

Abstract
Tenascin-C (TN-C) is induced in pulmonary vascular disease, where it colocalizes with proliferating smooth muscle cells (SMCs) and epidermal growth factor (EGF). Furthermore, cultured SMCs require TN-C for EGF-dependent growth on type I collagen. In this study, we explore the regulation and function of TN-C in SMCs. We show that a matix metalloproteinase (MMP) inhibitor (GM6001) suppresses SMC TN-C expression on native collagen, whereas denatured collagen promotes TN-C expression in a β3 integrin– dependent manner, independent of MMPs. Floating type I collagen gel also suppresses SMC MMP activity and TN-C protein synthesis and induces apoptosis, in the presence of EGF. Addition of exogenous TN-C to SMCs on floating collagen, or to SMCs treated with GM6001, restores the EGF growth response and “rescues” cells from apoptosis. The mechanism by which TN-C facilitates EGF-dependent survival and growth was then investigated. We show that TN-C interactions with αvβ3 integrins modify SMC shape, and EGF- dependent growth. These features are associated with redistribution of filamentous actin to focal adhesion complexes, which colocalize with clusters of EGF-Rs, tyrosine-phosphorylated proteins, and increased activation of EGF-Rs after addition of EGF. Cross-linking SMC β3 integrins replicates the effect of TN-C on EGF-R clustering and tyrosine phosphorylation. Together, these studies represent a functional paradigm for ECM-dependent cell survival whereby MMPs upregulate TN-C by generating β3 integrin ligands in type I collagen. In turn, αvβ3 interactions with TN-C alter SMC shape and increase EGF-R clustering and EGF-dependent growth. Conversely, suppression of MMPs downregulates TN-C and induces apoptosis.
Tenascin-c (TN-C)1 is an extracellular matrix (ECM) glycoprotein that is prominent in embryonic and adult tissues that are actively remodeling (Chiquet-Ehrismann et al., 1995), where it is often coexpressed with matrix metalloproteinases (MMPs), a family of zinc-dependent ECM-degrading enzymes (Hedin et al., 1991; Talhouk et al., 1991; Jones et al., 1995; Latijnhouwers et al., 1996; Vaalamo et al., 1996). For example, during postlactational involution of the mammary gland, increased levels of TN-C and stromelysin-1 directly contribute to the loss of tissue-specific gene expression (Sympson et al., 1994; Jones et al., 1995) and the onset of apoptosis (Boudreau et al., 1995a , 1996), which together characterize this stage of development (Strange et al., 1992). In mature blood vessels subjected to experimental balloon catheter injury, TN-C and MMP-2 expression are induced before the development of occlusive neointimal lesions (Mackie et al., 1992; Bendeck et al., 1994; Hedin et al., 1991; Zempo et al., 1994; Strauss et al., 1996), indicating that these proteins may also share a common function during vascular remodeling. In addition, since TN-C and MMP-2 each have the capacity to bind the αvβ3 integrin (Prieto et al., 1993; Sriramarao et al., 1993; Brooks et al., 1996) and their expression is sensitive to treatments that alter the integrity of the actin cytoskeleton (Aggeler et al., 1984; Chiquet-Ehrismann et al., 1994), it is possible that these proteins are regulated in an interdependent manner. Indeed, studies by Tremble et al. (1994) have demonstrated that TN-C–supplemented fibronectin matrices stimulate MMP expression in cultured rabbit synovial fibroblasts. Evidence that MMPs may regulate TN-C in a reciprocal manner, however, has not been forthcoming.
A functional role for TN-C in modulating vascular smooth muscle cell (SMC) proliferation has been suggested by studies demonstrating increased TN-C synthesis after wounding injury in vivo (Hedin et al., 1991), or by treatment with vasoactive peptides and growth factors in tissue culture (Mackie et al., 1992; Sharifi et al., 1992). Our recent studies in rat and human hypertensive pulmonary arteries (Jones and Rabinovitch, 1996; Jones et al., 1997), as well as those in vessels from spontaneously hypertensive rats and human vein grafts (Hahn et al., 1995; Chen et al., 1996), have shown that TN-C colocalizes with proliferating SMCs, as well as EGF (Jones et al., 1997). An additional link between TN-C expression and growth factor– dependent SMC proliferation was also provided by tissue culture studies that demonstrated that TN-C potentiates bFGF-dependent SMC growth and is critical for EGF-dependent SMC growth on type I collagen (Jones and Rabinovitch, 1996). Similarly, End et al. (1992) reported that TN-C collaborates with EGF to promote growth of several cell types, including fibroblasts and SMCs isolated from spontaneously hypertensive rats.
Although the mechanism by which TN-C cooperates with soluble growth factors to generate a mitogenic response in normal SMCs is not understood, recent studies with other cell types show that integrin and growth factor receptor signaling components may accumulate on the actin-based cytoskeletal scaffold to form a specialized focal adhesion complex (Miyamato et al., 1995a ,b; Plopper et al., 1995), where they collaborate to regulate activation of mitogen-activated protein kinases and receptor tyrosine kinases, including the EGF receptor (EGF-R) (Miyamoto et al., 1996). Mechanistically, epidermal growth factor receptors have been shown to directly interact with actin (den Hartigh et al., 1992), and from a functional standpoint, this association greatly enhances activation of the EGF-R and its downstream substrates (Diakonova et al., 1995; Gronowski and Bertics, 1995). Since TN-C has been shown to alter the integrity of the actin cytoskeleton (Murphy-Ullrich et al., 1991; Chung et al., 1996), it is therefore possible that this ECM component modulates EGF-R signaling through its effects on the actin cytoskeleton and associated focal adhesion components.
In this study, we first investigated the relationships between MMPs, TN-C, and EGF-dependent SMC survival. Using SMCs cultured on attached and floating collagen gels as a model system, we show that net MMP activity, TN-C expression, and SMC survival are suppressed on floating collagen gels. Furthermore, we demonstrate that β3 integrins support basal levels of TN-C expression on attached native type I collagen and that a specific inhibitor of MMPs (GM6001) suppresses this expression and limits cell survival. Moreover, addition of exogenous TN-C to SMC cultures in which MMP activity and TN-C production have been suppressed supports SMC survival and EGF-dependent growth. Culturing SMCs on denatured collagen obviates the requirement for MMPs and directly promotes TN-C expression in a β3 integrin–dependent manner. We next explored the mechanism by which TN-C facilitates this effect, and we show that TN-C binding to the αvβ3 integrin supports EGF-dependent SMC growth, and that this interaction leads to changes in cell morphology underscored by the reorganization of the actin cytoskeleton. This structural alteration is accompanied by clustering of EGF-Rs and accumulation of tyrosine-phosphorylated proteins at the cell periphery within focal adhesion complexes. Addition of EGF to these physically and biochemically primed cells leads to increased SMC EGF-R activation and growth. Together, these data provide a functional paradigm for the regulation of TN-C by MMPs in actively restructuring tissues and for how this ECM component interacts with β3 integrins to promote EGF-dependent growth and survival.
MATERIALS AND METHODS
Cell Culture
Vascular SMCs were isolated from pulmonary arteries of 300-g adult male Sprague-Dawley rats. Briefly, arteries were harvested from anaesthetized animals and placed in cold sterile PBS, pH 7.6. Endothelium was removed by gently scraping the lumenal surface with a scalpel blade, and the adventitia was also removed from the vessel. The medial layer was minced using scalpel blades and incubated at 37°C in medium 199 (M199; GIBCO BRL, Gaithersburg, MD) supplemented with 0.1% collagenase I (Sigma Chemical Co., St. Louis, MO.), and 0.1% BSA (Boehringer-Mannheim, Laval Canada) for 1 h with gentle rotation. Tissue was collected by centrifugation at 300 g, resuspended in M199/collagenase I solution, and incubated overnight at 37°C with gentle rotation. Smooth muscle cells were harvested by centrifugation and were routinely maintained in M199 containing 10% heat-inactivated FBS (Intergen, Purchase, NY), 10 U/ml penicillin G sodium, 10 mg/ml streptomycin sulfate, 0.25 mg/ml amphotericin B, and 0.1 mg/ml gentamicin sulfate (GIBCO BRL). Cells were passaged by trypsinization using 0.05% trypsin/EDTA (GIBCO BRL). Vascular SMCs were identified by their characteristic hills-and-valley morphology and immunohistochemical staining for α-smooth muscle actin.
Collagen gels were prepared based on methods that have been previously described (Jones and Rabinovitch., 1996). Briefly, 0.8 ml of a 3.1 mg/ ml solution of bovine dermal type I collagen (Vitrogen 100; Collagen Corp., CA), 0.1 ml of 0.1 M NaOH, and 0.1 ml of 10× PBS were mixed at 4°C for a final collagen concentration of 2.48 mg/ml. To determine the effect of TN-C on SMC behavior, neutralized collagen was supplemented with 15 μg/ml of human TN-C protein (Chemicon International, Temecula, CA). Fibrillogenesis was initiated overnight in a humid 5% CO2 environment at 37°C, and the gels were rinsed extensively (three times for 3 h) with M199 containing 0.1% BSA before use. Confluent cultures of rat PA SMCs were serum starved in M199/0.1% BSA for 48 h. Smooth muscle cells were collected by trypsinization and centrifugation, and cell number was determined using an improved Neubauer hemocytometer (American Optical, Buffalo, NY). Cell pellets were resuspended in M199/0.1% BSA plus 0.5% serum, and a 1-ml aliquot of SMCs was seeded onto the surface of each gel. 6 h later, cells were rinsed in M199 and were thereafter cultured in M199/0.1% BSA.
Smooth Muscle Growth on Collagen Gels
We used collagen gels and heat-denatured collagen to determine the interrelationships between TN-C, MMP activity, β3 integrins, and EGF-dependent SMC survival and growth. For these experiments, 2 × 104 cells were plated on collagen gels as described above. For anti-αvβ3 antibody blocking studies, SMCs were plated on collagen gels in the presence of 15 μg/ml LM609 monoclonal antibody (a kind gift from Dr. David Cheresh, Scripps Research Institute, La Jolla, CA) or with 15 μg/ml of control IgG (DAKO Corp., Carpenteria, CA). For β3 integrin studies on native and denatured collagen, SMCs were plated in the presence of 25 μg/ml anti-β3 monoclonal antibody (PharMingen, San Diego, CA) or with 25 μg/ml of control IgG (DAKO Corp.). For MMP inhibitor studies, 1–2 μm GM6001 (a kind gift from Dr. Stu Sweidler, Glycomed, Alameda, CA) in DMSO, or an equivalent volume of DMSO alone, was added to cultures 24 h after plating. GM6001 (N- [2 (R) - 2 - ( hydroxamidocarbonylmethyl ) - 4 - methylpentanoyl] - l - tryptophane methylamide) is a noncytotoxic synthetic inhibitor that is specific for MMPs. The hydroxamic group of GM6001 binds to the critical active site zinc atom present in MMPs. In addition, the isobutyl group and tryptophan side chain of GM6001 also binds to subsites on MMPs, which normally bind side chains of ECM proteins (Galardy, 1993; Boudreau et al., 1995a ; Strauss et al., 1996). For assessment of the effects of heat denatured collagen, cells were plated on 50 μg of type I collagen/35-mm-diam dish that had been boiled for 30 min with 0.02 M acetic acid, and then air dried to the bottom of each dish. Before plating cells, collagen substrates were rinsed extensively, (three times for 1 h) with M199/0.1% BSA. To “float” the collagen gels, we detached them from the plastic substratum 24 h after plating using a spatula. 24 h after plating, cells were cultured in M199/ 0.1% BSA either with or without 50 ng/ml EGF (GIBCO BRL). The number of cells retained on collagen gels was determined 72 h after plating. The dose of EGF chosen to be stimulatory to SMC growth was determined in pilot studies and previously published by our group (Jones and Rabinovitch, 1996). All experiments were performed in triplicate. Attachment efficiencies were determined by counting the number of cells in the medium using a hemocytometer or Coulter counter. The number of attached cells (number of cells seeded − number of cells in the medium) was expressed as a percentage of the number of cells seeded. To assess the number of cells retained on collagen, gels were digested with 1 mg/ml collagenase type II (Sigma Chemical Co.) for 1 h at 37°C. Free cells were pelleted by centrifugation at 4°C for 10 min at 300 g and suspended in PBS containing 0.05% trypsin, and cell number was determined by counting aliquots in triplicate using a hemocytometer.
Detection of Matrix Metalloproteinases
Conditioned medium was collected from cells maintained on attached or floating collagen gels for 4, 8, 12, and 24 h in M199 with 50 ng/ml EGF. For each culture and at every time point, SMCs were rinsed gently three times with culture medium and then reincubated with 1 ml of fresh medium. At the designated time points, conditioned medium was harvested from the cultures, and equal volumes of medium were lyophilized overnight. Conditioned medium was mixed 3:1 (vol/vol) with 4× sample buffer (10% SDS, 25% glycerol in 150 mM Tris-Cl, pH 6.8) and separated on nonreducing 10% polyacrylamide gels that contained 0.1% gelatin (Sigma Chemical Co.). After electrophoresis, gels were soaked in 2.5% Triton X-100 to remove SDS and incubated in substrate buffer (50 mM Tris-Cl, pH 7.5, 5 mM CaCl2) for 18 h at 37°C. The gels were then stained with Coomassie blue R250 (Bio-Rad Labs, Hercules, CA). Gelatinases appear as a clear zone on a blue background. To demonstrate the ability of GM6001 to inhibit MMPs, we incubated gelatin zymograms in substrate buffer containing 2 μM of this inhibitor. This treatment abolished MMP activity. For detection of the pro-form of MMP-2, 20 μg of total protein contained in conditioned medium harvested from SMCs cultured on attached and floating collagen for 24 h in serum-free medium (SFM) was subjected to SDS-PAGE electrophoresis. Proteins were electrophoretically transferred to nylon membranes, blocked for 1 h at 37°C in wash buffer (10 mM Tris, pH 7.5, 10 mM NaCl, and 0.1% Tween-20), containing 0.1% BSA and 1% normal goat serum (Sigma Chemical Co.), and then with an anti–MMP-2 rabbit polyclonal antisera (a kind gift from Drs. M. Silverman and M. Ailenburg, Department of Medicine, University of Toronto, Toronto, Canada) diluted 1:200 in wash buffer for 30 min at 37°C. To detect MMP-2, membranes were then incubated with a horseradish peroxidase–conjugated goat anti–rabbit secondary antibody (GIBCO BRL) diluted 1:3,000 in wash buffer, rinsed in wash buffer three times for 15 min, visualized by enhanced chemiluminescence (ECL kit; Amershan Corp., Arlington Heights, IL), and exposed to film (X-Omat; Kodak, Rochester, NY).
Immunofluorescent Detection of Phosphorylated Proteins, Actin, Epidermal Growth Factor Receptors, and Vinculin
For immunofluorescent studies, serum starved SMCs were cultured on glass coverslips (12 mm, No.1 thickness) coated with 2.48 mg/ml of neutralized type I collagen (+/− 15 μg/ml TN) in M199/0.1% BSA. For actin, EGF-Rs, and tyrosine-phosphorylated proteins, cells were exposed to 50 ng/ml EGF in M199/0.1% BSA for 30 min or subjected to control treatment with M199/0.1% BSA alone. To determine the effects of blocking the β3 integrin on EGF-R distribution, SMCs were preincubated with 25 μg/ml anti–rat β3 integrin monoclonal antibody (CD61; PharMingen) before plating on collagen- and TN-C–enriched substrates. At the appropriate time point, cells were rinsed in PBS and then fixed. For detection of F-actin, SMCs were fixed with extraction buffer (60 mM Pipes, 25 mM Hepes, 10 mM EDTA, 2 mM MgCl2, pH 6.9) supplemented with 0.2% Triton X-100 for 5 min at 4°C. Smooth muscle cells were then washed three more times for 5 min in extraction buffer without Triton X-100. Fixed cells were incubated with 2.5 U of rhodamine-phalloidin (Molecular Probes, OR) in PBS for 20 min at 37°C before rinsing briefly with PBS. To detect EGF-Rs, tyrosine-phosphorylated proteins, and vinculin, SMCs were fixed in 2% paraformaldehyde (Sigma Chemical Co.) for 10 min at room temperature and rinsed in PBS containing 0.1 M glycine for 30 min. For tyrosine-phosphorylated proteins and vinculin, SMCs were permeabilized with 0.2% Triton X-100 in PBS for 5 min and washed three more times. The fixed cells were incubated for 1 h at 37°C in wash buffer (PBS/ 1% BSA), supplemented with 10% normal goat serum for EGF-Rs, and then incubated for 1 h at 37°C with anti-EGF-R antisera (RK-2, diluted 1:100; a kind gift from Dr. Irit Lax, New York University Medical Center, New York), antiphosphotyrosine monoclonal antibody (clone 4G10; 5 μg/ ml; Upstate Biotechnology Inc., Lake Placid, NY), antivinculin monoclonal antibody (diluted 1:50; Sigma Chemical Co.), or appropriate preimmune sera and IgG controls in wash buffer. After being washed three times, coverslips were incubated for 1 h at 37°C either with fluorescein-conjugated goat anti–rabbit antibody (diluted 1:100; Sigma Chemical Co.) or with goat anti–mouse antibody (diluted 1:100; Sigma Chemical Co.). For double immunofluorescent staining experiments, all cells were fixed in 2% paraformaldehyde. For vinculin and EGF-Rs, cells were permeabilized in methanol for 2 min and were then air dried for 1 h at room temperature. For vinculin, tyrosine-phosphorylated proteins, and F-actin, fixed cells were washed in PBS containing 0.2% Triton X-100. All cells were then washed with PBS/0.1 M glycine and were sequentially incubated with appropriate antisera, antibodies, or rhodamine-phalloidin as described above. After washing, all coverslips were mounted onto glass slides using Antifade reagent (Molecular Probes). Observations and photomicrographs were obtained with a fluorescent microscope (Olympus Corp., Lake Success, NY) using epifluorescence.
Biochemical Analysis of Tyrosine-phosphorylated Proteins, Tenascin-C, and Epidermal Growth Factor Receptors
Tyrosine-phosphorylated proteins were analyzed by immunoblotting whole cell lysates, SMC membrane–enriched protein fractions, or EGF-R immunoprecipitates with a horseradish peroxidase–conjugated antiphosphotyrosine (RC20H; Transduction Labs, Lexington, KY). 4 × 106 SMCs cultured for 24 h on 10-cm-diam collagen gels (+/− TN-C) were treated with EGF (50 ng/ml) for 10 and 30 min or were further incubated in M199/ 0.1% BSA alone. Tenascin-C protein expression on collagen gels was assessed by immunoblotting equal numbers (2 × 104) of cell lysates with an anti–chicken TN-C rabbit polyclonal antisera (pK7; gift from Dr. M. Schachner, Swiss Federal Institute of Technology, Zurich, Switzerland). To determine the effect of the β3 integrin on TN-C–dependent EGF-R phosphorylation, SMCs were cultured on TN-C–supplemented collagen gels with 25 μg/ml of anti–rat β3 integrin monoclonal antibody (CD61; PharMingen) or with control IgG (DAKO Corp.).
To enrich for membrane proteins, SMCs were harvested at the appropriate time points in ice-cold buffer containing 50 mM Hepes, 1 mM benzamidine, 1 mM PMSF, 1 μg/ml aprotinin, 5 μg/ml leupeptin, 2 mM NaF, 2 mM Na3VO4, 1 mM MgCl2, and 0.25 M sucrose, pH 7.4 at 4°C. Cells were homogenized with a Dounce B glass homogenizer (60 strokes), and intact cells and nuclei were sedimented by centrifugation at 3,000 g for 5 min at 4°C. The resulting supernatants were centrifuged at 100,000 g for 1 h at 4°C, and the membrane-enriched pellet was solubilized by sonication in homogenization buffer supplemented with 1% Triton X-100. Protein content was determined using the Bio-Rad protein assay according to the manufacturer's instructions. For EGF-R immunoprecipitation experiments, 15 μg of membrane-enriched protein, resuspended in 0.5 ml RIPA lysis buffer (25 mM Tris, pH 7.2, 0.1% SDS, 1% Triton X-100, 1% sodium deoxycholate, 150 mM NaCl, and 1 mM EDTA) containing 1 mM benzamidine, 1 mM PMSF, 1 μg/ml aprotinin, 5 μg/ml leupeptin, 2 mM NaF, and 2 mM Na3VO4 was precleared with normal rabbit IgG and protein A–Sepharose (Sigma Chemical Co.) for 1 h at 4°C. Supernatants were then incubated for 1 h at 4°C with the rabbit polyclonal anti–EGF-R antibody diluted 1:400 in RIPA buffer and precipitated with protein A–Sepharose. Pellets were washed three times with ice-cold RIPA buffer and once in 50 mM Tris, pH 7.4. Membrane fractions (15 μg) and EGF-R immunoprecipitates were boiled in reducing sample buffer containing 0.2 M Tris-HCl, pH 8.8, 18% glycerol, 4% SDS, 0.01% bromophenol blue, and 10% β-mercaptoethanol. All samples were separated in 7% SDS–polyacrylamide gels. Proteins were transferred to Immobilon polyvinyl fluoride membranes (Millipore Corp., Milford, MA), and these were blocked for 1 h at 37°C in wash buffer (10 mM Tris, pH 7.5, 10 mM NaCl, and 0.1% Tween-20), supplemented with 0.1% BSA. To detect tyrosine-phosphorylated proteins, blots were incubated for 30 min with horseradish peroxidase–conjugated antiphosphotyrosine antibody, diluted 1:2,500 in wash buffer. To detect TN-C protein, membranes were sequentially incubated with anti–TN-C antisera and a horseradish peroxidase–conjugated goat anti–rabbit secondary antibody (GIBCO BRL) diluted 1:5,000 in wash buffer. Thereafter, all membranes were rinsed in wash buffer three times for 15 min, and tyrosine-phosphorylated and TN-C proteins were visualized by enhanced chemiluminescence (Amersham ECL kit) before exposure to film (X-Omat; Kodak).
β3 Integrin Cross-Linking Studies
To cluster β3 integrins, SMCs were first cultured on collagen gels and were then washed gently with medium and incubated with 150 μg/ml anti-β3 monoclonal antibody (PharMingen) diluted in M199/0.1% BSA for 60 min. The SMCs were then rinsed with medium and incubated either with 20 μg/ml of goat anti–mouse IgG F(ab′)2 fragment (ICN Biomedicals, Inc., Costa Mesa, CA) or with medium alone for 60 min at 37°C. For tyrosine-phosphorylation studies, SMCs were rinsed in medium and treated with 50 ng/ml EGF for 10 or 30 min. Control cultures were maintained in M199/0.1% BSA. Epidermal growth factor receptor immunoprecipitates were analyzed by immunoblotting with antiphosphotyrosine antibodies as described above.
In these studies, we also used immunofluorescence to demonstrate the clustering of β3 integrins. Accordingly, SMCs were rinsed in PBS and fixed in acetone at −20°C for 1 min. Cells were rehydrated in PBS, blocked in 1% PBSA for 1 h at 37°C, and incubated with a fluorescein-conjugated goat anti–mouse antibody diluted 1:100 in 1% PBSA for 30 min at 37°C, before being washed and visualized by epifluorescence. In these experiments, EGF-R distribution was also examined by immunofluorescence as described above.
Immunoprecipitation of Epidermal Growth Factor Receptors and Tenascin-C Protein
For immunoprecipitation of radiolabeled EGF-R and TN-C proteins, 2.5 × 104 serum-starved SMCs were cultured for 24 h on collagen gels in 35-mm-diam tissue culture dishes. To determine the effect of TN-C on EGF-R synthesis, collagen gels were supplemented with 15 μg/ml of TN-C. 24 h after plating, SMCs were rinsed twice and incubated in cysteine/methionine-free medium (ICN Biomedicals, Inc.) supplemented with 0.1% BSA for 3 h and then pulse-labeled with 100 μCi/ml [35S]methionine/cysteine translabel (ICN Biomedicals, Inc.) for 18 h. To determine the effects of MMP inhibition on TN-C protein synthesis, cells were treated with 1- or 2-μm GM6001 in DMSO or with DMSO alone. Tissue culture medium was collected in RIPA buffer and cleared by centrifugation. Total counts per minute were normalized by TCA precipitation and equal counts per minute (5 × 105 cpm/sample) were immunoprecipitated with 5 μl rabbit polyclonal antisera raised against rat EGF-Rs (RK-2) or human TN-C (GIBCO BRL). Immune complexes were precipitated with 25 μl protein A–Sepharose (Sigma Chemical Co.), washed in 1× RIPA buffer, 50 mM Tris, pH 7.4, and resuspended in 40 μl 2× SDS-PAGE sample buffer. Proteins were separated on 6% SDS–polyacrylamide gels, fixed, dried, and exposed to film (X-Omat; Kodak) for 3 d.
Apoptosis Assay
Internucleosomal DNA fragmentation is a characteristic of apoptosis and was used to assess programmed cell death in SMCs cultured on attached and floating type I collagen gels (+/− TN-C). 48 h after floating, cells were rinsed in PBS and incubated on ice with DNA lysis buffer (10 mM Tris, pH 8, 100 mM NaCl, 2 mM EDTA, and 0.5% SDS) for 5 min with rotation. Lysates were incubated with 1 μg/ml proteinase-K (Boehringer-Mannheim Corp.) at 37°C for 24 h with rotation. Genomic DNA was purified by sequential extraction with phenol/chloroform and was precipitated overnight in ethanol at −20°C. DNA pellets were treated with 10 μg/ml RNase A for 1 h at room temperature, and DNA was then quantified based on optical density. 10 μg of genomic DNA was analyzed by electrophoresis through 1.2% agarose gels and visualized with ethidium bromide.
Statistical Analyses
All statistical assessments were compared by one-way analysis of variance and Student Newman Keuls post hoc analysis. A P value of <0.05 was considered statistically significant. Mean values ±SEM are given in the figures.
RESULTS
Matrix Metalloproteinases and Tenascin-C Are Coordinately Expressed in Vascular Smooth Muscle Cells
When chick embryo fibroblasts are cultured on attached type I collagen gels, they express TN-C protein, whereas culture on floating collagen leads to suppression of TN-C expression (Chiquet-Ehrismann et al., 1994). Using this culture method as a model system, we first compared SMC morphology, TN-C production, and MMP activity and expression on attached and floating collagen gels. Within the first few hours of releasing attached collagen gels into the culture medium, SMCs lost their elongated morphology and became rounded (Fig. 1 A). Immunoprecipitation of radiolabeled TN-C protein from SMC conditioned medium after 24 h (Fig. 1 B) showed that synthesis of two TN-C protein isoforms, of apparent molecular masses 220 and 180 kD, was suppressed in SMCs cultured on floating collagen when compared to those cultured on attached collagen gels. No significant differences between the total number of TCA precipitable counts were noted between cells cultured on attached (1.10 × 106 cpm/culture, SEM ± 9.6 × 104, P < 0.05) versus floating collagen (1.13 × 106 cpm/culture, SEM ± 1.59 × 104, P < 0.05), indicating that despite suppression of TN-C protein synthesis, general protein synthesis at this time point was unaffected.
Figure 1.


Tenascin-C and matrix metalloproteinase-2 (MMP-2) expression in vascular smooth muscle cells. (A) Representative photomicrograph showing SMC morphology on attached type I collagen gels and on collagen gels that have been released for 2 h into the culture medium. (B) Autoradiograph (representative of two different experiments) showing immunoprecipitated TN protein from [35S]methionine/cysteine-labeled SMC lysates harvested from attached and floating type I collagen gels in SFM with 50 ng/ml EGF at 24 h. Synthesis of two TN-C protein isoforms of apparent molecular masses 220 and 180 kD, is suppressed in cells cultured on floating collagen. (C) Gelatin zymography was used to examine the levels and activity of gelatinases present in conditioned medium (CM) harvested from SMCs cultured on attached and floating collagen. Fresh SFM with 50 ng/ml EGF was added for 4 h before its collection at 4, 8, 12, and 24 h. Equal volumes of concentrated CM were then analyzed by gelatin substrate zymography in nonreducing 10% polyacrylamide gels containing 0.1% gelatin. A zymogram (representative of three different experiments) shows that on attached and floating collagen, SMCs secrete gelatinases with apparent molecular masses of 68 and 57 kD, and that on floating collagen, the activity is lower. (D) Densitometry of the 68- and 57-kD gelatinases shown in C. (E) Western immunoblot for the latent form of MMP-2 in conditioned medium harvested at 24 h from SMCs cultured on attached and floating collagen shows that expression of the 72-kD proform of this enzyme (equivalent to the 68-kD species under nonreducing conditions on the zymogram) is suppressed under floating conditions. Bar, 120 μm.
The production and activity of MMPs under these conditions was then examined by gelatin substrate zymography. Two proteins with approximate molecular masses of 68 and 57 kD were detected in conditioned medium harvested from SMCs cultured on attached and floating collagen (Fig. 1 C). Densitometric analysis of these gelatinases showed that, relative to attached collagen, the activities of the 68- and 57-kD gelatinase decreased with time on floating collagen (Fig. 1 D). Under the denaturing and nonreducing conditions used for zymography, the pro-form of MMP-2 has an apparent molecular mass of 68 kD, whereas the active form appears at ∼57 kD (Ailenburg et al., 1993). Thus, our data indicate that the gelatinases detected by zymography may represent MMP-2. Their identity as MMPs was substantiated by the abolishment of gelatinase activity after incubation of zymograms with EDTA or GM6001 (data not shown) and Western immunoblot analysis, which showed that expression of the pro-form of MMP-2 (which migrates at 72 kD under denaturing and reducing conditions) is suppressed on floating collagen (Fig. 1 E).
Matrix Metalloproteinases and β3 Integrins Positively Regulate Smooth Muscle Cell Tenascin-C Production
Previous studies have shown that interstitial collagenases, including MMP-2, are capable of degrading fibrillar native type I collagen to generate collagen fragments (Aimes and Quigley, 1995). At a functional level, this leads to exposure of cryptic Arg-Gly-Asp (RGD) sites within the native type I collagen molecule that bind the αvβ3 integrin receptor to promote cell survival (Davis, 1992; Montgomery et al., 1994). This integrin effect may also be achieved by culturing cells on heat-denatured collagen (Montgomery et al., 1994). Since increased SMC TN-C expression correlates with increased MMP expression and activity, we hypothesized that degradation of native type I collagen by MMPs may upregulate SMC TN-C expression through β3 integrin ligation.
To first establish whether MMPs regulate TN-C expression, we cultured SMCs on attached type I collagen gels in the presence of a specific MMP inhibitor, GM6001 (Galardy, 1993; Strauss et al., 1996). Immunoprecipitation of TN-C from radiolabeled cell lysates showed that inhibition of MMP activity results in suppression of TN-C protein synthesis (Fig. 2 A). In addition, the ability of this inhibitor to suppress TN-C protein synthesis is not due to cytotoxicity since there were no significant differences between the total number of TCA precipitable counts in radiolabeled control (3.18 × 106 cpm/culture, SEM ± 2.12 × 105, P < 0.05) and GM6001-treated cultures (3.75 × 106 cpm/culture, SEM ± 1.63 × 105, P < 0.05) at the time point examined.
Figure 2.

Regulation of tenascin-C protein expression by matrix metalloproteinases and β3 integrins. (A) Immunoprecipitation of TN-C protein from [35S]methionine/cysteine-labeled SMCs cultured on native type I collagen gels in serum-free control medium with 50 ng/ml EGF and 0.4% DMSO, or in control medium supplemented with 1 or 2 μM of GM6001, an MMP inhibitor. Inhibition of MMP activity with GM6001 leads to a marked decrease in TN-C protein synthesis. Positions of molecular mass standards in kD are indicated on the left. (B) Western immunoblotting for TN-C protein from SMCs cultured in SFM with 50 ng/ml EGF on native and heat-denatured type I collagen gels. A Western immunoblot shows that expression of two TN-C isoforms of apparent molecular masses 220 and 180 kD is increased in cells cultured on denatured collagen. (C) Western immunoblot of TN-C protein from SMCs cultured on heat-denatured type I collagen gels in control medium supplemented with either 0.4% DMSO or 2 μM of GM6001. Inhibition of MMP activity with GM6001 had no effect on TN-C protein expression. (D) Effect of β3 integrin blockade on TN-C protein expression on heat-denatured collagen. Immunoprecipitation of TN-C protein from [35S]methionine/cysteine- labeled SMCs cultured on native type I collagen gels in the presence of control IgG or with anti-β3 antibody shows that blocking β3 integrins inhibits TN-C protein expression on native type I collagen. (E) Effect of β3 integrin blockade on TN-C protein expression on heat-denatured collagen. Western immunoblot analysis of lysates derived from cells cultured on heat-denatured collagen in the presence of control IgG, or with anti-β3 integrin antibody shows that blocking β3 integrins inhibits TN-C protein expression on denatured collagen. Arrows indicate the presence of the 220-kD (upper band) and 180-kD (lower band) TN-C isoforms.
We next assessed TN-C protein expression on native versus heat-denatured collagen. Western immunoblot analysis showed that TN-C protein expression is upregulated on denatured collagen (Fig. 2 B), and that this substrate obviated the need for MMPs (Fig. 2 C). To address whether TN-C protein expression is regulated by β3 integrins, we used a function-blocking anti-β3 antibody. Western immunoblots showed decreased TN-C protein expression in the presence of the anti-β3 antibody when compared with IgG-treated cultures on both native (Fig. 2 D) and proteolyzed collagen. (Fig. 2 E).
Matrix Metalloproteinases and Tenascin-C Promote Smooth Muscle Cell Survival
Since MMPs regulate SMC TN-C protein synthesis on native type I collagen, and exogenous TN-C cooperates with EGF to facilitate SMC growth on this substrate (Jones and Rabinovitch, 1996), we next determined the functional consequences of suppressing MMPs on EGF-dependent SMC morphology, growth, and survival, in the presence and absence of exogenously added TN-C. Inhibition of MMPs with GM6001 in SMCs cultured on attached collagen resulted in cellular rounding (data not shown) and a significant decline in SMC number, whereas inclusion of exogenous TN-C in the collagen gels prevented this decrease (Fig. 3 A) and restored their characteristic elongated morphology (data not shown). Similarly, on floating collagen, in which MMPs and TN-C expression are suppressed, SMC survival is diminished (Fig. 3 B) via apoptosis as determined by oligonucleosomal DNA fragmentation assays (Fig. 3 C). In contrast, SMC numbers were unaffected (Fig. 3 B), and apoptosis was suppressed in cells cultured on floating collagen gels supplemented with exogenous TN-C protein (Fig. 3 C).
Figure 3.

Matrix metalloproteinases and tenascin-C act as vascular smooth muscle cell survival factors. (A) Smooth muscle cells (2 × 104 cells per dish) plated on type I collagen were maintained in serum-free control medium with 0.4% DMSO and 50 ng/ml EGF, or in control medium supplemented with 1 or 2 μM of GM6001 and EGF. GM6001 treatment resulted in a significant decline in SMC numbers by 48 h, whereas addition of exogenous human TN-C protein (15 μg/ml) to attached collagen substrates inhibited this effect. (B) Effect of exogenous TN-C on SMC numbers on floating collagen gels. A significant decline in SMC numbers is apparent on floating compared to attached collagen gels (P < 0.05), whereas addition of TN-C suppresses this effect. Values shown in C and D represent mean ±SEM derived from three experiments. The asterisk denotes a P < 0.05 difference from cell numbers recorded on collagen gels in EGF-containing medium. (C) Effect of TN-C on SMC apoptosis on floating collagen gels. Smooth muscle cells plated on attached type I collagen gels (2 × 104 cells per dish) were maintained in SFM with EGF (50 ng/ml) for 48 h or were floated in the same medium, either with or without addition of exogenous human TN-C (15 mg/ml). Genomic DNA was isolated from each culture and 10 μg per sample was analyzed on 1% agarose gels. DNA fragments comprised of ∼180-bp multimers, which are indicative of apoptosis, were apparent on floating collagen gels. In contrast, no evidence of DNA fragmentation was observed on either attached or TN-C–supplemented floating collagen. Positions of a standardized DNA ladder in kb are indicated on the right.
Tenascin-C Collaborates with the αvβ3 Integrin to Promote Epidermal Growth Factor–dependent Smooth Muscle Cell Proliferation
Tenascin-C has the capacity to bind and interact with multiple cell surface receptors, including the αvβ3 integrin (Prieto et al., 1993; Sriramarao et al., 1993). To determine whether this integrin mediates TN-C/EGF–dependent SMC growth, we evaluated the effect of a functional-blocking anti-αvβ3 integrin monoclonal antibody (LM609) on cell morphology and proliferation of SMC cultured on collagen and TN-C–enriched substrates in the presence of EGF. While LM609 had no effect on the stellate SMC morphology observed on collagen, the more elongated SMC morphology induced by TN-C was attenuated by this antibody. Instead, SMCs failed to spread and remained rounded (Fig. 4 A). LM609 had no significant effect on SMC attachment to TN-C–enriched substrates (Fig. 4 B) or on cell number after culture of SMC on collagen in the presence of EGF (Fig. 4 C). In contrast, TN-C–dependent growth in response to EGF was inhibited by LM609 (Fig. 4 C).
Figure 4.


Effect of blocking αvβ3 integrins on tenascin- C–dependent smooth muscle cell morphology, attachment efficiency, and survival. (A) Representative phase contrast photomicrographs of SMCs plated in SFM on collagen and TN-C–supplemented collagen gels with control IgG (15 μg/ml) or with an anti–αvβ3 integrin antisera (LM609; 15 μg/ml). IgG and LM609 treatment had no effect on the stellate morphology produced by the collagen substrate. In contrast, the more elongated SMC morphology observed on control-treated TN-C–enriched substrates was abrogated by inclusion of LM609, which prevented cells from spreading, resulting in a rounded morphology. (B) Effect of LM609 antisera on SMC attachment to TN-C–supplemented type I collagen gels. By 6 h after plating, no significant differences in attachment efficiency were noted between cells plated in SFM on TN-C–supplemented collagen gels in either the presence of control IgG or LM609 antisera. (C) Effect of blocking SMC αvβ3 integrins with LM609 antisera on EGF-dependent SMC cell growth on TN-C–supplemented collagen gels. Control and LM609 treatment produced no differences in SMC number after culture for 48 h on collagen gels in SFM with 50 ng/ml EGF. The significant (P < 0.05) increase in SMC growth observed on TN-supplemented collagen gels in the presence of EGF was attenuated by LM609 antisera. Values represent mean ±SEM from three different experiments. (The asterisk denotes P < 0.05 difference from IgG control level on type I collagen.) Bar, 150 μm.
Tenascin-C Promotes Accumulation of F-actin, Tyrosine-phosphorylated Proteins, and Epidermal Growth Factor Receptors to the Focal Adhesion Complex
The effects of TN-C–αvβ3 integrin interactions on SMC shape and EGF-dependent proliferation provided us with the rationale for investigating the organization of the filamentous actin cytoskeleton and the distribution of EGF-Rs and tyrosine-phosphorylated proteins. Accordingly, we used immunofluorescence microscopy to evaluate the distribution of F-actin in SMCs cultured on collagen gels supplemented with exogenous TN-C, and we determined whether this pattern was further altered by EGF (Fig. 5 A). Rhodamine-phalloidin staining revealed a longitudinal distribution pattern of F-actin stress fibers in SMCs cultured on collagen, which became more cortical after addition of EGF. On TN-C–supplemented collagen gels, however, high-intensity F-actin staining was observed at the cell periphery, and this pattern intensified after addition of EGF.
Figure 5.
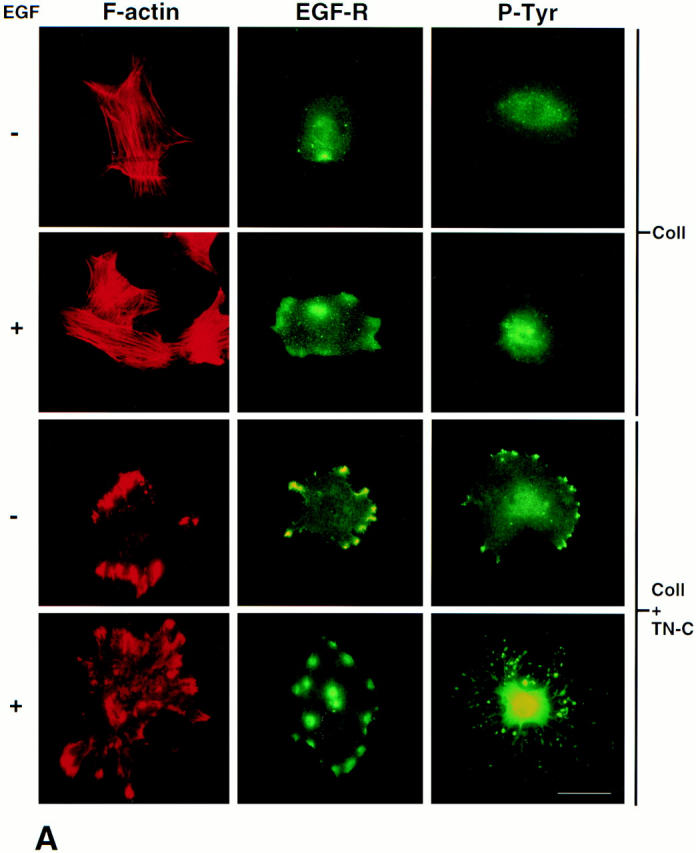
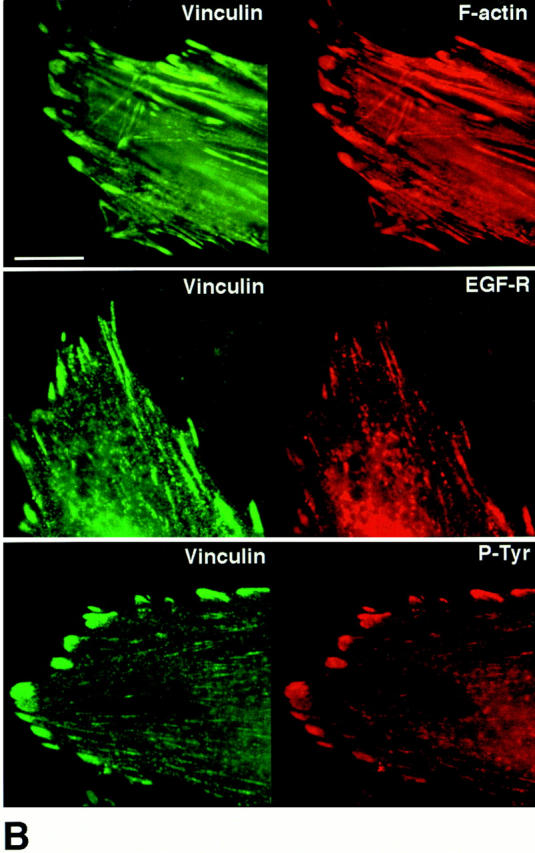
Tenascin-C modifies the patterns of distribution for filamentous actin, epidermal growth factor receptors, tyrosine-phosphorylated proteins, and vinculin. (A) Representative immunofluorescence photomicrographs (from two different experiments) showing distribution patterns for F-actin, EGF-Rs, and tyrosine-phosphorylated (P-Tyr) proteins in SMC cultured in SFM (+/- 50 ng/ml EGF for 30 min) on collagen alone or on TN-C (15 μg/ml) supplemented collagen substrates. Rhodamine-phalloidin staining of SMC on collagen revealed a longitudinal F-actin stress fiber pattern of distribution that was more cortical after treatment with EGF. Immunofluorescent staining for EGF-Rs and tyrosine-phosphorylated proteins was diffuse in SMC cultured on collagen alone and increased modestly after addition of EGF. In contrast, SMC cultured on TN-C–supplemented collagen gels showed high-intensity F-actin staining in regions that often overlapped with clusters of EGF-Rs and tyrosine-phosphorylated proteins. After addition of EGF to TN-C–treated cultures, the levels and distribution of EGF-Rs remained pronounced, and high levels of tyrosine-phosphorylated proteins were evident throughout the cell. (B) Representative double immunofluorescence photomicrographs showing codistribution of vinculin, F-actin, EGF-Rs, and tyrosine-phosphorylated (P-Tyr) proteins in SMC cultured in SFM on collagen supplemented with exogenous human TN-C protein. Bars: (A) 20 μm; (B) 5 μm.
Given that high-affinity EGF-Rs bind directly to actin (den Hartigh et al., 1992), we then assessed whether TN-C–dependent alterations in the organization of the F-actin cytoskeleton may be accompanied by EGF-R clustering, a prerequisite for EGF-dependent signaling, including ligand-dependent tyrosine phosphorylation (Heldin, 1995). Immunofluorescent studies revealed a diffuse pattern of EGF-R staining and tyrosine phosphorylation in SMCs cultured on collagen alone (Fig. 5 A). By contrast, in SMCs cultured on TN-C–enriched collagen gels, prominent EGF-R clustering was observed, especially at the cell periphery. After addition of EGF to collagen gels, a modest increase in both EGF-R clustering and tyrosine phosphorylation was apparent, whereas in TN-C–treated cultures, EGF-R clustering remained pronounced and high levels of tyrosine phosphorylation were evident within the nucleus and at a relatively lower level at the cell periphery. Furthermore, double immunostaining experiments with an antivinculin antibody established that TN-C–treated SMCs contained more focal adhesion complexes, which were larger than those produced on type I collagen alone (data not shown). Most strikingly, exogenous TN-C promoted accumulation of F-actin, EGF-Rs, and tyrosine-phosphorylated proteins within these focal adhesion sites (Fig. 5 B).
Tyrosine Phosphorylation of Epidermal Growth Factor Receptors Is Enhanced by Exogenous Tenascin-C
Having established that culturing SMCs on exogenous TN-C promotes EGF-R clustering, we next determined the effects of TN-C on EGF-R protein synthesis and tyrosine phosphorylation of this receptor. Immunoprecipitation of radiolabeled whole cell lysates with an anti–EGF-R antibody showed that the levels of receptor protein synthesis were identical on collagen and TN-C–supplemented gels (Fig. 6 A). Consistent with our immunofluorescent studies, Western immunoblotting of membrane-enriched fractions with an antiphosphotyrosine antibody revealed that there was increased tyrosine phosphorylation in the presence of exogenous TN-C (Fig. 6 B). In addition, on both type I collagen and TN-C–supplemented cultures, EGF treatment resulted in a transient increase in protein tyrosine phosphorylation that was enhanced by TN-C. Of particular interest was a 170-kD tyrosine-phosphorylated species that corresponds in molecular mass to the EGF-R. Also noted was an increase in an ∼125-kD species, which may represent focal adhesion kinase (Fig. 6 B). To specifically determine whether TN-C promotes EGF-R activation, EGF-Rs were immunoprecipitated from SMC membrane fractions, and their activation status was assessed using Western immunoblotting with an antiphosphotyrosine antibody. This confirmed that EGF-R phosphorylation occurs by 10 min after addition of EGF, and that this response is enhanced in TN-C–supplemented as opposed to SMC cultures on collagen gels alone (Fig. 6 C).
Figure 6.
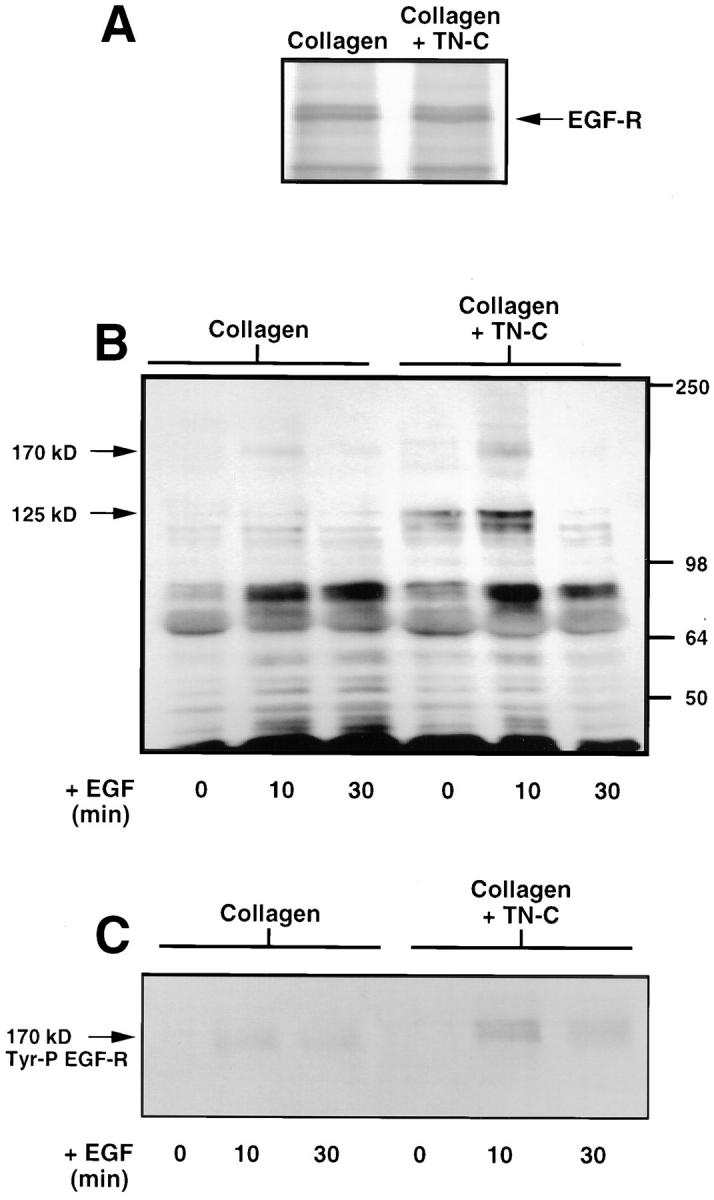
Tenascin-C potentiates ligand-dependent epidermal growth factor-receptor tyrosine phosphorylation. (A) Effect of TN-C on EGF-R protein synthesis. Representative autoradiograph (from two different experiments) showing immunoprecipitation of EGF-R protein from [35S]methionine-labeled SMCs cultured on native type I collagen gels (+/− 15 μg/ml TN-C) in SFM indicates that TN-C has no effect on the levels of EGF-R protein synthesis compared to SMCs cultured on collagen alone. (B) Effect of TN-C on tyrosine phosphorylation in SMCs cultured on collagen and TN-C–supplemented gels in SFM, or after addition of EGF (50 ng/ml) for 10 and 30 min. A Western immunoblot (representative of two different studies) was performed on membrane-enriched fractions (15 μg per sample) using an antibody against tyrosine-phosphorylated proteins. Note that relative to SMCs cultured on collagen alone, contact with exogenous TN-C led to an increase in basal levels of tyrosine phosphorylation. In response to EGF, qualitative and quantitative increases in tyrosine-phosphorylated proteins were observed on both collagen and TN-C, with changes being more pronounced on TN-C–enriched gels, including species at 170 and 125 kD. Positions of molecular mass standards in kD are indicated on the right. The 170-kD species may represent the EGF receptor, and the 125-kD species the focal adhesion kinase. (C) The 170-kD tyrosine-phosphorylated protein shown in B represents the EGF-R. Epidermal growth factor receptors were immunoprecipitated from 15 μg of membrane-enriched SMC fractions in cells cultured as in B. Epidermal growth factor immunoprecipitates were analyzed for evidence of tyrosine phosphorylation using Western immunoblotting with an antiphosphotyrosine antibody. After addition of EGF at 10 min, a transient increase in the levels of tyrosine-phosphorylated EGF-Rs occurs on collagen and TN-C–supplemented gels, with greater levels observed on TN-C–supplemented collagen substrates.
β3 Integrins Regulate Tenascin-C–dependent Epidermal Growth Factor Receptor Clustering and Ligand-dependent Tyrosine Phosphorylation
We next examined the effects of blocking TN-C–β3 integrin SMC interactions on EGF-R clustering and tyrosine phosphorylation. Smooth muscle cells were preincubated with an anti–β3 integrin antibody before plating on TN- C–supplemented collagen gels. As with the αvβ3 functional-blocking antibody experiments, the anti–β3 integrin antibody prevented cell spreading on TN-C (data not shown). We therefore used the anti–β3 integrin antibody in subsequent studies (although similar results would be expected with the αvβ3 antibody, LM609) and showed inhibition of TN-C–dependent EGF-R clustering (Fig. 7 A). Immunoprecipitation of SMC membrane–enriched fractions followed by Western immunoblotting with an antiphosphotyrosine antibody showed that upon addition of EGF, EGF-R activation on TN-C–enriched collagen substrates was abrogated in cells pretreated with the function-blocking anti–β3 integrin antibody (Fig. 7 B).
Figure 7.


The role of β3 integrins in tenascin-C–dependent epidermal growth factor receptor clustering and ligand-dependent receptor activation. (A) Representative immunofluorescence photomicrographs for EGF-R in SMCs cultured on TN-C (15 μg/ ml) supplemented collagen gels in the presence of control IgG or with an anti–β3 integrin antibody that prevented the formation of EGF-R clusters. (B) Effect of blocking β3 integrins on ligand-dependent EGF-R tyrosine phosphorylation in SMCs cultured on collagen gels supplemented with TN-C. Western immunoblot (representative of two different studies) of membrane preparations from SMCs treated with control IgG, with anti–β3 integrin antisera on TN-C–supplemented collagen in SFM alone, or in SFM with EGF (50 ng/ml) for 10 and 30 min. Epidermal growth factor receptors were immunoprecipitated from 15 μg of membrane-enriched SMC fractions and were analyzed for evidence of tyrosine phosphorylation using Western immunoblotting. Note that the anti–β3 integrin antibody prevents the transient increase in EGF-R tyrosine phosphorylation observed in TN-treated cultures after addition of EGF at 10 min. Bar, 17 μm.
β3 Integrin Cross-Linking Promotes Clustering and Activation of Epidermal Growth Factor Receptors
To determine whether clustering of β3 integrins on SMC surfaces, in the absence of exogenous TN-C, promotes EGF-R clustering and ligand-dependent phosphorylation of EGF-Rs, anti–β3 integrin antibody cross-linking studies were performed. Smooth muscle cells cultured on type I collagen gels were treated with either a combination of anti–β3 integrin and secondary goat anti–mouse IgG F(ab′)2 antibodies or with primary mouse monoclonal anti–β3 integrin antibody alone. Immunofluorescent microscopy revealed extensive β3 integrin clusters on SMC surfaces treated with primary and secondary antibodies, whereas a more diffuse pattern of β3 integrins was apparent in cultures treated with primary antibody alone (Fig. 8 A). Immunofluorescent microscopy with an anti–EGF-R antibody demonstrated that cross-linking the β3 integrin receptor leads to clustering of EGF-Rs, which appeared diffuse in SMCs treated with primary antibody alone (Fig. 8 A).
Figure 8.



Effect of cross-linking β3 integrins on epidermal growth factor receptor clustering. (A) Representative immunofluorescence micrographs for β3 integrins and EGF-Rs. Smooth muscle cells cultured on collagen substrates in SFM were preincubated for 60 min with an anti–β3 integrin antibody (150 μg/ml) and then for 60 min with SFM alone or with an anti-F(ab′)2 IgG (20 μg/ml) to promote cross-linking. Immunodetection of β3 integrins and EGF-Rs indicates that cross-linking β3 integrins promotes EGF-R clustering. (B) Effect of cross-linking β3 integrins on ligand-dependent tyrosine phosphorylation of EGF-Rs. Tyrosine phosphorylation of EGF-Rs in response to EGF was examined in SMC cultures treated with anti–β3 integrin antibody alone or in cultures incubated sequentially with an anti–β3 integrin antibody and anti-F(ab′)2 IgG. Epidermal growth factor receptor immunoprecipitates were analyzed by Western immunoblotting using an antiphosphotyrosine antibody, which indicated that cross-linking β3 integrins potentiates ligand-dependent EGF-R activation. (C) Densitometry of activated epidermal growth factor Western immunoblots (as shown in B) shows that a significant (*P < 0.05) increase in ligand-dependent tyrosine phosphorylation occurs after β3 integrin cross-linking. Values represent mean ±SEM from three different experiments. Bar, 25 μm.
To further assess the effect of β3 integrin–dependent EGF-R clustering on tyrosine phosphorylation of EGF-Rs, EGF was added to SMCs, which had been pretreated either with a combination of mouse anti–β3 integrin and secondary goat anti–mouse IgG F(ab′)2 antibodies, or with anti–β3 integrin antibody alone. Immunoprecipitation of EGF-Rs from SMC membrane fractions followed by Western immunoblotting with an antiphosphotyrosine antibody showed that a significant increase in EGF-dependent activation of EGF-Rs resulted from cross-linking β3 integrin receptors (Fig. 8, B and C).
DISCUSSION
A number of studies have described the induction of TN-C in remodeling vascular tissues, where it has been functionally linked to SMC proliferation (Hedin et al., 1991; Hahn et al., 1995; Chen et al., 1996; Jones and Rabinovitch, 1996; Jones et al., 1997). However, remarkably little was known about the nature of the factors that determine its expression in these tissues or the mechanism by which TN-C supports SMC growth. In this study, we demonstrate that MMPs positively regulate TN-C protein expression and that the mechanism likely involves exposure of cryptic β3 integrin–binding sites in type I collagen. We also show that TN-C rescues SMCs from apoptosis and promotes EGF-dependent SMC survival through interactions with the αvβ3 integrin receptor, which modulates cell shape and EGF-dependent tyrosine phosphorylation. These studies therefore provide new evidence to show how remodeling of the extracellular microenvironment by ECM-degrading proteinases may influence integrin growth factor receptor signaling functions and thus cell behavior. Fig. 9 shows a hypothetical model for the regulation and function of TN-C in SMCs based on our findings.
Figure 9.

Hypothetical model for the regulation and function of tenascin-C in vascular smooth muscle cells. (A) Vascular smooth muscle cells attach and spread on native type I collagen using β1 integrins. Under serum-free conditions, the cells withdraw from the cell cycle and become quiescent. (B) Degradation of native type I collagen by matrix metalloproteinases (MMPs) leads to the exposure of cryptic RGD sites that preferentially bind β3 subunit–containing integrins. In turn, occupancy and activation of β3 integrins signals the production of TN-C. (C) Incorporation of multivalent TN-C protein into the underlying substrate leads to further aggregation and activation of β3-containing integrins (αvβ3), and to the accumulation of tyrosine-phosphorylated (Tyr-P) signaling molecules and actin into a focal adhesion complex (FAC). Note that even in the absence of the EGF ligand, the TN-C–dependent reorganization of the cytoskeleton leads to clustering of actin-associated EGF-Rs. (D) Addition of EGF ligand to clustered EGF-Rs results in rapid and substantial tyrosine phosphorylation of the EGF-R and activation of downstream pathways culminating in the generation of nuclear signals leading to cell proliferation.
Recent studies by Chiquet-Ehrismann et al. (1994) demonstrated that chick embryo fibroblasts cultured on attached collagen gels express TN-C protein, whereas in floating cultures, TN-C synthesis is preferentially suppressed. This regulation occurs at the transcriptional level and was mapped to a putative “ECM response element” in the TN-C gene promoter that is distinct from the region important for induction by serum. However, the reason for this differential TN-C production on attached and floating collagen and the functional consequences of suppressing TN-C expression were not explored.
It is well established that specific cell–ECM interactions provide critical cues that regulate proliferation, migration, differentiation, and cell viability (Jones et al., 1993; Boudreau et al., 1995b ), and that these functions may depend upon proteolysis of ECM and cell surface molecules (Fujii and Imamaura, 1995; Brooks et al., 1996). Since MMPs and TN-C are often expressed at the same site within normal (Sympson et al., 1994; Jones et al., 1995) and injured remodeling tissues (Latijnhouwers et al., 1996; Vaalamo et al., 1996), including blood vessels (Bendeck et al., 1994; Strauss et al., 1996; Jones, P.L., J. Crack, and M. Rabinovitch. 1996. Mol. Biol. Cell. 7:418a), we examined the possibility that MMPs regulate SMC growth and viability through the induction of TN-C. Accordingly, we established a role for MMPs in regulating TN-C expression and SMC survival by showing that reducing MMP activity and expression on floating collagen gels, or with a specific MMP inhibitor, limits TN-C protein synthesis and SMC growth and survival.
Matrix metalloproteinase–mediated alterations in cell– ECM interactions have profound effects on receptor-mediated intracellular signaling and subsequent cell behavior (for review see Bausbaum and Werb, 1996). For example, MMP-2 exposes cryptic RGD ligands in native collagen, which may bind αvβ3 integrins to promote cell survival (Montgomery et al., 1994), an effect which can be mimicked by culturing cells on heat-denatured collagen (Davis, 1992; Montgomery et al., 1994). In the present study, we demonstrate that on native type I collagen, MMPs promote TN-C protein expression via β3 integrins, and using denatured collagen, we show that this substrate supports TN-C expression in a manner that still requires β3 integrins but is independent of MMPs. Thus, on attached type I collagen gels, activated MMPs may generate two RGD-containing ligands, i.e., denatured collagen and TN-C, and in this way, serve a dual function in cell survival. In addition, recent studies indicate that MMP-2 directly interacts with the αvβ3 integrin receptor to form a functional complex (Brooks et al., 1996). Since the appearance of TN-C expression within remodeling tissues is restricted in a precise spatial manner (Chiquet-Ehrismann et al., 1995), it is also tempting to speculate that induction of TN-C by MMPs serves to localize this ECM component with its cognate adhesion receptors, which include the αvβ3 integrin (Prieto et al., 1993; Sriramarao et al., 1993).
Considerable evidence demonstrates that regulation of cell shape and tissue organization by the ECM profoundly influences gene expression, differentiation, and growth behavior (Folkman and Moscona, 1978; Mooney et al., 1995; Ingber, 1993; Roskelley et al., 1994; Boudreau et al., 1996; Chen, 1997; Weaver et al., 1997). Our results show that suppression of endogenous TN-C occurs within a rounded SMC population, in which EGF-dependent growth and survival are limited, whereas addition of exogenous TN-C to these cultures is associated with the restoration of an elongated cell shape, the reestablishment of an EGF-dependent growth response, and the prevention of apoptosis. Also, using functional-blocking anti–β3 integrin antibodies, we show that EGF-R signaling is attenuated in SMCs that fail to spread on TN-C–enriched collagen substrates. These data strongly indicate that TN-C–dependent cell shape alterations may impact cell growth and survival functions in SMCs. Consistent with this idea, suppression of TN-C expression in mammary epithelial cells is associated with cellular rounding (Jones et al., 1995) and repression of growth-related genes including c-myc (Boudreau et al., 1996). Furthermore, we have shown that SMCs plated on nonadhesive substrates remain rounded and are unable to proliferate in response to EGF, whereas on tissue culture plastic, SMC spread and are able to grow (Jones, P.L., and M. Rabinovitch, unpublished observations).
Our studies comparing SMC growth and survival on attached and floating collagen gels add to previous work that suggests that mechanical factors are able to modulate growth factor–dependent functions, by relating this property to alterations in TN-C expression, and concomitant changes in cell shape and growth factor receptor activation. For example, fibroblasts in mechanically stressed collagen continue to proliferate in response to growth factors (Nakagawa et al., 1989), whereas cells in mechanically relaxed matrices become arrested in G0 and are less responsive to addition of serum or purified growth factors (Nishiyama et al., 1991). Mechanistically, autophosphorylation of PDGF and EGF-Rs in fibroblasts is decreased in mechanically relaxed collagen (Lin and Grinnell, 1993), whereas β1 integrin–dependent autophosphorylation of platelet derived growth factor (PDGF β) receptors is induced in fibroblasts in response to the application of external tension (Sundberg and Rubin, 1996). It is therefore possible that mechanical induction of TN-C and/or other ECM components might be important in the activation of a more extensive repertoire of growth factor receptors. Indeed, we have previously shown that TN-C cooperates with another soluble growth factor, bFGF, to facilitate SMC proliferation (Jones and Rabinovitch, 1996). Also, the cooperative effect between TN-C, EGF, and bFGF does not appear to be restricted to a single cell type, or cell surface receptor. Chung et al. (1996) showed that cell surface annexin II, which also acts as a TN-C receptor (Chung and Erickson, 1995), mediates TN-C/bFGF–dependent endothelial cell growth, whereas End et al. (1992) showed that TN-C and EGF stimulate proliferation of Swiss fibroblasts, NIH-3T3 cells and SMCs isolated from spontaneously hypertensive rats. Our present studies therefore confirm the idea that TN-C cooperates with soluble growth factors to promote cell proliferation and extend these studies by providing a mechanism for these cooperative effects that is based on ECM-dependent cell shape changes and cross-talk between integrins and growth factor signaling pathways.
Binding of EGF to its receptor leads to dimerization, which is essential to the subsequent activation of intrinsic tyrosine kinase activity (Schlessinger, 1988). In turn, EGF-R activation catalyzes the tyrosine phosphorylation of several protein substrates, including the receptor itself (Hunter and Cooper, 1988). In the present study, we show for the first time that the αvβ3 integrin mediates TN-C/EGF–dependent growth, and that this interaction leads to cell shape changes that are underscored by a redistribution of filamentous actin, clustering of EGF-Rs, and accumulation of tyrosine-phosphorylated proteins at the cell periphery within focal adhesion contacts, as well as increased activation of EGF-Rs after addition of EGF. Moreover, these TN-C–dependent effects on EGF-R clustering and tyrosine phosphorylation could be mimicked by cross-linking SMC β3 integrins. It is therefore possible that other ECM components that bind β3 integrins, e.g., vitronectin and proteolysed collagen, which have also been implicated in vascular pathobiology (Montgomery et al., 1994; Brooks et al., 1996), could function in a manner similar to TN-C. Indeed, a recent report shows that denatured monomeric collagen stimulates increased focal adhesion contact formation and PDGF-BB–dependent SMC proliferation (Koyama et al., 1996), whereas polymerized collagen inhibits growth by upregulating cyclin-dependent kinase inhibitors including p27kip1 and p21Cip1/Waf1.
Most integrins do not possess intrinsic kinase activity, and their ability to generate a signal after ligation to ECM appears to be dependent upon the formation of specialized structures comprised of multiple components, including clustered integrins, actin, growth factor receptor signaling molecules, and cytoskeletal-associated proteins such as vinculin and focal adhesion kinase (Miyamoto et al., 1995a; Plopper et al., 1995; Burridge and Chrzanowska-Woodnicka, 1996). In addition, a number of studies have provided evidence in favor of a direct role for the actin cytoskeleton in EGF-R signaling. For example, the EGF-R has been shown to be an actin-binding protein (den Hartigh et al., 1992), and from a functional standpoint, this association leads to the accumulation and activation of EGF-R substrates at the binding site (Gronowski and Bertics, 1993; Diakonova et al., 1995). However, the functional consequences of these types of interactions and the identity of cell surface receptors involved had not been explored. Our results strongly indicate that ligation of substrate-bound TN-C to SMC αvβ3 integrins leads to the formation of specialized cytoskeletal structures enriched with tyrosine-phosphorylated proteins that together favor the recruitment and clustering of actin-associated high-affinity EGF-Rs. Addition of EGF to these cells, which are now both physically and biochemically primed, would allow the generation of an optimal and ECM-specific mitogenic response.
In conclusion, our results represent a model for neointimal formation, whereby induction of MMPs upregulates TN-C expression, which in turn facilitates growth factor– dependent SMC proliferation. Based on the present studies, and our work that has linked increased TN-C expression and SMC proliferation in vivo (Jones and Rabinovitch, 1996; Jones et al., 1997), we suggest that TN-C and its receptors may be prime therapeutic targets for inhibiting SMC growth that is associated with vascular disease.
Acknowledgments
We would like to thank Drs. D. Cheresh, I. Lax, M. Silverman and M. Ailenburg, and Glycomed Inc. (GM6001 inhibitor) for providing reagents. We are also indebted to Claire Coulber, Joan Jowlabar, and Susy Taylor for their help in preparing this manuscript.
This study was supported by program grant T2229 from the Heart and Stroke Foundation of Canada.
Abbreviations used in this paper
- ECM
extracellular matrix
- EGF-R
epidermal growth factor receptor
- MMP
matrix metalloproteinase
- SFM
serum-free medium
- SMC
smooth muscle cell
- TN-C
tenascin-C
Footnotes
Address all correspondence to Marlene Rabinovitch, M.D., Division of Cardiovascular Research, The Hospital for Sick Children, 555 University Avenue, Toronto, Ontario, Canada MSG 1X8. Tel: (416) 813-5918. Fax: (416) 813-7480. e-mail: mr@sickkids.on.ca
REFERENCES
- Aimes RT, Quigley JP. Matrix metalloproteinase-2 is an interstitial collagenase. Inhibitor-free enzyme catalyzes the cleavage of collagen fibrils and soluble native type I collagen generating specific 3/4- and 1/4-length fragments. J Biol Chem. 1995;270:5872–5876. doi: 10.1074/jbc.270.11.5872. [DOI] [PubMed] [Google Scholar]
- Aggeler J, Frisch S, Werb Z. Changes in cell shape correlate with collagenase gene expression in rabbit synovial fibroblasts. J Cell Biol. 1984;98:1662–1671. doi: 10.1083/jcb.98.5.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ailenburg M, Weinstein T, Li I, Silverman M. Activation of procollagenase IV by cytochalasin D and concavalin A in cultured rat mesangial cells: linkage to cytoskeletal reorganization. J Am Soc Nephrol. 1993;4:1760–1770. doi: 10.1681/ASN.V4101760. [DOI] [PubMed] [Google Scholar]
- Bausbaum C, Werb Z. Focalized proteolysis: spatial and temporal regulation of extracellular matrix degradation at the cell surface. Curr Opin Cell Biol. 1996;8:731–738. doi: 10.1016/s0955-0674(96)80116-5. [DOI] [PubMed] [Google Scholar]
- Bendeck M, Zempo N, Clowes A, Galardy R, Reidy M. Smooth muscle cell migration and matrix metalloproteinase expression after arterial injury in the rat. Circ Res. 1994;75:539–545. doi: 10.1161/01.res.75.3.539. [DOI] [PubMed] [Google Scholar]
- Boudreau N, Sympson C, Werb Z, Bissell MJ. Suppression of ICE and apoptosis in mammary epithelial cells by extracellular matrix. Science (Wash DC) 1995a;267:891–893. doi: 10.1126/science.7531366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau N, Myers C, Bissell MJ. From laminin to lamin: regulation of tissue-specific gene expression by the ECM. Trends Cell Biol. 1995b;5:1–4. doi: 10.1016/s0962-8924(00)88924-2. [DOI] [PubMed] [Google Scholar]
- Boudreau N, Werb Z, Bissell MJ. Suppression of apoptosis by basement membrane requires three-dimensional tissue organization and withdrawal from the cell cycle. Proc Natl Acad Sci USA. 1996;93:3509–3513. doi: 10.1073/pnas.93.8.3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks P, Stromblad S, Sanders L, von Schalscha T, Aimes R, Stetler-Stevenson W, Quigley J, Cheresh D. Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with αvβ3 integrin. Cell. 1996;85:683–693. doi: 10.1016/s0092-8674(00)81235-0. [DOI] [PubMed] [Google Scholar]
- Burridge K., and M. Chrzanowska-Woodnicka. 1996. Focal adhesions, contractility and signaling. In The Cytoskeleton. James Spudich, editor. Annual Reviews Inc., Palo Alto, CA. 463–519. [DOI] [PubMed]
- Chen C, Ku D, Kikerki D, Lumsden A. Tenascin: a potential role in human arteriovenous PTFE graft failure. J Surg Res. 1996;60:409–416. doi: 10.1006/jsre.1996.0067. [DOI] [PubMed] [Google Scholar]
- Chen CS, Mrksich M, Huang S, Whitesides GM, Ingber DE. Geometric control of cell life and death. Science (Wash DC) 1997;276:1425–1428. doi: 10.1126/science.276.5317.1425. [DOI] [PubMed] [Google Scholar]
- Chiquet-Ehrismann R, Tannheimer M, Koch M, Brunner A, Spring J, Martin D, Baumgartner S, Chiquet M. Tenascin-C expression by fibroblasts is elevated in stressed collagen gels. J Cell Biol. 1994;127:2093–2101. doi: 10.1083/jcb.127.6.2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiquet-Ehrismann R, Hagios C, Schenk S. The complexity of regulating tenascins. Bioessays. 1995;17:873–878. doi: 10.1002/bies.950171009. [DOI] [PubMed] [Google Scholar]
- Chung C, Murphy-Ullrich J, Erickson H. Mitogenesis, cell migration, and loss of focal adhesions induced by tenascin-C interacting with its cell surface receptor annexin II. Mol Biol Cell. 1996;7:883–892. doi: 10.1091/mbc.7.6.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CY, Erickson HP. Cell surface annexin II is a high affinity receptor for the alternatively spliced segment of tenascin-C. J Cell Biol. 1994;126:539–548. doi: 10.1083/jcb.126.2.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GE. Affinity of integrins for damaged extracellular matrix: αvβ3 binds to denatured collagen type I through RGD sites. Biochem Biophys Res Commun. 1992;182:1025–1031. doi: 10.1016/0006-291x(92)91834-d. [DOI] [PubMed] [Google Scholar]
- den Hartigh J, van Bergen en Henegouwen P, Verkleij A, Boonstra J. The EGF receptor is an actin-binding protein. J Cell Biol. 1992;119:349–355. doi: 10.1083/jcb.119.2.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diakonova M, Payrastre B, van Velzen A, Hage W, van Bergen en Henegouwen P, Boonstra J, Cremers F, Humbel B. Epidermal growth factor induces rapid and transient association of phospholipase C-γ1 with EGF receptor and filamentous actin at membrane ruffles of A431 cells. J Cell Sci. 1995;108:2499–2509. doi: 10.1242/jcs.108.6.2499. [DOI] [PubMed] [Google Scholar]
- End P, Panayotou G, Entwistle A, Waterfield M, Chiquet M. Tenascin: a modulator of growth. Eur J Biochem. 1992;209:1041–1051. doi: 10.1111/j.1432-1033.1992.tb17380.x. [DOI] [PubMed] [Google Scholar]
- Folkman J, Moscona A. Role of cell shape in growth control. Nature (Lond) 1978;273:345–349. doi: 10.1038/273345a0. [DOI] [PubMed] [Google Scholar]
- Fujii K, Imamura S. Cell surface proteolysis by serine proteinases enhances RGD-sensitive melanoma cell adhesion on fibrinogen and vitronectin. Exp Cell Res. 1995;220:201–211. doi: 10.1006/excr.1995.1307. [DOI] [PubMed] [Google Scholar]
- Galardy R. Galardin. Drugs Future. 1993;18:1109–1111. [Google Scholar]
- Gronowski A, Bertics P. Modulation of epidermal growth factor receptor interaction with the detergent insoluble cytoskeleton and its effects on tyrosine kinase activity. Endocrinology. 1995;136:2198–2205. doi: 10.1210/endo.136.5.7720669. [DOI] [PubMed] [Google Scholar]
- Hahn A, Kern F, Jonas U, John M, Buhler F, Resink T. Functional aspects of vascular tenascin-C expression. J Vasc Res. 1995;32:162–174. doi: 10.1159/000159090. [DOI] [PubMed] [Google Scholar]
- Heldin C-H. Dimerization of cell surface receptors in signal transduction. Cell. 1995;80:213–223. doi: 10.1016/0092-8674(95)90404-2. [DOI] [PubMed] [Google Scholar]
- Hedin U, Holm J, Hansson G. Induction of tenascin-C in rat arterial injury: relationship to altered smooth muscle cell phenotype. Am J Pathol. 1991;139:649–656. [PMC free article] [PubMed] [Google Scholar]
- Hunter T, Cooper J. Protein tyrosine kinases. Annu Rev Biochem. 1985;54:897–930. doi: 10.1146/annurev.bi.54.070185.004341. [DOI] [PubMed] [Google Scholar]
- Ingber D. The riddle of morphogenesis: a question of solution chemistry or molecular cell engineering? . Cell. 1993;75:1249–1252. doi: 10.1016/0092-8674(93)90612-t. [DOI] [PubMed] [Google Scholar]
- Jones PL, Schmidhauser C, Bissell MJ. Regulation of gene expression and cell function by extracellular matrix. Crit Rev Eukaryotic Gene Expr. 1993;3:137–154. [PubMed] [Google Scholar]
- Jones PL, Rabinovitch M. Tenascin-C is induced with progressive pulmonary vascular disease in rats and is functionally related to increased smooth muscle cell proliferation. Circ Res. 1996;79:1131–1141. doi: 10.1161/01.res.79.6.1131. [DOI] [PubMed] [Google Scholar]
- Jones PL, Boudreau N, Myers C, Erickson HP, Bissell MJ. Tenascin-C inhibits extracellular matrix-dependent gene expression in mammary epithelial cells. J Cell Sci. 1995;108:519–527. doi: 10.1242/jcs.108.2.519. [DOI] [PubMed] [Google Scholar]
- Jones PL, Cowan K, Rabinovitch M. Tenascin-C, proliferation and subendothelial accumulation of fibronectin in progressive pulmonary vascular disease. Am J Pathol. 1997;150:1349–1360. [PMC free article] [PubMed] [Google Scholar]
- Koyama H, Raines EW, Bornfeldt KE, Roberts JM, Ross R. Fibrillar collagen inhibits arterial smooth muscle cell proliferation through regulation of cdk2 inhibitors. Cell. 1996;87:1069–1078. doi: 10.1016/s0092-8674(00)81801-2. [DOI] [PubMed] [Google Scholar]
- Latijnhouwers M, Bergers M, van Bergen B, Spruijt K, Andriessen M, Schalkwijk J. Tenascin expression during wound healing in human skin. J Pathol. 1996;178:30–35. doi: 10.1002/(SICI)1096-9896(199601)178:1<30::AID-PATH442>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Lin Y-C, Grinnell F. Decreased level of PDGF-stimulated receptor autophosphorylation by fibroblasts in mechanically relaxed collagen matrices. J Cell Biol. 1993;122:663–672. doi: 10.1083/jcb.122.3.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie E, Scott-Burden T, Hahn A, Kern F, Bernhardt J, Regenass J, Weller A, Buhler F. Expression of tenascin by vascular smooth muscle cells: alterations in hypertensive rats and stimulation by angiotensin II. Am J Pathol. 1992;141:377–388. [PMC free article] [PubMed] [Google Scholar]
- Miyamato S, Akiyama S, Yamada K. Synergistic roles for receptor occupancy and aggregation in integrin transmembrane function. Science (Wash DC) 1995a;267:883–885. doi: 10.1126/science.7846531. [DOI] [PubMed] [Google Scholar]
- Miyamato S, Teramoto H, Coso O, Gutkind J, Burbelo P, Akiyama S, Yamada K. Integrin function: molecular hierarchies of cytoskeletal and signaling molecules. J Cell Biol. 1995b;131:791–805. doi: 10.1083/jcb.131.3.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamato S, Teramoto H, Gutkind J, Yamada K. Integrins can collaborate with growth factors for phosphorylation of receptor tyrosine kinases and MAP kinase activation: roles of integrin aggregation and occupancy of receptors. J Cell Biol. 1996;135:1633–1642. doi: 10.1083/jcb.135.6.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery A, Reisfeld R, Cheresh D. Integrin αvβ3 rescues melanoma cells from apoptosis in three-dimensional dermal collagen. Proc Natl Acad Sci USA. 1994;91:8856–8860. doi: 10.1073/pnas.91.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooney D, Langer R, Ingber D. Cytoskeletal filament assembly and the control of cell spreading and function by extracellular matrix. J Cell Sci. 1995;108:2311–2320. doi: 10.1242/jcs.108.6.2311. [DOI] [PubMed] [Google Scholar]
- Murphy-Ullrich J, Lightner V, Aukhil I, Yan Y, Erickson H, Hook M. Focal adhesion integrity is downregulated by the alternatively spliced domain of human tenascin. J Cell Biol. 1991;115:1127–1136. doi: 10.1083/jcb.115.4.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa S, Pawelek P, Grinnell F. Extracellular matrix organization modulates fibroblast growth and growth factor responsiveness. Exp Cell Res. 1989;182:572–582. doi: 10.1016/0014-4827(89)90260-7. [DOI] [PubMed] [Google Scholar]
- Nishiyama T, Akutsu N, Horii I, Nakayama Y, Ozawa T, Hayashi T. Response to growth factors of human dermal fibroblasts in a quiescent state owing to cell-matrix contact inhibition. Matrix. 1991;11:71–75. doi: 10.1016/s0934-8832(11)80210-6. [DOI] [PubMed] [Google Scholar]
- Plopper G, McNamee H, Dike L, Bojanowski K, Ingber D. Convergence of integrin and growth factor receptor signaling pathways within the focal adhesion complex. Mol Biol Cell. 1995;6:1349–1365. doi: 10.1091/mbc.6.10.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto A, Edelman G, Crossin K. Multiple integrins mediate attachment to cytotactin/tenascin. Proc Natl Acad Sci USA. 1993;90:10154–10158. doi: 10.1073/pnas.90.21.10154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskelley C, Desprez P, Bissell MJ. Extracellular matrix-dependent tissue-specific gene expression in mammary epithelial cells requires both physical and biochemical signal transduction. Proc Natl Acad Sci USA. 1994;91:12378–12382. doi: 10.1073/pnas.91.26.12378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlessinger J. The epidermal growth factor as a multifunctional allosteric protein. Biochemistry. 1988;27:3119–3123. doi: 10.1021/bi00409a002. [DOI] [PubMed] [Google Scholar]
- Sharifi B, LaFleur D, Priola C, Forrester J, Fagin J. Angiotensin II regulates tenascin gene expression in vascular smooth muscle cells. J Biol Chem. 1992;267:23910–23915. [PubMed] [Google Scholar]
- Sriramarao P, Mendler M, Bourdon MA. Endothelial cell attachment and spreading on human tenascin is mediated by α2β1 and αvβ3 integrins. J Cell Sci. 1993;105:1001–1012. doi: 10.1242/jcs.105.4.1001. [DOI] [PubMed] [Google Scholar]
- Strange R, Li F, Saurer S, Friis R. Apoptotic cell death and tissue remodeling during mouse mammary gland involution. Development (Camb) 1992;115:49–58. doi: 10.1242/dev.115.1.49. [DOI] [PubMed] [Google Scholar]
- Strauss B, Robinson R, Batchelor W, Chisholm R, Ravi G, Natarajan M, Logan R, Mehta S, Levy D, Ezrin A, Keeley FW. In vivo collagen turnover following experimental balloon angioplasty injury and the role of matrix metalloproteinases. Circ Res. 1996;79:541–550. doi: 10.1161/01.res.79.3.541. [DOI] [PubMed] [Google Scholar]
- Sundberg C, Rubin K. Stimulation of β1 integrins on fibroblasts induces PDGF-independent tyrosine phosphorylation of PDGF-β receptors. J Cell Biol. 1996;132:741–752. doi: 10.1083/jcb.132.4.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sympson C, Talhouk R, Alexander C, Chin J, Clift S, Bissell MJ, Werb Z. Targeted expression of stromelysin-1 in mammary gland provides evidence for a role of proteinases in branching morphogenesis and the requirement for an intact basement membrane for tissue-specific gene expression. J Cell Biol. 1994;125:681–693. doi: 10.1083/jcb.125.3.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talhouk R, Chin J, Unemori E, Werb Z, Bissell MJ. Proteinases of the mammary gland: developmental regulation in vivo and vectorial secretion in culture. Development (Camb) 1991;112:439–449. doi: 10.1242/dev.112.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremble P, Chiquet-Ehrismann R, Werb Z. The extracellular matrix ligands fibronectin and tenascin collaborate in regulating collagenase gene expression in fibroblasts. Mol Biol Cell. 1994;5:439–453. doi: 10.1091/mbc.5.4.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaalamo M, Weckroth M, Puolakkainen P, Kere J, Saarinen P, Lauharanta J, Saarialho-Kere U. Patterns of matrix metalloproteinase and TIMP-1 expression in chronic and normally healing human cutaneous wounds. Br J Dermatol. 1996;135:52–59. [PubMed] [Google Scholar]
- Weaver, V.M., O.W. Petersen, F. Wang, C.A. Larabell, P. Briand, C. Damsky, and M.J. Bissell. 1997. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J. Cell Biol: 137:231–246. [DOI] [PMC free article] [PubMed]
- Zempo N, Kenagy R, Bendeck M, Clowes M, Reidy M. Matrix metalloproteinases of vascular wall cells are increased in balloon-injured rat carotid artery. J Vasc Surg. 1994;20:209–217. doi: 10.1016/0741-5214(94)90008-6. [DOI] [PubMed] [Google Scholar]