

Abstract
In a recently developed human breast cancer model, treatment of tumor cells in a 3-dimensional culture with inhibitory β1-integrin antibody or its Fab fragments led to a striking morphological and functional reversion to a normal phenotype. A stimulatory β1-integrin antibody proved to be ineffective. The newly formed reverted acini re-assembled a basement membrane and re-established E-cadherin–catenin complexes, and re-organized their cytoskeletons. At the same time they downregulated cyclin D1, upregulated p21cip,waf-1, and stopped growing. Tumor cells treated with the same antibody and injected into nude mice had significantly reduced number and size of tumors in nude mice. The tissue distribution of other integrins was also normalized, suggesting the existence of intimate interactions between the different integrin pathways as well as adherens junctions. On the other hand, nonmalignant cells when treated with either α6 or β4 function altering antibodies continued to grow, and had disorganized colony morphologies resembling the untreated tumor colonies. This shows a significant role of the α6/β4 heterodimer in directing polarity and tissue structure. The observed phenotypes were reversible when the cells were disassociated and the antibodies removed. Our results illustrate that the extracellular matrix and its receptors dictate the phenotype of mammary epithelial cells, and thus in this model system the tissue phenotype is dominant over the cellular genotype.
The extracellular matrix (ECM)1 modulates breast tissue homeostasis in vivo, and has been shown to regulate growth, differentiation, and apoptosis of normal murine and human mammary epithelial cells (MEC) in culture (Barcellos-Hoff et al., 1989; Petersen et al., 1992; Strange et al., 1992; Boudreau et al., 1995b ; for review see Damsky and Werb, 1992; Adams and Watt, 1993; Roskelley et al., 1995). Moreover, perturbations in cell-ECM interactions are a consistent feature of mammary tumors and cells in vivo and in culture (Petersen et al., 1992; Bernfield et al., 1993; Zutter et al., 1993; Sympson et al., 1994; Bergstraesser and Weitzman, 1994; Howlett et al., 1995). The mechanism by which ECM-mediated signal transduction events can lead to diverse changes in gene expression is now being unraveled. For example, it has been shown that the ECM mediates both biochemical and biomechanical signaling events (Roskelley et al., 1994), and that cell shape in turn can profoundly influence the phenotypic response of cells to the ECM (Dhawan and Farmer, 1994; Ingber et al., 1995). However, how alterations in these pathways can lead to mammary tumors is at present unknown. Because tumor cells have an altered response to the ECM and their microenvironment, we have argued that ECMsignaling pathways contain tumor suppressor checkpoints which impinge upon, and direct, cell and tissue architecture (Petersen et al., 1992; Howlett et al., 1994, 1995). If this were so, then the context and integration of adhesionderived signaling networks could dictate the final tissue phenotype (Bissell et al., 1982).
Epithelial tissue architecture and function are orchestrated and maintained through multimeric adhesion complexes known to interact directly with elements of the actin and intermediate filament cytoskeleton, as well as with kinases and phosphatases (Schmidt et al., 1993; Hodivala and Watt, 1994; for review see Gumbiner 1996; Drubin and Nelson, 1996; Giancotti and Mainiero, 1994; Dedhar, 1995; Ashkenas et al., 1996). The mechanism whereby these adhesive interactions control the spatio-temporal fate of cells, and direct higher order tissue structure is poorly understood, although a dynamic competition between integrins and cadherins for polymerized actin microfilaments, and a modulation of this interaction by tyrosine kinases appears likely (Adams and Watt, 1993; Drubin and Nelson, 1996). Thus, the coordinated regulation of integrins and adherens junctions has been described during keratinocyte differentiation (Hodivala and Watt, 1994; Braga et al., 1995; Hotchin et al., 1995), while cell–cell adhesion may be modulated by molecules such as fascin, an actin bundling/motility-associated protein (Tao et al., 1996). Furthermore, the importance of β1-integrins in directing cell–cell and cell-ECM polarity and differentiation has been shown by studies in kidney cells (Ojakian and Schwimmer, 1994; Schoenenberger et al., 1994) and keratinocytes (Symington et al., 1993; Watt et al., 1993). Also, the redistribution of α6/β4integrins has been reported to be associated with the establishment of epithelial cell polarity (Giancotti, 1996; Borradori and Sonnenberg, 1996), and integrin knockout studies have documented profound tissue degeneration and disorganization in β4-integrin null mice (Dowling et al., 1996). Thus, there is evidence which suggests that integrin and adherens receptor pathways direct the organization of epithelial tissue structure. However, how these events are coordinated, and whether there is a direct reciprocity or hierarchy in these interactions is not known.
A common feature of mammary epithelial tumors in vivo is a disruption of tissue organization and polarity. Consistent with their structural function, catenin-cadherin– cytoskeletal interactions have been shown to be frequently altered in breast cancers, and loss of cell–cell interactions is associated with altered tissue organization and increased tumor invasiveness (for review see Takeichi, 1993; Sommers, 1996; Alford and Taylor-Papadimitriou, 1996). Although cadherins and catenins are often downregulated or absent in invasive breast cancers, there are many examples of mammary carcinoma in situ and aggressive, metastatic breast tumors in vivo that express the full complement of adherens proteins. Instead the cells in these tissues have lost their ability to assemble adherens junctions (Birchmeier and Behrens, 1993; Moll et al., 1994; Sommers, 1996). While the precise cause(s) of this perturbation have yet to be determined, it has been shown that the assembly of adherens junctions can be disrupted by growth factors and oncogenes, and also modified by kinases and phosphatases (Warren and Nelson, 1987; D'Souza and Taylor-Papadimitriou, 1994; Hoschuetzky et al., 1994; Ochiai et al., 1994; Kinch et al., 1995). This suggests that connections exist between adherens junctions and signal transduction pathways and implies the potential for structural plasticity.
The disrupted tissue architecture observed in mammary adenocarcinoma is also frequently associated with alterations in integrin heterodimer profiles (D'Ardenne et al., 1991; Natali et al., 1992; Zutter et al., 1993; Gui et al., 1995; Howlett et al., 1995). Changes in β1-, β4-, α2-, α3, and α6integrins have been reported for mammary tumor cell lines and in tissue sections, and were shown to be associated with tissue disorganization, loss of polarity, increased tumor aggressiveness, and metastasis (Natali et al., 1992; Berdichevsky et al., 1994; Rossen et al., 1994; Gui et al., 1995; Zutter et al., 1995). A relationship between altered signal transduction via integrins and the adherens junction pathways, and its relevance to the origin of the tumor phenotype has not been directly examined. This is mainly due to the lack of appropriate model systems in which such changes can be studied (Weaver et al., 1995).
We have thus taken advantage of a unique epithelial cell model of breast cancer developed by Briand and coworkers (Briand et al., 1987, 1996). The HMT-3522 breast cancer series was established under chemically defined conditions from a breast biopsy of a woman with a nonmalignant breast lesion (Briand et al., 1987; Nielsen and Briand, 1989). A subline of these cells, generated from passage 118, became spontaneously tumorigenic after an additional 120 passages (passage 238), while the parental line remained nontumorigenic for greater than 400 passages (Nielsen et al., 1994; Briand et al., 1996). In this study we used nonmalignant S-1 cells at passage 50 (“normal”; S-1) and the tumorigenic progeny at passage 238 (T4-2; referred to as mt-1 in Briand et al., 1996). The latter cells were shown to have a trisomy of chromosome 7p in addition to other genetic alterations such as a p53 mutation and a c-myc amplification (Madsen et al., 1992; Moyret et al., 1994; Nielsen et al., 1994; Briand et al., 1996). These two cell lines, one originating from the other by spontaneous genetic events, therefore, provide a unique tool for addressing the specific mechanisms involved in malignant conversion in the breast. In this paper, we postulated that if there were a cause and effect relationship between perturbed tissue organization, loss of cell–cell interactions and altered ECMsignaling through integrins on the one hand, and tumor formation on the other hand, it should be possible to modify morphology and behavior of these malignant cells by altering cell-ECM interactions.
Here we show that modification of cell surface β1- and β4-integrins in a 3-dimensional (3-D) basement membrane (BM) assay (Petersen et al., 1992), influences mammary tissue morphogenesis and also regulates cell growth and signal transduction. Furthermore, cellular integrins, when normalized, promote the assembly of adherens junctions and influence the cytostructure of these cells, thereby implying that these two adhesion systems may be interconnected. Finally, our results suggest that growth as well as malignant behavior is regulated at the level of the tissue (acini) organization, i.e., the tissue structure appears to determine the phenotype which in turn overrides the cellular genotype.
Materials and Methods
Substrates and Antibodies
Commercially prepared EHS matrix (Matrigel, Collaborative Research, Waltham, MA) was used for reconstituted basement membrane assays, and Vitrogen (rat tail collagen 1), ∼3 mg/ml (Vitrogen 100, Celtrix Laboratories), was used for thinly coating the surfaces of culture dishes. Antibodies used for biochemical analysis and immunostaining studies were as follows: for immunostaining, immunoblotting, and immunoprecipitation of E-cadherin, α-catenin, and β-catenin, we used clones 36, 29, and 14, respectively (Transduction Laboratories, Lexington, NY); for immunostaining of type IV collagen we used clone PHM-12 (Biogenex, San Ramon, CA); for immunostaining of β1- and α6-integrins we used clones AIIB2 and J1B5 (C. Damsky); for immunostaining of α3-integrin we used clone P1B5; for immunostaining and immunoprecipitation of β4-integrin we used clone 3E1; for immunoblot analysis of β1-integrin we used clone DF5; for immunoblot analysis of β4-integrin we used polyclonal rabbit serum; for immunoprecipitation of β1-integrin we used polyclonal rabbit serum (all from Chemicon International, Temecula, CA); for immunostaining of Ki-67 we used clone MIB; for immunoblot analysis of cyclin D-1 we used clone 17A6-4; and for immunoblot analysis of p21cip,waf-1 we used clone EA10 (all from Oncogene Science, Uniondale, NY). Fluorescence and alkaline phosphatase–conjugated, unlabeled, and nonspecific rat and mouse IgG's were from Jackson Laboratories (West Grove, PA) and HRP-conjugated secondaries were from DAKO (Carpinteria, CA). Antibodies used for integrin function-altering studies within the 3-D reconstituted basement membrane assay were as follows: for β1-integrin functioninhibition we used clone AIIB2 (C. Damsky) and clone JB1a (Chemicon International); for β1-integrin function-stimulating we used clone TS2/16 (a kind gift of M. Hemler); for β4-integrin function-altering we used clone 3E1 (Chemicon International); and for α6-integrin function-blocking we used clone GoH3 (Chemicon International).
Cell Culture
The HMT-3522 mammary epithelial cells (Briand et al., 1987, 1996) were grown in H14 medium (for further description see Blaschke et al., 1994) consisting of DMEM:F12 medium (GIBCO BRL, St. Louis, MO), containing 250 ng/ml insulin (Boehringer Mannheim, Indianapolis, IN), 10 μg/ml transferrin (Sigma, St. Louis, MO), 2.6 ng/ml sodium selenite (Collaborative Research), 10−10 M estradiol (Sigma), 1.4 × 10−6 M hydrocortisone (Collaborative Research), and 5 μg/ml prolactin (Sigma). The S-1 cells were propagated as monolayers on plastic in the presence of 10 ng/ml epidermal growth factor (EGF; Collaborative Research); the T4-2 cells were grown as monolayers on dishes coated with collagen type I in the absence of EGF. Three dimensional (3D) cultures were prepared by growing S-1 and T4-2 cells to confluence as monolayers, followed by trypsinization and embedment into EHS matrices as single cells (8.5 × 105 cells/ml). Cultures were routinely grown for 10–12 d in serum-free medium as described above.
Indirect Immunofluorescence and Immunocytochemistry
EHS cultures were fixed in either 2% paraformaldehyde at ambient temperature for 20 min or in 1:1 methanol-acetone at −20°C for 2–3 min. Specimens were embedded in sucrose, frozen in Tissue-Tek OCT compound (Miles Laboratories, Elkhart, IN), and 5 μ frozen sections were prepared for immunostaining as described previously (Streuli et al., 1991). Sections were incubated with primary antibodies for 60 min followed directly by either FITC or Texas red–conjugated secondaries (45 min), or by biotinylated secondary antibodies (45 min), and either Texas red–conjugated streptavidin or HRP-conjugated streptavidin (30 min). Nuclei were counterstained with di-amino-phenyl-indole (DAPI, Sigma) or hematoxylin (5 min). Control sections were stained with second antibodies only.
Morphogenesis Criteria
Quantitative analysis of tumor cell conversion efficiency by β1-integrin function blocking antibodies was determined by calculating the percentage of colonies converted to a “nontumorigenic morphology.” Percent conversion was determined by directly examining the morphology, uniformity, and size of 2,000–4,000 colonies of S-1, T4-2, and β1-inhibitory antibodytreated T4-2 cells (T4-β1). Morphology was assessed in situ by examining the degree of colony organization visually by phase contrast microscopy, and by measuring colony diameter using an eyepiece equipped with a micrometer spindle. Colonies were deemed disorganized if they were both irregular in shape and the colony length exceeded the width by greater than twofold. Colony organization was also suggested by a lateral and radially distributed cytoarchitecture, which was visualized by immunostaining for the distribution of actin microfilaments and cytokeratin 18 intermediate filaments, and related to the distribution observed in acini formed by normal mammary epithelial cells. Polarity was indicated by the presence of a basally organized BM as determined by collagen IV and laminin immunostaining, and by basally immunolocalized α6-, β4-, and β1-integrins. Morphogenic reversion was scored positive by the deduced presence of adherens junctions as determined by colocalization of E cadherin and α- and β-catenins at cell–cell contacts.
Analysis of Cellular Growth and Apoptosis
The proliferative rate of cells in 3-D EHS was assessed by measuring the thymidine incorporation index. Cryosections (5 μ) of 3D cultures were pulse-labeled for 24 h with [3H]TdR (20 Ci/mmol, New England Nuclear Research Products, Dupont, Wilmington, DE), air dried, coated with Kodak NTB2 emulsion, developed, and scored visually (100–400 cells) for radiolabelled nuclei. Indices were calculated by expressing this value as a percentage of the total number of cells scored (Petersen et al., 1992). Cell growth was determined in monolayers or in 5 μ cryosections after a 12-h incubation with 10 μM BrdUrd followed by fixation in 70% ethanol, and staining with anti-BrdUrd using a kit (Boehringer Mannheim), and indices were calculated as above. Proliferative potential of 100–400 cells was assayed by immunostaining and calculating the Ki-67 labeling index in 5 μ cryosections of 3D EHS cultures. Proliferative status was determined also by counting and immunostaining the number of cells per colony as well as the average colony size. The colony size was measured directly in 3-D cultures by phase contrast microscopy using an eyepiece equipped with a micrometer spindle, which had been previously calibrated with a ruler. Cell number per colony was determined by counting the number of DAPI stained nuclei per colony in cryosections of cells in 3-D. Cell cycle status was determined by immunoblotting equal amounts of total cell protein lysates for cyclin D1 and p21cip,waf-1 protein levels after separating the proteins by reducing Laemmli gels. Apoptosis was assessed by detection of FITC-labeled 3'OH DNA ends using an in situ apoptosis kit (Boehringer Mannheim) in 5 μ cryosections (Boudreau et al., 1996). Cells were scored (150–200 cells) for the presence of DNA nicks and positively scored cells for each condition were expressed as a percentage of the total number of cells scored (apoptotic index).
Determination of α- and β-Catenin-E-Cadherin Adherens Junction Assembly
For the determination of total E-cadherin, α-catenin, and β-catenin levels, RIPA lysates (1% NP-40, 0.5% deoxycholate, 0.2% SDS, 150 mM sodium chloride, 50 mM Tris-HCl, pH 7.4 containing 20 mM sodium fluoride, and 1 mM sodium orthovanadate, 10 μg/ml leupeptin, 10 μg/ml pepstatin, 1 mM Pefabloc [Boehringer Mannheim], 10 μg/ml E64, 25 μg/ml aprotinin, and 0.5 mM benzamidine) were prepared from cells isolated from EHS by PBS/EDTA, separated on reducing Laemmli gels and immunoblotted. For the assessment of the soluble and cytoskeletal-associated E-cadherins, α-catenins and β-catenins, S-1, and T4-2 colonies were grown for 12 d in 3-D- reconstituted basement membranes and colonies were isolated using icecold PBS/EDTA (0.01 M sodium phosphate, 138 mM sodium chloride, 5 mM EDTA), homogenized and fractionated into Triton X-100 soluble and insoluble lysates essentially as described by Nathke et al. (1994). Lysates were separated on Laemmli reducing gels and immunoblotted for adherens junction proteins. Densitometric analysis of scanned enhanced chemiluminescence (ECL) film was used for the estimation and calculation of relative protein levels. For the assessment of the β-catenin-(or α-catenin)- E-cadherin interaction index, S-1, T4-2, and T4-β1 integrin-reverted cells were grown for 12 d in 3-D EHS. RIPA lysates of equal numbers of cells in EHS were prepared by directly homogenizing in RIPA, and were immunoprecipitated with anti–E-cadherin antibodies. Immunoprecipitants were separated on reducing Laemmli gels and immunoblotted for E-cadherin, α-catenin, and β-catenin proteins. ECL films were scanned and subjected to densitometric analysis for assessment of the relative protein levels. Interaction index values were calculated by dividing the densitometric value determined from the catenin protein coprecipitated with E-cadherin by the value calculated for the level of E-cadherin protein immunoprecipitated. For the assessment of in situ cytoskeletal-associated and soluble adherens junctions proteins, day 12 cultures of S-1 and T4-2 colonies were frozen (see above) without prior fixation, and fresh 8 μ cryosections were extracted with fresh CSK buffer (50 mM sodium chloride, 300 mM sucrose, 10 mM Pipes, pH 6.8, 3 mM MgCl2, 0.5% Triton X-100, containing a cocktail of protein inhibitors as described above). Sections were extracted for 40 min, fixed in 2% paraformaldehyde for 20 min and immunostained for E-cadherin, α-catenin, and β-catenin proteins.
Determination of Total Levels and Cell Surface Integrin Expression
For the assessment of total β1- and β4-integrin levels, cells were grown in 3D EHS cultures for 10–12 d, and colonies were isolated using ice-cold PBS/EDTA. Colonies were lysed in RIPA and equal amounts of lysate proteins were separated on reducing Laemmli gels and immunoblotted. ECL films were scanned and subjected to densitometric analysis for assessment of the relative protein levels. For the determination of colony surface β1- and β4-integrins, cells were grown in 3-D EHS cultures for 10– 12 d and colonies were then released from the matrix by 60–90 min incubation at 37°C with dispase (5,000 U/ml caseinolytic activity, Collaborative Research), washed three times in fresh DMEM:F12 medium and once in ice-cold PBS. Released colonies were incubated and rocked at 4°C for 60 min with 1 mg/ml sulfo-NHS-biotin (Pierce, Rockford, IL) in PBS to label the cell surface proteins, washed twice in ice-cold PBS containing 50 mM glycine, and incubated for 10–15 min on ice in PBS/glycine to stop the reaction. Colonies were washed three more times in ice-cold PBS/glycine and lysed in 1% NP-40 lysis buffer also containing 120 mM sodium chloride, 50 mM Tris-HCl and 10 μg/ml leupeptin, 10 μg/ml pepstatin, 1 mM Pefabloc (Boehringer Mannheim), 10 μg/ml E64, 25 μg/ml aprotinin, and 0.5 mM benzamidine. Equal numbers of cells (determined by counting the cells in parallel cultures) or equivalent amounts of extracted proteins, were immunoprecipitated with anti–β1- or β4-integrin monoclonal antibodies, separated on nonreducing Laemmli gels, and immunoblotted for the presence of biotinylated proteins using HRP-conjugated streptavidin antibody and ECL detection. Irrelevant mouse or rat IgG's served as nonspecific controls. Exposed films were scanned and subjected to densitometric analysis for the determination of intensity differences and relative quantification.
Preparation of Fab Fragments
Rat monoclonal AIIB2 IgG1's were isolated using Affi-Gel Protein A Maps II system (Bio-Rad Labs, Hercules, CA). After purification of rat IgG1, the material was put through a 10DG desalting column, concentrated by centrifugation and dialyzed against 100 mM sodium acetate buffer, pH 5.0. Fab fragments were generated after 20 h of papain digestion (20 mU enzyme/5–10 μg IgG protein, in 100 mM sodium acetate, pH 5.0, 50 mM cysteine, 1 mM EDTA) and reactions were terminated by addition of iodoacetamide. The presence of Fab fragments was assayed by Coomassie blue staining of acrylamide gels as well as by Western blotting using secondary antibodies. Completion of the reaction was monitored by the disappearance of high molecular weight intact antibody and the appearance of low molecular weight proteins comigrating with commercial Fab's. Western blotting with anti-[Fab]2 antibodies verified the absence of the latter fragments in the digest. Intact AIIB2 antibody was efficiently competed out by the Fab preparation, as shown by solution competition and sequential immunostaining of S-1 acini. Once the presence and activity of the Fab's was established, samples were treated with sodium azide and dialyzed into DMEM:F12 before cell culture experiments.
Integrin Function-altering Assays
Antibodies and Fab's and control non-immune mouse and rat IgG's were introduced into the cell-embedded substratum at the time of EHS gelation. For the crude ascites of β1- and β4-integrins, we used a range of 25–200 μg ascites protein/ml; for purified β1- and α6-integrins and purified mouse or rat IgG's we used 2–25 μg/ml, ∼10–150 nM. Cultures were scored visually after 12 d. 10–20 random fields were viewed by phase contrast microscopy and the number of normal spheres or tumor-like colonies were counted in each field and related to the behavior of control cultures either without antibodies or in the presence of irrelevant antibodies. To compare the reversion efficiency of various β1-integrin antibodies and Fab fragments on T4-2 cells, the diameter of 200 S-1 nonmalignant acini, as observed directly by phase contrast microscopy, ±SEM was calculated and the ratio of the tumor colony diameter to the nonmalignant S-1 acini diameter ±SEM was determined.
Reversion Assay
Equal densities of cells were seeded into 3-D basement membranes and were subjected to either β1-inhibitory integrin or mock-antibody (nonspecific rat IgG) treatment. After 12 d, cells were photographed, released from their matrices, and propagated as monolayers. They were then recultured within the 3-D basement membrane assay with or without the β1-integrin inhibitory antibody treatment. These steps were repeated two more times.
Tumor Formation In Vivo
Tumor cells were propagated as monolayers, grown for 3–4 d in a 3-D-reconstituted basement membrane in the absence of antibody, released by dispase (Collaborative Research), washed three times in DMEM:F12, and incubated with 100 μg/ml function blocking β1-integrin antibody (clone AIIB2), mock antibody (nonspecific rat IgG's), or no treatment (control group) for 3 h. Cell colonies were then washed three times in PBS and 1–4 × 106 cells were subcutaneously injected into the rear flanks of 4–6-wk-old BalbC nu/nu mice, without further antibody treatment. Nontreated, nonmalignant S-1 cells were also injected into nude mice as negative controls. Statistics were done by “unistat V 4.0 for windows.”
Experimental Subjects
In conducting research using animals, the investigators adhered to the “Guide for the Care and Use of Laboratory Animals,” prepared by the Committee on Care and Use of Laboratory Animals of the Institute of Laboratory Animal Resources, National Research Council (NIH publication no. 86-23, revised 1985). In the conduct of research where human specimens were used, the investigators adhered to the policies regarding the protection of human subjects as prescribed by 32 CFR 219 and subparts B, C, and D.
Results
S-1 and T4-2 Cells in a 3-D Basement Membrane Culture Assay Recapitulate the Phenotypic Characteristics of Normal and Malignant Breast Tissue In Vivo
Only small differences in morphology and growth rates could be observed between S-1 and T4-2 cells on tissue culture plastic (not shown), but profound differences became evident after only 4 d following their culture within a 3-D reconstituted basement membrane. Within 10 d, S-1 cells underwent morphogenesis and formed organized acini reminiscent of those formed by cells from reduction mammoplasty, while T4-2 cells formed large, loosely disorganized invasive colonies of cells similar to primary tumor cells tested in this assay previously (Fig. 1, a and a′; Petersen et al., 1992). In addition, S-1 cells were able to basally deposit and organize a basement membrane, as shown by immunostaining of type IV collagen (Fig. 1 b, and laminin, not shown), thereby demonstrating that the cells were able to form polarized structures. The irregular T4-2 colonies, while staining for basement membrane components had no discernible organized basement membrane (Fig. 1, b and b′). The failure of T4-2 cells to undergo morphogenesis was also indicated by their compromised cell–cell adhesion. This was shown by the absence of lateral E-cadherin immunostaining (Fig. 1, c and c′), an increase in cytoplasmic localization of E-cadherin (cell fractionation and extraction studies, not shown) and reduced interaction of α- and β-catenins with E-cadherin (co-immunoprecipitation, Fig. 1 d). Nevertheless, the two cell types expressed essentially the same levels of the three cell adhesion proteins (Fig. 1 e) indicating that the malignant conversion was associated with compromised assembly of adherens junctions rather than with down-regulation of these adhesion proteins. Coincident with the formation of acini, S-1 cells became growth arrested and exited the cell cycle, as demonstrated by negligible thymidine incorporation and low immunostaining for Ki-67. Consistent with their loss of structural organization, the tumorigenic T4-2 cells failed to growth arrest by these criteria (Fig. 1, f and g).
Figure 1.

Characterization of the HMT-3522 human breast cancer model. (a and a′) Phase contrast micrographs of nonmalignant S-1 cell colonies (a) and tumorigenic T4-2 cell colonies (a′) viewed directly inside EHS for morphology: S-1 cells formed spherical structures reminiscent of true acini (a), whereas T4-2 cells formed large irregular colonies (a′). (b and b′) Immunostaining for basement membrane components: S-1 acini (b) stained for basement membrane proteins at the cell-ECM junctions as expected, while basement membrane deposition in tumor colonies (b′) was clearly disorganized and no longer polarized. Comparable results were obtained for laminin (not shown). (c and c′) Confocal fluorescence microscopy of cryosectioned colonies immunostained for E-cadherin: S-1 acini (c) stained for E-cadherin primarily at the cell–cell junctions as is typically observed in normal breast sections. The T4-2 colonies (c′) showed punctate, dispersed membrane and intracellular staining. (d) The β-catenin–E-cadherin interaction ratio as a measure of adherens junction assembly: There was a 50% decrease in β-catenin protein coprecipitating with E-cadherin in T4-2 cells as compared to S-1 acini. Data are expressed as a ratio of β-catenin to E-cadherin densitometric measurements from duplicates of two separate experiments. Similar results were observed for α-catenin (not shown). (e) Immunoblots of total levels of E-cadherin, α-catenin, and β-catenin: There were comparable levels of all three adherens proteins in S-1 acini and T4-2 colonies. (f and g) Percent of thymidine and Ki-67 labeling in S-1 acini and T4-2 colonies, expressed as thymidine and Ki-67 labeling indices from 3–4 separate experiments: Greater than 90% of the S-1 acini were growth arrested (f), and had exited the cell cycle (g) whereas T4-2 colonies were still actively growing (f) and continued to cycle (g). All cultures were analyzed after 10–12 d inside EHS. Bars: (a and a′) 16 μm; (b and b′) 25 μm; (c and c′) 8 μm.
S-1 and T4-2 Cells Differ in the Localization and Expression of ECM Integrin Receptors
We have postulated previously that tumor cells have lost their ability to respond correctly to ECM-induced signals (Petersen et al., 1992; Howlett et al., 1995). To determine if the loss of structural organization and growth control exhibited by the tumor cells was related to an alteration in their integrins, we characterized the integrin receptors for laminin in the two cell lines. While both S-1 and T4-2 cells expressed integrins β1, β4, α3, and α6, their distribution patterns were radically different in 3-D cultures (Fig. 2, a and a′ through d and d′). S-1 acini had basally distributed β1-, β4-, and α6-integrins and basolateral α3-integrin, consistent with their polarized phenotype. In contrast, all of these integrins were found to be randomly distributed and disorganized in the T4-2 colonies. Western blot analysis of the tumor colonies showed over-expression of both β1- and β4-integrins relative to the levels observed in the S-1 acini (Fig. 3, a and b). Cell surface labeling and immunoprecipitation experiments revealed that the surface levels of β1-integrins were slightly higher (12.5%) in the T4-2 cells (Fig. 3 c) whereas the surface β4-integrin levels were much lower (60%) in T4-2 than in S-1 colonies (Fig. 3 d). However, the ratio of β1- to β4-integrins at the cell surface was increased by more than 2.8-fold in the T4-2 cell colonies than in the S-1 acini (Fig. 3 e).
Figure 2.
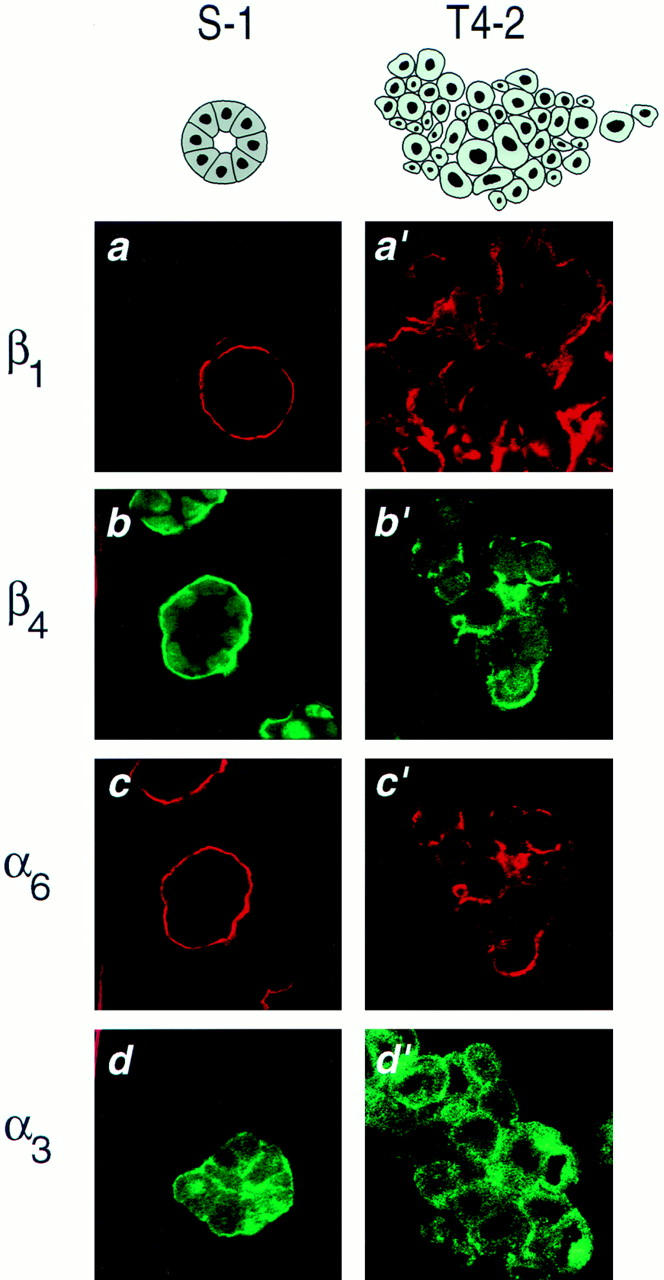
Immunofluorescence characterization of integrins in the HMT-3522 cells in 3-dimensional cultures. (a–d) Cryosections of S-1 acini and (a′–d′) T4-2 colonies, immunostained and examined by confocal fluorescence microscopy for localization of β1- (a and a′), β4- (b and b′), α6- (c and c′), and α3-integrin (d and d′) localization: β1-, β4-, and α6-integrins were targeted to the cell-ECM junction in the S-1 acini (a–c), in contrast, in T4-2 colonies (a′–c′) this polarized-basal distribution was lost. S-1 acini exhibited basolateral α-3 integrins (d), whereas T4-2 colonies (d) demonstrated disorganized plasma membrane and cytosolic expression of this integrin. All cultures were analyzed after 10–12 d inside EHS. Bars: (a–d and a′–d′) 16 μm.
Figure 3.


Biochemical characterization of integrins in the HMT3522 cells after culture in 3-dimensions. (a and b) Western analysis of β1- and β4-integrins: Total cell lysates of S-1 and T4-2 colonies showed elevated expression of β1- (a) and β4-integrins (b) in the tumor cell colonies. (c and d) Cell surface expression of β1- and β4-integrin heterodimers using biotinylation: Tumor colonies had 12.5% higher plasma membrane levels of β1- (c) but 60% lower levels of β4-integrin heterodimers (d) than S-1 acini. (e) Relative scan units (RSU) showing relative colony surface levels of β1- and β4-integrin heterodimers: There was a 2.8-fold relative increase in the ratio of β1-integrins to β4-integrin on the colony cell surface of T4-2 colonies as compared to the S-1 acini. All cells were analyzed after 10–12 d inside EHS.
Function Blocking β1-Integrin Antibodies Cause Dramatic Phenotypic Reversion of the T4-2 Cells
Since T4-2 cells had both a higher total level and an elevated ratio of cell surface β1- to β4-integrins, we wondered whether the aberrant malignant behavior may be a reflection of the changes in these integrins. Accordingly, we examined the consequences of treatment in 3-D with varying concentrations of a previously characterized rat monoclonal β1-integrin antibody (clone AIIB2) which has been shown to inhibit ligand binding (Werb et al., 1989). This antibody caused massive apoptosis in S-1 cells, as shown previously (Howlett et al., 1995), while T4-2 cells were refractory (Fig. 4). Remarkably however, in addition to resistance to apoptosis, almost all the antibody-treated T4-2 tumor cells assumed a morphology which was indistinguishable from that observed in S-1 cultures and was discernible as early as 4 d after incubation. When examined by light microscopy after 12 d, these cultures appeared as if they had truly reverted to a “nonmalignant” phenotype. We therefore cryosectioned the colonies and examined their morphology by immunofluorescence confocal microscopy. As markers of normal acinar formation, we examined both cytoskeletal organization and superimposition and distribution of cadherins and catenins. Sections of S-1 acini revealed uniform and polarized nuclei (stained with propidium iodide; red), well-organized filamentous actin (FITC phalloidin; green) (Fig. 5 a), and uniformly superimposed E-cadherin and β-catenin at the lateral cell–cell junctions (Fig. 5 b). In contrast, untreated or IgG-treated tumor cells had polymorphic nuclei and a grossly disorganized actin cytoskeleton, visualized as random, hatched bundles (Fig. 5 a′). Additionally, E-cadherin and β-catenin were no longer colocalized (Fig. 5 b′). In contrast, β1-treated T4-2 cells (referred to as T4-β1) revealed striking rearrangements of cytoarchitecture as demonstrated by their well-organized actin (Fig. 5 a′′), and cytokeratin 18 intermediate filament (not shown) networks. Furthermore, organized adherens junctions became evident in T4-β1 acini (Fig. 5 b′′) and were accompanied by the re-establishment of E-cadherin–catenin complexes (not shown). These changes were shown to occur in greater than 95% of the tumor colonies treated with blocking antibody, as quantified by analyzing the numbers of disorganized vs organized spheroids in relation to the S-1 and the mock-treated T4-2 cells (Fig. 5 c). Viability and growth assays conducted on cells grown as monolayers ruled out toxicity (not shown). Interestingly, while treatment with the inhibitory β1-integrin antibody reduced cell adhesion and retarded the rate of cell proliferation when T4-2 cells were grown in two-dimension, culture in a three-dimensional reconstituted basement membrane was required for complete expression of the reverted phenotype (not shown).
Figure 4.

Apoptosis induction in HMT-3522 cells by inhibitory β1-integrin function blocking antibodies. Apoptotic labeling indices calculated for S-1 and T4-2 cultures treated with inhibitory β1-integrin antibodies for 4 d in 3-dimensional cultures: Greater than 70% of S-1 cells were induced to undergo apoptosis within 4 d of incubation with β1-integrin function blocking antibody (S-1 β1) while the level of basal apoptosis was less than 5% in the isotype controls (S-1 IgG). In contrast T4-2 cells similarly treated (T4 β1) were refractory and had an apoptosis rate comparable to basal levels (T4-2 IgG). Results are the mean and SE of 3–6 separate experiments of duplicates.
Figure 5.

β1-inhibitory antibody treatment of tumor cells leads to the formation of reverted acini. (a–a′′) Confocal fluorescence microscopy images of F actin: Both the S-1 (a) and T4-β1 reverted acini (a′′) showed basally localized nuclei (propidium iodide) and organized filamentous F-actin (FITC), while T4-2 mock-treated colonies (T4-2 IgG) had disorganized, hatched bundles of actin and pleiomorphic nuclei (a′). (b–b′′) Confocal immunofluorescence microscopy images of E-cadherin (FITC) and β-catenin (Texas red): In S-1 (b) and T4-β1 reverted acini (b′′), E-cadherin and β-catenins were colocalized and superimposed at the cell–cell junctions. (c) Quantitative analysis of tumor cell conversion efficiency by β1-integrin function blocking antibodies: Greater than 95% of S-1 and T4-β1 colonies were scored as organized, while 97% of T4-2 IgG colonies were considered disorganized. Other reversion criteria, such as scoring for actin and cadherin organization also yielded comparable results (not shown). (d) Cell number per colony in S-1 acini, T4-2 IgG colonies and T4β1 acini from 3–5 experiments: Both S-1 and T4-β1-revertant acini contained 6–8 cells, while T4-2 IgG tumor colonies contained 18–22 cells when scored after 10–12 d (d). (e) Percent of thymidine-labeled cells in S-1, T4-2 IgG, and T4-β1 colonies: While a high percentage of T4-2 IgG colonies (greater than 30%) were still actively growing, the T4-2 β1 revertant acini had a greatly reduced growth rate similar to that observed in the S-1 acini. (f) Immunoblot of cyclin D1 levels in S-1, T4-2, T4-2 IgG, and T4-β1 colonies: The level of D1 cyclin was clearly decreased to the level of S-1 acini after β1 inhibitory antibody treatment. (g and g′) Collagen IV deposition as an indicator of basement membrane organization: T4-β1 reverted acini (g′) deposited an organized collagen IV-containing BM at the cell-ECM junctions, similar to that observed in S-1 acini (see Fig. 1 b). Note the contrast with T4-2 mock–treated tumor colonies (g). (Comparable results were obtained for laminin, not shown.) All cultures were analyzed after 10–12 d inside EHS. Bars: (a–a′′and b–b′′) 16 μm; (g and g′) 25 μm.
An examination of markers of proliferation and cell cycle status in T4-β1 cells revealed that they were growth arrested as indicated by both a decrease in thymidine incorporation and the size of the acini which was now composed of only 6–8 cells, similar to that observed for S-1 cells (Fig. 5, d and e). This was in marked contrast to the average of 18–22 cells observed for nontreated T4-2 tumor colonies and the high mean number of T4-2 nuclei which incorporated [3H]thymidine (Fig. 5, d and e). T4-β1 colonies also had dramatically decreased cyclin D-1 levels (Fig. 5 f) again comparable to that seen in S-1 cultures and markedly reduced Ki-67 levels (not shown), suggesting that most reverted cells had exited the cell cycle and therefore had a reduced propensity to proliferate.
Cryosections of T4-β1 colonies incubated with antibodies against either collagen IV (Fig. 5 g′) or laminin (not shown) revealed deposition of a basally distributed, almost continuous basement membrane, with characteristics similar to that observed in the S-1 acini (Fig. 1 b). In contrast, punctate and inversely polarized collagen IV (Fig. 5 g) and laminin immunostaining (not shown) were observed in the mock-treated tumor colonies. Thus, these tumor cells had retained the ability to deposit a basement membrane and to form polarized structures if the correct structural cues could be received.
The Inhibitory β1-Integrin Activity Is Necessary for Phenotypic Reversion
The AIIB2 monoclonal antibody (mAb) recognizes a region connecting the two putative extracellular ligand binding sites of the β1-integrin receptor, thereby inducing a conformational change consistent with inhibition of receptor activity (Takada and Puzon, 1993). To distinguish between integrin binding per se or a requirement for inhibition of signaling, we examined whether the T4-2 cells would phenotypically revert upon treatment with a nonfunctional (neither activating nor inhibiting) anti–β1-integrin mAb (clone DF5; Korhonen et al., 1991), an alternate inhibitory anti–β1-integrin mAb (clone JB1a or J10; Stupack et al., 1991), and an activating anti–β1-integrin mAb (clone TS2/16; Weitzman et al., 1995), whose epitope was mapped to the same region as the AIIB2 antibody (Takada and Puzon, 1993). Only the β1-integrin function blocking antibodies were found to be capable of inducing phenotypic reversion of the T4-2 cells (Table I), implying that overexpression or altered signaling through a β1-integrin–specific pathway is at least in part responsible for the malignant phenotype of these tumor cells. To investigate the possibility that the inhibitory antibodies may have facilitated reversion by inducing clustering of the integrin receptors by divalent mAb-mediated cross-bridging, we purified the rat IgG1 AIIB2 mAb and made AIIB2 Fab fragments (Miyamoto et al., 1995a ,b). Significantly, both the purified AIIB2 mAb and its Fab fragments were capable of inducing the same phenotypic reversion of the T4-2 tumor cells (Table I), indicating that it was the β1-integrin function at the cell surface that needed to be reduced for reversion to occur, rather than augmentation of integrin signaling by receptor clustering per se.
Table I.
Comparison of Various β1-Integrin Antibody Treatments on Colony Size in 3-Dimensional Culture
| Description | Mean colony size | Relative size ratio of T4-2/S-1 colonies* | ||
|---|---|---|---|---|
| μm | ||||
| S-1 50 cells | 24.5 ± 0.7 | N/A | ||
| Control T4-2 cells (nonspecific rat IgG's)‡ | 74.3 ± 2.4 | 3.03 ± 0.03 | ||
| Inhibitory β1-antibody-treated cells, clone AIIB2 crude ascites | 24.1 ± 1.0 | 0.98 ± 0.10 | ||
| Inhibitory β1-antibody-treated cells, clone AIIB2 purified IgG1 | 28.2 ± 1.1 | 1.15 ± 0.04 | ||
| Inhibitory β1-antibody-treated cells, clone AIIB2 Fab fragments | 24.5 ± 1.4 | 1.04 ± 0.04 | ||
| Inhibitory β1-antibody-treated cells, clone JB1a crude ascites (alternate inhibitory mAb) | 32.3 ± 1.4 | 1.32 ± 0.06 | ||
| Activating β1-antibody-treated cells, clone TS2/16 crude ascites (stimulatory mAb) | 61.7 ± 3.3 | 2.52 ± 0.13 |
The diameter of 200 S-1 acini as observed directly by phase contrast microscopy, were measured after 12 d in 3-D culture using an eyepiece equipped with a micrometer spindle. The same measurements were made on 100–400 T4-2 colonies in the presence of 100 μg antibody ascites protein or 25 μg purified IgG/ml EHS matrix of various β1-integrin antibodies (listed above). The average measurements of each of these groups ± SEM were calculated and the ratio of T4-2/S-1 diameters ± SEM were determined.
Comparable results were also obtained with nonfunction altering antibodies α6- (clone- J1B5) and β1-integrin (clone DF5), which are not shown.
The Reversion Is Phenotypic and Reversible
To distinguish between a reversible, microenvironmentally induced change as opposed to selection of possible nonmalignant contaminants or genetically altered mutants, we undertook a series of “reversion rescue” studies. Despite two rounds of reversion, rescue, monolayer propagation, and reculturing in 3-D, these cells were able to resume their original tumorigenic phenotypes when cultured in the absence of antibody (Fig. 6).
Figure 6.
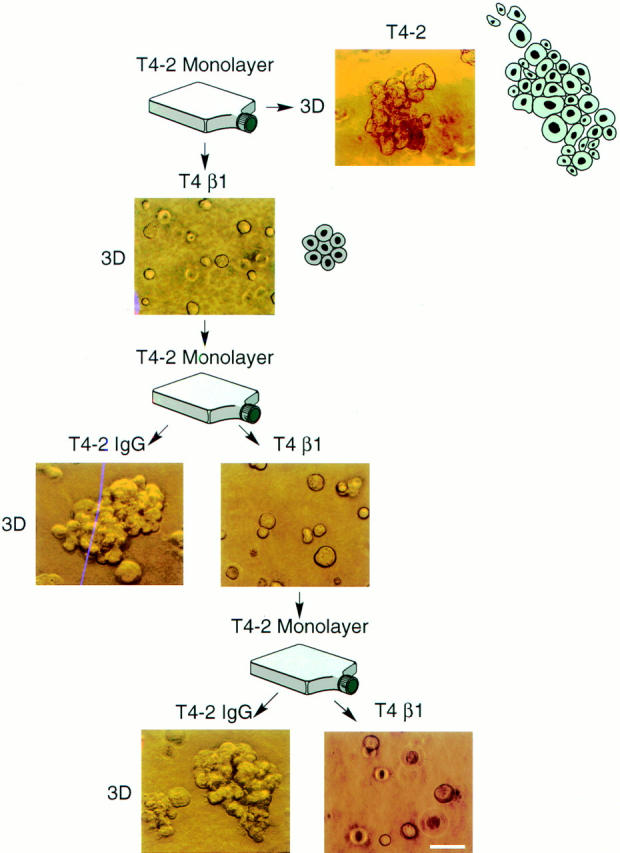
Phenotypic reversion as opposed to selection. Phase contrast micrographs of T4-2 cells grown in the presence of anti–β1-integrin function blocking antibody (T4 β1), mock antibody (nonspecific IgG's) (T4-2 IgG) or no antibodies (T4-2) viewed directly inside EHS: Despite two rounds of treatment, these antibody reverted cells were able to resume their original tumorigenic phenotypes when cultured in the absence of antibody. All cultures were analyzed after 10–12 d inside EHS. Bar, 50 μm.
Treatment with β1-Inhibitory Antibody Is Sufficient to Significantly Reduce Malignancy In Vivo
To find out whether phenotypic reversion of tumor cells would be sufficient to reduce tumorigenicity in vivo, we injected tumor cells treated in suspension with β1-integrin blocking mAb, mock mAb, or no treatment for 3 h, as well as S-1 cells into nude mice. Within two weeks small nodules were observed in all injected sites including the S-1 controls (not shown). Whereas these nodules regressed rapidly in the S-1 and T4-β1 groups, actively growing tumors were observed in greater than 75–90% of the mock mAb or vehicle-treated T4-2 mice. Upon sacrifice we observed both a significantly reduced tumor number and tumor size in the T4-β1 group (Table II). These data suggest that “normalization” of the tumor cell phenotype in culture has a counterpart in vivo where the malignant potential is reduced or lost.
Table II.
Effect of β1-Inhibitory Antibody Induced Phenotype Reversion on Tumorigenicity In Vivo
| Treatment description | Total number of tumors per group | Tumor size | ||||||
|---|---|---|---|---|---|---|---|---|
| Large tumors* | Small tumors‡ | Mice with no tumors | ||||||
| Control | ||||||||
| nontreated | ||||||||
| T4-2 cells | 15/16 | 14/16 | 1/16 | 1/16 | ||||
| Non-immune | ||||||||
| rat IgG-treated | ||||||||
| T4-2 cells | 14/16 | 11/16 | 3/16 | 2/16 | ||||
| β1-integrin | ||||||||
| function blocking | ||||||||
| antibody-treated T4-2 cells | 7/16 | 5/16§ | 2/16 | 9/16§ | ||||
tumors >5 <300 mm3.
tumors <5 mm3.
P < 0.005 by X2 test.
Malignant Conversion Is Associated with Alterations in α6- and β4-Integrins
The ability of T4-β1 cells to re-form a basally organized basement membrane (Fig. 5 g′) indicated the re-establishment of acinar polarity. Polarity has been shown to be associated with a basal localization of α6/β4-integrin heterodimers in keratinocytes (DeLuca et al., 1994). T4-β1 reverted acini had polar, basally localized α6- and β4-integrins (compare Fig. 7, a–7 a′), increased cell surface β4-integrin heterodimers (Fig. 7 b), and higher p21cip,waf-1 levels (Fig. 7 c). These findings showed that the β1-integrin signaling pathway was not only interconnected with cateninE-cadherin adherens junction assembly, but was also connected to the α6/β4-integrin signaling pathway.
Figure 7.

Alteration of α6/β4-integrin signaling in S-1 cells leads to the formation of disorganized colonies. (a and a′) Confocal fluorescence microscopy of double immunostaining for α6- (Texas red) and β4-integrins (FITC): T4-β1 revertant acini (a′) showed basally polarized α6- and β4-integrins, while T4-2 mock–treated (T-4 IgG) colonies (a) demonstrated disorganized, nonpolarized expression. (b) Cell surface expression of β4-integrin heterodimers using biotinylation: Tumor colonies had lower cell surface levels of β4-integrin heterodimers (60% lower) relative to the S-1 acini (see Fig. 3 d), which were restored in the T4-β1 reverted acini (40% higher) relative to the T4-2 colonies. (c) Immunoblot of p21cip,waf-1 levels in T4-2 IgG and T4-β1 colonies: The level of p21 protein was clearly increased in the T4-β1 reverted acini. (d and d′) Phase contrast micrographs of S-1 nonmalignant acini (d) and tumorigenic T4-2 colonies (d′) viewed directly inside EHS: S-1 cells formed spherical structures (d), whereas T4-2 cells formed large, irregular colonies (d′). (e and e′) Phase contrast micrographs of S-1 nonmalignant cells treated with function altering β4-integrin antibodies (e) and tumorigenic T4-2 cells treated with function blocking β1-integrin antibodies (e′) viewed directly inside EHS: S-1 cells treated with β4-integrin antibodies formed large, irregular colonies (e), while T4-2 cells treated with β1-integrin function-blocking antibody (e′) formed spherical structures similar to S-1 acini (see Fig. 1 a). All cultures were analyzed after 10–12 d inside EHS. Bar, 16 μm.
We therefore examined whether an alteration of α6/β4 signaling in the nonmalignant cells might lead to abrogation of growth control, disruption of morphogenesis and loss of polarity. We used clone 3E1 (Hessle et al., 1984), a β4-integrin mAb that was shown to influence the function of colon adenoma and prostatic carcinoma cells (Jewell et al., 1995), and a protocol similar to that described for the β1-integrin inhibitory antibody studies. Treatment of S-1 cells with this antibody led to the abrogation of normal morphogenesis and loss of growth control. Thus, β4-integrin mAb-treated S-1 cells formed disorganized, large structures similar to T4-2 colonies (Fig. 7 e vs 7 d′). Interestingly the treatment had little or no effect on T4-2 colonies (not shown). The heterodimer partner for β4 is the α6-integrin (Sonnenberg et al., 1988a ). Treatment of nonmalignant cells with GoH3 (Sonnenberg et al., 1988b ), an α6-integrin function-blocking antibody, led to similar results (not shown). These findings suggest that the α6/β4 pathway not only influences morphogenesis but is also involved in growth regulation in nonmalignant MEC's. Furthermore, these results support the notion that the tumorigenic conversion of the HMT-3522 cells may be associated with an alteration in the α6/β4 signaling pathway. If this were so, then treatment of T4-β1 acini with β4 function-altering antibodies (or simultaneous treatment with β1 and β4 function-altering antibodies) should not alter the T4-β1 reverted phenotype. This in fact was found to be the case (not shown).
Discussion
The use of a 3-D BM assay and two related human MEC lines, one phenotypically “normal” and the other malignant, have allowed us to examine the fundamental role of integrins and their intimate relationship to adherens junctions in the regulation of growth and tissue morphogenesis. The results described in this paper demonstrate not only that misregulation of integrins may play a causal role in the expression of malignancy in human epithelial breast cells, but also that manipulations from the cell surface (such as modification of the transmembrane signaling receptors), can restore tissue form and function and reduce tumorigenicity in vivo. Thus, analogous to the effects following retinoid treatment of leukemias, cell surface ECM-receptor manipulations may offer a plausible alternative therapeutic modality for solid epithelial tumors.
Our results showed that the T4-2 tumorigenic cells have increased β1-integrin expression and cell surface levels which are associated with loss of growth control and perturbed morphogenesis. Moreover, a reduction of β1- cell surface integrin activity was found to be sufficient to revert this tumor phenotype. These observations are consistent with the reciprocal relationship shown to exist between proliferation and differentiation and changes in levels of β1-integrins in keratinocyte cells (Jones et al., 1995), and the anchorage-free growth described for kidney cells overexpressing a downstream integrin signaling protein (Frisch et al., 1996). Overexpression and perturbed signaling through β1-integrin heterodimers have previously been reported in primary and metastatic cancers of the breast, colon, skin, and prostate (Cress et al., 1995; Fujita et al., 1995; Leppa et al., 1995; Mortarini et al., 1995; Shaw et al., 1996). Enhanced β1-integrin expression was also shown to be associated with increased tumor aggressiveness, invasiveness, and metastatic potential, and was found to correlate with decreased patient survival (Friedrichs et al., 1995). However, a direct effect of the altered integrin expression on the tumor phenotype was not demonstrated in these studies, although our results may offer a plausible explanation for this relationship. Furthermore, the reported inhibition of invasion and metastasis of bladder and gastric carcinoma cells, by inhibitory β1-integrin antibodies or ArgGly-Asp peptides may be by a mechanism similar to that described in this paper (Saiki et al., 1990; Fujita et al., 1992).
Despite dramatic differences in morphology, both the nonmalignant and tumorigenic HMT-3522 cell lines expressed comparable levels of cell–cell adhesion proteins, but the ability to form cytoskeletal-associated adherens complexes was found to be compromised in the T4-2 cells. This is a condition which has been shown to characterize mammary carcinoma in situ and has been described for several aggressive metastatic breast cancers in vivo and cell lines in culture (Takeichi, 1993; Birchmeirer and Behrens, 1994; Sommers, 1996). By inhibiting tumor cell β1-integrins we were able to rescue adherens junction assembly and revert the tumor phenotype of the T4-2 cells. These results are in agreement with the metastasis-suppressor/epithelial-promoting role of enhanced cell–cell interactions previously shown in metastatic MB-435S/1 breast cells (Frixen et al., 1991) and in HeLa cells (Doyle et al., 1995) transfected with cell adhesion molecules. These observations also emphasize the plasticity of cell–cell junction perturbations, and establish a definite link between integrin signaling, adherens junction assembly and the coordinated formation of a differentiated tissue structure, as has been previously implied by studies in keratinocytes and kidney cells (Hodivala et al., 1994; Ojakian and Schwimmer, 1994; Schoenenberger et al., 1994; Braga et al., 1995). While our findings suggest an integrated mechanism, the key intermediates still need to be identified, although perturbed adherens junction assembly can be restored by kinase inhibitors suggesting down-stream signal transduction molecules are involved (Kinch et al., 1995). The possibility that cytoskeletal proteins such as fascin, a new β-catenin–binding protein which competes against junction assembly, and is thought to interact with the actin cytoskeleton to promote filament bundling and enhance cell motility, may play a role is intriguing and is under investigation (Tao et al., 1996). Also, whether a manipulation of the catenin-E-cadherin pathway would reciprocally affect integrins, or whether direct restoration of adherens assembly would be sufficient to lead to phenotypic reversion and inhibition of tumor formation in this system remain important questions.
Polarity is associated with a basal localization of α6/β4integrin heterodimers in keratinocytes (DeLuca 1994), which has been shown to direct intermediate filament organization (Spinardi et al., 1993; Guo et al., 1995), while its activity correlates with an increase in the level of p21cip,waf-1 (Clarke et al., 1995), a gene important in growth arrest (Harper et al., 1993). Our data showing reorganized intermediate filaments, increased levels of p21cip,waf-1 and polarized α6- and β4-integrins in T4-β1 acini, are consistent with these findings and emphasize the existence of coordinate β1–β4-integrin pathway interactions. Since we showed that α6- and β4-integrin perturbing antibodies were able to disrupt normal MEC cell morphogenesis, this also strongly implicates α6/β4-integrins in establishing mammary gland morphogenesis and tissue organization, as has been previously reported for kidney cells (Sorokin et al., 1990; Matter and Laurie, 1994) and in β4-integrin knockout mice (Dowling et al., 1996). How these interactions occur has yet to be determined although a role for actin and intermediate filament-associated proteins, interacting with the β4-integrin cytoplasmic tail, such as that described for neural BPAG1 should be considered (Yang et al., 1996). Finally, because we were unable to perturb the morphogenesis induced by β1-integrin inhibition in the T4-β1 reverted acini either by sequential or simultaneous incubation with function-altering β4-integrin antibodies, this indicates to us that a genetic defect in a facet of α6/β4-integrin signaling pathway may indeed be responsible for the observed loss of growth control and tissue organization observed in the T4-2 cells. Further experiments will be required to clarify the validity of this argument.
We used a well studied parameter of cell cycle progression, cyclin D1, and of quiescence, p21cip,waf-1, in addition to standard DNA replication and Ki-67 assays, to determine how the reverted tumor cells compared to functionally normal acini. By all these criteria, T4-β1 revertant colonies behaved like growth-arrested S-1 acini. It was also clear from these studies that the formation of a BM accompanied the cessation of growth, confirming our previous findings of an inverse relationship between the presence of a BM and proliferation (Petersen et al., 1992; Howlett et al., 1994; Desprez et al., 1995). It is therefore plausible that the repression of tumorigenicity in vivo, as documented in these studies may have been directly related to the observed growth arrest in culture. Thus, it is reasonable to postulate that it is the balance of signals transduced by the β1- and β4-integrin receptors (or the corresponding α-integrin subunits) that allows these breast cells to sense their microenvironment appropriately and drive tissue organization and function. Therefore, integrins must function in concert with growth factor signaling pathways to determine the decision to grow or to undergo morphogenesis. Very recently, Sastry et al. (1996) overexpressed chimeric α5- and α6/β1-integrins in quail muscle cells and concluded that it is the cytoplasmic domain of these integrins which determines proliferation or growth arrest. Clearly then both direct and indirect evidence in the literature points to the fact that integrins, much like other biological parameters, are cell and tissue context specific. While the detailed molecular mechanism of β1-integrin signaling in breast cells remains to be elucidated, it is safe to conclude that a change in the surface-associated β1-integrin sets into motion global changes in growth and higher order tissue organization. The model system described here offers the opportunity to elucidate how β1- and α6/β4-integrin heterodimers, catenin-cadherin adherens junction proteins, and growth factor pathways communicate within and between epithelial cells in the 3-D tissue.
The demonstrated plasticity of T4-2 cells, their ability to revert to a near normal phenotype and remain quiescent for up to one month in culture, as well as their reduced tumorigenicity in vivo is reminiscent of the “reversion” of mouse teratocarcinoma cells by a normal embryonic microenvironment. In these seminal experiments, embryonal carcinoma cells which were fused with normal blastocysts were able to give rise to phenotypically normal, genetically mosaic mice (Mintz and Illmensee, 1975; for review see Mintz and Fleischman, 1981). While the conclusion of these studies was that the teratocarcinoma cells did not possess cancer-associated genetic defects, we show in our system that despite the presence of genetic defects they could be restrained by the normal tissue structure. A similar dominant, tumor suppressor role of the embryonic microenvironment was shown by our previous work in which active pp60src constructs attached to a lacZ gene, packaged in a replication-defective virus, when injected into stage 24 chick embryos appeared as apparently normal blue cells in a well formed background of feathers and tissues (Stoker et al., 1990). Significantly, once these cells were removed from the embryos, trypsinized and placed in culture, they became rapidly transformed (Boudreau et al., 1995a ). These findings may also provide a possible mechanistic explanation for the poorly understood phenomenon of both spontaneous breast tumor reversion, tumor cell dormancy, and the sporadic occurrence of an auxiliary nodal metastasis in which a true ductal carcinoma in situ (DCIS), complete with a central necrosis and a surrounding intact basement membrane is recapitulated (Barsky et al., 1994).
In conclusion, our results show that changes in tissue structure are critical for the expression of the malignant phenotype and that ECM receptors are important modulators of this effect. Furthermore, these data underscore the concept that it is the relative distribution and proportion of cell surface integrins (undoubtedly in cooperation with other receptors at the cell surface) and their integrated down-stream events which maintain the structural homeostasis of breast and possibly other epithelial tissues. Therefore, tumor cells need not have lost their ability to respond to ECM-generated signals, but rather alterations in the level of the integrated signals can lead to aberrant behavior. Our results may also have a bearing on why some tumor cells retain strong cell-ECM interactions: integrin switching may promote enhanced survival in alternate microenvironments encouraging metastasis.
Most fundamentally, these studies show that despite a number of prominent mutations, amplifications, and deletions, signaling events which are linked to the maintenance of normal tissue architecture are sufficient to abrogate malignancy and to repress the tumor phenotype. It is thus fair to state that cellular and tissue architecture act as the most dominant tumor-suppressor of all, and that the phenotype can—and does—override the genotype as long as the tissue architecture is maintained. These results may explain why breast cancer takes so long to develop even in individuals with inherited susceptibility. Finally, these studies should further aid in the development of therapeutic peptides and antibodies for the treatment of breast and other solid tissue malignancies.
Acknowledgments
We thank S. Dedhar, Z. Werb, G. Riethmueller, D.M. Bissell, J. Ashkenas, C. Roskelley, and J. Muschler for helpful discussions and critical reading of the manuscript and K. Yamada for helpful suggestions. We thank S. Clarke, J. Zhou, and N. Bailey for their technical assistance and B. Johansen for his administrative support, C. Garbarsch for help with the statistics, and R.S. Schwartz for technical and computer advice and support.
Footnotes
This work is based on ideas and research supported by the Office of Health and Environmental Research of the U.S. Department of Energy (currently under contract DE-AC03-SF00098, to M.J. Bissell). More recent efforts have been partially funded by a grant from the National Institutes of Health (CA-64786, to M.J. Bissell). V.M. Weaver was briefly supported by a grant from the U.S. Department of Defense (94MM4558) and subsequently by the Canadian Medical Research Council and is presently funded by the Breast Cancer Fund of the State of California (BCRP University of California-IFB-0400). O.W. Petersen is funded by the Danish Cancer Society (#95-100-44), the Thaysen Foundation, the Novo Foundation, and the Danish Medical Research Council (#9503681).
Please address all correspondence to Dr. Mina J. Bissell, Ernest Orlando Lawrence Berkeley National Laboratory, Mailstop 83-101, One Cyclotron Road, Berkeley, CA 94720. Tel.: (510) 486-4365. Fax: (510) 486-5586.
1. Abbreviations used in this paper: BM, basement membrane; MEC, mammary epithelial cell; 3-D, 3-dimensional.
References
- Adams JC, Watt FM. Regulation of development and differentiation by the extracellular matrix. Development. 1993;117:1183–1198. doi: 10.1242/dev.117.4.1183. [DOI] [PubMed] [Google Scholar]
- Alford D, Taylor-Papadimitriou J. Cell adhesion molecules in the normal and cancerous mammary gland. J Mamm Gland Biol Neo. 1996;1:207–218. doi: 10.1007/BF02013644. [DOI] [PubMed] [Google Scholar]
- Ashkenas J, Muschler J, Bissell MJ. The extracellular matrix in epithelial biology: Shared molecules and common themes in distant phyla. Dev Biol. 1996;180:433–444. doi: 10.1006/dbio.1996.0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcellos-Hoff MH, Aggeler J, Ram TG, Bissell MJ. Functional differentiation and alveolar morphogenesis of primary mammary cultures on reconstituted basement membrane. Development. 1989;105:223–235. doi: 10.1242/dev.105.2.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsky, S.H., S.A. Doberneck, D.A. Grossman, and S.M. Love. 1994. Recapitulation of carcinoma in situ in axillary nodal breast carcinoma metastasis: clinical and biological implications. Breast Cancer Res.Treat. 32(Suppl): 8.
- Berdichevsky F, Wetzels R, Shearer M, Maringnone S, Ramaekers FCS, Taylor-Papdimitriou J. Integrin expression in relation to cell phenotype and malignant change in the human breast. Mol Cell Diff. 1994;2:255–274. [Google Scholar]
- Bergtraesser LM, Weitzman SA. Alterations in integrin and basement membrane protein expression by malignant breast cells. Int J Oncology. 1994;4:915–930. doi: 10.3892/ijo.4.4.915. [DOI] [PubMed] [Google Scholar]
- Bernfield, M., M.T. Hinkes, and R.L. Gallo. 1993. Developmental expression of the syndecans: possible function and regulation. Development. (Suppl):205–212. [PubMed]
- Birchmeier W, Behrens J. Cadherin expression in carcinomas: role in the formation of cell junctions and the prevention of invasiveness. Biochem Biophys Acta. 1993;1198:11–26. doi: 10.1016/0304-419x(94)90003-5. [DOI] [PubMed] [Google Scholar]
- Bissell MJ, Hall HG, Parry G. How does extracellular matrix direct gene expression? . J Theor Biol. 1982;99:31–68. doi: 10.1016/0022-5193(82)90388-5. [DOI] [PubMed] [Google Scholar]
- Blashke RJ, Howlett AR, Desprez P-Y, Petersen OW, Bissell MJ. Cell differentiation by extracellular matrix components. Methods Enzymol. 1994;245:535–556. doi: 10.1016/0076-6879(94)45027-7. [DOI] [PubMed] [Google Scholar]
- Borradori L, Sonnenberg A. Hemidesmosomes: roles in adhesion, signaling and human diseases. Curr Opin Cell Biol. 1996;8:647–656. doi: 10.1016/s0955-0674(96)80106-2. [DOI] [PubMed] [Google Scholar]
- Boudreau N, Reddy ST, Stoker AW, Fairman C, Bissell MJ. The embryonic environment and the extracellular matrix suppress oncogenic transformation by rous sarcoma virus in the chick embryo. Mol Cell Diff. 1995a;3:261–274. [Google Scholar]
- Boudreau N, Sympson CJ, Werb Z, Bissell MJ. Suppression of ICE and apoptosis in mammary epithelial cells by extracellular matrix. Science (Wash DC) 1995b;267:891–893. doi: 10.1126/science.7531366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau N, Werb Z, Bissell MJ. Suppression of apoptosis by basement membrane requires three-dimensional tissue organization and withdrawal from the cell cycle. Proc Natl Acad Sci USA. 1996;93:9309–9313. doi: 10.1073/pnas.93.8.3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braga VMM, Hodivala KJ, Watt FM. Calcium-induced changes in distribution and solubility of cadherins, integrins and their associated cytoplasmic proteins in human keratinocytes. Cell Adhes Commun. 1995;3:201–215. doi: 10.3109/15419069509081287. [DOI] [PubMed] [Google Scholar]
- Briand P, Petersen OW, van Deurs B. A new diploid nontumorigenic human breast epithelial cell line isolated and propagated in chemically defined medium. In Vitro Cell Dev Biol. 1987;23:181–188. doi: 10.1007/BF02623578. [DOI] [PubMed] [Google Scholar]
- Briand P, Nielsen KV, Madsen MW, Petersen OW. Trisomy 7p and malignant transformation of human breast epithelial cells following epidermal growth factor withdrawal. Cancer Res. 1996;56:2039–2044. [PubMed] [Google Scholar]
- Clarke AS, Lotz MM, Chao C, Mercurio AM. Activation of the p21 pathway of growth arrest and apoptosis by the β4 integrin cytoplasmic domain. J Biol Chem. 1995;270:22673–22676. doi: 10.1074/jbc.270.39.22673. [DOI] [PubMed] [Google Scholar]
- Cress AE, Rabinovitz I, Zhu W, Nagle RB. The α6β1 and α6β4 integrins in human prostate cancer progression. Cancer Metastasis Rev. 1995;14:219–228. doi: 10.1007/BF00690293. [DOI] [PubMed] [Google Scholar]
- D'Ardenne AJ, Richman PI, Horton MA, McAuley AE, Jordon S. Co-ordinate expression of the α6 integrin laminin receptor sub-unit and laminin in breast cancer. J Pathol. 1991;165:213–220. doi: 10.1002/path.1711650304. [DOI] [PubMed] [Google Scholar]
- D'Souza B, Taylor-Papadimitriou J. Overexpression of ERBB2 in human mammary epithelial cells signals inhibition of transcription of the E-cadherin gene. Proc Natl Acad Sci USA. 1994;91:7202–7206. doi: 10.1073/pnas.91.15.7202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damsky CH, Werb Z. Signal transduction by integrin receptors for extracellular matrix: cooperative processing of extracellular information. Curr Opin Cell Biol. 1992;4:772–781. doi: 10.1016/0955-0674(92)90100-q. [DOI] [PubMed] [Google Scholar]
- De Luca M, Pellegrini G, Zambruno G, Marchisio PC. Role of integrins in cell adhesion and polarity in normal keratinocytes and human skin pathologies. J Derm. 1994;21:821–828. doi: 10.1111/j.1346-8138.1994.tb03296.x. [DOI] [PubMed] [Google Scholar]
- Dedhar S. Integrin mediated signal transduction in oncogenesis: an overview. Cancer Metastasis Rev. 1995;24:165–172. doi: 10.1007/BF00690289. [DOI] [PubMed] [Google Scholar]
- Desprez P-Y, Hara E, Bissell MJ, Campisi J. Suppression of mammary epithelial cell differentiation by the helix-loop-helix protein Id-1. Mol Cell Biol. 1995;15:3398–3404. doi: 10.1128/mcb.15.6.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhawan J, Farmer SR. Induction of collagen synthesis in response to adhesion and TGF β is dependent on the actin-containing cytoskeleton. Adv Exp Med Biol. 1994;358:159–168. doi: 10.1007/978-1-4615-2578-3_15. [DOI] [PubMed] [Google Scholar]
- Dowling J, Yu QC, Fuchs E. β4 integrin is required for hemidesmosome formation, cell adhesion and cell survival. J Cell Biol. 1996;134:559–572. doi: 10.1083/jcb.134.2.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle JP, Stempak JG, Cowin P, Colman DR, D'Urso D. Protein zero, a nervous system adhesion molecule, triggers epithelial reversion in host carcinoma cells. J Cell Biol. 1995;131:465–482. doi: 10.1083/jcb.131.2.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drubin DG, Nelson WJ. Origins of cell polarity. Cell. 1996;84:335–344. doi: 10.1016/s0092-8674(00)81278-7. [DOI] [PubMed] [Google Scholar]
- Friedrichs K, Ruiz P, Franke F, Gille I, Terpe H-J, Imhof BA. High expression level of α6 integrin in human breast carcinoma is correlated with reduced survival. Cancer Res. 1995;55:901–906. [PubMed] [Google Scholar]
- Frisch SM, Vuori K, Ruoslahti E, Chan-Hui PY. Control of adhesion-dependent cell survival by focal adhesion kinase. J Cell Biol. 1996;134:793–799. doi: 10.1083/jcb.134.3.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frixen UH, Behrens M, Sachs M, Eberle G, Voss B, Warda A, Lochner D, Birchmeier W. E-Cadherin-mediated cell–cell adhesion prevents invasiveness. J Cell Biol. 1991;113:173–185. doi: 10.1083/jcb.113.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita S, Suzuki H, Kinoshita M, Hirohashi S. Inhibition of cell attachment, invasion and metastasis of human carcinoma cells by anti-integrin β1 subunit antibody. Jpn J Cancer Res. 1992;83:1317–1326. doi: 10.1111/j.1349-7006.1992.tb02764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita S, Watanabe M, Kubota T, Teramoto T, Kitajima M. Alteration of expression in integrin β1-subunit correlates with invasion and metastasis in colorectal cancer. Cancer Letters. 1995;91:145–149. doi: 10.1016/0304-3835(95)03735-f. [DOI] [PubMed] [Google Scholar]
- Giancotti FG. Signal transduction by the α6β4 integrin: charting the path between laminin binding and nuclear events. J Cell Sci. 1996;109:1165–1172. doi: 10.1242/jcs.109.6.1165. [DOI] [PubMed] [Google Scholar]
- Giancotti FG, Mainiero F. Integrin-mediated adhesion and signaling in tumorigenesis. Biochem Biophys Acta. 1994;1198:47–64. doi: 10.1016/0304-419x(94)90005-1. [DOI] [PubMed] [Google Scholar]
- Gui GPH, Wells CA, Browne PD, Yeomans P, Jordan S, Puddefoot JR, Vinson GP, Carpenter R. Integrin expression in primary breast cancer and its relation to axillary nodal status. Surgery. 1995;117:102–108. doi: 10.1016/s0039-6060(05)80236-3. [DOI] [PubMed] [Google Scholar]
- Gumbiner BM. Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell. 1996;84:345–357. doi: 10.1016/s0092-8674(00)81279-9. [DOI] [PubMed] [Google Scholar]
- Guo L, Degenstein L, Dowling J, Yu QC, Wollman R, Perman B, Fuchs E. Gene targeting of BPAG1: abnormalities in mechanical strength and cell migration in stratified epithelia and neurologic degeneration. Cell. 1995;81:233–243. doi: 10.1016/0092-8674(95)90333-x. [DOI] [PubMed] [Google Scholar]
- Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- Hessle H, Sakai LY, Hollister DW, Burgeson RE, Engvall E. Basement membrane diversity detected by monoclonal antibodies. Differentiation. 1984;26:49–54. doi: 10.1111/j.1432-0436.1984.tb01372.x. [DOI] [PubMed] [Google Scholar]
- Hodivala KJ, Watt FM. Evidence that cadherins play a role in the downregulation of integrin expression that occurs during keratinocyte terminal differentiation. J Cell Biol. 1994;124:589–600. doi: 10.1083/jcb.124.4.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoschuetzky H, Aberle H, Kemler R. β-Catenin mediates the interaction of the cadherin-catenin complex with epidermal growth factor receptor. J Cell Biol. 1994;127:1375–1380. doi: 10.1083/jcb.127.5.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotchin NA, Gandarillas A, Watt FM. Regulation of cell surface β1 integrin levels during keratinocyte terminal differentiation. J Cell Biol. 1995;128:1209–1219. doi: 10.1083/jcb.128.6.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett AR, Petersen OW, Steeg PS, Bissell MJ. A novel function for the nm23-H1 gene: overexpression in human breast carcinoma cells leads to the formation of basement membrane and growth arrest. J Natl Cancer Inst. 1994;86:1838–1844. doi: 10.1093/jnci/86.24.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett AR, Bailey N, Damsky C, Petersen OW, Bissell MJ. Cellular growth and survival are mediated by β1 integrins in normal human breast epithelium but not in breast carcinoma. J Cell Sci. 1995;108:1945–1957. doi: 10.1242/jcs.108.5.1945. [DOI] [PubMed] [Google Scholar]
- Ingber DE, Prusty D, Sun Z, Betensky H, Wang N. Cell shape, cytoskeletal mechanics, and cell cycle control in angiogenesis. J Biomech. 1995;28:1471–1484. doi: 10.1016/0021-9290(95)00095-x. [DOI] [PubMed] [Google Scholar]
- Jewell K, Kapron-Bras C, Jeevaratnam P, Dedhar S. Stimulation of tyrosine phosphorylation of distinct proteins in response to antibodymediated ligation and clustering of α3 and α6 integrins. J Cell Sci. 1995;108:1165–1174. doi: 10.1242/jcs.108.3.1165. [DOI] [PubMed] [Google Scholar]
- Jones PH, Harper S, Watt FM. Stem cell patterning and fate in human epidermis. Cell. 1995;80:83–93. doi: 10.1016/0092-8674(95)90453-0. [DOI] [PubMed] [Google Scholar]
- Kinch MS, Clark GJ, Der CJ, Burridge K. Tyrosine phosphorylation regulates the adhesions of Ras-transformed breast epithelia. J Cell Biol. 1995;130:461–471. doi: 10.1083/jcb.130.2.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korhonen MJY, Laitenen L, Cooper HM, Quaranta V, Virtanen I. Distribution of the α1-α6 integrin subunits in human developing and term placenta. Lab Invest. 1991;65:347–356. [PubMed] [Google Scholar]
- Leppa S, Heino J, Jalkanen M. Increased glycosylation of β1 integrins affects the interaction of transformed S115 mammary epithelial cells with laminin-1. Cell Growth Diff. 1995;6:853–861. [PubMed] [Google Scholar]
- Madsen MW, Lykkesfeldt AE, Laursen I, Nielsen KV, Briand P. Altered gene expression of c-myc, epidermal growth factor receptor, transforming growth factor-α,and c-erb-B2 in an immortalized human breast epithelial cell line, HMT-3522, is associated with decreased growth factor requirements. Cancer Res. 1992;52:1210–1217. [PubMed] [Google Scholar]
- Matter ML, Laurie GW. A novel laminin E8 cell adhesion site required for lung alveolar formation in vitro. J Cell Biol. 1994;124:1083–1090. doi: 10.1083/jcb.124.6.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyoshi N, Hamaguchi M, Taniguchi S, Nagafuchi A, Tsukita S, Takeichi M. Cadherin-mediated cell–cell adhesion is perturbed by v-src tyrosine phosphorylation in metastatic fibroblasts. J Cell Biol. 1992;118:703–714. doi: 10.1083/jcb.118.3.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintz B, Illmensee K. Normal genetically mosaic mice produced from malignant teratocarcinoma cells. Proc Natl Acad Sci USA. 1975;72:3585–3589. doi: 10.1073/pnas.72.9.3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintz B, Fleischman RA. Teratocarcinomas and other neoplasms as developmental defects in gene expression. Adv Cancer Res. 1981;34:211–278. doi: 10.1016/s0065-230x(08)60243-2. [DOI] [PubMed] [Google Scholar]
- Miyamoto S, Tieramoto H, Coso OA, Gutkind JS, Burbelo PD, Akiyama SK, Yamada KM. Integrin function: molecular hierarchies of cytoskeletal and signaling molecules. J Cell Biol. 1995a;131:791–805. doi: 10.1083/jcb.131.3.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto S, Akiyama SK, Yamada KM. Synergistic roles for receptor occupancy and aggregation in integrin trans-membrane function. Science (Wash DC) 1995b;267:883–885. doi: 10.1126/science.7846531. [DOI] [PubMed] [Google Scholar]
- Moll R, Mitze M, Frixen UH, Birchmeier W. Differential loss of E-cadherin expression in infiltrating ductal and lobular breast carcinomas. Am J Pathol. 1994;143:1731–1742. [PMC free article] [PubMed] [Google Scholar]
- Mortarini R, Gismondi A, Maggioni A, Santoni A, Herlyn M, Anichini A. Mitogenic activity of laminin on human melanoma and melanocytes: difference signal requirements and role of β1 integrins. Cancer Res. 1995;55:4702–4710. [PubMed] [Google Scholar]
- Moyret C, Madsen MW, Cooke J, Briand P, Theillet C. Gradual selection of a cellular clone presenting a mutation at Codon 179 of the p53 gene during establishment of the immortalized human breast epithelial cell line HMT-3522. Exp Cell Res. 1994;215:380–385. doi: 10.1006/excr.1994.1355. [DOI] [PubMed] [Google Scholar]
- Natali PG, Nicotra MR, Botti C, Mottolese M, Bigotti A, Segatto O. Changes in expression of α6β4 integrin heterodimer in primary and metastatic breast cancer. Br J Cancer. 1992;66:318–322. doi: 10.1038/bjc.1992.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathke IS, Hinck L, Swedlow JR, Papkoff J, Nelson WJ. Defining interactions and distributions of cadherin and catenin complexes in polarized epithelial cells. J Cell Biol. 1994;125:1341–1352. doi: 10.1083/jcb.125.6.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen KV, Briand P. Cytogenic analysis of in vitro karyotype evolution in a cell line established from nonmalignant human mammary epithelium. Cancer Genet Cytogenet. 1989;39:103–118. doi: 10.1016/0165-4608(89)90236-7. [DOI] [PubMed] [Google Scholar]
- Nielsen KV, Madsen MW, Briand P. In vitro karyotype evolution and cytogenetic instability in the non-tumorigenic human breast epithelial cell line HMT-3522. Cancer Genet Cytogenet. 1994;78:189–198. doi: 10.1016/0165-4608(94)90089-2. [DOI] [PubMed] [Google Scholar]
- Ochiai A, Akimoto S, Kanai Y, Shibata T, Oyama T, Hirohashi S. c-erb B-2 gene product associates with catenins in human cancer cells. Biochem Biophys Res Commun. 1994;205:73–78. doi: 10.1006/bbrc.1994.2631. [DOI] [PubMed] [Google Scholar]
- Ojakian GK, Schwimmer R. Regulation of epithelial cell surface polarity reversal by β1 integrins. J Cell Sci. 1994;107:561–576. [PubMed] [Google Scholar]
- Petersen OW, Ronnov-Jessen L, Howlett AR, Bissell MJ. Interaction with basement membrane serves to rapidly distinguish growth and differentiation pattern of normal and malignant human breast epithelial cells. Proc Natl Acad Sci USA. 1992;89:9064–9068. doi: 10.1073/pnas.89.19.9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskelley CD, Desprez PY, Bissell MJ. Extracellular matrix-dependent tissue-specific gene expression in mammary epithelial cells requires both physical and biochemical signal transduction. Proc Natl Acad Sci USA. 1994;91:12378–12382. doi: 10.1073/pnas.91.26.12378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskelley CD, Srebrow A, Bissell MJ. A hierarchy of ECM-mediated signalling regulates tissue-specific gene expression. Curr Opin Cell Biol. 1995;7:736–747. doi: 10.1016/0955-0674(95)80117-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossen K, Dahlstrom KK, Mercurio AM, Wewer UM. Expression of the α6β4 integrin by squamous cell carcinomas and basal cell carcinomas: possible relation to invasive potential? . Acta Dermato-Venereologica. 1994;74:101–105. doi: 10.2340/0001555574101105. [DOI] [PubMed] [Google Scholar]
- Saiki T, Murata J, Matsuno K, Ogawa R, Nishi N, Tokura S, Azuma I. Anti-metastatic and anti-invasive effects of polymeric Arg-Gly-Asp (RGD) peptide, poly(RGD), and its analogues. Jpn J Cancer Res. 1990;81:660–667. doi: 10.1111/j.1349-7006.1990.tb02624.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastry SK, Lakonishok M, Thomas DA, Muschler J, Horwitz AF. Integrin α subunit ratios, cytoplasmic domains, and growth factor synergy regulate muscle proliferation and differentiation. J Cell Biol. 1996;133:169–184. doi: 10.1083/jcb.133.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt JW, Piepenhagen PA, Nelson WJ. Modulation of epithelial morphogenesis and cell fate by cell-to-cell signals and regulated cell adhesion. Semin Cell Biol. 1993;4:161–173. doi: 10.1006/scel.1993.1020. [DOI] [PubMed] [Google Scholar]
- Schoenenberger C-A, Zuk A, Zinkl GM, Kendall D, Matlin KS. Integrin expression and localization in normal MDCK cells and transformed MDCK cells lacking apical polarity. J Cell Sci. 1994;107:527–541. doi: 10.1242/jcs.107.2.527. [DOI] [PubMed] [Google Scholar]
- Shaw LM, Chao C, Wewer UM, Mercurio AM. Function of the integrin α6β1 in metastatic breast carcinoma cells assessed by expression of a dominant-negative receptor. Cancer Res. 1996;56:959–963. [PubMed] [Google Scholar]
- Sommers CL. The role of cadherin-mediated adhesion in breast cancer. J Mamm Gland Biol Neo. 1996;1:219–229. doi: 10.1007/BF02013645. [DOI] [PubMed] [Google Scholar]
- Sonnenberg A, Hogervorst F, Osterop A, Veltman FE. Identification and characterization of a novel antigen complex on mouse mammary tumor cells using a monoclonal antibody against platelet glycoprotein Ic. J Biol Chem. 1988a;263:14030–14038. [PubMed] [Google Scholar]
- Sonnenberg A, Modderman PW, Hogervorst F. Laminin receptor on platelets is the integrin VLA-6. Nature (Lond) 1988b;336:487–489. doi: 10.1038/336487a0. [DOI] [PubMed] [Google Scholar]
- Sorokin L, Sonnenberg A, Aumailley M, Timpl R, Ekblom P. Recognition of the laminin E8 cell-binding site by an integrin possessing the α6 subunit is essential for epithelial polarization in developing kidney tubules. J Cell Biol. 1990;111:1265–1273. doi: 10.1083/jcb.111.3.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinardi L, Ren Y-L, Sander R, Giancotti FG. The β4 subunit cytoplasmic domain mediates the interaction of α6β4 integrin with the cytoskeleton of hemidesmosomes. Mol Biol Cell. 1993;4:871–884. doi: 10.1091/mbc.4.9.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strange R, Li F, Saurer S, Burkhardt A, Friis RR. Apoptotic cell death and tissue remodelling during mouse mammary gland involution. Development. 1992;115:49–58. doi: 10.1242/dev.115.1.49. [DOI] [PubMed] [Google Scholar]
- Streuli CH, Bailey N, Bissell MJ. Control of mammary epithelial differentiation: the separate roles of cell-substratum and cell–cell interaction. J Cell Biol. 1991;115:1383–1395. doi: 10.1083/jcb.115.5.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoker AW, Hatier C, Bissell MJ. The embryonic environment strongly attenuates v-src oncogenesis in mesenchymal and epithelial tissues, but not in endothelial. J Cell Biol. 1990;111:217–228. doi: 10.1083/jcb.111.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupack DG, Stewart S, Carter WG, Wayner EA, Wilkins JA. B lymphocyte fibronectin receptors: expression and utilization. Scand J Immunology. 1991;34:761–769. doi: 10.1111/j.1365-3083.1991.tb01601.x. [DOI] [PubMed] [Google Scholar]
- Symington BE, Takada Y, Carter WG. Interaction of integrins α3β1 and α2β1: potential role in keratinocyte intercellular adhesion. J Cell Biol. 1993;120:523–535. doi: 10.1083/jcb.120.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sympson CJ, Talhouk RS, Alexander CM, Chin JR, Clift SM, Bissell MJ, Werb Z. Targeted expression of stromelysin-1 in mammary gland provides evidence for a role of proteinases in branching morphogenesis and the requirement for an intact basement membrane for tissue-specific gene expression. J Cell Biol. 1994;125:681–693. doi: 10.1083/jcb.125.3.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada Y, Puzon W. Identification of a regulatory region of integrin β1 subunit using activating and inhibiting antibodies. J Biol Chem. 1993;268:17597–17601. [PubMed] [Google Scholar]
- Takeichi M. Cadherins in cancer: implications for invasion and metastasis. Curr Opin Cell Biol. 1993;5:806–811. doi: 10.1016/0955-0674(93)90029-p. [DOI] [PubMed] [Google Scholar]
- Tao YS, Edwards RA, Tubb B, Wang S, Bryan J, McCrea PD. β-Catenin associates with the actin-bundling protein fascin in a noncadherin complex. J Cell Biol. 1996;134:1271–1281. doi: 10.1083/jcb.134.5.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren SL, Nelson WJ. Nonmitogenic morphoregulatory action of pp60(v-src) on multicellular epithelial structures. Mol Cell Biol. 1987;7:1326–1337. doi: 10.1128/mcb.7.4.1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt FM, Kubler MD, Hotchin NA, Nicholson LJ, Adams JC. Regulation of keratinocyte terminal differentiation by integrin-extracellular matrix interactions. J Cell Sci. 1993;106:175–182. doi: 10.1242/jcs.106.1.175. [DOI] [PubMed] [Google Scholar]
- Weaver VM, Howlett AR, Langton-Webster B, Petersen OW, Bissell MJ. The development of a functionally relevant cell culture model of progressive human breast cancer. Semin Cancer Biol. 1995;6:175–184. doi: 10.1006/scbi.1995.0021. [DOI] [PubMed] [Google Scholar]
- Weitzman JB, Chen A, Hemler ME. Investigation of the role of β1 integrins in cell–cell adhesion. J Cell Sci. 1995;108:3635–3644. doi: 10.1242/jcs.108.11.3635. [DOI] [PubMed] [Google Scholar]
- Werb Z, Tremble PM, Behrendtsen O, Crowley E, Damsky CH. Signal transduction through the fibronectin receptor induces collagenase and stromelysin gene expression. J Cell Biol. 1989;109:877–889. doi: 10.1083/jcb.109.2.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Dowling J, Yu Q-C, Kouklis P, Cleveland DW, Fuchs E. An essential cytoskeletal linker protein connecting actin microfilaments to intermediate filaments. Cell. 1996;86:655–665. doi: 10.1016/s0092-8674(00)80138-5. [DOI] [PubMed] [Google Scholar]
- Zutter MM, Krigman HR, Santoro SA. Altered integrin expression in adenocarcinoma of the breast. Analysis by in situ hybridization. Am J Pathol. 1993;142:1439–1448. [PMC free article] [PubMed] [Google Scholar]
- Zutter MM, Santoro SA, Staatz WD, Tsung YL. Re-expression of the α2β1 integrin abrogates the malignant phenotype of breast carcinoma cells. Proc Natl Acad Sci USA. 1995;92:7411–7415. doi: 10.1073/pnas.92.16.7411. [DOI] [PMC free article] [PubMed] [Google Scholar]