

Abstract
In Xenopus embryos, β-catenin has been shown to be both necessary and sufficient for the establishment of dorsal cell fates. This signaling activity is thought to depend on the binding of β-catenin to members of the Lef/Tcf family of transcription factors and the regulation of gene expression by this complex. To test whether β-catenin must accumulate in nuclei to establish dorsal cell fate, we constructed various localization mutants that restrict β-catenin to either the plasma membrane, the cytosol, or the nucleus. When overexpressed in Xenopus embryos, the proteins localize as predicted, but surprisingly all forms induce an ectopic axis, indicative of inducing dorsal cell fates. Given this unexpected result, we focused on the membrane-tethered form of β-catenin to resolve the apparent discrepancy between its membrane localization and the hypothesized role of nuclear β-catenin in establishing dorsal cell fate. We demonstrate that overexpression of membrane-tethered β-catenin elevates the level of free endogenous β-catenin, which subsequently accumulates in nuclei. Consistent with the hypothesis that it is this pool of non–membrane-associated β-catenin that signals in the presence of membrane-tethered β-catenin, overexpression of cadherin, which binds free β-catenin, blocks the axis-inducing activity of membrane- tethered β-catenin. The mechanism by which ectopic membrane-tethered β-catenin increases the level of endogenous β-catenin likely involves competition for the adenomatous polyposis coli (APC) protein, which in other systems has been shown to play a role in degradation of β-catenin. Consistent with this hypothesis, membrane-tethered β-catenin coimmunoprecipitates with APC and relocalizes APC to the membrane in cells. Similar results are observed with ectopic plakoglobin, casting doubt on a normal role for plakoglobin in axis specification and indicating that ectopic proteins that interact with APC can artifactually elevate the level of endogenous β-catenin, likely by interfering with its degradation. These results highlight the difficulty in interpreting the activity of an ectopic protein when it is assayed in a background containing the endogenous protein. We next investigated whether the ability of β-catenin to interact with potential protein partners in the cell may normally be regulated by phosphorylation. Compared with nonphosphorylated β-catenin, β-catenin phosphorylated by glycogen synthase kinase-3 preferentially associates with microsomal fractions expressing the cytoplasmic region of N-cadherin. These results suggest that protein–protein interactions of β-catenin can be influenced by its state of phosphorylation, in addition to prior evidence that this phosphorylation modulates the stability of β-catenin.
β-catenin is a multifunctional protein involved in both the regulation of intercellular adhesion and cell signaling during development (for reviews see Miller and Moon, 1996; Peifer, 1995). These functions are dependent on interactions between β-catenin and various protein partners, including members of the cadherin superfamily of cell adhesion molecules (for review see Kemler, 1993; Peifer, 1995), the adenomatous polyposis coli (APC)1 protein (Rubinfeld et al., 1993; Su et al., 1993), and members of the Lef/Tcf family of transcription factors (Behrens et al., 1996; Molenaar et al., 1996). Each of these interactions occurs in a distinct subcellular compartment and reflects different activities of β-catenin in each compartment of the cell.
The binding of β-catenin to Lef/Tcf transcription factors and their translocation into the nucleus may play an essential role in controlling the transcription of genes required for specification of cell fate during early development of Xenopus (Behrens et al., 1996; Molenaar et al., 1996; Larabell et al., 1997) and Drosophila (Brunner et al., 1997; Riese et al., 1997; van de Wetering et al., 1997). In Xenopus embryos, recent studies demonstrate that there are dorso–ventral asymmetries in both the levels and subcellular localization of β-catenin in cleavage stage blastomeres (Larabell et al., 1997). Since an amino-terminal site of β-catenin is required for its maximal in vitro phosphorylation by glycogen synthase kinase 3 (GSK-3) and mutation of this site stabilizes β-catenin, it has been hypothesized that the dorso–ventral differences in β-catenin levels arise from lower GSK-3 activity on the prospective dorsal side of the embryo, resulting in an increase in the pool of dorsal β-catenin (Yost et al., 1996; Larabell et al., 1997). The mechanism underlying this process may also involve the stabilization of interactions between β-catenin and APC that has been shown to occur in response to Wnt signals in cultured cells (Papkoff et al., 1996). Since the interaction of APC with β-catenin is strongly linked to the regulation of steady-state β-catenin levels (Munemitsu et al., 1995; Papkoff et al., 1996; Hayashi et al., 1997), stabilization of β-catenin/APC complexes might saturate a rate-limiting step in a degradative pathway and result in the accumulation of newly synthesized β-catenin in the cytosol on the dorsal side of the embryo (Larabell et al., 1997). The elevated dorsal pool of β-catenin would then undergo translocation into the nucleus, perhaps via interactions with Lef/Tcf transcription factors (Behrens et al., 1996; Molenaar et al., 1996). The presence of β-catenin in nuclei of dorsal but not ventral blastomeres presages and likely contributes directly to the activation of dorsal-specific genes at the onset of zygotic transcription (Larabell et al., 1997), and the products of these genes then work in a combinatorial manner with other signal transduction pathways to specify dorsal cell fate (for review see Kimelman et al., 1992).
Given this proposed model of the role of β-catenin in the establishment of dorsal cell fates, we sought to test a prediction of this model: Specification of dorsal cell fate requires the accumulation of β-catenin in nuclei rather than in the cytoplasm or at membranes. Without such information one cannot exclude the possibility that β-catenin induces dorsal cell fate by acting in other cellular compartments in which it is found, such as in the cytoplasm or at the plasma membrane (for review see Miller and Moon, 1996). Testing this relationship has recently gained importance for a second reason. Ectopic expression of plakoglobin in Xenopus embryos leads to its accumulation in nuclei and the induction of dorsal cell fate as observed with β-catenin. Surprisingly, a form of plakoglobin that cannot enter nuclei is also active in promoting dorsal cell fate, and the authors conclude that it functions by anchoring XTcf-3 or a related factor from acting as a repressor of dorsal cell fate (Merriam et al., 1997). By analyzing the signaling activities of localization mutants of β-catenin, their affects on steady-state levels of endogenous β-catenin, and their effects on the subcellular localization of endogenous β-catenin, APC, and XTcf-3, we have reached an alternative conclusion. Our results are most consistent with the hypothesis that β-catenin functions in the specification of dorsal cell fate in Xenopus in a manner dependent upon its entry into the nucleus.
MATERIALS AND METHODS
Expression Constructs
All green fluorescent protein (GFP) and myc-tagged β-catenin constructs were produced by PCR amplification of full-length wild-type β-catenin and subcloning of the resulting PCR product into the CS2+ vector upstream from, and in frame with, either a c-myc epitope (Evan et al., 1985) or GFP (S65T mutant; Heim et al., 1995). Both the nuclear exclusion sequence (NES) and nuclear localization sequence (NLS) localization mutants were produced by subcloning sequences encoding either the nuclear exclusion sequence of rabbit pkI (Wen et al., 1994, 1995) or the nuclear localization sequence of the SV-40 large T-antigen (Kalderon et al., 1984; Lanford and Butel, 1984) in frame with the NH2 terminus of β-catenin. Both the NES and NLS oligos contained a consensus Kozak sequence (Kozak, 1984) followed by an AUG translation initiation site. The transmembrane β-catenin mutant was produced by subcloning a fragment of Xenopus N-cadherin (Detrick et al., 1990) that encodes the translation start site, signal sequence, and transmembrane (TM) domain in frame with the NH2 terminus of wt–β-catenin–GFP. TM–β-catenin 1–9 was produced by PCR amplifying repeats 1–9 of β-catenin and subcloning this fragment in frame to the N-cadherin transmembrane. TM–β-catenin 1-myc was produced by subcloning the N-cadherin TM domain in frame into an XhoI site of wt–β-catenin–myc.
APC–GFP was produced by PCR amplifying the central portion of a human APC cDNA (amino acids 1013–2026; full-length human APC cDNA was kindly provided by Ray White, University of Utah, Salt Lake City, UT) and subcloning this fragment into a CS2+/GFP vector resulting in a fusion protein with GFP at the carboxy terminus.
The myc-tagged human plakoglobin construct was kindly provided by Michael Klymkowsky (University of Colorado, Boulder, CO) (Merriam et al., 1997) and the HA-tagged XTcf-3 construct was generously provided by Olivier Destree (Hubrecht Laboratory, Utrecht, The Netherlands) (Molenaar et al., 1996).
Microinjection of Synthetic RNAs and Embryo Culture
Capped synthetic RNAs encoding each of the constructs used in this study were prepared with the Message Machine kit (Ambion, Inc., Austin, TX). For axis duplication experiments, 250 pg of each RNA was injected into the two ventral blastomeres at the four-cell stage, and embryos were reared to stage 40, at which time they were scored for the presence or absence of a secondary dorsal axis. For Western blot and immunoprecipitation experiments, 1–2 ng of the indicated RNAs were injected into all four blastomeres at the four-cell stage, and embryos were cultured at room temperature to stage 7. For intracellular localization experiments, 1–2 ng of the indicated RNAs were injected into the animal pole of blastomeres at the two- to four-cell stage, and embryos were cultured to either stage 9 or 10. Where indicated, 10,000–mol wt Oregon Green dextran (1 mg/ml final concentration; Molecular Probes, Eugene, OR) was coinjected as a lineage tracer.
Animal Cap Explants and Confocal Microscopy
Animal cap explants were fixed in 4% paraformaldehyde–PBS for 1–2 h at room temperature followed by two washes in PBS + 0.2% Triton X-100 (PBT). After fixation, explants to be stained for endogenous β-catenin were washed in MeOH, bleached in 50% MeOH, 30% H2O2, 20% DMSO overnight at room temperature, and washed twice in MeOH. GFP-labeled explants were not bleached because the bleaching process eliminates GFP fluorescence. Antibody staining was performed in PBT supplemented with 10% goat serum. Antihuman c-myc 9E10 monoclonal supernatant was used at a 1:20 dilution, and anti–β-catenin polyclonal antibodies (Yost et al., 1996) were used at a 1:500 dilution. After incubation with the primary antibody, explants were washed three times in PBT and incubated overnight at 4°C with a 1:250 dilution of Cy-3–conjugated secondary antibody (Jackson ImmunoResearch, West Grove, PA) in PBT supplemented with 10% goat serum. After three washes in PBT, explants were mounted in Vectashield (Vector Laboratories, Burlingame, CA).
Confocal microscopy was performed with a scan head (model MRC-600; Bio-Rad Labs, Hercules, CA) attached to a microscope (model Optiphot-2; Nikon, Inc., Melville, NY). All images were collected with a 60× 1.4 NA PlanApo objective. Multicolor images were collected sequentially with the appropriate filter blocks to ensure that there was no bleed through between channels. Images were processed using Adobe Photoshop software (San Jose, CA).
Western Blot and Immunoprecipitation
To compare differences in the steady-state levels of endogenous β-catenin protein in response to injection of control RNA (GFP) or membrane-tethered β-catenin RNA, 25 injected embryos at stage 7 were homogenized in RIPA buffer (10 mM Hepes, pH 7.4, 150 mM NaCl, 2 mM EGTA, 2 mM EDTA, 1% Triton X-100, 0.5% Na deoxycholate, 0.1% SDS, 1 mM PMSF, 1 μg/ml leupeptin) at 4°C. Lysates were cleared by spinning for 15 min at 15,000 rpm in a microcentrifuge. An aliquot of this lysate was collected and represents total (T) protein. The remainder of the cleared lysate was incubated with ConA-Sepharose beads (Pharmacia LKB Biotech., Piscataway, NJ) for 1 hour at 4°C to remove all cadherin-bound β-catenin. The resulting supernatant contains the soluble (S), cytoplasmic or nuclear, pool of β-catenin. Protein samples were analyzed by SDS-PAGE, and blots were probed with anti–β-catenin (1:1,000) and anti–α spectrin (1:1,000; Giebelhaus et al., 1987) antibodies. Immunoblots were then probed with a HRP-conjugated goat anti–rabbit secondary antibody (1:10,000; Jackson ImmunoResearch). The HRP signal was visualized by enhanced chemiluminescence (Amersham Corp., Arlington Heights, IL). Signals for β-catenin and α-spectrin were quantitated from scanned images using NIH Image. Endogenous β-catenin levels (total and soluble) from control-injected and TM–β-catenin–injected embryos were compared after normalization to α-spectrin. Levels of β-catenin in controls was set to 1.0 and levels in TM–β-catenin–injected embryos are expressed relative to this scale.
To determine if the ectopic TM–β-catenin interacts with endogenous APC and a cadherin fraction, we performed immunoprecipitation and ConA precipitation analyses. Stage 7 embryos previously injected with 1.25 ng TM–β-catenin 1-myc were homogenized in lysis buffer (10 mM Tris, pH 7.4, 150 mM NaCl, 2 mM EDTA, 0.5% NP-40, 1 mM PMSF, 1 μg/ml leupeptin) at 4°C. A cleared lysate was prepared by spinning the crude homogenates at 15,000 rpm for 15 min in a microcentrifuge. For APC immunoprecipitation, the cleared lysate was precleared for 1 h at 4°C with protein A–Sepharose beads to eliminate proteins that bind nonspecifically to the beads. The lysates were then incubated with anti-APC polyclonal antibodies (Näthke et al., 1996) bound to protein A–Sepharose beads or control protein A–Sepharose beads for 1 h at 4°C. After five washes in lysis buffer, samples were resuspended in SDS sample buffer. For ConA precipitation, cleared lysates were incubated with ConA-Sepharose beads or control Sepharose beads for 1 h at 4°C. After five washes, the bound proteins were solubilized with SDS sample buffer. Protein samples were analyzed by SDS-PAGE, and blots were probed with anti-myc antibodies (1:50) and HRP-conjugated goat anti–mouse secondary antibodies (Jackson ImmunoResearch) to detect the presence of TM–β-catenin 1-myc in the samples. To determine levels of endogenous β-catenin present in APC and ConA precipitates, we probed blots with anti–β-catenin antibodies as described above.
Rabbit Reticulocyte Lysate Assays
RNAs encoding β-catenin and Xenopus N-cadherin (Detrick et al., 1990) were transcribed in vitro using a Message Machine kit (Ambion, Inc.). A rabbit reticulocyte lysate was used with RNAs translated individually for 60 min at 30°C. The β-catenin reactions were supplemented with [35S]methionine and then centrifuged (4°C) after translation for 15 min. Xenopus N-cadherin RNA was translated using nonradioactive methionine in the presence of canine pancreatic microsomes (Promega Corp., Madison, WI). To test β-catenin binding to N-cadherin, 25-μl aliquots of translations of microsomes with or without N-cadherin were mixed with 15-μl reactions of β-catenin translations, followed by addition of ATP to 0.4 mM. Samples were incubated for 3.5 h at (4°C) on a rotator to allow binding to membranes. Samples were centrifuged for 15 min at 20,000 g (4°C) and then pelleted microsomes were carefully rinsed with reticulocyte lysate lacking labeled protein. Pellets and aliquots of the supernatant were diluted with SDS gel sample buffer.
To determine whether phosphorylated β-catenin would interact with microsomes in the presence of N-cadherin, RNAs were translated as described above except that nonradioactive methionine replaced the [35S]methionine. 60 μl of microsome translations conducted in the presence or absence of N-cadherin RNA were mixed with 50 μl of translations of precentrifuged β-catenin. Assay tubes were then supplemented with 50 μCi of [γ32P]ATP, and ∼0.1 U of recombinant Xenopus glycogen synthase kinase-3 (Xgsk-3) where indicated. After 20 min at 30°C to facilitate phosphorylation, samples were rotated for 3 h at 4°C to allow binding to microsomes and then centrifuged for 20 min at 20,000 g. The supernatants and pellets were subjected to immunoprecipitation with a rabbit polyclonal antibody to β-catenin, followed by separation on SDS 10% polyacrylamide gels and autoradiography.
RESULTS
Intracellular Distribution and Signaling Activity of β-Catenin Localization Mutants
β-catenin is a multifunctional protein that is localized to several intracellular compartments, including the plasma membrane, cytosol, and nucleus (for review see Miller and Moon, 1996). The localization of β-catenin to each of these compartments is thought to reflect the interaction of β-catenin with various protein partners and the different functions of β-catenin in cell adhesion and signal transduction. Recent studies have suggested that the signaling function of β-catenin may be linked to accumulation of β-catenin–Lef/Tcf complexes in nuclei (Behrens et al., 1996; Molenaar et al., 1996). Although these studies suggest that the signaling function of β-catenin is carried out in the nucleus, we sought to directly test where in the cell β-catenin is required to have signaling activity. We therefore constructed a series of β-catenin localization mutants that contain additional sequences that target the resulting fusion proteins to a specific subcellular compartment (Fig. 1 A). Each fusion protein was tagged with GFP (S65T mutant; Heim et al., 1995) so that its intracellular localization could be easily determined by confocal microscopy. The wild-type (WT)–β-catenin–GFP construct was prepared as a control and was expected to mimic both the localization and function of endogenous β-catenin in various subcellular compartments. NES–β-catenin–GFP is a mutant that contains the nuclear exclusion sequence of rabbit pkI at the NH2 terminus (Wen et al., 1994, 1995). This mutant was expected to mimic the functions of cytosolic and membrane-bound β-catenin and would help test the requirement of nuclear localization for the signaling function of β-catenin. NLS–β-catenin–GFP is a mutant that contains the nuclear localization signal of the SV-40 large T-antigen (Kalderon et al., 1984; Lanford and Butel, 1984) fused to the NH2 terminus of β-catenin. This protein was expected to localize exclusively to nuclei and was produced to test the requirement for either cytosolic or membrane-localized β-catenin in signal transduction. Finally, TM–β-catenin is a mutant that contains the signal sequence and transmembrane domain of Xenopus N-cadherin fused to the NH2 terminus of β-catenin. This construct was produced to restrict β-catenin to membranes and, similar to the NES–β-catenin construct, would test whether nuclear localization is required for the signaling function of β-catenin.
Figure 1.

Schematic representation of constructs used in this study. (A) Diagrams depicting the structure of wild-type and localization mutant β-catenin proteins. Some constructs were tagged at the COOH terminus with either GFP (S65T mutant; Heim et al., 1995) or a c-myc epitope (Evan et al., 1985). Shadowed boxes represent the 13 Arm repeats with a nonrepeat sequence between repeats 10 and 11. Sequences directing β-catenin to specific intracellular compartments were added to the NH2 terminus of both wild-type and truncated forms of β-catenin. Wild-type human plakoglobin possesses an overall structure identical to that of β-catenin and is tagged at the NH2 terminus with a c-myc epitope (Merriam et al., 1997). (B) Linear representation of wild-type human APC protein showing conserved motifs, including the oligomerization domain, Arm repeats, 15– and 20–amino acid repeats, microtubule binding domain (MT binding), and discs large binding domain (Dlg binding). The central portion of APC, which is sufficient for β-catenin binding and downregulation (Munemitsu et al., 1995), was tagged at the COOH terminus with GFP (S65T mutant; Heim et al., 1995).
To determine the intracellular localization of each mutant construct, Xenopus animal cap cells expressing each GFP-tagged form of β-catenin were examined by confocal microscopy. WT–β-catenin–GFP accumulates at the plasma membrane, in the cytosol, and in the nucleus (Fig. 2 A). This pattern is identical to that seen for endogenous β-catenin in animal cap cells (Yost et al., 1996), demonstrating that the GFP tag does not influence the subcellular localization of the β-catenin fusion protein. NES–β-catenin–GFP is found predominantly in the cytoplasm and at the plasma membrane, although low levels of GFP fluorescence are detected in the nucleus (Fig. 2 B). The NLS–β-catenin–GFP fusion protein is localized almost exclusively to nuclei, although faint fluorescence is detected in association with the plasma membrane (Fig. 2 C). TM–β-catenin– GFP is localized to intracellular vesicles and organelles predictive of its association with the endoplasmic reticulum and Golgi apparatus (Fig. 2 D). TM–β-catenin–GFP was never detected in the nucleus.
Figure 2.
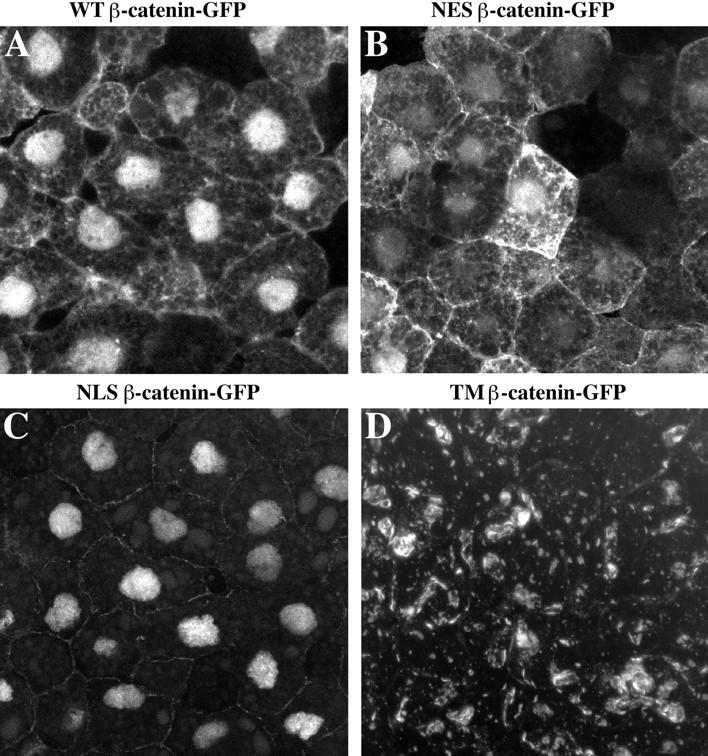
Localization of wild-type and mutant β-catenin–GFP proteins in animal cap cells. The intracellular distribution of each mutant was determined by confocal microscopy (A–D). The WT– β-catenin–GFP protein (A) is localized to the plasma membrane, cytosol, and nucleus in a pattern indistinguishable from that seen for endogenous β-catenin protein (Yost et al., 1996). NES–β-catenin–GFP (B) is present at plasma membrane and at high levels in the cytoplasm. Low levels of fluorescence are detected in the nucleus, which likely reflects the fact that the NES domain does not inhibit nuclear entry but instead promotes the rapid export of tagged proteins from the nucleus. NLS–β-catenin–GFP (C) is predominantly found in the nucleus, and very little fluorescence is observed in the cytoplasm or in association with the plasma membrane. The TM–β-catenin–GFP mutant (D) localizes to intracellular vesicles and organelles in apparent association with the endoplasmic reticulum and Golgi apparatus. TM–β-catenin–GFP fluorescence was never detected in the nucleus.
The intracellular distribution observed for each localization mutant of β-catenin matched our predictions and, therefore, allowed us to test whether the signaling activity of β-catenin is dependent on its subcellular localization. The signaling activity of β-catenin can be assayed by determining its ability to induce a secondary axis after overexpression in ventral blastomeres (Funayama et al., 1995), as also observed with other components of the Wnt-1 signaling pathway (for review see Moon et al., 1997). Therefore, synthetic RNAs encoding each localization mutant were injected into the marginal zone of each ventral blastomere at the four-cell stage, and embryos were scored at stage 40 for the presence or absence of a secondary embryonic axis. Given the recent demonstration that β-catenin interacts with members of the Lef/Tcf family of transcription factors (Behrens et al., 1996; Molenaar et al., 1996), we predicted that both the NES and TM mutants, which do not accumulate in nuclei, would be inactive in the axis duplication assay, whereas the WT and NLS mutants would cause axis duplication. In contrast to these predictions, we found that overexpression of any of the localization mutants resulted in embryos with secondary axes (Table I) suggesting that the translocation of ectopic β-catenin into the nucleus may not be required for the establishment of dorsal cell fates.
Table I.
Frequency of Axis Duplication by β-Catenin Localization Mutants
| Construct | Percentage of embryos with axis duplication (n) | |
|---|---|---|
| WT–β-catenin–GFP | 98.2 (55) | |
| NES–β-catenin–GFP | 97.1 (69) | |
| NLS–β-catenin–GFP | 98.3 (59) | |
| TM–β-catenin–GFP | 96.8 (62) |
Embryos were injected ventrally at the four-cell stage with 250 pg RNA encoding each β-catenin protein, and axis duplication was scored at stage 40 by the presence of a secondary dorsal axis.
The Signaling Activity of the TM–β-Catenin Mutant Is Suppressed by Overexpression of Cadherin
One interpretation of the axis duplication results is that the signaling activity of β-catenin is independent of its localization in the cell, consistent with the observation that a membrane-tethered form of plakoglobin is also able to induce an axis duplication (Merriam et al., 1997). Another possible interpretation, however, is that the ectopic β-catenin competes with endogenous β-catenin for access to the machinery that regulates steady-state levels of β-catenin. This hypothetical competition could lead to the elevation of endogenous β-catenin levels that could engage in signaling activities. To distinguish between these possibilities, we took advantage of the observation that overexpression of C-cadherin can suppress the signaling activity of β-catenin by anchoring β-catenin to the membrane (Fagotto et al., 1996). Therefore, we prepared a deletion mutant of TM–β-catenin that lacks the amino terminus, Arm repeats 10–13, and the carboxy terminus (Fig. 1 A, TM–β-catenin 1–9). This mutant lacks the region necessary for C-cadherin binding but retains the region required for APC binding (Fagotto et al., 1996). The rationale for this experiment is that if the TM–β-catenin 1–9 mutant is directly involved in establishing dorsal cell fates, then overexpression of C-cadherin should not be able to suppress its signaling activity. However, if the TM–β-catenin 1–9 mutant is functioning indirectly by elevating a free, signaling pool of endogenous β-catenin, overexpression of C-cadherin should be able to suppress axis duplication by sequestering endogenous β-catenin to the plasma membrane. Therefore, RNA encoding TM–β-catenin 1–9 was injected into ventral blastomeres in the presence or absence of C-cadherin RNA, and embryos were scored at stage 40 for duplication of the embryonic axes (Table II). Overexpression of TM–β-catenin 1–9 in ventral blastomeres resulted in the induction of secondary axes at very high frequency (Table II). In contrast, coinjection of C-cadherin completely suppressed the ability of TM–β-catenin 1–9 to induce secondary axes. This result demonstrates that the TM–β-catenin mutant lacks signaling activity, defined as an ability to induce an ectopic embryonic axis, under conditions (overexpression of C-cadherin) where endogenous β-catenin is sequestered to the plasma membrane.
Table II.
C-cadherin Inhibits the Ability of TM–β-Catenin to Induce a Secondary Dorsal Axis
| TM–β-catenin 1–9 | TM–β-catenin 1–9 + C-cadherin | |||
|---|---|---|---|---|
| Percentage of embryos with duplicated axis (n) | 94.8 (77) | 0 (77) |
Embryos were injected ventrally at the four-cell stage with either 50 pg TM–β-catenin 1–9 RNA or a combination of 50 pg TM–β-catenin 1–9 RNA and 1.25 ng C-cadherin RNA. Axis duplication was scored at stage 40 by the presence of a secondary dorsal axis.
Overexpression of the TM–β-Catenin Mutant Results in Stabilization of Endogenous β-Catenin
To test whether the overexpression of TM–β-catenin causes a stabilization of endogenous β-catenin in the cytosol and nucleus, we extracted protein from embryos injected with control GFP RNA or TM–β-catenin RNA and performed immunoblot analyses to determine the relative levels of endogenous β-catenin in both total (T) and soluble (S) protein fractions (Fig. 3). Soluble fractions represent lysates that have been incubated with ConA-Sepharose beads, which bind many membrane glycoproteins, including all cadherins (Fagotto et al., 1996). Thus, β-catenin present in the supernatant after incubation with ConA beads represents the soluble, non–cadherin-bound pool of β-catenin in the cell. This procedure allowed us to estimate the relative distribution of endogenous β-catenin in total homogenates (Fig. 3, T) and soluble, non–cadherin-bound pools (Fig. 3, S). Protein samples were analyzed by SDS-PAGE, and blots were probed with anti–β-catenin and anti–α-spectrin antibodies. We found that overexpression of either TM–β-catenin 1-myc or TM–β-catenin 1–9 results in an approximate twofold increase in total (T) and an approximate three- to fourfold increase in soluble (S) levels of endogenous β-catenin after normalizing β-catenin to levels of α-spectrin (numbers below each lane represent relative levels of β-catenin in TM–β-catenin–injected embryos compared to GFP-injected controls). The ability of both TM–β-catenin mutants to stabilize endogenous β-catenin was confirmed in a second, independent experiment (data not shown). Thus, overexpression of TM–β-catenin results in the elevation of both total and soluble pools of endogenous β-catenin.
Figure 3.

Overexpression of TM–β-catenin results in the stabilization of endogenous β-catenin. Injection of 1.25 ng of control RNA (GFP), TM–β-catenin 1-myc RNA, or TM–β-catenin 1–9 RNA demonstrates overexpression of both forms of TM–β-catenin results in an increase in the steady-state levels of endogenous β-catenin in both total (T) and soluble (S, non–cadherin-bound) lysates. In this experiment, two-cell stage embryos were injected with RNA at four sites, followed by protein extraction at stage 7. To control for protein loading, all endogenous β-catenin bands were normalized to α-spectrin signals from the same Western blot. Numbers below each lane represent the relative level of endogenous β-catenin in each sample (control levels were assigned a value of 1.0). Molecular mass markers indicated are 113 and 75 kD.
Overexpression of the TM–β-Catenin Mutant Results in Accumulation of Endogenous β-Catenin in Nuclei
Given that the TM–β-catenin mutant appears to signal by elevating a free, signaling pool of endogenous β-catenin, we sought to determine whether overexpression of TM– β-catenin results in the accumulation of endogenous β-catenin in nuclei in a manner similar to that seen after inhibition of GSK-3 (Yost et al., 1996) and activation of Wnt signaling (Larabell et al., 1997). Therefore, we overexpressed several mutant β-catenin constructs and examined the distribution of endogenous β-catenin in animal caps by confocal microscopy. Since our anti–β-catenin antibody recognizes the NH2-terminal domain (Yost et al., 1996), we used TM–β-catenin mutants that lack the NH2-terminal domain to distinguish endogenous β-catenin protein from ectopic β-catenin proteins. We found that overexpression of TM–β-catenin 1–9 resulted in the increased accumulation of endogenous β-catenin in nuclei (Fig. 4, A–F). Specifically, after injection of TM–β-catenin we observed that a subset of animal cap cells show elevated levels of endogenous β-catenin in nuclei (Fig. 4, A and D, arrowheads). Since a fluorescent lineage tracer (Oregon green dextran [OGDx]; Fig. 4, B and E) was coinjected with the RNA, simultaneous analysis of cells possessing elevated nuclear β-catenin and the lineage tracer (Fig. 4, C and F) reveals a direct correspondence between the presence of TM–β-catenin and accumulation of endogenous β-catenin in the nucleus. Neighboring cells not receiving TM–β-catenin 1–9 RNA do not show high levels of endogenous β-catenin in the nucleus (Fig. 4, A, C, D, and F, arrows). In addition, a small percentage of cells (∼10%) that received TM–β-catenin RNA also does not show high levels of endogenous β-catenin in the nucleus. The reason for this result is unclear, but it may simply represent differences in the responsiveness of individual cells to TM–β-catenin expression, variations in the levels of TM–β-catenin RNA received by each cell, or cell cycle differences in the levels of endogenous β-catenin in the nucleus. The effect of overexpression of TM–β-catenin on the localization of endogenous β-catenin was confirmed in four separate experiments in which ∼15 animal cap explants were analyzed. Identical results to those presented in Fig. 4 were also obtained with a TM–β-catenin ΔN mutant, which only lacks the amino-terminal domain (data not shown). In contrast to the effect of TM–β-catenin expression on nuclear β-catenin levels, injection of a control RNA encoding β-catenin ΔN-9 (Fig. 1 A), a mutant that lacks the NH2-terminal domain and Arm repeats 1–9 and is not active in the axis duplication assay (data not shown), does not affect levels of endogenous β-catenin in the nucleus (Fig. 4, G–I). Thus, ectopic expression of β-catenin causes an unsuspected accumulation of endogenous β-catenin in nuclei, similar to overexpression of activators of the Wnt pathway (Schneider et al., 1996; Yost et al., 1996; Larabell et al., 1997). The simplest explanation is that ectopic β-catenin present in the cytoplasm or even at membranes competes with endogenous β-catenin for access to the degradative machinery, resulting in the stabilization and accumulation of endogenous β-catenin in the nucleus, a hypothesis tested below.
Figure 4.

Overexpression of TM–β-catenin results in the accumulation of endogenous β-catenin in the nucleus. RNA encoding TM– β-catenin 1–9 (A–F) was coinjected with Oregon green dextran (OGDx; B, E, and H) as a lineage tracer to determine the effect of TM– β-catenin overexpression on the distribution of endogenous β-catenin (A, C, D, and F). Merged images (C and F) demonstrate that cells overexpressing TM–β-catenin 1–9 possess high levels of endogenous β-catenin in nuclei (arrowheads) in contrast to that observed in nonexpressing cells (arrows). Overexpression of ΔN-9 β-catenin (G–I), that does not promote axis duplication, does not affect the levels of endogenous β-catenin in the nucleus (G and I; arrowheads mark nuclei of cells expressing the ΔN-9 mutant and arrows mark nuclei of nonexpressing cells).
TM–β-Catenin Competes with Endogenous β-Catenin for Interactions With Endogenous APC and a Cadherin Fraction
Both APC and cadherin are potential regulators of the signaling function of β-catenin (for review see Miller and Moon, 1996) and, therefore, are candidates to play a role in the stabilization of endogenous β-catenin after overexpression of TM–β-catenin. To test whether TM–β-catenin binds endogenous APC and cadherin, protein lysates extracted from embryos injected with TM–β-catenin 1-myc RNA were subjected to immunoprecipitation with anti-APC antibodies (Fig. 5 A, APC-IP) and ConA precipitation (Fig. 5 A, ConA, represents cadherin-bound β-catenin). The association of TM–β-catenin 1-myc with APC and the ConA fraction containing cadherins was then assayed by immunoblotting with anti-myc antibodies. We found that TM–β-catenin 1-myc coimmunoprecipitated with APC (Fig. 5 A, APC-IP) and was also present in the ConA precipitates that contain cadherins (Fig. 5 A, ConA). TM–β-catenin 1-myc is not detected in lysates incubated with beads alone, demonstrating the specificity of both precipitations (Fig. 5 A, beads). Thus, ectopic TM– β-catenin interacts with endogenous APC and is present in a fraction that contains cadherins. Given this result, we asked whether the binding of TM–β-catenin to APC and its association with a fraction that contains cadherins reduces the levels of endogenous β-catenin associated with APC and the cadherin fraction. Protein extracts from embryos injected with either a control RNA or TM–β-catenin 1-myc were subjected to immunoprecipitation with anti-APC antibodies (Fig. 5 B) or ConA precipitation (Fig. 5 C), and the levels of endogenous β-catenin were subsequently determined by immunoblotting with anti–β-catenin antibodies. We observed that overexpression of TM– β-catenin 1-myc results in a decrease in the steady-state levels of endogenous β-catenin associated with APC (Fig. 5 B) and a cadherin fraction (Fig. 5 C). The levels of endogenous β-catenin that coimmunoprecipitate with APC after overexpression of TM–β-catenin 1-myc were found to decrease approximately threefold relative to control levels in two experiments and decreased approximately 1.2-fold in a third experiment. Levels of endogenous β-catenin associated with a cadherin fraction after overexpression of TM–β-catenin 1-myc were found to decrease to ∼0.6 of control levels in each of three experiments. These data suggest that the binding of ectopic β-catenin to both APC and cadherin results in a decrease in the binding of endogenous β-catenin to each protein and the accumulation of a free, signaling pool of endogenous β-catenin in the cell.
Figure 5.

Ectopic TM–β-catenin competes with endogenous β-catenin for binding to endogenous APC and a cadherin fraction. (A) Protein extracted from embryos injected with 1.25 ng of TM–β-catenin 1-myc RNA was subjected to APC immunoprecipitation (APC-IP) or ConA precipitation (ConA, represents cadherin-bound fraction) followed by immunoblotting with anti-myc antibodies. Control lysates were incubated with beads alone (beads). These experiments show that TM–β-catenin 1-myc binds endogenous APC and is present in ConA-bound fractions, indicating an association with cadherin. (B) To determine the effect of overexpression of TM–β-catenin 1-myc on the levels of endogenous β-catenin associated with APC, protein extracts from control embryos or embryos injected with 1.25 ng TM–β-catenin 1-myc were subjected to immunoprecipitation with anti-APC antibodies followed by immunoblotting with anti–β-catenin antibodies. These analyses show that overexpression of TM–β-catenin 1-myc causes a decrease relative to controls in the levels of endogenous β-catenin associated with APC. (Relative levels of endogenous β-catenin are shown below each lane with controls set to 1.0.) (C) Changes in the levels of endogenous β-catenin associated with a cadherin fraction were examined by preparing ConA precipitates from protein extracts prepared from control or TM–β-catenin 1-myc RNA–injected embryos followed by immunoblotting with anti–β-catenin antibodies. The levels of endogenous β-catenin present in ConA fractions decreased to 0.6 of control levels, suggesting that ectopic TM–β-catenin competes with endogenous β-catenin for binding to cadherin. (Relative levels of endogenous β-catenin are shown below each lane.) Molecular mass markers indicated in A are 198, 113, and 75 kD and those indicated in B and C are 113 and 75 kD.
Overexpression of the TM–β-Catenin Mutant Results in the Relocalization of APC
To further examine the ability of TM–β-catenin mutants to compete for interactions with APC, a protein thought to be involved in regulating the stability of β-catenin in cells (Munemitsu et al., 1995; Papkoff et al., 1996), we investigated whether overexpression of TM–β-catenin can affect the localization of APC in the cell. We attempted to examine the effect of TM–β-catenin expression on the localization of endogenous APC but were unsuccessful in obtaining any specific staining in animal cap cells using the anti-APC antibodies from two different sources (Rubinfeld et al., 1993; Näthke et al., 1996). Therefore, to perform this experiment we constructed an APC deletion mutant that contains the β-catenin–binding sites that are necessary and sufficient to promote the rapid degradation of β-catenin in cultured cells (Munemitsu et al., 1995) and fused this mutant to GFP to facilitate visualization of the fusion protein by confocal microscopy (Fig. 1 B, APC–GFP). Analysis of cells expressing APC–GFP demonstrated that this protein is distributed uniformly throughout the cytoplasm and is absent from the nucleus (Fig. 6 A). Coexpression of TM– β-catenin 1–9 resulted in the dramatic redistribution of APC–GFP (Fig. 6 B) to a pattern similar to that seen for TM–β-catenin GFP (see Fig. 2 D). Overexpression of TM–β-catenin 1–9 did not affect the localization of control GFP protein (data not shown), demonstrating the specificity of its affect on the distribution of APC–GFP. Experiments using an myc-tagged TM–β-catenin construct (Fig. 1 A, TM–β-catenin 1-myc) further demonstrate that the distribution of the membrane-tethered β-catenin protein (Fig. 6 D) and that of APC–GFP (Fig. 6 C) completely overlap (yellow represents overlapping staining; Fig. 6 E). These data strongly suggest that ectopic β-catenin mutants interact with APC, a component of the degradative machinery. Such interactions may reduce the ability of endogenous β-catenin to interact with endogenous or ectopic APC, resulting in its stabilization and increased availability for signaling.
Figure 6.

Overexpression of TM–β-catenin results in the redistribution of APC–GFP in animal cap cells. Intracellular distribution of APC–GFP in the absence (A) or presence (B) of TM–β-catenin 1–9. Coexpression of TM–β-catenin 1–9 causes a dramatic redistribution of APC–GFP from a diffuse cytosolic pattern (A) to an apparent association with the secretory apparatus (B). The interaction between TM–β-catenin and APC–GFP was further demonstrated by coexpressing an myc-tagged TM–β-catenin mutant (TM–β-catenin 1-myc) with APC–GFP and examining the pattern of each protein in animal cap cells. Both TM–β-catenin 1-myc (C) and APC–GFP (D) proteins display an overlapping intracellular distribution seen in the merged image (E; arrows in C–E).
Overexpression of Plakoglobin Results in the Accumulation of Endogenous β-Catenin in the Nucleus and the Relocalization of APC–GFP in the Cytoplasm
Plakoglobin is closely related to β-catenin, and like β-catenin, ectopic plakoglobin accumulates in nuclei and can induce a secondary axis when overexpressed in Xenopus embryos (Karnovsky and Klymkowsky, 1995). Given that plakoglobin interacts with many of the same protein partners as β-catenin, we hypothesized that ectopic plakoglobin may compete with endogenous β-catenin for interactions with these protein partners resulting in the formation of a free, signaling pool of endogenous β-catenin in a manner similar to that seen after overexpression of the membrane-tethered β-catenin. To test this possibility, we investigated whether overexpression of plakoglobin (Fig. 1 A, h-plakoglobin) alters the distribution of endogenous β-catenin in animal cap cells. We injected RNA encoding myc-tagged plakoglobin into the animal pole of blastomeres at the four-cell stage and examined the distribution of both endogenous β-catenin (Fig. 7, A and C) and ectopic plakoglobin (Fig. 7, B and C) in animal cap cells. Ectopic myc-tagged plakoglobin accumulated in the cytoplasm of animal cap cells but was not seen in nuclei (Fig. 7, B and C). Visualization of endogenous β-catenin in the same cells (Fig. 7, A and C) revealed that elevated levels of endogenous β-catenin were present in nuclei of cells that possessed ectopic plakoglobin (Fig. 7, A and C, arrowheads), whereas no such elevation was apparent in cells that do not possess ectopic plakoglobin (Fig. 7, A and C, arrows). Therefore, overexpression of plakoglobin results in the accumulation of endogenous β-catenin in nuclei. These data are consistent with the idea that the observed signaling activity of plakoglobin may be attributable, at least in part, to the formation of a free, signaling pool of endogenous β-catenin that accumulates in the nucleus to affect target gene expression. Importantly, we observe the accumulation of endogenous β-catenin in the nucleus at levels of injected plakoglobin RNA (1–2 ng) that are less than was used by Karnovsky and Klymkowsky (1995) to establish the axis-inducing activity of plakoglobin.
Figure 7.

Overexpression of plakoglobin causes the accumulation of endogenous β-catenin in the nucleus and the redistribution of APC–GFP. Human plakoglobin was overexpressed in animal cap cells, and the distribution of both endogenous β-catenin (A and C) and myc-tagged plakoglobin (B and C) was determined by confocal microscopy. Cells expressing ectopic plakoglobin possess high levels of endogenous β-catenin in the nucleus (arrowheads in A and C) when compared to cells not expressing ectopic plakoglobin (arrows in A and C). Overexpression of plakoglobin also results in the redistribution of APC–GFP within the cell to sites of plakoglobin accumulation. The localization of APC–GFP (D) and ectopic plakoglobin (E) in the absence of other RNAs demonstrates the different localization patterns of the two ectopic proteins. Coinjecting APC–GFP and plakoglobin, however, results in the redistribution of APC–GFP (F) to a pattern indistinguishable from that seen for ectopic plakoglobin (G). Overlapping APC–GFP (F) and plakoglobin (G) staining appears as yellow staining in the merged image (H). Arrows mark examples of APC–GFP and plakoglobin colocalization.
The effect of overexpression of plakoglobin on the intracellular distribution of endogenous β-catenin prompted us to ask whether ectopic plakoglobin can sequester APC as we had shown for membrane-tethered β-catenin. Coexpression of myc-tagged plakoglobin and APC–GFP resulted in a dramatic change in the localization of APC–GFP (compare Fig. 7 F to control localization shown in Fig. 7 D) to a pattern identical to that of plakoglobin (Fig. 7 G). Superimposing the individual images confirms the colocalization of plakoglobin and APC–GFP in cells (arrows mark overlapping staining that appears yellow in Fig. 7 H). These data strongly suggest that ectopic plakoglobin interacts with APC, an important regulator of β-catenin stability. This situation may result in competition between ectopic plakoglobin and endogenous β-catenin for binding to APC, leading to an increase in a free, signaling pool of β-catenin that accumulates in the nucleus.
Different Phosphorylation Isoforms of β-Catenin Compete for Interactions With Cadherin
The above experiments suggest that ectopic β-catenin competes with endogenous β-catenin, leading to the accumulation of endogenous β-catenin in nuclei. This result raises the question of whether competition exists normally between endogenous pools of β-catenin in the cell. We hypothesized that besides promoting β-catenin/Armadillo degradation (Peifer et al., 1994; Yost et al., 1996), GSK-3 might also modulate the interaction of β-catenin with cadherins at the plasma membrane. Consistent with this idea, both tyrosine- and serine/threonine-phosphorylated forms of Armadillo preferentially associate with membrane fractions compared to soluble, cytosolic fractions in Drosophila embryos (Peifer et al., 1994). To address this hypothesis, we investigated whether phosphorylation of β-catenin by Xgsk-3 changes the association of β-catenin with N-cadherin in vitro. In control experiments, we found that 35S-labeled β-catenin specifically bound to microsomal membranes containing N-cadherin (compare Fig. 8 A, lanes 2 and 3) in reticulocyte lysates. In these experiments, we also determined that the unbound β-catenin present in the supernatant (Fig. 8 A, lane 1) reflected a 12–18-fold excess relative to that bound to the N-cadherin microsomes in the pellet (Fig. 8 A, lane 2).
Figure 8.

Phosphorylation of β-catenin by Xgsk-3 enhances association with N-cadherin in vitro. (A) β-catenin was translated in vitro in the presence of [35S]methionine and, in separate reactions, microsomal membranes were incubated in rabbit reticulocyte lysate with unlabeled methionine in the presence or absence of N-cadherin RNA. The β-catenin reactions and microsome reactions were then mixed, and the samples were incubated to allow binding of β-catenin to the microsomal membranes. The samples were then centrifuged to generate microsomal pellets and supernatants. Lane 1, 35S–β-catenin present in the supernatant after centrifugation. Adjusting for volumes, these lanes contain 40% of the total supernatant. Lane 2, 35S–β-catenin associated with the pelleted microsomes containing N-cadherin. Lane 3, 35S–β-catenin associated with control microsomes. The data show that a small percentage of the total 35S-labeled β-catenin binds microsomal membranes in a cadherin-dependent manner (compare lanes 1 and 2). (B) β-catenin and Xgsk-3 RNAs, as well as microsomal membranes with or without N-cadherin RNAs, were translated in vitro with nonradioactive methionine and then incubated with or without recombinant Xgsk-3 protein in the presence of [γ32P]ATP. After centrifugation, the samples were immunoprecipitated with an anti–β-catenin antibody. Lanes 1 and 5, immunoprecipitation of β-catenin from the supernatant (lane 1) and microsome/N-cadherin pellet (lane 5) after cotranslation with Xgsk-3 RNA demonstrates that most of the phosphorylated β-catenin associates with the microsomal pellet. Lanes 2 and 6, immunoprecipitation of β-catenin from the supernatant (lane 2) and pellet (lane 6) of β-catenin translations incubated with recombinant Xgsk-3 protein confirms the conclusion of lanes 1 and 5. Lanes 3 and 7, omission of N-cadherin RNA demonstrates that most phosphorylated β-catenin is immunoprecipitated from the supernatant (lane 3) and not the microsomal pellet lacking N-cadherin (lane 7). Lanes 4 and 8, omission of exogenous Xgsk-3 protein and RNA demonstrates basal phosphorylation of β-catenin by kinases present in the lysate, that is substantially lower than in samples supplemented with Xgsk-3, and this phosphorylated β-catenin accumulates primarily in the supernatant (lane 4) rather than the pellet (lane 8), even though the microsomes contained N-cadherin.
We then exploited the excess of β-catenin in the supernatant relative to that bound to cadherin in the reticulocyte lysate binding assay to directly test whether the phosphorylation of β-catenin by Xgsk-3 altered its ability to bind microsomes expressing N-cadherin. The assays were conducted in the presence of [γ32P]ATP, which was added to the reticulocyte lysate to facilitate detection of phosphoproteins, and all samples were immunoprecipitated with an anti–β-catenin antibody to specifically monitor phosphorylated β-catenin. When β-catenin RNA is cotranslated with Xgsk-3 RNA, then bound to microsomes expressing N-cadherin, most of the phosphorylated β-catenin is associated with the microsomal pellet (Fig. 8 B, lane 5) rather than the supernatant (Fig. 8 B, lane 1), which contains the majority of the total β-catenin in this assay (Fig. 8 A). The same results were obtained when recombinant Xgsk-3 protein was added to translations of β-catenin (Fig. 8 B, lane 6, pellet, and lane 2, supernatant). The preferential association of phosphorylated β-catenin with N-cadherin containing microsomes is a specific interaction since the amount of phosphorylated β-catenin in the microsomal pellet was reduced and the amount in the supernatant increased, by omission of N-cadherin from the assay (Fig. 8 B, lane 7, pellet, and lane 3, supernatant). Omission of Xgsk-3 from the assay (Fig. 8 B, lane 8, pellet, and lane 4, supernatant) also results in a decrease in the N-cadherin– bound β-catenin relative to that present in the supernatant, demonstrating that phosphorylation of β-catenin by Xgsk-3, and not kinases that may be present in reticulocyte lysates, is responsible for the observed increase in the association of phosphorylated β-catenin with N-cadherin. Since phosphorylated β-catenin is primarily associated with the microsomal membranes in a cadherin-dependent manner under conditions where the majority of β-catenin remains in the postmicrosomal supernatant (Fig. 8 A), these data strongly suggest that phosphorylation of β-catenin by Xgsk-3 enhances its association with the cytoplasmic domain of cadherins. In Fig. 8 and in independent experiments, we observed that the total level of phosphorylated β-catenin (estimated by the combined levels in both the supernatant and pellet lanes) was somewhat greater in the presence relative to the absence of N-cadherin (data not shown). This raises the questions of whether the phosphorylation of β-catenin occurs before, during, or after association with cadherins and whether the interaction of β-catenin with cadherin facilitates the process of phosphorylation or decreases the degradation of the phosphorylated β-catenin.
In independent experiments, we asked if recombinant β-catenin, phosphorylated in vitro by recombinant Xgsk-3, preferentially associates with N-cadherin–GST fusion protein bound to glutathione beads. Consistent with the conclusions reported here, we find that under conditions where the majority of the β-catenin is not phosphorylated and is not bound to the cadherin beads (i.e., β-catenin is present in excess) the phosphorylated form of β-catenin is preferentially bound to the cadherin beads (Torres, M., and R.T. Moon, unpublished data).
DISCUSSION
The function of β-catenin in specifying dorsal cell fate in Xenopus embryos may be dependent on interactions with Lef/Tcf transcription factors and the accumulation of β-catenin–Lef/Tcf complexes in the nucleus, where they positively regulate expression of dorsal-specific genes (for review see Moon et al., 1997). In support of this hypothesis, β-catenin is detected in nuclei of dorsal but not ventral blastomeres at the 16–32-cell stages of Xenopus (Larabell et al., 1997) and after zygotic transcription has commenced at the late blastula stage (Schneider et al., 1996). Moreover, several recent studies have demonstrated that both β-catenin and Armadillo can function as a transcriptional activator when complexed to Lef/Tcf transcription factors (Molenaar et al., 1996; Korinek et al., 1997; Morin et al., 1997; van de Wetering et al., 1997). Taken together, these data have led to the hypothesis that the signaling function of β-catenin in establishing the dorsal axis in Xenopus embryos is dependent on its nuclear localization. Although many lines of evidence support this idea, several observations raise questions regarding this proposed model of β-catenin signaling. Overexpression of β-catenin increases gap junction permeability in the absence of transcription (Guger and Gumbiner, 1995), suggesting it is modulating intercellular communication in a manner independent of de novo transcription. In addition, overexpression of a membrane-tethered form of plakoglobin that is unable to enter nuclei results in the induction of an ectopic dorsal axis (Merriam et al., 1997), suggesting that the nuclear accumulation of endogenous β-catenin is not required to induce the expression of dorsal-specific genes. Given these discrepancies in the proposed model of how β-catenin functions to establish dorsal cell fate, we have directly examined this issue and conclude that the signaling function of β-catenin in axis specification is dependent on its localization to the nucleus. Our studies also present a clear cautionary note to all studies in which mutant proteins are overexpressed in a wild-type background, as we show that an unsuspected competition exists between ectopic and endogenous proteins, resulting in functional activation of the endogenous protein.
Is Nuclear β-Catenin Required for the Induction of Dorsal Cell Fate?
We targeted ectopic forms of β-catenin to membranes, the cytoplasm, or the nucleus to test whether the accumulation of β-catenin in nuclei is required for its role in specifying the dorso–ventral axis in Xenopus embryos. We show that all of the localization mutants tested are active in inducing an ectopic axis, suggesting at first glance that the signaling activity of ectopically expressed β-catenin is not dependent on its localization to a specific subcellular compartment. The axis-inducing activity of the TM–β-catenin mutant was particularly perplexing given the proposed role of nuclear β-catenin–Lef/Tcf complexes (Behrens et al., 1996; Huber et al., 1996; Molenaar et al., 1996; Brunner et al., 1997; Korinek et al., 1997; Morin et al., 1997; Riese et al., 1997; van de Wetering et al., 1997). Therefore, we examined the mechanism underlying the signaling activity of the membrane-tethered β-catenin and found that it acts indirectly to specify dorsal cell fate by elevating the steady-state levels of endogenous β-catenin, resulting in the accumulation of a signaling pool of β-catenin in the nucleus. Several lines of evidence support this conclusion: (a) Overexpression of C-cadherin completely blocks the ability of the TM–β-catenin 1–9 to induce an ectopic axis (Table II) despite the fact that the presence of Arm repeats 1–9 is not sufficient for C-cadherin binding (Fagotto et al., 1996). This result suggests that overexpression of C-cadherin suppresses the axis-inducing activity of TM– β-catenin 1–9 by sequestering endogenous β-catenin to the plasma membrane where it is unable to signal. (b) Overexpression of various TM–β-catenin mutants results in an approximate three- to fourfold increase in the steady-state levels of endogenous β-catenin in non–cadherin-bound fractions (Fig. 3). This increase in the levels of soluble β-catenin is manifest by the accumulation of endogenous β-catenin in the nucleus (Fig. 4).
The mechanism by which overexpression of TM–β-catenin causes the observed stabilization of endogenous β-catenin may be the result of competition between ectopic and endogenous β-catenin for binding to various protein partners, including APC and cadherin. Both APC and cadherin have been implicated in regulating the signaling activity of β-catenin and Armadillo (Munemitsu et al., 1995; Cox et al., 1996; Fagotto et al., 1996; Papkoff et al., 1996; Sanson et al., 1996; Korinek et al., 1997; Morin et al., 1997; Rubinfeld et al., 1997) and, therefore, are candidates to play a role in the stabilization of endogenous β-catenin after overexpression of TM–β-catenin. We show that ectopic TM–β-catenin 1-myc protein interacts with endogenous APC and with a cell fraction that contains cadherin and that these interactions result in a decrease in the levels of endogenous β-catenin associated with APC and cadherin (Fig. 5). Moreover, overexpression of various TM–β-catenin mutants dramatically alters the subcellular distribution of ectopic APC (Fig. 6). This interaction with APC is likely to be the most important factor in predicting the ability of ectopic β-catenin to stabilize endogenous β-catenin and induce dorsal cell fate. Previous studies have shown that the ability of various β-catenin deletion mutants to induce a secondary axis coincides with the ability of each of these mutants to bind APC, while several of these active mutants do not bind C-cadherin (Fagotto et al., 1996). Together, these results are consistent with the idea that the ectopic β-catenin competes with endogenous β-catenin for binding to various protein partners, and in particular APC. As APC is then unavailable for interactions with endogenous β-catenin to promote its degradation, endogenous β-catenin accumulates in the cell and is available for signaling.
The data presented here, taken together with evidence that β-catenin interacts with Lef/Tcf transcription factors (Behrens et al., 1996; Huber et al., 1996; Korinek et al., 1997; Molenaar et al., 1996; Morin et al., 1997; Rubinfeld et al., 1997; van de Wetering et al., 1997) and that endogenous β-catenin accumulates in nuclei of dorsal but not ventral blastomeres of cleavage stage Xenopus embryos (Larabell et al., 1997), strongly argue that the signaling activity of β-catenin is dependent on its translocation into the nucleus. In the nucleus, it is likely that Lef/Tcf bind to the promoters of specific genes, and β-catenin interacts with these promoters indirectly through its binding to Lef/ Tcf, resulting in altered DNA bending and modulation of gene expression (Behrens et al., 1996). A likely target gene of β-catenin–Lef/Tcf complexes is the dorsal regulatory gene siamois, whose expression is induced in response to Wnt (Carnac et al., 1996; Yang-Snyder et al., 1996) and β-catenin (Brannon and Kimelman, 1996; Brannon et al., 1997).
Given the preponderance of evidence indicating that β-catenin mediates its signaling activity through interactions with Lef/Tcf transcription factors, one may question the importance of our demonstrating that nuclear accumulation is necessary for the signaling activity of β-catenin. A recent study by Merriam et al. (1997), however, demonstrates that overexpression of a membrane-tethered plakoglobin mutant in Xenopus embryos results in the induction of an ectopic secondary axis. In contrast to our conclusion regarding the function of TM–β-catenin, these authors argue that the membrane-tethered plakoglobin induces an ectopic dorsal axis by anchoring in the cytoplasm a negative regulator of dorsal cell fate, hypothesized to be XTcf-3 or a related factor, thereby blocking its inhibitory function in the nucleus (Merriam et al., 1997). This interpretation suggests that the result of Wnt signaling through β-catenin or its ortholog plakoglobin is to inactivate an inhibitor of dorsal cell fate instead of a model where β-catenin or plakoglobin functions in concert with Lef/Tcf transcription factors to activate downstream target genes. Our data are not consistent with the hypothesis of Merriam and colleagues since we find that ectopic membrane-tethered β-catenin, and by extension membrane-tethered plakoglobin, function indirectly by elevating levels of endogenous β-catenin in the cell. Similarly, we find that overexpression of plakoglobin in Xenopus animal cap cells results in the accumulation of endogenous β-catenin in the nucleus (Fig. 7) and that ectopic plakoglobin alters the distribution of APC (Fig. 7), a regulator of β-catenin stability and β-catenin–Lef/Tcf transcriptional activity (Munemitsu et al., 1995; Papkoff et al., 1996; Hayashi et al., 1997; Korinek et al., 1997; Morin et al., 1997). These data are most consistent with the idea that ectopic plakoglobin saturates available APC-binding sites, reducing the interaction of endogenous β-catenin with APC, resulting in the stabilization of endogenous β-catenin. Finally, we have found that TM–β-catenin does not sequester XTcf-3, a potential regulator of dorsal-specific gene expression (Molenaar et al., 1996), in the cytoplasm (data not shown). Thus, it seems unlikely that the related form of membrane-tethered plakoglobin (Merriam et al., 1997) would be active in axis induction by anchoring XTcf-3 or a related inhibitory factor to the plasma membrane. Our data, however, do not rule out the possibility that XTcf-3 normally acts as a transcriptional repressor when bound to DNA on its own and that the binding of β-catenin to XTcf-3 relieves this inhibition and instead promotes transcription (Brannon et al., 1997). In summary, we suggest that gain-of-function assays with plakoglobin are difficult to interpret because of the unanticipated effects on endogenous β-catenin and APC described above. These data taken together with the recent observation that depletion of plakoglobin transcripts in early Xenopus embryos does not inhibit the development of dorsal cell fates (Kofron et al., 1997) suggest that plakoglobin is not a necessary contributor to the signaling events involved in axis specification.
Implications of Competition between Ectopic and Endogenous β-Catenin on Interpretations of Overexpression Studies in Xenopus Embryos
Our analyses of the function of TM–β-catenin raise a very important caveat of overexpression analyses in Xenopus embryos. We demonstrate that overexpression of TM– β-catenin causes the elevation of a free, signaling pool of endogenous β-catenin, and it is this endogenous pool that is active in inducing an ectopic dorsal axis. Given this result, we hypothesize that the signaling activity of various β-catenin mutants (Fagotto et al., 1996) or plakoglobin mutants (Merriam et al., 1997) may be indirect, functioning by elevating levels of endogenous β-catenin in the cell (Fig. 9). In fact, a discrepancy exists in the literature regarding the signaling activity of various β-catenin mutants (Funayama et al., 1995; Fagotto et al., 1996) and similar mutants in Armadillo (Orsulic and Peifer, 1996), the Drosophila homologue of β-catenin. For example, a mutant form of β-catenin lacking both the amino and carboxyl terminus induces a secondary dorsal axis when overexpressed in Xenopus embryos (Funayama et al., 1995), yet a similar mutant form of Armadillo is unable to transduce the Wingless signal in Drosophila embryos (Peifer and Wieschaus 1990; Orsulic and Peifer 1996). Moreover, a recent study has demonstrated that the carboxy-terminal domain of β-catenin and Armadillo constitutes a transactivation domain that is required for Lef/Tcf-dependent transcriptional activation (van de Wetering et al., 1997). Although β-catenin and Armadillo possess very similar functional domains, it remains possible that this disparity in the signaling activity of various β-catenin and Armadillo mutants may be an actual difference between the functional domains required for this activity. On the other hand, this difference could be resolved by arguing that ectopically expressed β-catenin acts indirectly by competing with endogenous β-catenin for interactions with APC, a potential component of the degradative machinery. This competition would result in the stabilization of a free, signaling pool of endogenous β-catenin. In fact, examination of the published data regarding the axis-inducing activity of various β-catenin mutants shows that the ability of a given ectopically expressed β-catenin mutant to induce dorsal cell fate corresponds with its ability to bind APC (Funayama et al., 1995; Fagotto et al., 1996). This conclusion calls into question the interpretation of many β-catenin overexpression studies performed in Xenopus embryos and underscores the importance of determining the effects of overexpression on the endogenous protein and protein partners with which the overexpressed protein may interact.
Figure 9.

Model of the Wnt signaling pathway showing competition between endogenous and ectopic β-catenin for interactions with several protein partners, including cadherin, APC, and Lef/ Tcf. Overexpression of various β-catenin mutants that possess the ability to interact with APC (TM–β-catenin is shown) is predicted to increase the stability of endogenous β-catenin. The accumulation of endogenous β-catenin via activation of the Wnt pathway or expression of ectopic β-catenin both result in the increase of “free” β-catenin capable of interacting with members of the Lef/Tcf family of transcription factors. This β-catenin transcription factor complex translocates into the nucleus, where it regulates the expression of target genes responsible for establishing dorsal cell fate. Furthermore, phosphorylation of β-catenin by XGSK-3 may not only regulate β-catenin stability but may also result in competition between phosphorylated and nonphosphorylated β-catenin isoforms for binding to the cytoplasmic domain of cadherin.
Implications of Competition between Different Phosphorylation Isoforms of β-Catenin on the Regulation of β-Catenin Distribution and Function
Given that ectopic and endogenous β-catenin can compete for binding to different protein partners, we asked whether different phosphorylation isoforms of wild-type β-catenin may also compete for interactions with one such protein partner, N-cadherin. We demonstrate that phosphorylation of β-catenin by Xgsk-3 increases the ability of phosphorylated relative to nonphosphorylated β-catenin to bind to microsomes containing the cytoplasmic domain of N-cadherin. This result shows that competition between different phosphorylation isoforms of β-catenin can occur in vitro. Therefore, differential phosphorylation and competition may be an important mechanism by which cells regulate the interaction of β-catenin with different partners, thereby determining its function and subcellular distribution. Consistent with this idea, β-catenin and Armadillo are often highly phosphorylated in cells, and the regulation of tyrosine phosphorylation of β-catenin may regulate adherens junction organization and cadherin function (for review see Miller and Moon, 1996). Furthermore, both tyrosine- and serine/threonine-phosphorylated forms of Armadillo preferentially associate with membrane fractions compared to soluble, cytosolic fractions (Peifer et al., 1994), consistent with the results presented here and the evidence that cytoplasmic phosphorylated β-catenin is quickly targeted for degradation (Yost et al., 1996). In addition to promoting interactions with cadherin, phosphorylation of β-catenin by Xgsk-3 or other intracellular kinases may also modulate the association of β-catenin with other protein partners, thereby regulating both β-catenin function and its subcellular distribution. Further experiments will likely uncover possible connections between β-catenin phosphorylation and their effects on its binding interactions with different protein partners.
Acknowledgments
We thank Cheng-Jung Lai for constructing the TM–β-catenin–GFP construct. We also thank Ray White for the human APC cDNA, Michael Klymkowsky (University of Colorado, Boulder, CO) for the myc-tagged plakoglobin construct, Inke Näthke (Stanford University, Stanford CA), W. James Nelson (Stanford University), and Paul Polakis (Onyx Pharmaceuticals) for anti-APC antibodies, Olivier Destree (Hubrecht Laboratory) for the HA-tagged Xtcf-3 construct, and Cynthia Yost and David Kimelman for Xgsk-3 protein and assistance in the analysis of the axis-inducing activity of TM–β-catenin. We thank Monica Torres for permission to cite her unpublished data.
Abbreviations used in this paper
- APC
adenomatous polyposis coli
- GFP
green fluorescent protein
- NES
nuclear exclusion sequence
- NLS
nuclear localization sequence
- TM
transmembrane
- WT
wild-type
- Xgsk-3
Xenopus glycogen synthase kinase-3
Footnotes
Address all correspondence to Randall T. Moon, Howard Hughes Medical Institute, Box 35370, Room K536C HSB, University of Washington, Seattle, WA 98195. Tel.: (206) 543-1722. Fax: (206) 616-4230. e-mail: rtmoon@u.washington.edu
J.R. Miller is an associate and R.T. Moon an Investigator of the Howard Hughes Medical Institute, which supported this research.
REFERENCES
- Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. Functional interaction of β-catenin with the transcription factor LEF-1. Nature (Lond) 1996;382:638–642. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- Brannon M, Kimelman D. Activation of Siamoisby the Wnt pathway. Dev Biol. 1996;180:344–347. doi: 10.1006/dbio.1996.0306. [DOI] [PubMed] [Google Scholar]
- Brannon M, Gomperts M, Sumoy L, Moon RT, Kimelman D. A β-catenin/XTcF-3 complex binds to the siamois promoter to regulate dorsal axis specification in Xenopus. . Genes Dev. 1997;11:2359–2370. doi: 10.1101/gad.11.18.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner E, Peter O, Schweizer L, Basler K. pangolin encodes a Lef-1 homologue that acts downstream of Armadillo to transduce the Wingless signal in Drosophila. . Nature (Lond) 1997;385:829–833. doi: 10.1038/385829a0. [DOI] [PubMed] [Google Scholar]
- Carnac G, Kodjabachian L, Gurdon JB, Lemaire P. The homeobox gene Siamois is a target of the Wnt dorsalisation pathway and triggers organiser activity in the absence of mesoderm. Development (Camb) 1996;122:3055–3065. doi: 10.1242/dev.122.10.3055. [DOI] [PubMed] [Google Scholar]
- Cox RT, Kirkpatrick C, Peifer M. Armadillo is required for adherens junction assembly, cell polarity, and morphogenesis during Drosophilaembryogenesis. J Cell Biol. 1996;134:133–148. doi: 10.1083/jcb.134.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detrick RJ, Dickey D, Kintner CR. The effects of N-cadherin misexpression on morphogenesis in Xenopusembryos. Neuron. 1990;4:493–506. doi: 10.1016/0896-6273(90)90108-r. [DOI] [PubMed] [Google Scholar]
- Evan GI, Lewis GK, Ransay G, Bishop JM. Isolation of monoclonal antibodies specific for human c-mycprotooncogene product. Mol Cell Biol. 1985;5:3610–3616. doi: 10.1128/mcb.5.12.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagotto F, Funayama N, Gluck U, Gumbiner BM. Binding to cadherins antagonizes the signaling activity of β-catenin during axis formation in Xenopus. . J Cell Biol. 1996;132:1105–1114. doi: 10.1083/jcb.132.6.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagotto F, Guger K, Gumbiner BM. Induction of the primary dorsalizing center in Xenopusby the Wnt/GSK/β-catenin signaling pathway, but not by Vg1, Activin or Noggin. Development (Camb) 1997;124:453–460. doi: 10.1242/dev.124.2.453. [DOI] [PubMed] [Google Scholar]
- Funayama N, Fagotto F, McCrea P, Gumbiner BM. Embryonic axis induction by the armadillo repeat domain of β-catenin: evidence for intracellular signaling. J Cell Biol. 1995;128:959–968. doi: 10.1083/jcb.128.5.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giebelhaus DH, Zelus BD, Henchman SK, Moon RT. Changes in expression of α-fodrin during embryonic development of Xenopus laevis. . J Cell Biol. 1987;105:843–853. doi: 10.1083/jcb.105.2.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guger KA, Gumbiner BM. β-Catenin has Wnt-like activity and mimics the Nieuwkoop signaling center in Xenopusdorsal-ventral patterning. Dev Biol. 1995;172:115–125. doi: 10.1006/dbio.1995.0009. [DOI] [PubMed] [Google Scholar]
- Hayashi S, Rubinfeld B, Souza B, Polakis P, Wieschaus E, Levine AJ. A Drosophilahomolog of the tumor suppressor gene adenomatous polyposis coli down-regulates β-catenin but its zygotic expression is not essential for the regulation of Armadillo. Proc Natl Acad Sci USA. 1997;94:242–247. doi: 10.1073/pnas.94.1.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim R, Cubitt AB, Tsien RY. Improved green fluorescence. Nature (Lond) 1995;373:663–664. doi: 10.1038/373663b0. [DOI] [PubMed] [Google Scholar]
- Huber O, Korn R, McLaughlin J, Ohsugi M, Herrmann BG, Kemler R. Nuclear localization of β-catenin by interaction with transcription factor LEF-1. Mech Dev. 1996;59:3–10. doi: 10.1016/0925-4773(96)00597-7. [DOI] [PubMed] [Google Scholar]
- Kalderon D, Roberts BL, Richardson WD, Smith AE. A short amino acid sequence able to specify nuclear location. Cell. 1984;39:499–509. doi: 10.1016/0092-8674(84)90457-4. [DOI] [PubMed] [Google Scholar]
- Karnovsky A, Klymkowsky MW. Anterior axis duplication in Xenopusinduced by the overexpression of the cadherin-binding protein plakoglobin. Proc Natl Acad Sci USA. 1995;92:4522–4526. doi: 10.1073/pnas.92.10.4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemler R. From cadherins to catenins: cytoplasmic protein interactions and regulation of cell adhesion. Trends Genet. 1993;9:317–321. doi: 10.1016/0168-9525(93)90250-l. [DOI] [PubMed] [Google Scholar]
- Kimelman D, Christian JL, Moon RT. Synergistic principles of development: overlapping patterning systems in Xenopusmesoderm induction. Development (Camb) 1992;116:1–9. doi: 10.1242/dev.116.Supplement.1. [DOI] [PubMed] [Google Scholar]
- Kofron M, Spagnuolo A, Klymkowsky M, Wylie C, Heasman J. The roles of maternal α-catenin and plakoglobin in the early Xenopusembryo. Development (Camb) 1997;124:1553–1560. doi: 10.1242/dev.124.8.1553. [DOI] [PubMed] [Google Scholar]
- Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H. Constitutive transcriptional activation by a β-catenin-Tcf complex in APC−/− colon carcinoma. Science (Wash DC) 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- Kozak M. Compilation and analysis of sequences upstream from the translational start site in eukaryotic mRNAs. Nucleic Acids Res. 1984;12:857–872. doi: 10.1093/nar/12.2.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanford RE, Butel JS. Constructions and characterization of an SV40 mutant defective in nuclear transport of T antigen. Cell. 1984;39:801–813. doi: 10.1016/0092-8674(84)90415-x. [DOI] [PubMed] [Google Scholar]
- Larabell CA, Torres M, Rowning BA, Yost C, Miller JR, Wu M, Kimelman D, Moon RT. Establishment of the dorso-ventral axis in Xenopusembryos is presaged by early asymmetries in β-catenin that are modulated by the Wnt signaling pathway. J Cell Biol. 1997;136:1123–1136. doi: 10.1083/jcb.136.5.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merriam JM, Rubenstein AB, Klymkowsky MW. Cytoplasmically-anchored plakoglobin induces a WNT-like phenotype in Xenopus. . Dev Biol. 1997;185:67–81. doi: 10.1006/dbio.1997.8550. [DOI] [PubMed] [Google Scholar]
- Miller JR, Moon RT. Signal transduction through β-catenin and specification of cell fate during embryogenesis. Genes Dev. 1996;10:2527–2539. doi: 10.1101/gad.10.20.2527. [DOI] [PubMed] [Google Scholar]
- Molenaar M, van de Wetering M, Oosterwegel M, Peterson J, Maduro, Godsave S, Korinek V, Roose J, Destree O, Clevers H. XTcf-3 transcription factor mediates β-catenin-induced axis formation in Xenopusembryos. Cell. 1996;86:391–399. doi: 10.1016/s0092-8674(00)80112-9. [DOI] [PubMed] [Google Scholar]
- Moon RT, Brown JD, Torres M. Wnts modulate cell fate and behavior during vertebrate development. Trends Genet. 1997;13:157–162. doi: 10.1016/s0168-9525(97)01093-7. [DOI] [PubMed] [Google Scholar]
- Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science (Wash DC) 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- Munemitsu S, Albert I, Souza B, Rubinfeld B, Polakis P. Regulation of intracellular β-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proc Natl Acad Sci USA. 1995;92:3046–3050. doi: 10.1073/pnas.92.7.3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Näthke IS, Adams CL, Polakis P, Sellin JH, Nelson WJ. The adenomatous polyposis coli tumor suppressor protein localizes to plasma membrane sites involved in active cell migration. J Cell Biol. 1996;134:165–179. doi: 10.1083/jcb.134.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsulic S, Peifer M. An in vivo structure-function study of Armadillo, the β-catenin homologue, reveals both separate and overlapping regions of the protein required for cell adhesion and for Wingless signaling. J Cell Biol. 1996;134:1283–1300. doi: 10.1083/jcb.134.5.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papkoff J, Rubinfeld B, Schryver B, Polakis P. Wnt-1 regulates free pools of catenins and stabilizes APC-catenin complexes. Mol Cell Biol. 1996;16:2128–2134. doi: 10.1128/mcb.16.5.2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peifer M. Cell adhesion and signal transduction: the Armadillo connection. Trends Cell Biol. 1995;5:224–229. doi: 10.1016/s0962-8924(00)89015-7. [DOI] [PubMed] [Google Scholar]
- Peifer M, Wieschaus E. The segment polarity gene armadillo encodes a functionally modular protein that is the Drosophilahomolog of human plakoglobin. Cell. 1990;63:1167–1176. doi: 10.1016/0092-8674(90)90413-9. [DOI] [PubMed] [Google Scholar]
- Peifer M, Pai L-M, Casey M. Phosphorylation of the Drosophilaadherens junction protein Armadillo: roles for Wingless signal and Zeste-white 3 kinase. Dev Biol. 1994;166:543–556. doi: 10.1006/dbio.1994.1336. [DOI] [PubMed] [Google Scholar]
- Peifer M, Sweeton D, Casey M, Wieschaus E. Wingless signal and Zeste-white 3 kinase trigger opposing changes in the intracellular distribution of Armadillo. Development (Camb) 1994;120:369–380. doi: 10.1242/dev.120.2.369. [DOI] [PubMed] [Google Scholar]
- Riese J, Yu X, Munnerlyn A, Eresh S, Hsu S-C, Grosschedl R, Bienz M. LEF-1, a nuclear factor coordinating signaling inputs from wingless and decapentapalegic. . Cell. 1997;88:777–787. doi: 10.1016/s0092-8674(00)81924-8. [DOI] [PubMed] [Google Scholar]
- Rubinfeld B, Souza B, Albert I, Muller O, Chamberlain SH, Masiarz FR, Munemitsu S, Polakis P. Association of the APC gene product with β-catenin. Science (Wash DC) 1993;262:1731–1734. doi: 10.1126/science.8259518. [DOI] [PubMed] [Google Scholar]
- Rubinfeld B, Robbins P, El-Gamil M, Albert I, Porfiri E, Polakis P. Stabilization of β-catenin by genetic defects in melanoma cell lines. Science (Wash DC) 1997;275:1790–1792. doi: 10.1126/science.275.5307.1790. [DOI] [PubMed] [Google Scholar]
- Sanson B, White P, Vincent JP. Uncoupling cadherin-based adhesion from winglesssignaling in Drosophila. Nature (Lond) 1996;383:627–630. doi: 10.1038/383627a0. [DOI] [PubMed] [Google Scholar]
- Schneider S, Steinbeisser H, Warga RM, Hausen P. β-Catenin translocation into nuclei demarcates the dorsalizing centers of frog and fish embryos. Mech Dev. 1996;57:191–198. doi: 10.1016/0925-4773(96)00546-1. [DOI] [PubMed] [Google Scholar]
- Su LK, Vogelstein B, Kinzler KW. Association of the APC tumor suppressor protein with catenins. Science (Wash DC) 1993;262:1734–1737. doi: 10.1126/science.8259519. [DOI] [PubMed] [Google Scholar]
- Torres MA, Yang JA, Snyder, Purcell SM, DeMarais AA, McGrew LL, Moon RT. Activities of the Wnt-1 class of secreted signaling factors are antagonized by the Wnt-5A class and by a dominant negative cadherin in early Xenopusdevelopment. J Cell Biol. 1996;133:1123–1137. doi: 10.1083/jcb.133.5.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Wetering M, Cavallo R, Dooijes D, van Beest M, van Es J, Loureiro J, Ypma A, Hursh D, Jones T, Bejsovec A, et al. Armadillo coactivates transcription driven by the product of the Drosophila segment polarity gene dTCF. . Cell. 1997;88:789–799. doi: 10.1016/s0092-8674(00)81925-x. [DOI] [PubMed] [Google Scholar]
- Wen W, Harootunian AT, Adams SR, Feramisco J, Tsien R, Meinkoth JL, Taylor SS. Heat-stable inhibitors of cAMP-dependent protein kinase carry a nuclear export signal. J Biol Chem. 1994;269:32214–32220. [PubMed] [Google Scholar]
- Wen W, Meinkoth JL, Tsien RY, Taylor SS. Identification of a signal for rapid export of proteins from the nucleus. Cell. 1995;82:463–473. doi: 10.1016/0092-8674(95)90435-2. [DOI] [PubMed] [Google Scholar]
- Yang-Snyder J, Miller JR, Brown JD, Lai C-J, Moon RT. A frizzledhomolog functions in a vertebrate Wnt signaling pathway. Curr Biol. 1996;6:1302–1306. doi: 10.1016/s0960-9822(02)70716-1. [DOI] [PubMed] [Google Scholar]
- Yost C, Torres M, Miller JR, Huang E, Kimelman D, Moon RT. The axis-inducing activity, stability, and subcellular distribution of β-catenin is regulated in Xenopusembryos by glycogen synthase kinase 3. Genes Dev. 1996;10:1443–1454. doi: 10.1101/gad.10.12.1443. [DOI] [PubMed] [Google Scholar]