

Abstract
Hypertrophic cardiomyopathy is a human heart disease characterized by increased ventricular mass, focal areas of fibrosis, myocyte, and myofibrillar disorganization. This genetically dominant disease can be caused by mutations in any one of several contractile proteins, including β cardiac myosin heavy chain (βMHC). To determine whether point mutations in human βMHC have direct effects on interfering with filament assembly and sarcomeric structure, full-length wild-type and mutant human βMHC cDNAs were cloned and expressed in primary cultures of neonatal rat ventricular cardiomyocytes (NRC) under conditions that promote myofibrillogenesis. A lysine to arginine change at amino acid 184 in the consensus ATP binding sequence of human βMHC resulted in abnormal subcellular localization and disrupted both thick and thin filament structure in transfected NRC. Diffuse βMHC K184R protein appeared to colocalize with actin throughout the myocyte, suggesting a tight interaction of these two proteins. Human βMHC with S472V mutation assembled normally into thick filaments and did not affect sarcomeric structure. Two mutant myosins previously described as causing human hypertrophic cardiomyopathy, R249Q and R403Q, were competent to assemble into thick filaments producing myofibrils with well defined I bands, A bands, and H zones. Coexpression and detection of wild-type βMHC and either R249Q or R403Q proteins in the same myocyte showed these proteins are equally able to assemble into the sarcomere and provided no discernible differences in subcellular localization. Thus, human βMHC R249Q and R403Q mutant proteins were readily incorporated into NRC sarcomeres and did not disrupt myofilament formation. This study indicates that the phenotype of myofibrillar disarray seen in HCM patients which harbor either of these two mutations may not be directly due to the failure of the mutant myosin heavy chain protein to assemble and form normal sarcomeres, but may rather be a secondary effect possibly resulting from the chronic stress of decreased βMHC function.
A sarcomere, the functional unit of muscle, is composed of a precise arrangement of at least 20 known proteins ordered in nearly crystalline fashion. Complex interactions of a few of these proteins, mainly actin and myosin, produce the force necessary for muscular contraction (for review see Squire, 1986). The association of actin and myosin is tightly regulated by the troponin complex, tropomyosin, and flux of Ca2+ ions. Productive muscular contraction is dependent upon proper spatial relationships of muscle structural proteins within the sarcomere. Genetic analysis in Drosophila and Caenorhabditis has shown that mutations in several of the sarcomeric proteins disrupt myofibrillar organization and affect muscle function (for review see Epstein and Bernstein, 1992). However, for most of these mutations the underlying biochemical/functional defect has not been elucidated. Thick and thin filaments can form independently of one another, yet apparently equal stoichiometric quantities of actin and myosin are required for proper sarcomeric order (Beall et al., 1989).
The discovery that mutations in sarcomeric proteins cause an inherited form of human heart disease has brought significant attention towards understanding muscle protein function. Familial hypertrophic cardiomyopathy (HCM)1 is an autosomal dominant, genetically heterogeneous heart disease with variable penetrance and an assortment of clinical phenotypes ranging from mild syncope to sudden death. The hallmark of this disease is unexplained left ventricular hypertrophy in which dilation of the ventricular chamber is absent (Maron, 1988) and an increase in myocyte disarray associated with abnormal myofilament structure in hearts with HCM is apparent. Genetic mapping has identified a minimum of six loci which cosegregate with the disease, any of which can be responsible for the dominant phenotype. The genes identified as responsible for HCM all encode different structural components of the sarcomere, β myosin heavy chain (βMHC; Geisterfer-Lowrance et al., 1990), cardiac troponin T, α tropomyosin (Thierfelder et al., 1994), C protein (Bonne et al., 1995; Watkins et al., 1995a ) as well as the cardiac essential and regulatory myosin light chains (Poetter et al., 1996). Interestingly, mutations in the myosin light chains produce skeletal muscle defects and are associated with a rare form of HCM which displays midventricular hypertrophy. However, the connection between mutations in sarcomeric proteins and the pathogenesis of HCM is not understood.
Mutations in the βMHC gene are present in ∼30% of the HCM families currently characterized (Watkins et al., 1995b ). Nearly all of the βMHC mutations reported are in the S1 head domain and occur at amino acids that are highly conserved throughout MHC phylogeny (Warrick and Spudich, 1987; Mornet et al., 1989). The crystal structure of S1 shows that many of the HCM mutations contribute to functional or structural domains within the globular myosin head (Rayment et al., 1995). Reduced function has been demonstrated for a few of these mutations (Cuda et al., 1993; Sweeney et al., 1994; Lankford et al., 1995), yet it remains to be elucidated how decreased myosin function may lead to the morphological disarray observed in HCM hearts. The possibility exists that the mutations in βMHC are directly responsible for the myofibrillar disarray causing defective assembly of the mutant protein and/or interference with the assembly of the wild-type proteins by a dominant negative function. In this regard, a class of missense mutations in the S1 portion of Caenorhabditis muscle MHC displays dominant phenotypes that dramatically disrupt sarcomere organization (Bejsovec and Anderson, 1988, 1990). Although none of the C. elegans mutations correspond to those described in HCM, it is possible that the human βMHC mutant proteins have a similar deleterious affect on myofibril assembly and/or structure.
To address this question, we have developed an in vivo competition assay that will allow the direct evaluation of the ability of an epitope tagged mutant human cardiac βMHC protein to effectively compete with a wild-type βMHC protein for assembly into endogenous sarcomeric units in living myocardial cells. In this paper we report the cloning and construction of full length human βMHC cDNAs encoding wild-type and mutant proteins. Expression of these clones either individually or in combination within transiently transfected primary cultures of neonatal rat ventricular cardiomyocytes (NRC) was used to analyze the effect of βMHC mutations on subcellular localization and myofibril assembly. A missense mutation in the consensus ATP binding sequence dramatically affected sarcomere structure, disrupting myofibril assembly at an early stage. However, βMHC with either of two HCM mutations, R249Q or R403Q, assemble normally. This indicates the myofibrillar disarray observed in HCM patients may not be the direct result of mutant contractile proteins altering myofilament assembly. Furthermore, the data presented here support previous information suggesting that biochemical defects in the human βMHC proteins produce physiological dysfunction and pathological alterations in the heart.
Materials and Methods
Isolation of Human βMHC cDNA Clones
A human heart (ventricular) specific cDNA library (Stratagene, La Jolla, CA) was screened with end labeled oligonucleotide probes derived from the βMHC published sequence (Jaenicke et al., 1990; Liew et al., 1990). End labeling and library screening were performed as per standard protocols (Sambrook et al., 1989). Selected cDNAs were sequenced using cDNAsequence–specific primers (synthesized on a Pharmacia LKB Gene Assembler Plus; Pharmacia Biotech, Piscataway, NJ) in conjunction with the Sequenase kit (United States Biochemicals, Cleveland, OH). Restriction enzymes used for mapping and subcloning were obtained from GIBCO BRL (Gaithersburg, MD). Site-directed mutagenesis was performed with the Sculptor kit (Amersham Corp., Arlington Heights, IL) or by PCR as described by Higuchi et al. (1988). A small portion of the final PCR product was excised from the full length product by treatment with appropriate restriction enzymes and subsequent gel purification (GeneClean; Bio 101, La Jolla, CA). The small fragment was sequenced in its entirety to confirm that the only change(s) present were the desired sequence insertion or mutation. Correct subclones were then used to replace the wild-type sequence in the full length βMHC cDNA. To eliminate the possibility of a second mutation affecting the K184R phenotype, this construct was made independently a second time and subsequently tested. There was no difference in the phenotype observed for both clones with this mutation.
Tissue Culture and Transfections
Neonatal rat cardiac cells were isolated from hearts of 1–2-d-old neonatal rats, purified on a discontinuous percoll gradient (Zhu et al., 1991), and were plated on chamber slides (NUNC, Naperville, IL) coated with laminin (Sigma Chemical Co., St. Louis, MO). After ∼24 h the cells were transiently transfected using a modified calcium phosphate method (Chen and Okayama, 1988) in DME plus 15% serum (10% horse serum, 5% fetal calf serum). Approximately 3 μg of DNA was used per well for a two-well chamber slide. The precipitate was allowed to be in contact with the cells for 12–20 h. After transfection the cells were washed with PBS, changed to DME with 1% horse serum, 10−4 M phenylephrine, and were subsequently cultured for 2–5 d. Alternatively, the washed cells were cultured in 15% serum (10% horse serum, 5% fetal calf serum) for 2–5 d. No difference was observed between these two culture conditions.
Nonmuscle cells (CHO) were grown in two-well chamber slides and were transfected as described for the neonatal rat ventricular myocytes. 48 h after transfection the cells were fixed in 3% paraformaldehyde and were immunostained.
Antibodies, Immunostaining, and Microscopy
For indirect immunofluorescence staining, the cells were washed briefly in relaxation buffer A (0.1 M KCl, 5 mM EDTA, 1 mM EGTA), fixed in fresh 3% paraformaldehyde (made in buffer A), neutralized with 50 mM NH4Cl, and immunostained with the appropriate antibodies. Sequential staining using two mouse monoclonals as primary antibodies was performed as follows: after incubation with the first primary antibody and subsequent washing with buffer A, the immunostained cells were incubated with an FITC conjugated goat anti–mouse affinity purified F(ab) fragment diluted 1:50 for 40 min. Washing in buffer A was followed by the second primary antibody, applied for 1 h. The second secondary was lissamine rhodamine (LRSC) AffiniPure donkey anti–mouse diluted 1:100. The anti–rat myosin light chain 2v antibody was produced in our lab by Dr. S. Kubalak, as previously described (Kubalak et al., 1996). The epitope specific glu–glu (EE) antibody, derived from polyoma virus medium T antigen, was a generous gift of Dr. G. Walter (University of California, San Diego, CA) and was used at a 1:50 dilution of hybridoma supernatant. βMHC, 4H3, monoclonal antibody was a generous gift of Dr. J. Leger (Institut National de la Santé et de la Recherche Médicale, Montpellier, France) and was diluted 1:20. Anti-HA antibody was purchased from Boehringer Mannheim (Indianapolis, IN) and was diluted 1:10. Antibody to actin (A20) was a generous gift of Dr. J. Lin (University of Iowa, Iowa City, IA) and was diluted 1:20. Phalloidin–rhodamine was purchased from Molecular Probes Inc. (Eugene, OR) and was diluted 1:1,000. Secondary antibodies conjugated to FITC or LRSC (1:100 to 1:200 dilution) were purchased from Jackson Laboratories (Bar Harbor, ME). Stained cells were mounted using gelvitol with 2.5% DABCO (1,4-diazabicyclo-[2.2.2] octane). Laser scanning confocal microscopy was performed using the Bio-Rad MRC1024 equipped with LaserSharp software. All confocal pictures were derived from a z-series which has been projected to produce the final image shown. Composite images were given pseudocolor and merged in Adobe Photoshop.
Results
Cloning and Characterization of Wild-type and Mutant Myosin cDNAs
A full-length human βMHC cDNA was constructed from the overlapping clones using restriction enzyme sites endogenous to the βMHC sequence (Fig. 1). The reconstructed full-length cDNA was completely sequenced on both strands to ensure that no mutations occurred in the encoded wildtype protein. Small portions of the wild-type βMHC cDNA were used as templates to generate nucleotide changes encoding desired missense mutations as well as the insertion of epitope tags. These fragments were sequenced to ensure the only change present was the preferred mutation and were subsequently exchanged into the full length cDNA. The resulting clones were sequenced across any restriction sites used during construction to ensure the desired mutation was the only change present. The open reading frame of all the βMHC clones was confirmed via in vitro transcription and translation (data not shown).
Figure 1.

Cloning and construction of human βMHC cDNA expression plasmids. (A) The three overlapping cDNA clones (λcDNA4, 13, and 10) used for construction of the 6-kbp, fulllength human βMHC cDNA are shown below the representation of the full-length cDNA (βMHC cDNA). The restriction sites SmaI (S), BamHI (B), and AatII (A) were used to assemble the full-length clone. Subclones KS5′, c13-5′, and c13-M were used to insert the epitope tag sequences or for generation of the mutant codons. Also listed are the full-length, epitope tagged clones used. (B) Single letter amino acid sequence of wild-type myosin from several species are compared. The numbering is based upon the published human βMHC sequence (Liew et al., 1990). The underlined area is involved in ATP binding. 1, human βMHC; 2, chick sarcomeric MHC (Molina et al., 1987); 3, rat embryonal sarcomeric MHC (Strehler et al., 1986); 4, Ceanorhabditis elegans MHC A (Karn et al., 1983); 5, chick smooth muscle MHC (Yanagisawa et al., 1987); 6, Dictyostelium discoideum (Warrick et al., 1986); 7, Acathamoeba castellanii myosin II (Hammer et al., 1987).
An epitope tag was inserted in the NH2-terminal region of human βMHC to facilitate detection of exogenous protein in transfected NRC. Exogenous βMHC proteins containing either haemaglutinin (HA-1 = YPYDVPDYA; Wilson et al., 1984) or glu–glu (EE = EEYMPME; Grussenmeyer et al., 1985) epitopes were specifically recognized by their respective monoclonal antibodies in transiently transfected NRC (Fig. 2). Each epitope was positioned between amino acids 3 and 4 of the βMHC molecule, a domain with no ascribed function. As noted in previous studies, the crystal structure of the myosin S1 head reveals that the NH2 terminus is free in solution, existing as a random coil on the surface molecule (Rayment et al., 1993a ,b), making this region a good target for the insertion of short sequences. The exogenous βMHC protein was incorporated into myofibrils in a pattern indistinguishable from the endogenous MLC2v (data not shown), a molecule known to bind MHC.
Figure 2.

Epitope tagged wild-type human βMHC protein assembles into normal myofibrils in transiently transfected NRC. Cells were transfected with plasmids containing the wild-type human βMHC cDNA tagged with either the EE (A–C) or HA (D–F). The cells were stained for the presence of actin filaments using rhodamine–phalloidin (A and C) and were costained with the anti-EE (B) or anti-HA (E) specific antibodies. Sarcomeres containing human βMHC have well defined A bands (a), I bands (i), and M lines (m). The composite images (C and F) show that the exogenous βMHC (green) fills the A band, except the H zone as expected, in a pattern that is complementary and partially overlapping with F-actin (red, C and F). Bar, 20 μm (inset enlarged 3×).
Four missense mutations were generated in the βMHC coding sequence via PCR (see Materials and Methods). Two of the βMHC mutations generated were based upon corresponding mutants that remove functions of the Dictyostelium MHC II. The lysine at amino acid 185 in Dictyostelium MHC is in the ATP binding pocket (Fisher et al., 1995), and mutation of this amino acid to arginine blocks ATP exchange of Dictyostelium MHC resulting in a “rigorlike” molecule (Spudich, J., personal communication). The second mutation is a serine to valine change at amino acid 472 in human βMHC. Dictyostelium myosin with this mutation has slightly higher than normal actin activated ATPase yet in an in vitro motility assay this myosin can move F-actin at only 10% of the rate for wild-type MHC (Ruppel and Spudich, 1996). If actin motility is a requirement for sarcomeric assembly, then βMHC protein with this mutation may be expected to disrupt myofilament structure. The other two changes, R249Q and R403Q, are representative of the class of βMHC mutations found associated with human hypertrophic cardiomyopathy (Geisterfer-Lowrance et al., 1990; Rosenzweig et al., 1991). In families with these two mutations the disease has high penetrance and a high incidence of sudden death (Epstein et al., 1992; Watkins et al., 1992). Furthermore, the R403Q mutant protein has been shown to display defective contractile function in a variety of in vitro assays (Cuda et al., 1993; Sweeney et al., 1994; Lankford et al., 1995).
It is possible that insertion of an epitope tag or mutations at specific amino acids may render the βMHC molecule unstable once expressed in cultured cells. However, transiently transfected cultures of COS cells immunostained with either 4H3 (anti-βMHC) or the epitope specific antibodies show detectable levels of βMHC protein in ∼25% of the cells for all constructs (data not shown). Furthermore, the frequency of transfection in NRC is the same for all of the βMHC expression plasmids (⩽1% of NRC was transfected). Taken together, these studies indicate that the epitope tagged wild-type and mutant proteins were synthesized at comparable levels.
Epitope Tagged Wild-type Human βMHC Assembles in a Normal Sarcomeric Pattern in Transfected Neonatal Rat Cardiomyocytes
Transfected NRC display two distinct classes of phenotype relevant to the organization of the myofibrils in immunostained cells. One third of the cells display highly organized myofibrils having well defined sarcomeres identifiable by clear H zones, A bands, and I bands. The remaining transfected cells were not well developed or were too faint to be analyzed. Both classes of cellular phenotype were observed in nontransfected as well as in transiently transfected NRC fixed and stained for either endogenous or exogenous myofibrillar proteins. NRC cultures exposed to calcium phosphate transfection with the backbone vector pCB6 produce ∼40% of the cells filled with highly organized myofibrils. Cells were only scored if some level of sarcomeric organization was present. This eliminated contaminating fibroblasts from the data set.
Human βMHC protein accumulates and was detectable in NRC transiently transfected with the expression constructs containing the βMHC epitope tagged cDNA. In transfected NRC, the human wild-type βMHC protein tagged with either EE or HA epitopes assembled into highly ordered sarcomeres in cells with organized myofilaments (Fig. 2). Ordered myofibrils containing exogenous human βMHC have defined H zones in addition to A band periodicity complementary to thin filament staining within the same cell. Clearly defined H zones visible in cells immunostained for human βMHC showed that the globular head of the tagged myosin was excluded from the H zone which indicated that these thick filaments have normal structure. This result was similar to the previously described distribution of epitope tagged myosin light chain (Soldati and Perriard, 1991), which decorated the A band and was excluded from the H zone, a region devoid of myosin heads. The thick filament marker myomesin was found at the M line in mature myofibrils containing wild-type human βMHC (data not shown). Furthermore, the staining pattern of human βMHC was coincident with endogenous MHC as well as exogenous epitope tagged myosin light chain (data not shown). Similar to the control cultures, endogenous thin and thick filament markers detected in transfected cells displayed the same degree of organization observed for the exogenous wild-type protein (Table I). These data indicate that the exogenous human βMHC protein assembled normally into sarcomeres in transfected NRC, and the presence of the glu–glu or HA epitope tag between amino acids 3 and 4 did not inhibit this process nor disrupt sarcomeric organization.
Table I.
Morphology of Transfected NRC
| Transfected DNA* | Percentage of cells with organized myofibrils | |||||
|---|---|---|---|---|---|---|
| Epitope | Actin | Myomesin | ||||
| pCB6 | n.a.‡ | 40§ | 40§ | |||
| Wild-type‖ | 33 | 100 | 100 | |||
| K184R | 0 | 1.1 | 0.8 | |||
| R249Q | 32 | 100 | 100 | |||
| R403Q | 34 | 100 | 100 | |||
| S472V | 33 | 100 | 100 | |||
For all DNAs transfected, at least 2,000 cells were scored over several different transfected cultures.
n.a., not available.
Since transfected cells are not identifiable for the control backbone vector, pCD6, NRC were randomly scored for endogenous F-actin (via rhodamine–phalloidin) or myomesin (B4 antibody).
For the epitope tagged constructs only immunopositive cells were counted. The numbers are presented as percentage of transfected NRC, which display fully organized myofilaments as detected by indirect immunofluorescence. Epitope specific antibodies identify the exogenous βMHC protein. In transfected NRC with highly organized myofilaments, the percentage of highly ordered endogenous myofibril markers is also given.
The K184R Mutation Disrupts Sarcomeric Structure in Transfected NRC
The K184R mutation in βMHC resulted in a severe alteration in the subcellular localization of this mutant protein in transfected cardiomyocytes (Fig. 3). The distribution of K184R protein was visualized as diffuse and somewhat uniform throughout the cell or occurred as cable-like structures. Alternatively, K184R accumulated as brightly staining globules of various size. NRC with large amounts of K184R protein (i.e., stain very brightly for this molecule) were small and irregularly shaped with many pointed lamellipodia (data not shown). As seen in Fig. 3 B, K184R mutant protein formed cable-like structures similar to those described as the premyofibril (Rhee et al., 1994). In this same cell, endogenous MHC (Fig. 3 A) displayed nearly complete loss of sarcomeric organization. The presence of K184R βMHC in cardiomyocytes caused disorganization of endogenous myomesin (Fig. 3, C and D), indicating that thick filament structure is disturbed. Thin filament order was also disrupted in cells expressing K184R (Fig. 3, E and F). In a series of experiments, >2,000 individual cells successfully transfected with K184R were analyzed by immunostaining. In more than half of these cells sarcomeric organization of thick or thin filament markers was completely absent. The remaining K184R positive cells contained few sarcomeres, except for rare cells in which the entire cell volume was filled with highly organized myofibrils (Table I). In NRC with a few recognizable myofilaments, K184R βMHC sometimes appeared to decorate thin filaments or occurred in A band regions, albeit in a sporadic pattern that never filled the entire sarcomere. Even though organized phalloidin staining was lost in many cardiomyocytes with K184R, G actin was readily detectable (Fig. 4). The scattered distribution of K184R protein within the cell appears to colocalize with G actin, indicating that these molecules may be in close association with one another.
Figure 3.
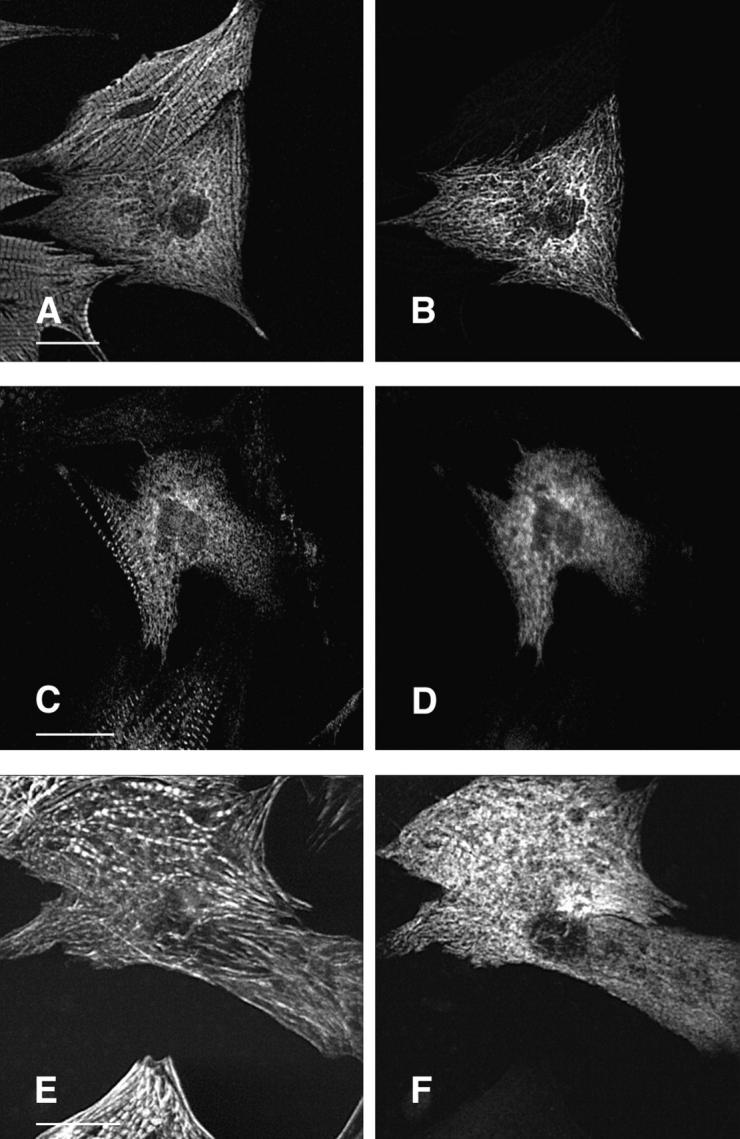
Expression of K184R βMHC in NRC disrupts myofibril assembly. NRC transfected with the human βMHC K184R plasmid were stained for with the anti-EE epitope specific antibody (B, D, and F). These cells were costained with anti-βMHC (A), anti-myomesin (C), or rhodamine–phalloidin (E). Sequential staining with two primary monoclonal antibodies is described in Materials and Methods. Bar, 20 μm.
Figure 4.



K184R protein colocalizes with actin. A transfected NRC expressing K184R was costained with monoclonal antibody A20 (anti-actin, A) and the epitope specific antibody (anti-EE, B). The composite image (C) shows the high degree of colocalization of these two proteins throughout the cell. Bar, 20 μm.
Mutations at Amino Acids 249, 403, and 472 Do Not Alter MHC Assembly
There was no detectable difference in the distribution of phenotypes comparing expression of wild-type βMHC with the three βMHC mutations R249Q, R403Q, or S472V in NRC. All three of these mutant proteins assembled into extremely organized myofibrils (Fig. 5) at the same frequency as observed for the wild-type βMHC protein (Table I). R249Q, R403Q, and S472V mutant proteins assembled into A band regions as defined via costaining for endogenous myofilament proteins (Figs. 5 and 6). This result indicated that the three mutants are readily incorporated into thick filaments, and such inclusion did not produce an observable defect in sarcomere organization. Unlike the K184R mutant there was little excess accumulation or abnormal subcellular localization of the R249Q, R403Q, or S472V molecules even in cells with little sarcomeric structure. These three mutant proteins always colocalized with the endogenous MHC in transfected cells.
Figure 5.
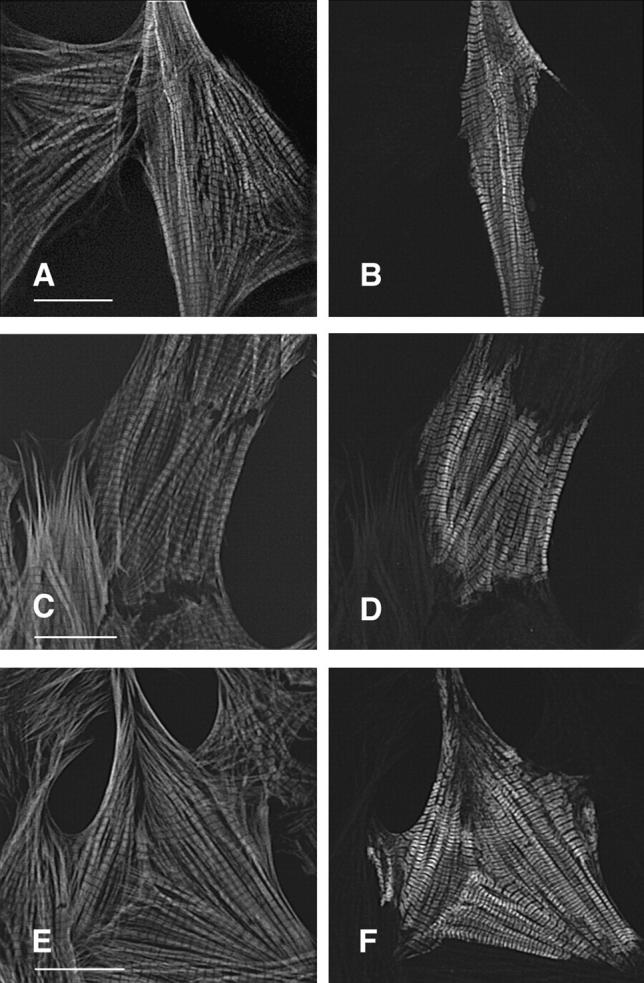
S472V as well as hypertrophic cardiomyopathy mutants R249Q and R403Q assemble normally and do not disrupt myofibril structure. Cells transfected with R249Q (A and B), R403Q (C and D), or S472V (E and F) were stained for the presence of exogenous myosin with the epitope specific antibody (B, D, and F) and with rhodamine–phalloidin (A, C, and E). Bar, 20 μm.
Figure 6.

Thick filaments containing R403Q have normal organization within the sarcomere. NRC transfected with human R403Q plasmid were costained for exogenous βMHC (B) with the epitope specific antibody and myomesin (A). The merged image (C) shows that myomesin staining is completely localized to the M-line, indicating normal thick filament and sarcomere structure. Bars: (A) 20 μm; (inset), 5 μm.
Coexpression of Mutant and Wild-type βMHC in the Same Cell
Expressing both mutant and wild-type βMHC within the same transfected cell provided a sensitive measure of detecting minor differences in subcellular localization of the two proteins. Utilization of two different epitopes inserted at the same location within the βMHC sequence facilitated immunostaining of both mutant and wild-type βMHC proteins within the same cell. However, the two epitopes that were useful in the βMHC context were both recognized by mouse monoclonal antibodies. Therefore a sequential staining procedure was used to detect signal specific to each primary antibody (see Materials and Methods). Fig. 7, A and B shows that little or no bleed through signal was detected in NRC expressing only one wild-type βMHC construct that was stained with both epitope specific antibodies. In cotransfected cells, both wild-type constructs were observed, and as would be expected, the subcellular localization was identical for the two wild-type proteins present within the same cell (Fig. 7, C and D). Both proteins were coincorporated into A bands in a pattern that was complementary to thin filament staining.
Figure 7.
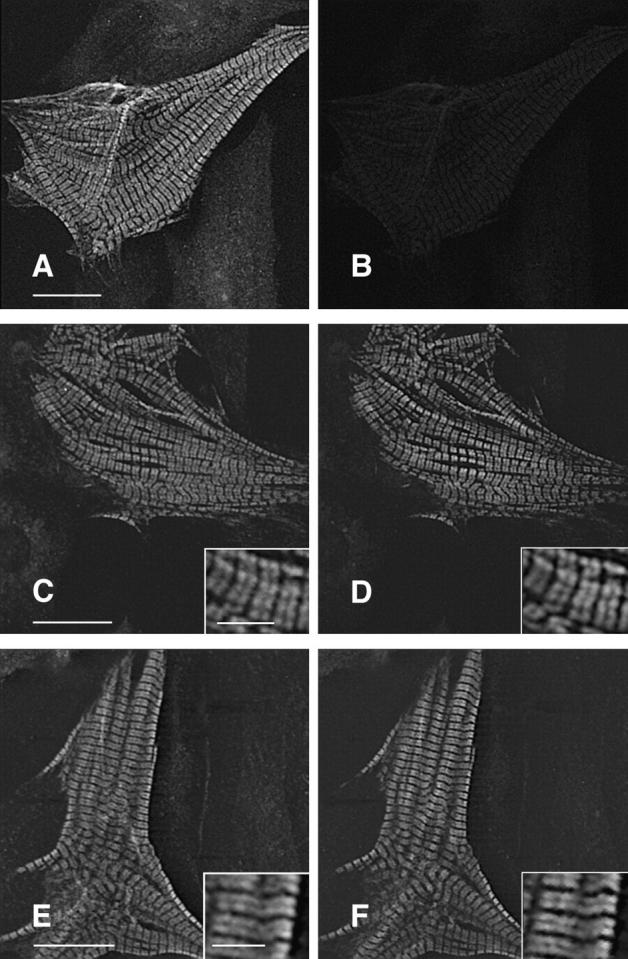
Coexpression of both mutant and wild-type βMHC molecules detects no differences in subcellular localization of the two exogenous proteins. NRC were cotransfected with both wildtype and mutant expression plasmids, each tagged with a different epitope. Sequential staining (see Materials and Methods) allows detection of each protein within the same cell. A and B show a cardiomyocyte transfected with Hnwt only. This cell was immunostained with anti-HA, goat anti–mouse–FITC (F(ab)), and anti- EE and then donkey anti–mouse–LRSC. A shows the anti-HA specific staining (FITC channel), and B shows the LRSC channel, indicating that the anti-EE antibody does not crossreact, and minimal signal bleed through occurs under these conditions. Using anti-EE to detect Tnwt, the same lack of crossreactivity is observed when costaining with anti-HA (data not shown). Expression and detection of both Hnwt (C) and Tnwt (D) in the same cell shows identical subcellular localization for both proteins. The TnR403Q mutant (F) also shows identical distribution to Hnwt βMHC (E) when coexpressed in the same cell. Similar results are obtained when coexpressing either R249Q or S472V with wildtype βMHC. The type of epitope does not affect the outcome of this experiment. Bars: (A, C, and E) 20 μm; (E and inset) 5 μm.
Coexpression of wild-type βMHC with either R249Q, R403Q, or S472V showed that the assembly characteristics of the mutant proteins are equivalent to the wild-type molecule. All three mutant proteins coassembled into thick filaments with the same localization as the wild-type βMHC within the same cell (Fig. 7, E and F). There was no abnormal distribution or accumulation of mutant in relation to wild-type βMHC in NRC expressing these human proteins. In contrast, coexpression of K184R disrupts the sarcomeric organization of wild-type human βMHC within the same cell (data not shown).
Discussion
In this paper we describe an in vivo system that should prove useful in the molecular dissection of structure–function relationships between specific residues in contractile proteins and their role in myofibrillar assembly in cardiac muscle cells. The in vivo competition assay can determine the sorting characteristics of individual contractile protein isoforms, elucidating subtle differences in subcellular localization within the cardiomyocyte (Komiyama et al., 1996). As an initial model system we have focused on human cardiac βMHC proteins since point mutations in this protein are implicated as causative in hypertrophic cardiomyopathy. Furthermore, since MHC are highly conserved, insights from studies of invertebrate and nonmuscle myosins can be analyzed for effects in mammalian cells.
Full length cDNAs expressed βMHC protein when transiently transfected into primary cultures of neonatal rat ventricular cardiomyocytes. The distribution of the wildtype human protein is not the result of sticking to myofilaments, based upon the observed highly ordered nature of the immuno-stained sarcomeres containing human βMHC. Thick filaments containing human βMHC are normal in structure based upon the A band localization and the distribution of myomesin staining. Furthermore, the lack of any fluorescent signal found within the H zone strongly suggests the exact incorporation of human βMHC into thick filaments. Previous studies have shown that a truncated form of MHC containing the S1 and S2 domains does not incorporate into sarcomeres in a defined pattern (Johnson et al., 1988), making it unlikely that a premature stop codon is present in the βMHC cDNAs expressed in the current studies.
A critical aspect of this in vivo, dual epitope competition assay is that the presence of the EE or HA epitopes does not affect the assembly of the human βMHC molecule into normal sarcomeres of neonatal rat cardiomyocytes. Slightly more than one-third of the NRC transfected with either wild-type epitope tagged construct is filled with highly ordered myofibrils which have incorporated the exogenous protein throughout their length. This is nearly the same number of highly organized cells observed in NRC cultures transfected with the backbone vector. NRC transfected with the epitope tagged myosin light chain construct, MLC3f-vsv, also show a similar pattern in which approximately one third of the transfected cells have perfectly ordered myofibrils throughout the cell (data not shown). It should be noted that previous studies have demonstrated that the epitope tag on MLC3f-vsv does not affect subcellular localization of this molecule in cultures of micro-injected adult rat (Soldati and Perriard, 1991) or in transfected NRC (Komiyama et al., 1996).
A point mutation in the ATP binding domain of human β cardiac MHC acts as a dominant negative on myofilament structure. The subcellular localization of human βMHC protein in transfected NRC is dramatically affected by the K184R mutation. The corresponding mutant in Dictyostelium MHC (K185R) binds actin “normally” but will not release in the presence of ATP (Spudich, J., personal communication). In vivo the Dictyostelium mutant can form thick filaments, however the mutant-containing cells have the same phenotype as cells in which the myosin gene is deleted. The K184R mutation is located in the consensus nucleotide binding sequence (GlyXXXXGlyLys; Walker et al., 1982). The lysine in this sequence contributes to nucleotide binding via neutralization of one of the phosphate groups as proposed from the crystal structure of two GTP binding proteins, EF-Tu and Ha-ras (Jurnak, 1985; Pai et al., 1989). The inability to hydrolyze or bind nucleotide has been described for mutations in other amino acids within the ATP binding consensus in several proteins such as ras (Clanton et al., 1987), herpes virus thymidine kinase (Liu and Summers, 1988), and Escherichia coli adenylate kinase (Reinstein et al., 1988). Since the nucleotide binding sequence is so highly conserved it would be predicted that the K184R mutation in the human βMHC protein is a “rigor-like” molecule which binds strongly to actin, resulting in the inability to complete the energy transduction cycle. This is consistent with the observed mutant myosin distribution as well as the colocalization of human K184R and endogenous actin. In vitro analysis should elucidate the biochemical characteristics of this mutant myosin.
Human K184R βMHC may be incorporated into thick filaments in transfected NRC however sarcomeric organization is disrupted in these cells. A similar effect occurs when cytoplasmic actins are over expressed in cardiomyocytes leading to the decay of thin filaments (von Arx et al., 1995). In contrast to the K184R mutant, cardiac cells containing cytoplasmic actin maintain the framework of the myofibril including Z lines, A bands, and M lines. Some sarcomeric assemblages can be found in K184R expressing NRC, however the predominant phenotype for K184Rpositive cardiomyocytes is the lack of thick and thin filament organization (Figs. 3 and 4). This suggests a dominant negative action for the mutant K184R myosin. Mutations in the glycine residues of the consensus ATP binding sequence in C. elegans MHC B are dominant, disrupting thick filament assembly and muscle function (Bejsovec and Anderson, 1988, 1990). Thin filament number and organization are normal in these animals. Only small amounts of the mutant MHC B protein accumulates in the body wall muscles, which may account for the presence of thin filaments. To our knowledge this is the first report of a myosin mutation, K184R, that destroys the structure of both thick and thin myofilaments.
Sarcomeric disorder in NRC expressing K184R may be the result of blocking nascent myofilament assembly. K184R can form short strings of cable-like structures (Fig. 3) similar to the phenotype of loosely aligned MHC filaments found in developing sarcomeres (Lu et al., 1992). Myomesin, which is found only in mature myofibrils (Lin et al., 1994), is not associated with K184R βMHC protein outside of organized sarcomeres. In cells costained for epitope tagged βMHC and actin, the K184R mutant colocalizes with actin throughout the cell (Fig. 4). This suggests that premyofibril thin filaments (Rhee et al., 1994), or I-Z-I structures (Schultheiss et al., 1990), are present in association with the mutant myosin that may be irreversibly bound to actin, and this aggregation blocks the maturation of the myofibril. Alternatively, K184R may destroy the cytoarchitecture after incorporation into sarcomeric thick filaments. It is possible that both nascent assembly is blocked and the destruction of myofilaments is occurring in the presence of K184R. The abnormal distribution of K184R in transfected NRC validates the use of this system to explore sequence-specific requirements on myosin assembly through single amino acid changes created in vitro.
The S472V change does not disrupt assembly of human βMHC, documenting the specificity of the effect of the K184R mutation on sarcomeric assembly. The corresponding Dictyostelium mutant myosin, S465V, has normal actin binding and actin activated ATPase activities yet moves actin filaments at only 10% of the rate of wild-type MHC (Ruppel and Spudich, 1996). This amino acid is part of a random coil that is highly conserved in all myosins and which connects the upper and lower domains of the 50-kD fragment in the myosin head (Rayment et al., 1993a ). It is thought that movement of these two domains occurs during the transition from weak to strong actin binding, which may decrease affinity for the products of ATP hydrolysis thus initiating the power stroke (Rayment et al., 1993b ). Dictyostelium myosin with this mutation partially rescues the myosin null phenotype and has increased basal ATPase activity yet has severely impaired ability to mobilize actin filaments in vitro (Ruppel and Spudich, 1996). It is plausible that a similar effect on βMHC function is produced by the S472V human βMHC mutation, which would suggest that productive acto–myosin interaction is not required for sarcomere assembly. Alternatively, mutation of this highly conserved amino acid does not alter function in the human βMHC protein.
In contrast to the K184R ATP binding site mutation, hypertrophic cardiomyopathy mutations, R249Q and R403Q, in human βMHC display normal assembly and have no dominant negative effects on sarcomeric structure. The subcellular localization of these two proteins is coincident with endogenous thick filament markers myomesin, MHC and myosin light chain. In the cotransfection assay HCM mutant MHCs colocalized with wild-type human βMHC, and both proteins assembled into structurally normal myofilaments. This is consistent with the observation for αMHC R403Q/+ mice in which mutant αMHC accumulates normally and cardiac dysfunction precedes histopathologic changes (Geisterfer-Lowrance et al., 1996). It is unclear, however, why Marian et al. (1995) have observed sarcomere disarray when expressing human βMHC R403Q in adult feline cardiomyocytes. These authors used an adenoviral vector to infect cultured adult cardiomyocytes and described sarcomere disorganization in cells 5 d after being exposed to high MOI of virus containing βMHC cDNA under the control of the CMV promoter. Marian et al. (1995) are unable to show that βMHC protein is incorporated into sarcomeres of infected cells. Furthermore, adult rat cardiomyocytes dedifferentiate with prolonged exposure to culture conditions (Messerli et al., 1993), and this may also be the case for the feline system. Prolonged culture of NRC transfected with our human βMHC constructs does not change the observations presented and, expression of our R403Q construct in regenerating adult rat cardiomyocytes does not interfere with normal myofilament structure (data not shown). However, it is possible that myofibrils in adult feline myocytes are more sensitive to disruption by the R403Q mutant protein. In addition, the NRC system may not accurately mimic the mechanical load found in vivo which may be necessary to produce disarray.
The data presented in this paper support the interpretation that a decrease in βMHC function is the primary defect in the etiology of hypertrophic cardiomyopathy. These results are consistent with the idea that MHC functional insufficiency is the primary stimulus for the hypertrophic cardiomyopathy phenotype. Therefore, compensatory hypertrophy and sudden death may be secondary responses to chronic decreased contractile function. R403Q βMHC protein purified from HCM patients has impaired ability to mobilize actin filaments in vitro (Cuda et al., 1993), and skeletal muscle fibers from such patients have reduced power output as well as decreased unloaded maximal velocity of shortening (Lankford et al., 1995). Decreased functional capabilities have also been observed for other HCM βMHC mutations. Future studies employing analysis of single cell function in live transfected NRC should address the relationship between the biochemical defect in human βMHC and its effect on contractility.
It is not necessary to propose a poison effect of the βMHC molecule on the thick filament, as a functional insufficiency would not interfere with the wild-type βMHC present in HCM patients, yet could still present the stimulus for hypertrophy, which is the hallmark of this disease. Transgenic mice overexpressing oncogenic ras in the heart have ventricular hypertrophy, selective diastolic dysfunction (Hunter et al., 1995), increased systolic function, myocyte disarray, fibrosis, and increased juvenile mortality (Gottshall et al., 1997). This suggests that chronic stimulation of a second messenger system in the heart produces all the overt characteristics of hypertrophic cardiomyopathy in humans caused by mutations in sarcomeric proteins. The data presented in this paper suggest that cellular dysmorphology is also secondary to biochemical dysfunction of the sarcomere producing decreased heart function.
Abbreviations used in this paper
- HCM
hypertrophic cardiomyopathy
- MHC
myosin heavy chain
- NRC
neonatal rat cardiomyocytes
Footnotes
The authors are very much indebted to Mahmoud Itani for excellent technical assistance in preparation of neonatal rat cardiomyocytes. The authors thank the Institute for Biomedical Engineering Confocal and Imaging Core for the use of the confocal microscope. We also thank Drs. Sanford Bernstein, Pieter Doevendans, Peter Gruber, Kirk Knowlton, and Howard Rockman for discussions and critical reading of the manuscript.
The Core facility is supported by the Whittaker Foundation Development Award (Grants Section). K.D. Becker is supported by an individual National Research Service Award from the National Institutes of Health. This work was supported by funds to K.R. Chien from the National Institutes of Health/National Heart, Lung, and Blood Institute (1 RO1 HL51549; PO1 HL46345; HL53773; 1 RO1 Hl 55926) and American Heart Association (HL 91-022170).
Please address all correspondence to K. David Becker, Department of Medicine, 0613-C, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92093. Tel.: (619) 534-8207; Fax.: (619) 534-8081; E-mail: dbecker@ucsd.edu
References
- Beall CJ, Sepanski MA, Fyrberg EA. Genetic dissection of Drosophilamyofibril formation: effects of actin and myosin heavy chain null alleles. Genes Dev. 1989;3:131–140. doi: 10.1101/gad.3.2.131. [DOI] [PubMed] [Google Scholar]
- Bejsovec A, Anderson P. Myosin heavy-chain mutations that disrupt Caenorhabditis elegansthick filament assembly. Genes Dev. 1988;2:1307–1317. doi: 10.1101/gad.2.10.1307. [DOI] [PubMed] [Google Scholar]
- Bejsovec A, Anderson P. Functions of the myosin ATP and actin binding sites are required for C. elegansthick filament assembly. Cell. 1990;60:133–140. doi: 10.1016/0092-8674(90)90723-r. [DOI] [PubMed] [Google Scholar]
- Bonne G, Carrier L, Bercovici J, Cruaud C, Richard P, Hainque B, Gautel M, Labeit S, James M, Beckmann J, et al. Cardiac myosin binding protein-C gene splice acceptor site mutation is associated with familial hypertrophic cardiomyopathy. Nat Genet. 1995;11:438–440. doi: 10.1038/ng1295-438. [DOI] [PubMed] [Google Scholar]
- Chen CA, Okayama H. Calcium phosphate-mediated gene transfer: a highly efficient transfection system for stably transforming cells with plasmid DNA. Biotech. 1988;6:632–638. [PubMed] [Google Scholar]
- Clanton DJ, Lu YY, Blair DG, Shih TY. Structural significance of the GTP-binding domain of ras p21 studied by site-directed mutagenesis. Mol Cell Biol. 1987;7:3092–3097. doi: 10.1128/mcb.7.9.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuda G, Fananapazir L, Zhu W-S, Sellers JR, Epstein ND. Skeletal muscle expression and abnormal function of β-myosin in hypertrophic cardiomyopathy. J Clin Invest. 1993;91:2861–2865. doi: 10.1172/JCI116530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein HF, Bernstein SI. Genetic approaches to understanding muscle development. Dev Biol. 1992;154:231–244. doi: 10.1016/0012-1606(92)90064-n. [DOI] [PubMed] [Google Scholar]
- Epstein ND, Cohn GM, Cyran F, Fananapazir L. Differences in clinical expression of hypertrophic cardiomyopathy associated with two distinct mutations in the β-myosin heavy chain gene. J Clin Invest. 1992;86:345–352. doi: 10.1161/01.cir.86.2.345. [DOI] [PubMed] [Google Scholar]
- Fisher AJ, Smith CA, Thoden J, Smith R, Sutoh K, Holden HM, Rayment I. Structural studies of myosin/nucleotide complexes: A revised model for the molecular basis of muscle contraction. Biophys J. 1995;68:19s–28s. [PMC free article] [PubMed] [Google Scholar]
- Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, Seidman JG. A molecular basis for familial hypertrophic cardiomyopathy: a β cardiac myosin heavy chain gene missense mutation. Cell. 1990;62:999–1006. doi: 10.1016/0092-8674(90)90274-i. [DOI] [PubMed] [Google Scholar]
- Geisterfer-Lowrance AAT, Christe M, Conner DA, Ingwall JS, Schoen FJ, Seidman CE, Seidman JG. A mouse model of familial hypertrophic cardiomyopathy. Science (Wash DC) 1996;272:731–734. doi: 10.1126/science.272.5262.731. [DOI] [PubMed] [Google Scholar]
- Gottshall, K.R., F.F. Hunter, N. Tanaka, K.D. Becker, J. Ross, Jr., and K.R. Chien. 1997. Ras dependent pathways induce obstructive hypertrophy in echo-selected mice. Proc. Natl. Acad. Sci. USA. In press. [DOI] [PMC free article] [PubMed]
- Grussenmeyer T, Scheidtmann HT, Hutchinson MA, Eckhart W, Walter G. Complexes of polyoma virus medium T antigen and cellular proteins. Proc Natl Acad Sci USA. 1985;82:7952–7954. doi: 10.1073/pnas.82.23.7952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer JA, Bowers B, Paterson BM, Korn ED. Complete nucleotide sequence and deduced polypeptide sequence of a nonmuscle myosin heavy chain gene from Acanthamoeba: evidence of a hinge in the rodlike tail. J Cell Biol. 1987;105(2):913–925. doi: 10.1083/jcb.105.2.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi R, Krummel B, Saiki RK. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 1988;16:7351–7367. doi: 10.1093/nar/16.15.7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter JJ, Tanaka N, Rockman HA, Ross J, Jr, Chien KR. Ventricular expression of a MLC-2v-ras fusion gene induces cardiac hypertrophy and selective diastolic dysfunction in transgenic mice. J Biol Chem. 1995;270:23173–23178. doi: 10.1074/jbc.270.39.23173. [DOI] [PubMed] [Google Scholar]
- Jaenicke T, Diederich KW, Haas W, Schleich J, Lichter P, Pfordt M, Bach A, Vosberg HP. The complete sequence of the human β-myosin heavy chain gene and a comparative analysis of its product. Genomics. 1990;8:194–206. doi: 10.1016/0888-7543(90)90272-v. [DOI] [PubMed] [Google Scholar]
- Johnson DS, McKenna NM, Wang Y. Association of microinjected myosin and its subfragments with myofibrils in living muscle cells. J Cell Biol. 1988;107:2213–2221. doi: 10.1083/jcb.107.6.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurnak F. Structure of the GDP domain of EF-Tu and location of the amino acids homologous to ras oncogene proteins. Science (Wash DC) 1985;230:32–36. doi: 10.1126/science.3898365. [DOI] [PubMed] [Google Scholar]
- Karn J, Brenner S, Barnett L. Protein structural domains in the Caenorhabditis elegans unc-54myosin heavy chain gene are not separated by introns. Proc Natl Acad Sci USA. 1983;80:4253–4257. doi: 10.1073/pnas.80.14.4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komiyama M, Soldati T, von Arx P, Perriard J-C. The intracompartmental sorting of myosin alkali light chain isoproteins reflects the sequence of developmental expression as determined by double epitope tagging competition. J Cell Sci. 1996;109:2089–2099. doi: 10.1242/jcs.109.8.2089. [DOI] [PubMed] [Google Scholar]
- Kubalak, S.W., P.A. Doevendans, H.A. Rockman, J.J. Hunter, N. Tanaka, J. Ross, Jr., and K.R. Chien. 1996. Molecular analysis of cardiac muscle diseases based on mouse genetics. In Methods in Molecular Genetics, Human Molecular Genetics; K.W. Adolph, editor. Academic Press, San Diego, CA. 8:470–487.
- Lankford EB, Epstein ND, Fananapazir L, Sweeney HL. Abnormal contractile properties of muscle fibers expressing β-myosin heavy chain gene mutations in patients with hypertrophic cardiomyopathy. J Clin Invest. 1995;95:1409–1414. doi: 10.1172/JCI117795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liew CC, Sole MJ, Yamauchi-Takihara K, Kellam B, Anderson DH, Lin LP, Liew JC. Complete sequence and organization of the human cardiac β-myosin heavy chain gene. Nucleic Acids Res. 1990;18:3647–3651. doi: 10.1093/nar/18.12.3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Lu M-H, Choi J, Holtzer S, DiLullo C, Fischman DA, Holtzer H. Sequential appearance of muscle-specific proteins in myoblasts as a function of time after cell division: Evidence for a conserved myoblast differentiation program is skeletal muscle. Cell Motil Cytoskeleton. 1994;29:1–19. doi: 10.1002/cm.970290102. [DOI] [PubMed] [Google Scholar]
- Liu QY, Summers WC. Site-directed mutagenesis of a nucleotidebinding domain in HSV-1 thymidine kinase: effects on catalytic activity. Virology. 1988;163:638–642. doi: 10.1016/0042-6822(88)90308-x. [DOI] [PubMed] [Google Scholar]
- Lu M-H, DiLullo C, Schultheiss T, Holtzer S, Murray JM, Choi J, Fischman DA, Holtzer H. The vinculin/sarcomeric–alpha-actinin/alpha actin nexus in cultured cardiac myocytes. J Cell Biol. 1992;117:1007–1022. doi: 10.1083/jcb.117.5.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marian AJ, Yu Q-T, Mann DL, Graham FL, Roberts R. Expression of a mutation causing hypertrophic cardiomyopathy disrupts sarcomere assembly in adult feline cardiac myocytes. Circ Res. 1995;77:98–106. doi: 10.1161/01.res.77.1.98. [DOI] [PubMed] [Google Scholar]
- Maron, B.J. 1988. Historical perspective, nomenclature, and definition. In Cardiomyopathy Update 2: Hypertrophic Cardiomyopathy. University of Tokyo Press. Tokyo. pp. 390.
- Messerli JM, Eppenberger ME, Rutishauser B, Schwarb P, von Arx P, Koch-Schneidemann S, Eppenberger HM, Perriard J-C. Remodeling of cardiomyocyte cytoarchitecture visualized by 3D confocal microscopy. Histochemistry. 1993;100:193–202. doi: 10.1007/BF00269092. [DOI] [PubMed] [Google Scholar]
- Mornet D, Bonet AA, Audemard EE, Bonicel J. Functional sequences of the myosin head. J Musc Res Cell Motil. 1989;10:10–24. doi: 10.1007/BF01739853. [DOI] [PubMed] [Google Scholar]
- Pai EF, Kabsch W, Krengel U, Holmes KC, John J, Wittinghofer A. Structure of the guanine-nucleotide-binding domain of the Ha-ras oncogene product p21 in the triphosphate conformation. Nature (Lond) 1989;341:209–214. doi: 10.1038/341209a0. [DOI] [PubMed] [Google Scholar]
- Poetter K, Jiang H, Hassanzadeh S, Master SR, Chang A, Dalakas MC, Rayment I, Sellers JR, Fananapazir L, Epstein ND. Mutations in either the essential or regulatory light chains of myosin are associated with a rare myopathy in human heart and skeletal muscle. Nat Genet. 1996;13:63–69. doi: 10.1038/ng0596-63. [DOI] [PubMed] [Google Scholar]
- Rayment I, Rypniewski WR, Schmidt-Base K, Smith R, Tomchick DR, Benning MM, Winkelmann DA, Wesenberg G, Holden HM. Three-dimensional structure of myosin subfragment-1: A molecular motor. Science (Wash DC) 1993a;261:50–58. doi: 10.1126/science.8316857. [DOI] [PubMed] [Google Scholar]
- Rayment I, Holden HM, Whittaker M, Yohn CB, Lorenz M, Holmes KC, Milligan RA. Structure of the actin-myosin complex and its implications for muscle contraction. Science (Wash DC) 1993b;261:58–65. doi: 10.1126/science.8316858. [DOI] [PubMed] [Google Scholar]
- Rayment I, Holden HM, Sellers JR, Fananapazir L, Epstein ND. Structural interpretation of the mutations in the β-cardiac myosin that have been implicated in familial hypertrophic cardiomyopathy. Proc Natl Acad Sci USA. 1995;92:3864–3868. doi: 10.1073/pnas.92.9.3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinstein J, Brune M, Wittinghofer A. Mutations in the nucleotide binding loop of adenylate kinase of Escherichia coli. . Biochemistry. 1988;27:4712–4720. doi: 10.1021/bi00413a020. [DOI] [PubMed] [Google Scholar]
- Rhee D, Sanger JM, Sanger JW. Ther premyofibril: Evidence for its role in myofibrillogenesis. Cell Motil Cytoskeleton. 1994;28:1–24. doi: 10.1002/cm.970280102. [DOI] [PubMed] [Google Scholar]
- Rosenzweig A, Watkins H, Hwang DS, Miri M, McKenna W, Traill TA, Seidman JG, Seidman CE. Preclinical diagnosis of familial hypertrophic cardiomyopathy by genetic analysis of blood lymphocytes. N Engl J Med. 1991;325:635–647. doi: 10.1056/NEJM199112193252501. [DOI] [PubMed] [Google Scholar]
- Ruppel KM, Spudich JA. Structure-function studies of the myosin motor domain: importance of the 50-kDa cleft. Mol Biol Cell. 1996;7:1123–1136. doi: 10.1091/mbc.7.7.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory, Cold Harbor, NY.
- Schultheiss T, Lin Z-X, Lu MH, Murray J, Fischman DA, Weber K, Masaki T, Imamura M, Holtzer H. Differential distribution of subsets of myofibrillar proteins in cardiac nonstriated and striated myofibrils. J Cell Biol. 1990;110:1159–1172. doi: 10.1083/jcb.110.4.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldati T, Perriard J-C. Intracompartmental sorting of essential myosin light chains: molecular dissection and in vivo monitoring by epitope tagging. Cell. 1991;66:277–289. doi: 10.1016/0092-8674(91)90618-9. [DOI] [PubMed] [Google Scholar]
- Strehler EE, Strehler-Page MA, Perriard J-C, Periasamy M, NadalGinard B. Complete nucleotide and encoded amino acid sequence of a mammalian myosin heavy chain gene. Evidence against intron-dependent evolution of the rod. J Mol Biol. 1986;190:291–317. doi: 10.1016/0022-2836(86)90003-3. [DOI] [PubMed] [Google Scholar]
- Squire, J.M. 1986. Muscle: design, diversity and disease. Benjamin/Cummings Publishing Co., Menlo Park, CA.
- Sweeney HL, Straceski AJ, Leinwand LA, Tikunov BA, Faust L. Heterologous expression of a cardiomyopathic myosin that is defective in its actin interaction. J Biol Chem. 1994;269:1603–1605. [PubMed] [Google Scholar]
- Thierfelder L, Watkins H, MacRae C, Lamas R, McKenna W, Vosberg HP, Seidman JG, Seidman CE. Alpha-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell. 1994;77:701–712. doi: 10.1016/0092-8674(94)90054-x. [DOI] [PubMed] [Google Scholar]
- von Arx P, Bantle S, Soldati T, Perriard J-C. Dominant negative effect of cytoplasmic actin isoproteins on cardiomyocyte cytoarchitecture and function. J Cell Biol. 1995;6:1759–1773. doi: 10.1083/jcb.131.6.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JE, Saraste M, Runswick MJ, Gay NJ. Distantly related sequences in the alpha and beta subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO (Eur Mol Biol Organ) J. 1982;1(8):945–951. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrick HM, Spudich JA. Myosin structure and function in cell motility. Annu Rev Cell Biol. 1987;3:379–421. doi: 10.1146/annurev.cb.03.110187.002115. [DOI] [PubMed] [Google Scholar]
- Warrick HM, De Lozanne A, Leinwand LA, Spudich JA. Conserved protein domains in a myosin heavy chain gene from Dictyostelium discoideum. . Proc Natl Acad Sci USA. 1986;83:9433–9437. doi: 10.1073/pnas.83.24.9433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins H, Rosenzweig T, Hwang DS, Levi T, McKenna W, Seidman CE, Seidman JG. Characteristics and prognostic implication of myosin missense mutations in familial hypertrophic cardiomyopathy. N Engl J Med. 1992;326:1106–1114. doi: 10.1056/NEJM199204233261703. [DOI] [PubMed] [Google Scholar]
- Watkins H, Conner D, Thierfelder L, Jarcho JA, MacRae C, McKenna WJ, Maron BJ, Seidman JG, Seidman CE. Mutations in the cardiac myosin binding protein-C gene on chromosome 11 cause familial hypertrophic cardiomyopathy. Nature Genet. 1995a;11:434–437. doi: 10.1038/ng1295-434. [DOI] [PubMed] [Google Scholar]
- Watkins H, McKenna WJ, Thierfelder L, Suk HJ, Anan R, O'Donoghue A, Spirito P, Matsumori A, Moravec CS, Seidman JG, Seidman CE. Mutations in the genes for cardiac troponin T and alpha tropomyosin in hypertrophic cardiomyopathy. N Engl J Med. 1995b;332:1058–1064. doi: 10.1056/NEJM199504203321603. [DOI] [PubMed] [Google Scholar]
- Wilson IA, Niman HL, Houghten RA, Cherenson AR, Connolly ML, Lerner RA. The structure of an antigenic determinant in a protein. Cell. 1984;37:767–778. doi: 10.1016/0092-8674(84)90412-4. [DOI] [PubMed] [Google Scholar]
- Yanagisawa M, Hamada Y, Katsuragawa Y, Imamura M, Mikawa T, Masaki T. Complete primary structure of vertebrate smooth muscle myosin heavy chain deduced from its complementary DNA sequence. Implications on topography and function of myosin. J Mol Biol. 1987;198:143–157. doi: 10.1016/0022-2836(87)90302-0. [DOI] [PubMed] [Google Scholar]
- Zhu H, Garcia AV, Ross RS, Evans SM, Chien KR. A conserved 28-base-pair element (HF-1) in the rat cardiac myosin light-chain-2 gene confers cardiac-specific and alpha-adrenergic-inducible expression in cultured neonatal rat myocardial cells. Mol Cell Biol. 1991;11:2273–2281. doi: 10.1128/mcb.11.4.2273. [DOI] [PMC free article] [PubMed] [Google Scholar]