

Abstract
The receptor tyrosine kinase p185c-neu can be constitutively activated by the transmembrane domain mutation Val664→ Glu, found in the oncogenic mutant p185neu. This mutation is predicted to allow intermolecular hydrogen bonding and receptor dimerization. Understanding the activation of p185c-neu has assumed greater relevance with the recent observation that achondroplasia, the most common genetic form of human dwarfism, is caused by a similar transmembrane domain mutation that activates fibroblast growth factor receptor (FGFR) 3. We have isolated novel transforming derivatives of p185c-neu using a large pool of degenerate oligonucleotides encoding variants of the transmembrane domain. Several of the transforming isolates identified were unusual in that they lacked a Glu at residue 664, and others were unique in that they contained multiple Glu residues within the transmembrane domain. The Glu residues in the transforming isolates often exhibited a spacing of seven residues or occurred in positions likely to represent the helical interface. However, the distinction between the sequences of the transforming clones and the nontransforming clones did not suggest clear rules for predicting which specific sequences would result in receptor activation and transformation. To investigate these requirements further, entirely novel transmembrane sequences were constructed based on tandem repeats of simple heptad sequences. Activation was achieved by transmembrane sequences such as [VVVEVVA]n or [VVVEVVV]n, whereas activation was not achieved by a transmembrane domain consisting only of Val residues. In the context of these transmembrane domains, Glu or Gln were equally activating, while Lys, Ser, and Asp were not. Using transmembrane domains with two Glu residues, the spacing between these was systematically varied from two to eight residues, with only the heptad spacing resulting in receptor activation. These results are discussed in the context of activating mutations in the transmembrane domain of FGFR3 that are responsible for the human developmental syndromes achondroplasia and acanthosis nigricans with Crouzon Syndrome.
The neu oncogene was initially isolated from a rat ethylnitrosourea-induced neuro/glioblastoma and encodes a receptor tyrosine kinase belonging to the epidermal growth factor receptor (EGFR)1 family; the oncogenic mutant, referred to as p185neu, is closely related to its wild-type cellular homologue, referred to as p185c-neu (Shih et al., 1981; Schechter et al., 1984; Bargmann et al., 1986a , b; Dougall et al., 1994). Like other members of the EGFR family, p185c-neu consists of an extracellular ligandbinding domain, a transmembrane domain, and an intracellular tyrosine kinase domain (Drebin et al., 1984; Ullrich and Schlessinger, 1990). As for other receptor tyrosine kinases, ligand binding to the wild-type p185c-neu receptor induces receptor dimerization, leading to tyrosine kinase activation and subsequent downstream signaling events (Peles et al., 1991; Dougall et al., 1994; Stein et al., 1994; for review see Heldin, 1995).
Activation of p185c-neu arises from structural changes, such as the Val664→ Glu mutation in the transmembrane domain (Bargmann et al., 1986b ) or deletions in the extracellular juxtamembrane region (Siegel et al., 1994). Activated p185neu resembles a ligand-stimulated receptor, as evidenced by its increased tyrosine phosphorylation of other proteins, as well as elevated levels of autophosphorylation (Bargmann and Weinberg, 1988a ; Segatto et al., 1988; Stern et al., 1988; Weiner et al., 1989a ; Peles et al., 1991; Ben-Levy et al., 1992; Cao et al., 1992; Qian et al., 1995). In addition, p185neu is primarily found in an aggregated form while wild-type p185c-neu is monomeric (Weiner et al., 1989b ). The mutation Val664→ Glu may facilitate dimerization by allowing intermolecular hydrogen bonding between the side chain of Glu664 in one receptor and the carbonyl oxygen of Ala661 in another receptor, thereby stabilizing the interaction between the two α-helical transmembrane domains (Sternberg and Gullick, 1989). In the initial characterization of p185neu, various mutations at Val664 were examined. Both Glu and Gln were found to be equally activating for transformation, with other potential hydrogen bond-forming residues such as Asp and Tyr exhibiting low activity (Bargmann and Weinberg, 1988b ).
Prior mutagenesis studies have suggested that the primary structure in the vicinity of Glu664 must play a significant role in activation of p185neu. For example, substitution of Glu at the neighboring positions 663 or 665 does not result in activation (Bargmann and Weinberg, 1988b ). The importance of the subdomain surrounding Glu664 has been further examined by the mutations Val663→ Gly and Gly665→ Val, which both abolished transforming activity (Cao et al., 1992). This study also found that the lateral position and rotational orientation of Glu in the transmembrane domain did not correlate with transformation.
A subdomain containing a loosely defined sequence motif has been noted in the transmembrane domains of many other receptor tyrosine kinases (Sternberg and Gullick, 1990). This motif consists of a five-residue segment: position 0 (P0), corresponding to Ala661 in p185c-neu, exhibits a small side chain such as Gly, Ala, Ser, or Thr; position P3 displays an aliphatic side chain like Ala, Val, Leu, or Ile, but is occupied by Glu664 in activated p185neu; and position P4 exhibits either Gly or Ala. The presence of this motif in many different receptor tyrosine kinases suggests an important role in facilitating ligand-induced receptor dimerization.
For many years, p185neu represented the sole example of a receptor tyrosine kinase that is activated by mutation within its transmembrane domain. Recently, however, it has become clear that some human developmental abnormalities are due to transmembrane domain mutations in fibroblast growth factor receptor (FGFR) 3. Achondroplasia, the most common genetic form of human dwarfism, results from a single amino acid change in the transmembrane domain of FGFR3, Gly380→ Arg (Rousseau et al., 1994; Shiang et al., 1994). As shown in work from this laboratory, this mutation results in constitutive activation of FGFR3, not unlike the constitutive activation of p185neu by the mutation Val664→ Glu (Webster and Donoghue, 1996). Achondroplasia is not the only developmental syndrome arising from transmembrane domain mutations in FGFR3, as Meyers et al. (1995) recently identified a novel substitution mutation in FGFR3 that is responsible for two distinct developmental syndromes: acanthosis nigricans in association with Crouzon Syndrome. The existence of these human developmental abnormalities arising from activating mutations in the transmembrane domain of a receptor tyrosine kinase suggests that the paradigm of receptor activation provided by p185neu is relevant to human disease and well worth probing in molecular detail.
In this study, we have used pools of long oligonucleotides encoding the p185c-neu transmembrane domain to target degeneracies to specific positions. This represents a powerful approach for the simultaneous mutation of multiple residues, facilitating the isolation of transforming and nontransforming clones from a large pool of different mutants. In an effort to understand the differences between the transforming and nontransforming clones, we constructed completely novel transmembrane domains and demonstrated that some of these, such as a tandem repeat of [VVVEVVV]n, are able to induce efficient receptor activation as evidenced by transformation of NIH3T3 cells. These results demonstrate that an apparently complex transmembrane domain can be substituted by simple sequences of repeated residues, provided that amino acids capable of hydrogen bonding, such as Glu or Gln, are included at appropriate positions. Significantly, receptor activation by the tandem repeats is accomplished without any specific sequence contribution from the native p185c-neu transmembrane domain. The experiments described here provide a basis for understanding the role of the transmembrane domain in the activation of p185neu, or for the activation of other receptor tyrosine kinases such as FGFR3.
Materials and Methods
Construction of Degenerate Pools and Additional Mutants
Oligonucleotides were synthesized encoding the p185c-neu transmembrane domain with degenerate codons targeted to the presumptive α-helical positions designated “a,” “d,” and “e,” shown in Fig. 1. In Pool 1, codons for Val, Ala, Gly, and Glu were targeted to each of these positions. In Pool 2, the codons for Val, Ala, or Gly, but not Glu, were targeted to these positions. The degenerate oligonucleotides were synthesized on an oligonucleotide synthesizer (Appiled Biosystems Inc., Foster City, CA) and then amplified by PCR. The degenerate pools were ligated into a pSV2-derived vector encoding p185c-neu (pSV2neuNheI/SacI), replacing the wild-type transmembrane domain at silent NheI and SacI restriction sites (Webster and Donoghue, 1996). The NheI site corresponds to bases 1973–1978, and the SacI site corresponds to bases 2114–2119, in the published nucleotide sequence encoding p185c-neu (Bargmann et al., 1986a ).
Figure 1.

Isolates recovered from pools with degenerate transmembrane domains. (A) The sequences of wild-type p185c-neu and oncogenic p185neu (with the mutation Val664→ Glu) are shown. The locations of the presumptive heptad repeats of the transmembrane region are shown, along with the letters indicating the heptad positions. Vertical lines designate the probable borders of the transmembrane domain. The activating Glu residue of p185neu is boldfaced. The five-residue sequence motif found in many receptor tyrosine kinase transmembrane domains (Sternberg and Gullick, 1990), referred to as P0-P4, corresponds to residues 661–665. The location of P0 and P3 are shown. (B) Oligonucleotides coding for degenerate transmembrane domains were synthesized, amplified, and cloned into pSV2neuNheI/SacI as described in Materials and Methods. In the first pool of degenerate oligos, codons for either Ala, Val, Gly, or Glu were randomly targeted to the “a,” “d,” and “e” positions. In Pool 2, codons for Ala, Val, or Gly, but not Glu, were targeted to the same positions. Asterisks denote the positions at which these degeneracies were targeted. (C) Pool 1 was transfected into NIH3T3 cells; individual foci were expanded and used for PCR- mediated recovery of the unique transmembrane domain present in each focus. Each recovered transmembrane domain was ligated into pSV2neuNheI/SacI to ensure that transformation was due to a unique transmembrane domain rather than a mixture of sequences. The sequences of these transforming isolates are shown, with dashes designating the residues that remained unchanged from the parent p185c-neu. Glu residues are boldfaced. (D) For comparison, nontransforming isolates from Pool 1 were also characterized. The sequences of some of these isolates are shown. For A, C, and D, transformation by each isolate was quantitated as a percentage of p185neu. Results represent the average values from three independent experiments, normalized by cotransfection with pSV2neo, and presented as follows: −, 0–5% of p185neu; +, 6–40% of p185neu; ++ , 41–100% of p185neu. For B, transformation by DNA representing the entire pool was recorded as follows: −, 0 foci per 10 μg of DNA; ++, ∼35 foci per 10 μg of DNA.
The synthetic oligonucleotide sequence used to generate the degenerate pools is shown below. Silent restriction sites for NheI (GCTAGC) and SacI (GAGCTC) embedded within the sequence are shown in bold. Extra residues were added at both ends to facilitate restriction digestion of the PCR-amplified double-stranded product. The first and last triplet codons shown correspond to Ala653 (GCT of NheI site) and Leu701 (CTC of SacI site), respectively, in the published amino acid sequence of p185c-neu (Bargmann et al., 1986a ): 5′-GCAGAGA.GCT.AGC.CCG.GTG.ACA. TTC.ATC.ATT.GXA.ACT.GTA .GXA.GXA .GTC.CTG.GXA .TTC.CTG. GXA.GXA.GTG.GTG.GXA.GTT.GGA .GXA.GXA. ATC. AAA.CGA. AGG. AGA.CAG. AAG. ATC.CGG. AAG.TAT.ACG.ATG.CGT. AGG. CTG.CTG.CAG.GAA.ACT.GAG.CTC.GTGGAGCC−3′. Degeneracies were encoded as follows: for Pool 1, X = A, G, C, or T; and for Pool 2, X = G, C, or T. The pools of single-stranded degenerate oligonucleotides were PCR amplified using standard conditions with the following primers: a sense strand primer, corresponding to the 5′-end of the degenerate oligonucleotide sequence shown above and spanning the NheI site, 5′-GCAGAGAGCTAGCCCGGTGAC-3′; and an antisense strand primer, corresponding to the complement of the 3′-end of the degenerate oligonucleotide sequence shown above and spanning the SacI site, 5′-GGCTCCACGAGCTCAGTTTCC-3′.
Additional mutants described in the text were constructed by synthesizing pairs of single-stranded oligonucleotides to create double-stranded DNA fragments with NheI and SacI cohesive overhangs, for ligation into pSV2neuNheI/SacI (Webster and Donoghue, 1996).
Focus Assays
NIH3T3 cells were transfected using a modified calcium phosphate transfection protocol (Chen and Okayama, 1987), as described previously (Maher et al., 1993). Transfection frequencies were determined by cotransfecting with 0.1 μg of pSV2neo, and half of the cells were subsequently split into media containing G418 (Maher et al., 1993). For each DNA sample, the number of foci was normalized with respect to the number of Neoresistant colonies and then expressed as a percentage of the transformation efficiency obtained with pSV2neuNT, encoding p185neu with the mutation Val664→ Glu. All mutants were assayed at least three times in independent experiments for transformation efficiency.
Isolation of Transmembrane Domains from Degenerate Pools
At 9 d after transfection of degenerate Pool 1 into NIH3T3 cells, individual foci of transformed cells were expanded and RNA was isolated. cDNA was prepared using the following primer: 5′-ATACGCTTCATCTAGAATTTCTTTG-3′, complementary to bases 2323–2347 in the published nucleotide sequence encoding p185c-neu (Bargmann et al., 1986a ). This cDNA was used for PCR with primers described above, and the products digested with NheI and SacI. For the mutants designated DEG.1 through DEG.5, each recovered transmembrane domain was ligated into pSV2neuNheI/SacI and sequenced, and its ability to activate p185c-neu to a transforming phenotype was reconfirmed by transfection into NIH3T3 cells. This protocol ensured that transformation was due to a unique sequence, containing a single transmembrane domain, rather than a mixture of sequences. The nontransforming isolates, DEG.6 through DEG.10, were recovered in the same way but were negative for NIH3T3 transformation when assayed as individual clones.
Indirect Immunofluorescence
Transiently transfected Cos-1 cells (Chen and Okayama, 1987) were fixed in 3% paraformaldehyde/PBS for 10 min and then permeabilized in 0.5% Triton X-100/PBS for 5 min (Lee and Donoghue, 1992). Cells were incubated with mouse monoclonal antibody 7.16.4 to an extracellular epitope of p185c-neu (c-neu [AB-4] clone 7.16.4 from Oncogene Science Inc., Manhasset, NY) followed by FITC-conjugated goat anti–mouse antiserum (Boehringer Mannheim Corp., Indianapolis, IN). To detect cell surface expression of p185c-neu and derivatives, cells were fixed with paraformaldehyde and incubated without permeabilization, as described previously (Hannink and Donoghue, 1986; Lee and Donoghue, 1992).
For double-label experiments to detect both intracellular and cell surface expression, cells were fixed as described above and then treated with mouse mAb 7.16.4 (1:50 dilution) against an extracellular epitope of p185c-neu, followed by FITC-conjugated goat anti–mouse antiserum (Boehringer Mannheim Corp.) at 1:1,500 dilution. The same cells were then permeabilized with 1% Triton X-100/PBS and treated with a rabbit polyclonal p185c-neu C-18 antibody (Santa Cruz Biotechnology, Santa Cruz, CA) at 1:3,000 dilution, which was detected with rhodamine-conjugated goat anti–rabbit antiserum (Boehringer Mannheim Corp.) at 1:4,000 dilution.
Immunoprecipitation, In Vitro Kinase, and Dimerization Assays
Cos-1 cells were split at a density of ∼2 × 105 cells per 60-mm plate and transfected with 10 μg of DNA the next day (Chen and Okayama, 1987). After 2 d, the transfected cells were labeled for ∼7 h with 100 μCi of [35S]Cys and [35S]Met each per ml in DME lacking Cys and Met. The cells were rinsed with TS buffer and lysed in 0.5 ml NP-40 lysis buffer (20 mM Tris, pH 7.5, 137 mM NaCl, 1% NP-40, 1 mM sodium orthovanadate, 5 mM EDTA, 10 μg/ml aprotinin, 10% glycerol), and clarified lysates were prepared (Maher et al., 1993). Immunoprecipitation was carried out using mAb 7.16.4, and immune complexes were collected with protein A–Sepharose beads. After immunoprecipitation, the Sepharose beads were washed in NP-40 lysis buffer, and the samples were split in half. Samples for the kinase assay were washed twice in kinase buffer (20 mM Tris, pH 7.5, 10 mM MnCl2, 5 mM MgCl2), resuspended in 20 μl kinase buffer containing 10 μCi γ-[32P]ATP, incubated at room temperature for 10 min, washed twice with NP-40 lysis buffer, and analyzed by 7.5% SDS-PAGE and autoradiography. The other half of the samples was analyzed by 7.5% SDSPAGE, and the 35S-labeled proteins were visualized by fluorography.
To examine dimerization, transfected cells were lysed in RIPA (10 mM sodium phosphate, pH 7.0, 1% Triton X-100, 0.1% SDS, 1% DOC, 150 mM NaCl, 2 mM EDTA, 50 mM NaF, 1% aprotinin, 1 mM sodium orthovanadate, 100 μM PMSF, and 10 mM iodoacetamide) and immunoprecipitated as described above. 2× nonreducing sample buffer (4% SDS, 10 mM sodium phosphate, pH 7.0, 20% glycerol, 0.08% bromophenol blue) was added to the protein A–Sepharose beads and then boiled. For the reducing samples, an aliquot was removed and 2-mercaptoethanol was added to 0.7 M and DTT was added to 20 mM. Samples were boiled again and run on a 4–12% gradient gel. Proteins were then transferred to nitrocellulose, blotted with a rabbit polyclonal p185c-neu C-18 antiserum, and detected by enhanced chemiluminescence (ECL).
To examine phosphotyrosine incorporation in immunoprecipitated receptors, reduced samples prepared as above were run on a 4–12% gradient gel, transferred to nitrocellulose, blotted with mouse antiphosphotyrosine mAb 4G10 (Upstate Biotechnology, Inc., Lake Placid, NY) and detected by ECL. The membrane was then reprobed with polyclonal p185c-neu C-18 antiserum to examine receptor expression, which was again detected by ECL.
Results
Pools of Mutants with Degenerate Transmembrane Domains
The model proposed by Sternberg and Gullick (1989) postulated the transmembrane domain of p185neu as an α-helical sequence in which Glu664 promotes activation by creating a hydrogen bond between two receptor molecules. To further characterize these interactions, we constructed pools of degenerate oligonucleotides that permitted variability in the transmembrane domain at positions P0, P3, and P4 in the Sternberg and Gullick notation, which are predicted to be of greatest structural importance based on a comparison of transmembrane domains from many growth factor receptors (Sternberg and Gullick, 1990). The repeating heptad motif present in interacting α-helical domains can also be designated (abcdefg)n, in which the “a” and “d” positions are occupied by the residues at the interface where the two α-helical domains interact.
In degenerate Pool 1 (Fig. 1 B), we randomly targeted codons for Val, Ala, Gly, or Glu to heptad positions “a,” “d,” and “e.” This choice allowed us to examine the potential effects of multiple Glu residues targeted to a variety of positions. The pool of degenerate oligonucleotides was amplified by PCR and then ligated into a pSV2-derived vector encoding p185c-neu (pSV2neuNheI/SacI), replacing the wild-type transmembrane domain. The DNA was transfected into NIH3T3 cells, and the resulting foci were expanded. Subsequently, to recover individual transmembrane domains from foci of transformed cells, RNA was prepared from expanded cells and subjected to reverse transcriptase–PCR as described in Materials and Methods, and the resulting sequences were then subcloned as unique plasmids. Individual transmembrane domains recovered in this fashion were next ligated back into pSV2neuNheI/ SacI and reassayed for transformation of NIH3T3 cells. This ensured that transformation would be due to a unique sequence, rather than a mixture of transmembrane sequences.
The sequences of the transforming mutants isolated from this pool, designated DEG.1 through DEG.5 (Fig. 1 C), were completely distinct in comparison with other mutants previously described. Interestingly, none of the activated mutants except DEG.1 exhibited a Glu at residue 664. Moreover, all of these mutants, except for DEG.2, exhibited multiple Glu residues spaced at intervals of seven residues, or multiples of seven residues. These results were intriguing considering that Glu residues are rare in hydrophobic transmembrane domains. These transforming isolates provided clear evidence that multiple Glu residues do not disrupt the ability of the transmembrane domain to insert properly into the membrane.
Approximately 15 nontransforming mutants were also isolated from Pool 1, five of which, DEG.6 through DEG.10, are described in Fig. 1 D. The distinctions between the transforming and nontransforming isolates were not entirely obvious. Double-label indirect immunofluorescence was used to determine whether the lack of transforming activity of these mutants might reflect an abnormal subcellular localization. This was accomplished by using an antibody against an extracellular epitope to detect surface expression in cells before permeabilization, followed by a second antibody against the COOH terminus to detect intracellular expression in the same cell after permeabilization (Fig. 2). Fig. 2 B shows the immunofluorescence observed for control cells under identical conditions, with mock-transfected cells negative for both intracellular and surface staining (Fig. 2 B, A and B), whereas cells expressing p185neu exhibit readily detectable staining both intracellularly and also at the cell surface (Fig. 2 B, C and D). As shown in Fig. 2 A, the mutants DEG.7, DEG.9, and DEG.10 were expressed intracellularly but failed to reach the cell surface (Fig. 2 A, D, H, and J), suggesting that their lack of biological activity might be due to aggregation or interaction with some component of the ER/Golgi. In contrast, the mutants DEG.6 and DEG.8 were expressed at the cell surface (Fig. 2 A, B and F), suggesting that their transmembrane domains fail to activate these receptors, despite localizing normally to the plasma membrane.
Figure 2.
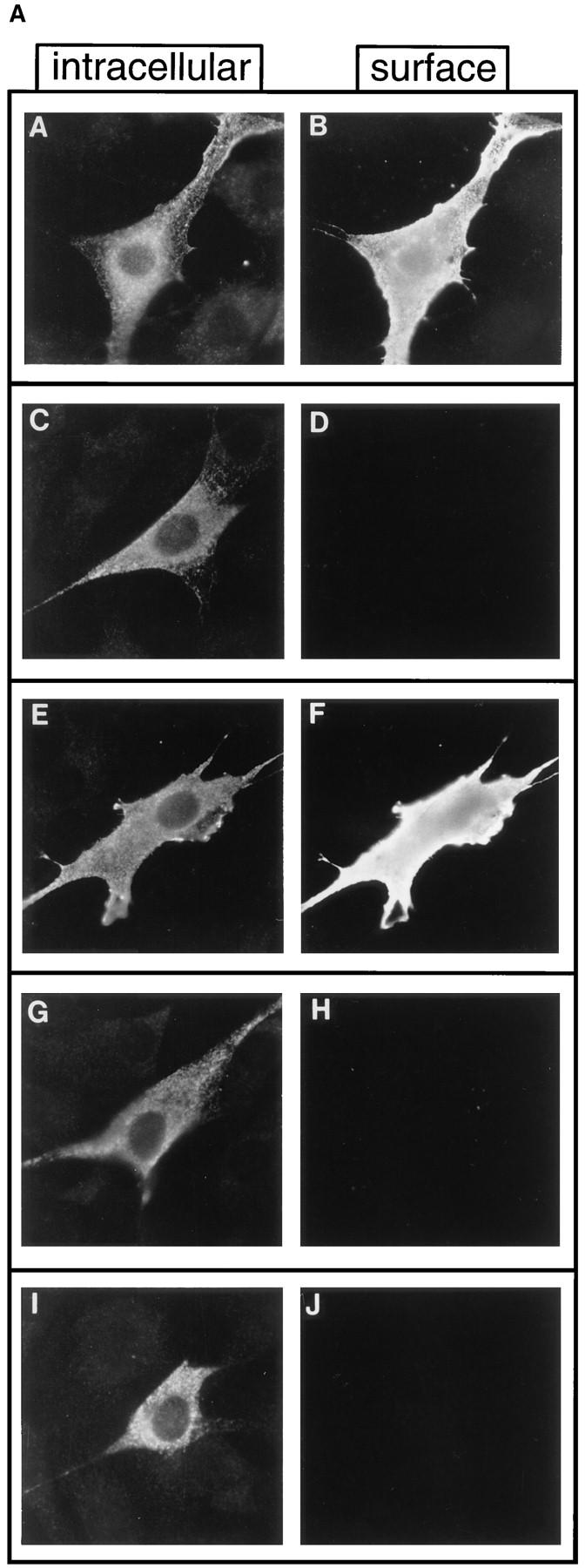

Immunofluorescence of nontransforming isolates DEG.6–DEG.10. Double-label indirect immunofluorescence was used to detect either cell surface expression of p185c-neu-related proteins (right) or, after permeabilization of cells, intracellular expression (left). Conditions for the double-label immunofluorescence are described in Materials and Methods. (A) (A and B) mutant DEG.6; (C and D) mutant DEG.7; (E and F) mutant DEG.8; (G and H) mutant DEG.9; (I and J) mutant DEG.10. (B) (A and B) Mock transfected cells; (C and D) p185neu.
Importantly, no transforming mutants were isolated from degenerate Pool 2 (Fig. 1 B), in which Val, Ala, or Gly, but not Glu, were targeted to heptad positions “a,” “d,” and “e.” Clearly, the presence of Glu residues, which can potentially satisfy a requirement for hydrogen bonding, is a key element in receptor activation.
Design of Transmembrane Domains with Consensus Heptad Repeats
The transmembrane domains of the transforming mutants DEG.1–DEG.5 suggested that there are many allowed positions for Glu residues, yet the distinction between these sequences and those of the nontransforming mutants DEG.6–DEG.10 did not suggest clear rules for predicting which specific sequences will result in receptor activation and transformation. We therefore designed simple consensus repeats, containing a centrally located Glu residue, with most of the other residues substituted by Val, such as [VVVEVVA]n, [VVVEVVG]n, [VVVEVVV]n, or [AVVEGVL]n (designated CONS.A, CONS.B, CONS.C, and CONS.D, respectively). Mutants were then constructed with the transmembrane domain composed entirely of these consensus sequences, repeated over the entire transmembrane domain of ∼25 residues. Surprisingly, these constructs were transforming, with the CONS.A and CONS.D constructs exhibiting the greatest activity (Fig. 3 B). These results suggest that these simple repeating sequences contain all the information necessary for receptor activation. Evidently, there is little sequence specificity necessary for constitutive activation within the p185neu transmembrane domain itself.
Figure 3.

Consensus sequences and derivatives. (A) The p185c-neu and p185neu sequences are shown. The locations of the presumptive heptad repeats, along with the letters indicating the heptad positions, are specified. Vertical lines designate the probable borders of the transmembrane domain. Glu664 is in boldface. (B) Consensus sequence mutants were constructed as described in the text. The placement of the Glu residues in each mutant is identical. Based on the variations found in the transforming isolates from degenerate Pool 1, the heptad position g in CONS.B and CONS.C was mutated from Ala in CONS.A to Gly and Val, respectively. The repeating heptad in CONS.D is heptad 2 from p185neu with Thr662 changed to Val. The mutant CONS.CE→ V has a transmembrane domain composed entirely of Val residues and was constructed as a negative control. Glu residues are boldfaced. (C) Derivatives of CONS.A mutant. Derivatives were designed in which the Glu residues were substituted by other residues capable of hydrogen bonding, such as Lys, Gln, Ser, or Asp. For example, CONS.AE→ Q is identical to CONS.A except that the Glu residues are substituted by Gln. Transformation by each isolate was quantitated as a percentage of p185neu. Results represent the average values from three independent experiments, normalized by cotransfection with pSV2neo, and presented as −, +, or ++ as described in Fig. 1. Surface expression was determined by indirect immunofluorescence, as described in text.
It was also important to demonstrate that transformation was due to specific interactions involving Glu residues. Towards this end, we constructed a derivative of the CONS.C sequence in which the central Glu of each repeat was changed to Val. This mutant transmembrane domain, consisting entirely of Val residues, was designated CONS.CE→ V, and was completely devoid of transforming activity (Fig. 3 B).
Design of Derivative Transmembrane Domains Based on Consensus Heptad Repeats
We also tested whether other residues could substitute for Glu in the context of the CONS.A heptad repeat (Fig. 3 C). We replaced Glu with Lys, Gln, Ser, and Asp, creating the mutants CONS.AE→ K, CONS.AE→ Q, CONS.AE→ S, and CONS.AE→ D. These mutants were designed to test whether other hydrogen-bonding residues could substitute in place of Glu. Only substitution with Gln allowed significant transforming activity, whereas Lys, Ser, and Asp were unable to activate the receptor. The observation that Gln can substitute for Glu in the mutant CONS.AE→ Q is consistent with the original characterization of p185neu showing that Val664→ Glu and Val664→ Gln were equally effective at activation (Bargmann and Weinberg, 1988b ).
As shown in Fig. 4, A–L, indirect immunofluorescence demonstrated that most of the mutants exhibited cell surface expression. The ability of these altered receptors to reach the surface indicates that the mutant transmembrane domains did not interfere with the proper localization of the protein. The only exception was CONS.AE→ D, which exhibited little or no cell surface staining, even though the protein is clearly being translated, as shown by the intracellular staining of permeabilized cells (Fig. 4, O). However, its reduced surface expression may be due to aggregation or interaction with another protein that is retained in the ER and/or Golgi, as discussed previously for some of the nontransforming mutants, DEG.7, DEG.9, and DEG.10.
Figure 4.
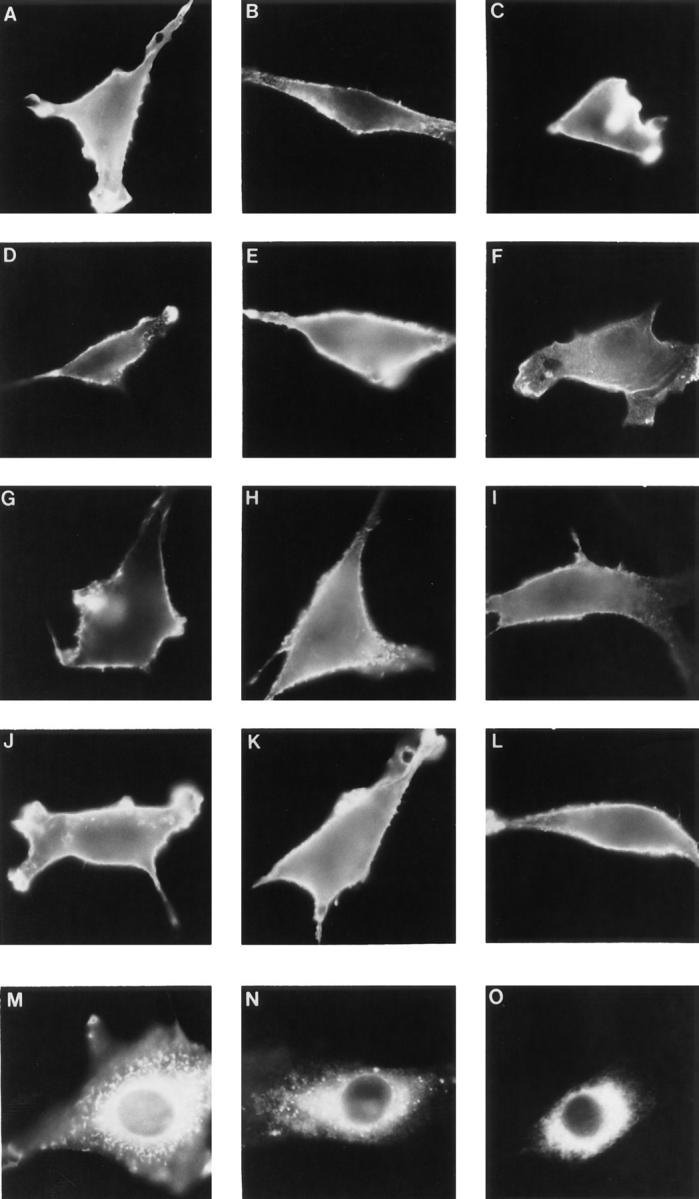
Indirect immunofluorescence of consensus mutants and derivatives. Indirect immunofluorescence using the monoclonal antibody 7.16.4 directed against the extracellular region of p185c-neu and a fluorescein-conjugated goat anti– mouse secondary antibody revealed both cell surface and intracellular protein expression. A–L show nonpermeabilized cells to examine cell surface expression. (A) p185c-neu; (B) p185neu; (C) CONS.A; (D) CONS.B; (E) CONS.C; (F) CONS.D; (G) CONS.AE2; (H) CONS.AE4; (I) CONS.AE→ K; (J) CONS.AE→ Q; (K) CONS. AE→ S; (L) CONS.CE→ V. M–O show cells after permeabilization to examine intracellular expression. (M) p185c-neu; (N) p185neu; (O) CONS.AE→ D.
Spacing and Number of Glu Residues
The design of the degenerate pools of transmembrane domains, as well as the design of the subsequent consensus repeat clones described above, assumed without any real evidence that there would exist a fundamental heptad structural motif resulting from the α-helical transmembrane domain. To directly examine this premise, we constructed a series of clones in which we varied the spacing between Glu residues. As the parental clone for this series, we chose a clone with two Glu residues spaced seven residues apart in a transmembrane domain composed otherwise of only Val residues, designated CONS.C2xE. The spacing between the two Glu residues was then varied from two to eight residues, as shown in Fig. 5 A. The results of transformation assays with these clones revealed a dramatic effect of spacing, with the heptad spacing yielding significantly greater transforming activity than any of the other clones (Fig. 5 B). When cells expressing these constructs were examined by indirect immunofluorescence, all constructs exhibited cell surface expression (data not shown), indicating that the lack of transformation was not due to a defect in surface localization. This experiment provides compelling evidence for a basic heptad structural motif in the transmembrane domain of activated forms of p185c-neu.
Figure 5.


Importance of the spacing of Glu residues. (A) A series of clones, based on the mutant CONS.C2xE, were constructed varying the spacing between two Glu residues, from a minimum spacing of two residues (diad mutant) to a maximum spacing of eight residues (octad mutant). (B) Transformation by each isolate shown in (A) was quantitated as a percentage of p185neu. Numerical results of transformation assays are presented graphically and represent the average values from two independent experiments, normalized by cotransfection with pSV2neo.
Most of the transforming DEG mutants described earlier contained multiple Glu residues (Fig. 1 C), as did the transforming mutants CONS.A–CONS.D, which all contained three Glu residues (Fig. 3 B). The occurrence of multiple Glu residues is in contrast to p185neu, which contains only a single Glu residue in the transmembrane domain at position 664. We therefore examined whether multiple Glu residues are in fact required for activity.
This issue was addressed through two different sets of mutants, one constructed in the background of CONS.A, [VVVEVVA], and another set in the background of CONS.C, [VVVEVVV]. These two sets of mutants yielded somewhat different results. As shown in Fig. 6 A, “singleGlu” derivatives of the CONS.A sequences were designed, each possessing only a single Glu residue located at different positions, and designated CONS.AE2, CONS.AE3, and CONS.AE4. None of these mutants was transforming, indicating that within the context of the CONS.A heptad repeat, a single-Glu residue is not sufficient to provide activation. This result is in contrast to the single Glu664 residue that leads to activation of p185neu, suggesting that there may be sequence information within the transmembrane domain, in addition to the Val664→ Glu mutation, that stabilizes dimerization (Cao et al., 1992). We considered the possibility that these mutant proteins might fail to reach the cell surface and that this defect might explain their inactivity in transformation assays. However, all of the single-Glu proteins were expressed at the cell surface, as shown by indirect immunofluorescence in Fig. 4, G and H, for CONS.AE2 and CONS.AE4.
Figure 6.

Importance of the number of Glu residues. (A) Derivatives of CONS.A. Three derivatives were constructed, each having a single Glu residue. Depending upon the position of the Glu residue, these mutants were designated CONS.AE2, CONS.AE3, and CONS.AE4. (B) Derivatives of CONS.C. Derivatives were constructed in the background of CONS.C having from one to four Glu residues. These clones are designated CONS.C1xE, CONS.C2xE, CONS.C3xE, and CONS.C4xE. Note that CONS.C3xE is the parental CONS.C mutant presented in Fig. 3. Transformation by each isolate was quantitated as a percentage of p185neu. Results represent the average values from three independent experiments, normalized by cotransfection with pSV2neo, and presented as −, +, or ++ as described in Fig. 1. For transformation results shown in B, numerical values are also shown in parentheses as the percentage of p185neu.
Several mutants were also designed in the CONS.C background that had either one, two, three, or four Glu residues, designated CONS.C1xE–CONS.C4xE. The clone containing three Glu residues, CONS.C3xE, is the original CONS.C clone. As shown in Fig. 6 B , the mutants with either two or four Glu residues, designated CONS.C2xE and CONS.C4xE, exhibited the greatest activity, almost twice as much as the mutant with three Glu residues. However, in this series of mutants, even the presence of a single Glu residue, as in CONS.C1xE, led to significant transformation above background.
It is curious that a single Glu residue is activating in the mutant CONS.C1xE (Fig. 6 B), but not in the very similar mutant CONS.AE2 (Fig. 6 A). The active mutant has a background CONS.C motif of [VVVEVVV], whereas the inactive mutant has a background CONS.A motif of [VVVEVVA]; these sequences differ only by the Val→ Ala substitution in the last position of the repeat motif. This suggests that the ability of a single Glu residue to activate a particular transmembrane domain, as occurs in the case of p185neu, may be critically dependent upon other specific details of the transmembrane domain that are not immediately obvious. These results may help to explain the negative results of earlier studies in which repositioning of the Glu664 residue to other locations in the transmembrane domain did not result in transformation (Cao et al., 1992).
Kinase Activity of Mutants
Transformation by the neu oncogene is dependent upon p185neu having a functional kinase domain, and p185neu exhibits increased turnover, autophosphorylation, tyrosine phosphorylation of intracellular substrates, and activation of PLC-γ (Bargmann and Weinberg, 1988a ; Stern et al., 1988; Weiner et al., 1989a ,b; Peles et al., 1991; Cao et al., 1992; Brown et al., 1994). Moreover, transformation and SH2-dependent signaling by p185neu requires intermolecular receptor association that is mediated by the transmembrane domain, as demonstrated by functional complementation between truncated kinase-active p185neu and full-length kinase-inactive p185neu (Qian et al., 1994, 1995). Another indication of receptor dimerization and aggregation is provided by measurement of ligand-binding using chimeras consisting of the ligand-binding domain of EGFR substituted into either p185c-neu or p185neu; the Val→ Glu mutation in the transmembrane domain of such EGFR/Neu chimeras results in the conversion of low-affinity ligandbinding sites into high-affinity binding sites, consistent with an oligomerized state of the oncogenic receptor (Ben-Levy et al., 1992).
To confirm kinase activation of the mutants described here, selected mutants were examined for receptor activation using an immunoprecipitation/kinase assay of transfected Cos-1 cells. As shown in Fig. 7 A, p185neu exhibited approximately threefold greater autophosphorylation than p185c-neu (lanes 3 and 2, respectively). The mutants CONS.CE→ V and CONS.C were examined, as they represent an interesting pair of closely related mutants. The first of these is inactive in transformation assays, whereas the latter mutant is active. In the immunoprecipitation/kinase assay, the biologically active mutant CONS.C exhibited a similar increase in autophosphorylation compared with the inactive receptor CONS.CE→ V (lanes 5 and 4, respectively). We also examined another pair of closely related mutants, CONS.AE→ S and CONS.AE→ Q, where once again the first mutant is inactive in transformation assays, but the latter mutant is active. Once again, in the immunoprecipitation/kinase assay, the biologically active mutant CONS.AE→ Q exhibited an increase in autophosphorylation compared with the inactive mutant CONS.AE→ S (lanes 7 and 6, respectively). In this experiment, similar levels of protein expression were achieved for p185c-neu, p185neu, and the various mutants examined, as demonstrated by 35S-metabolically labeled/immunoprecipitated proteins from the same lysates (Fig. 7 B). Thus, consistent with prior experimental results from other laboratories (Bargmann and Weinberg, 1988a ; Stern et al., 1988; Weiner et al., 1989a ,b; Cao et al., 1992), biologically active derivatives constructed in this work exhibited increased levels of kinase activity, as determined by receptor autophosphorylation in immunoprecipitation/kinase assays.
Figure 7.
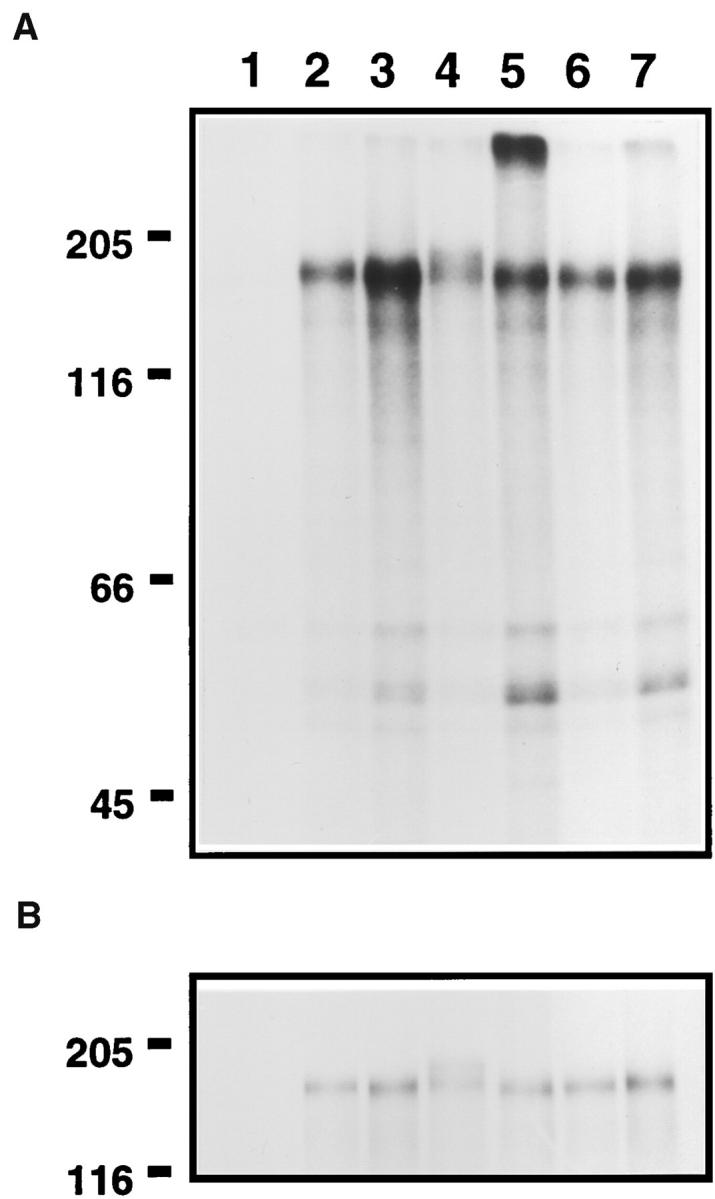
Immunoprecipitation/kinase assay of consensus mutants. Lysates from transfected cells, labeled metabolically with [35S]Cys and [35S]Met, were subjected to immunoprecipitation using monoclonal antibody 7.16.4, as described in Materials and Methods. (A) Kinase assay. Immunoprecipitated lysates were subjected to in vitro kinase reactions using γ-[32P]ATP. 32P-labeled proteins were detected by SDS-PAGE and autoradiography. Exposure time was 21 h. (B) Expression. To demonstrate equivalent levels of protein expression for different mutants, identical aliquots of immunoprecipitated lysates as used in A were analyzed by SDS-PAGE followed by fluorography to detect 35S-labeled proteins. Exposure time was 3 d. The samples shown in lanes 2, 4, and 6 represent nontransforming clones, while the samples shown in lanes 3, 5, and 7 represent transforming derivatives. Lane 1, mock; lane 2, p185c-neu; lane 3, p185neu; lane 4, CONS.CE→ V; lane 5, CONS.C; lane 6, CONS.AE→ S; lane 7, CONS.AE→ Q.
Immunoprecipitated samples were also examined for the presence of phosphotyrosine by immunoblotting. Fig. 8 A demonstrates that two transforming mutants, CONS.C (lane 5) and CONS.AE→ Q (lane 7), exhibit significant incorporation of phosphotyrosine into immunoprecipitated receptors, as did the positive control, p185neu (lane 3). Little or no phosphotyrosine was associated with the nontransforming mutants, CONS.CE→ V (lane 4) and CONS.AE→ S (lane 6), or with the nontransforming control, p185c-neu (lane 2). As a control for receptor expression levels, B demonstrates approximately equivalent levels of receptor expression, determined by immunoblotting using polyclonal p185c-neu C-18 antiserum.
Figure 8.

Detection of phosphotyrosine in immunoprecipitated receptors. Lysates were prepared from transfected cells and were subjected to immunoprecipitation using monoclonal antibody 7.16.4, as described in Materials and Methods. Immunoprecipitated lysates were analyzed on a 4–12% gradient gel under reducing conditions, transferred to nitrocellulose, probed with monoclonal phosphotyrosine antiserum 4G10 (A) or with polyclonal p185c-neu C-18 antiserum (B), and specific proteins were detected by ECL. The samples shown in lanes 2, 4, and 6 represent nontransforming clones, while the samples shown in lanes 3, 5, and 7 represent transforming derivatives. A shows detection of phosphotyrosine, and B shows detection of receptor expression. Samples are: lane 1, mock; lane 2, p185c-neu; lane 3, p185neu; lane 4, CONS.CE→ V; lane 5, CONS.C; lane 6, CONS.AE→ S; lane 7, CONS.AE→ Q.
These same mutants were also examined for receptor dimerization using gradient gel electrophoresis under nonreducing conditions (Weiner et al., 1989b ; Burke et al., 1997). Fig. 9 B presents these results, while the same lysates were also analyzed under reducing conditions as a control, shown in A. Two transforming mutants, CONS.C (lane 5) and CONS.AE→ Q (lane 7), exhibited significant bands of dimeric receptors, as did the positive control, p185neu (lane 3). Importantly, no dimerization was observed for the nontransforming mutants analyzed here, CONS.CE→ V (lane 4) and CONS.AE→ S (lane 6), or for the nontransforming control, p185c-neu (lane 2). The ability to detect receptor dimerization for the transforming mutants, but not for the biologically inactive mutants, indicates that the consensus sequences described here promote receptor dimerization, and that in this respect they are similar to the parental transforming clone, p185neu.
Figure 9.

Dimerization assay of consensus mutants. Lysates were prepared from transfected cells in RIPA containing 10 mM iodoacetamide and were subjected to immunoprecipitation using monoclonal antibody 7.16.4, as described in Materials and Methods. Immunoprecipitated lysates were analyzed on a 4–12% gradient gel and transferred to nitrocellulose, probed with a polyclonal p185c-neu C-18 antiserum, and specific proteins were detected by ECL. The samples shown in lanes 2, 4, and 6 represent nontransforming clones, while the samples shown in lanes 3, 5, and 7 represent transforming derivatives. A shows samples analyzed under reducing gel electrophoresis conditions. B shows samples analyzed under nonreducing gel electrophoresis conditions. Samples are: Lane 1, mock; lane 2, p185c-neu; lane 3, p185neu; lane 4, CONS.CE→ V; lane 5, CONS.C; lane 6, CONS.AE→ S; lane 7, CONS.AE→ Q.
Discussion
Function of the Transmembrane Domain
The results presented here demonstrate that activation of p185c-neu can be achieved by a transmembrane domain with very little sequence specificity. Glu residues in the appropriate positions of a simplified transmembrane domain, such as [VVVEVVV]n, lead to receptor activation as demonstrated by biological transformation (Fig. 3), increased kinase activity (Fig. 7), increased incorporation of phosphotyrosine (Fig. 8), and receptor dimerization (Fig. 9). Receptor activation is conferred by as few as one or as many as four Glu residues within some of these repeats. Moreover, a periodic heptad spacing seems to be critical for activation; this was demonstrated by systematically varying the spacing between two Glu residues from two to eight, with only the heptad spacing resulting in activation (Fig. 5 B). Thus, an apparently complex system can be reduced to a repeated sequence motif. Furthermore, in these simplified transmembrane domains, Gln is able to substitute efficiently for Glu, as shown by the mutant CONS.AE→ Q. This is consistent with previous observations (Bargmann and Weinberg, 1988b ) that Gln, but not Ser, Lys, or Asp, was able to substitute for Glu at position 664 in receptor activation (Fig. 3).
From the transforming isolates of degenerate Pool 1, it is clear that the location of Glu at position 664 is not critical for activation (Fig. 1), in contrast to prior results suggesting that the primary structure around Glu664 plays an important role. For instance, mutants with Glu663 or Glu665 were inactive (Bargmann and Weinberg, 1988b ). More recently, Cao et al. (1992) analyzed the requirements for activation of the p185neu transmembrane domain; all of their transforming mutants retained Glu at position 664, and a mutant (designated lil670VEG) with the motif VEG seven residues COOH-terminal, was nontransforming.
Our isolation of the transforming mutants DEG.2, DEG.3, DEG.4, and DEG.5, all of which lack a Glu at position 664, would suggest that there exists considerable flexibility with regard to the allowed placement of the Glu residue(s) that may mediate activation. The isolation of these transforming mutants also demonstrates that the construction of a degenerate pool of oligonucleotides encoding transmembrane domains provides a powerful approach to identifying and isolating those rare sequences that may allow receptor activation.
Our data show that activation of p185c-neu can be mediated by Glu residues spaced seven amino acids apart in a repeating heptad. Analysis of the sequences of the isolates from Pool 1 (Fig. 1) is consistent with this proposal. Most of the transforming isolates contain Glu residues separated by multiples of seven amino acids. In the nontransforming isolate, DEG.6, which was expressed at the cell surface (in contrast to DEG.7 and DEG.10), there are Glu residues spaced seven amino acids apart, but there is also a Glu that does not follow that pattern. This may suggest that the presence of a Glu in a nonheptad motif may cause a disruption in the interaction between receptor molecules that prevents transforming ability, although it is difficult to infer absolute rules because of the other sequence variations exhibited by the DEG mutants.
The Val→ Glu mutation that activates p185neu is also activating when introduced into the corresponding position of the Drosophila EGFR homologue (DER), resulting in increased kinase activity (Wides et al., 1990). Although it was initially reported that the corresponding Val627→ Glu mutation does not activate the human EGFR transmembrane domain (Kashles et al., 1988), a more recent study suggests that this mutation is activating (Beguinot et al., 1995). Additionally, a Val→ Glu mutation at position 659 in c-erbB-2, the human Neu homologue that is overexpressed in many breast cancers, results in elevated tyrosine kinase activity (Segatto et al., 1988). These examples provide further evidence for activation of EGFR family members by appropriate introduction of a strongly polar residue in their transmembrane domains.
The hydrophobic environment of the membrane creates an energetic need to shield polar side chains such as that of Glu (even if uncharged), which can be accomplished by appropriate hydrogen bonding. Previously, it was proposed that there may be a requirement for Ala661 in p185neu to allow hydrogen bonding between the Glu664 side chain of one receptor with the carbonyl oxygen of Ala661 in the other. There is clearly no strict requirement for Ala at position 661, however, as shown by the fact that Val is tolerated at position 661 in many of the biologically active mutants described here. The presence of the branched side chain at Val661, which might interfere with side chain– backbone hydrogen bonding, argues against this model but does not exclude it. More likely, however, would be a model of side chain–side chain hydrogen bonding between the Glu of one subunit with the corresponding Glu of the other subunit. The choice between these two models will require further biophysical characterization. While the results presented in this work are certainly consistent with a coiled-coil arrangement for the interacting transmembrane domains of p185neu, other structural models cannot be excluded at the present time, such as one proposed for the homodimerizing transmembrane domain of glycophorin A (Lemmon et al., 1992; Treutlein et al., 1992).
Relevance of p185neu Activation to Other Systems
As summarized in Fig. 10, prior studies have demonstrated that substitutions at position 664 of p185neu exhibit the following pattern of biological activity: Glu, Gln > Asp, Tyr ≫ Val, Lys, Gly, His (Bargmann and Weinberg, 1988b ). These results are generally consistent with a role of polar or hydrophilic residues in promoting activation, although the failure of Lys and His to allow for activation remains to be explained. The E5 oncoprotein of bovine papilloma virus (BPV) provides another example of the importance of the transmembrane domain for activation, since this 44residue integral membrane protein requires dimerization and disulfide bond formation for its biological activity (Horwitz et al., 1988, 1989; Goldstein et al., 1992). A key requirement of its hydrophobic transmembrane domain is the presence of Gln17, which appears to participate in interhelical hydrogen bonding and may be substituted by either Glu or Lys, and to a lesser extent by His (Meyer et al., 1994). Significantly, the transmembrane domain of E5 can be largely replaced by the membrane-spanning region from activated p185neu (Meyer et al., 1994).
Figure 10.

Oncoproteins and receptors activated by mutations in the transmembrane domain. Amino acids are shown that allow activation of p185c-neu when substituted at residue 664, activation of BPV-E5 when substituted at residue 17, and activation of FGFR3 when substituted at residue 380, as discussed in the text. Those substitutions that allow activation in these three different systems share the property that they are strongly polar in an otherwise hydrophobic membrane environment, and thus share the ability to participate in hydrogen bond formation that may stabilize dimer formation.
The transmembrane domain of the T cell receptor α chain (TCRα) also appears to play a key role in complex formation between TCRα and the CD3γ chain, mediated by two basic residues in the TCRα transmembrane domain (Cosson et al., 1991). As another relevant example, the basic residues within the transmembrane domain of gp41 in human immunodeficiency virus type 1 (HIV-1) have been suggested to play a role in oligomerization of the HIV-1 envelope glycoprotein (Owens et al., 1994).
Transmembrane Domain Mutations in Human Developmental Syndromes
Transmembrane-mediated receptor activation clearly plays a fundamental role in several human developmental syndromes. Achondroplasia, the most common genetic form of human dwarfism, results from a single amino acid change in the transmembrane domain of FGFR3, Gly380→ Arg (Rousseau et al., 1994; Shiang et al., 1994). This mutation results in constitutive activation of FGFR3, leading to abnormal development at the growth plate of long bones (Webster and Donoghue, 1996). As depicted in Fig. 10, we have previously demonstrated that mutations at residue 380 of FGFR3 exhibit the following pattern of activation: Arg, Glu, Asp > Gln, His ≫ Lys (Webster and Donoghue, 1996). Recently, Meyers et al. (1995) characterized an autosomal dominant mutation in FGFR3 that is manifested as two distinct developmental syndromes presented together: first, acanthosis nigricans, a hyperplastic epithelial proliferation syndrome resulting in thickened hyperpigmented skin; and second, Crouzon Syndrome, characterized by craniosynostosis or premature fusion of the cranial sutures, resulting in cranial malformation and ocular proptosis. The mutation responsible for these developmental anomalies maps to the transmembrane domain of FGFR3 and, surprisingly, involves a single substitution mutation creating a Glu residue (Ala391→ Glu). Using FGFR3/Neu chimeras, we have recently demonstrated that the Ala391→ Glu mutation also leads to constitutive receptor activation (Webster, M.K., and D.J. Donoghue, unpublished data).
It is clear that transmembrane domains play a more substantial role than serving as structural elements for membrane anchoring. The examples discussed above underscore the importance of understanding the molecular details of activating mutations in the transmembrane domains of receptors. The principles elucidated here are likely to be generally relevant to understanding activation and signal transduction by other receptor tyrosine kinases in addition to p185neu.
Acknowledgments
We thank Robert Weinberg (Massachusetts Institute of Technology, Cambridge, MA) for providing the pSV2neuN and pSV2neuNT plasmids. We thank Laura Castrejon for excellent editorial assistance, Brendan Galvin for excellent technical support and advice, and all lab members for their many valuable comments and suggestions concerning experimental design and preparation of the manuscript. We also thank David Stern and Chris Burke (Yale University, New Haven, CT) for advice about the dimerization assay.
This work was supported by grant CA 40573 from the National Institutes of Health, grant 1RB-0318 from the University of California Breast Cancer Research Program, and grants 3RT-0242 and 4FT-0193 (to M.K. Webster) from the University of California Tobacco Related Disease Research Program.
Footnotes
1. Abbreviations used in this paper: BPV, bovine papilloma virus; ECL, enhanced chemiluminescence; EGFR, epidermal growth factor receptor; FGFR, fibroblast growth factor receptor; TCRα, T cell receptor α chain.
Address all correspondence to Daniel J. Donoghue, Department of Chemistry and Biochemistry, and Center for Molecular Genetics, University of California, San Diego, La Jolla, California 92093-0367. Tel.: (619) 534-2167. Fax: (619) 534-7481.
References
- Bargmann CI, Weinberg RA. Increased tyrosine kinase activity associated with the protein encoded by the activated neuoncogene. Proc Natl Acad Sci USA. 1988a;85:5394–5398. doi: 10.1073/pnas.85.15.5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargmann CI, Weinberg RA. Oncogenic activation of the neuencoded receptor protein by point mutation and deletion. EMBO (Eur Mol Biol Organ) J. 1988b;7:2043–2052. doi: 10.1002/j.1460-2075.1988.tb03044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargmann CI, Hung M-C, Weinberg RA. The neuoncogene encodes an epidermal growth factor receptor-related protein. Nature (Lond) 1986a;319:226–230. doi: 10.1038/319226a0. [DOI] [PubMed] [Google Scholar]
- Bargmann CI, Hung M-C, Weinberg RA. Multiple independent activations of the neuoncogene by a point mutation altering the transmembrane domain of p185. Cell. 1986b;45:649–657. doi: 10.1016/0092-8674(86)90779-8. [DOI] [PubMed] [Google Scholar]
- Beguinot, L., W.C. Vass, and M. Miloso. 1995. SHC is constitutively activated by a transforming EGF receptor mutant with a point mutation in the transmembrane domain. J. Cell Biochem. 19A(Suppl.):11–11.
- Ben-Levy R, Peles E, Goldman-Michael R, Yarden Y. An oncogenic point mutation confers high affinity ligand binding to the neureceptor. J Biol Chem. 1992;267:17304–17313. [PubMed] [Google Scholar]
- Brown VI, Shah N, Smith R, Hellman M, Jarett L, Mikami Y, Cohen E, Qian X, Greene MI. Demonstration by two-color flow cytometry that tyrosine kinase activity is required for down-modulation of the oncogenic neu receptor. DNA Cell Biol. 1994;13:193–209. doi: 10.1089/dna.1994.13.193. [DOI] [PubMed] [Google Scholar]
- Burke CL, Lemmon MA, Coren BA, Engelman DM, Stern DF. Dimerization of the p185neu transmembrane domain is necessary but not sufficient for transformation. Oncogene. 1997;14:687–696. doi: 10.1038/sj.onc.1200873. [DOI] [PubMed] [Google Scholar]
- Cao H, Bangalore L, Bormann BJ, Stern DF. A subdomain in the transmembrane domain is necessary for p185neu*activation. EMBO (Eur Mol Biol Organ) J. 1992;11:923–932. doi: 10.1002/j.1460-2075.1992.tb05131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Okayama H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol. 1987;7:2745–2752. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosson P, Lankford SP, Bonifacino JS, Klausner RD. Membrane protein association by potential intramembrane charge pairs. Nature (Lond) 1991;351:414–416. doi: 10.1038/351414a0. [DOI] [PubMed] [Google Scholar]
- Dougall WC, Qian X, Greene MI. Interaction of the neu/p185 and EGF receptor tyrosine kinases: implications for cellular transformation and tumor therapy. J Cell Biochem. 1993;53:61–73. doi: 10.1002/jcb.240530108. [DOI] [PubMed] [Google Scholar]
- Dougall WD, Qian X, Peterson NC, Miller MJ, Samanta A, Greene MI. The neu-oncogene: signal transduction pathways, transformation mechanisms, and evolving therapies. Oncogene. 1994;9:2109–2123. [PubMed] [Google Scholar]
- Drebin JA, Stern DF, Link VC, Weinberg RA, Greene MI. Monoclonal antibodies identify a cell-surface antigen associated with an activated cellular oncogene. Nature (Lond) 1984;312:545–548. doi: 10.1038/312545a0. [DOI] [PubMed] [Google Scholar]
- Goldstein DJ, Kulke R, DiMaio D, Schlegel R. A glutamine residue in the membrane-associating domain of the bovine papillomavirus type 1 E5 oncoprotein mediates its binding to a transmembrane component of the vacuolar H+-ATPase. J Virol. 1992;66:405–413. doi: 10.1128/jvi.66.1.405-413.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannink M, Donoghue DJ. Cell surface expression of membraneanchored v-sisgene products: glycosylation is not required for cell surface transport. J Cell Biol. 1986;103:2311–2322. doi: 10.1083/jcb.103.6.2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldin CH. Dimerization of cell surface receptors in signal transduction. Cell. 1995;80:213–223. doi: 10.1016/0092-8674(95)90404-2. [DOI] [PubMed] [Google Scholar]
- Horwitz BH, Burkhardt AL, Schlegel R, DiMaio D. 44-aminoacid E5 transforming protein of bovine papillomavirus requires a hydrophobic core and specific carboxyl-terminal amino acids. Mol Cell Biol. 1988;8:4071–4078. doi: 10.1128/mcb.8.10.4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz BH, Weinstat DL, DiMaio D. Transforming activity of a 16-amino-acid segment of the bovine papillomavirus E5 protein linked to random sequences of hydrophobic amino acids. J Virol. 1989;63:4515–4519. doi: 10.1128/jvi.63.11.4515-4519.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashles O, Szapary D, Bellot F, Ullrich A, Schlessinger J, Schmidt A. Ligand-induced stimulation of epidermal growth factor receptor mutants with altered transmembrane regions. Proc Natl Acad Sci USA. 1988;85:9567–9571. doi: 10.1073/pnas.85.24.9567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BA, Donoghue DJ. Intracellular retention of membraneanchored v-sisprotein abrogates autocrine signal transduction. J Cell Biol. 1992;118:1057–1070. doi: 10.1083/jcb.118.5.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon MA, Flanagan JM, Treutlein HR, Zhang J, Engelman DM. Sequence specificity in the dimerization of transmembrane α-helices. Biochemistry. 1992;31:12719–12725. doi: 10.1021/bi00166a002. [DOI] [PubMed] [Google Scholar]
- Maher DW, Strawn LM, Donoghue DJ. Alanine mutagenesis of conserved residues in the platelet-derived growth factor family: identification of residues necessary for dimerization and transformation. Oncogene. 1993;8:533–541. [PubMed] [Google Scholar]
- Meyer AN, Xu Y-F, Webster MK, Smith AE, Donoghue DJ. Cellular transformation by a transmembrane peptide—structural requirements for the bovine papilloma virus E5 oncoprotein. Proc Natl Acad Sci USA. 1994;91:4634–4638. doi: 10.1073/pnas.91.11.4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers GA, Orlow SJ, Munro IR, Przylepa KA, Jabs EW. Fibroblast growth factor receptor 3 (FGFR3) transmembrane mutation in Crouzon syndrome with acanthosis nigricans. Nat Genet. 1995;11:462–464. doi: 10.1038/ng1295-462. [DOI] [PubMed] [Google Scholar]
- Owens RJ, Burke C, Rose JK. Mutations in the membrane-spanning domain of the human immunodeficiency virus envelope glycoprotein that affect fusion activity. J Virol. 1994;68:570–574. doi: 10.1128/jvi.68.1.570-574.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peles E, Levy RB, Or E, Ullrich A, Yarden Y. Oncogenic forms of the neu/HER2 tyrosine kinase are permanently coupled to phospholipase Cγ. EMBO (Eur Mol Biol Organ) J. 1991;10:2077–2086. doi: 10.1002/j.1460-2075.1991.tb07739.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Dougall WC, Hellman ME, Greene MI. Kinase-deficient neu proteins suppress epidermal growth factor receptor function and abolish cell transformation. Oncogene. 1994;9:1507–1514. [PubMed] [Google Scholar]
- Qian X, Dougall WC, Fei Z, Greene MI. Intermolecular association and trans-phosphorylation of different neu-kinase forms permit SH2dependent signaling and oncogenic transformation. Oncogene. 1995;10:211–219. [PubMed] [Google Scholar]
- Rousseau F, Bonaventure J, Legeai-Mallet L, Pelet A, Rozet J-M, Maroteaux P, Le Merrer M, Munnich A. Mutations in the gene encoding fibroblast growth factor receptor-3 in achondroplasia. Nature (Lond) 1994;371:252–254. doi: 10.1038/371252a0. [DOI] [PubMed] [Google Scholar]
- Schechter AL, Stern DF, Vaidyanathan L, Decker SJ, Drebin JA, Greene MI, Weinberg RA. The neu oncogene: an erb-B-related gene encoding a 185,000 Mrtumour antigen. Nature (Lond) 1984;312:513–516. doi: 10.1038/312513a0. [DOI] [PubMed] [Google Scholar]
- Segatto O, King CR, Pierce JH, Di Fiore PP, Aaronson SA. Different structural alterations upregulate in vitro tyrosine kinase activity and transforming potency of the erbB-2 gene. Mol Cell Biol. 1988;8:5570–5574. doi: 10.1128/mcb.8.12.5570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiang R, Thompson LM, Zhu Y-Z, Church DM, Feilder TJ, Bocian M, Winokur ST, Wasmuth JJ. Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell. 1994;78:335–342. doi: 10.1016/0092-8674(94)90302-6. [DOI] [PubMed] [Google Scholar]
- Shih C, Padhy LC, Murray M, Weinberg RA. Transforming genes of carcinomas and neuroblastomas introduced into mouse fibroblasts. Nature (Lond) 1981;290:261–264. doi: 10.1038/290261a0. [DOI] [PubMed] [Google Scholar]
- Siegel PM, Dankort DL, Hardy WR, Muller WJ. Novel activating mutations in the neu proto-oncogene involved in induction of mammary tumors. Mol Cell Biol. 1994;14:7068–7077. doi: 10.1128/mcb.14.11.7068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein D, Wu J, Fuqua SAW, Roonprapunt C, Yajnik V, D'Eustachio P, Moskow JJ, Buchberg AM, Osborne CK, Margolis B. The SH2 domain protein GRB-7 is co-amplified, overexpressed and in a tight complex with HER2 in breast cancer. EMBO (Eur Mol Biol Organ) J. 1994;13:1331–1340. doi: 10.1002/j.1460-2075.1994.tb06386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern DF, Kamps MP, Cao H. Oncogenic activation of p185neustimulates tyrosine phosphorylation in vivo. Mol Cell Biol. 1988;8:3969–3973. doi: 10.1128/mcb.8.9.3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg MJE, Gullick WJ. Neu receptor dimerization. Nature (Lond) 1989;339:587. doi: 10.1038/339587a0. [DOI] [PubMed] [Google Scholar]
- Sternberg MJE, Gullick WJ. A sequence motif in the transmembrane region of growth factor receptors with tyrosine kinase activity mediates dimerization. Protein Eng. 1990;3:245–248. doi: 10.1093/protein/3.4.245. [DOI] [PubMed] [Google Scholar]
- Treutlein HR, Lemmon MA, Engelman DM, Brunger AT. The glycophorin A transmembrane domain dimer: sequence-specific propensity for a right-handed supercoil of helices. Biochemistry. 1992;31:12726–12733. doi: 10.1021/bi00166a003. [DOI] [PubMed] [Google Scholar]
- Ullrich A, Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell. 1990;61:203–212. doi: 10.1016/0092-8674(90)90801-k. [DOI] [PubMed] [Google Scholar]
- Webster MK, Donoghue DJ. Constitutive activation of fibroblast growth factor receptor 3 by the transmembrane domain point mutation found in achondroplasia. EMBO (Eur Mol Biol Organ) J. 1996;15:520–527. [PMC free article] [PubMed] [Google Scholar]
- Weiner DB, Kokai Y, Wada T, Cohen JA, Williams WV, Greene MI. Linkage of tyrosine kinase activity with transforming ability of the p185neu oncoprotein. Oncogene. 1989a;4:1175–1183. [PubMed] [Google Scholar]
- Weiner DB, Liu J, Cohen JA, Williams WV, Greene MI. A point mutation in the neuoncogene mimics ligand induction of receptor aggregation. Nature (Lond) 1989b;339:230–231. doi: 10.1038/339230a0. [DOI] [PubMed] [Google Scholar]
- Wides RJ, Zak NB, Shilo B-Z. Enhancement of tyrosine kinase activity of the Drosophilaepidermal growth factor receptor homolog by alterations of the transmembrane domain. Eur J Biochem. 1990;189:637–645. doi: 10.1111/j.1432-1033.1990.tb15532.x. [DOI] [PubMed] [Google Scholar]