag0,0) agrin was prepared from transiently transfected COS cells as previously described by Ferns et al. (1993), and concentrations were determined by comparison to purified agrin (Sugiyama et al., 1994) on Western blots.
Production of the MuSK−/− Cell Line
Mice heterozygous for a mutant MuSK allele (DeChiara et al., 1996) were crossed with transgenic mice that were hemizygous for a temperature-sensitive large T antigen transgene under the control of a γ interferon-inducible MHC promoter (Jat et al., 1991). The resulting double heterozygotes were crossed to obtain mice that were homozygous for the mutant MuSK allele and hemizygous for the large T transgene. Hindlimb muscles from both limbs were dissected at birth, placed into 1 ml of Dulbecco's modified minimum Eagle's medium (DMEM) supplemented with l-glutamine and penicillin/streptomycin, and placed on ice. Collagense B (Sigma Chemical Co.) was added to a final concentration of 0.25% and incubated at 37°C for 30 min. The tissue was washed several times in DMEM and then incubated in 0.25% trypsin for 30 min at 37°C with several gentle agitations. The tissue was disrupted by pipeting 7–10 times with a narrow tip pipette; the cell suspension was filtered through sterile nylon mesh to remove debris. The filtrate was diluted in DMEM containing 10% fetal bovine serum, l-glutamine, penicillin/streptomycin, and 10 ng/ml γ interferon (Biosource International, Camarillo, CA), and then plated onto a 10-cm tissue culture dish to allow the fibroblasts to attach. After 40 min at 30°C, the myoblast-enriched supernatant was harvested and replated in 96-well plates. The cells were grown in the presence of 10 ng/ml γ interferon for ∼10 d. Individual clones were expanded and tested for their ability to form myotubes when incubated at the nonpermissive temperature of 37°C in DMEM containing 2% horse serum, l-glutamine, and penicillin/ streptomycin without γ interferon.
Muscle Cell Culture
C2C12 mouse muscle cells were cultured as described by Gordon and Hall (1989). Myoblasts were grown in DMEM containing 20% fetal calf serum, 0.5% chick embryo extract (GIBCO BRL), 2 mM glutamine, and penicillin/streptomycin. When the cells reached confluence, the growth medium was replaced with DMEM containing 5% horse serum and 2 mM glutamine to induce myotube differentiation. MuSK−/− myotube cultures were grown, as described above, on glass chamberslides coated with fibronectin.
AChR Clustering Assay
C2 myotubes were grown on Permanox-treated 8-well chamberslides (Nunc, Inc., Naperville, IL), and MuSK−/− myotubes were grown on 8-well glass chamberslides coated with fibronectin. Controlled experiments showed that the use of fibronectin did not affect laminin-1–induced AChR clustering. Mature myotubes were treated with soluble neural agrin (C-Ag4,8) or with laminin-1 for 18 h (unless otherwise mentioned), and the cells were subsequently stained with rhodamine-conjugated α-bungarotoxin (Molecular Probes, Inc.) for 1 h at 37°C to visualize AChR clusters. The cells were then rinsed, fixed in cold methanol for 5 min, and mounted in glycerol containing p-phenylenediamine. AChR clusters were counted in random fields (chosen under phase optics) under 400× (C2 cultures) or 200× (MuSK−/− cultures). Within each experiment (n = 1), 10 fields/treatment were counted and averaged. To measure the intensity of rhodamine-labeled AChR clusters, random fields (10 fields/treatment per experiment) under 600× were scanned into a MetaMorph image analysis program, and an average of the measured intensity was taken after subtracting the background intensity. To measure the length of rhodamine-labeled AChR clusters, within an experiment, all clusters within 10 random fields (randomly chosen under phase optics) were measured under 400× and averaged.
MuSK Extraction and Isolation
Mature C2 myotubes were incubated with neural agrin (C-Ag4,8), laminin-1, or with both for 45 min at 37°C. After the treatments, the cells were rinsed with Ca2+/Mg2+-free PBS and scraped up into an extraction buffer (1% Nonidet P-40 or 1% digitonin; 10 mM Tris, pH 7.5; 150 mM NaCl; 5 mM each EDTA and EGTA; 1 mM sodium orthovanadate; 1 mM each PMSF, benzamidine, N-ethylmaleimide, sodium tetrathionate; and 20 μg/ml each leupeptin and aprotinin). After removing insoluble material by centrifugation at 18,000 g for 10 min, the C2 extracts were incubated with a polyclonal anti-MuSK serum followed by protein A coupled to Sepharose. The Sepharose beads were washed in extraction buffer and proteins bound to the beads were removed by boiling in SDS sample buffer. The immunoprecipitated proteins were then separated by SDS-PAGE, transferred to nitrocellulose sheets, and the blots incubated with a mixture of phosphotyrosine antibodies, mAbs 4G10 and PY20, followed by incubation with an HRP-conjugated anti–mouse secondary antibody. Immunolabeled bands were visualized by enhanced chemiluminescence (ECL; Amersham Corp., Arlington Heights, IL). The blots were stripped with 0.2 M glycine, pH 2.5, and 0.1% Tween20 and reprobed with the MuSK polyclonal antiserum to confirm that similar amounts of total MuSK were isolated.
AChR Extraction and Isolation
C2 myotubes were treated and extracted as described above. AChRs were isolated by incubating the C2 extracts with α-bungarotoxin coupled to Sepharose beads, followed by washing the beads in extraction buffer and boiling in SDS sample buffer. Western blotting was performed as described above. The blots were stripped and reprobed with an antibody against the β subunit, mAb 124, to confirm that similar amounts of total AChR β subunit were isolated on Separose beads.
RESULTS
Laminin-1 Induces AChR Cluster Formation on C2 Myotubes
Previous experiments have shown that laminin-1 can induce AChR clustering on primary rat myotubes or on G8-1 clonal rat muscle cells (Vogel et al., 1983). We compared laminin-1 and agrin-induced AChR clustering on C2 myotubes using laminin-1 isolated from the basement membrane of the mouse EHS tumor, and a soluble COOH-terminal truncated agrin fragment (Ferns et al., 1993). When cultures of C2 mouse myotubes were treated with either 40 nM (33 μg/ml) of laminin-1 or 150 pM soluble neural agrin (C-Ag4,8), the number of AChR clusters observed after labeling with rhodamine-conjugated α-bungarotoxin was dramatically increased compared to the small number of spontaneous AChR clusters seen in untreated C2 myotubes (Fig. 1, A–C). These results were further analyzed by counting the number of AChR clusters induced by various concentrations of laminin-1. The effects of laminin-1 were detectable at concentrations as low as 12 nM and reached maximal levels at concentrations of ∼120 nM (Fig. 2 A). When saturating concentrations were used, laminin-1 was nearly as effective in inducing the formation of AChR clusters as agrin (compare Figs. 2, A and B). Clusters produced by laminin-1, however, appeared more intensely stained than those produced by agrin, suggesting that the density of the AChRs in the laminin-1–induced clusters may be higher. When the fluorescence intensity of rhodamine-labeled AChR clusters was quantitated using an image analysis program, we found that the AChR clusters induced by 40 nM laminin-1 were over two times more brightly stained than those induced by 15 pM agrin (Fig. 3 A). In addition, the length of the laminin-1–induced AChR clusters appeared to be greater than those induced by higher concentrations (1.5 nM) of agrin (Fig. 3 B).
Figure 1.

Laminin-1 induces AChR clustering and increases agrin-induced AChR clustering in an additive manner. C2 myotubes were untreated (A), treated with 150 pM soluble neural agrin (B), 40 nM laminin-1 (C), or treated with both 150 pM neural agrin and 40 nM laminin-1 (D). Cultures were then stained with rhodamine-conjugated α-bungarotoxin. Neural agrin and laminin-1 induce the formation of AChR clusters, while treatment with both further increases the number of AChR clusters. Bar, 25 μm.
Figure 2.


(A) Laminin-1 induces AChR aggregation in a dose-dependent manner. C2 myotubes were treated with concentrations of laminin-1 ranging from 1.2 to 400 nM. Laminin-1 begins to induce AChR clustering at 12 nM and peaks at 120 nM. (B) Laminin-1 and neural agrin act additively to induce AChR clustering. C2 myotubes were treated with varying concentrations of neural agrin ranging from 0.15 pM to 15 nM, without (closed squares) or with (open circles) 40 nM of laminin-1. Treatment with both laminin-1 and agrin results in an increased number of AChR clusters over that induced by neural agrin alone. For each treatment, the number of AChR clusters was drawn from three experiments by counting and averaging clusters within 10 fields/ experiment and is shown as means ± SEM (n = 3).
Figure 3.


(A) Laminin-1–induced AChR clusters are more intensely stained than agrin-induced AChRs. C2 myotubes were treated with 15 pM neural agrin, 40 nM laminin-1, or with the two together, and the fluorescence intensity was measured as described in Materials and Methods. The laminin-1–induced clusters are more intensely stained than the agrin-induced AChRs, while treatment with both agrin and laminin-1 produced brightly stained clusters more similar to those induced by laminin-1. Data are averaged from two different experiments (10 fields/experiment) and shown as means ± SD (n = 2). (B) AChR clusters induced by high concentrations of agrin are shorter than those induced by low concentrations of agrin or by laminin-1. C2 myotubes were treated with 15 pM or 1.5 nM neural agrin, 12 or 120 nM laminin-1, or with both 1.5 nM neural agrin and 120 nM laminin-1. Clusters induced by high (1.5 nM) concentrations of agrin are smaller than those induced by low (15 pM) concentrations. In contrast, both concentrations of laminin- 1–induced large AChR clusters to form. Treatment with high concentrations of both agrin and laminin-1 produced large clusters, similar in size to the laminin-induced clusters. Data are averaged from three different experiments (10 fields/experiment) and shown as means ± SEM (n = 3). Asterisks denote significant difference (*P < 0.05 or **P < 0.01) between high concentrations of agrin and other treatments according to analysis of variance (ANOVA) and a Student-Newman-Keuls test.
When C2 myotubes were treated with both neural agrin and laminin-1, a greater number of AChR clusters was observed than with either treatment alone (Fig. 1 D). Some AChR clusters that formed in response to treatment with both neural agrin and laminin-1 also appeared to be longer and more organized along the myotube edge (Fig. 1 D) compared to clusters induced by laminin-1 or neural agrin alone. To test whether laminin-1 and neural agrin act through the same pathway or through independent pathways to produce AChR clusters, we determined whether or not their effects were additive. C2 myotubes were treated with various concentrations of neural agrin (0.15 pM to 15 nM) in the presence or absence of 40 nM of laminin-1. At all concentrations of agrin tested, including concentrations that produced a maximal effect, the addition of laminin-1 induced a strictly additive response (Fig. 2 B). These observations suggest that the two ligands do not compete for the same receptor and that the pathways by which they act do not share limiting steps.
To determine whether the laminin effect on AChR clustering is specfic for particular isoforms, we tested whether other laminins expressed in muscle could cause AChRs to aggregate. Accordingly, C2 myotubes were treated with either laminin-1, -2 (merosin), or -11 (a synapse-specific isoform containing the β2 chain). We found that only the laminin-1 isoform could induce AChR clustering (Fig. 4). Neither laminin-2 nor -11 was active, even at concentrations of 120 nM (the concentration of laminin-1 that produced a maximal clustering response). Both laminin-2 and -11 used in these experiments were shown to be active in other biological assays (Kleinman, H., and A. Chiu, personal communication). The failure of laminin-11 to induce clustering is surprising since it is localized at the adult neuromuscular junction (Patton, B.L., and J.R. Sanes, personal communication). Induction of AChR clustering on myotubes is thus not a general property of laminins, but appears to be specific for laminin-1 (α1β1γ1). As α1 is the only chain of laminin-1 that is not shared with the other two isoforms, it appears to confer specificity for clustering.
Figure 4.

AChR clustering activity is specific for the laminin-1 isoform. C2 myotubes were untreated (A) or treated with 60 nM of laminin-2 (B), laminin-11 (C), or laminin-1 (D). Cultures were stained with rhodamine-conjugated α-bungarotoxin and examined under 400×. Unlike laminin-1, laminin-2 and -11 were both unable to induce AChR aggregation over background levels. Bar, 25 μm.
Laminin-1 and Agrin Cluster AChRs with Different Time Courses
Agrin acts quickly to aggregate surface AChRs; after the addition of agrin to C2 myotubes, AChR clusters are first apparent after 3 h, and their number reaches a peak within 6 to 8 h (Ferns et al., 1996). To determine if laminin-1 acts with the same time course, we treated C2 myotubes with either neural agrin (150 pM) or laminin-1 (120 nM) for 4, 12, or 18 h. We then stained the myotubes with rhodamine– α-bungarotoxin to visualize the receptor clusters and counted the number of AChR clusters in random fields (Fig. 5). After 4 h of treatment, agrin-treated cultures exhibited a large increase in the number of AChR clusters, whereas laminin-1–treated myotubes displayed no increase over the background level. After 12 h, however, C2 cultures treated with laminin-1 exhibited roughly a twofold increase in AChR cluster number, and after 18 h of treatment, the number of laminin-1–induced clusters approached the number of clusters induced by 150 pM of neural agrin. Thus, the induction of AChR clusters by laminin-1 occurs significantly more slowly than by neural agrin.
Figure 5.

Agrin and laminin-1 induce AChR aggregation over different time courses. C2 myotubes were treated with 150 pM soluble neural agrin (solid bars) or with 120 nM laminin-1 (striped bars) for 4, 12, or 18 h. Whereas agrin quickly induces receptor aggregation within 4 h and has a peak effect by 12 h, laminin-1 acts with a slower time course, and half-maximal clustering does not occur until ∼12 h. Data averaged from three (Laminin-1) or four (Agrin) experiments are shown as means ± SEM (n = 3 or 4). Asterisks denote significant difference between untreated and agrin-treated (**P < 0.001) or laminin-treated (*P < 0.01) cultures according to analysis of variance (ANOVA) and a Student-Newman-Keuls test.
Laminin-1 Induces AChR Clustering in MuSK−/− Myotubes
Neural agrin requires MuSK, a receptor tyrosine kinase, to induce the formation of AChR clusters. Mice with a targeted disruption of the gene encoding MuSK do not form neuromuscular synapses (DeChiara et al., 1996), and myotubes from such mice fail to cluster AChRs in response to neural agrin (Glass et al., 1996). To determine whether MuSK is also required for laminin-1 clustering activity, we treated immortalized myotube cultures made from MuSK−/− mouse muscle with either laminin-1 or neural agrin and then stained them with rhodamine–α-bungarotoxin. When treated with laminin-1, the MuSK−/− myotubes produced AChR clusters of varying sizes, ranging from short, dense clusters on the myotube surface to long clusters along the edge of the myotube, similar to those seen on C2 myotubes (Fig. 6). Quantitation of these results by counting clusters in random fields showed that only myotubes treated with laminin-1 were able to produce AChR clusters (Fig. 7). The number of AChR clusters induced by laminin-1 on the MuSK−/− myotubes, however, was not as great as those observed on C2 myotubes (∼20-fold fewer clusters compared to C2 myotubes), even when concentrations as high as 120 nM of laminin-1 were used. In general, the MuSK−/− myotubes differed in appearance and were thinner than the C2 myotubes. Thus, the reduced number of AChR aggregates on the mutant myotubes could be the result of inherent differences between C2 myotubes and the immortalized MuSK−/− myotubes, or it could be a specific consequence of the absence of MuSK. As expected, we did not observe AChR clustering on MuSK−/− myotubes treated with neural agrin, even at concentrations up to 15 nM. In the absence of laminin-1, occasional bright spots and faint edge staining were observed on the MuSK−/− myotubes but did not resemble AChR clusters. This nonspecific staining accounted for the nominal clustering values recorded for untreated and agrin-treated MuSK−/− myotubes (Fig. 7).
Figure 6.
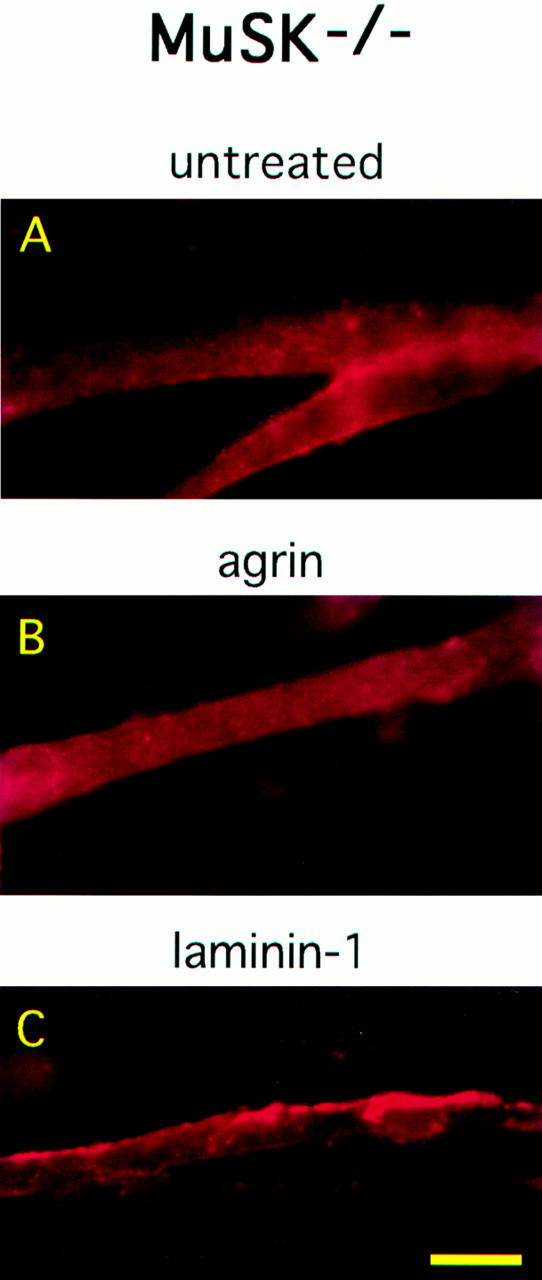
Laminin-1 induces AChR clustering on MuSK−/− myotubes. MuSK−/− myotubes were untreated (A) or treated with 150 pM neural agrin (B) or 120 nM laminin-1 (C). Cultures were stained with rhodamine-conjugated α-bungarotoxin and visualized under 400×. Only laminin-1 treatment induces AChR aggregation on MuSK−/− myotubes. Bar, 25 μm.
Figure 7.

Quantitation of laminin-1–induced AChR clustering on MuSK−/− myotubes. AChR clusters observed on MuSK−/− myotubes (untreated or treated with 120 nM laminin-1 or 150 pM neural agrin) were counted under 200×, and an average from four experiments (10 fields/experiment) is shown as means ± SEM. (n = 4). Note that there are somewhat fewer MuSK−/− myotubes per field compared to C2 myotubes, and the magnification used for MuSK−/− cluster quantitation is different from that used for C2 cluster quantitation in Fig. 2 A (400×); thus, the number of clusters per field on MuSK−/− and C2 myotubes cannot be directly compared.
Laminin-1 Does Not Induce MuSK Tyrosine Phosphorylation
Since MuSK activation is thought to be an early event in the agrin signaling pathway, we next tested whether laminin-1 activates MuSK. C2 myotubes were treated for 45 min with either 4, 12, 40, or 120 nM of laminin-1, 1.5 pM to 15 nM of soluble neural agrin (Ag4,8), or with 15 nM of soluble muscle agrin (Ag0,0). Proteins were then extracted in a detergent-containing buffer and divided into two groups. Extracts from one group were immunoprecipitated with a MuSK polyclonal serum, and the remaining extracts were treated to isolate AChRs as described below. After separating proteins by SDS-PAGE and transferring them to nitrocellulose, the blots were probed with a mixture of the phosphotyrosine antibodies, mAb 4G10 and mAb PY20. We found that under these conditions, treatment of C2 myotubes with neural agrin, even at concentrations of 15 pM, induced MuSK tyrosine phosphorylation. Thus, the concentrations of neural agrin that induce AChR clustering and those that induce MuSK tyrosine phosphorylation are similar. Laminin-1, however, did not induce MuSK phosphorylation even at high concentrations (Fig. 8) or after 5, 8, 12, or 18 h of treatment (Sugiyama, J.E., and Z.W. Hall, unpublished observations), indicating that it is probably not a functional ligand for MuSK. The blots were later reprobed for total MuSK to confirm that equivalent amounts of MuSK had been immunoprecipitated. The lower bands in the MuSK immunoblot are most likely nonspecific bands recognized by the antiserum.
Figure 8.

Laminin-1 does not induce MuSK phosphorylation. C2 myotubes were incubated with either laminin-1 (4 to 120 nM) or with neural agrin (1.5 pM to 15 nM) for 45 min at 37°C. MuSK was isolated from cell extracts by immunoprecipitation, and isolated proteins on nitrocellulose blots were probed with anti-phosphotyrosine antibodies. Laminin-1 does not induce MuSK tyrosine phosphorylation, even at concentrations in excess of what is required to induce AChR clustering. In contrast, strongly labeled phosphotyrosine bands corresponding to MuSK were observed in lanes containing extracts treated with 0.15 to 15 nM neural agrin. No phosphotyrosine signal was observed in lanes containing untreated or muscle agrin-treated extracts. The blots were stripped and reprobed with a polyclonal antiserum against MuSK to show that the amount of MuSK in each lane is similar. The arrowhead indicates the MuSK-reactive band that corresponds to the single band in the phosphotyrosine blot above; the other nonspecific bands are due to antibody crossreactions.
Laminin-1 Does Not Induce Phosphorylation of the AChR β Subunit
Because tyrosine phosphorylation of the β subunit of the AChR is an early response to agrin that precedes, and may be required for, agrin-induced AChR clustering, we were interested to determine whether laminin-1 also caused tyrosine phosphorylation of the AChR. C2 myotubes were incubated with either 4, 12, 40, or 120 nM of laminin-1, 1.5 pM to 15 nM of soluble neural agrin (Ag4,8), or 15 nM of soluble muscle agrin (Ag0,0) as described above, and detergent extracts were prepared. The extracts were then incubated with α-bungarotoxin conjugated to Sepharose beads to isolate AChRs. Bound AChR was removed from the toxin beads with SDS and the subunits separated by SDS-PAGE and transferred to nitrocellulose blots. The blots were assayed for tyrosine phosphorylation of the AChR β subunit as described above and then reprobed with mAb 124, an antibody against the β subunit. Immunoblots with mAb 124 showed that equal amounts of AChR had been isolated on the α-bungarotoxin beads. Whereas treatment with neural agrin induced β subunit phosphorylation, neither laminin-1 treatment, at any concentration tested, nor muscle agrin treatment caused tyrosine phosphorylation of the β subunit (Fig. 9). Thus, tyrosine phosphorylation of the β subunit is not required for AChR clustering.
Figure 9.

Laminin-1 does not induce tyrosine phosphorylation of the AChR β subunit. C2 myotubes were treated with neural agrin (1.5 pM to 15 nM) or with laminin-1 (4 to 120 nM) for 45 min at 37°C. AChRs were isolated on α-bungarotoxin Sepharose beads, and nitrocellulose blots containing AChRs were probed with anti-phosphotyrosine antibodies. Laminin-1 did not induce β subunit phosphorylation at any concentration. The same concentrations of neural agrin that induced both AChR clustering and MuSK phosphorylation also induced β subunit phosphorylation. Untreated and muscle agrin-treated cultures did not exhibit β subunit phosphorylation. The nitrocellulose blot was stripped and reprobed with an antibody against the β subunit (mAb 124) to ensure that similar amounts of β subunit were present in each sample.
DISCUSSION
Laminin-1 Induces AChR Aggregation on C2 Myotubes
Vogel et al. (1983) first demonstrated that laminin-1 purified from EHS tumors could induce AChR aggregation in a rat clonal muscle cell line and in primary rat myotubes. We have confirmed these results with cultured C2 myotubes, showing that soluble laminin-1 induces AChR clusters in a dose-dependent manner. At its optimal concentration (120 nM), laminin-1 induced an approximately fivefold increase in cluster number over background. The increase in AChR cluster number induced by laminin-1 was similar to that induced by soluble neural agrin, and the size of the clusters produced by low concentrations of both ligands was similar. The laminin-1–induced AChR clusters appeared by eye to be more intensely stained, and quantitative analysis of the fluorescence intensity confirmed this impression. The increased intensity of AChR staining most likely represents an increase in receptor density, although increased folding of the membrane could be responsible.
Laminin-1 and Agrin Use Different Pathways to Cluster the AChR
Although laminin-1 and agrin both induce AChR clusters, the two ligands use separate signaling pathways. This conclusion rests on four observations: (a) the number of AChR clusters induced by laminin-1 and by agrin are additive, even at saturating concentrations of agrin (Fig. 2 b); (b) although agrin-induced AChR clustering requires MuSK (DeChiara et al., 1996; Glass et al., 1996), laminin-1–induced clustering does not (Figs. 6–8); (c) agrin- and laminin-1–induced clustering occur with different time courses (Fig. 5); and (d) agrin induces tyrosine phosphorylation of the AChR, whereas laminin-1 does not (Fig. 9). We examine each of these points in turn.
If laminin-1 and agrin shared a signaling pathway, then a saturating response to one would not be further increased by treatment with the other. This is clearly not the case, as 40 nM of laminin-1 increased the agrin response by a constant amount at all concentrations of agrin tested, indicating that the effects are strictly additive (Fig. 2 b). Although this result does not exclude the possibility that the two pathways may share nonlimiting, downstream steps, the limiting steps of each pathway, and those preceding them, must be separate.
Neural agrin activates MuSK, a muscle-specific receptor tyrosine kinase that is thought to be part of the agrin receptor complex (DeChiara et al., 1996; Glass et al., 1996). This activation appears to be the first step in the pathway of agrin-induced AChR clustering. Thus, myotubes from mice that are homozygous for a disruption of the MuSK gene do not respond to agrin by clustering AChRs (Glass et al., 1996). In contrast, laminin-1 did induce AChR clustering in MuSK−/− myotubes (Figs. 6 and 7), while treatment of C2 myotubes with laminin-1 did not induce tyrosine phosphorylation of MuSK (Fig. 8). Thus, laminin-1–induced clustering of the AChR must occur by a pathway that is independent of MuSK activation.
The time courses of laminin-1- and of agrin-induced AChR clustering are quite different. Whereas agrin-induced AChR clustering occured within a few hours (Fig. 5; Ferns et al., 1996), AChR clusters were not detected until at least 12 hours after laminin-1 treatment (Fig. 5). In addition to laminin-1 and agrin, other agents have been reported to induce AChR clustering, but those that have been tested all do so with a time course similar to that of agrin. Thus, treatment with either VVA-B4 lectin (Martin and Sanes, 1995) or polystyrene beads (Baker et al., 1992), caused clustering that was detectable within 4 h and reached a peak well before 8 h. The slower course of laminin-induced AChR clustering suggests that it occurs by a mechanism that is distinct from that used by other agents that cluster surface AChRs.
Agrin treatment of C2 myotubes not only causes AChR clustering, but also tyrosine phosphorylation of the AChR β subunit (Wallace et al., 1991; Qu and Huganir, 1994; Wallace, 1994; Ferns et al., 1996). Several lines of evidence indicate that phosphorylation of the AChR β subunit may be required for agrin-induced AChR aggregation. Agrin-induced phosphorylation occurs before clustering (Ferns et al., 1996), and under a variety of conditions, agrin-induced AChR clustering and tyrosine phosphorylation are associated. Similar concentrations of agrin induce both clustering and tyrosine phosphorylation of the AChR, and tyrosine kinase inhibitors inhibit both agrin-induced AChR clustering and β subunit phosphorylation (Wallace, 1994; Ferns et al., 1996). Furthermore, treatment of C2 myotubes with phosphatase inhibitors decreases the extractability of AChRs within the membrane (Meier et al., 1995). Thus, while circumstantial evidence links the two events, direct experimental proof is lacking. In the case of laminin-1–induced AChR clustering, however, no evidence was found for tyrosine phosphorylation of the AChR (Fig. 9). Although it may be required for agrin-induced cluster formation or for cluster stability within the synaptic membrane, AChR clustering in myotubes does not require tyrosine phosphorylation of the β subunit, as aggregation can occur in its absence.
The Molecular Mechanism of Laminin -1–Induced AChR Clustering
The molecular mechanism of AChR clustering induced by laminin, as in the case of agrin, is unknown. The clusters presumably represent the redistribution of AChRs on the myotube surface, rather than the insertion of newly synthesized AChR clusters, since surface AChRs pre-labeled with rhodamine–α-bungarotoxin are clustered by laminin (Vogel et al., 1983). Preliminary observations showing that the appearance of the laminin-1–induced clusters is not inhibited by cycloheximide (Sugiyama, J.E., and Z.W. Hall, unpublished observations) are consistent with this interpretation. Neither the receptor nor the signaling pathway activated by laminin-1, which results in AChR clustering, is known. Laminin-1–binding proteins in muscle that could potentially mediate its effect include integrins (Venstrom and Reichardt, 1993) and α-dystroglycan (Ervasti and Campbell, 1993; Gee et al., 1993). The integrins known to be present in myotubes include those containing α7, which is concentrated at synapses, α1, 3-7, 9, αV, and β1 (Martin et al., 1996). In addition, full length muscle agrin, which has been shown to induce AChR clusters (Ferns et al., 1992), also binds laminin (Sugiyama, J.E., and Z.W. Hall, unpublished results) and may thus play a role in laminin-induced AChR clustering. Further investigation will be required to determine which, if any, of these proteins mediate the AChR clustering effect of laminin-1 on myotubes.
Tyrosine phosphorylation appears to play a role in the signaling pathway for AChR clustering. Immunofluorescence experiments show that phosphotyrosine staining colocalizes with AChR clusters early in development (Qu and Huganir, 1994), and the dispersal of agrin-induced AChR clusters by tyrosine kinase inhibitors occurs at a time when AChR phosphorylation has declined, suggesting that phosphorylation of other proteins may be required for cluster maintenance (Ferns et al., 1996). Experiments with cells heterologously expressing the AChR also suggest that tyrosine phosphorylation of proteins other than the AChR could be required for clustering (Gillespie et al., 1996). Initial experiments to test the effects of tyrosine kinase inhibitors on laminin-1–induced AChR clustering have as yet yielded ambiguous results (Sugiyama, J.E., and Z.W. Hall, unpublished observations). Thus although AChR β subunit tyrosine phosphorylation is clearly not required for laminin-1–induced AChR clustering, the possibility remains that tyrosine phosphorylation of other proteins may play a role.
An interesting recent observation shows that low concentrations of laminin-1 (30 pM to 6 nM) cause the clustering of α-dystroglycan (Cohen et al., 1997). This finding is of potential interest for the role of laminin in synaptogenesis, as α-dystroglycan is localized at synapses in vivo and occurs in association with agrin-induced AChR clusters in cultured myotubes (Campanelli et al., 1994; Gee et al., 1994; Sugiyama et al., 1994; Cohen et al., 1995). As laminin-1 is known to bind α-dystroglycan (Ervasti and Campbell, 1993; Gee et al., 1993), self-association between laminin-1 molecules (Yurchenco and Cheng, 1994) is postulated to be the mechanism responsible for the clustering of α-dystroglycan. This mechanism is unlikely to cause laminin-induced AChR clustering, however, as laminin-1 is not known to bind the AChR directly. As the laminin-1–induced α-dystroglycan clusters were not associated with AChR clusters, their significance for synaptogenesis is unclear.
What Is the Physiological Role of Laminin-1–induced AChR Clustering?
Although in vitro laminin-1 induces AChR clusters by a pathway that is clearly distinct from that used by agrin, how does this pathway function in vivo? In mice that lack agrin, few AChR clusters are associated with sites of nerve–muscle contact, indicating that agrin-induced clustering is the major mechanism for concentrating AChRs beneath nerve terminals (Gautam et al., 1996). A few nerve-associated AChR clusters remain, however, leading Gautam et al. (1996) to conclude that additional pathways must mediate nerve-induced clustering. The laminin-1 pathway could provide one explanation for the AChR clusters that appear in agrin-deficient animals. One problem with this hypothesis is that AChR clusters are not seen in the muscles of MuSK-deficient animals, implying that the agent responsible for AChR clustering in agrin-deficient animals requires MuSK, which laminin-1 does not. An attractive possibility is that during synaptogenesis, the laminin-1 pathway is not normally used to initiate AChR clusters but builds on agrin-initiated AChR clusters in a way that uses MuSK as a structural element. Laminin-1–induced clustering would thus be facilitated by MuSK, but would not activate it or require it. This idea is consistent with recent experiments suggesting that MuSK is part of the primary synaptic scaffold to which AChRs are added (Apel et al., 1997). Under this interpretation, in MuSK−/− animals, the laminin-1 pathway would not be strongly enough activated during synaptogenesis to initiate AChR clustering by itself, but under in vitro conditions, AChR clustering could be elicited even in the absence of MuSK by high concentrations of laminin-1. Such an interpretation might explain the lower level of AChR clusters observed after laminin-1 addition to MuSK−/− myotubes, as compared to C2 myotubes.
The idea that laminin-1 acts to increase the size of agrin-induced AChR clusters is consistent with its slower time course, and with observations of the properties of AChR clusters formed by the combination of laminin-1 and agrin. At high concentrations of agrin (1.5 or 15 nM), the AChR clusters that formed were smaller and less intensely stained than those seen after treatment with low concentrations of agrin (15 or 150 pM). In contrast, all laminin-1–induced clusters, regardless of the concentration used, were large, more condensed, and intensely stained. The clusters that formed when myotubes were treated with laminin-1 and agrin together (using either low or high concentrations of agrin) appeared as highly condensed clusters along the edges of the myotubes, similar to those observed upon treatment with laminin-1 alone. Laminin-1 may thus act to further concentrate or organize the agrin-induced AChR clusters.
If the laminin-1 pathway is found to be used, what is the agent that activates it in vivo? Of the three isoforms that we have examined, only laminin-1 is active, but other isoforms may be active as well. It will be of particular interest to examine the activity of a recently identified synaptic form of laminin containing the α4 chain. Our results showing that laminin-1 (α1β1γ1) is active, whereas laminin-2 (α2β1γ1) and laminin-11 (α5β2γ1) are not, suggest that the clustering activity of each laminin isoform arises from the identity of the α chain. Only incomplete information is currently available about the distribution of laminin isoforms during muscle development (Sanes et al., 1990). Recent immunocytochemical experiments indicate that the α1 chain is present very early in muscle development, but that it may not be localized at developing synapses (Patton, B.L., and J.R. Sanes, personal communication). The α5 chain, in contrast, is concentrated at adult endplates, but at least within the context of β2 and γ1, is inactive in AChR clustering. Available data thus suggests that although laminin-1 activates the pathway that we have described, it may not be the functional ligand during synaptogenesis.
The fact that laminin and other agents, in addition to neural agrin, can cause AChR clustering suggests that auxiliary pathways of clustering may operate during maturation of the postsynaptic membrane. One attractive hypothesis is that AChR clustering is initiated by neural agrin, present in limiting amounts, and that clustering may be amplified during development of the synapse by other, less active but more abundant agents present in muscle, such as laminin or muscle agrin (Godfrey et al., 1988; Fallon and Gelfman, 1989). It will be of interest to elucidate the molecular mechanism of laminin-induced AChR clustering and to determine the in vivo factor which modulates this alternative pathway.
Acknowledgments
We thank Dr. Arlene Chiu for the generous gift of purified laminin-11 and for her critical analysis of the manuscript and Dr. Hynda Kleinman for sharing both the laminin-2 and her knowledge of the laminin field. We appreciate Dr. Josh Sanes' insightful comments and also thank Dr. Christian Fuhrer for preparing the α-bungarotoxin–Sepharose beads and for many helpful discussions. Finally, we are grateful to Drs. Matt Daniels, Brian Lowe, and Robin Taylor for their help with MetaMorph image analysis and to Dr. Vikram Ramanathan for his statistical expertise.
This work was supported by the National Institutes of Health.
Footnotes
Address all correspondence to Dr. Zach Hall, Office of the Director, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD 20892. Tel.: (301) 496-9746; Fax: (301) 496-0296.
1. Abbreviation used in this paper: AChR, acetylcholine receptor.
REFERENCES
- Apel ED, Glass DJ, Moscoso LM, Yancopoulos GD, Sanes JR. MuSK forms a synaptic scaffold in the absence of rapsyn, but requires rapsyn to signal and recruit additional synaptic components. Neuron. 1997;18:623–635. doi: 10.1016/s0896-6273(00)80303-7. [DOI] [PubMed] [Google Scholar]
- Baker LP, Chen Q, Peng HB. Induction of acetylcholine receptor clustering by native polystyrene beads. J Cell Sci. 1992;102:543–555. doi: 10.1242/jcs.102.3.543. [DOI] [PubMed] [Google Scholar]
- Bayne EK, Anderson MJ, Fambrough DM. Extracellular matrix organization in developing muscle: correlation with acetylcholine receptor aggregates. J Cell Biol. 1984;99:1486–1501. doi: 10.1083/jcb.99.4.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen DC, Sugiyama J, Ferns M, Hall ZW. Neural agrin activates a high-affinity receptor in C2 muscle cells that is unresponsive to muscle agrin. J Neurosci. 1996;16:3791–3797. doi: 10.1523/JNEUROSCI.16-12-03791.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burden SJ, Sargent PB, McMahan UJ. Acetylcholine receptors in regenerating muscle accumulate at original synaptic sites in the absence of the nerve. J Cell Biol. 1979;82:412–425. doi: 10.1083/jcb.82.2.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgeson RE, Chiquet M, Duetzmann R, Ekblom P, Engel J, Kleinman H, Martin GR, Meneguzzi G, Paulsson M, Sanes J, et al. A new nomenclature for the laminins. Matrix Biol. 1994;14:209–211. doi: 10.1016/0945-053x(94)90184-8. [DOI] [PubMed] [Google Scholar]
- Campanelli JT, Roberds SL, Campbell KP, Scheller RH. A role for dystrophin-associated glycoproteins and utrophin in agrin induced acetylcholine receptor clustering. Cell. 1994;77:663–674. doi: 10.1016/0092-8674(94)90051-5. [DOI] [PubMed] [Google Scholar]
- Chiu AY, Sanes JR. Development of basal lamina in synaptic and extrasynaptic portions of embryonic rat muscle. Dev Biol. 1984;103:456–467. doi: 10.1016/0012-1606(84)90333-6. [DOI] [PubMed] [Google Scholar]
- Cohen MW, Jacobson C, Godfrey EW, Campbell KP, Carbonetto S. Distribution of α dystroglycan during embryonic nerve-muscle synaptogenesis. J Cell Biol. 1995;129:1093–1101. doi: 10.1083/jcb.129.4.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MW, Jacobson C, Yurchenco PD, Morris GE, Carbonetto S. Laminin-induced clustering of dystroglycan on embryonic muscle cells: comparison with agrin-induced clustering. J Cell Biol. 1997;136:1047–1058. doi: 10.1083/jcb.136.5.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels MP, Vigny M, Sonderegger P, Bauer HC, Vogel Z. Association of laminin and other basement membrane components with regions of high acetylcholine receptor density on cultured myotubes. Int J Dev Neurosci. 1984;2:87–99. doi: 10.1016/0736-5748(84)90063-7. [DOI] [PubMed] [Google Scholar]
- DeChiara TM, Bowen DC, Valenzuela DM, Simmons MV, Poueymirou WT, Thomas S, Kinetz E, Compton DL, Rojas E, Park JS, et al. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell. 1996;85:501–512. doi: 10.1016/s0092-8674(00)81251-9. [DOI] [PubMed] [Google Scholar]
- Ervasti JM, Campbell KP. A role for the dystrophin–glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol. 1993;122:809–823. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallon JR, Gelfman CE. Agrin-related molecules are concentrated at acetylcholine receptor clusters in normal and aneural developing muscle. J Cell Biol. 1989;108:1527–1535. doi: 10.1083/jcb.108.4.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferns M, Hoch W, Campanelli JT, Rupp F, Hall ZW, Scheller RH. RNA splicing regulates agrin-mediated acetylcholine receptor clustering activity on cultured myotubes. Neuron. 1992;8:1079–1086. doi: 10.1016/0896-6273(92)90129-2. [DOI] [PubMed] [Google Scholar]
- Ferns MJ, Campanelli JT, Hoch W, Scheller RH, Hall Z. The ability of agrin to cluster AChRs depends on alternative splicing and on cell surface proteoglycans. Neuron. 1993;11:491–502. doi: 10.1016/0896-6273(93)90153-i. [DOI] [PubMed] [Google Scholar]
- Ferns M, Deiner M, Hall Z. Agrin-induced acetylcholine receptor clustering in mammalian muscle requires tyrosine phosphorylation. J Cell Biol. 1996;132:937–944. doi: 10.1083/jcb.132.5.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fertuck HC, Salpeter MM. Quantitation of junctional and extrajunctional acetylcholine receptors by electron microscope autoradiography after 125I-α bungarotoxin binding at mouse neuromuscular junctions. J Cell Biol. 1976;69:144–158. doi: 10.1083/jcb.69.1.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam M, Noakes PG, Moscoso L, Rupp F, Scheller RH, Merlie JP, Sanes JR. Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell. 1996;85:525–535. doi: 10.1016/s0092-8674(00)81253-2. [DOI] [PubMed] [Google Scholar]
- Gee SH, Blacher RW, Douville PJ, Yurchenco PD, Carbonetto S. LBP120 from brain is closely related to the dystrophin-associated glycoprotein, dystroglycan, and binds with high affinity to the major heparin binding domain of laminin. J Biol Chem. 1993;268:14972–14980. [PubMed] [Google Scholar]
- Gee SH, Montanaro F, Lindenbaum MH, Carbonetto S. Dystroglycan-α, a dystrophin-associated glycoprotein, is a functional agrin receptor. Cell. 1994;77:675–686. doi: 10.1016/0092-8674(94)90052-3. [DOI] [PubMed] [Google Scholar]
- Gillespie SKH, Balasubramanian S, Fung ET, Huganir RL. Rapsyn clusters and activates the synapse-specific receptor tyrosine kinase MuSK. Neuron. 1996;16:953–962. doi: 10.1016/s0896-6273(00)80118-x. [DOI] [PubMed] [Google Scholar]
- Glass DJ, Bowen DC, Stitt TN, Radziejewski C, Bruno J, Ryan TE, Gies DR, Shah S, Mattsson K, Burden SJ, et al. Agrin acts via a MuSK receptor complex. Cell. 1996;85:513–523. doi: 10.1016/s0092-8674(00)81252-0. [DOI] [PubMed] [Google Scholar]
- Godfrey EW, Siebenlist RE, Wallskog PA, Walters LM, Bolender DL, Yorde DE. Basal lamina components are concentrated in premuscle masses and at early acetylcholine receptor clusters in chick embryo hindlimb muscles. Dev Biol. 1988;130:471–486. doi: 10.1016/0012-1606(88)90343-0. [DOI] [PubMed] [Google Scholar]
- Gordon H, Hall ZW. Glycosaminoglycan variants in the C2 muscle cell line. Dev Biol. 1989;135:1–11. doi: 10.1016/0012-1606(89)90152-8. [DOI] [PubMed] [Google Scholar]
- Gordon H, Lupa M, Bowen D, Hall Z. A muscle cell variant defective in glycosaminoglycan biosynthesis forms nerve-induced but not spontaneous clusters of the acetylcholine receptor and the 43 kDa protein. J Neurosci. 1993;13:586–595. doi: 10.1523/JNEUROSCI.13-02-00586.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Hall ZW. Characterization of acetylcholine receptor subunits in developing and in denervated mammalian muscle. J Biol Chem. 1988;263:12878–12885. [PubMed] [Google Scholar]
- Hall, Z.W., and J.R. Sanes. 1993. Synaptic structure and development: the neuromuscular junction. Neuron. 10(Suppl.):99–121. [DOI] [PubMed]
- Hoch W, Ferns M, Campanelli JT, Hall ZW, Scheller RH. Developmental regulation of highly active alternatively spliced forms of agrin. Neuron. 1993;11:479–490. doi: 10.1016/0896-6273(93)90152-h. [DOI] [PubMed] [Google Scholar]
- Hunter DD, Shah V, Merlie JP, Sanes JR. A laminin-like adhesive protein concentrated in the synaptic cleft of the neuromuscular junction. Nature (Lond) 1989;338:229–234. doi: 10.1038/338229a0. [DOI] [PubMed] [Google Scholar]
- Jat PS, Noble MD, Ataliotis P, Tanaka Y, Yannoutsos N, Larsen L, Kioussis D. Direct derivation of conditionally immortal cell lines from an H-2Kb-tsA58 transgenic mouse. Proc Natl Acad Sci USA. 1991;88:5096–5100. doi: 10.1073/pnas.88.12.5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leivo I, Engvall E. Merosin, a protein specific for basement membranes of Schwann cells, striated muscle, and trophoblast, is expressed late in nerve and muscle development. Proc Natl Acad Sci USA. 1988;85:1544–1548. doi: 10.1073/pnas.85.5.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lentz SI, Miner JH, Sanes JR, Snider WD. Distribution of the ten known laminin chains in the pathways and targets of developing sensory axons. J Comp Neurol. 1997;378:547–561. doi: 10.1002/(sici)1096-9861(19970224)378:4<547::aid-cne9>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Martin PT, Sanes JR. Role for a synapse-specific carbohydrate in agrin-induced clustering of acetylcholine receptors. Neuron. 1995;14:743–754. doi: 10.1016/0896-6273(95)90218-x. [DOI] [PubMed] [Google Scholar]
- Martin PT, Ettinger AJ, Sanes JR. Synaptic localization domain in the synaptic cleft protein laminin β2 (s-laminin) Science (Wash DC) 1995;269:413–416. doi: 10.1126/science.7618109. [DOI] [PubMed] [Google Scholar]
- Martin PT, Kaufman SJ, Kramer RH, Sanes JR. Synaptic integrins in developing, adult, and mutant muscle: selective association of α1, α7A, and α7B integrins with the neuromuscular junction. Dev Biol. 1996;174:125–139. doi: 10.1006/dbio.1996.0057. [DOI] [PubMed] [Google Scholar]
- McMahan, U.J. 1990. The agrin hypothesis. In Cold Spring Harbor Symposia on Quantitative Biology. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York. 407–418. [DOI] [PubMed]
- Mundegar RR, van Oertzen J, Zierz S. Increased laminin A expression in regenerating myofibers in neuromuscular disorders. Muscle Nerve. 1995;18:992–999. doi: 10.1002/mus.880180911. [DOI] [PubMed] [Google Scholar]
- Meier T, Perez GM, Wallace BG. Immobilization of nicotinic acetylcholine receptors in mouse C2 myotubes by agrin-induced protein tyrosine phosphorylation. J Cell Biol. 1995;131:441–451. doi: 10.1083/jcb.131.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miner JH, Patton BL, Lentz SI, Gilbert DJ, Snider WD, Jankins NA, Copeland NG, Sanes JR. The laminin α chains: expression, developmental transitions, and chromosomal locations of α1–5, identification of heterotrimeric laminins 8–11, and cloning of a novel α3 isoform. J Cell Biol. 1997;137:685–701. doi: 10.1083/jcb.137.3.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nastuk MA, Lieth E, Ma J, Cardasis CA, Moynihan EB, McKechnie BA, Fallon JR. The putative agrin receptor binds ligand in a calcium-dependent manner and aggregates during agrin-induced acetylcholine receptor clustering. Neuron. 1991;7:807–818. doi: 10.1016/0896-6273(91)90283-6. [DOI] [PubMed] [Google Scholar]
- Nitkin RM, Rothschild TC. Agrin-induced reorganization of extracellular matrix components on cultured myotubes: relationship to AChR aggregation. J Cell Biol. 1990;111:1161–1170. doi: 10.1083/jcb.111.3.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng HB, Baker LP, Chen Q. Induction of synaptic development in cultured muscle cells by basic fibroblast growth factor. Neuron. 1991;6:237–246. doi: 10.1016/0896-6273(91)90359-8. [DOI] [PubMed] [Google Scholar]
- Peng HB, Afshan A, Ali, Dai Z, Daggett DF, Raulo E, Rauvala H. The role of heparin-binding growth-associated molecule (HB-GAM) in the postsynaptic induction in cultured muscle cells. J Neurosci. 1995;15:3027–3038. doi: 10.1523/JNEUROSCI.15-04-03027.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Z, Huganir RL. Comparison of innervation and agrin-induced tyrosine phosphorylation of the nicotinic acetylcholine receptor. J Neurosci. 1994;14:6834–6841. doi: 10.1523/JNEUROSCI.14-11-06834.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruegg MA, Tsim KWK, Horton SE, Kroger S, Escher G, Gensch EM, McMahan UJ. The agrin gene codes for a family of basal lamina proteins that differ in function and distribution. Neuron. 1992;8:691–699. doi: 10.1016/0896-6273(92)90090-z. [DOI] [PubMed] [Google Scholar]
- Rupp F, Payan DG, Magill-Solc C, Cowan DM, Scheller RH. Structure and expression of a rat agrin. Neuron. 1991;6:811–823. doi: 10.1016/0896-6273(91)90177-2. [DOI] [PubMed] [Google Scholar]
- Sanes, J.R., E. Engvall, R. Butkowski, and D.D. Hunter. 1990. Molecular heterogeneity of basal laminae: isoforms of laminin and collagen IV at the neuromuscular junction and elsewhere. 111:1685–1699. [DOI] [PMC free article] [PubMed]
- Sewry CA, Chevallay M, Tomé FMS. Expression of laminin subunits in human fetal skeletal muscle. Histochem J. 1995;27:497–504. [PubMed] [Google Scholar]
- Sugiyama J, Bowen DC, Hall ZW. Dystroglycan binds nerve and muscle agrin. Neuron. 1994;13:103–115. doi: 10.1016/0896-6273(94)90462-6. [DOI] [PubMed] [Google Scholar]
- Timpl R, Rhode H, Risteli L, Ott U, Gehron P, Robey, Martin GR. Laminin. Methods Enzymol. 1982;82:831–839. doi: 10.1016/0076-6879(82)82104-6. [DOI] [PubMed] [Google Scholar]
- Tsim KWK, Ruegg MA, Escher G, Kroger S, McMahan UJ. cDNA that encodes active agrin. Neuron. 1992;8:677–689. doi: 10.1016/0896-6273(92)90089-v. [DOI] [PubMed] [Google Scholar]
- Valenzuela DM, Stitt TN, DiStefano PS, Rojas E, Mattsson K, Compton DL, Nunez L, Park S, Stark JL, Gies D, et al. Receptor tyrosine kinase specific for the skeletal muscle lineage: expression in embryonic muscle, at the neuromuscular junction, and after injury. Neuron. 1995;15:573–584. doi: 10.1016/0896-6273(95)90146-9. [DOI] [PubMed] [Google Scholar]
- Venstrom KA, Reichardt LF. Extracellular matrix 2: role of extracellular matrix molecules and their receptors in the nervous system. FASEB J. 1993;7:996–1003. doi: 10.1096/fasebj.7.11.8370483. [DOI] [PubMed] [Google Scholar]
- Vogel Z, Christian CN, Vigny M, Bauer HC, Sonderegger P, Daniels MP. Laminin induces acetylcholine receptor aggregation on cultured myotubes and enhances the receptor aggregation activity of a neuronal factor. J Neurosci. 1983;3:1058–1068. doi: 10.1523/JNEUROSCI.03-05-01058.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace BG. Agrin-induced specializations contain cytoplasmic, membrane, and extracellular matrix-associated components of the postsynaptic apparatus. J Neurosci. 1989;9:1294–1302. doi: 10.1523/JNEUROSCI.09-04-01294.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace BG. Staurosporine inhibits agrin-induced acetylcholine receptor phosphorylation and aggregation. J Cell Biol. 1994;125:661–668. doi: 10.1083/jcb.125.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace BG, Qu Z, Huganir RL. Agrin induces phosphorylation of the nicotinic acetylcholine receptor. Neuron. 1991;6:869–878. doi: 10.1016/0896-6273(91)90227-q. [DOI] [PubMed] [Google Scholar]
- Yurchenco PD, Cheng Y-S. Laminin self-assembly: a three-arm interaction hypothesis for the formation of a network in basement membranes. Contrib Nephrol. 1994;107:47–56. doi: 10.1159/000422960. [DOI] [PubMed] [Google Scholar]