

Abstract
Just before bud emergence, a Saccharomyces cerevisiae cell forms a ring of chitin in its cell wall; this ring remains at the base of the bud as the bud grows and ultimately forms part of the bud scar marking the division site on the mother cell. The chitin ring seems to be formed largely or entirely by chitin synthase III, one of the three known chitin synthases in S. cerevisiae. The chitin ring does not form normally in temperature-sensitive mutants defective in any of four septins, a family of proteins that are constituents of the “neck filaments” that lie immediately subjacent to the plasma membrane in the mother-bud neck. In addition, a synthetic-lethal interaction was found between cdc12-5, a temperature-sensitive septin mutation, and a mutant allele of CHS4, which encodes an activator of chitin synthase III. Two-hybrid analysis revealed no direct interaction between the septins and Chs4p but identified a novel gene, BNI4, whose product interacts both with Chs4p and Cdc10p and with one of the septins, Cdc10p; this analysis also revealed an interaction between Chs4p and Chs3p, the catalytic subunit of chitin synthase III. Bni4p has no known homologues; it contains a predicted coiled-coil domain, but no other recognizable motifs. Deletion of BNI4 is not lethal, but causes delocalization of chitin deposition and aberrant cellular morphology. Overexpression of Bni4p also causes delocalization of chitin deposition and produces a cellular morphology similar to that of septin mutants. Immunolocalization experiments show that Bni4p localizes to a ring at the mother-bud neck that lies predominantly on the mother-cell side (corresponding to the predominant site of chitin deposition). This localization depends on the septins but not on Chs4p or Chs3p. A GFP-Chs4p fusion protein also localizes to a ring at the mother-bud neck on the mother-cell side. This localization is dependent on the septins, Bni4p, and Chs3p. Chs3p, whose normal localization is similar to that of Chs4p, does not localize properly in bni4, chs4, or septin mutant strains or in strains that accumulate excess Bni4p. In contrast, localization of the septins is essentially normal in bni4, chs4, and chs3 mutant strains and in strains that accumulate excess Bni4p. Taken together, these results suggest that the normal localization of chitin synthase III activity is achieved by assembly of a complex in which Chs3p is linked to the septins via Chs4p and Bni4p.
The “neck filaments” of the yeast Saccharomyces cerevisiae were originally identified by electron microscopy (Byers and Goetsch, 1976; Byers, 1981). These filaments are ∼10 nm in diameter and lie immediately subjacent to the plasma membrane in the mother-bud neck; they appear just before or coincident with bud emergence and disappear during cytokinesis. Temperature-sensitive mutants defective in any of four genes, CDC3, CDC10, CDC11, and CDC12, fail to form the neck filaments at restrictive temperature (Byers, B., and L. Goetsch. 1976. J. Cell Biol. 70:35a; Byers, 1981; Adams, 1984). These mutants also exhibit hyperpolarized bud growth and fail to complete cytokinesis (Hartwell, 1971a ; Adams and Pringle, 1984; Slater et al., 1985). Because the mutants are not blocked in DNA replication or nuclear division, they arrest at restrictive temperature with multiple elongated buds and multiple nuclei.
Sequence analysis revealed that Cdc3p, Cdc10p, Cdc11p, and Cdc12p are members of a family of related proteins, now known as septins (Sanders and Field, 1994; Longtine et al., 1996; Cooper and Kiehart, 1996). Immunofluorescence studies showed that the septins localize to the mother-bud neck ∼15 min before bud emergence and behave as if they are constituents of the neck filaments (Haarer and Pringle, 1987; Ford and Pringle, 1991; Kim et al., 1991). Localization of all four septins to the neck is lost when any one of the four temperature-sensitive mutants is shifted to restrictive temperature (Ford and Pringle, 1991; Kim et al., 1991; Longtine et al., 1996), and several other lines of genetic and biochemical evidence also suggest that there are direct interactions among these proteins (Longtine et al., 1996).
In addition to Cdc3p, Cdc10p, Cdc11p, and Cdc12p, S. cerevisiae also contains three other septins. Two of these, Spr3p and Spr28p, are expressed only in sporulating cells and appear to play roles in the extension of the forespore membrane and/or in the deposition of spore-wall components (De Virgilio et al., 1996; Fares et al., 1996). The function of the seventh septin (revealed by the genome-sequencing project) is currently under investigation. Septins have also been identified in a variety of other fungi and animals (reviewed by Longtine et al., 1996); they appear to be generally involved in cytokinesis, and protein localization data suggest that they may also play other roles in the organization of the cell surface. All of the known septins contain a predicted nucleotide binding site that apparently can bind GTP (Field et al., 1996; Longtine, M., and J.R. Pringle, unpublished results), although the role of nucleotide binding remains unclear. In addition, all but a few of the known septins contain predicted coiled-coil domains, which might be involved in the assembly of these proteins into filaments or other higher-order structures (Field et al., 1996; Longtine et al., 1996). Interestingly, although both immunolocalization and cell-fractionation experiments suggest that the septins are associated with the plasma membrane, motifs that suggest how these proteins might interact with the membrane have not been identified.
The localization and timing of septin assembly suggest that these proteins might play a role in the organization of cell wall assembly at the bud site and, in particular, in the formation of the chitin ring. This ring forms at the presumptive bud site just before bud emergence. It remains at the base of the bud as the bud grows and ultimately forms part of the bud scar marking the division site on the mother cell. The chitin ring appears to be formed largely or entirely through the action of chitin synthase III, one of the three known chitin synthases in S. cerevisiae (Shaw et al., 1991; Bulawa, 1993; Orlean, 1997). In contrast, chitin synthase II appears to be involved in the synthesis of the chitin in the primary septum that separates mother and daughter cells during cell division (Shaw et al., 1991; Bulawa, 1993; Orlean, 1996), whereas chitin synthase I may be involved in cell wall repair (Cabib et al., 1989, 1992). Chitin synthase III synthesizes ∼90% of the total cellular chitin (Bulawa, 1993; Orlean, 1996), and is also largely or entirely responsible for the synthesis of the chitosan (modified chitin) of the spore wall (Briza et al., 1990, 1994; Pammer et al., 1992; Bulawa, 1993).
Genetic screens have identified several genes that are required for chitin synthesis by chitin synthase III (Roncero et al., 1988; Valdivieso et al., 1991; Bulawa, 1992, 1993; Pammer et al., 1992; Kawamoto et al., 1992; Cabib, 1994; Orlean, 1997). These include CHS3 (also known as CSD2, CAL1, CAL4, CAL5, DIT101, or KTI2), CHS4 (also known as CSD4, CAL2, or SKT5), CHS5 (also known as CAL3), and CHS6 (also known as CSD3). Chs3p is a transmembrane protein that is thought to be the catalytic subunit of chitin synthase III (Bulawa, 1993; Chuang and Shekman, 1996), and Chs5p may be involved in the delivery of Chs3p to the budding site (Santos and Snyder, 1997). The function of Chs6p has yet to be determined. Chs4p appears to be an activator of chitin synthase III, as indicated by both in vivo and in vitro analyses (Roncero et al., 1988; Bulawa, 1992, 1993; Trilla et al., 1997). The predicted amino acid sequence of Chs4p (GenBank/ EMBL/DDBJ accession number Z23261, ORF YBL0519; Saccharomyces Genome Database ORF YBL061C; Scherens et al., 1993; Bulawa, 1993; Trilla et al., 1997; Greene, J., and B. DiDomenico, personal communication; Bulawa, C., personal communication) contains a potential E-F hand loop calcium-binding domain and a COOH-terminal CAAX motif preceded by a cluster of basic amino acids, as in the domains responsible for the membrane localization of some p21ras proteins (Hancock et al., 1990).
In this study, we have investigated the role of the septins in the spatial localization of chitin deposition. The results suggest that Chs3p is normally localized by its assembly into a complex that involves the septins, Chs4p, and a novel protein, Bni4p.
MATERIALS AND METHODS
Strains, Growth Conditions, and Genetic and DNA Methods
The yeast strains used in this study are listed in Table I. Yeast media (YM-P rich liquid medium, YPD rich solid medium, synthetic complete [SC]1 medium, SC lacking specific nutrients, and sporulation medium) have been described previously (Lillie and Pringle, 1980; Guthrie and Fink, 1991). Yeast strains were grown at 30°C except where noted. Escherichia coli strains DH5α and DH12S (GIBCO BRL, Gaithersburg, MD) and standard media and methods (Sambrook et al., 1989) were used for plasmid propagation except where noted. E. coli strain JMB9r−m+ Δ-trpF (Sterner et al., 1995) was used to rescue TRP1-containing plasmids from yeast strain EGY48R; transformants were plated directly onto Vogel and Bonner medium (Davis et al., 1980) supplemented with 0.2% glucose, 0.5% casaminoacid hydrolysate, 0.01 M FeCl3, and 75 mg/liter ampicillin.
Table I.
S. cerevisiae Strains Used in This Study
| Name | Relevant Genotype* | Source | ||
|---|---|---|---|---|
| LH104-HO1 | a /α cdc3-1/cdc3-1 | Adams and Pringle, 1984 | ||
| LH17012-HO1 | a /α cdc10-1/cdc10-1 | Adams and Pringle, 1984 | ||
| JPT194-HO1 | a /α cdc11-6/cdc11-6 | Adams and Pringle, 1984 | ||
| JPTA1493-HO1 | a /α cdc12-6/cdc12-6 | Adams and Pringle, 1984 | ||
| 314D5 | a /α cdc4-1/cdc4-1 | Hartwell, 1971b | ||
| CS02A | α cdc12-6 leu2 ura3 | This study‡ | ||
| 124Y03A | a cdc12-5 “sl124” ade2 ade3 leu2 lys2 ura3 [pB12G] | This study§ | ||
| DDY156 | a cdc12-5 “sl124” ade2 ade3 leu2 lys2 ura3 [YEp24(CDC12)N] | This study§ | ||
| JF2 | a cdc12-5 ade2 ade3 his7 leu2 ura3 | See footnote 2 | ||
| 70.2A | α cscl4-3::LEU2 ade2-101 leu2-Δ1 lys2-801 trp1-Δ63 ura3-52 | C. Bulawa‖ | ||
| EGY48R | α his3 trp1 ura3-52 leu2::pLEU2-lexAop6 [pSH18-34] | Note ¶ | ||
| YEF473 | a /α his3-Δ200/his3-Δ200 leu2-Δ1/leu2-Δ1 lys2-801/lys2-801 | Bi and Pringle, 1996 | ||
| trp1-Δ63/trp1-Δ63 ura3-52/ura3-52 | ||||
| DDY172 | as YEF473 except CHS4/chs4-Δ1 | This study** | ||
| DDY172-2A | a chs4-Δ1 his3-Δ200 leu2-Δ1 lys2-801 trp1-Δ63 ura3-52 | Segregant from DDY172 | ||
| DDY172-2AX | as DDY172-2A except [p326] | See text | ||
| DDY173 | as YEF473 except BNI4/bni4-Δ1 | This study** | ||
| DDY174 | as YEF473 except chs4-Δ1/chs4-Δ1 | This study†† | ||
| DDY175 | as YEF473 except bni4-Δ1/bni4-Δ1 | This study†† | ||
| DDY176 | as YEF473 except CHS4-Δ1/chs4-Δ1 BNI4/bni4-Δ1 | This study†† | ||
| DDY179 | as YEF473 except chs4-Δ1/chs4-Δ1 bni4-Δ1/bni4-Δ1 | This study†† | ||
| DDY181 | as YEF473 except CHS3/chs3-Δ1 | This study** | ||
| DDY185 | as YEF473 except CDC10/cdc10-Δ1 | This study** | ||
| DDY185-1A | a cdc10-Δ1 his3-Δ200 leu2-Δ1 lys2-801 trp1-Δ63 ura3-52 | Segregant from DDY185 | ||
| DDY186 | as YEF473 except chs3-Δ1/chs3-Δ1 | This study§§ | ||
| DDY197 | as YEF473 except chs4-Δ1/chs4-Δ1 [p326] | This study‖ ‖ | ||
| DDY212 | as YEF473 except chs4-Δ1/chs4-Δ1 bni4-Δ1/BNI4 [p326] | This study¶ ¶ | ||
| DDY213 | a /α cdc12-6/CDC12-6 CHS4/chs4-Δ1 HIS3/his3-Δ200 leu2/leu2-Δ1 | This study** | ||
| LYS2/lys2-801 TRP1/trp1-Δ63 ura3/ura3-52 [p326] | ||||
| DDY217 | as YEF473 except chs4-Δ1/chs4-Δ1 bni4-Δ1/bni4-Δ1 [p326] | This study†† ‡ | ||
| DDY218 | a /α cdc12-6/cdc12-6 chs4-Δ1/chs4-Δ1 his3-Δ200/HIS3 Leu− Trp+Ura− [p326] | This study§§ § | ||
| DDY242 | as YEF473 except CHS4/chs4-Δ1 CHS3 / chs3-Δ1 [p326] | This study‖ ‖ ‖ | ||
| DDY244 | as YEF473 except chs4-Δ1/chs4-Δ1 chs3-Δ1/chs3-Δ1 [p326] | This study¶ ¶ ¶ |
Plasmids are indicated in brackets and are described in Table II.
Constructed by several crosses from the original cdc12-6 isolate (Adams and Pringle, 1984).
“sl124” refers to the mutation producing synthetic lethality in combination with cdc12-5. Strain 124Y03A is a segregant from a cross of the original synthetic-lethal mutant to strain JF12. Strain DDY156 was derived from 124Y03A as described in the text.
csd4-3::LEU2 has LEU2 inserted at the NheI site (at nucleotide 1011 from the start of the CHS4 ORF) in the csd4-3 mutant allele of CHS4.
Strain EGY48 (Zervos et al., 1993) containing reporter plasmid pSH18-34 (Gyuris et al., 1993).
YEF473 transformed with the appropriate PCR product (Materials and Methods). For chs4-Δ1 and bni4-Δ1, the selectable marker was TRP1; for chs3-Δ1 and cdc10-Δ1, the selectable marker was HIS3.
Constructed by mating appropriate segregants from DDY172, DDY173, and/or DDY176.
Constructed by mating appropriate segregants from DDY181.
∥
∥ Constructed by mating DDY172-2AX to an appropriate segregant from DDY172.
¶
¶ Constructed by mating DDY172-2AX to an appropriate segregant from DDY176.
Constructed by mating CS02A to DDY172-2AX.
‡‡
‡Constructed by mating appropriate segregants from DDY212.
§§
§ Constructed by mating appropriate segregants from DDY213; phenotypes are shown where the precise genotypes are not known.
∥
∥
∥ Constructed by mating DDY172-2AX to an appropriate segregant from DDY181.
¶
¶
¶ Constructed by mating appropriate segregants from DDY242.
Standard methods were used for DNA manipulations and yeast genetics (Sambrook et al., 1989; Guthrie and Fink, 1991) except where noted. Most plasmids used in this study are listed in Table II; others are described where appropriate below. Filters containing λ clones covering the yeast genome (Riles et al., 1993) were purchased from the American Type Culture Collection (Rockville, MD). Vent DNA polymerase (New England Biolabs, Inc., Beverly, MA) was used in the PCR synthesis of DNA fragments for cloning; Taq DNA polymerase (Promega Corp., Madison, WI) was used in other PCR applications. The PCR method of Baudin et al. (1993) was used to generate cassettes for deletion of the complete coding regions of CHS4, CDC10, and CHS3, and for deletion of all but the first 40 nucleotides of the BNI4 coding region. pRS303 and pRS304 (Sikorski and Hieter, 1989) were used as templates for the HIS3 and TRP1 markers used in these cassettes. The primers used are listed in Table III. The amplified cassettes were used to transform strain YEF473, and PCR and/or DNA blot-hybridization analysis was performed to identify transformants that contained the deletions.
Table II.
Plasmids Used in This Study
| Name | Description* | Source or reference | ||
|---|---|---|---|---|
| YEp13 | 2μ, LEU2 | Guthrie and Fink, 1991 | ||
| pRS315 | CEN, LEU2 | Sikorski and Hieter, 1989 | ||
| pRS316 | CEN, URA3 | Sikorski and Hieter, 1989 | ||
| pRS425 | 2μ, LEU2 | Christianson et al., 1992 | ||
| YEp351 | 2μ, LEU2 | Hill et al., 1986 | ||
| YCplac111 | CEN, LEU2 | Gietz and Sugino, 1988 | ||
| pB12G | 2μ, LEU2, CDC12, ADE3 | See footnote 2 | ||
| YEp24 (CDC12)N | 2μ, URA3, CDC12 | See footnote 2 | ||
| pJG4-5 | 2μ, TRP1, transcriptional AD | Ausubel et al., 1993 | ||
| pJG4-5PL | 2μ, TRP1, transcriptional AD | This study‡ | ||
| pEG202 | 2μ, HIS3, lexA DBD | Ausubel et al., 1993 | ||
| pSH18-34 | 2μ, URA3, lexOp-lacZ | Gyuris et al., 1993 | ||
| p196 | CEN, LEU2, ∼12 kb of chromosome II including CHS4 | See text | ||
| p197 | CEN, LEU2, ∼12 kb of chromosome V | See text | ||
| p206 | CEN, LEU2, chs4Δ678 | See text | ||
| p241 | 2μ, HIS3, DBD-CHS4 | See text | ||
| p267 | CEN, URA3, CHS4 | See text | ||
| p279 | 2μ, TRP1, AD-bni4E § | See text | ||
| p326 | CEN, URA3, GFP-CHS4 | See text | ||
| p348 | 2μ, HIS3, DBD-chs4C693S | See text | ||
| p356 | 2μ, LEU2, BNI4 | See text | ||
| p357 | CEN, URA3, chs4Δ610 | See text | ||
| p361 | CEN, URA3, chs4C693S | See text | ||
| p365 | 2μ, LEU2, BNI4 | See text | ||
| p366 | CEN, LEU2, BNI4 | See text | ||
| p367 | 2μ, LEU2, bni4Δ508 | See text | ||
| p368 | CEN, LEU2, bni4Δ508 | See text | ||
| p372 | 2μ, LEU2, BNI4 | See text | ||
| p374 | 2μ, LEU2, bni4Δ20 | See text | ||
| p376 | CEN, LEU2, chs4Δ560 | See text | ||
| p377 | CEN, LEU2, chs4Δ79 | See text | ||
| p408 | CEN, LEU2, CHS3 | See text | ||
| p422 | CEN, LEU2, CDC12 | M.S. Longtine‖ |
2μ indicates high-copy plasmids; CEN indicates low-copy plasmids.
pJG4-5PL is identical to pJG4-5 except that its polylinker has been replaced with that of pEG202.
bni4E encodes amino acids 65 to ∼730 of Bni4p. This plasmid was a primary isolate from the two-hybrid screen (see text).
Contains CDC12 on an ∼2.0-kb XbaI/PstI fragment ligated to XbaI/PstI-digested YCplac111.
Table III.
Oligonucleotide Primers Used for PCR-mediated Gene Deletion and Cloning
| Name of primer | Sequence of primer | |
|---|---|---|
| CHS4-5′-TRP1* | CGTCTTTTTTGGTGATAAAGTTAAAATAAAAGGATAAAAAGTACAATCTTGATCCGGAGC | |
| CHS4-3′-TRP1* | GTTGCACCTATAAAGAATGAAAACAATCTAGTATGTGTACCTCCTTACGCATCTGTGCGG | |
| CDC10-5′-HIS3* | CAAGCCCCACGGTTACTACAAGCACTGTATAAATATATTAGATTGTACTGAGAGTGCACC | |
| CDC10-3′-HIS3* | AGGAGAGAAGCAAGTTACGGTCTTTTCATATAGAATAATTCTGTGCGGTATTTCACACCG | |
| CHS3-5′-HIS3* | GGTCCTGTTTAGACTATCCGCAGGAAAGAAATTAGAAAGAGCTTTGGTGAGCGCT | |
| CHS3-3′-HIS3* | GTAAGTATCACAGTAAAAATATTTTCATACTGTCTATCCGTCGAGTTCAAGAGA | |
| BNI4-5′-TRP1* | ATGTCGGATAGTATTTCAGATTCAAAGTCCTCAGAACTTTGTACAATCTTGATCCGGAGC | |
| BNI4-3′-TRP1* | ATTTGATTCATTTCCATTTCTCCCAGTTTTCTGCTCTAATCTCCTTACGCATCTGTGCGG | |
| CHS4-5′‡ | CGCGGATCCAAATGGCAAGTTCACCGC | |
| CHS4-3′‡ | CGCGGATCCCAGTCCAAAAACCCCAG | |
| CHS4-3′C693S§ | ACAATCCTCGAGGTGTACTTACATAATTACAGAGTCTTTTTTTTTTTTACCTTG | |
| CHS3F5′‖ | CGGCGGCCATGGATGGGCAAGTTCACCGCAGGTACATCC | |
| CHS3MR¶ | GGGCTCGAGCGTTGCCCGAGCCCTTAATTAAACCATCACAAAC |
Primers used to generate deletion cassettes for the indicated genes. Nucleotides corresponding to the flanking regions of the gene to be deleted are in bold face; the remaining nucleotides correspond to regions upstream and downstream of the selectable marker gene.
BamHI sites and the first ATG of CHS4 are underlined.
A XhoI site and the missense mutation that substitutes serine for cysteine are underlined.
A NcoI site and the first ATG of CHS3 are underlined.
A XhoI site is underlined.
Cloning of CHS4 and CHS3
A plasmid shuffle was performed in strain 124Y03A (Table I) to replace plasmid pB12G with plasmid YEp24(CDC12)N (Table II), thus creating strain DDY156. This strain was transformed with a yeast genomic DNA library in plasmid YCp50-LEU2 (kindly provided by F. Spencer and P. Hieter, Johns Hopkins University, Baltimore, MD), and the transformants were replica-plated to SC-Leu supplemented with 0.1% 5-fluoroorotic acid (5-FOA) to select against plasmid YEp24(CDC12)N. A complementing plasmid, p196, contained CHS4 (see Results). A 3.4-kb XbaI–HindIII fragment, which encodes amino acids 1–678 of CHS4 (Scherens et al., 1993; see Fig. 2 A), was subcloned from p196 into pRS315 (Table II), thus creating p206. Unidirectional deletions of DNA were made in this plasmid using appropriate restriction enzymes and a modified exonuclease III procedure (Henikoff, 1984; Beltzer et al., 1986), thus creating a series of plasmids containing CHS4 truncated to different extents at the 3′ end of the coding region. Included in this series of plasmids are p377 and p376, which contain alleles chs4Δ79 and chs4Δ560, which encode the NH2-terminal 79 and 560 amino acids of Chs4p, respectively. The complete CHS4 gene was subcloned from p196 into pRS316 as a BglII–EcoRV fragment that contained ∼1 kb on either side of the CHS4 open reading frame (ORF; see Fig. 2 A), thus creating p267. p267 was digested with EcoRI (one site in CHS4 and the other in vector sequences) and religated to remove the 3′ 259 nucleotides of the CHS4 ORF and all of the cloned DNA downstream of CHS4; the resulting plasmid, p357, contains the chs4Δ610 allele, which encodes amino acids 1–610 of Chs4p. p361 was constructed by simultaneously ligating a 2.7-kb XbaI–EcoRI fragment of p267 (XbaI site from vector sequences) and a 270-bp EcoRI–XhoI fragment of p348 (see below) into XbaI/XhoI-digested pRS316; this plasmid contains the chs4C693S allele.
Figure 2.

(A) Physical maps of the CHS4 and BNI4 regions and structures of the chs4-Δ1 and bni4-Δ1 deletion alleles. Open boxes represent open reading frames, and stippled boxes represent the TRP1 gene plus flanking sequences. Arrows indicate the directions of transcription. Solid black bars represent the fragments used to probe DNA blots. Lines beneath the CHS4 map represent segments of DNA assayed (in the indicated low-copy plasmids) for complementation of the synthetic lethality of strain DDY156 or 124Y03A (SL) and of the chitin synthesis defect of the chs4-Δ1 strain DDY174 (CS); AA indicates the COOH-terminal Chs4p amino acid encoded by the corresponding DNA. Restriction enzymes: G, BglII; H, HindIII; N, NsiI; R, EcoRI; S, SalI; Sa, Sau3AI; Sp, SphI; V, EcoRV; X, XbaI. There are other Sau3AI sites in the BNI4 region; those indicated represent the ends of the insert in plasmid p356 (see text). (B) DNA–DNA blot-hybridization analyses of chs4-Δ1 and bni4-Δ1 strains. Genomic DNAs were digested with HindIII. Lanes 1–6 were probed with a 2.9-kb BglII–HindIII fragment that contains most of CHS4 and some upstream DNA (see A). Lanes 7–12 were probed with a 2-kb EcoRV fragment that contains a portion of BNI4 and some downstream DNA (see A) together with a portion of the tetR gene of YEp13. Lanes 1 and 7, YEF473 (wild-type); lanes 2 and 8, DDY176 (chs4-Δ1/CHS4 bni4-Δ1/BNI4); lanes 3–6 and 9–12, the four segregants from one tetratype tetrad from DDY176. Fragments of the predicted sizes were visualized in all lanes (note the presence of a HindIII site in TRP1); their molecular weights are indicated in kb.
To construct plasmids for two-hybrid analysis, CHS4 was amplified by PCR using primers CHS4-5′ and CHS4-3′ (Table III) and plasmid p196 as template. The BamHI-digested PCR product was ligated into BamHI- digested pEG202 (Table II), thus fusing full-length CHS4 to the lexA DNA-binding domain (DBD) sequences and creating p241. In addition, an EcoRI fragment of p241 was subcloned into EcoRI-digested pEG202; the resulting plasmid contains the DBD sequences fused to the chs4Δ610 allele (see above). To construct the chs4C693S allele (encoding Chs4p with Ser substituted for Cys at amino acid 693), CHS4 DNA was amplified by PCR using primers CHS4-5′ and CHS4-3′C693S and p196 as template. The BamHI/XhoI-digested PCR product was ligated into BamHI/XhoI- digested pEG202, thus creating p348, which contains the DBD sequences fused to the chs4C693S allele. Finally, a fragment encoding amino acids 1–700 of the 1165–amino acid Chs3p (Bulawa, 1992) was amplified by PCR using primers CHS3F5′ and CHS3MR and genomic DNA as template. The NcoI/XhoI-digested PCR product was ligated into NcoI/XhoI-digested pEG202 and pJG4-5PL, thus creating plasmids containing chs3Δ700 fused to the DBD sequences and activation domain (AD) sequences, respectively.
To construct a fusion gene encoding full-length Chs4p fused to the green fluorescent protein (GFP) (Chalfie et al., 1994), a fragment containing GFP-encoding sequences was amplified by PCR using primers that contained both EcoRI and NotI restriction sites and pS65T-C1 (Clontech, Palo Alto, CA) as template. The PCR product was digested with EcoRI and ligated into EcoRI-digested pUC18, thus creating pUC18GFP. CHS4 was subcloned from p267 in an XbaI–KpnI fragment (both sites from the polylinker) into XbaI/KpnI-digested pALTER (Promega Corp.), and a NotI site was introduced immediately upstream of the CHS4 ORF using the procedures recommended by Promega Corp., thus creating pANCHS4. The NotI fragment containing GFP-encoding sequences was isolated from pUC18GFP and ligated into NotI-digested pANCHS4, thus fusing the GFP and CHS4 open reading frames in plasmid pANGFPCHS4. The XbaI–KpnI fragment that contained the GFP–CHS4 fusion was isolated from pANGFPCHS4 and ligated into XbaI/KpnI- digested pRS316. The ligation mixture was transformed directly into strain DDY172-2A (chs4-Δ1; Table I), and Ura+ transformants were screened for normal chitin deposition by Calcofluor staining. One such transformant, DDY172-2AX, was considered to contain the desired GFP-CHS4 plasmid, p326. Attempts to recover this plasmid in E. coli were unsuccessful.
To construct plasmid p408, the CHS3-containing plasmid pHV7 (Valdivieso et al., 1991) was digested with EcoRI and SalI (both sites from vector sequences). The CHS3-containing fragment was ligated into EcoRI/SalI-digested pBSKS+ (Stratagene Inc., La Jolla, CA), thus creating pBSKSCHS3. This plasmid was digested with SalI and NotI, and the CHS3-containing fragment was ligated into SalI/NotI-digested pRS315. The ligation mixture was transformed directly into strain DDY244 (chs3-Δ1/chs3-Δ1 chs4-Δ1/chs4-Δ1 [p326]; Table I). Leu+ Ura+ transformants were selected and screened by Calcofluor staining. Several transformants displayed normal Calcofluor staining and were thus considered to contain p408. Attempts to recover this plasmid in E. coli were unsuccessful.
Cloning of BNI4
A yeast genomic DNA library was constructed by ligating BamHI- digested YEp13 to partially Sau3AI-digested yeast genomic DNA from strain JC01A (α cdc12-5 met1; S288C background) and transforming the ligation mixture into E. coli. Approximately 5.5 × 104 transformants were obtained, of which ∼95% contained cloned DNA. A DNA fragment from p279 (Table II) was used to screen this library by colony hybridization. A BNI4-containing plasmid, p356, was isolated (see Fig. 2 A). BNI4 was subcloned from p356 by digesting with SphI (one site in the vector sequences, one site downstream of BNI4), treating with T4 DNA polymerase, and ligating into SmaI-digested pRS425 and pRS315, thus creating high-copy (p365) and low-copy (p366) BNI4 plasmids. p367 and p368 were constructed by digesting p365 and p366, respectively, with BglII (which cuts uniquely in the BNI4 ORF), treating with T4 DNA polymerase, and religating; the resulting bni4Δ508 allele encodes amino acids 1–508 of Bni4p followed by 12 missense amino acids. In addition, a BNI4-containing HindIII–SacI fragment (both sites from the polylinker) was subcloned from p365 into YEp351, thus creating p372. This plasmid was digested with SalI and religated, thus creating p374, in which the BNI4 open reading frame is truncated after codon 20.
Two-hybrid Analyses
Two-hybrid analysis (Fields and Sternglanz, 1994) was performed using strain EGY48R and the system described by Gyuris et al. (1993). Screening was performed using CHS4 plasmid p241 as the bait, and a yeast genomic DNA library in pJG4-5. Leu+ transformants were screened for galactose-dependent β-galactosidase activity by filter assays (Shapira et al., 1983), and quantitative β-galactosidase assays were then performed in duplicate or triplicate on promising candidates essentially as described previously (Ausubel et al., 1993). Directed two-hybrid analyses were quantitated similarly. Fusions of full-length CDC3, CDC10, CDC11, and CDC12 to the AD sequences in pJG4-5 and the DBD sequences in pEG202 have been described previously (De Virgilio et al., 1996).
Antibodies and Protein Procedures
Cdc11p-specific and Chs3p-specific antibodies have been described previously (Ford and Pringle, 1991; Chuang and Schekman, 1996). To prepare Bni4p-specific antibodies, a malE–BNI4 fusion was constructed by subcloning an EcoRI–SalI fragment encoding Bni4p amino acids 65–497 (see Fig. 2 A; the EcoRI site was in the vector) from p279 into pMAL-c2 (New England Biolabs). The fusion protein produced by the resulting plasmid was largely degraded upon induction (data not shown). Therefore, this plasmid was digested with NsiI and SalI (see Fig. 2 A), treated with T4 DNA polymerase, and religated, thus creating plasmid p353, which encodes MalE fused to amino acids 65 to 288 of Bni4p. This fusion protein was not degraded extensively during induction or extraction (data not shown). In addition, the same fragment of BNI4 was subcloned from p353 (using sites from the vector) into pGEX1 (Pharmacia LKB Biotechnology Inc., Piscataway, NJ), thus creating p363, which encodes glutathione-S-transferase (GST) fused to amino acids 65–288 of Bni4p. MalE-Bni4p was extracted from E. coli containing p353 as described by Maina et al. (1988), purified on an amylose resin column (New England Biolabs), and used to raise antibodies by standard procedures (Cocalico Biologicals, Reamstown, PA). To affinity-purify Bni4p-specific antibodies, GST-Bni4p was extracted from E. coli containing p363 as described by Ausubel et al. (1993), purified on a glutathione-agarose column (Thiol Picky Gel; Molecular Probes, Inc., Eugene, Oregon), electrophoresed on SDS gels (Laemmli, 1970), and blotted to nitrocellulose. Affinity purification was then performed as described by Ford and Pringle (1991).
Yeast proteins were isolated for Western analysis by boiling cells in 2.5× Laemmli buffer (Laemmli, 1970). Western analysis was performed as described previously (Haarer and Pringle, 1987). Blots were stained with Ponceau S to ensure equal protein loading.
Morphological Observations
Cells to be observed were grown in liquid media except for those containing GFP-Chs4p (see below). Overall cell morphologies were observed using an oil-immersion ×60 objective with differential-interference-contrast (DIC) optics. Immunofluorescence and visualization of chitin deposition by Calcofluor staining were performed as described previously (Pringle et al., 1989). The localization of GFP-Chs4p was visualized by growing cells containing plasmid p326 on SC-Ura or SC-Ura-Leu solid medium for 16– 20 h at 23°C. Cells were scraped from the plates into ice-cold 70% ethanol and incubated for 10 min on ice. The cells were then collected by centrifugation, resuspended in water, and observed by epifluorescence microscopy using an FITC filter set. The localization of GFP-Chs4p was visualized similarly in temperature-sensitive mutant cells, except that some plates were shifted to 37°C for 20 min before collecting the cells in 70% ethanol. All microscopy was performed using a Microphot SA microscope (Nikon Inc., Melville, NY).
RESULTS
Delocalization of Chitin Deposition in Septin Mutants
The septin proteins form a ring at the presumptive bud site shortly before the chitin ring is formed in the overlying cell wall (see above). These observations suggested that the septins might be involved in the localization of chitin deposition. To address this possibility, temperature-sensitive septin mutants (cdc3, cdc10, cdc11, and cdc12) were shifted from permissive to restrictive temperature, and cells were examined by fluorescence microscopy after staining with Calcofluor. In each mutant, the chitin rings and bud scars formed during growth at permissive temperature appeared normal (Fig. 1, A–D), and their appearance did not change after the shift to restrictive temperature (Fig. 1, E–H). The first buds formed during growth at restrictive temperature, however, did not have discrete chitin rings at their bases. Instead, the Calcofluor-stainable material formed a diffuse band whose intensity decreased with distance from the mother-bud neck (Fig. 1, E–H). In contrast, several other temperature-sensitive cell-cycle mutants (cdc7, cdc13, cdc21, cdc31) displayed apparently normal chitin rings at the bases of the buds formed during cell-cycle arrest at restrictive temperature (data not shown). Of particular interest was the cdc4 mutant, which forms abnormally elongated buds like those of the septin mutants (Hartwell, 1971b ) but maintains its septin organization (Byers, B., and L. Goetsch. 1976. J. Cell Biol. 70:35a; Haarer and Pringle, 1987; Kim et al., 1991) and forms normal-looking chitin rings (Fig. 1 I) at restrictive temperature. Thus, it appears that the delocalization of chitin deposition in the septin mutants results specifically from the loss of septin function and is not simply a consequence of cell-cycle arrest or abnormal bud shape.
Figure 1.
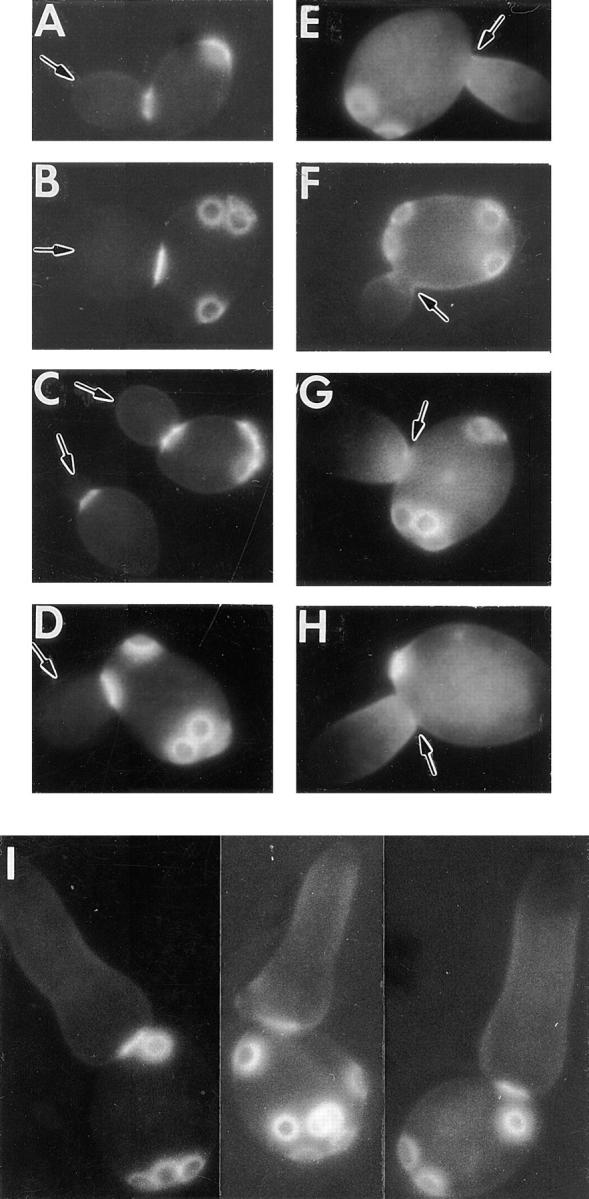
Delocalization of chitin deposition in septin mutants. Cells were examined during growth at 23°C (A–D) or after a shift to 37°C for 80–120 min (E–I). (A and E) cdc3 strain LH104-HO1; (B and F) cdc10 strain LH17012-HO1; (C and G) cdc11 strain JPT194-HO1; (D and H) cdc12 strain JPTA1493-HO1; (I) cdc4 strain 314D5. Arrows in A–D indicate growing buds; arrows in E–H indicate the mother-bud necks lacking normal chitin rings; bud scars formed during earlier growth at permissive temperature are also visible on these cells.
Synthetic Lethality Between cdc12-5 and chs4
A genetic link between the septins and chitin synthesis was uncovered during a screen for mutations that are synthetically lethal with the cdc12-5 septin mutation. This screen identified several complementation groups,2 one of which, defined by strain 124Y03A, required at least two additional mutations in combination with cdc12-5 to produce the synthetic-lethal phenotype. To identify the genes defined by these mutations, strain DDY156 (a derivative of 124Y03A containing a URA3-marked CDC12 plasmid; see Materials and Methods) was transformed with a genomic DNA library in a low-copy, LEU2-marked vector, and candidate plasmids were selected on the basis of their ability to allow growth on medium containing 5-FOA, which selects against the URA3-marked plasmid. Two of the ∼8,000 transformants screened contained plasmids that complemented the synthetic lethality. One plasmid, p196, complemented well, and the other, p197, complemented poorly.
To begin characterizing p196 and p197, portions of the cloned DNAs were used as probes for DNA–DNA blot-hybridization analyses. The p196 and p197 DNAs did not hybridize to each other or to CDC12 DNA. In addition, the p196 DNA hybridized to DNA from the left arm of chromosome II (λ′ clone ATCC numbers 70693, 70310, and 71168), whereas the p197 DNA hybridized to DNA from the left arm of chromosome V (λ′ clone ATCC numbers 70355 and 70842). CDC12 is on the right arm of chromosome VIII (Mortimer and Schild, 1980). Thus, p196 and p197 contain different genes, and neither contains CDC12. Because p196 allows DDY156 to grow relatively well in the presence of 5-FOA (i.e., in the absence of plasmid-borne CDC12), it presumably complements the mutation that is most deleterious in combination with cdc12-5. This plasmid was therefore chosen for further characterization.
To identify the gene of interest in p196, various fragments were subcloned into pRS315 (low copy, LEU2) or pRS316 (low-copy, URA3) and transformed into strain DDY156 or 124Y03A. A 3.4-kb XbaI–HindIII fragment (Fig. 2 A, plasmid p206) was sufficient for complementation of the synthetic lethality. Partial sequence analysis of this fragment (data not shown) and comparison to the full sequence of this chromosome region (Scherens et al., 1993; accession number Z23261) revealed that the fragment contains two truncated ORFs. One of these contains all but the COOH-terminal 18 codons of YBL0519 or CHS4/ CSD4/CAL2/SKT5 (Bulawa, 1992, 1993; Kawamoto et al., 1992; Trilla et al. 1997; Greene, J., and B. DiDomenico, personal communication; Bulawa, C., personal communication), a gene involved in chitin synthesis (see introduction), and the other contains the NH2-terminal 272 codons of YBL0517, which encodes a 687–amino acid protein of unknown function. A 3.8-kb BglII–EcoRV fragment (Fig. 2 A, plasmid p267), which contains all of CHS4 but only 103 codons of YLB0517, was also sufficient for complementation of the synthetic lethality, suggesting that CHS4 was the gene responsible for the complementation. To confirm this conclusion, restriction enzyme digestion and an exonuclease procedure (see Materials and Methods) were used to truncate CHS4 further. Plasmids that contained as little as 560 codons of CHS4 still complemented well (Fig. 2 A, plasmid p376), but a plasmid that contained only 79 codons of CHS4 (together with the same portion of YBL0517) failed to complement (Fig. 2 A, plasmid p377). Thus, CHS4 indeed appears to be responsible for the observed complementation. Because the two reported sequences for the CHS4 region differ (Kawamoto et al., 1992; Scherens et al., 1993), we resequenced the regions of discrepancy (nucleotides −20–80 and 1655–1755 of YBL0519). Our sequence agrees with that of Scherens et al. (1993), which predicts that CHS4 encodes a protein of 696 amino acids that contains a putative calcium-binding domain (amino acids 237–250) and terminates in a probable prenylation site (CAAX motif) (Fig. 3 A).
Figure 3.

Amino acid sequences of Chs4p and Bni4p. Asterisks represent the termination codons. (A) Portions of the predicted amino acid sequence of Chs4p that include the possible calcium-binding domain (amino acids 237–250; underlined) and the COOH terminus including the CAAX motif (amino acids 693– 696; underlined). (B) Predicted sequence of Bni4p. The predicted coiled-coil domain (amino acids 106–139) is underlined, and the first residue translated from the BNI4-containing AD fusion clone is overlined.
Because of the complicated genetics of strain 124Y03A, we asked if cdc12-5 and a mutant allele of CHS4 (csd4-3:: LEU2; Table I, note ∥) were synthetically lethal in the absence of other known mutations. Strain JF2 (cdc12-5) was mated to strain 70.2A (csd4-3::LEU2), and random spores from the resulting diploid were analyzed at 23°C. From a total of 100 viable segregants recovered, 42 were Leu+, and thus contained the csd4-3::LEU2 mutation. Of these 42, 20 were unable to grow at 37°C, indicating that they also contained the cdc12-5 mutation. Of these 20, 8 were also inviable at 30°C, a temperature at which strain JF2 can grow. In addition, at 23°C, all of the putative double mutants displayed the morphological abnormalities characteristic of both single mutants when grown at 37°C (i.e., elongated buds for cdc12-5 [Adams and Pringle, 1984] and high vacuole content for csd4-3::LEU2 [Bulawa, C., personal communication]). Thus, cdc12-5 and csd4-3::LEU2 do have a synthetic phenotype, but whether the double mutants are viable or not depends on the growth temperature and the genetic background, consistent with the observations on strain 124Y03A itself.
Two-hybrid Analysis of Chs4p
The data described above suggested that Chs4p might interact directly with Cdc12p or one of the other septins. To test this possibility, we performed directed two-hybrid analysis using LexA DBD fusions of full-length Chs4p, a truncated Chs4p that contains amino acids 1–610 (the product of chs4Δ610), and Chs4p with an altered CAAX motif (the product of chs4C693S), together with AD fusions of full-length Cdc3p, Cdc10p, Cdc11p, and Cdc12p (see Materials and Methods). None of the combinations displayed significant interaction (data not shown). These results suggested that the Chs4p-septin interaction might be indirect. Therefore, to identify other potentially relevant proteins, a two-hybrid screen was performed using DBD-Chs4p (from plasmid p241) as the bait (see Materials and Methods). 200 Leu+ colonies were picked from a total of ∼8 × 106 transformants; of these 200, 38 proved to be galactose-dependent for both leucine prototrophy and β-galactosidase activity, indicating that the plasmids in these strains encoded proteins that interacted with DBD-Chs4p. Sequencing of the vector-insert junctions revealed that the 38 plasmids defined 10 genes. To identify genes whose products might be involved in septin-dependent chitin deposition, representative plasmids were tested in two- hybrid assays against DBD fusions of full-length Cdc3p, Cdc10p, Cdc11p, and Cdc12p. One plasmid, p279, containing a portion (denoted bni4E) of a gene we named BNI4 (bud neck involved), showed interaction with DBD-Cdc10p as well as with DBD-Chs4p (Table IV), and was thus chosen for further analysis (see below). Other Chs4p interactors will be described elsewhere.
Table IV.
Two-hybrid Interactions among Chs4p, Bni4p, the Septins, and Chs3p
| DBD fusion* | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AD fusion* | Chs4p | Chs4pC693S | Chs4pΔ610 | Cdc3p | Cdc10p | Cdc11p | Cdc12p | Chs3pΔ700 | Control§ | |||||||||
| Bni4pE‡ | 238 | 2224 | 3 | 9 | 1006 | 8 | 0 | 2 | 2 | |||||||||
| Chs3pΔ700 | 617 | 568 | 57 | 2 | 2 | 0 | 0 | 123 | 3 | |||||||||
| Control§ | 20 | 18 | 25 | 14 | 38 | 14 | 3 | 3 | ND | |||||||||
Shown for each pair of plasmids are the units of β-galactosidase activity, measured as described in Materials and Methods. Interactions considered to be positive (β-galactosidase ativities at least fivefold greater than background and more than 100 U) are in italics.
See Materials and Methods.
Primary isolate p279 from the library in pJG4-5 (see text); the bni4E fragment encodes amino acids 65 to ∼730 of Bni4p.
Control plasmids are either pJG4-5 (AD fusion vector) or pEG202 (DBD fusion vector).
Because Chs4p does not require its COOH-terminal region to complement the chitin synthesis defect caused by the chs4-Δ1 allele (see below), we also tested DBD-Chs4pΔ610 and DBD-Chs4pC693S against the AD-Bni4pE isolate. A strong signal was observed with DBD-Chs4pC693S, but no interaction was observed with DBD-Chs4pΔ610 (Table IV). Therefore, the two-hybrid interaction between DBD-Chs4p and AD-Bni4pE appears to require the COOH-terminal 86 amino acids of Chs4p, but not the CAAX motif. (The increased two-hybrid signal with DBD-Chs4pC693S, relative to DBD-Chs4p, probably reflects a lack of prenylation that allows more of the protein to enter the nucleus.)
Because Chs4p is an activator of chitin synthase III (Bulawa, 1993), we also performed two-hybrid analyses using the Chs4p, Bni4p, and septin constructs together with DBD and AD fusions to a fragment containing the NH2-terminal 700 amino acids of Chs3p (the catalytic subunit of chitin synthase III). The Chs3p fragment interacted with Chs4p, Chs4pC693S, and itself, but not (or only very weakly) with Chs4pΔ610, Bni4pE, or any of the septins (Table IV).
Taken together, the two-hybrid results suggest that Chs4p may interact directly with Chs3p and indirectly with the septins through Bni4p.
Cloning, Mapping, and Sequence Analysis of BNI4
The BNI4 fragment from the original two-hybrid clone was used to obtain the full-length gene by colony hybridization of E. coli harboring a yeast genomic-DNA library in plasmid YEp13 (see Materials and Methods). One plasmid, p356, was found to contain the complete BNI4 ORF plus at least 0.6 kb of DNA on either side (Fig. 2 A). A BNI4-containing fragment extending from the SphI site of YEp13 to the SphI site immediately downstream of BNI4 (Fig. 2 A) was subcloned from p356 into the low-copy vector pRS315. The resulting plasmid (p366) complemented the bni4-Δ1 allele (see below). BNI4 (accession number Z69381, ORF N1146 [Pandolfo et al., 1996]; Saccharomyces Genome Database ORF YNL233W) has an open reading frame of 892 codons predicted to encode a 100,588-Mr protein with a net charge of −44 (Fig. 3 B). Bni4p is predicted (Lupas et al., 1991) to contain a coiled-coil domain (probability >90%) from amino acids 106–139 (Fig. 3 B); no other motifs or homologies have been identified. The region upstream of BNI4 (nt −290 to −285) contains an MluI restriction site, suggesting that this gene might be regulated in a cell-cycle–dependent fashion (Andrews and Mason, 1993; Koch and Nasmyth, 1994; Breeden, 1996). Hybridization to a λ′ clone (ATCC number 70396; Riles et al., 1993) agreed with the sequence analysis in placing BNI4 on the left arm of chromosome XIV between ZWF1 and MDG1.
Genetic Analysis of CHS4 and BNI4
To examine the phenotypes resulting from the loss of CHS4 and/or BNI4 function, these ORFs were deleted (Fig. 2 A) in the same genetic background. DNA–DNA blot-hybridization analyses confirmed the success of these deletions (Fig. 2 B). For both DDY172 (CHS4/chs4-Δ1) and DDY173 (BNI4/bni4-Δ1) (Table I), viability segregated 4+:0−, and TRP1 segregated 2:2 at 22, 30, 37, and 39°C, indicating that neither CHS4 nor BNI4 is essential for viability. chs4-Δ1 and bni4-Δ1 haploids were mated, and the resulting diploid (DDY176) was subjected to tetrad analysis. Segregants that contained both mutations were viable at temperatures from 22–37°C and had growth rates similar to those of their CHS4 BNI4 siblings. DIC microscopy revealed that some chs4-Δ1 cells had enlarged bud necks and that many had protuberances at previous division sites (Fig. 4 B; cf. wild-type cells in Fig. 4 A), as noted previously for chs1 chs3 mutants by Shaw et al. (1991). Similarly, the majority of bni4-Δ1 cells had enlarged bud necks, and some had protuberances at previous division sites (Fig. 4 C), and the majority of chs4-Δ1 bni4-Δ1 cells had both enlarged bud necks and protuberances (Fig. 4 G). The double-mutant cells also were extensively vacuolated.
Figure 4.
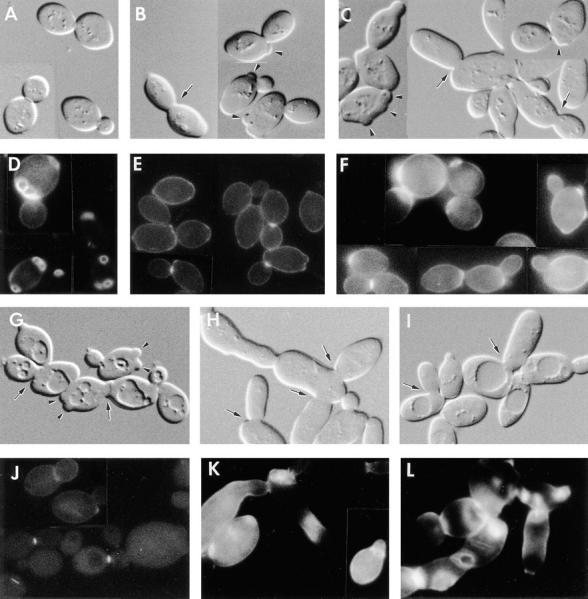
Phenotypic analysis of chs4 and bni4 mutants and of a BNI4-overexpressing strain. Shown are DIC micrographs (A–C, G–I) and fluorescence micrographs of Calcofluor-stained cells (D–F, J–L) of strains (A and D) YEF473 (wild-type); (B and E) DDY174 (chs4-Δ1/chs4-Δ1); (C and F) DDY175 (bni4-Δ1/bni4-Δ1); (G and J) DDY179 (chs4-Δ1/chs4-Δ1 bni4-Δ1/bni4-Δ1); (H and K) DDY175 containing plasmid p356 (high-copy BNI4); and (I and L) DDY185-1A (cdc10-Δ1). All strains were grown at 30°C except DDY185-1A, which was grown at 23°C. Arrowheads indicate protuberances at previous division sites; arrows indicate larger-than-normal bud necks.
Consistent with previous evidence that Chs4p is essential for normal chitin synthase III activity (Roncero et al., 1988; Bulawa, 1992, 1993), Calcofluor staining showed that the chs4-Δ1 mutants and chs4-Δ1 bni4-Δ1 double mutants lacked bud scars, although some cells did display Calcofluor-staining material at the narrowest point of constriction in the mother-bud neck (Fig. 4, E and J; cf. Fig. 4 D and Fig. 1, A–D), presumably due to chitin synthesized by chitin synthase I and/or II (Bulawa, 1993; Orlean, 1997). In contrast, examination of bni4-Δ1 strains revealed that Calcofluor-staining material was abundant but partially delocalized (Fig. 4 F). This phenotype and those described below were complemented by a low-copy plasmid containing full-length BNI4 (p366) but not by a plasmid that contained BNI4 truncated after codon 508 (p368) (data not shown). Thus, Bni4p appears to be required for normal localization of chitin deposition, and the COOH-terminal portion of Bni4p is required for its function. In addition, because the Calcofluor-staining material seen in the mother-bud necks of chs4-Δ1 bni4-Δ1 double-mutant cells (Fig. 4 J) is indistinguishable from that seen in chs4-Δ1 cells (Fig. 4 E), it appears that Bni4p acts specifically in the chitin synthase III pathway.
Several truncated alleles of CHS4 complemented the synthetic lethality between cdc12-5 and chs4 (see above and Fig. 2 A). Therefore, we determined if these truncated alleles and chs4C693S could also complement the chitin synthesis defect of a chs4-Δ1 strain (DDY174). Interestingly, plasmids p361 (chs4C693S), p206 (chs4Δ678), p357 (chs4Δ610), and p376 (chs4Δ560) all rescued the chitin synthesis defect as assayed by Calcofluor staining, although p377 (chs4Δ79) did not (Fig. 2 A). In the cases of chs4C693S, chs4Δ678, and chs4Δ610, the chitin synthesized was localized normally, but in the case of chs4Δ560, chitin was delocalized (data not shown).
To determine the effects of Bni4p overexpression, a high-copy BNI4 plasmid (p356) was transformed into BNI4 (YEF473) and bni4-Δ1 (DDY175) strains. The resulting transformants were viable at 23, 30, and 37°C. However, the majority of cells grew with multiple, elongated buds with enlarged bud necks and displayed delocalized chitin deposition (Fig. 4, H and K). This phenotype was similar to that of cdc10-Δ1 mutants (Fig. 4, I and L; Flescher et al. [1993]). Immunoblot analysis (see Fig. 5) confirmed that Bni4p indeed accumulates to higher-than-normal levels when plasmid p356 is present. To confirm that the abnormal morphology was indeed due to BNI4 overexpression (and not to some other element in p356), a high-copy BNI4 plasmid (p372) and a derivative that contained the same DNA except for a fragment internal to the BNI4 ORF (p374) were constructed and transformed into wild-type strain YEF473. Transformants that carried p372 had the abnormal morphology, whereas those that carried p374 had normal morphology (data not shown). In addition, a high-copy plasmid that encodes only the NH2-terminal 508 amino acids of Bni4p (p367) caused the same morphology as did the plasmids carrying full-length BNI4, although Bni4pΔ508 was unable to complement the bni4-Δ1 allele when expressed from either low-copy (p368) or high-copy (p367) plasmids (data not shown). Thus, the abnormal morphology seen in strains carrying multiple copies of BNI4 is in fact due to overaccumulation of Bni4p, which suggests that excess Bni4p can titrate a factor or factors required for normal chitin deposition and cytokinesis. The NH2-terminal 508 amino acids of Bni4p appear to be sufficient for this titration.
Figure 5.

Immunoblot analysis of Bni4p-specific antibodies. Extracts of bni4-Δ1/bni4-Δ1 strain DDY175 (lane 1); wild-type strain YEF473 (lane 2); and strain YEF473 containing the high-copy BNI4 plasmid p356 (lane 3) were analyzed using affinity-purified antibodies to Bni4p (see Materials and Methods). The mobilities of molecular mass markers are indicated.
Localization of Bni4p in Wild-type and Mutant Strains
To determine the intracellular localization of Bni4p, we raised antibodies against a MalE-Bni4p fusion protein and affinity-purified them using a GST-Bni4p fusion protein (see Materials and Methods). In extracts of wild-type cells, the purified antibodies recognized primarily a polypeptide of approximately the Mr predicted for Bni4p (Fig. 5, lane 2). This polypeptide was absent in extracts of a bni4-Δ1 strain (Fig. 5, lane 1) and present at a higher level (along with several probable breakdown products) in extracts of a strain containing a high-copy BNI4 plasmid (Fig. 5, lane 3), confirming that the antibodies are specific for Bni4p.
In immunofluorescence experiments on wild-type cells grown in rich medium, Bni4p was detected as a single ring on most unbudded cells and as a single ring on the mother-cell side of the mother-bud neck in nearly all small-budded cells and most medium-budded cells (Fig. 6 A). In some medium-budded and many large-budded cells, Bni4p appeared as a double ring in the mother-bud neck, although with the signal on the bud side generally weaker than that on the mother-cell side (Fig. 6 A, arrowheads). The similarity in appearance between most of the Bni4p rings seen on unbudded cells and those seen at the necks of small-budded cells suggests that the former represent sites of incipient bud emergence. However, some of the Bni4p rings on unbudded cells appear to represent residual Bni4p at the preceding division site, a hypothesis supported by the observation of some cells with Bni4p rings near both poles (Fig. 6 A, arrows). In wild-type cells grown in synthetic complete medium, the Bni4p signal was also asymmetric, but the signal on the bud side typically appeared earlier in the cell cycle and subsequently reached an intensity apparently equivalent to that on the mother-cell side (Fig. 6 B). No signal was observed in a bni4-Δ1 strain (Fig. 6 C), confirming the specificity of the staining. Thus, Bni4p appears to localize to a ring at the presumptive bud site before bud emergence and is present asymmetrically on the mother-cell side of the neck early in the cell cycle, a pattern that correlates well with the localization and timing of bud-scar chitin deposition. Later in the cell cycle, Bni4p appears to accumulate also on the bud side of the neck, and a symmetric distribution of Bni4p across the neck was typical even in cells with small buds in a Bni4p-overproducing strain (Fig. 6 D). The overproducing cells also appeared to have Bni4p distributed throughout the cell or plasma membrane, and some unbudded cells displayed an intensely staining disc or ring (Fig. 6 D, arrow). Localization of Bni4p to the mother-bud neck is consistent with the two-hybrid data showing its interactions with Cdc10p and Chs4p, two other proteins that localize to this region (see introduction and below).
Figure 6.
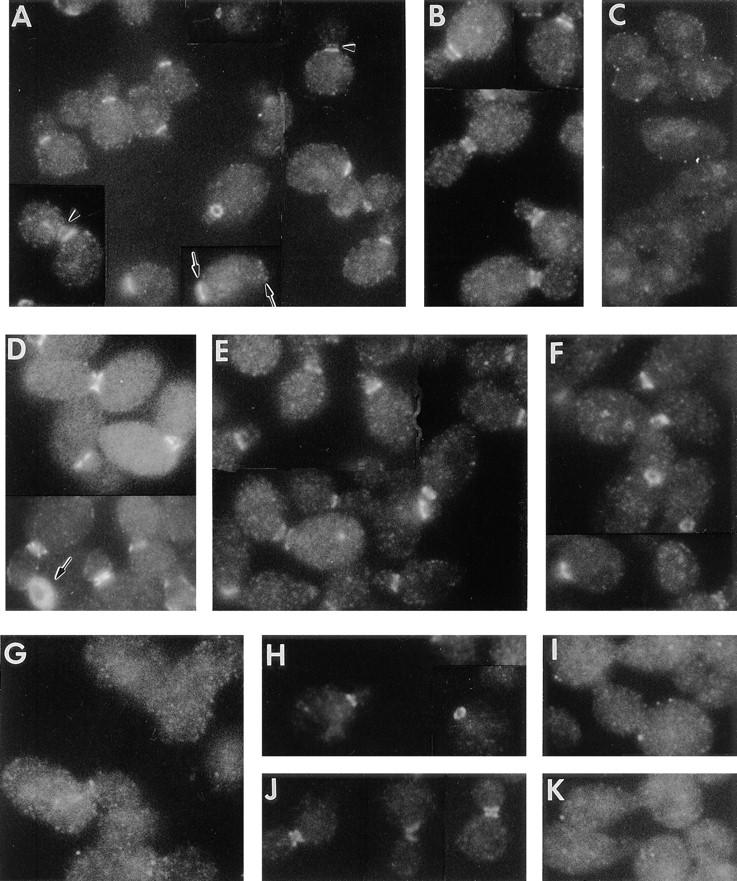
Localization of Bni4p. Cells were grown in rich (YM-P) medium except for those shown in B, which were grown in SC medium. (A–G) Exponentially growing cells of strains (A and B) YEF473 (wild-type); (C) DDY175 (bni4-Δ1/bni4-Δ1); (D) DDY175 containing plasmid p356 (high-copy BNI4); (E) DDY174 (chs4-Δ1/chs4-Δ1); (F) DDY186 (chs3-Δ1/chs3-Δ1); and (G) DDY185-1A (cdc10-Δ1) were examined by immunofluorescence microscopy using Bni4p-specific antibodies. (H–K) Cells of strain JPTA1493-H01 (cdc12-6/ cdc12-6) growing exponentially at 23°C (H and J) or collected 5 min after a shift to 37°C (I and K) were examined by immunofluorescence microscopy using Bni4p-specific antibodies (H and I) or Cdc11p-specific antibodies (J and K). Arrows and arrowheads indicate structures discussed in the text.
To determine if mutations in functionally related genes affect Bni4p localization, we examined chs4-Δ1, chs3-Δ1, cdc10-Δ1, and cdc12-6 mutants. (The cdc12-6 allele was used because of its rapid loss of the septin ring upon shift to nonpermissive temperature [Adams, 1984; Haarer and Pringle, 1987; Chant et al., 1995].) Bni4p localization appeared normal in the chs4-Δ1 and chs3-Δ1 strains (Fig. 6, E and F), but the immunofluorescence signal was completely or almost completely absent in the cdc10-Δ1 strain (Fig. 6 G). In addition, Bni4p localized normally in a cdc12-6 strain grown at 23°C (Fig. 6 H) but was undetectable after shift to nonpermissive temperature for 5 or 60 min (Fig. 6 I, and data not shown), paralleling the loss in septin localization (Fig. 6, J and K). Taken together, these results show that Bni4p localization depends upon the septins but not upon Chs4p or Chs3p.
Localization of Chs4p in Wild-type and Mutant Strains
To localize Chs4p, we constructed plasmid p326, a low-copy plasmid encoding a GFP-Chs4p fusion protein. chs4-Δ1 strains harboring this plasmid (DDY172-2AX and DDY197) displayed normal Calcofluor staining and normal morphology (data not shown), indicating that the GFP-Chs4p fusion protein can supply Chs4p function. Examination of DDY197 cells by fluorescence microscopy revealed that GFP-Chs4p localized to a patch or ring at the presumptive bud site in many unbudded cells (Fig. 7 A, small arrows), to a ring at the base of the bud in most (if not all) cells with tiny buds (Fig. 7 A, arrowheads), and to both sides of the neck in many cells with large buds (Fig. 7 A, asterisk); localized signal was rarely detectable in cells with medium-sized buds (Fig. 7 A, large arrow). Some of the GFP-Chs4p fluorescence on unbudded cells appeared to represent residual protein at the preceding division site, a hypothesis supported by the observation that some cells displayed patches or rings of fluorescence near both poles (Fig. 7 A and G, small arrows). The localization observed for GFP-Chs4p is very similar to that observed for Chs3p (Chuang and Schekman, 1996; Santos and Snyder, 1997; also see below) in that both proteins are localized to the mother-bud neck early and late in the cell cycle but appear to be absent from the neck during intermediate stages of the cell cycle.
Figure 7.

Localization of GFP-Chs4p. Cells were grown, fixed, and observed as described in Materials and Methods. (A–F) Cells of the indicated strains were collected during growth at 23°C: (A) DDY197 (chs4-Δ1/chs4-Δ1 [p326]); (B) DDY217 (chs4-Δ1/chs4-Δ1 bni4-Δ1/ bni4-Δ1 [p326]) containing pRS315 (control plasmid); (C) DDY217 containing p366 (low-copy, BNI4); (D) DDY197 containing p365 (high-copy, BNI4); (E) DDY244 (chs4-Δ1/chs4-Δ1 chs3-Δ1/chs3-Δ1 [p326]); and (F) DDY244 containing p408 (low-copy, CHS3). (G–J) Cells of strain DDY218 (chs4-Δ1/chs4-Δ1 cdc12-6/cdc12-6 [p326]) containing either (G and H) YCplac111 (control plasmid) or (I and J) p422 (low-copy, CDC12) were collected during growth at 23°C (G and I) or 20 min after a shift to 37°C (H and J). Small arrows indicate GFP-Chs4p at presumptive bud sites or previous sites of cell division; small arrowheads indicate rings of Chs4p at the bases of tiny buds; large arrows indicate mother-bud necks lacking GFP-Chs4p fluorescence; large arrowheads indicate mislocalized Chs4p; the asterisk indicates Chs4p on either side of the mother-bud neck (see text for details).
To explore the roles of other proteins in Chs4p localization, we examined chs4-Δ1, p326-containing cells that were also bni4-Δ1, chs3-Δ1, or cdc12-6, or that overproduced Bni4p. bni4-Δ1 cells with or without a control plasmid showed little or no localization of GFP-Chs4p to presumptive bud sites or the necks of tiny buds (Fig. 7 B; Table V; and data not shown). As expected, introduction of a low-copy BNI4 plasmid restored normal GFP-Chs4p localization (Fig. 7 C; Table V). In contrast, overexpression of Bni4p from a high-copy plasmid led to mislocalization of GFP-Chs4p to bud tips, to the daughter-cell side of the neck in small-budded cells, and throughout growing buds (Fig. 7 D). A chs3-Δ1 strain also displayed little or no normal localization of GFP-Chs4p (Fig. 7 E; Table V), although this protein was occasionally mislocalized to the tips of tiny buds (Fig. 7 E, arrowhead). As expected, introduction of a low-copy CHS3 plasmid restored normal GFP-Chs4p localization (Fig. 7 F; Table V). Finally, shift of a cdc12-6 strain from 23 to 37°C led to a loss of normal GFP-Chs4p localization (Fig. 7, G and H; Table V). As expected, when a low-copy CDC12 plasmid was introduced, GFP-Chs4p remained localized after shift to 37°C (Fig. 7, I and J; Table V). Taken together, these results suggest that normal Chs4p localization depends upon the septins, Bni4p, and Chs3p.
Table V.
Localization of GFP-Chs4p in Wild-type and Mutant Strains*
| Relevant genotype | Growth temperature | Percent of cells with localized GFP-Chs4p | ||
|---|---|---|---|---|
| “Wild-type”‡ | 23°C | 36 | ||
| bni4-Δ1 [control plasmid] | 23°C | 8 | ||
| bni4-Δ1 [BNI4 plasmid] | 23°C | 30 | ||
| chs3-Δ1 § | 23°C | 4 | ||
| chs3-Δ1 [CHS3 plasmid] | 23°C | 34 | ||
| cdc12-6 [control plasmid] | 23°C | 33 | ||
| cdc12-6 [control plasmid] | 37°C | 2‖ | ||
| cdc12-6 [CDC12 plasmid] | 23°C | 33 | ||
| cdc12-6 [CDC12 plasmid] | 37°C | 32 |
Unbudded and tiny-budded cells from the experiments described in Fig. 7 were scored for the presence or absence of a patch or ring of fluorescence at the presumptive bud site or at the base of the tiny bud. 200 cells were scored for each strain.
DDY197 (chs4-Δ1/chs4-Δ1 [p326]).
Introduction of a control plasmid into this strain did not detectably affect GFP-Chs4p localization (data not shown).
In addition, a few cells (<1%) with tiny buds had concentrations of GFP-Chs4p fluorescence in the vicinity of the bud but not in the normal band at the base of the bud (see Fig. 7 H).
Localization of Chs3p in chs4, bni4, and Septin Mutants
The evidence presented above suggests that the localization of Chs3p (and hence of chitin deposition) may depend upon Chs4p, Bni4p, and the septins. To test this hypothesis, Chs3p-specific antibodies (Chuang and Schekman, 1996) were used to localize this protein in wild-type, chs4-Δ1, bni4-Δ1, chs4-Δ1 bni4-Δ1, Bni4p-overexpressing, and septin mutant strains. As found previously (Chuang and Schekman, 1996; Santos and Snyder, 1997), Chs3p was observed in wild-type cells as a patch (presumably at the incipient bud site) in many unbudded cells (Fig. 8 A, arrow), as a distinct ring on the mother-cell side of the mother-bud neck in many tiny-budded cells (Fig. 8 A, arrowheads), and as a band on both sides of the neck in some cells with large buds (data not shown). In addition, punctate staining, which was also seen (although to a lesser extent) in chs3-Δ1 control cells (Fig. 8 B), was visible throughout the cells. In contrast, in chs4-Δ1, bni4-Δ1, and chs4-Δ1 bni4-Δ1 strains, Chs3p was typically found to be localized diffusely throughout the bud and in the vicinity of the mother-bud neck in tiny-budded cells, but it did not form a distinct ring at the neck like those seen in wild-type cells (Fig. 8, C–E). In addition, some unbudded cells of the mutant strains displayed patches of staining resembling, but typically less distinct than, those seen in wild-type cells (Fig. 7, C–E, arrows). In Bni4p-overexpressing and cdc10-Δ1 strains, Chs3p was observed in a ring in a few cells (Fig. 8, F and G, arrowheads) but was more commonly found diffusely throughout the cells. In a cdc12-6 strain grown at 23°C, Chs3p localized normally (Fig. 8 H), but normal localization was lost within 5 min after a shift to restrictive temperature (Fig. 8 I), paralleling the loss of septin localization (Fig. 8, J and K). Taken together, the data suggest that the normal localization of Chs3p to its major site of action (i.e., the site of chitin-ring formation) is dependent upon Chs4p, Bni4p, and the septins, and that the localization of Chs3p and Chs4p is interdependent (see also above). In the absence of Chs4p and/or Bni4p, the cell appears to recruit Chs3p to the correct general location but fails to assemble it into the well-defined ring seen in wild-type cells.
Figure 8.

Immunolocalization of Chs3p in wild-type and mutant strains. (A–G) Exponentially growing cells of strains (A) YEF473 (wild-type); (B) DDY186 (chs3-Δ1/chs3-Δ1); (C) DDY174 (chs4-Δ1/chs4-Δ1); (D) DDY175 (bni4-Δ1/bni4-Δ1); (E) DDY179 (chs4-Δ1/ chs4-Δ1 bni4-Δ1/bni4-Δ1); (F) DDY175 containing plasmid p356 (high-copy BNI4); and (G) DDY185-1A (cdc10-Δ1) were examined by immunofluorescence microscopy using Chs3p-specific antibodies. (H–K) Cells of strain JPTA1493-HO1 (cdc12-6/cdc12-6) growing exponentially at 23°C (H and J) or collected 5 min after a shift to 37°C (I and K) were examined by immunofluorescence microscopy using Chs3p-specific antibodies (H and I) or Cdc11p-specific antibodies (J and K). Arrowheads in some panels indicate rings at the bases of tiny buds; arrows indicate patches of staining at presumptive bud sites.
Localization of Septins in chs4, bni4, and chs3 Mutants
The results presented above indicate that the localization of Bni4p, Chs4p, and Chs3p depends upon the septins. To determine if the localization of the septins depends upon these other proteins, chs4-Δ1, bni4-Δ1, chs4-Δ1 bni4-Δ1, chs3-Δ1, and Bni4p-overexpressing strains were examined by immunofluorescence using Cdc11p-specific antibodies. Cdc11p localized nearly normally in the mutant strains (Fig. 9). However, the band of septin staining appeared somewhat narrower (not extending as far into either mother cell or bud) in the mutants (compare Fig. 9, B–E, to Fig. 9 A). This effect was not apparent in the Bni4p-overexpressing strain (Fig. 9 F). It is not clear whether the altered appearance of the septin band is a real change in the structure of the septin assembly or a visual artifact resulting from a change in neck shape and/or dimensions. In any case, it appears that Chs4p, Bni4p, and Chs3p have, at most, a modest influence on septin organization and are not required to localize the septins to the mother-bud neck.
Figure 9.

Immunolocalization of Cdc11p in wild-type and mutant strains. Exponentially growing cells of strains (A) YEF473 (wild-type); (B) DDY174 (chs4-Δ1/chs4-Δ1); (C) DDY175 (bni4-Δ1/bni4-Δ1); (D) DDY179 (chs4-Δ1/chs4-Δ1 bni4-Δ1/bni4-Δ1); (E) DDY186 (chs3-Δ1/chs3-Δ1); and (F) DDY175 containing plasmid p356 (high-copy BNI4) were examined by immunofluorescence microscopy using Cdc11p-specific antibodies.
DISCUSSION
A Model for the Spatial Localization of Chitin Synthase III Activity
The results described above suggest a model in which a hierarchic assembly of proteins based on the septins is responsible for the spatial localization of cell-wall chitin deposition by chitin synthase III. In this model (Fig. 10 A), the septins are recruited to and anchored at the presumptive bud site and mother-bud neck by mechanisms independent of the other proteins considered here. The septin complex then localizes Bni4p through the interaction of this protein with Cdc10p, and Bni4p, in turn, localizes the chitin synthase III complex (including Chs3p and Chs4p) through its interaction with Chs4p.
Figure 10.

Model for the interactions among the chitin synthase III complex, Bni4p, and the septins. (A) The transmembrane protein Chs3p synthesizes chitin, which is deposited in the cell wall (solid arrows). Chs4p, anchored to the cytoplasmic face of the plasma membrane by its (presumably) prenylated COOH terminus, interacts both with Chs3p and with Bni4p, which is tethered to the septin complex by its interaction with Cdc10p. The box with the question mark indicates a postulated protein that is sufficient to anchor the septin complex to the cell membrane in the absence of Bni4p, Chs4p, and/or Chs3p. (B) In the absence of Bni4p or the septin complex, the chitin synthase III complex diffuses through the lipid bilayer (dashed arrows), resulting in delocalized chitin delocalization.
Several lines of evidence support the central role of the septins in this model. The septins form a ring at the presumptive bud site ∼15 min before bud emergence (Ford and Pringle, 1991; Kim et al., 1991), and the chitin ring, formed by chitin synthase III (Shaw et al., 1991; Bulawa, 1993; Orlean, 1997), appears in the immediately overlying cell wall somewhat later (just before bud emergence) (Hayashibe and Katohda, 1973; Kim et al., 1991). Thus, the septins are in the right place at the right time to play a role in localizing chitin synthase III activity. In addition, conventional electron microscopic observations on the septin-associated filaments (Byers and Goetsch, 1976; Byers, 1981), immunofluorescence and immunoelectron microscopy (Haarer and Pringle, 1987; Kim et al., 1991; Ford and Pringle, 1991; Mulholland, J., D. Preuss, and D. Botstein, personal communication), and cell-fractionation experiments (Healy, A., M. Longtine, and J.R. Pringle, unpublished results) all suggest that the septins are closely associated with the plasma membrane, so that a role in the localization of the plasma membrane–associated chitin synthase III complex is plausible. Moreover, temperature-sensitive septin mutants are unable to form normal chitin rings at restrictive temperature (Fig. 1). Although a variety of cell-cycle mutants display delocalized chitin deposition after extended incubations at restrictive temperature (Sloat et al., 1981; Roberts et al., 1983; Shaw et al., 1991), the failure to form a chitin ring at the base of the first bud produced at restrictive temperature was unique to the septin mutants among those examined, suggesting that this effect results specifically from the loss of septin function. Finally, the septin mutants are also defective in the localization of Bni4p, Chs4p, and Chs3p (Figs. 6–8).
The mechanisms by which the septins themselves are recruited to and anchored at the presumptive bud site and mother-bud neck remain obscure. Septin localization appears to depend on a signal from the Cdc42p GTPase, but the effectors of this signal are not known (Pringle et al., 1995). Although it initially seemed possible that the transmembrane protein Chs3p or the (presumably) prenylated protein Chs4p might be involved in anchoring the septins to the plasma membrane, deletion of CHS3, CHS4, or BNI4 has little or no effect on septin localization, at least as judged by immunofluorescence. As the septins themselves have no motifs that suggest how membrane association might occur (Longtine et al., 1996), there is presumably at least one protein involved in this association that has yet to be identified.
Several lines of evidence also suggest that Bni4p provides a link between the septins and the chitin synthase III complex. The two-hybrid data show that Bni4p can interact with both Cdc10p and Chs4p. In addition, except for its asymmetric concentration on the mother-cell side of the neck (as considered further below), Bni4p colocalizes with the septins, and Bni4p localization is lost in septin mutants. (In this regard, the residual Bni4p localization observed in some cdc10-Δ1 cells probably just reflects residual septin function in this severely compromised but viable strain.) Interaction of Bni4p with the septins is further supported by the observations that bni4 mutant cells often have enlarged bud necks and that overproduction of Bni4p (or a Bni4p fragment) causes a phenotype similar to that of septin mutants, presumably because excess Bni4p titrates some factor from the neck that is required for septin function. Finally, either a loss of Bni4p or its overproduction causes a partial delocalization of chitin deposition, which parallels an inability to achieve normal localization of either Chs4p or Chs3p. In contrast, the localization of Bni4p itself is not obviously affected by the loss of either Chs4p or Chs3p. Bni4p appears to function specifically in the chitin synthase III pathway, because the amount and localization of residual chitin (presumably synthesized by chitin synthase I and/or II) appear the same in a chs4 bni4 strain as in a chs4 BNI4 strain.
Several lines of evidence also suggest that Chs4p links Chs3p to Bni4p, and hence to the septins. The two-hybrid data show that Chs4p can interact with both Bni4p and Chs3p. In addition, Chs4p and a fraction of the Chs3p colocalize with Bni4p and the septins for at least a portion of the cell cycle, and this localization is lost in both bni4 and septin mutants. Although the loss of chitin synthase III activity in a chs4 mutant makes it impossible to ask directly where chitin would be deposited, immunofluorescence observations show that the normal localization of Chs3p is also lost in a chs4 mutant. Importantly, the data also suggest that Chs3p is not just a passive passenger but instead plays a key role in the assembly of the complex of which it is a part. In particular, the two-hybrid data show that Chs3p can interact with itself as well as with Chs4p, and the localization of Chs4p is lost in a chs3 mutant.
An attractive feature of the model described above is that it helps to explain how the chitin ring can form asymmetrically on the mother-cell side of the neck. In particular, although the ring of septin proteins (and septin-associated filaments) appears to be symmetric across the neck (Byers and Goetsch, 1976; Byers, 1981; Haarer and Pringle, 1987; Kim et al., 1991; Ford and Pringle, 1991), Bni4p is concentrated asymmetrically on the mother-cell side of the neck, especially early in the cell cycle. This presumably restricts the chitin synthase III complex (and hence chitin deposition) to the mother-cell side of the neck, as indeed observed experimentally using GFP-Chs4p and antibodies to Chs3p. It is not yet clear how Bni4p itself becomes asymmetrically localized, but one possibility is suggested by the presence in the BNI4 upstream region of an MluI restriction site (and hence a potential MCB-type transcription-control element), which suggests that BNI4 may be transcribed specifically early in the cell cycle (Andrews and Mason, 1993; Koch and Nasmyth, 1994; Breeden, 1996), as indeed are a variety of other genes whose products are involved in cell-wall synthesis (Igual et al., 1996). Thus, most Bni4p may be synthesized and become bound to the septins before bud emergence, when the septins exist only as a ring at the surface of the mother cell. Subsequent low-level synthesis of Bni4p could account for its gradual accumulation in association with the septins on the bud side of the neck; however, by this point, chitin synthase III components may have been retrieved from the plasma membrane by endocytosis (Chuang and Schekman, 1996) or otherwise inactivated, so that no chitin deposition on the bud side results.
The analyses reported here leave several interesting issues unresolved. For instance, the model of Fig. 10 A does not address the mechanisms involved in the initial delivery of the chitin synthase components to the presumptive bud site or their later retrieval from the plasma membrane (see, however, Chuang and Schekman, 1996; Santos and Snyder, 1997). In this regard, it is interesting that Chs3p showed some localization to the incipient bud site in bni4, chs4, and septin mutants, although it was not organized into the normal tight band at the base of the emerging bud. Similarly, GFP-Chs4p also sometimes appeared concentrated at the incipient bud site in bni4, chs3, and septin mutants, although the localization rarely (if ever) appeared normal. These observations parallel the findings that chitin deposition is not completely delocalized in bni4 and septin mutants. Thus, it appears that there is a mechanism for the delivery of Chs3p and other chitin synthase III components to the general region of the presumptive bud site that is independent of the septins, Bni4p, and Chs4p. This mechanism might simply be the polarization of the actin cytoskeleton and secretory system, which is expected to result in the polarized delivery to the cell surface of integral membrane proteins such as Chs3p (Drubin and Nelson, 1996). In the absence of the restraint offered by the septin/Bni4p/Chs4p complex, Chs3p and associated proteins might simply diffuse through the membrane away from their site of initial insertion (Fig. 10 B), resulting (when Chs4p and other components necessary for chitin synthase activity are present) in the delocalized chitin deposition that is observed.
In addition, some questions remain about the interactions among Chs4p, Bni4p, and Chs3p. The truncated Chs4p encoded by the chs4Δ610 allele failed to show interaction in two-hybrid tests either with Bni4p or with Chs3p, although Chs4pΔ610 could still support not only chitin synthase III activity but also the normal localization of the chitin synthesized. Although these observations appear at first sight to violate the model of Fig. 10, we note that negative results in two-hybrid tests can be misleading (for example, if the fusion proteins are awkwardly folded) and that the tests in question were performed with truncated alleles of both BNI4 and CHS3, whereas the analysis of chitin deposition was performed in a strain that contained chs4Δ610but was wild-type for BNI4 and CHS3. Thus, given the weight of other evidence supporting the model of Fig. 10, we think it most likely that Chs4pΔ610 does interact with full-length Bni4p and Chs3p under normal circumstances in vivo. This interpretation is supported by the observation that Chs4pΔ610 does not support chitin synthesis in a bni4-Δ1 strain (DeMarini, D.J., unpublished observations).
Finally, it should be noted that the model of Fig. 10 addresses specifically the localization of the proteins of interest in the period just before and during bud emergence; the mechanisms involved in the apparent relocalization of Chs3p, Chs4p, and (perhaps) associated proteins to the mother-bud neck late in the cell cycle may be different.
The Roles of Chs4p and Chs3p
Our results taken together with those of previous studies (Roncero et al., 1988; Bulawa, 1992, 1993) suggest that Chs4p has at least two roles in relation to chitin synthase III. One role is to activate Chs3p, and the second is to anchor Chs3p to the septins via Bni4p. Interestingly, these roles appear to be separable, as judged from the occurrence of delocalized chitin deposition in a bni4 mutant but not in a bni4 chs4 double mutant, the ability of a CHS4 plasmid to restore (delocalized) chitin deposition to the latter strain, and the ability of a chs4Δ560 plasmid to restore chitin synthesis, but not normal chitin localization, to a chs4 mutant strain. The mechanism by which Chs4p activates Chs3p remains unclear, although the two-hybrid data suggest that the two proteins may interact directly.
A surprising result is that the presumed membrane- attachment domain of Chs4p (the CAAX motif and preceding cluster of basic amino acids) is apparently not required for either of the roles just discussed. Presumably, protein–protein interactions are sufficient to localize Chs4p appropriately in the absence of direct membrane attachment. It is possible that the membrane-attachment domain is simply unnecessary for any aspect of Chs4p function, but it also seems possible that this domain is important for some as-yet-undefined role of the protein. In this regard, it is of interest to ask why Chs4p and Chs3p reappear at the mother-bud neck late in the cell cycle, given that chitin synthase II appears to synthesize the chitin of the primary septum (Shaw et al., 1991; Bulawa, 1993; Orlean, 1997). One possibility is that chitin synthase III may function as a redundant system for the synthesis of septal chitin. This possibility is supported by the observations that a chs2 strain is viable and produces an abnormally thick, chitin-based septum and that chs2 and chs3 mutations are synthetically lethal (Shaw et al., 1991). However, it also seems possible that Chs4p and/or Chs3p are involved in the recruitment and/or spatial organization of other proteins that participate in cell-wall synthesis or other events at the division site. There are several other observations consistent with this possibility. For example, it is not clear that either the formation of protuberances at previous division sites by chs4 and bni4 mutants or the synthetic lethality of chs4 and cdc12 mutations can be explained in terms of the role of Chs4p in chitin synthase III activity. In addition, we observed that mutation of the putative Ca2+-binding site of Chs4p (a double D238R, D240R mutation; see Fig. 3 A) had no obvious effect on chitin synthase III activity, as judged by Calcofluor staining (DeMarini, D.J, unpublished observations). Finally, it is intriguing that Fks1p, one of the catalytic subunits of the enzyme that synthesizes the major cell-wall constituent 1,3-β-d-glucan (Douglas et al., 1994), also localizes to the mother-bud neck just before cytokinesis (Qadota et al., 1996).
Except for the apparent noninvolvement of the putative membrane-attachment and Ca2+-binding domains in the action of chitin synthase III at the beginning of the cell cycle, little can be said as yet about structure–function relationships in Chs4p. However, the observation that the chs4Δ560 allele could support chitin synthesis, but not the proper localization of the chitin synthesized, does suggest that the COOH-terminal portion of Chs4p is required for its interaction with Bni4p.
A General Model for Septin Function
Proteins in the septin family have now been identified in a wide variety of fungal and animal cells (Cooper and Kiehart, 1996; Longtine et al., 1996). In every cell type examined to date, the septins appear to be involved in cytokinesis and/or septum formation, but the precise role of the septins in these processes remains obscure, a problem compounded by the fact that the mechanisms of cytokinesis in fungal and animal cells otherwise appear quite different. In addition, the septins are found localized in ways that suggest that they have important roles in processes distinct from cytokinesis. For example, in mating yeast cells, the septins are concentrated near the bases of mating projections (Ford and Pringle, 1991; Kim et al., 1991) and appear to interact with other proteins involved in the polarized morphogenesis that occurs in response to mating pheromones (Konopka et al., 1995). In sporulating yeast cells, the septins appear to be concentrated at the leading edge of the extending forespore membrane during the early stages of spore formation and subsequently are found surrounding the entire developing spore before spore-wall deposition (De Virgilio et al., 1996; Fares et al., 1996). In Drosophila, septins are highly concentrated at the leading edge of the furrows during cellularization of the embryo, in specific surface domains of polarized epithelial cells, and in neurons of the embryonic central nervous system (Neufeld and Rubin, 1994; Fares et al., 1995). A major challenge is to understand what general principles of septin function may explain their roles in all these different contexts.
In this study, an amalgam of genetic, two-hybrid, and protein-localization data have led us to a model for septin function in localizing the action of chitin synthase III, an enzyme responsible for the synthesis of a particular component of the cell wall. In particular, as discussed in detail above, it appears that the septins provide a template upon which the proteins of the chitin synthase III complex assemble. Previous studies had suggested a similar role for the septins in the function of Bud3p and Bud4p, two proteins that are required for the selection of bud sites in the axial pattern (Chant et al., 1995; Sanders and Herskowitz, 1996). In particular, Bud3p and Bud4p assemble at the mother-bud neck in a septin-dependent fashion during each cell cycle; they then remain at the division sites on mother and daughter cells long enough to direct the assembly of a new bud site to an adjacent location in the next cell cycle. Extrapolation from these examples and consideration of the patterns of septin localization described above suggest that the septins may function generally in the spatial organization of cell surface proteins that are involved in signaling, localized membrane insertion, or the synthesis of components of the cell wall or extracellular matrix. In this context, it is interesting to note that chitin synthase III is also responsible for the synthesis of the chitosan layer of the S. cerevisiae spore wall (Pammer et al., 1992; Bulawa, 1993; Orlean, 1997) and that a homologue of Chs4p, but not Chs4p itself, is expressed in sporulating cells (Bulawa, 1993). If this general view of septin function is correct, then progress in understanding septin function in particular contexts will depend on identifying the particular proteins with which the septins interact. Both genetic and biochemical approaches, in appropriate systems, should contribute to such identification.
Acknowledgments
We thank C. Bulawa, B. Osmond, and P. Robbins for strains, helpful discussions, and the communication of unpublished results; B. DiDomenico and J. Greene for communicating unpublished results; members of the Pringle laboratory, especially E. Bi and M. Longtine, for antibodies, strains, plasmids, and valuable discussions; P. Watts for the library of AD fusions; C. Roncero for plasmid pHV7; and S. Whitfield for providing outstanding photographic services.
This work was supported by National Institutes of Health Grant GM31006 (to J.R. Pringle), funds from the RJEG Trust, fellowships from the L. and Th. LaRoche Stiftung and the Ciba-Geigy-Jubilaeums-Stiftung (to C. De Virgilio), and a Howard Hughes Medical Institute predoctoral fellowship (to J. Chuang).
Abbreviations used in this paper
- 5-FOA
5-fluoroorotic acid
- AD
activation domain
- DBD
DNA-binding domain
- DIC
differential-interference-contrast
- GFP
green fluorescent protein
- GST
glutathione-S-transferase
- ORF
open reading frame
- SC
synthetic complete
Footnotes
The present address of Douglas J. DeMarini is Department of Comparative Genetics, Smithkline Beecham Pharmaceuticals, King of Prussia, PA 19406.
The present address of Hanna Fares is Department of Biochemistry, Columbia University, New York, NY 10032.
The present address of Claudio De Virgilio is Botanisches Institut der Universität Basel, Basel, Switzerland, CH-4056.
Address all correspondence to John R. Pringle, Department of Biology, CB #3280 Coker Hall, University of North Carolina, Chapel Hill, NC 27599-3280. Phone: 919-962-2293; FAX: 919-962-0320; E-mail: jpringle @email.unc.edu
2. Fares, H., M.S. Longtine, and J.R. Pringle, manuscript in preparation.
REFERENCES
- Adams, A.E.M. 1984. Cellular morphogenesis in the yeast Saccharomyces cerevisiae. Ph.D. thesis. The University of Michigan, Ann Arbor, MI. 179 pp.
- Adams AEM, Pringle JR. Relationship of actin and tubulin distribution to bud growth in wild-type and morphogenetic-mutant Saccharomyces cerevisiae. . J Cell Biol. 1984;98:934–945. doi: 10.1083/jcb.98.3.934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews BJ, Mason SW. Gene expression and the cell cycle: a family affair. Science (Wash DC) 1993;261:1543–1544. doi: 10.1126/science.8372349. [DOI] [PubMed] [Google Scholar]
- Ausubel, F.M., R. Brent, R.E. Kingston, D.D. Moore, J.G. Seidman, J.A. Smith, and K. Struhl. 1995. Current Protocols in Molecular Biology. John Wiley and Sons Ltd., New York.
- Baudin A, Ozier-Kalogeropoulos O, Denouel A, Lacroute F, Cullin C. A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae. . Nucleic Acids Res. 1993;21:3329–3330. doi: 10.1093/nar/21.14.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltzer JP, Chang L-FL, Hinkkanen AE, Kohlhaw GB. Structure of yeast LEU4. . J Biol Chem. 1986;261:5160–5167. [PubMed] [Google Scholar]
- Bi E, Pringle JR. ZDS1 and ZDS2, genes whose products may regulate Cdc42p in Saccharomyces cerevisiae. . Mol Cell Biol. 1996;16:5264–5275. doi: 10.1128/mcb.16.10.5264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breeden L. Start-specific transcription in yeast. Curr Top Microbiol Immunol. 1996;208:95–125. doi: 10.1007/978-3-642-79910-5_5. [DOI] [PubMed] [Google Scholar]
- Briza P, Breitenbach M, Ellinger A, Segall J. Isolation of two developmentally regulated genes involved in spore wall maturation in Saccharomyces cerevisiae. . Genes Dev. 1990;4:1775–1789. doi: 10.1101/gad.4.10.1775. [DOI] [PubMed] [Google Scholar]
- Briza P, Eckerstorfer M, Breitenbach M. The sporulation-specific enzymes encoded by the DIT1 and DIT2genes catalyze a two-step reaction leading to a soluble LL-dityrosine-containing precursor of the yeast spore wall. Proc Natl Acad Sci USA. 1994;91:4524–4528. doi: 10.1073/pnas.91.10.4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulawa CE. CSD2, CSD3, and CSD4, genes required for chitin synthesis in Saccharomyces cerevisiae: the CSD2 gene product is related to chitin synthases and to developmentally regulated proteins in Rhizobium species and Xenopus laevis. . Mol Cell Biol. 1992;12:1764–1776. doi: 10.1128/mcb.12.4.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulawa CE. Genetics and molecular biology of chitin synthesis in fungi. Annu Rev Microbiol. 1993;47:505–534. doi: 10.1146/annurev.mi.47.100193.002445. [DOI] [PubMed] [Google Scholar]
- Byers, B. 1981. Cytology of the yeast life cycle. In The Molecular Biology of the Yeast Saccharomyces: Life Cycle and Inheritance. J.N. Strathern, E.W. Jones, and J.R. Broach, editors. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 59–96.
- Byers B, Goetsch L. A highly ordered ring of membrane-associated filaments in budding yeast. J Cell Biol. 1976;69:717–721. doi: 10.1083/jcb.69.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabib, E. 1994. Nomenclature of genes related to chitin synthesis. In Yeast: A Newsletter for Persons Interested in Yeast, Volume 43. M.-A. LaChance, editor. 58.
- Cabib E, Sburlati A, Bowers B, Silverman SJ. Chitin synthase 1, an auxiliary enzyme for chitin synthesis in Saccharomyces cerevisiae. . J Cell Biol. 1989;108:1665–1672. doi: 10.1083/jcb.108.5.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabib E, Silverman SJ, Shaw JA. Chitinase and chitin synthase 1: counterbalancing activities in cell separation of Saccharomyces cerevisiae. . J Gen Microbiol. 1992;138:97–102. doi: 10.1099/00221287-138-1-97. [DOI] [PubMed] [Google Scholar]
- Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science (Wash DC) 1994;263:802–804. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- Chant J, Mischke M, Mitchell E, Herskowitz I, Pringle JR. Role of Bud3p in producing the axial budding pattern of yeast. J Cell Biol. 1995;129:767–778. doi: 10.1083/jcb.129.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson TW, Sikorski RS, Dante M, Shero JH, Hieter P. Multifunctional yeast high-copy-number shuttle vectors. Gene (Amst) 1992;110:119–122. doi: 10.1016/0378-1119(92)90454-w. [DOI] [PubMed] [Google Scholar]
- Chuang JS, Schekman RW. Differential trafficking and timed localization of two chitin synthase proteins, Chs2p and Chs3p. J Cell Biol. 1996;135:597–610. doi: 10.1083/jcb.135.3.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JA, Kiehart DP. Septins may form a ubiquitous family of cytoskeletal filaments. J Cell Biol. 1996;134:1345–1348. doi: 10.1083/jcb.134.6.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, R.W., D. Botstein, and J.R. Roth. 1980. Advanced Bacterial Genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 251 pp.
- De Virgilio C, DeMarini DJ, Pringle JR. SPR28, a sixth member of the septin gene family in Saccharomyces cerevisiaethat is expressed specifically in sporulating cells. Microbiology (Reading) 1996;142:2897–2905. doi: 10.1099/13500872-142-10-2897. [DOI] [PubMed] [Google Scholar]
- Douglas CM, Foor F, Marrinan JA, Morin N, Nielsen JB, Dahl AM, Mazur P, Baginsky W, Li W, El-Sherbeini M, et al. The Saccharomyces cerevisiae FKS1 (ETG1) gene encodes an integral membrane protein which is a subunit of 1,3-β-d-glucan synthase. Proc Natl Acad Sci USA. 1994;91:12907–12911. doi: 10.1073/pnas.91.26.12907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drubin DG, Nelson WJ. Origins of cell polarity. Cell. 1996;84:335–344. doi: 10.1016/s0092-8674(00)81278-7. [DOI] [PubMed] [Google Scholar]
- Fares H, Peifer M, Pringle JR. Localization and possible functions of Drosophilaseptins. Mol Biol Cell. 1995;6:1843–1859. doi: 10.1091/mbc.6.12.1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fares H, Goetsch L, Pringle JR. Identification of a developmentally regulated septin and involvement of the septins in spore formation in Saccharomyces cerevisiae. . J Cell Biol. 1996;132:399–411. doi: 10.1083/jcb.132.3.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field CM, Al-Awar O, Rosenblatt J, Wong ML, Alberts B, Mitchison TJ. A purified Drosophilaseptin complex forms filaments and exhibits GTPase activity. J Cell Biol. 1996;133:605–616. doi: 10.1083/jcb.133.3.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields S, Sternglanz R. The two-hybrid system: an assay for protein-protein interactions. Trends Genet. 1994;10:286–292. doi: 10.1016/0168-9525(90)90012-u. [DOI] [PubMed] [Google Scholar]
- Flescher EG, Madden K, Snyder M. Components required for cytokinesis are important for bud site selection in yeast. J Cell Biol. 1993;122:373–386. doi: 10.1083/jcb.122.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford SK, Pringle JR. Cellular morphogenesis in the Saccharomyces cerevisiae cell cycle: localization of the CDC11gene product and the timing of events at the budding site. Dev Genet. 1991;12:281–292. doi: 10.1002/dvg.1020120405. [DOI] [PubMed] [Google Scholar]
- Gietz RD, Sugino A. New yeast-Escherichia colishuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene (Amst) 1988;74:527–534. doi: 10.1016/0378-1119(88)90185-0. [DOI] [PubMed] [Google Scholar]
- Guthrie, C., and G.R. Fink, editors. 1991. Guide to yeast genetics and molecular biology. Methods Enzymol. 194:1–933. [PubMed]
- Gyuris J, Golemis E, Chertkov H, Brent R. Cdi1, a human G1 and S phase protein phosphatase that associates with Cdk2. Cell. 1993;75:791–803. doi: 10.1016/0092-8674(93)90498-f. [DOI] [PubMed] [Google Scholar]
- Haarer BK, Pringle JR. Immunofluorescence localization of the Saccharomyces cerevisiae CDC12gene product to the vicinity of the 10-nm filaments in the mother-bud neck. Mol Cell Biol. 1987;7:3678–3687. doi: 10.1128/mcb.7.10.3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock JF, Paterson H, Marshall CJ. A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21rasto the plasma membrane. Cell. 1990;63:133–139. doi: 10.1016/0092-8674(90)90294-o. [DOI] [PubMed] [Google Scholar]
- Hartwell LH. Genetic control of the cell division cycle in yeast. IV. Genes controlling bud emergence and cytokinesis. Exp Cell Res. 1971a;69:265–276. doi: 10.1016/0014-4827(71)90223-0. [DOI] [PubMed] [Google Scholar]
- Hartwell LH. Genetic control of the cell division cycle in yeast. II. Genes controlling DNA replication and its initiation. J Mol Biol. 1971b;59:183–194. doi: 10.1016/0022-2836(71)90420-7. [DOI] [PubMed] [Google Scholar]
- Hayashibe M, Katohda S. Initiation of budding and chitin-ring. J Gen Appl Microbiol. 1973;19:23–39. [Google Scholar]
- Henikoff S. Unidirectional digestion with exonuclease III creates targeted breakpoints for DNA sequencing. Gene (Amst) 1984;28:351–359. doi: 10.1016/0378-1119(84)90153-7. [DOI] [PubMed] [Google Scholar]
- Hill JE, Myers AM, Koerner TJ, Tzagoloff A. Yeast/E. colishuttle vectors with multiple unique restriction sites. Yeast. 1986;2:163–167. doi: 10.1002/yea.320020304. [DOI] [PubMed] [Google Scholar]
- Igual JC, Johnson AL, Johnston LH. Coordinated regulation of gene expression by the cell cycle transcription factor SWI4 and the protein kinase C MAP kinase pathway for yeast cell integrity. EMBO J. 1996;15:5001–5013. [PMC free article] [PubMed] [Google Scholar]
- Kawamoto S, Nomura M, Ohno T. Cloning and characterization of SKT5, a Saccharomyces cerevisiae gene that affects protoplast regeneration and resistance to killer toxin of Kluyveromyces lactis. . J Ferment Bioeng. 1992;4:199–208. [Google Scholar]
- Kim HB, Haarer BK, Pringle JR. Cellular morphogenesis in the Saccharomyces cerevisiae cell cycle: localization of the CDC3gene product and the timing of events at the budding site. J Cell Biol. 1991;112:535–544. doi: 10.1083/jcb.112.4.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch C, Nasmyth K. Cell cycle regulated transcription in yeast. Curr Opin Cell Biol. 1994;6:451–459. doi: 10.1016/0955-0674(94)90039-6. [DOI] [PubMed] [Google Scholar]
- Konopka JB, DeMattei C, Davis C. AFR1 promotes polarized apical morphogenesis in Saccharomyces cerevisiae. . Mol Cell Biol. 1995;15:723–730. doi: 10.1128/mcb.15.2.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (Lond) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lillie SH, Pringle JR. Reserve carbohydrate metabolism in Saccharomyces cerevisiae: responses to nutrient limitation. J Bacteriol. 1980;143:1384–1394. doi: 10.1128/jb.143.3.1384-1394.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, DeMarini DJ, Valencik ML, Al-Awar OS, Fares H, De Virgilio C, Pringle JR. The septins: roles in cytokinesis and other processes. Curr Opin Cell Biol. 1996;8:106–119. doi: 10.1016/s0955-0674(96)80054-8. [DOI] [PubMed] [Google Scholar]
- Lupas A, Van Dyke M, Stock J. Predicting coiled coils from protein sequences. Science (Wash DC) 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- Maina CV, Riggs PD, Grandea AG, III, Slatko BE, Moran LS, Tagliamonte JA, McReynolds LA, di Guan C. An Escherichia colivector to express and purify foreign proteins by fusion to and separation from maltose-binding protein. Gene (Amst) 1988;74:365–373. doi: 10.1016/0378-1119(88)90170-9. [DOI] [PubMed] [Google Scholar]
- Mortimer RK, Schild D. Genetic map of Saccharomyces cerevisiae. . Microbiol Rev. 1980;44:519–571. doi: 10.1128/mr.44.4.519-571.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeld TP, Rubin GM. The Drosophila peanutgene is required for cytokinesis and encodes a protein similar to yeast putative bud neck filament proteins. Cell. 1994;77:371–379. doi: 10.1016/0092-8674(94)90152-x. [DOI] [PubMed] [Google Scholar]
- Orlean, P. 1996. Biogenesis of yeast wall and surface components. In The Molecular and Cellular Biology of the Yeast Saccharomyces. Cell Cycle and Cell Biology. J.R. Pringle, J.R. Broach, and E.W. Jones, editors. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 229–362.
- Pammer M, Briza P, Ellinger A, Schuster T, Stucka R, Feldmann H, Breitenbach M. DIT101 (CSD2, CAL1), a cell cycle-regulated yeast gene required for synthesis of chitin in cell walls and chitosan in spore walls. Yeast. 1992;8:1089–1099. doi: 10.1002/yea.320081211. [DOI] [PubMed] [Google Scholar]
- Pandolfo D, De Antoni A, Lanfranchi G, Valle G. The DNA sequence of cosmid 14-5 from chromosome XIV reveals 21 open reading frames including a novel gene encoding a globin-like domain. Yeast. 1996;12:1071–1076. doi: 10.1002/(SICI)1097-0061(199609)12:10B%3C1071::AID-YEA4%3E3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Pringle JR, Preston RA, Adams AEM, Stearns T, Drubin DG, Haarer BK, Jones EW. Fluorescence microscopy methods for yeast. Methods Cell Biol. 1989;31:357–435. doi: 10.1016/s0091-679x(08)61620-9. [DOI] [PubMed] [Google Scholar]
- Pringle JR, Bi E, Harkins HA, Zahner JE, De Virgilio C, Chant J, Corrado K, Fares H. Establishment of cell polarity in yeast. Cold Spring Harbor Symp Quant Biol. 1995;60:729–744. doi: 10.1101/sqb.1995.060.01.079. [DOI] [PubMed] [Google Scholar]
- Qadota H, Python CP, Inoue SB, Arisawa M, Anraku Y, Zheng Y, Watanabe T, Levin DE, Ohya Y. Identification of yeast Rho1p GTPase as a regulatory subunit of 1,3-β-glucan synthase. Science (Wash DC) 1996;272:279–281. doi: 10.1126/science.272.5259.279. [DOI] [PubMed] [Google Scholar]
- Riles L, Dutchik JE, Baktha A, McCauley BK, Thayer EC, Leckie MP, Braden VV, Depke JE, Olson MV. Physical maps of the six smallest chromosomes of Saccharomyces cerevisiaeat a resolution of 2.6 kilobase pairs. Genetics. 1993;134:81–150. doi: 10.1093/genetics/134.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RL, Bowers B, Slater ML, Cabib E. Chitin synthesis and localization in cell division cycle mutants of Saccharomyces cerevisiae. . Mol Cell Biol. 1983;3:922–930. doi: 10.1128/mcb.3.5.922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roncero C, Valdivieso MH, Ribas JC, Durán A. Isolation and characterization of Saccharomyces cerevisiaemutants resistant to Calcofluor White. J Bacteriol. 1988;170:1950–1954. doi: 10.1128/jb.170.4.1950-1954.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Sanders SL, Field CM. Septins in common? . Curr Biol. 1994;4:907–910. doi: 10.1016/s0960-9822(00)00201-3. [DOI] [PubMed] [Google Scholar]
- Sanders SL, Herskowitz I. The Bud4 protein of yeast, required for axial budding, is localized to the mother/bud neck in a cell cycle-dependent manner. J Cell Biol. 1996;134:413–427. doi: 10.1083/jcb.134.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos B, Snyder M. Targeting of chitin synthase 3 to polarized growth sites in yeast requires Chs5p and Myo2p. J Cell Biol. 1997;136:95–110. doi: 10.1083/jcb.136.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherens B, El M, Bakkoury, Vierendeels F, Dubois E, Messenguy F. Sequencing and functional analysis of a 32,560 bp segment on the left arm of yeast chromosome II. Identification of 26 open reading frames, including the KIP1 and SEC17genes. Yeast. 1993;9:1355–1371. doi: 10.1002/yea.320091210. [DOI] [PubMed] [Google Scholar]
- Shapira SK, Chow J, Richaud FV, Casadaban MJ. New versatile plasmid vectors for expression of hybrid proteins coded by a cloned gene fused to lacZgene sequences encoding an enzymatically active carboxy-terminal portion of β-galactosidase. Gene (Amst) 1983;25:71–82. doi: 10.1016/0378-1119(83)90169-5. [DOI] [PubMed] [Google Scholar]
- Shaw JA, Mol PC, Bowers B, Silverman SJ, Valdivieso MH, Durán A, Cabib E. The function of chitin synthases 2 and 3 in the Saccharomyces cerevisiaecell cycle. J Cell Biol. 1991;114:111–123. doi: 10.1083/jcb.114.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. . Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater ML, Bowers B, Cabib E. Formation of septum-like structures at locations remote from the budding sites in cytokinesis-defective mutants of Saccharomyces cerevisiae. . J Bacteriol. 1985;162:763–767. doi: 10.1128/jb.162.2.763-767.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloat BF, Adams A, Pringle JR. Roles of the CDC24 gene product in cellular morphogenesis during the Saccharomyces cerevisiaecell cycle. J Cell Biol. 1981;89:395–405. doi: 10.1083/jcb.89.3.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterner R, Dahm A, Darimont B, Ivens A, Liebl W, Kirschner K. (βα)8-barrel proteins of tryptophan biosynthesis in the hyperthermophile Thermotoga maritima. . EMBO J. 1995;14:4395–4402. doi: 10.1002/j.1460-2075.1995.tb00118.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trilla JA, Cos T, Durán A, Roncero C. Characterisation of CHS4 (CAL2), a gene of Saccharomyces cerevisiae involved in chitin biosynthesis and allelic to SKT5 and CSD4. . Yeast. 1997;13:795–807. doi: 10.1002/(SICI)1097-0061(199707)13:9<795::AID-YEA139>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Valdivieso MH, Mol PC, Shaw JA, Cabib E, Durán A. CAL1, a gene required for activity of chitin synthase 3 in Saccharomyces cerevisiae. . J Cell Biol. 1991;114:101–109. doi: 10.1083/jcb.114.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zervos AS, Gyuris J, Brent R. Mxi1, a protein that specifically interacts with Max to bind Myc-Max recognition sites. Cell. 1993;72:223–232. doi: 10.1016/0092-8674(93)90662-a. [DOI] [PubMed] [Google Scholar]