

Abstract
Tumor necrosis factor–α, interleukin-1, and endotoxin stimulate the expression of vascular endothelial cell (EC) adhesion molecules. Here we describe a novel pathway of adhesion molecule induction that is independent of exogenous factors, but which is dependent on integrin signaling and cell–cell interactions. Cells plated onto gelatin, fibronectin, collagen or fibrinogen, or anti-integrin antibodies, expressed increased amounts of E-selectin, vascular cell adhesion molecule–1, and intercellular adhesion molecule–1. In contrast, ECs failed to express E-selectin when plated on poly-l-lysine or when plated on fibrinogen in the presence of attachment-inhibiting, cyclic Arg-Gly-Asp peptides. The duration and magnitude of adhesion molecule expression was dependent on EC density. Induction of E-selectin on ECs plated at confluent density was transient and returned to basal levels by 15 h after plating when only 7 ± 2% (n = 5) of cells were positive. In contrast, cells plated at low density displayed a 17-fold greater expression of E-selectin than did high density ECs with 57 ± 4% (n = 5) positive for E-selectin expression 15 h after plating, and significant expression still evident 72 h after plating. The confluency-dependent inhibition of expression of E-selectin was at least partly mediated through the cell junctional protein, platelet/endothelial cell adhesion molecule–1 (PECAM-1). Antibodies against PECAM-1, but not against VE-cadherin, increased E-selectin expression on confluent ECs. Co– culture of subconfluent ECs with PECAM-1– coated beads or with L cells transfected with full-length PECAM-1 or with a cytoplasmic truncation PECAM-1 mutant, inhibited E-selectin expression. In contrast, untransfected L cells or L cells transfected with an adhesion-defective domain 2 deletion PECAM-1 mutant failed to regulate E-selectin expression. In an in vitro model of wounding the wound front displayed an increase in the number of E-selectin–expressing cells, and also an increase in the intensity of expression of E-selectin positive cells compared to the nonwounded monolayer. Thus we propose that the EC junction, and in particular, the junctional molecule PECAM-1, is a powerful regulator of endothelial adhesiveness.
The endothelial lining of the vascular system normally displays a nonactivated, nonadhesive phenotype. Stimulation with agents such as tumor necrosis factor-α (TNF-α)1, interleukin-1 (IL-1), or lipopolysaccharide (LPS) are known to induce the expression of proteins on the endothelial surface that mediate coagulation (Bevilacqua et al., 1986), leukocyte adhesion (Bevilacqua et al., 1985; Gamble et al., 1985; Pober et al., 1986b ; Doherty et al., 1989), and leukocyte transendothelial migration (Furie et al., 1989; Moser et al., 1989). The endothelial antigens that are important for the adhesion of leukocytes are members of the selectin family, E- and P-selectin, and the immunoglobulin gene superfamily, vascular cell adhesion molecule–1 (VCAM-1) and intercellular adhesion molecule–1 (ICAM-1) (Carlos and Harlan, 1994; Litwin et al., 1995).
The induction of E-selectin expression on endothelial cells (ECs) in vitro after cytokine stimulation is transient and independent of the continued presence of the stimulant (Pober et al., 1986a ). Previous studies have shown that E-selectin mRNA and protein levels peak between 2 and 4 h, respectively, after treatment with an agonist, returning to near basal levels by 24 h (Bevilacqua et al., 1989; Read et al., 1994). VCAM-1 (Osborn et al., 1989) and ICAM-1 (Pober et al., 1986b ) are maximal 6 and 12 h, respectively, after stimulation.
In contrast to the transiency of E-selectin and VCAM expression demonstrated by the in vitro data, these antigens have been detected on venular endothelium in chronic inflammatory lesions, such as the synovium in rheumatoid arthritis (Koch et al., 1991), and the skin in psoriasis (Petzelbauer et al., 1994). E-selectin expression is also detected on angiogenic vessels in human hemangiomas, a noninflammatory angiogenic disease (Kraling et al., 1996). Moreover, the architecture and anatomic localization of capillary loops influence the pattern of endothelial expression of E-selectin and VCAM-1, independently of the availability of cytokines (Petzelbauer et al., 1994). Thus it is likely that alternate control mechanisms exist to allow prolonged, locality-based expression of adhesion molecules on the endothelium. At least one of these alternate mechanisms may be flow, since increased shear stress has been shown to selectively modulate adhesion molecule expression, upregulating ICAM-1 but not E-selectin or VCAM-1 (Nagel et al., 1994).
Since sites of inflammation are often associated with morphological changes including cell retraction of the endothelium (Schumacher, 1973), we hypothesized that cell contacts may be important in the regulation of endothelial phenotype. We describe here the central role of the junctional protein, platelet/endothelial cell adhesion molecule–1 (PECAM-1), through the formation of cell–cell interactions, in the maintenance of the functional integrity of the endothelial monolayer. Furthermore, we demonstrate a novel pathway for the induction of adhesion molecules on endothelial cells that is independent of exogenous addition of cytokines, but is related to integrin- and cell shape–associated signaling events.
MATERIALS AND METHODS
Cell Culture
ECs were extracted from human umbilical veins by collagenase treatment, according to a modified method of Wall et al. (1978). Cells were grown in 25-cm2, gelatin-coated Costar flasks (Costar Corp., Cambridge, MA), maintained with endotoxin-free medium 199 (Cytosystems, Sydney, Australia), 20% FCS (Commonwealth Serum Laboratories, Melbourne, Australia), 20 mM Hepes, 2 mM glutamine, 1 mM sodium pyruvate, 1% nonessential amino acids (Cytosystems), 0.225% sodium bicarbonate and antibiotics in the absence of exogenously added heparin or basic fibroblast growth factor, at 37°C in a humidified, 5% CO2 (in air) atmosphere. After 2–5 d of culture, the cells were harvested by trypsin-EDTA treatment (Cytosystems) and replated at specified cell densities on 1.9-cm2, gelatin- and fibronectin-coated multiwell dishes (Nunc, Roskilde, Denmark) for flow cytometry experiments, or on fibronectin-coated glass or Permanox® chamber slides (Labtek, Nunc, Roskilde, Denmark) for immunofluorescence and adhesion assays. The preparation of PECAM-1–transfected L cells has been reported previously (DeLisser et al., 1993). The cells were maintained in RPMI 1640 (GIBCO BRL, Gaithersburg, MD) 10% FCS with 0.5 mg/ml G418.
Antibodies
Mouse mAbs directed against E-selectin (49-1B11), VCAM-1 (51-10C9), PECAM-1 (51-9H6, 51-6F6, 55-3D2), VE-cadherin (55-7H1), integrin β1 (61-2C4), colony-stimulating factor (CSF), common βc chain (3D7, supplied by Q. Sun, Hanson Centre for Cancer Research [HCCR], Adelaide, Australia), and Keyhole Limpet Hemacyanin (23-1F11) were raised at the HCCR. The anti–PECAM-1 mAbs were functional in an assay of neutrophil transendothelial migration and anti–VE-cadherin antibody was functional in an assay of EC aggregation. For coating of wells with antibody, plates were coated with rabbit anti–mouse Ig (50 μg/ml) for 18 h, blocked with 1% BSA, and then anti-integrin or anti-CSFβc chain–purified antibodies added for 1 h at 37°C. Wells were then washed and further blocked before addition of cells.
Flow Cytometry
Flow cytometric analysis of in situ endothelial monolayers was performed as previously described (Gamble et al., 1993). EC monolayers were blocked in 5% sheep serum, and then were stained with primary antibody for 30 min at 37°C, washed twice with RPMI 1640 containing 2.5% FCS, and then stained with FITC-conjugated, anti–mouse Ig (Fab2, DAF; Silenus Laboratories, Hawthorn, Australia) for 30 min at room temperature. Cells were washed twice, removed by trypsin-EDTA treatment, and then fixed in 1% formaldehyde, 0.02% azide, and 0.02% glucose. In experiments involving endothelial pretreatment with mAbs, E-selectin was detected using a single layer, FITC-conjugated, anti–E-selectin mAb (49-1B11).
A minimum of 1,000 events per test was analyzed using an EPICS Profile II (Coulter Immunology, Hialeah, FL). Results of individual EC lines are expressed either as a plot of frequency versus log fluorescence, or as the mean fluorescence channel number, subtracting the accompanying value for the negative control Ig. When results from multiple EC lines have been pooled, the mean fluorescence intensity (MFI) represents n cell lines.
In cocultures of ECs and L cells prepared for flow cytometric analysis, the cells were stained with anti–VE-cadherin, detected with phycoerythrin (PE)-conjugated anti–mouse F(ab′)2 (DDAPE; Silenus Laboratories, Hawthorn, Australia) and simultaneously stained with goat anti–E-selectin detected with FITC–conjugated, anti–goat antibody (Silenus Laboratories). L cells were negative for VE-cadherin, EC were 100% positive for VE-cadherin and were selected for analysis of E-selectin (FITC staining) using a second fluorescence detector. The flow cytometer was calibrated using single PE- or FITC-stained cells.
Immunofluorescence Confocal Microscopy
Confocal microscopy was performed on ECs cultured on fibronectin-coated glass or Permanox® chamber slides. A staining three layer method was used in wounding assays and involved initial fixation in ice-cold methanol for 5 min and then acetone 1 min and washing in M199-containing 2.5% FCS. Cells were stained with saturating amounts of mAb for 30 min at 25°C. After two washes, cells were incubated with biotin-conjugated, affinity-purified anti–mouse Ig (Vector Labs, Burlingame, CA), washed twice, and incubated with avidin-FITC (Dako Corp., Carpinteria, CA). Slides were mounted using 2% propylgallate in glycerol as an anti-fade agent. Images were captured with a laser scanning confocal microscope (MRC600; Bio Rad Laboratories, Hercules, CA). Comparison images were subjected to equivalent amounts of contrast enhancement.
Neutrophil–Endothelial Adhesion
Peripheral blood from normal volunteers was sedimented on dextran, followed by density-gradient centrifugation on Lymphoprep (Nycomed, Oslo, Norway) at 450 g. Contaminating erythrocytes were then lysed by hypotonic 0.2% sodium chloride. Cells were resuspended in RPMI-1640 with 2.5% FCS and yielded a purity of >98%. 5 × 105 neutrophils were added in 125 μl medium to human umbilical vein endothelial cells (HUVECs), which had been plated 16 h earlier onto fibronectin-coated chamber slides at cobblestone and subconfluent densities. After 25 min at 37°C in a humidified 5% CO2 in air atmosphere, the wells were washed three times removing unattached neutrophils. The slides were fixed in 0.5% glutaraldehyde, examined by confocal microscopy, and the number of neutrophils attached per EC counted. At least 160 ECs were assessed.
Preparation of Protein-coupled Beads
Tosyl-activated paramagnetic beads (Dynabeads M-450; DYNAL A.S., Oslo, Norway) were coated with purified platelet PECAM-1 as previously described (Plopper and Ingber, 1993). Essentially 99% of beads were coated with PECAM-1 as assessed by flow cytometry using polyclonal anti–PECAM-1 antibody staining.
Endotoxin Assay
A quantitative, photometric assay (Coatest; Kabi Diagnostica, Stockholm, Sweden) based upon activation of a proenzyme in limulus amoebocyte lysate was used, which detected endotoxin at 0.1–1.2 EU/ml.
Cytokines and Cytokine Antagonists
TNF-α (lot S9010AX; sp act 6.27 × 107 U/mg), TGF-β (lot 8987-53), and a monoclonal anti-TNF were gifts from Genentech, Inc. (South San Francisco, CA) IL-1β (108 thymocyte mitogenesis U/mg) was kindly supplied by Immunex (Seattle, WA). IL-1ra was a gift from Synergen (Boulder, CO). All cytokines contained <3 U/ml of LPS.
Reagents and Peptides
Polymyxin B sulfate (Sigma Chemical Co., St. Louis, MO) was used at 10 μg/ml. When added at plating, it effectively abolished induction of E-selectin by LPS on ECs. Soluble PECAM-1 protein was purified from platelet and was used at 0.01–100 μg/ml. Cyclic RGD and RAD peptides (EMD66203, 67679, 69601) were kindly supplied by A. Jonczyk from Merck KGaA (Darmstadt, Germany). These peptides were identical to those used by Brooks et al. (1994) in inhibiting αvβ3-dependent angiogenesis.
Enumeration of EC Contacts
Multiple photomicrographs of low power, phase contrast fields (see Fig. 1) were obtained of ECs plated at 0.25 and 105 cells per cm2. The number of cell contacts made with adjoining ECs were counted for 10 ECs per field.
Figure 1.

Phase contrast photomicrographs of EC monolayers 15 h after seeding at confluent 1.0 (a), subconfluent 0.25 (b), and sparse 0.05 × 105 cells per cm2 densities (c). Bar, 40 μm.
Wounding EC Monolayers
ECs plated at cobblestone density on fibronectin-coated chamber slides were wounded by scraping with the tip of a 1,000-μl pipette. The wells were washed three times with medium. All wounds consisted of a clearly demarcated cross in the center of a monolayer and healed as an advancing front of elongated, flattened cells. At specified times after wounding, cells were stained for immunofluorescence confocal microscopy as described above. Phase contrast microscopy with ×100 magnification was used to select fields at wound fronts and on areas of cellular monolayer, at least two fields were removed from a wound. Selected fields were then examined with the laser confocal microscope at ×200 magnification and the images stored. Images were retrieved in SETCOL format (COMOS 7.0; Bio Rad Laboratories, Hercules, CA), which displays fluorescence intensity on a color scale (green being minimal, red maximal), and were then examined for the number of ECs positive and negative for E-selectin per ×200 field. An EC was determined to be positive if any part of its surface, >5 mm in diam was red. At least 1,700 ECs were counted at wounds or monolayers per well and positivity was expressed as a percentage of all cells counted. The proportion of ECs expressing E-selectin in the top 50% range of expression intensity was assessed by analysis of pixel intensity in areas of monolayer and wound front and calculated using the histogram format.
Statistics
The statistical significance of results was assessed using the two-tailed Student's t test with either paired or unpaired groups of data as indicated. The frequency histograms of neutrophil adherence to ECs were compared by the Kolmogorov-Smirnov test and the effects of PECAM-1–coated beads on E-selectin expression by the analysis of variance (ANOVA) test.
RESULTS
Confluency-dependent Expression of Endothelial Adhesion Molecules
HUVECs were seeded onto gelatin-coated tissue culture plates at varying numbers (Fig. 1). 15 h after plating, the EC surface expression of E-selectin was measured by flow cytometry. Confluent, cobblestone cultures as seen in Fig. 1 a, showed negligible E-selectin expression (Fig. 2 a). This is in agreement with published work demonstrating a lack of E-selectin on unstimulated ECs (Pober et al., 1986b ; Bevilacqua et al., 1989). However, subconfluent ECs (Fig. 1 b) displayed substantial expression of E-selectin (Fig. 2 a). A comparison between subconfluent and confluent density cells using five separate EC lines showed a 17-fold greater induction in subconfluent ECs, (6.4 ± 1.0 vs. 0.37 ± 0.19 mean fluorescence intensity units ± SEM, P = 0.0003, unpaired t test). In these five experiments, 57 ± 4% of ECs at subconfluent density were positive for E-selectin, as opposed to 7 ± 2% of cells in a cobblestone monolayer. The confluency-dependent expression of E-selectin was evident over a range of EC densities (Fig. 2 b) and was seen on HUVECs extracted from their original monolayer culture by treatment with trypsin-EDTA or EDTA alone (data not shown).
Figure 2.
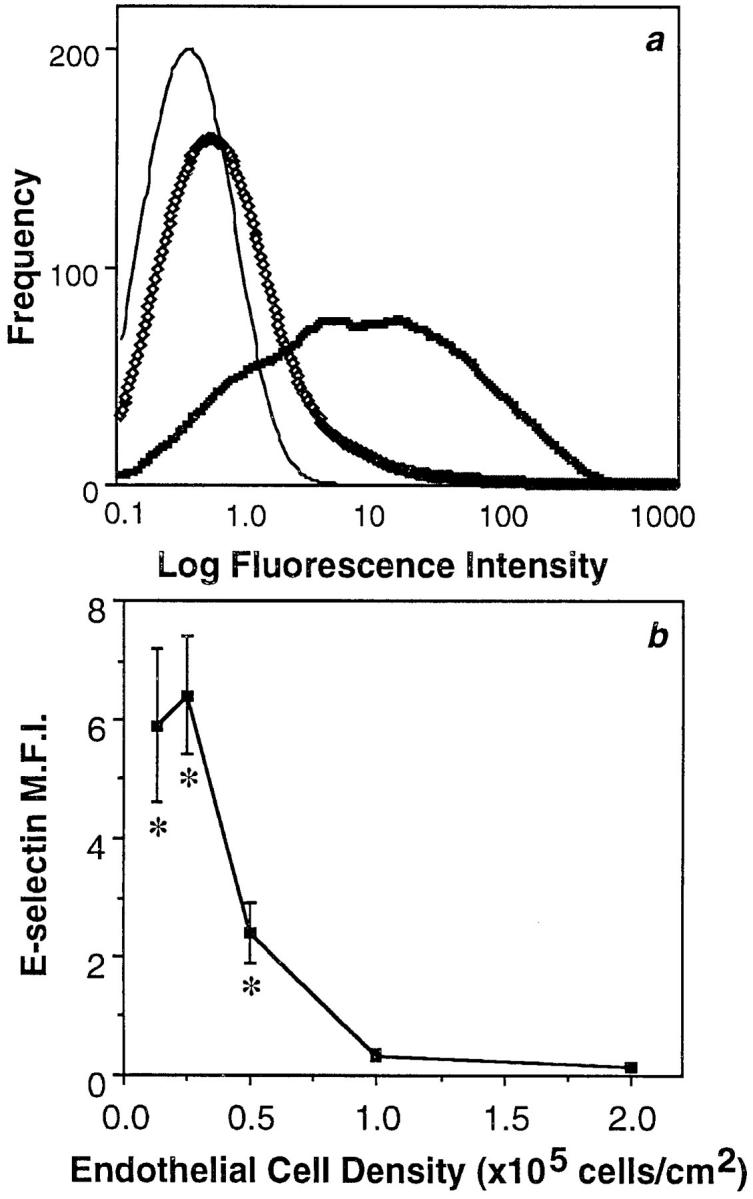
E-selectin expression varies with EC density. (a) Flow cytometry profile of a representative EC line stained for E-selectin 15 h after plating. Cells were plated at cobblestone (⋄⋄⋄, 105 cells per cm2) or subconfluent densities (thick line; 0.25 × 105 cells per cm2). (thin line) Nonbinding control immunoglobulin, which gave a similar profile for confluent or subconfluent cells. (b) ECs were cultured for 15 h after plating at cell densities ranging from sparse to confluent (0.125– 2.0 × 105 cells per cm2). The MFI ± SEM of E-selectin expression is shown for five EC lines, except for values at densities 0.5 and 2.0 × 105 cells per cm2, which are triplicates. Asterisks indicate values significantly different (P < 0.003) from confluent density ECs (105 cells per cm2) by unpaired t test.
Time Course of E-selectin Expression
The expression of E-selectin was measured on ECs at varying times after plating. Fig. 3 shows that cells plated at high and low densities both displayed significant levels of E-selectin early after plating that is within 4 to 12 h. However, expression of E-selectin on high density ECs was fourfold less at its maximum 8 h after plating, was transient and returned to basal levels by 24 h. In contrast, E-selectin expression on ECs plated at low density peaked at 12 h after plating and was still evident at 32 h. ECs at very sparse densities, such that single cells were maintained over the course of the experiment, displayed a persistent and significant expression of E-selectin even 72 h after plating (data not shown). There was considerable variation in the absolute levels of E-selectin between different EC preparations, the reason for which is not known. However, the fold induction calculated between high and low density cells was relatively consistent (21.1 ± 3.6%, n = 30) when measured 16–20 h after plating.
Figure 3.

Time course of E-selectin expression after EC plating. ECs were plated at confluent (▵, 105 cells per cm2) and subconfluent (▪, 0.25 × 105 cells per cm2) densities. The expression of E-selectin was assayed by flow cytometry at specified times after plating. The MFI (± SEM) of three to five cell lines is shown but values at 2, 32, and 72 h after plating are singlicates. Asterisks denote values significantly different between the two cell densities (P < 0.03) by paired t test.
VCAM-1 and ICAM-1 (as well as E-selectin) demonstrated EC density–dependent expression, but the expression of PECAM-1 (or CD31), a non-inducible adhesion protein was not altered (Fig. 4). Increased expression of VCAM-1, ICAM-1, and E-selectin was independent of cell size, as identical forward scatter gates of high and low density cells were always used in the FACS® analysis. Furthermore, the change in adhesion molecule expression was observed, whether the cells were stained in situ before detachment, or after extraction from matrix (data not shown).
Figure 4.

Expression of E-selectin, VCAM-1, and ICAM-1 are confluency dependent. ECs plated at subconfluent density (stripes, 0.25 × 105 cells per cm2) and confluent density (solid, 105 cells per cm2) were stained 20 h later for a, E-selectin, b, VCAM-1, c, ICAM-1, or d, PECAM-1. Expression is shown as the MFI of 23, 11, 7, and 7 EC lines, respectively. Error bars represent the SEM. Asterisks denote values significantly different from cells plated at cobblestone density (P < 0.03) by paired t test.
E-selectin Expression by Subconfluent ECs Is Independent of Cytokines and Supports Neutrophil Adhesion
The time course of expression seen in Fig. 2 suggested two phases in the regulation of E-selectin expression: an induction phase, and a maintenance phase. Induction of the expression of adhesion molecules by subconfluent ECs occurred in the absence of exogenous cytokines. As shown in Fig. 5 a, IL-1 receptor antagonist (IL-1ra) or blocking anti–TNF-α antibody potently and specifically inhibited IL-1 or TNF-α–mediated induction of E-selectin, respectively, but these agents were ineffective on the induction of E-selectin by subconfluent ECs (Fig. 5 b). Furthermore, conditioned medium taken from subconfluent ECs or from cells multiply wounded such that the majority of cells were undergoing migration, did not induce E-selectin expression on confluent density ECs (Table I). Thus a role for endogenous endothelial cytokine production, or the release of some other stimulant from the EC, appeared unlikely.
Figure 5.

The induction of E-selectin on subconfluent ECs is not mediated through TNF-α or IL-1. (a) ECs plated at subconfluent density (0.25 × 105 cells per cm2) were stimulated with 10 U/ml TNF-α (TNF) or 1 ng/ml IL-1β (IL-1) in the presence of anti–TNF-α (TNF + anti-TNF) (1:1,000), or IL-1ra (IL-1 + IL-1ra) (100 ng/ml), respectively. Inhibitors and agonists were added immediately after EC plating and E-selectin expression was assessed 12 h later. (b) ECs were plated at subconfluent density (stripes, 0.25 × 105 cells per cm2) and confluent density (solid, 105 cells per cm2). Anti–TNF-α or IL-1ra were added at plating. E-selectin expression was assessed 12 h later. Results shown are of one representative experiment of four performed.
Table I.
E-selectin Expression Is Not Stimulated by Supernatant from Migrating Endothelial Cells
| Addition | MFI | |
|---|---|---|
| Unwounded SN | 0.01 ± 0.01 | |
| 6 h SN | 0.04 ± 0.04 | |
| 24 h SN | 0.02 ± 0.02 | |
| TNF (10 U/ml) | 20.5 ± 5.9 |
A confluent EC monolayer was multiply wounded such that all cells in the well were induced to migrate. The cells were washed twice to remove debris and replaced with fresh medium. Supernatants (SNs) were collected at 6 and 24 h thereafter and added to other wells containing confluent EC. TNF (10 U/ml) was added to some of these wells as a control, and E-selectin expression measured 4 h later. The results are given as the MFI ± SEM of two experiments. Neither the 6 nor the 24 SN gave significant increases in E-selectin expression compared to SN from unwounded cells.
Further, the induction of E-selectin is not related to cell proliferation. Rapamycin, an inhibitor of the G1-S transition (Kato et al., 1994) showed no effects on E-selectin induction in subconfluent ECs (data not shown) although TGF-β, another inhibitor of cell proliferation (Heimark et al., 1986), which we have shown previously to inhibit cytokine-induced E-selectin expression (Gamble et al., 1993) did inhibit the cytokine-independent induction of E-selectin (data not shown). Moreover, E-selectin induction was independent of cell cycling since no difference in the level of E-selectin expression was seen in cells in G0/G1 with respect to S/G2/M at either cell density, both up to 20 h and even at 72 h after EC plating (data not shown).
Low doses of TNF-α were able to increase E-selectin expression on both low and high density ECs (Fig. 6 a) suggesting that the cytokine and non-cytokine pathways of induction are at least additive. Although the level of E-selectin expression induced by the cytokine-independent mechanism is less than that induced by TNF or IL-1 (Figs. 5 and 6), it is however, functionally relevant. Assessment of the number of neutrophils adherent per EC showed that there was an increase in the number of neutrophils attached per EC in cells plated at low versus high density EC (Fig. 6 b). In three experiments, the percentage of EC supporting the adhesion of two or more neutrophils was 24.3% ± 14% for low density EC and 1.4% ± 1.0% for high density EC (mean ± SEM; P = 0.02).
Figure 6.

The non–cytokine-mediated pathway of E-selectin expression is additive with TNF-induced E-selectin expression and supports neutrophil adherence. (a) E-selectin expression on ECs plated at cobblestone density (105 cells per cm2) or subconfluent densities (0.25 × 105 cells per cm2) 15 h after plating (black). In some groups 1 U/ml of TNF-α was added 4 h before analysis (gray). Results shown are the mean ± SEM of four EC lines. Differences between non–cytokine-induced expression and expression after TNF stimulation, were significant by paired t test (P = 0.04, confluent density ECs and P = 0.02 subconfluent density ECs). (B) 15 h after plating EC at either confluent (solid, 105 cells per cm2) or subconfluent densities (stripes, 0.25 × 105 cells per cm2) neutrophils were added. 30 min later, attached neutrophils were removed by gentle washing and the number of neutrophils adherent per EC were counted using microscopy. A plot of the distribution frequency is shown. 176 ECs at cobblestone density and 168 ECs at subconfluent density were counted in the experiment shown, which is representative of at least three experiments. The two groups were statistically different (P = 0.01) by the Kolmogorov-Smirnov test.
E-selectin Induction Is Dependent on Integrin Engagement
ECs plated on collagen 1, gelatin, fibronectin, fibrinogen, and laminin in BSA-containing media resulted in an upregulation of E-selectin when measured 6 h after plating (Fig. 7 a). The level of induction on these matrices in the absence of FCS in the media was never as high as in the presence of FCS. E-selectin expression was not induced on ECs plated on poly-l-lysine (Fig. 7 a) although they responded to TNF stimulation, suggesting that the poly- l-lysine was not toxic (data not shown). The induction was seen whether cells were plated at subconfluent density (Fig. 7) or at high density (data not shown). Cells plated on anti–β1 integrin-coated surfaces, attached, spread, and induced E-selectin. In contrast, cells plated on an antibody to the common β chain (βc) of GM-CSF, IL-3, and IL-5 receptor, which is also expressed on EC (Korpelainen et al., 1993), attached but failed to spread and also did not result in E-selectin induction (data not shown). Cells plated on fibrinogen in the presence of cyclic RGD peptide also failed to induce E-selectin (Fig. 7 b) although the peptide had no effect on E-selectin expression on cells plated on collagen (data not shown). These results suggest integrin engagement and possibly cell shape changes also are important in E-selectin induction.
Figure 7.

Integrin engagement is critical for E-selectin induction. (a) ECs were plated onto multiwell dishes coated with 0.2% gelatin (Gel.) or 50 μg/ml of fibrinogen (Fg.), collagen 1 (Col. 1), fibronectin (Fn.), laminin (Ln.), or poly-l-lysine (P-L-L) for 2 h. The cells were plated at a density of 0.25 × 105 cells per cm2 in 1% BSA without added FCS. Cells were stained with anti–E-selectin or control antibody 6 h later and results given as the mean ± SEM of triplicate determinations from one experiment, which is representative of at least three similar experiments. The background level of staining of cells on each matrix, using a nonbinding antibody has been subtracted. Asterisks denote values significantly different from control group, which was stained with nonbinding antibody (P < 0.002). (b) EC at subconfluent density (0.25 × 105 cells per cm2) were plated onto fibrinogen-coated wells in the presence of cyclic RGD (Arg-Gly-Asp) or RAD (Arg-Ala-Asp) peptides (final concentration 10 μM) in 1% BSA-containing media. E-selectin expression was measured 6 h after plating and is given as the mean ± SEM of a representative experiment where each group was performed in triplicate. Asterisks denote values significantly different from control peptide (P < 0.01).
PECAM-1 Regulates the Maintenance of Endothelial Adhesion Molecules
Although E-selectin is induced on all cells, only subconfluent EC maintain this expression. The chief difference between these density phenotypes lies in the number of cell– cell contacts and their establishment rate. ECs plated at confluent density (105 cells per cm2) were rapidly surrounded by other cells. Within 1 h they had established 5 ± 1 contacts with adjoining cells (mean ± SEM; n = 3). This number remained constant over the following 24 h. By contrast, subconfluent ECs (0.25 × 105 cells per cm2) had an average of only 1 ± 1 contact (n = 3) at 1 h after plating and continued to form new associations such that 24 h later they had 4 ± 1 contacts (n = 6).
Two molecules known to be concentrated in cell–cell contacts and implicated in establishment of some of the junctional properties of endothelial cell monolayers are PECAM-1 (Albelda et al., 1991; DeLisser et al., 1994) and VE-cadherin (Lampugnani et al., 1992; Ayalon et al., 1994). To determine whether PECAM-1 was involved in the cytokine-independent regulation of E-selectin, three independent methods were used. Firstly, confluent density EC monolayers were exposed to functional antibodies directed to PECAM-1 or VE-cadherin. Monoclonal anti– VE-cadherin antibody (antibody 7H1) had no effect. Polyclonal anti–PECAM-1 resulted in a twofold increase in E-selectin expression (Fig. 8, a and b). Addition of both anti–PECAM-1 and anti–VE-cadherin antibodies produced no further increase than with anti–PECAM-1 antibody alone (data not shown). Although two mAb directed to domain one of PECAM-1 consistently and significantly enhanced E-selectin expression their activity was always less than that seen with the polyclonal anti–PECAM-1 antibody (Fig. 8 b) suggesting the involvement of multiple domains. 55-3D2, an mAb directed to domain two-thirds of PECAM-1 was without function in these assays although it inhibits neutrophil transendothelial cell migration (Yan et al., 1995). The anti–PECAM-1 antibody effect was dose dependent (maximal efficacy at 5 μg/ml) and not due to contaminating endotoxin as polymyxin B did not abrogate the enhancement, the antibodies did not have detectable endotoxin and boiling the antibody abolished its potency (Fig. 8 a). VCAM-1 was also upregulated on cobblestone ECs by polyclonal anti–PECAM-1 antibody by 1.7 ± 0.19-fold (mean ± SEM, n = 3, P = 0.03, paired t test).
Figure 8.

Anti–PECAM-1 antibody upregulates E-selectin on ECs plated at cobblestone density. (a) ECs at cobblestone density (105 cells per cm2) were treated with Ig-purified rabbit polyclonal antibody to PECAM-1 (5 μg/ml) or a control nonimmune rabbit Ig at the time of plating. E-selectin expression was assessed by flow cytometry 12 h later. The MFI (± SEM ) of two to six experiments is given where untreated confluent density ECs have been normalized to 1.0. Subconfluent ECs showed a 5.1 ± 1.4-fold increase in E-selectin expression relative to confluent EC. Asterisks denote a significant difference (P < 0.05) compared with control rabbit Ig by unpaired t test. (b) ECs at confluent density were treated with 5 μg/ml Ig-purified mAbs to PECAM-1 or VE-cadherin at the time of plating or polyclonal anti–PECAM-1 antibody. E-selectin expression was measured 12 h later. The MFI (± SEM) of three experiments is given where untreated confluent density ECs have been normalized to 1.0. Subconfluent ECs showed a 4.0 ± 0.7-fold relative increase in E-selectin expression relative to confluent EC. Asterisks denote a significant difference (P < 0.05) compared with cobblestone EC group.
The second approach used platelet purified PECAM-1 immobilized on beads. E-selectin expression on EC plated at low density in the presence of PECAM-1–coated beads was inhibited when measured 18 h after plating (inhibition was 61 ± 20%; n = 3). BSA-coated beads had no effect (Fig. 9). Interestingly, neither purified, soluble PECAM-1, nor its immobilization on plastic was able to regulate E-selectin expression (data not shown) suggesting that valency, concentration, or microenvironment problems may be operating.
Figure 9.

Adhesion through PECAM-1–coated beads downregulates E-selectin. ECs at subconfluent density (0.25 × 105 cells per cm2) were plated in the absence or presence of beads (20 beads per cell) coated either with purified PECAM-1 or the blocking agent (BSA) alone. Cells at high density (105 cells per cm2) were also plated. E-selectin expression was measured 18 h after plating. The data represents three experiments where each group was performed in triplicate and is presented relative to the E-selectin expression on subconfluent ECs, which is shown as 100%. (The mean MFI at 100% was 2.01 ± 0.09.) Asterisk indicates where PECAM-1–coated beads significantly decreased E-selectin (P < 0.001 by ANOVA) compared to BSA-coated beads.
The third approach used PECAM-1 transfectants. EC were plated at subconfluent density in the presence of L cells expressing full-length PECAM-1, PECAM-1 lacking domain 2, or lacking the cytoplasmic tail. Both the full length and the truncation of the cytoplasmic tail inhibited E-selectin expression, whereas the mutant lacking domain 2 had no effect (Fig. 10 a). Untransfected L cells (Fig. 10 b) or L cells expressing the L1 adhesion glycoprotein (a member of the Ig superfamily expressed by neural cells and lymphocytes (Hubbe et al., 1993) failed to regulate E-selectin expression.
Figure 10.

PECAM-1–transfected L cells regulate E-selectin expression. (a) EC at subconfluent density (0.2 × 105 cells per cm2) were plated in the presence or absence of various concentrations of PECAM-1–transfected L cells (○, full-length PECAM-1; □, domain 2 deletion mutant; ▵, untransfected L cells). EC were also plated at high density (♦). A ratio of 1 EC to 8 L cells would be equivalent in cell number to a high density culture of EC. 18 h later, cells were harvested and stained with goat anti–E-selectin polyclonal antibody and detected using an anti–goat labeled polyclonal antibody together with a mouse anti–VE-cadherin antibody and detected with an anti–mouse, FITC-conjugated antibody. Cells were analyzed by two color analysis, and only the fluorescein positive (i.e., EC) were analyzed for E-selectin expression. The results are given as the MFI of one representative experiment of at least four performed where a similar trend was seen in each. (b) Subconfluent EC were plated as in a but either the full-length, PECAM-1 L cell transfectants (○), or cytoplasmic tail deletion mutant (X). Analysis was as for a and is given as the MFI of a representative experiment of at least four performed.
E-selectin Is Upregulated at Endothelial Wound Edges Whereas the Cell Junctional Molecule PECAM-1 Is Diminished
An in vitro wound assay was established as an in vivo correlate of EC migration (Schimmenti et al., 1992; Taylor and Alexander, 1993). ECs were plated and allowed to come to confluence. At variable times thereafter wounds were made and the cells stained 11 and 27 h later for E-selectin and PECAM-1. As seen in Fig. 11 b, the cells at the wound front displayed a spread, motile, morphology, and had advanced beyond the wound edge. These migrating cells had less PECAM-1 staining at the cell-to-cell borders. Significantly more ECs at the wound front and immediately behind the front expressed E-selectin in comparison to the nonwounded areas (Fig. 11, c and d), and this was substantiated by direct counts of the number of E-selectin expressing cells (Table II). The proportion of ECs expressing E-selectin in the top 50% range of intensity of expression was 1.7 ± 0.7% in the cell monolayer compared to 16 ± 5.0% at the wound front (mean ± SEM of three separate experiments at 16 h after wounding; P = 0.05).
Figure 11.

Immunofluorescence confocal microscopy of wounded ECs 40 h after plating and 11 h after wounding. a and c are cobblestone monolayers distant from the wound front. b and d are ECs at wound edges. a and b are stained for PECAM-1, whereas c and d are stained for E-selectin. b and d are not the same area but are of representative areas along the migrating front. The wound front is marked with a vertical bar. Bar, 40 μm.
Table II.
E-selectin Expression by ECs at Wound Edges
| Time after wounding | E-selectin–positive ECs | Mean increase in number of E-selectin–positive ECs (n = 3) | ||||
|---|---|---|---|---|---|---|
| % | ||||||
| Monolayer | Wound front | |||||
| 11 h | 4.1 | 13.5 | 2.2 ± 0.5 | |||
| 27 h | 4.3 | 10.4 | 2.4 ± 0.5 | |||
Confluent EC monolayers were wounded by scraping with a pipette tip at either 13 or 29 h after plating. The cells were stained for E-selectin 27 or 11 h thereafter, respectively (i.e., 40 h after plating). An average of 2,600 cells was counted in the confluent monolayer, and 1,700 at the wound edge for each experiment, and the E-selectin–positive cells was calculated as described in Materials and Methods. The percent of positive cells at either the wound front or in the monolayer in a single experiment (n = 1) is shown. Also shown is the mean increase in the number of positive cells at the wound front compared to the monolayer for three experiments where all experiments showed the same trend.
DISCUSSION
In this article, we have demonstrated a novel mechanism for the induction and expression of adhesion molecules on ECs. The induction is initiated through an integrin-dependent process and is associated with cell spreading. The intensity and duration of expression of the adhesion molecules is inversely proportional to cell density, and is mediated at least in part through the junctional protein, PECAM-1. This phenomenon does not involve the known cytokines and differs from the cytokine-dependent pathways in a number of ways: (a) it is dependent on integrin engagement and cell shape; (b) it is regulated by cell junctions; and (c) it can result in prolonged cell surface expression of adhesion molecules (Table III).
Table III.
Mechanisms Controlling Expression of Endothelial Adhesion Molecules
| Type I | Type II | |||
|---|---|---|---|---|
| Induction | Cytokine, endotoxin | Integrin, cell shape | ||
| Peak | 4–6 h | 8–12 h | ||
| Duration | Transient | Prolonged | ||
| Regulation | Receptor mediated | Cell junctions |
Since the phenomena described in this paper represent a fundamentally new mechanism underlying important pathological processes, we suggest nomenclature where type I induction refers to the classic cytokine-dependent process, and type II induction refers to the new cytokine-independent pathway. Although the level of E-selectin induced in type II conditions is less than that seen for maximal doses of cytokine-induced E-selectin, it is functional as evidenced by increased neutrophil attachment, and is at least additive with low doses of TNF. Such situations of low cytokine concentrations may be relevant during early phases of inflammatory lesions, suggesting the potential importance of both these induction processes in determining endothelial phenotype.
Type II induction of E-selectin described herein is independent of cell size, cell proliferation, and cell cycle, but is initially integrin mediated and seen in cells plated at both high and low density. Cells plated onto ligands that facilitated integrin-mediated attachment induce E-selectin expression (Fig. 7). However if the initial attachment is blocked, as with the addition of cyclic RGD peptides to cells on fibrinogen, E-selectin induction is inhibited. Binding of RGD to cells per se is insufficient to inhibit E-selectin induction since RGD peptides have no effect on cells plated onto collagen.
The intensity and duration of induction of type II responses was related to the density of cell plating. Cells plated at confluent density expressed less E-selectin when measured 8–48 h after plating, compared to cells plated at subconfluent density (Fig. 3). Morphologically, cells plated at high density form rapid cell–cell interactions in contrast to low density cells. Thus, junctional control of EC adhesion molecule expression was considered. Two molecules have been described that are known to be important in endothelial junctional integrity (Albelda et al., 1991; Lampugnani et al., 1992; Ayalon et al., 1994; DeLisser et al., 1994): PECAM-1, a transmembrane glycoprotein belonging to the Ig gene superfamily containing six extracellular, Ig-like domains; and VE-cadherin, an endothelium-specific member of the cadherin family of cell junctional molecules. The involvement of PECAM-1 in the control of E-selectin expression was shown by three independent methods. Firstly, anti–PECAM-1 but not anti–VE-cadherin antibodies induced E-selectin expression on confluent ECs (Fig. 8). Although the increases achieved with the antibodies were not to the level seen on low density cells, they were consistent. This may suggest that molecules other than PECAM-1 could exert secondary events that influence the level of expression of E-selectin. Secondly, PECAM-coated beads, when added at plating, inhibited the level of E-selectin expression on low density cells compared to BSA-coated beads (Fig. 9). Thirdly, transfectants expressing full-length PECAM-1, or those expressing the extracellular domains but lacking the cytoplasmic domain, inhibited E-selectin expression on subconfluent ECs (Fig. 10). In contrast, L cells expressing the domain 2 deletion mutant failed to effect E-selectin expression, suggesting a critical involvement of domain 2 in this regulation. These results are in agreement with other studies showing the importance of domain 2 in cell aggregation (Sun et al., 1996). Together with our antibody studies showing that antibodies to domain 1 increased the level of E-selectin expression on high density cells, our results suggest that both domains 1 and 2 of PECAM-1 are critical in regulation of E-selectin expression.
Cell–cell interactions are known to regulate a number of events, including cell proliferation (Gradl et al., 1995), release of bFGF from astrocytes (Murphy et al., 1988), responsiveness of fibroblasts to TGF-β (Paulsson et al., 1988), and the regulation of intracellular pH (Galkina et al., 1995). The mechanisms that underlie such regulation however have not been elucidated, although autocrine release of growth inhibitory factors (Antonelli-Orlidge et al., 1989; Gradl et al., 1995) and gap junction changes (Chen et al., 1995) have been proposed. As described here, a cell surface junctional protein, PECAM-1, plays a central role in the density-dependent regulation of endothelial E-selectin expression. The possibility of a signaling role for PECAM-1 has been raised previously in a number of different systems. These include activation of integrins on T cells (Tanaka et al., 1992), natural killer cells (Berman et al., 1996), monocytes and neutrophils (Berman and Muller, 1995), and on CD34+ hematopoietic progenitor cells (Leavesley et al., 1994), inhibition of EC proliferation (Fawcett et al., 1995), and platelet aggregation (Newman et al., 1992). The association of the tyrosine phosphatase-SHP2 with aggregated platelets (demonstrated recently by Jackson et al. [1997]) has put credence to a signaling pathway associated with PECAM-1 itself. Of interest to our study here is the recent observation by Lu et al. (1996) that integrin engagement and cell spreading results in PECAM-1 dephosphorylation in EC. Thus, the likely cross-talk between integrin and PECAM-1 is further strengthened.
Our in vitro model of cell wounding displays similar features to that described in the density-dependent regulation of adhesion molecules. Firstly, the induction of E-selectin expression is restricted to the migrating front and is associated with a change in cell shape and PECAM-1 redistribution away from the cell junction. Secondly, the E-selectin expression on these migrating cells is maintained at least up to 30 h after the wound signal. Thus we would suggest that this model may reflect identical signaling pathways as those operating in our plating experiments, and may therefore be appropriate as a model for in vivo endothelial regulation at wound sites.
Pathological tissue inflammation is characterized by increased and chronic expression of endothelial adhesion molecules (Koch et al., 1991; Petzelbauer et al., 1994; Kraling et al., 1996), changes in endothelial morphology and angiogenesis (Fitzgerald et al., 1991). These morphological changes include the formation of high endothelial venules (Freemont et al., 1983) and cell retraction with associated intercellular gaps (Nagel et al., 1994). In balloon angioplasty, expression of adhesion molecules at the wound front associated with morphological changes has been observed in contrast to the lack of expression in the area behind the wound edge (Tanaka et al., 1993). More recently, a role for E-selectin in angiogenesis has been postulated (Nguyen et al., 1993; Koch et al., 1995; Kraling et al., 1996). The results reported here, demonstrate: (a) a cytokine-independent mechanism of adhesion molecule expression, and (b) that cell–cell interactions influence the duration and magnitude of this expression, may explain some of these in vivo observations. Furthermore, the observation that restoration of PECAM-1 interactions can downregulate adhesion molecule expression on ECs offers the promise that manipulation of EC junctional molecules may permit the development of novel therapeutics.
Acknowledgments
We thank Y. Khew-Goodall for helpful discussions and M. Walker for manuscript preparation. We express our gratitude to the staff at the delivery wards of the Women's and Children's Hospital (Adelaide, South Australia), and the Burnside War Memorial Hospital (Adelaide, South Australia) for collection of umbilical cords.
This work was supported by the National Health and Medical Research Council (Australia), the Anti-Cancer Foundation of the Universities of South Australia, and the National Heart Foundation of Australia.
Abbreviations used in this paper
- CSF
colony stimulating factor
- EC
endothelial cell
- HUVEC
human umbilical vein endothelial cells
- ICAM-1
intercellular adhesion molecule–1
- IL-1 and IL-1ra
interleukin-1 and IL-1 receptor agonist
- LPS
lipopolysaccharide
- MFI
mean fluorescence intensity
- PECAM
platelet/endothelial cell adhesion molecule
- TNF-α
tumor necrosis factor-α
- VCAM-1
vascular cell adhesion molecule–1
Footnotes
M. Vadas and J. Gamble contributed equally to this paper.
Address all correspondence to Jennifer Gamble, Division of Human Immunology, Hanson Centre for Cancer Research, Institute of Medical and Veterinary Science, P.O. Box 14, Rundle Mall, Adelaide, South Australia, 5000 Australia. Tel. 6188-232-4092. Fax: 6188-232-4092. e-mail: jgamble@immuno.imvs
REFERENCES
- Albelda SM, Muller WA, Buck CA, Newman PJ. Molecular and cellular properties of PECAM-1 (endoCAM/CD31): a novel vascular cell– cell adhesion molecule. J Cell Biol. 1991;114:1059–1068. doi: 10.1083/jcb.114.5.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonelli-Orlidge A, Saunders KB, Smith SR, D'Amore PA. An activated form of transforming growth factor β is produced by cocultures of endothelial cells and pericytes. Proc Natl Acad Sci USA. 1989;86:4544–4548. doi: 10.1073/pnas.86.12.4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayalon O, Sabanai H, Lampugnani M-G, Dejana E. Spatial and temporal relationships between cadherins and PECAM-1 in cell–cell junctions of human endothelial cells. J Cell Biol. 1994;126:247–258. doi: 10.1083/jcb.126.1.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman ME, Muller WA. Ligation of platelet/endothelial cell adhesion molecule-1 (PECAM-1, CD31) on monocytes and neutrophils increases binding capacity of leukocyte CR3 (CD11b/CD18) J Immunol. 1995;154:299–307. [PubMed] [Google Scholar]
- Berman ME, Xie Y, Muller WA. Roles of platelet/endothelial cell adhesion molecule-1 (PECAM-1, CD31) in natural killer cell transendothelial migration and β2 integrin activation. J Immunol. 1996;156:1515–1524. [PubMed] [Google Scholar]
- Bevilacqua MP, Pober JS, Wheeler ME, Cotran RS, Gimbrone MA., Jr Interleukin 1 acts on cultured human vascular endothelium to increase the adhesion of polymorphonuclear leukocytes, monocytes and related leukocyte lines. J Clin Invest. 1985;76:2003–2009. doi: 10.1172/JCI112200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevilacqua MP, Pober JS, Majeau GR, Fiers W, Cotran RS, Gimbrone MA., Jr Recombinant tumor necrosis factor induces procoagulant activity in cultured human vascular endothelium: characterisation and comparison with the actions of interleukin 1. Proc Natl Acad Sci USA. 1986;83:4533–4537. doi: 10.1073/pnas.83.12.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevilacqua MP, Stengelin S, Gimbrone MA, Jr, Seed B. Endothelial leukocyte adhesion molecule-1: an inducible receptor for neutrophils related to complement regulatory proteins and lectins. Science (Wash DC) 1989;243:1160–1165. doi: 10.1126/science.2466335. [DOI] [PubMed] [Google Scholar]
- Brooks PC, Montgomery AMP, Rosenfeld M, Reisfeld T, Hu G, Klier G, Cheresh DA. Integrin αvβ3antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell. 1994;79:1157–1164. doi: 10.1016/0092-8674(94)90007-8. [DOI] [PubMed] [Google Scholar]
- Carlos TM, Harlan JM. Leukocyte-endothelial adhesion molecules. Blood. 1994;84:2068–2101. [PubMed] [Google Scholar]
- Chen SC, Pelletier DB, Ao P, Boynton AL. Connexin 43 reverses the phenotype of transformed cells and alters their expression of cyclin/cyclin-dependent kinases. Cell Growth Diff. 1995;6:681–690. [PubMed] [Google Scholar]
- DeLisser HM, Yan HC, Newman PJ, Muller WA, Buck CA, Albelda SM. Platelet/endothelial cell adhesion molecule-1 (CD31)-mediated cellular aggregation involves cell surface glycosaminoglycans. J Biol Chem. 1993;168:16037–16046. [PubMed] [Google Scholar]
- DeLisser HM, Newman PJ, Albelda SM. Molecular and functional aspects of PECAM-1/CD31. Immunol Today. 1994;15:490–495. doi: 10.1016/0167-5699(94)90195-3. [DOI] [PubMed] [Google Scholar]
- Doherty DE, Zagarella L, Henson PM, Worthen GS. Lipopolysaccharide stimulates monocyte adherence by effects on both the monocyte and the endothelial cell. J Immunol. 1989;143:3673–3679. [PubMed] [Google Scholar]
- Fawcett J, Buckley C, Holness CL, Bird IN, Spragg JH, Saunders J, Harris A, Simmons DL. Mapping the homotypic binding sites in CD31 and the role of CD31 adhesion in the formation of interendothelial cell contacts. J Cell Biol. 1995;128:1229–1241. doi: 10.1083/jcb.128.6.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald O, Soden M, Yanni G, Robinson R, Bresnihan B. Morphometric analysis of blood vessels in synovial membranes obtained from clinically affected and unaffected knee joints of patients with rheumatoid arthritis. Ann Rheum Dis. 1991;50:792–796. doi: 10.1136/ard.50.11.792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freemont AJ, Jones CJP, Bromley M, Andrews P. Changes to vascular endothelium related to lymphocyte collections in diseased synovia. Arthritis Rheum. 1983;26:1427–1433. doi: 10.1002/art.1780261203. [DOI] [PubMed] [Google Scholar]
- Furie MB, McHugh DD. Migration of neutrophils across endothelial monolayers is stimulated by treatment of the monolayers with interleukin-1 or tumor necrosis factor-α. J Immunol. 1989;143:3309–3317. [PubMed] [Google Scholar]
- Galkina SI, Sud'ina GF, Dergacheva GB, Margolis LB. Regulation of intracellular pH by cell–cell adhesive interactions. FEBS (Fed Eur Biol Soc) Lett. 1995;374:17–20. doi: 10.1016/0014-5793(95)00969-g. [DOI] [PubMed] [Google Scholar]
- Gamble JR, Harlan JM, Klebanoff SJ, Vadas MA. Stimulation of the adherence of neutrophils to umbilical vein endothelium by human recombinant tumor necrosis factor. Proc Natl Acad Sci USA. 1985;82:8667–8671. doi: 10.1073/pnas.82.24.8667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamble JR, Khew-Goodall Y, Vadas MA. Transforming growth factor-β inhibits E-selectin expression on human endothelial cells. J Immunol. 1993;150:4494–4503. [PubMed] [Google Scholar]
- Gradl G, Faust D, Oesch F, Wieser RJ. Density-dependent regulation of cell growth by contactinhibin and the contactinhibin receptor. Curr Biol. 1995;5:526–535. doi: 10.1016/s0960-9822(95)00105-9. [DOI] [PubMed] [Google Scholar]
- Heimark RL, Twardzik DR, Schwartz SM. Inhibition of endothelial regeneration by type-β transforming growth factor from platelets. Science (Wash DC) 1986;233:1078–1080. doi: 10.1126/science.3461562. [DOI] [PubMed] [Google Scholar]
- Hubbe M, Kowitz A, Schirrmacher V, Schachner M, Altevogt P. L1 adhesion molecule on mouse leukocytes: regulation and involvement in endothelial cell binding. Eur J Immunol. 1993;23:2927–2931. doi: 10.1002/eji.1830231130. [DOI] [PubMed] [Google Scholar]
- Jackson DE, Ward CM, Wang R, Newman PJ. The protein-tyrosine phosphatase SHP-2 binds platelet/endothelial cell adhesion molecule-1 (PECAM-1) and forms a distinct signaling complex during platelet aggregation. J Biol Chem. 1997;272:6986–6993. doi: 10.1074/jbc.272.11.6986. [DOI] [PubMed] [Google Scholar]
- Kato J, Matsuoka M, Polyak K, Massague J, Sherr CJ. Cyclic AMP-induced G1 phase arrest mediated by an inhibitor (p27KIP1) of cyclin-dependent kinase 4 activation. Cell. 1994;79:487–496. doi: 10.1016/0092-8674(94)90257-7. [DOI] [PubMed] [Google Scholar]
- Koch AE, Burrows JC, Haines GK, Carlos TM, Harlan JM, Leibovich SJ. Immunolocalisation of endothelial and leukocyte adhesion molecules in human rheumatoid and osteoarthritic synovial tissues. Lab Invest. 1991;64:313–320. [PubMed] [Google Scholar]
- Koch AE, Halloren MM, Haskell CJ, Shah MR, Polverini PJ. Angiogenesis is mediated by soluble forms of E-selectin and vascular cell adhesion molecule-1. Nature (Lond) 1995;376:517–519. doi: 10.1038/376517a0. [DOI] [PubMed] [Google Scholar]
- Korpelainen EI, Gamble JR, Smith WB, Goodall GJ, Qiyu S, Woodcock JM, Dottore M, Vadas MA, Lopez AF. The receptor for interleukin 3 is selectively induced in human endothelial cells by tumor necrosis factor α and potentiates interleukin 8 secretion and neutrophil transmigration. Proc Natl Acad Sci USA. 1993;90:11137–11141. doi: 10.1073/pnas.90.23.11137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraling BM, Razon MJ, Boon LM, Zurakowski D, Seachord C, Darveau RP, Mulliken JB, Corless CL, Bischoff J. E-selectin is present in proliferating endothelial cells in human hemangiomas. Am J Pathol. 1996;148:1181–1191. [PMC free article] [PubMed] [Google Scholar]
- Lampugnani MG, Resnati M, Raiteri M, Pigott R, Piscane A, Houen G, Ruco LP, Dejana E. A novel endothelial-specific membrane protein is a marker of cell–cell contacts. J Cell Biol. 1992;118:1511–1522. doi: 10.1083/jcb.118.6.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leavesley DI, Oliver JM, Swart BW, Berndt MC, Haylock DN, Simmons PJ. Signals from platelet/endothelial cell adhesion molecule enhance the adhesive activity of the very late antigen-4 integrin of human CD34+ hemopoietic progenitor cells. J Immunol. 1994;153:4673–4683. [PubMed] [Google Scholar]
- Litwin, M.S., J.R. Gamble, and M.A. Vadas. 1995. Role of cytokines in endothelial cell functions. In Human Cytokines: Their Role in Disease and Therapy. B.B. Aggarawal, and R.K. Puri, editors. Blackwell Science, Cambridge, Massachusetts. 101–130.
- Lu TT, Yan LG, Madri JA. Integrin engagement mediates tyrosine dephosphorylation on platelet-endothelial cell adhesion molecule 1. Proc Natl Acad Sci USA. 1996;93:11808–11813. doi: 10.1073/pnas.93.21.11808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser R, Schleiffenbaum B, Groscurth P, Fehr J. Interleukin 1 and tumor necrosis factor stimulate human vascular endothelial cells to promote transendothelial neutrophil passage. J Clin Invest. 1989;83:444–455. doi: 10.1172/JCI113903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy PR, Sato R, Sato Y, Friesen HG. Fibroblast growth factor messenger ribonucleic acid expression in a human astrocytoma cell line: regulation by serum and cell density. Mol Endocrinol. 1988;2:591–598. doi: 10.1210/mend-2-7-591. [DOI] [PubMed] [Google Scholar]
- Nagel T, Resnick N, Atkinson WJ, Dewey CF, Gimbrone MA. Shear stress selectively upregulates intercellular adhesion molecule expression in cultured human vascular endothelial cells. J Clin Invest. 1994;94:885–891. doi: 10.1172/JCI117410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman PJ, Hillery CA, Albrecht R, Parise LV, Berndt MC, Mazurov AV, Dunlop LC, Zhang J, Rittenhouse SE. Activation-dependent changes in human platelet PECAM-1: phosphorylation, cytoskeletal association, and surface membrane redistribution. J Cell Biol. 1992;119:239–246. doi: 10.1083/jcb.119.1.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen M, Strubel NA, Bischoff J. A role for sialyl lewis-x/a glycoconjugates in capillary morphogenesis. Nature (Lond) 1993;365:267–269. doi: 10.1038/365267a0. [DOI] [PubMed] [Google Scholar]
- Osborn L, Hession C, Tizard R, Vassallo C, Luhowskyj S, Chi-Rosso G, Lobb R. Direct expression cloning of Vascular Cell Adhesion Molecule 1, a cytokine-induced endothelial protein that binds to lymphocytes. Cell. 1989;59:1203–1211. doi: 10.1016/0092-8674(89)90775-7. [DOI] [PubMed] [Google Scholar]
- Paulsson Y, Beckman M, Westermark B, Heldrin C-H. Density-dependent inhibition of cell growth by transforming growth factor β1 in normal human fibroblasts. Growth Factors. 1988;1:19–27. doi: 10.3109/08977198809000243. [DOI] [PubMed] [Google Scholar]
- Petzelbauer P, Pober JS, Keh A, Braverman IM. Inducibility and expression of microvascular endothelial adhesion molecules in lesional, perilesional and uninvolved skin of psoriatic patients. J Invest Dermatol. 1994;103:300–305. doi: 10.1111/1523-1747.ep12394720. [DOI] [PubMed] [Google Scholar]
- Plopper G, Ingber DE. Rapid induction of focal adhesion complexes. Biochem Biophys Res Commun. 1993;193:571–578. doi: 10.1006/bbrc.1993.1662. [DOI] [PubMed] [Google Scholar]
- Pober JS, Bevilacqua MP, Mendrick DL, Lapierre LA, Fiers W, Gimbrone MA., Jr Two distinct monokines, interleukin 1 and tumor necrosis factor, each independently induce biosynthesis and transient expression of the same antigen on the surface of cultured human vascular endothelial cells. J Immunol. 1986a;136:1680–1687. [PubMed] [Google Scholar]
- Pober JS, Gimbrone MA, Jr, Lapierre LA, Mendrick DL, Fiers W, Rothlein R, Springer TA. Overlapping patterns of activation of human endothelial cells by interleukin 1, tumor necrosis factor and immune interferon. J Immunol. 1986b;137:1893–1896. [PubMed] [Google Scholar]
- Read MA, Whitley MZ, Williams AJ, Collins T. NF-kB and IkBa: an inducible regulatory system in endothelial activation. J Exp Med. 1994;179:503–512. doi: 10.1084/jem.179.2.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimmenti LA, Yan H-C, Madri JA, Albelda SM. Platelet endothelial cell adhesion molecule, PECAM-1, modulates cell migration. J Cell Physiol. 1992;153:417–428. doi: 10.1002/jcp.1041530222. [DOI] [PubMed] [Google Scholar]
- Schumacher HR. Joint involvement in progressive systemic sclerosis (scleroderma) Am J Clin Pathol. 1973;60:593–600. doi: 10.1093/ajcp/60.5.593. [DOI] [PubMed] [Google Scholar]
- Sun J, Williams J, Yan H-C, Amin KM, Albelda SM, DeLisser HM. Platelet endothelial cell adhesion molecule-1 (PECAM-1) homophilic adhesion is mediated by immunoglobulin-like domains 1 and 2 and depends on the cytoplasmic domain and the level of surface expression. J Biol Chem. 1996;271:18561–18570. doi: 10.1074/jbc.271.31.18561. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Albelda SM, Horgan KJ, van Seventer GA, Shimizu Y, Newman W, Hallam J, Newman PJ, Buck CA, Shaw S. CD31 expressed on distinctive T cell subsets is a preferential amplifier of β1integrin-mediated adhesion. J Exp Med. 1992;176:245–253. doi: 10.1084/jem.176.1.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H, Sukhova GK, Swanson SJ, Clinton SK, Ganz P, Cybulsky MI, Libby P. Sustained activation of vascular cells and leukocytes in the rabbit aorta after balloon injury. Circulation. 1993;88:1788–1803. doi: 10.1161/01.cir.88.4.1788. [DOI] [PubMed] [Google Scholar]
- Taylor WR, Alexander RW. Autocrine control of wound repair by insulin-like growth factor I in cultured endothelial cells. Am J Physiol. 1993;265:801–805. doi: 10.1152/ajpcell.1993.265.3.C801. [DOI] [PubMed] [Google Scholar]
- Wall RT, Harker LA, Quadricci LJ, Striker GE. Factors influencing endothelial cell proliferation in vitro. J Cell Physiol. 1978;96:203–213. doi: 10.1002/jcp.1040960209. [DOI] [PubMed] [Google Scholar]
- Yan H-C, Pilewski JM, Zhang Q, DeLisser HM, Romer L, Albelda SM. Localization of multiple functional domains on human PECAM-1 (CD31) by monoclonal antibody epitope mapping. Cell Adhes Commun. 1995;3:45–66. doi: 10.3109/15419069509081277. [DOI] [PubMed] [Google Scholar]