

Abstract
Lysosomes are considered to be a terminal degradative compartment of the endocytic pathway, into which transport is mostly unidirectional. However, specialized secretory vesicles regulated by Ca2+, such as neutrophil azurophil granules, mast cell–specific granules, and cytotoxic lymphocyte lytic granules, share characteristics with lysosomes that may reflect a common biogenesis. In addition, the involvement of Ca2+ transients in the invasion mechanism of the parasite Trypanosoma cruzi, which occurs by fusion of lysosomes with the plasma membrane, suggested that lysosome exocytosis might be a generalized process present in most cell types.
Here we demonstrate that elevation in the intracellular free Ca2+ concentration of normal rat kidney (NRK) fibroblasts induces fusion of lysosomes with the plasma membrane. This was verified by measuring the release of the lysosomal enzyme β-hexosaminidase, the appearance on the plasma membrane of the lysosomal glycoprotein lgp120, the release of fluid-phase tracers previously loaded into lysosomes, and the release of the lysosomally processed form of cathepsin D. Exposure to the Ca2+ ionophore ionomycin or addition of Ca2+containing buffers to streptolysin O–permeabilized cells induced exocytosis of ∼10% of the total lysosomes of NRK cells. The process was also detected in other cell types such as epithelial cells and myoblasts. Lysosomal exocytosis was found to require micromolar levels of Ca2+ and to be temperature and ATP dependent, similar to Ca2+-regulated secretory mechanisms in specialized cells.
These findings highlight a novel role for lysosomes in cellular membrane traffic and suggest that fusion of lysosomes with the plasma membrane may be an ubiquitous form of Ca2+-regulated exocytosis.
Lysosomes are acidic organelles delimited by a single membrane, containing a characteristic set of acid hydrolases (Novikoff, 1961). They receive newly synthesized lysosomal enzymes from the biosynthetic pathway and are responsible for the degradation of internalized material from the endocytic and autophagic pathways. Therefore, lysosomes are generally considered to be a terminal degradative compartment, the final destination of unidirectionally transported soluble macromolecules (DeDuve, 1963; Kornfeld and Mellman, 1989).
Extracellular release of lysosomal contents, however, has been described in several cell types, such as hepatocytes (LeSage et al., 1993), activated platelets (Silverstein and Febbraio, 1992), pancreatic acinar cells (Hirano et al., 1991), macrophages (Tapper and Sundler, 1990), and osteoclasts (Baron et al., 1990). In neutrophils, fusion of lysosomes with incompletely formed phagosomes also results in extracellular leakage of their contents (Pryzwansky et al., 1979). Based on the presence of hydrolytic enzymes and lysosomal membrane glycoproteins, a common biogenesis has been proposed between lysosomes and specialized Ca2+-regulated secretory vesicles, such as azurophil granules in neutrophils (Borregaard et al., 1993), specific granules in mast cells (Jamur et al., 1986), and lytic granules in cytotoxic T lymphocytes (Burkhardt et al., 1990; Peters et al., 1991).
Despite these similarities, conventional lysosomes of nonspecialized cells have not been generally described as organelles that fuse with the plasma membrane upon stimulation. Recent studies of cell infection with the protozoan parasite Trypanosoma cruzi have, however, provided evidence for the existence of a lysosomal exocytic pathway in fibroblasts and epithelial cells. During T. cruzi invasion, lysosomes are recruited to the parasite entry site and gradually fuse with the plasma membrane (Tardieux et al., 1992; Rodríguez et al., 1996). Intracellular free Ca2+ concentration ([Ca2+]i)1 transients, triggered in host cells by a trypanosome-soluble factor (Burleigh and Andrews, 1995), cause reversible disassembly of the cortical actin cytoskeleton of normal rat kidney (NRK) fibroblasts (Rodríguez et al., 1995), similar to what has been described in regulated exocytosis. The T. cruzi–triggered [Ca2+]i transients appear to be required for the lysosome-mediated invasion process, since buffering or depletion of host cell intracellular Ca2+ blocks parasite entry (Tardieux et al., 1994; Rodríguez et al., 1995).
Taken together, the observations described above suggested that increases in [Ca2+]i might be sufficient to trigger lysosomal exocytosis in most cell types, in a similar fashion to the well-characterized process of Ca2+-regulated exocytosis in specialized secretory cells. In this work we provide evidence in favor of this hypothesis, demonstrating that conventional lysosomes can be induced to fuse with the plasma membrane in a Ca2+, temperature- and ATP-dependent fashion.
Materials and Methods
Materials
BSA, bombesin, and gold chloride were obtained from Sigma Chemical Co. (St. Louis, MO); ionomycin and colchicine were from CalbiochemNovabiochem Corp. (La Jolla, CA). Lucifer yellow and FITC-transferrin (human) were from Molecular Probes, Inc. (Eugene, OR). Reduced streptolysin O was from Murex Diagnostics (Dartford, UK), and hexokinase was from Boehringer Mannheim Biochemicals (Indianapolis, IN). 3H-dextran (mol wt 70,000) was obtained from Amersham Corp. (Arlington Heights, IL). Human diferric 125I-transferrin was obtained from DuPont (Wilmington, DE). Purified rabbit antibodies against cathepsin D were from Biodesign Intl. (Kennebunk, ME). Trypanosome-soluble fraction (TSF) was prepared from the infective stages of T. cruzi as described in Rodríguez et al. (1995).
Cell Culture
All cells were grown at 37°C with 5% CO2. Cultures of primary human fibroblasts (NIGMS; Coriell Institute for Medical Research, Camden, NJ), NRK, J774, IMR-90, L6E9, and LLC-MK2 cell lines were grown in DME containing 10% FBS. CHO cells were grown in α-MEM with 5% FBS. Confluent monolayers containing 6 × 104 cells per cm2 were used for all experiments.
Ionomycin Treatment
Confluent monolayers of NRK cells in 60-mm culture dishes were washed with PBS and incubated with 0.5 ml of either PBS or 10 μM ionomycin in PBS for the indicated times. The incubation buffer was collected and centrifuged at 11,000 g for 5 min before performing β-hexosaminidase, 3H-dextran, lucifer yellow, or cathepsin D detection assays. Total cell extracts were obtained by incubation of culture dishes with 0.5 ml of PBS 1% NP-40 (NP-40), followed by a 5-min centrifugation of the extract at 11,000 g.
N-Acetyl-β-d-Glucosaminidase (β-Hexosaminidase) Activity Assay
For each sample, 350 μl of the incubation buffer was incubated for 15 min at 37°C with 50 μl of 6 mM 4-methyl-umbellyferyl-N-acetyl-β-d-glucosaminide in sodium citrate-phosphate buffer, pH 4.5. The reaction was stopped by addition of 100 μl of 2 M Na2CO3 and 1.1 M glycine, and the fluorescence was measured in an F-2000 spectrofluoremeter (Hitachi Instruments, Inc., Danbury, CT) at excitation 365 nm/emission 450 nm. To determine the total cellular content of β-hexosaminidase, cell extracts were diluted 1:10 and 350 μl was used for enzyme detection. In the TSF experiment, background levels of β-hexosaminidase in TSF (<0.2% of the total content of control NRK cells) were subtracted from each sample.
Lactate Dehydrogenase Activity Assay
For each point, 100 μl of the incubation buffer or 1:10 dilutions of NP-40 extracts prepared as described above were incubated with 900 μl of reaction buffer (0.23 mM NADH, 1 mM sodium pyruvate, 0.1% Triton X-100, 0.2 M Tris-HCl, pH 7.3). Decrease in absorbance at 340 nm was measured in the spectrophotometer for either 2 min (permeabilized cells) or 5 min (nonpermeabilized cells).
Lucifer Yellow and 3H-Dextran Loading of Lysosomes and Detection
Cells were incubated for 4 h at 37°C in DME containing 10 mg/ml lucifer yellow or for 1 h with 100 μCi/ml 3H-dextran, washed extensively with PBS, and chased for another 2 h in DME 10% FCS before performing the ionomycin or permeabilization treatments. The incubation buffer was collected and centrifuged for 5 min at 11,000 g. Lucifer yellow was detected in each sample of supernatant by measuring the fluorescence at excitation 428 nm/emission 540 nm; 3H-dextran was measured in a scintillation counter.
Staining for Surface lgp120
After the different treatments, NRK cells were incubated at 4°C for 30 min with culture supernatant from a mouse hybridoma line (Ly1C6) producing antibodies to rat lgp120 (kindly provided by I. Mellman, Yale University School of Medicine, New Haven, CT). Cells were then fixed with 2% paraformaldehyde for 15 min at 4°C, washed in PBS, and incubated with rhodamine-conjugated anti–mouse IgG antibodies (Boehringer Mannheim Biochemicals) for 30 min at room temperature.
Flow Cytometry
Confluent NRK cells were trypsinized and washed twice with PBS before incubation with PBS or 10 μM ionomycin in PBS for 5 min at 37°C. 106 cells for each assay were centrifuged and resuspended in Ly1C6 supernatant (mAb to rat lgp120) or FITC-transferrin (0.5 mg/ml) at 4°C for 30 min. Cells were washed in PBS and fixed in 2% paraformaldehyde for 15 min. Anti-lgp–treated cells were further incubated with FITC-conjugated anti–mouse antibodies (Boehringer Mannheim Biochemicals) for 30 min at room temperature. Finally, cells were washed, resuspended in 0.5 ml PBS, and analyzed on a FACS® Vantage Flow Cytometer (Becton Dickinson & Co., Mountain View, CA). Cells were illuminated at 488 nm and emission was detected at 525 nm. Forward angle scatter, right angle scatter, and fluorescence intensity were recorded from 10,000 cells whose forward angle scatter fell above a threshold used to distinguish intact from damaged cells.
BSA–Gold Labeling of Lysosomes and EM
Lysosomes were labeled by the internalization of BSA adsorbed to 5 nm gold particles, prepared according to published procedures (Slot and Geuze, 1985). Cells were incubated with BSA–gold (OD 4.0 at 255 nm) for 3 h at 37°C, washed, and chased for 2 h in DME 10% FBS before addition of 10 μM ionomycin for 2.5 and 10 min at 37°C. For resin embedding, cells were fixed in 2.5% glutaraldehyde in 100 mM sodium cacodylate (pH 7.4), postfixed in 1% aqueous osmium tetroxide, en bloc stained in 1% uranyl acetate in 50 mM sodium maleate (pH 5.2), dehydrated in methanol, and embedded in Epon. Sections were contrasted with uranyl acetate and lead citrate. For immunocytochemistry, cells were fixed in 4% paraformaldehyde in 200 mM Hepes buffer, pH 7.2, scraped from the dish, pelleted, and embedded in 10% gelatin. Embedded pellets were infiltrated with 2.1 M sucrose in PBS and frozen by immersion in liquid nitrogen. Cryosectioning was performed using established methods, as described previously (Webster et al., 1994). Thawed sections were labeled with affinity-purified rabbit anti–lgp120-tail antibodies (Rodríguez et al., 1996) or mouse anti– rat lysosomal membrane glycoprotein (lgp120) mAbs (Ly1C6). The mAb was followed by application of a rabbit anti–mouse bridging antibody. Binding of antibodies was visualized using protein A–gold probes (purchased from Department of Cell Biology, University of Utrecht, The Netherlands).
Streptolysin O Permeabilization
Confluent monolayers of NRK cells in 60-mm culture dishes were washed twice with 1 ml ice-cold buffer A (20 mM Hepes, 110 mM NaCl, 5.4 mM KCl, 0.9 mM Na2HPO4, 10 mM MgCl2, 2 mM CaCl2, and 11 mM glucose, pH 7.4) and incubated for 10 min at 4°C with streptolysin O (SLO) at 0.5 U/ml in buffer A. Cells were then washed once with 1 ml ice-cold buffer B (20 mM Hepes, 100 mM K-glutamate, 40 mM KCl, and 5 mM EGTA, pH 7.2) containing 2 mM MgATP and 5 mM free Mg2+ (added as MgCl2). 0.5 ml of buffer B with MgATP and Mg2+, or buffer B with MgATP, Mg2+, and Ca2+/EGTA at the indicated concentrations, was added to the cells at 37°C for the indicated time periods. The concentrations of free Mg2+ and Ca2+ were maintained using a Ca2+ or Mg2+/EGTA buffering system, calculated using the software developed by Foehr and Warchol (Ulm, Germany). Incubation buffer was collected for each sample and centrifuged for 5 min at 11,000 g before performing enzyme assays. Total extracts were obtained by incubation of culture dishes with 0.5 ml of PBS 1% NP-40, followed by a 5-min centrifugation of the extract at 11,000 g. In the ATP depletion experiment, cell permeabilization was performed in cells in suspension, which were previously trypsinized and washed before SLO permeabilization as described above. 106 cells were used for each assay. After permeabilization, cells were incubated with hexokinase (150 U/ml) and glucose 5 mM in buffer B with Mg2+ for 15 min at 37°C, before performing the Ca2+-induced exocytosis assay.
Detection of Cathepsin D
Confluent IMR-90 cells in 150-mm culture dishes were either treated with PBS or 10 μM ionomycin or permeabilized with SLO and incubated with a 0 or 1 μM Ca2+ buffer as described above. The supernatants of these cells (3 ml) were collected after 5 min and concentrated with a Centricon10 (Amicon Corp., Beverly, MA) to 50 μl (25 μg of total protein for PBS and ionomycin samples and 80 μg for permeabilized cell samples). The total extract was obtained by addition of 1 ml of lysis buffer (150 mM NaCl, 50 mM Tris, pH 8.6, 1% NP-40) to the cells, and 15 μl (20 μg) was used for detection. 4× concentrated SDS-PAGE loading buffer (62.5 mM Tris, pH 6.8, 10% glycerol, 2% SDS, and 5% β-mercaptoethanol) was added to the samples, which were heated to 95°C for 4 min before electrophoresis in a 10% SDS-polyacrylamide gel. Proteins were then transferred to Nytran filters by semidry electroblotting (Schleicher & Schuell, Keene, NH). Blots were probed with rabbit anti–cathepsin D antibodies (dilution 1:1,000 of a 12.5 mg/ml stock solution), followed by peroxidase-conjugated goat anti– rabbit immunoglobulin G and enhanced chemiluminescence detection (ECL; Amersham Intl., Buckinghamshire, UK).
Transferrin Release Assay
125I-transferrin release from SLO-permeabilized cells was performed essentially as described by Galli et al. (1994). Confluent NRK cells plated at 2 × 104/cm2 24 h before were trypsinized and washed in buffer K (20 mM Hepes, 128 mM NaCl, 3 mM KCl, 1 mM Na2HPO4, 1.2 mM MgSO4, 2.7 mM CaCl2, and 11 mM glucose, pH 7.4, with NaOH) before incubation of 1.5 × 105 cells per μl in buffer K containing 20 μCi/ml human 125I-transferrin for 1 h at 37°C. Cells were washed once with ice-cold buffer K and twice with buffer A, and permeabilization was performed as described above. Cells were resuspended in buffer B and divided into 100-μl aliquots containing 106 cells each for determining transferrin release. Supernatants were collected after incubation at 37°C for 20 min or the indicated times, and the amount of 125I-transferrin was determined in a scintillation counter. In each experiment, samples in triplicate were pelleted at the beginning of the 37°C incubation (defined as time 0), the amount of 125I-transferrin in the supernatant was determined, and this value was subtracted from each sample. Cell extracts to quantify the total amount of internalized 125I-transferrin were obtained for each sample by incubation of pellets with 100 μl of PBS 1% NP-40.
Results
Ionomycin Induces the Release of Lysosomal Contents into the Extracellular Medium
To test if an increase in [Ca2+]i would stimulate exocytosis of lysosomes, the calcium ionophore ionomycin was added to NRK fibroblasts in a Ca2+-containing buffer (PBS with 1 mM CaCl2). Cells were incubated at 37°C with 10 μM ionomycin, and release of the lysosomal enzyme β-hexosaminidase was measured in the incubation buffer. This enzyme is frequently used as a marker for lysosomes, since 90% of the total β-hexosaminidase in NRK cells is located in this compartment (Griffiths et al., 1990). A continuous release of β-hexosaminidase, typically reaching ∼10% of the total enzyme content of the cells after 10 min, was specifically triggered by ionomycin (Fig. 1 a).
Figure 1.

Ionomycin induces exocytosis of β-hexosaminidase and lysosomal fluid-phase tracers but not LDH from intact NRK fibroblasts. (a and b) NRK cells were incubated at 37°C with either PBS (open circles) or 10 μM ionomycin in PBS (black circles). At the indicated time points, the incubation buffer was collected and assayed for β-hexosaminidase and LDH activity. The amount of enzyme released at each point is expressed as a percentage of the total content of enzyme in control cells. (c and d) Lysosomes of NRK cells were loaded with lucifer yellow or 3H-dextran by fluidphase endocytosis followed by a 2-h chase, before treatment with PBS or 10 μM ionomycin for 10 min. The incubation buffer was collected, lucifer yellow was detected by reading the fluorescence, and 3H-dextran was detected by scintillation counting. The amount of lucifer yellow or 3H-dextran released in each sample is expressed as a percentage of the total amount of tracer present in control cells. The data represent the average of triplicate determinations ±SD.
To verify if cell integrity was affected by the exposure to ionomycin, activity of the cytoplasmic enzyme lactate dehydrogenase (LDH) was measured in the incubation buffer of treated and untreated cells. Fig. 1 b shows that LDH levels in the incubation buffer were low in both conditions, indicating that the observed release of β-hexosaminidase induced by the ionophore was not due to cell lysis.
Lysosomes can be specifically labeled by incubation of cells with an endocytosis fluid-phase marker, followed by a chase of several hours (Adams et al., 1982). To confirm that the β-hexosaminidase release described above was indeed a consequence of lysosome exocytosis, similar experiments were performed after loading lysosomes with the soluble fluorescent tracer lucifer yellow or 3H-dextran. Release into the supernatant of ∼10% of the total amount of the tracers taken up by NRK cells was detected after 10min exposure to 10 μM ionomycin (Fig. 1, c and d). This corresponds to an approximate increase of 6–10-fold over the levels observed during the same period in PBS-treated control cells.
Another fluid-phase tracer, BSA–gold, was used for specific visualization of lysosomes by EM. NRK cell lysosomes were loaded with BSA–gold, and then treated with 10 μM ionomycin for different periods of time. In PBStreated control NRK cells, a population of vesicles containing BSA–gold and with the characteristic morphology and dimensions of lysosomes (Holtzman, 1989) was observed, mostly clustered in the perinuclear area (Fig. 2, A and B). In contrast, cells exposed to ionomycin showed a more dispersed distribution of gold-loaded vesicles, with accumulation in the proximity of the plasma membrane (Fig. 2, C–F). Images that strongly suggested exocytic events, with extracellular release of the gold complexes and other membranous lysosomal contents, were observed at both 2.5 and 10 min after exposure to ionomycin (Fig. 2, D and F). No similar images were detected in control cells (Fig. 2, A and B).
Figure 2.
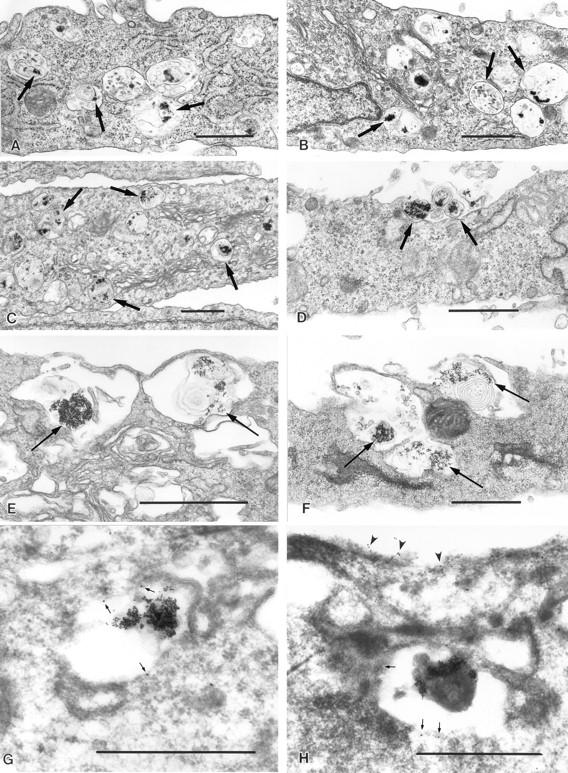
Exocytosis of BSA–gold-loaded lysosomes is triggered by [Ca2+]i elevation. NRK cells loaded with 5 nm BSA–gold complexes for 4 h followed by a chase of 2 h were incubated with PBS (A and B) or 10 μM ionomycin (C–H) for 2.5 (C and D) or 10 min (E–H). Transmission EM sections show gold-loaded vesicles (arrows), observed in close proximity to the plasma membrane in ionomycintreated cells (C, E, and F). Exocytosed BSA–gold was also detected after exposure to ionomycin (D and F). Labeling with antibodies against lgp120 (10 nm gold) was detected on BSA–gold-containing vesicles (G and H). Small arrows indicate anti-lgp120 labeling on lysosomes; arrowheads indicate anti-lgp120 labeling on the plasma membrane after ionomycin treatment. Bars, 1 μM.
To confirm that the BSA–gold-loaded vesicles consisted of lysosomes, labeling with antibodies against the abundant lysosomal membrane glycoprotein lgp120 (Kornfeld and Mellman, 1989) was performed on cryosections. Specific lgp120 label was detected on the membrane of vesicles accumulated next to the plasma membrane in ionomycin-treated cells (Fig. 2, G and H). Anti-lgp120 labeling was also detected on the plasma membrane of ionomycintreated cells (Fig. 2 H), an observation that was confirmed by immunofluorescence in live cells, as described below (Fig. 3, a–d).
Figure 3.
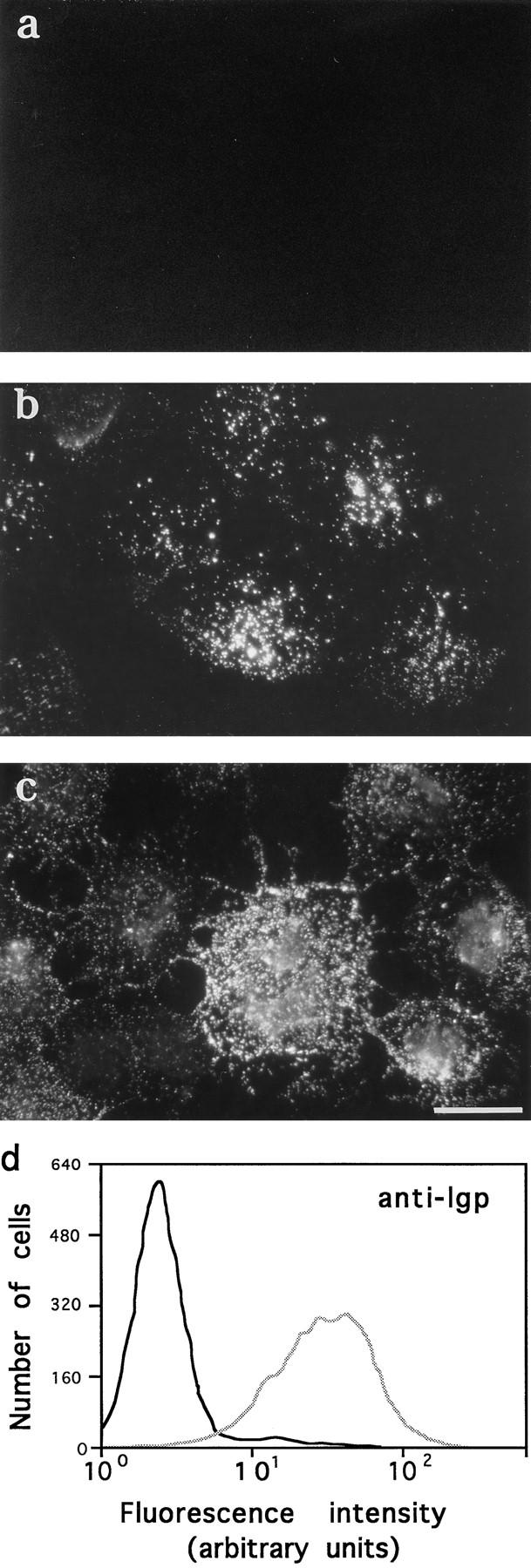
Ionomycin induces appearance of lgp120 on the plasma membrane. NRK cells, either attached to coverslips (a–c) or in suspension (d), were incubated with PBS or 10 μM ionomycin in PBS at 37°C, followed by immunofluorescent surface labeling with an mAb to a luminal domain of lgp 120. (a) lgp120 surface staining 10 min after exposure to PBS; (b) lgp120 surface staining 3 min after exposure to ionomycin; (c) lgp120 surface staining 10 min after exposure to ionomycin. (d) FACS® analysis of NRK cells in suspension treated for 5 min with PBS (black line) or 10 μM ionomycin in PBS (gray line). Cells were incubated with antilgp120 mAbs for 30 min at 4°C before fixation. Bar, 5 μm.
Ionomycin Induces the Appearance of lgp120 on the Plasma Membrane
Additional evidence for Ca2+-dependent lysosomal fusion with the plasma membrane was provided by exposure of the luminal domain of lgp120 on the cell surface. After treating NRK cells with 10 μM ionomycin, surface staining was performed in live cells at 4°C with an mAb recognizing the luminal domain of lgp120. A gradual increase in extracellularly exposed lgp120 was detected over time in ionomycin-treated cells, but not in control PBS-treated cells (Fig. 3, a–c). As a control, surface staining of ionomycin-treated cells with an mAb for the cytosolic protein kinesin was performed, showing no detectable fluorescence under the same conditions (not shown).
To quantify the percentage of the ionomycin-treated cell population that presented lgp120 on the plasma membrane, a similar experiment was performed with cells in suspension. NRK fibroblasts were trypsinized, washed, and incubated with 10 μM ionomycin for 5 min before incubation at 4°C with the anti-lgp120 mAb, followed by fixation. Cells were analyzed for surface staining by flow cytometry. An increase in membrane staining for anti-lgp120 was observed in 95% of ionomycin-treated cells (Fig. 3 d).
Agonists That Mobilize Ca2+ from Intracellular Stores Induce a Lower Level of Lysosomal Exocytosis
The results described above indicated that ionomycinmediated influx of Ca2+ from the extracellular medium induced fusion of lysosomes with the plasma membrane. To verify if Ca2+ mobilized from intracellular stores by receptor-mediated agonists had the same effect, release of β-hexosaminidase was measured in the supernatant of cells treated with several concentrations of the signaling peptide bombesin. As shown in Fig. 4 a, bombesin also induced lysosomal exocytosis in a dose-dependent way, but to a much lesser extent than ionomycin (a 1.5-fold maximum increase). When tested for induction of translocation to the plasma membrane of the lysosomal protein lgp120, bombesin was found to induce levels detectable by immunofluorescence (Fig. 4, c and d).
Figure 4.

Ca2+ agonists induce a low level of β-hexosaminidase release and appearance of lgp120 in the plasma membrane. (a) NRK cells were incubated with different concentrations of bombesin for 5 min, and the incubation buffer was assayed for β-hexosaminidase activity. The amount of enzyme released at each point is expressed as a percentage of the total content of enzyme in control cells. The data represent the average of triplicate determinations ± SD. (b) Same as a, except that cells were incubated for 5 min with PBS, TSF, or heat-inactivated TSF. (c) lgp120 surface staining 5 min after exposure to PBS; (d) lgp120 surface staining of NRK cells 5 min after exposure to 10 μM bombesin. Bar, 5 μM.
The protozoan parasite Trypanosoma cruzi induces fusion of lysosomes with the plasma membrane at its invasion site (Tardieux et al., 1992; Rodríguez et al., 1996). It also triggers IP3 formation and release of Ca2+ from intracellular stores in host cells, through a signaling factor present in soluble extracts of the parasites (Burleigh and Andrews, 1995; Burleigh et al., 1997). To verify if the trypanosome-soluble factor induced lysosome exocytosis, release of β-hexosaminidase was measured in the supernatant of cells treated with native or heat-inactivated TSF. As shown in Fig. 4 b, a low level of stimulated β-hexosaminidase exocytosis, of ∼1.2-fold over the background levels, was induced in NRK cells by native TSF in a 5-min assay. Heat inactivation of TSF (95°C, 5 min) resulted in the loss of Ca2+-agonist activity (not shown) and of the ability to induce lysosomal exocytosis (Fig. 4 b).
Ca2+ Concentrations between 1 and 5 μM Are Sufficient for Optimal Exocytosis of Lysosomes in Permeabilized Cells
To further characterize the process of Ca2+-dependent lysosome exocytosis, we next performed experiments in fibroblasts permeabilized with SLO (Miller and Moore, 1991), a system that allows precise control of the intracellular Ca2+ concentration.
Initially, to determine the efficiency of the permeabilization procedure under our experimental conditions, SLOtreated NRK cells were stained with propidium iodide, a fluorescent nuclear stain that does not penetrate intact cells. Virtually all cells were found to be permeabilized, when observed under a fluorescence microscope (not shown). The release of β-hexosaminidase was then measured in permeabilized and nonpermeabilized cells, in the absence or presence of 1 and 5 μM Ca2+. As shown in Fig. 5 a, in a 5-min assay, an approximate fivefold increase in the release of β-hexosaminidase from permeabilized cells was triggered by addition of 1 or 5 μM Ca2+.
Figure 5.

Release of β-hexosaminidase and LDH from intact and SLO-permeabilized NRK cells. Intact cells (black columns) and SLO-permeabilized cells (white columns), were incubated in permeabilization buffer containing either 0, 1, or 5 μM Ca2+ for 5 min at 37°C. Supernatants were assayed for (a) β-hexosaminidase and (b) LDH activity. The amount of enzyme released in each sample is expressed as a percentage of the total enzyme content of control cells. The data represent the average of triplicate determinations ±SD.
The optimal concentration of SLO for the permeabilization of NRK cells was determined for each batch of toxin used and was found to vary between 0.2 and 0.5 U/ml, as determined by titrations using the Ca2+-dependent β-hexosaminidase release assay described above. Concentrations of SLO lower or higher than the optimal one resulted in decreased Ca2+-induced release of β-hexosaminidase (not shown).
Release of the cytosolic enzyme LDH from SLO-permeabilized cells was measured as a further control for permeabilization. Fig. 5 b shows that a significantly higher level of the enzyme was released when the cells were permeabilized (Fig. 5 b). The total amount of LDH released (5–7% of the total cellular content) reflects the relative loss of cytosolic components occurring during the 5-min period of the assay. Such minor cytosolic loss has been shown not to interfere significantly with secretory functions in previously characterized systems (Pimplikar et al., 1994).
To determine the optimal Ca+ concentration inducing exocytosis of lysosomes in the permeabilized cell system, we performed the β-hexosaminidase, lucifer yellow, and 3H-dextran release assays in buffers with Ca2+ concentrations ranging from 0–5 μM. Optimal release of lysosomal markers, reaching 7–10-fold above the levels observed in the absence of Ca2+, was found to require 1–5 μM Ca2+, although exocytosis was already detectable at 0.1 μM Ca2+ (Fig. 6, a–c).
Figure 6.

Ca2+-dependent release of β-hexosaminidase, lucifer yellow, and 3H-dextran from permeabilized NRK cells. SLO-permeabilized cells were incubated for 5 min at 37°C in permeabilization buffer containing different Ca2+ concentrations. (a) β-hexosaminidase activity released. (b) Lucifer yellow released from previously loaded cells. (c) 3H-Dextran released from previously loaded cells. Values are expressed as a percentage of the total content of either β-hexosaminidase, lucifer yellow, or 3H-dextran in control cells. The data represent the average of triplicate determinations ±SD.
Elevated [Ca2+]i Induces Secretion of the Lysosomally Processed Form of Cathepsin D
To confirm the lysosomal origin of the Ca2+-triggered exocytic products, we examined the lysosomal protease cathepsin D released into the supernatant of cells with elevated [Ca2+]i. Cathepsin D is processed into a mature polypeptide of 31 kD only after being transported to lysosomes (Gieselmann et al., 1983). Elevation of [Ca2+]i in the human fibroblast cell line IMR-90, by treatment with ionomycin or by addition of 1 μM Ca2+ buffer to SLO-permeabilized cells, induced the appearance in the supernatant of the 31-kD mature form of cathepsin D (Fig. 7, lanes 3 and 5). A band of identical migration was also detected in whole cell extracts (Fig. 7, lane 1). The intermediate (47 kD) and precursor (53 kD) forms of the protein (Gieselmann et al., 1983) were detected in the supernatant at similar levels in each condition, probably reflecting the constitutive extracellular release of unprocessed cathepsin D that can occur before reuptake and targeting to lysosomes (von Figura and Hasilik, 1986; Kornfeld and Mellman, 1989). The higher molecular mass bands also appearing in Fig. 7 are due to nonspecific cross-reaction of the antibodies.
Figure 7.

Elevation in [Ca2+]i induces secretion of the 31-kD lysosomally processed form of cathepsin D. Rabbit anti–cathepsin D antibodies were used to probe a Western blot containing a lysate and concentrated supernatants of IMR-90 human fibroblasts. (Lane 1) total lysate; (lane 2) concentrated supernatant of cells treated with PBS for 5 min; (lane 3) concentrated supernatant of cells treated with 10 μM ionomycin for 5 min; (lanes 4 and 5) concentrated supernatants of SLO-permeabilized cells incubated for 5 min in buffers containing 0 or 1 μM Ca2+, respectively. Different ECL exposures were performed for each treatment to allow visualization of the secreted mature 31-kD cathepsin D band.
Elevated [Ca2+]i Has a Minor Stimulatory Effect on the Recycling of Transferrin Receptor–containing Endosomes
To verify if the elevation in [Ca2+]i triggering secretion of lysosomes also affected the exocytosis of other elements of the endocytic pathway, we measured the release of previously internalized 125I-labeled transferrin using the same SLO-permeabilized cell system. As described previously (Galli et al., 1994), permeabilized cells released 125I-transferrin linearly for ∼20 min after permeabilization (Fig. 8 a). Confirming previous reports that describe a small stimulation in the exocytosis of transferrin receptor–containing vesicles upon treatment with ionophores or Ca2+-mobilizing agonists (Buys et al., 1984; Wiley and Kaplan, 1984), there was a minor increase (1.5-fold) in the extracellular release of transferrin at increasing Ca2+ concentrations (Fig. 8 b). The release of apotransferrin into the medium after recycling to the cell surface allows the use of externally added, labeled transferrin to estimate the number of receptors on the surface at a given time. Using this method, we found that the number of receptors available for binding of FITC-transferrin on the surface of NRK cells was also slightly increased after exposure to 10 μM ionomycin (Fig. 8 c).
Figure 8.

Effect of elevated [Ca2+]i on exocytosis of transferrin receptor–containing endosomes. (a) Kinetics of release of preinternalized 125I-transferrin from SLOpermeabilized NRK cells in the absence of Ca2+. (b) Release of preinternalized 125Itransferrin from SLO-permeabilized NRK cells at different Ca2+ concentrations. Values are expressed as a percentage of the total 125Itransferrin present in each sample (cell associated plus released). The data represent the average of triplicate determinations ±SD. (c) FACS® analysis of NRK cells in suspension treated for 5 min with PBS (black line) or 10 mM ionomycin in PBS (gray line). Cells were incubated with FITC-transferrin for 30 min at 4°C before fixation.
Characterization of Lysosome Exocytosis in Permeabilized Cells
The presence of Ca2+-dependent lysosomal exocytosis was tested in different cell types. Cells were permeabilized and assayed for β-hexosaminidase release for 5 min at 37°C. To control for permeabilization efficiency in each cell type, LDH release was measured during the 5-min assay time. Levels of LDH release ranging from 10–20% confirmed that cells were efficiently permeabilized (not shown). In LLC-MK2 and L6E9 cells, a preincubation of 3 min at 37°C in the absence of Ca2+ was necessary to allow pore formation before addition of the 1 μM Ca2+ buffer. As shown in Table I, Ca2+-dependent lysosomal exocytosis was detected in various cell types, including a primary culture of human fibroblasts as well as epithelial and myoblast cell lines. Enhancement in β-hexosaminidase release over background levels varied from 2–12-fold among the various cell types. The lowest Ca2+-induced lysosome exocytosis levels in this group were observed with the macrophage cell line J774. Interestingly, the fusion of lysosomes with phagosomes in J774 and monocyte-derived macrophages was recently reported not to be regulated by Ca2+ (Zimmerli et al., 1996).
Table I.
Ca2+-dependent β-Hexosaminidase Secretion in Different Cell Types
| Species | Cell type | No Ca2+ | 1 μM Ca2+ | |||||
|---|---|---|---|---|---|---|---|---|
| NRK | rat | fibroblast | 0.9 ± 0.5* | 11.1 ± 0.5 | ||||
| Human fibroblasts | human | fibroblast | 0.4 ± 0.09 | 4.4 ± 0.6 | ||||
| IMR-90 | human | fibroblast | 1.1 ± 0.2 | 4.7 ± 1.6 | ||||
| J774 | mouse | monocyte | 2.4 ± 0.1 | 4.8 ± 0.1 | ||||
| CHO | hamster | epithelial- | 0.8 ± 0.01 | 6.6 ± 0.8 | ||||
| fibroblastoid | ||||||||
| LCC-MK2 | monkey | epithelial | 0.9 ± 0.4 | 5.0 ± 1.5 | ||||
| L6E9 | rat | myoblast | 0.4 ± 0.06 | 4.3 ± 0.09 |
The incubation buffer was assayed after a 5-min incubation, as described in Materials and Methods. Values shown correspond to the percentage of the total cellular content of β-hexosaminidase released ±SD (average of triplicate determinations).
The kinetics of Ca2+-induced lysosomal secretion in NRK cells, as measured by release of β-hexosaminidase, showed a rapid increase in the first 5 min that did not significantly change with longer incubation times (Fig. 9 a). Conversely, there was a continuous increase in the extracellular accumulation of LDH, due to cytoplasmic leakage from the permeabilized cells. As mentioned before, however, the degree of cytosol loss that occurred during the first 5 min after permeabilization is not sufficient to account for the sharp decrease in β-hexosaminidase secretion; as shown in Fig. 8 a, transferrin recycling, a process known to depend on cytosolic factors (Podbilewicz and Mellman., 1990; Galli et al., 1994), proceeds in a linear fashion for at least 20 min, under the same conditions.
Figure 9.

Characterization of Ca2+-induced lysosome exocytosis in permeabilized cells. Release of β-hexosaminidase from SLOpermeabilized NRK cells was measured after addition of 0 or 1 μM Ca2+ buffers. (a) Kinetics of release of β-hexosaminidase (black circles) and LDH (white circles) from NRK cells after addition of 1 μM Ca2+ at 37°C. (b) Sensitivity of β-hexosaminidase release to microtubule depolymerization. Cells were treated with 10 μM colchicine for 1 h before addition of the Ca2+ buffers for 5 min at 37°C. (c) Temperature dependence of β-hexosaminidase release. Cells were incubated for 5 min at 37°C to allow SLO pore formation, and then the secretion assay was performed at 4° or 37°C for 5 min. (d) Sensitivity of β-hexosaminidase release to ATP depletion. After permeabilization, cells were incubated in the presence of 2 mM MgATP or an ATP-depleting system (5 mM glucose, 150 U/ml hexokinase) for 15 min at 37°C, before performing the 5-min secretion assay. In all experiments, the amount of enzyme released is expressed as a percentage of the total enzyme content of control cells. The data represent the average of triplicate determinations ±SD.
To investigate the role of microtubules in the Ca2+-regulated lysosome exocytosis, NRK cells were treated with 10 μM colchicine for 1 h in the absence of serum. This is a condition that induces extensive microtubule depolymerization, as visualized by immunofluorescence with antitubulin antibodies (not shown). A small reduction of ∼15% in lysosomal exocytosis, reproducible in several experiments, was observed (Fig. 9 b).
We next investigated whether β-hexosaminidase release from SLO-permeabilized NRK cells exhibited the properties expected for an exocytic process. The release was found to be temperature dependent; when cells previously shifted for 5 min to 37°C to allow SLO pore formation were returned to ice, Ca2+-dependent β-hexosaminidase release was almost totally blocked (Fig. 9 c). β-Hexosaminidase release was also found to be dependent on the presence of MgATP. Cells were first permeabilized and then exposed or not to an ATP-depleting system (hexokinase 150 U/ml, glucose 5 mM) for 15 min at 37°C. A significant inhibition of Ca2+-triggered β-hexosaminidase secretion was observed in ATP-depleted cells (Fig. 9 d).
Discussion
In this work we demonstrate that increase in [Ca2+]i triggers exocytosis of lysosomes, in NRK fibroblasts and other cell types not considered “professional” Ca2+-regulated secretory cells. To confirm the lysosomal nature of the secretory vesicles in our experiments, we used four different approaches, taking advantage of molecular markers and functional characteristics of lysosomes.
First, we measured the release into the extracellular medium of the lysosomal enzyme β-hexosaminidase. About 90% of the total β-hexosaminidase of NRK cells is located in dense lysosomes (Griffiths et al., 1990). Therefore, the release of ∼10% of the total cellular content of this enzyme into the medium upon increase in [Ca2+]i strongly suggested the occurrence of lysosomal exocytosis. This was observed in both ionomycin-treated and in SLO-permeabilized cells, in the presence of Ca2+-containing buffers.
Second, we examined the extracellular release of tracers previously loaded into lysosomes by fluid-phase endocytosis. A chase of 2 h after the uptake period was used to ensure delivery of the endocytic tracer to lysosomes (Adams et al., 1982; Rodríguez et al., 1996). The release into the medium of lucifer yellow or 3H-dextran previously loaded into lysosomes was measured after elevating the [Ca2+]i with ionomycin or by exposing permeabilized cells to Ca2+-containing buffers. The results were essentially identical to those obtained for β-hexosaminidase secretion, with maximum release values for fluid-phase tracers reaching between 10 and 20% of the total cellular content. Another fluid-phase tracer, BSA–gold, was used to visualize the distribution of lysosomes by EM after elevation in [Ca2+]i. In contrast to normal cells, after [Ca2+]i elevation, a large number of BSA–gold-containing vesicles, with the size and morphological characteristics of lysosomes, was observed closely associated with the plasma membrane. In several instances, lysosomal contents were detected in direct contact with the extracellular medium, strongly suggesting the occurrence of exocytosis.
Third, we monitored the appearance on the plasma membrane of a luminal epitope from the lysosomal integral membrane glycoprotein lgp120. This protein is highly enriched in lysosomes and is not detectable on the surface of NRK cells under normal conditions (Harter and Mellman, 1992). The appearance of lgp120 on the surface of the large majority of ionomycin-treated NRK cells, as demonstrated by flow cytometry, indicates that lysosomal exocytosis occurred homogeneously in the majority of cells of the population.
Fourth, we studied the extracellular release of the protease cathepsin D, frequently used as a marker for lysosomes. This enzyme is synthesized in the ER as a 53-kD precursor, which is subsequently cleaved into a 47-kD intermediate form, and then to an active 31-kD mature polypeptide. The last processing event that generates the 31-kD mature form occurs after transport to dense lysosomes (Gieselmann et al., 1983). We observed that after elevation of [Ca2+]i, in both ionomycin-treated and SLOpermeabilized cells, a 31-kD band specifically recognized by anti–cathepsin D antibodies appeared in the extracellular medium.
The results obtained with these four different approaches, combining the use of ionomycin and of SLOpermeabilized cells, indicate that elevation in [Ca2+]i triggers exocytosis of lysosomes in fibroblasts. We cannot, however, rule out the participation of late endosomes, since in some cell types these organelles, in addition to containing hydrolases and lgp120 (Griffiths et al., 1988), appear to retain soluble tracers even after long chase periods (Rabinowitz et al., 1992).
Parallel experiments measuring the recycling of transferrin receptor–containing endosomes to the plasma membrane showed that lysosomes are the main vesicle population that undergoes exocytosis upon [Ca2+]i elevation. A small stimulation in transferrin release and in the number of transferrin receptors on the cell surface was observed upon elevation of [Ca2+]i, confirming previous findings (Buys et al., 1984; Wiley and Kaplan, 1984). However, the increase in exocytosis triggered by Ca2+ in NRK cells was several fold higher for lysosomes than for early endosomes, under the same conditions. Furthermore, transferrin receptor–containing vesicles fused efficiently with the plasma membrane in the absence of Ca2+, while lysosomes did not. Constitutive transport from the TGN to the plasma membrane is also known to occur at similar rates in the absence or presence of Ca2+ up to 1 μM (Miller and Moore, 1991).
Increases in [Ca2+]i regulate many secretory events, such as neurotransmitter release (Smith and Augustine, 1988) and exocytosis of secretory granules (Holz et al., 1991). Intracellular constitutive fusion mechanisms, such as ER to Golgi transport (Beckers and Balch, 1989) or fusion of nuclear membrane vesicles (Sullivan et al., 1993), require basal levels of Ca2+ (∼100 nM); regulated exocytosis fusion events in specialized secretory cells (Holz et al., 1991) and lysosome–phagosome fusion in neutrophils (Jaconi et al., 1990) require higher [Ca2+]i, ∼0.5–5 μM in average. Significantly higher Ca2+ concentrations, reaching hundreds of micromolar in microseconds, are involved in triggering synaptic vesicle fusion at the neuron terminal (Heidelberger et al., 1994). Using the SLO-permeabilized system, which allows the equilibration of intracellular and extracellular Ca2+ concentrations (Ahnert-Hilger et al., 1989), we were able to define the optimal [Ca2+]i required to trigger lysosomal exocytosis in NRK cells as ∼1 μM.
The similar [Ca2+]i levels necessary to trigger lysosome exocytosis in fibroblasts and regulated secretion in specialized cells add evidence in support of a common biogenesis between lysosomes and Ca2+-regulated secretory granules (Borregaard et al., 1993; Jamur et al., 1986; Burkhardt et al., 1990; Peters et al., 1991). The temperature and MgATP dependence we observed for lysosomal exocytosis is also consistent with what is known for other regulated secretory systems. Kinetic studies of exocytosis in neuroendocrine cells revealed the existence of multiple Ca2+-activated steps and have shown that secretion can be dissociated into MgATP-dependent and -independent stages, requiring distinct cytosolic factors (Bittner and Holz, 1992; Neher and Zucker, 1993; Hay and Martin, 1992; Chamberlain et al., 1995). Involvement of components of the NSF-SNAPSNARE putative fusion machinery (Sollner et al., 1993) in several Ca2+-dependent exocytic processes has been reported, but the precise events in which they participate are presently unclear (DeBello et al., 1995; Chamberlain et al., 1995; Bi et al., 1995).
The kinetics of Ca2+-dependent lysosome exocytosis in SLO-permeabilized NRK cells showed no significant increase after the first 5 min. This does not appear to be due to depletion of essential cytosolic components, since recycling of transferrin receptor–containing vesicles, a process known to require cytosolic factors, proceeds in permeabilized cells at increasing rates for at least 20 min (Fig. 8 a, compare with Fig. 9 a). Furthermore, after a significantly longer period of permeabilization (15 min), the Ca2+induced β-hexosaminidase release measured in a 5-min assay was only reduced in ∼40% (Fig. 9 d, compare with 9 b). The fact that only 10–20% of the lysosome population (as defined by the total content of β-hexosaminidase or fluidphase tracers) appears to be capable of Ca2+-triggered exocytosis might reflect heterogeneity in spatial distribution and/or functional characteristics. Similar results have been described for chromaffin cells, where the Ca2+-triggered release of catecholamines is limited to 20–30% of the total cellular content (Dunn and Holz, 1983). Consistent with this interpretation, we observed only a very minor inhibition in lysosome secretion after microtubule depolymerization, suggesting that most of the lysosomes that undergo exocytosis are not transported for long distances, but are already located close to the plasma membrane before the Ca2+ stimulation. Microtubule-based movement was actually shown not to be required for exocytosis in several systems, being only critical for delivery of newly synthesized material to specific locations (Kelly, 1990).
A recent report, based on capacitance measurements, showed that elevation in [Ca2+]i triggers massive exocytosis in CHO cells and 3T3 fibroblasts, resulting in a surface area increase of 20–30% in 4 min (Coorsen et al., 1996). The Ca2+ concentration range described in that study to trigger exocytosis is the same we determined for lysosome exocytosis in NRK cells, from 0.1 to 4 μM. Interestingly, the rate of exocytosis observed by those authors was found to increase strongly with [Ca2+]i, while the amount did not. This finding is consistent with our observation that only 10–20% of the lysosome population is capable of Ca2+triggered exocytosis, with no significant increase after 5 min. Our present study reinforces the suggestion that Ca2+-regulated exocytosis is not restricted to specialized secretory cells, and it strongly suggests that lysosomes are the source of the large amount of exocytosed membrane detected in that previous study (Coorsen et al., 1996).
Another earlier observation consistent with our present results is the Ca2+-dependent quantal release of acetylcholine loaded by endocytosis in CHO cells (Morimoto et al., 1995). Although the loading and chase periods used in that study were short (∼15 min), it is conceivable that endocytosed acetylcholine reached lysosomes before exocytosis was triggered by Ca2+. Recently, Ca2+-regulated secretion of glycosaminoglycan chains was detected in CHO fibroblasts (Chavez et al., 1996). Although the Ca2+ concentration range for maximal secretion (1–5 μM) is the same as the one required for lysosomal exocytosis, the glycosaminoglycan-containing organelles described in that study were found to be less dense than lysosomes, and did not colocalize with lgp on immunofluorescence assays.
Our data indicate that optimal secretion of lysosomes in NRK cells occurs at Ca2+ concentrations of ∼1 μM. Therefore, a more significant level of exocytosis was initially expected in our experiments with the agonists bombesin and TSF. These agents were previously shown to induce [Ca2+]i transients reaching low micromolar levels in NRK cells (Burleigh and Andrews, 1995). However, there is increasing evidence that IP3-mediated mobilization of Ca2+ from intracellular stores can occur in a highly localized fashion, giving rise to a heterogeneous pattern of [Ca2+]i elevation in cells (Hahn et al., 1992; Paradiso et al., 1995). It is conceivable that in our agonist treatment experiments, only lysosomes in the immediate vicinity of areas with elevated Ca2+ were able to fuse with the plasma membrane. It is interesting to note, in this context, that the mobilization of lysosomes to the plasma membrane by the parasite T. cruzi was recently shown to involve only lysosomes located at a distance of ∼12 μm from the parasite invasion site (Rodríguez et al., 1996).
Regulated secretion of lysosomal components has been described previously mostly as part of specialized processes. In osteoclasts, Ca2+ regulation of lysosomal enzyme release was suggested (Davidson et al., 1994), and, in neutrophils, both secretion of the lysosome-related azurophil granules (Sengelov et al., 1993) and lysosome–phagosome fusion (Jaconi et al., 1990) are Ca2+-dependent processes. Release of lysosomal contents from platelets and pancreatic acinar cells is triggered by specific agonists, suggesting a possible Ca2+-regulated pathway (Febbraio and Silverstein, 1990; Hirano et al., 1991; Grondin and Beaudoin, 1996). Certain carcinoma cells present lgps in their surface (Saitoh et al., 1992; Garrigues et al., 1994) and secrete lysosomal enzymes (Sloane et al., 1986). A model of secretory lysosomes fusing directly with the plasma membrane was proposed for mediating this process in colon carcinoma cells (Saitoh et al., 1992). Therefore, it is conceivable that the Ca2+-regulated lysosomal exocytosis we describe here corresponds to a ubiquitous mechanism that might be upregulated in certain pathological conditions.
A physiological function for a ubiquitous, Ca2+-dependent exocytosis of lysosomes is unclear. One intriguing possibility, strongly suggested by our data, is that lysosomes are the intracellular organelles recently proposed to be responsible for membrane resealing in wounded cells (Bi et al., 1995; Miyake and McNeil, 1995). Membrane resealing was initially shown to require Ca2+-regulated exocytosis, in 3T3 fibroblasts and sea urchin embryos (Steinhardt et al., 1994). More recently, the exocytic vesicles observed to fuse with the plasma membrane at sites of injury, in 3T3 fibroblasts and bovine endothelial cells, were proposed to belong to the endosomal/lysosomal compartment, based on their labeling with lipophilic fluorescent dyes (Miyake and McNeil, 1995). In light of our present results, it is conceivable that a localized [Ca2+]i elevation, caused by Ca2+ influx through damaged membranes, triggers the fusion of nearby lysosomes with the plasma membrane. Lysosome exocytosis may therefore be at least one of the mechanisms underlying the essential process of plasma membrane repair in all cells.
Acknowledgments
We thank H. Tan and B. Burleigh for photography, B. Burleigh and A. Sinai for helpful discussions and comments on the manuscript, R. Carbone for FACS® analysis, and A. Ma for excellent technical assistance.
This work was supported by the National Institutes of Health grant RO1AI34867 to N.W. Andrews.
Abbreviations used in this paper
- [Ca2+]i
intracellular free Ca2+ concentration
- LDH
lactate dehydrogenase
- lgp
lysosomal membrane glycoprotein
- NRK
normal rat kidney
- SLO
streptolysin O
- TSF
trypanosomesoluble fraction
Footnotes
Please address all correspondence to Norma W. Andrews, Department of Cell Biology, Yale University School of Medicine, 333 Cedar Street, New Haven, CT 06520. Tel.: (203) 785-4314. Fax: (203) 785-7226.
References
- Adams CJ, Maurey KM, Storrie B. Exocytosis of pinocytic contents by Chinese hamster ovary cells. J Cell Biol. 1982;93:632–637. doi: 10.1083/jcb.93.3.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahnert-Hilger G, Mach W, Fohr KJ, Gratzl M. Poration by α-toxin and streptolysin-O: an approach to analyze intracellular processes. Methods Cell Biol. 1989;31:63–90. doi: 10.1016/s0091-679x(08)61602-7. [DOI] [PubMed] [Google Scholar]
- Baron R, Neff L, Brown W, Louvard D, Courtnoy PJ. Selective internalization of the apical plasma membrane and rapid redistribution of lysosomal enzymes and mannose-6-phosphate receptors during osteoclast inactivation by calcitonin. J Cell Sci. 1990;97:439–447. doi: 10.1242/jcs.97.3.439. [DOI] [PubMed] [Google Scholar]
- Beckers CJM, Balch WE. Calcium and GTP: essential components in vesicular trafficking between the endoplasmic reticulum and Golgi apparatus. J Cell Biol. 1989;108:1245–1256. doi: 10.1083/jcb.108.4.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi G-Q, Alderton JM, Steinhardt RA. Calcium-regulated exocytosis is required for cell membrane resealing. J Cell Biol. 1995;131:1747–1758. doi: 10.1083/jcb.131.6.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittner MA, Holz RW. Kinetic analysis of secretion from permeabilized adrenal chromaffin cells reveals distinct components. J Biol Chem. 1992;267:16219–16225. [PubMed] [Google Scholar]
- Borregaard N, Lollike K, Kjeldsen L, Sengelov H, Bastholm L, Nielsen MH, Baiton DF. Human neutrophil granules and secretory vesicles. Eur J Haematol. 1993;51:187–198. doi: 10.1111/j.1600-0609.1993.tb00629.x. [DOI] [PubMed] [Google Scholar]
- Burkhardt JK, Hester S, Lapham CK, Argon Y. The lytic granules of natural killer cells are dual function organelles combining secretory and prelysosomal compartments. J Cell Biol. 1990;111:2327–2340. doi: 10.1083/jcb.111.6.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burleigh BA, Andrews NW. A 120 KDa alkaline peptidase from Trypanosoma cruzi is involved in the generation of a novel Ca2+-signaling factor for mammalian cells. J Biol Chem. 1995;270:5172–5180. doi: 10.1074/jbc.270.10.5172. [DOI] [PubMed] [Google Scholar]
- Burleigh BA, Caler EV, Webster P, Andrews NW. A cytosolic serine endopeptidase from Trypanosoma cruzi is required for the generation of Ca2+signaling in mammalian cells. J Cell Biol. 1997;136:609–620. doi: 10.1083/jcb.136.3.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buys SS, Keogh EA, Kaplan J. Fusion of intracellular membrane pools with cell surface of macrophages stimulated by phorbol esters and calcium ionophores. Cell. 1984;38:569–576. doi: 10.1016/0092-8674(84)90511-7. [DOI] [PubMed] [Google Scholar]
- Chamberlain L, Roth D, Morgan A, Burgoyne RD. Distinct effects of α-SNAP, 14-3-3 proteins, and calmodulin on priming and triggering of regulated exocytosis. J Cell Biol. 1995;130:1063–1070. doi: 10.1083/jcb.130.5.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez RA, Miller SG, Moore HH. A biosynthetic regulated secretory pathway in constitutive secretory cells. J Cell Biol. 1996;133:1177–1191. doi: 10.1083/jcb.133.6.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coorsen JR, Schmitt H, Almers W. Ca2+triggers massive exocytosis in Chinese hamster ovary cells. EMBO (Eur Mol Biol Organ) J. 1996;15:3787–3791. [PMC free article] [PubMed] [Google Scholar]
- Davidson I, Shankar G, Horton MA, Mason WT. Integrin-dependent mobilization of intracellular calcium ions in osteoclasts. Ann NY Acad Sci. 1994;710:287–300. doi: 10.1111/j.1749-6632.1994.tb26636.x. [DOI] [PubMed] [Google Scholar]
- DeBello WM, O'Connor V, Dresbach T, Whiteheart SW, Wang SH, Schweizer FE, Betz H, Rothman JE, Augustine GJ. SNAP-mediated protein-protein interactions essential for neurotransmitter release. Nature (Lond) 1995;373:626–630. doi: 10.1038/373626a0. [DOI] [PubMed] [Google Scholar]
- DeDuve, C. 1963. The lysosome concept. In Lysosomes. Ciba Foundation Symposium. A.V.S. de Reuck and M.P. Cameron, editors. Churchill, London. 1–35.
- Dunn LA, Holz W. Chatecholamine secretion from digitonintreated adrenal medullary chromaffin cells. J Biol Chem. 1983;258:4989–4993. [PubMed] [Google Scholar]
- Febbraio M, Silverstein R. Identification and characterization of LAMP-1 as an activation-dependent platelet surface glycoprotein. J Biol Chem. 1990;265:18531–18537. [PubMed] [Google Scholar]
- Galli T, Chilcote T, Mundigl O, Binz T, Niemann H, De Camilli P. Tetanus toxin-mediated cleavage of cellubrevin impairs exocytosis of transferrin receptor-containing vesicles in CHO cells. J Cell Biol. 1994;125:1015–1024. doi: 10.1083/jcb.125.5.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrigues J, Anderson J, Hellstrom KE, Hellstrom I. Anti-tumor antibody BR96 blocks cell migration and binds to a lysosomal membrane glycoprotein on cell surface microspikes and ruffled membranes. J Cell Biol. 1994;125:129–142. doi: 10.1083/jcb.125.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gieselmann V, Pohlmann R, Hasilik A, von Figura K. Biosynthesis and transport of cathepsin D in cultured human fibroblasts. J Cell Biol. 1983;97:1–5. doi: 10.1083/jcb.97.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths G, Hoflack B, Simons K, Mellman I, Kornfeld S. The mannose 6-phosphate receptor and the biogenesis of lysosomes. Cell. 1988;52:329–341. doi: 10.1016/s0092-8674(88)80026-6. [DOI] [PubMed] [Google Scholar]
- Griffiths G, Matteoni R, Back R, Hoflack B. Characterization of the cation-independent mannose 6-phosphate receptor-enriched prelysosomal compartment in NRK cells. J Cell Sci. 1990;95:441–461. doi: 10.1242/jcs.95.3.441. [DOI] [PubMed] [Google Scholar]
- Grondin G, Beaudoin AR. Immunocytochemical and cytochemical demonstration of a novel selective lysosomal pathway (SLP) of secretion in the exocrine pancreas. J Histochem Cytochem. 1996;44:357–368. doi: 10.1177/44.4.8601695. [DOI] [PubMed] [Google Scholar]
- Hahn K, DeBiasio R, Taylor DL. Patterns of elevated free calcium and calmodulin activation in living cells. Nature (Lond) 1992;359:736–738. doi: 10.1038/359736a0. [DOI] [PubMed] [Google Scholar]
- Harter C, Mellman I. Transport of the lysosomal membrane glycoprotein lgp120 (lgpA) to lysosomes does not require appearance on the plasma membrane. J Cell Biol. 1992;117:311–325. doi: 10.1083/jcb.117.2.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay JC, Martin TFJ. Resolution of regulated secretion into sequential MgATP-dependent and calcium-dependent stages mediated by distinct cytosolic proteins. J Cell Biol. 1992;119:139–151. doi: 10.1083/jcb.119.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberger R, Heinemann C, Neher E, Matthews G. Calcium dependence of the rate of exocytosis in a synaptic terminal. Nature (Lond) 1994;371:513–515. doi: 10.1038/371513a0. [DOI] [PubMed] [Google Scholar]
- Hirano T, Saluja A, Ramarao P, Lerch MM, Saluja M, Steer ML. Apical secretion of lysosomal enzymes in rabbit pancreas occurs via a secretagogue regulated pathway and is increased after pancreatic duct obstruction. J Clin Invest. 1991;87:865–869. doi: 10.1172/JCI115091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman, E. 1989. Autophagy and related phenomena. In Lysosomes. P. Siekevitz, editor. Plenum Press, New York. 243–316.
- Holz RW, Senyshyn J, Bittner MA. Mechanisms involved in calcium-dependent exocytosis. Ann NY Acad Sci. 1991;635:382–392. doi: 10.1111/j.1749-6632.1991.tb36506.x. [DOI] [PubMed] [Google Scholar]
- Jaconi MEE, Lew DP, Carpentier JL, Magnusson KE, Sjogren M, Stendahl O. Cytosolic free calcium elevation mediates the phagosome– lysosome fusion during phagocytosis in human neutrophils. J Cell Biol. 1990;110:1555–1564. doi: 10.1083/jcb.110.5.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamur MC, Vugman I, Hand AR. Ultrastructural and cytochemical studies of acid phosphatase and trimetaphosphatase in rat peritoneal mast cells developing in vitro. Cell Tissue Res. 1986;244:557–563. doi: 10.1007/BF00212533. [DOI] [PubMed] [Google Scholar]
- Kelly RB. Microtubules, membrane traffic, and cell organization. Cell. 1990;61:5–7. doi: 10.1016/0092-8674(90)90206-t. [DOI] [PubMed] [Google Scholar]
- Kornfeld S, Mellman I. The biogenesis of lysosomes. Annu Rev Cell Biol. 1989;5:483–525. doi: 10.1146/annurev.cb.05.110189.002411. [DOI] [PubMed] [Google Scholar]
- LeSage GD, Robertson WE, Baumgart MA. Bile acid-dependent vesicular transport of lysosomal enzymes into bile in the rat. Gastroenterology. 1993;105:889–900. doi: 10.1016/0016-5085(93)90909-v. [DOI] [PubMed] [Google Scholar]
- Miller SG, Moore H-PH. Reconstitution of constitutive secretion using semi-intact cells: regulation by GTP but not calcium. J Cell Biol. 1991;112:39–54. doi: 10.1083/jcb.112.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake K, McNeil PL. Vesicle accumulation and exocytosis at sites of plasma membrane disruption. J Cell Biol. 1995;131:1737–1745. doi: 10.1083/jcb.131.6.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto T, Popov S, Buckley KM, Poo MM. Calcium-dependent transmitter secretion from fibroblasts: modulation by synaptotagmin I. Neuron. 1995;15:689–696. doi: 10.1016/0896-6273(95)90156-6. [DOI] [PubMed] [Google Scholar]
- Neher E, Zucker S. Multiple calcium-dependent processes related to secretion in bovine chromaffin cells. Neuron. 1993;10:21–30. doi: 10.1016/0896-6273(93)90238-m. [DOI] [PubMed] [Google Scholar]
- Novikoff, A.B. 1961. Lysosomes and related particles. In The Cell. Vol. II. Cells and Their Component Parts. J. Brachet and A.E. Mirsky, editors. Academic Press, New York. 423–488.
- Paradiso AM, Mason SJ, Lazarowski ER, Boucher RC. Membrane restricted regulation of Ca2+release and influx in polarized epithelia. Nature (Lond) 1995;377:643–646. doi: 10.1038/377643a0. [DOI] [PubMed] [Google Scholar]
- Peters PJ, Borst J, Oorschot V, Fukuda M, Krahenbuhl O, Tschopp J, Slot JW, Geuze HJ. Cytotoxic T lymphocyte granules are secretory lysosomes, containing both perforin and granzymes. J Exp Med. 1991;173:1099–1109. doi: 10.1084/jem.173.5.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimplikar SW, Ikonen E, Simmons K. Basolateral protein transport in streptolysin O–permeabilized MDCK cells. J Cell Biol. 1994;125:1025–1035. doi: 10.1083/jcb.125.5.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podbilewicz B, Mellman I. ATP and cytosol requirements for transferrin recycling in intact and disrupted MDCK cells. EMBO (Eur Mol Biol Organ) J. 1990;9:3477–3487. doi: 10.1002/j.1460-2075.1990.tb07556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryzwansky KB, MacRae EK, Spitznagel JK, Cooney MH. Early degranulation of human neutrophils: immunocytochemical studies of surface and intracellular phagocytic events. Cell. 1979;18:1025–1033. doi: 10.1016/0092-8674(79)90215-0. [DOI] [PubMed] [Google Scholar]
- Rabinowitz S, Horstmann H, Gordon S, Griffiths G. Immunocytochemical characterization of the endocytic and phagolysosomal compartments in peritoneal macrophages. J Cell Biol. 1992;116:95–112. doi: 10.1083/jcb.116.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez A, Rioult MG, Ora A, Andrews NW. A trypanosomesoluble factor induces IP3 formation, intracellular Ca2+mobilization and microfilament rearrangement in host cells. J Cell Biol. 1995;129:1263–1273. doi: 10.1083/jcb.129.5.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez A, Samoff E, Rioult MG, Chung A, Andrews NW. Host cell invasion by trypanosomes requires lysosomes and microtubule/kinesin-mediated transport. J Cell Biol. 1996;134:349–362. doi: 10.1083/jcb.134.2.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh O, Wang WC, Lotan R, Fukuda M. Differential glycosylation and cell surface expression of lysosomal membrane glycoproteins in sublines of a human colon cancer exhibiting distinct metastatic potentials. J Biol Chem. 1992;267:5700–5711. [PubMed] [Google Scholar]
- Sengelov H, Kjeldsen L, Borregaard N. Control of exocytosis in early neutrophil activation. J Immunol. 1993;150:1535–1543. [PubMed] [Google Scholar]
- Silverstein RL, Febbraio M. Identification of lysosome-associated membrane protein-2 as an activation-dependent platelet surface glycoprotein. Blood. 1992;80:1470–1475. [PubMed] [Google Scholar]
- Sloane BF, Rozhin J, Johndon K, Taylor H, Crissman JD, Honn KV. Cathepsin B: association with plasma membrane in metastasic tumors. Proc Natl Acad Sci USA. 1986;83:2483–2487. doi: 10.1073/pnas.83.8.2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slot JW, Geuze HJ. A new method of preparing gold probes for multiple-labeling cytochemistry. Eur J Cell Biol. 1985;38:87–93. [PubMed] [Google Scholar]
- Smith SJ, Augustine GJ. Calcium ions, active zones and synaptic transmitter release. Trends Neurosci. 1988;11:458–464. doi: 10.1016/0166-2236(88)90199-3. [DOI] [PubMed] [Google Scholar]
- Sollner T, Whiteheart SW, Brinner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman JE. SNAP receptors implicated in vesicle targeting and fusion. Nature (Lond) 1993;363:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- Steinhardt RA, Guoqiang B, Alderton JM. Cell membrane resealing by a vesicular mechanism similar to neurotransmitter release. Science (Wash DC) 1994;263:390–393. doi: 10.1126/science.7904084. [DOI] [PubMed] [Google Scholar]
- Sullivan KMC, Busa WB, Willson KL. Calcium mobilization is required for nuclear vesicle fusion in vitro: implications for membrane traffic and IP3 receptor function. Cell. 1993;73:1411–1422. doi: 10.1016/0092-8674(93)90366-x. [DOI] [PubMed] [Google Scholar]
- Tapper H, Sundler R. Role of lysosomal and cytosolic pH in the regulation of macrophage lysosomal enzyme secretion. Biochem J. 1990;272:407–414. doi: 10.1042/bj2720407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardieux I, Webster P, Ravensloot J, Boron W, Lunn JA, Heuser JE, Andrews NW. Lysosome recruitment and fusion are early events required for trypanosome invasion of mammalian cells. Cell. 1992;71:1117–1130. doi: 10.1016/s0092-8674(05)80061-3. [DOI] [PubMed] [Google Scholar]
- Tardieux I, Nathanson M, Andrews NW. Role in host cell invasion of Trypanosoma cruzi-induced cytosolic-free Ca2+transients. J Exp Med. 1994;179:1017–1022. doi: 10.1084/jem.179.3.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Figura K, Hasilik A. Lysosomal enzymes and their receptors. Annu Rev Biochem. 1986;55:167–193. doi: 10.1146/annurev.bi.55.070186.001123. [DOI] [PubMed] [Google Scholar]
- Webster P, Vanacore L, Nairn A, Marino C. Subcellular localization of CFTR to endosomes in a ductal epithelium. Am J Physiol. 1994;267:c340–348. doi: 10.1152/ajpcell.1994.267.2.C340. [DOI] [PubMed] [Google Scholar]
- Wiley HS, Kaplan J. Epidermal growth factor rapidly induces redistribution of transferrin receptor pools in human fibroblasts. Proc Natl Acad Sci USA. 1984;81:7456–7460. doi: 10.1073/pnas.81.23.7456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerli S, Maajeed M, Gustavson M, Stendahl O, Sanan DA, Ernst JD. Phagosome–lysosome fusion is a calcium-independent event in macrophages. J Cell Biol. 1996;132:49–61. doi: 10.1083/jcb.132.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]