

Abstract
Integrins α3β1 and α6β4 are abundant receptors on keratinocytes for laminin-5, a major component of the basement membrane between the epidermis and the dermis in skin. These integrins are recruited to distinct adhesion structures within keratinocytes; α6β4 is present in hemidesmosomes, while α3β1 is recruited into focal contacts in cultured cells. To determine whether differences in localization reflect distinct functions of these integrins in the epidermis, we studied skin development in α3β1-deficient mice. Examination of extracellular matrix by immunofluorescence microscopy and electron microscopy revealed regions of disorganized basement membrane in α3β1-deficient skin. Disorganized matrix was first detected by day 15.5 of embryonic development and became progressively more extensive as development proceeded. In neonatal skin, matrix disorganization was frequently accompanied by blistering at the dermal-epidermal junction. Laminin-5 and other matrix proteins remained associated with both the dermal and epidermal sides of blisters, suggesting rupture of the basement membrane itself, rather than detachment of the epidermis from the basement membrane as occurs in some blistering disorders such as epidermolysis bullosa. Consistent with this notion, primary keratinocytes from α3β1-deficient skin adhered to laminin-5 through α6 integrins. However, α3β1-deficient keratinocytes spread poorly compared with wild-type cells on laminin-5, demonstrating a postattachment requirement for α3β1 and indicating distinct roles for α3β1 and α6β4. Our findings support a novel role for α3β1 in establishment and/or maintenance of basement membrane integrity, while α6β4 is required for stable adhesion of the epidermis to the basement membrane through hemidesmosomes.
Integrins are heterodimeric, transmembrane proteins consisting of an α and a β subunit that are receptors for cell adhesion to the extracellular matrix (ECM)1 or to other cells (Hynes, 1992). In the epidermis, basal keratinocytes adhere to the basement membrane through integrins. Human keratinocytes express a number of integrins, including α2β1, α3β1, α5β1, α6β4, and αvβ5 (reviewed in Watt and Hertle, 1994). Integrin expression is normally restricted to the basal, proliferative cell layer, both in the epidermis and in stratified cultures of keratinocytes (Peltonen et al., 1989; Carter et al., 1990a ,b; Hertle et al., 1991; Adams and Watt, 1991). When keratinocytes of the basal layer withdraw from the cell cycle and become committed to terminal differentiation, they detach from the basement membrane and migrate into the suprabasal layers. This process appears to involve both changes in the ligandbinding activities of certain integrins and changes in expression of integrins on the cell surface (Adams and Watt, 1990; Hotchin and Watt, 1992), and may involve complex interactions between integrins and other classes of adhesion receptors, such as cadherins (Hodivala and Watt, 1994). Changes in integrin expression and function may also be important for keratinocytes during wound healing (Clark, 1990; Cavani et al., 1993), psoriasis (Pelligrini et al., 1992; Carroll et al., 1995), and tumorigenesis (Tennenbaum et al., 1992).
The integrins α3β1 and α6β4 are abundant in keratinocytes and function as cell adhesion receptors for laminin-5 (Niessen et al., 1994; Delwel et al., 1994), a member of the laminin family of ECM proteins that was originally described as kalinin (Rousselle et al., 1991), nicein (Verrando et al., 1988), and a component of epiligrin (Carter et al., 1991). Laminin-5 consists of three distinct, multidomain chains designated α3, β3, and γ2 (Burgeson et al., 1994), and is a major component of the epidermal basement membrane. Keratinocyte adhesion to laminin-5 is critical for maintenance of the dermal-epidermal junction in the skin, since mutations in laminin-5 present in inherited junctional epidermolysis bullosa (JEB), and anti-laminin-5 antibodies present in acquired autoimmune diseases such as cicatricial pemphigoid cause blistering of the epidermis from the basement membrane (Domloge-Hultsch, 1992; Aberdam et al., 1994; Pulkkinen et al., 1994a ,b; Uitto et al., 1994; Kivirikko et al., 1995; Kirtschig et al., 1995).
Despite their common functions as laminin-5 receptors, α3β1 and α6β4 are recruited to distinct cell adhesion structures. α6β4 is a component of hemidesmosomes (Stepp et al., 1990; Jones et al., 1991), the adherens junctions that anchor basal keratinocytes of the epidermis to the basement membrane; α6β4 thereby links laminin-5 anchoring filaments outside the cell with the keratin filament network inside the cell. The distribution of α6β4 to hemidesmosomes is reflected in keratinocytes cultured on laminin-5, where α6β4 localizes to hemidesmosome-like structures called stable anchoring contacts (Carter et al., 1990b , 1991). In contrast, α3β1 is recruited to focal contacts in keratinocytes and other cells in culture, and thereby links the ECM to components of the actin cytoskeleton (Carter et al., 1990b ; Grenz et al., 1993; DiPersio et al., 1995). These differences in localization between α3β1 and α6β4 appear to reflect differences in adhesion-related functions. For example, α6β4 mediates stable anchorage of keratinocytes to the substrate, while α3β1 appears to function in cell spreading and migration (Carter et al., 1990b ; Xia et al., 1996). Furthermore, cell adhesion through α6β4 or α3β1 results in tyrosine phosphorylation of distinct sets of cellular proteins (Kornberg et al., 1991; Mainiero et al., 1995; Jewell et al., 1995; Xia et al., 1996), indicating the activation of distinct signal transduction pathways via these integrins.
In addition to its function as a receptor for laminin-5, α3β1 has been implicated as a weak receptor for a variety of other ECM proteins, including fibronectin, collagens, laminin-1, entactin/nidogen, and thrombospondin (Wayner and Carter, 1987; Gehlsen et al., 1988; Tomaselli et al., 1990; Elices et al., 1991; Dedhar et al., 1992; Wu et al., 1995; DeFreitas et al., 1995; DiPersio et al., 1995). Although the physiological significance of these weaker interactions is not known, some of these ECM proteins are present in the epidermal basement membrane (Martin, 1987; Timpl, 1989) or in the provisional matrix during wound healing (Raugi et al., 1987; Clark, 1990; Cavani et al., 1993) and, therefore, may be ligands for α3β1 in skin.
Mutation or altered expression of components of hemidesmosomes and the keratin filament network have been shown to be important for cell adhesion and/or mechanical integrity at the dermal-epidermal junction. These include either subunit of integrin α6β4 (Georges-Labouesse et al., 1996; van der Neut et al., 1996; Dowling et al., 1996), bullous pemphigoid antigen 1 (BPAG1; Guo et al., 1995), BPAG2 (McGrath et al., 1995), and keratin 14 (Coulombe et al., 1991). In contrast, the role of integrin α3β1 in skin has been unclear. We have studied the skin of α3β1-deficient mice generated by null mutation of the gene encoding the α3 integrin subunit (Kreidberg et al., 1996). Mice homozygous for the α3-null mutation die shortly after birth, possibly due to defects in kidney and/or lung organogenesis (Kreidberg et al., 1996). In this report, we used immunofluorescence and ultrastructural analysis to show that the epidermal basement membrane becomes disorganized during skin development in α3-null mice. By birth, matrix disorganization was correlated with blistering of the epidermis from the dermis. Surprisingly, all basement membrane proteins tested localized to both the epidermal and dermal sides of these blisters, identifying a novel form of epidermal blistering caused by rupture of the basement membrane itself rather than by detachment of the epidermis from basement membrane ligands. Consistent with this blistering mechanism, primary keratinocytes from α3-null, neonatal mice could still attach to laminin-5 through α6 integrins; however, α3β1 was required for postattachment spreading of keratinocytes on laminin-5. Our results support distinct but overlapping roles, rather than redundant functions, for α3β1 and α6β4 in keratinocytes and reflect distinct roles for these integrins in the epidermis.
Materials and Methods
Antibodies
Rabbit antisera against the cytoplasmic domains of the human β1 subunit and the chicken α3A subunit were prepared as described (Marcantonio and Hynes, 1988; Hynes et al., 1989; DiPersio et al., 1995). Rabbit antiserum against the cytoplasmic domain of the α6A subunit, and the monoclonal antibody 346-11A against the β4 subunit, were kindly provided by V. Quaranta (Scripps Research Institute, La Jolla, CA) and Stephen Kennel (Oak Ridge National Laboratory, Oak Ridge, TN), respectively. The monoclonal antibody GoH3 against the α6 integrin subunit was purchased from Immunotech (Westbrook, ME). Rabbit antisera against laminin-5 and type VII collagen were gifts from R. Burgeson (Massachusetts General Hospital, Charlestown, MA). Rabbit antiserum against entactin was a gift from A. Chung (University of Pittsburgh, Pittsburgh, PA). Rabbit antiserum against rat plasma fibronectin was prepared as described (Mautner and Hynes, 1977). Rabbit antiserum specific for the EIIIB segment of fibronectin was raised against an EIIIB-GST fusion protein and immunopurified, as described (Peters et al., 1995).
Genotyping of Mice by PCR
Embryonic or neonatal offspring were collected from matings of mice heterozygous for a null mutation of the α3 integrin gene (Kreidberg et al., 1996), and their tails were removed and digested overnight at 56°C in 0.2% SDS, 100 mM Tris, pH 6.8, 200 mM NaCl, 5 mM EDTA, and 0.1 mg/ ml proteinase K. DNA was precipitated with isopropanol, dissolved in 50 μl–100 μl of 10 mM Tris (pH 7.5) and 0.1 mM EDTA. PCR analysis was carried out using 1 μl of template DNA and the following oligonucleotide primers: wild-type primer (from α3 gene): 5′-CCGTCTATGTCTTCATGAACC-3′; α3-knockout primer (neomycin-resistance gene): 5′-GGGGAACTTCCTGACTAG-3′; and common primer (α3 gene): 5′-GGAATCCATCCTGGTTGATGTC. PCR reaction conditions were as follows: denaturation at 94°C for 30 s; extension at 55°C for 40 s; and annealing at 72°C for 30 s. Thirty amplification cycles were performed. The wild-type and common primers amplified a 130-bp fragment from the wild-type α3 gene; and the α3-knockout and common primers amplified a 285-bp fragment from the targeted allele. Amplified PCR products were separated by electrophoresis on a 1.5% agarose gel and visualized by staining with ethidium bromide.
Preparation of Frozen Sections and Immunofluorescence
Limbs were removed from neonatal mice after sacrifice by CO2 narcosis, and then embedded and frozen in O.C.T. compound and stored at −80°C. Embryos were collected by Cesarean section at days 11.5, 15.5, or 17.5 postcoitum, and limbs or torso skin (day 17.5) or whole embryos (days 11.5 and 15.5) were embedded as described above. 8-μm sections were cut on a Reichert-Jung cryostat (model 2800 Frigocut-E), and were either stained with hematoxylin and eosin or prepared for immunofluorescence. For immunofluorescence, frozen sections were hydrated in PBS plus 0.9 mM CaCl2 and 0.5 mM MgCl2 (PBS/Ca2+Mg2+) for 5 min. Since recognition of fibronectin by the anti-EIIIB serum is inhibited by N-linked oligosaccharides, sections to be stained with anti-EIIIB were incubated at 37°C for 2 h with N-glycanase (PNGase F, New England Biolabs, Beverly, MA) at 50,000 U/ml in reaction buffer provided by the manufacturer, as described previously (Peters and Hynes, 1996). Sections were fixed in 3.7% formaldehyde for 10 min and then incubated in blocking buffer (0.1% BSA, 0.2% Triton X-100, 0.1% glycine in PBS/ Ca2+Mg2+) for at least 30 min. Sections were stained with rabbit antisera (diluted in blocking buffer at 70 μg/ml for immunopurified anti-EIIIB, or at 1:100 for other antisera) for at least 1 h, followed by biotin-conjugated goat anti–rabbit Ig (1:200 dilution; TAGO Biosource, Camarillo, CA) for 30 min and then avidin-FITC (1:100 dilution; Sigma Chemical Co., St. Louis, MO) for 30 min. For double-label immunofluorescence, a monoclonal antibody against either the α6 integrin subunit (GoH3) or the β4 integrin subunit (346-11A) was included in the incubation with primary antibodies, followed by Texas red–conjugated goat anti–rat IgG (Oncogene Science, Manhasset, NY) at a concentration of 1 μg/ml. Representative fields were photographed on a Zeiss Axiophot microscope (Thornwood, NY). Control experiments showed that neither bleed-through of fluorescein to Texas red nor bleed-through in the opposite direction occurs using our conditions.
Ultrastructural Analysis of the Epidermal Basement Membrane
Limbs from neonatal mice were prepared as described above, and feet were removed and prepared for electron microscopy as follows: samples were fixed in 2.5% glutaraldehyde/2.5% paraformaldehyde in 0.1 M cacodylate buffer, pH 7.2, for 1 h at 4°C, washed in 0.1 M cacodylate buffer, postfixed in 2% osmium tetroxide in cacodylate buffer for 2 h at 4°C, and then washed in cacodylate buffer. Samples were then dehydrated in ethanol and propylene oxide and embedded in Spurr resin for 48 h at 60°C. Ultrathin sections (70 nm) were cut on a Leica microtome, mounted onto 200 mesh copper grids, stained with uranyl acetate and lead citrate, and viewed on a JEOL 1200EX electron microscope (Peabody, MA).
Isolation and Culture of Primary Mouse Keratinocytes
Epidermal keratinocytes were prepared from neonatal mice essentially as described previously (Dlugosz et al., 1995). Briefly, newborn mice were sacrificed by CO2 narcosis, washed in 10% iodine solution (Mallinckrodt, Paris, KY) in PBS for 10 min, rinsed with PBS, washed in 70% ethanol for 10 min, and then rinsed in PBS. Tails were removed and used for genotyping by PCR; limbs were removed and used to prepare frozen skin sections (see above). Skins were removed from the torso and head, and then floated on 0.25% trypsin solution (GIBCO BRL, Gaithersburg, MD) overnight at 4°C, with the epidermis facing upward. Skins were then transferred to a dry, sterile surface with the epidermis facing down, and the dermis was separated from the epidermis and discarded. The epidermis was minced, suspended in growth medium (see below), and agitated to release keratinocytes. Suspensions were passed through a sterile, 70-μm nylon filter (Becton Dickinson, Mountain View, CA) to remove cornified sheets. Keratinocytes were seeded onto tissue culture plates coated with 30 μg/ml denatured rat tail collagen (Collagen Corp., Palo Alto, CA) at a density of ∼2–4 × 105 cells/cm2. To prevent differentiation of keratinocytes, cultures were grown in low calcium medium consisting of Eagle's Minimum Essential Medium (EMEM; BioWhittaker, Walkersville, MD) supplemented with 4% FBS (Intergen, Purchase, NY) from which Ca2+ had been removed by chelation (Brennen et al., 1982), 0.05 mM CaCl2, “HICE” mix (0.5 μg/ml hydrocortisone [Calbiochem-Novabiochem Corp., La Jolla, CA], 5 μg/ml insulin [Sigma], 10−10 M cholera toxin [ICN Biomedicals, Inc., Costa Mesa, CA], 10 ng/ml epidermal growth factor [Upstate Biotechnology, Lake Placid, NY], 2 × 10−9 M T3 [Sigma]), 100 U/ml penicillin and 100 μg/ml streptomycin (GIBCO BRL). Mouse keratinocytes were cultured at 34°C, 7.5% CO2 for 5–7 d before use in experiments.
Iodination and Immunoprecipitation of Integrins
Mouse keratinocyte cultures were grown for 7 d, and monolayers were surface-labeled with 0.5 mCi/10-cm plate of Na[125I] (New England Nuclear, Boston, MA) using the lactoperoxidase-glucose oxidase method (Hynes, 1973). Cells were washed four times with 50 mM NaI in PBS/ Ca2+Mg2+ and lysed for 15 min on ice in 1 ml of a detergent buffer containing 200 mM octyl-β-d-glucopyranoside (Calbiochem), 50 mM Tris, pH 7.5, 150 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, 2 mM PMSF (Sigma), 0.02 mg/ml aprotinin (Sigma), and 0.0125 mg/ml leupeptin (Calbiochem). Lysates were sedimented for 10 min at 10,000 g. Supernatants were preincubated with 100 μl of protein A–Sepharose (1:1 slurry; Pharmacia LKB, Piscataway, NJ) for 1 h and the beads sedimented for 2 min at 10,000 g. Protein concentrations of supernatants were determined using a Bio-Rad kit, and equal amounts of protein were immunoprecipitated with antiintegrin antibodies as described (Marcantonio and Hynes, 1988). Briefly, BSA was added to lysates (180 μg total protein) to a final concentration of ∼3 mg/ml, followed by 5–10 μl of antiserum. After incubation at 4°C for 1 h, 50 μl of protein A–Sepharose (1:1 slurry preabsorbed with 10 mg/ml BSA in lysis buffer) was added to reactions. Reactions were incubated overnight at 4°C. Samples were washed four times with cold lysis buffer plus protease inhibitors, and samples were suspended in sample buffer (2% SDS, 80 mM Tris-HCl, pH 6.8, 2 mM EDTA, 10% glycerol, and bromophenol blue) and boiled for 5 min. Nonreducing SDS-PAGE was performed by the method of Laemmli (1970) using 5% acrylamide and a 3% stacking gel.
Preparation of Laminin-5–rich Extracellular Matrix from Keratinocytes
To prepare laminin-5–rich ECM, human epidermal keratinocytes (HEKs) were either prepared from neonatal foreskin as described (Rheinwald and Green, 1975) or purchased from Clonetics (San Diego, CA) and grown, respectively, in either FAD medium (1:3 mix of Ham's F12 and DMEM), 1.8 × 10−4 M adenine, 10% FBS, HICE mix, 100 U/ml penicillin, and 100 μg/ml streptomycin) or serum-free Keratinocyte Growth Medium (GIBCO BRL) supplemented with EGF and bovine pituitary extract, as directed by the manufacturer. Cells were grown on 35-mm tissue culture plates (Becton Dickinson) for several days, removed with 0.05% trypsin, 1 mM EDTA in PBS, and discarded. Surfaces coated with HEK-secreted ECM were treated with 0.5 mg/ml soybean trypsin inhibitor (Sigma) in PBS and then blocked with 1 mg/ml BSA in PBS.
Cell Spreading Assays
Purified human laminin-5 was kindly provided by R. Burgeson (Massachusetts General Hospital, Charlestown, MA). 96-well tissue culture plates (Costar Corp.,Cambridge, MA) were coated overnight at 4°C with laminin-5 dissolved in PBS at a concentration of 20 μg/ml; in some cases rat fibronectin (GIBCO BRL) was included at a concentration of 20 μg/ ml. Coated plates were blocked with 2 mg/ml heat-denatured BSA in PBS for at least 1 h. Cultures of primary mouse keratinocytes were split with 0.05% trypsin and subcultured onto coated plates. For subculture onto laminin-5–rich, HEK-secreted matrix, mouse keratinocytes were seeded at a density of 105 cells/cm2, allowed to adhere for 1.5 h at 34°C, and then photographed. For spreading assays on purified laminin-5, mouse keratinocytes were seeded at a density of 3 × 104 cells/well, allowed to adhere for 1 h at 34°C, and nonattached cells were washed gently away with PBS. In some cases, the monoclonal antibody GoH3, which specifically blocks adhesion through α6 integrins, was added to cells before plating at a concentration of 2 μg/ml. Adhered cells were then fixed with 4% paraformaldehyde, stained with 0.02% Giemsa (Sigma), and photographed.
Results
Formation of Skin Blisters in Mice Deficient for α3β1 Integrin
Frozen sections were prepared from the limbs of α3-null or wild-type mice and stained by immunofluorescence with an antiserum against the cytoplasmic domain of the α3 subunit (DiPersio et al., 1995). In wild-type mice, α3 expression was restricted to the basal layer of keratinocytes in the epidermis (Fig. 1, A and B, arrows), as expected for this integrin (Peltonen et al., 1989; Carter et al., 1990a ,b; Hertle et al., 1991). Preimmune serum did not stain the epidermis (Fig. 1, C and D). In α3-null mice, stratification of the epidermis appeared normal (Figs. 1 E and 2 B). However, staining of the basal keratinocytes of α3-null epidermis with anti-α3 (Fig. 1 F, arrow) was comparable to background staining with preimmune serum (Figs. 1, G and H). The absence of α3β1 from α3-null skin was confirmed by Western blotting (data not shown). α3 staining intensity of basal keratinocytes in mice that were heterozygous for the α3 mutation was intermediate (data not shown).
Figure 1.

α3β1 integrin is absent from the skin of α3-null mice. Frozen skin sections were prepared from the limbs of wild-type (A–D) or α3-null (E–H) mice and stained by immunofluorescence with an antiserum against the cytoplasmic domain of α3 (B and F), or with the preimmune serum (D and H). The corresponding phase contrast (A, C, E, and G) is shown to the left of each immunofluorescence panel. e, epidermis; d, dermis. Arrowheads point to basal keratinocytes of the epidermis. Bar, 50 μm.
Figure 2.
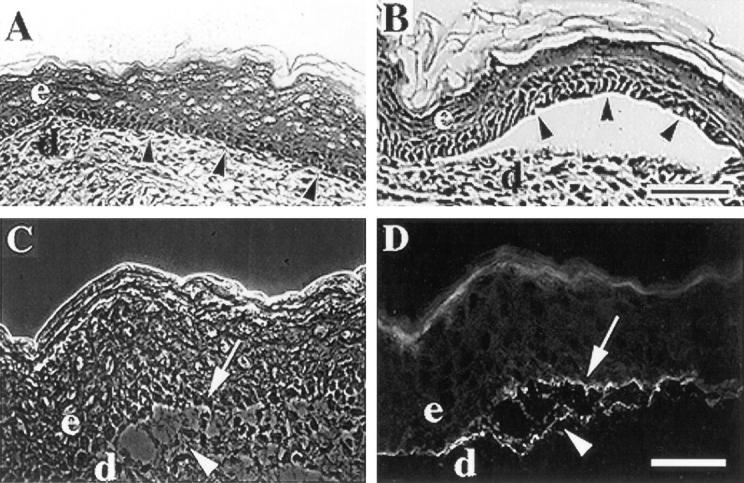
α3-null mice form skin blisters. (A and B) Frozen skin sections from wildtype (A) or α3-null (B) mice were stained with hematoxylin and eosin and the epidermal-dermal junctions were compared. Arrowheads point to basal keratinocytes of the epidermis. (C and D) A frozen skin section from an α3null mouse showing a blister viewed by phase contrast (C) or stained by immunofluorescence with an antiserum against laminin-5 (D). Arrowheads and arrows point to areas of laminin-5 staining at the dermal and epidermal sides of the blister, respectively. e, epidermis; d, dermis. Bars, 50 μm.
Examination of frozen skin sections from α3-null mice revealed occasional blisters where the epidermis was separated from the dermis (Fig. 2 B). Basal keratinocytes of the epidermis were intact over blistered regions (Fig. 2 B, arrowheads). Blisters appeared to be concentrated on the legs and footpads, possibly due to physical trauma to these regions within the first several hours after birth, and were never found in skin sections from wild-type mice (Fig. 2 A) or heterozygous mice (not shown). Epidermal blistering in the α3-null mice is reminiscent of more severe blistering phenotypes associated with loss of α6β4 integrin through null mutations of either the α6 (Georges-Labouesse et al., 1996) or the β4 subunit (van der Neut et al., 1996; Dowling et al., 1996) and with human blistering diseases caused by defects in laminin-5 (Domloge-Hultsch, 1992; Baudoin et al., 1994; Kirtschig et al., 1995), the ligand for both α6β4 and α3β1. In these cases, extreme blistering appears to result from detachment of basal keratinocytes from the basement membrane due either to the absence of α6β4 integrin or to deficiencies in its ligand laminin-5. Indeed, in α6-null or β4-null skin, laminin-5 remains associated with the dermal sides of blisters (Georges-Labouesse et al., 1996; van der Neut et al., 1996; Dowling et al., 1996). Therefore, the blistering phenotype in the α3-null epidermis appeared consistent with a role for α3β1 as a receptor for laminin-5 or other basement membrane ligands.
To determine the distribution of laminin-5 within blisters of α3-null skin, we stained frozen skin sections from neonatal mice with a polyclonal antiserum against laminin-5 (Marinkovich et al., 1992). Surprisingly, laminin-5 was detected at both the epidermal and dermal sides of blisters, and occasionally in what appeared to be remnants of matrix extending through the blisters (Fig. 2, C and D), suggesting that the split occurred within the basement membrane rather than between the basal keratinocytes and the basement membrane. Therefore, skin blisters in mice lacking α3β1 are distinct from those seen in mice lacking α6β4.
Defects in Basement Membrane Organization in α3-Null Mice
Given the rupture of the basement membrane in blistered regions of α3-null skin, we also examined basement membrane organization in nonblistered regions. As expected, laminin-5 in wild-type skin was restricted to the basement membrane directly below the basal keratinocytes of the epidermis (Fig. 3, A and C, arrowheads); the basement membrane appeared intact throughout each section examined. In marked contrast, staining of skin from α3-null mice revealed extensive regions of matrix disorganization, where laminin-5 was also detected below the plane of the basement membrane (Fig. 3 D, arrowheads). Phase contrast of the same field showed that the epidermis and dermis were intact through these regions (Fig. 3 B). In some cases, dense concentrations of laminin-5, resembling continuous remnants of basement membrane, extended down into the dermal regions (for example, see Fig. 4 F). Laminin-5 staining of the basement membrane appeared normal in mice heterozygous for the α3-null mutation, where α3β1 levels were reduced but still easily detectable (not shown). Based on the disorganized matrix/blistering phenotype, we were routinely able to identify skin sections from α3-null mice before confirmation of genotype by PCR or Southern blot analysis.
Figure 3.
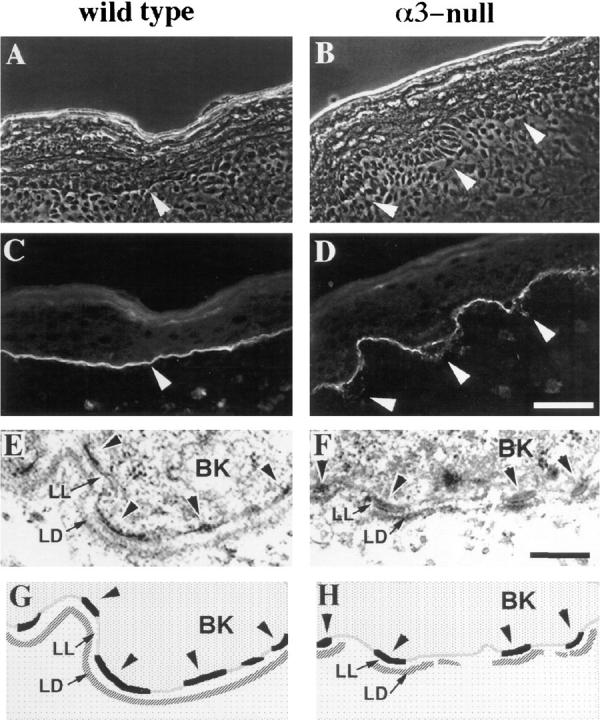
The basement membrane in α3-null skin is disorganized. (A–D) Frozen skin sections from wild-type (A and C) or α3-null (B and D) mice were viewed by phase contrast (A and B) or stained by immunofluorescence with an antiserum against laminin-5 (C and D). Arrowheads point to areas of laminin-5 staining at the basement membrane in the wild-type skin (A and C) or at regions of disorganized basement membrane in α3-null skin (B and D). (E and F) Electron micrographs comparing ultrastructure of the basement membrane zone in wild-type (E) and α3-null (F) skin. (G and H) Schematic illustration of relevant structures seen in E and F. BK, basal keratinocyte; LL, lamina lucida; LD, lamina densa; arrowheads point to hemidesmosomes along the basal aspect of the plasma membranes in the basal keratinocytes. Bars: (D) 50 μm; (F) 200 nm.
Figure 4.

Entactin and type VII collagen codistribute with laminin-5 over regions of disorganized basement membrane and to both dermal and epidermal sides of blisters in α3-null skin. Frozen sections from neonatal skin were stained by double-label immunofluorescence with GoH3 monoclonal antibody against the α6 integrin subunit and antiserum against laminin-5 (A, E, I, and B, F, J, respectively), or by immunofluorescence with antisera against type VII collagen (C, G, and K) or entactin (D, H, and L). (A–D) Representative fields from wild-type skin. (E–H) Adjacent sections through a region of disorganized basement membrane in α3-null skin; arrows point to areas in the dermis, outside the basement membrane zone as defined by α6-staining (E) where basement membrane proteins are detected (F–H). (I–L) Adjacent sections through a blister in α3-null skin; arrowheads and arrows point to the dermal and epidermal sides of the blister, respectively. Bar, 50 μm.
We also examined the ultrastructure of the dermal-epidermal junction. The results of these analyses, shown in Fig. 3, E and F, are illustrated schematically in Fig. 3, G and H, respectively. In neonatal skin from wild-type mice (Fig. 3, E and G), the electron-dense hemidesmosomes (arrowheads) were detected along the basal surfaces of keratinocytes adjacent to the lamina lucida (LL) of the basement membrane zone, and the lamina densa (LD) of the basement membrane was visible as a continuous, electrondense zone below the lamina lucida. In neonatal skin from α3-null mice (Fig. 3, F and H), hemidesmosomes were present on the basal keratinocytes and were of comparable size and frequency to those seen in wild-type skin. However, in contrast with wild-type skin, the lamina densa of α3-null skin appeared discontinuous in sections from several animals; electron-dense material was associated with regions directly beneath and adjacent to hemidesmosomes but was reduced in or absent from regions between hemidesmosomes. Therefore, disorganization of the basement membrane is also evident at the ultrastructural level in α3-null skin.
Since hemidesmosomes appeared normal at the ultrastructural level in α3-null skin, we also examined the expression and distribution of α6β4, a component of hemidesmosomes (Stepp et al., 1990; Jones et al., 1991). The α6 subunit also associates with the β1 subunit in some cells (Hynes, 1992). However, most or all α6 in the epidermis appears to be associated with β4 (Watt and Hertle, 1994), and little or no α6 was detected in skin from β4-null mice (van der Neut et al., 1996; Dowling et al., 1996). In addition, we detected no α6β1 in primary mouse keratinocytes (discussed below), and the distributions of α6 and β4 were identical in wild-type and α3-null skin. Double-label immunofluorescence of wild-type skin, using anti–laminin-5 serum and the anti-α6 monoclonal antibody GoH3 (Sonnenberg et al., 1987), showed that α6 subunit was concentrated at the basal surfaces of keratinocytes (Fig. 4 A) adjacent to laminin-5 in the basement membrane (Fig. 4 B), as expected for α6β4 (Carter et al., 1990b ). Although α6 showed the same distribution in α3-null epidermis, it did not codistribute with laminin-5 when the latter was detected below the plane of the basement membrane in regions of disorganized matrix (compare Fig. 4, E and F). Therefore, double-label immunofluorescence, with antibodies against either the α6 or the β4 integrin subunit and laminin-5, was used in all subsequent analyses to confirm the presence of disorganized basement membrane.
In contrast with laminin-5, α6 remained confined to the basal surfaces of basal keratinocytes on the epidermal sides of blisters (compare Fig. 4, I and J) and occasionally showed a discontinuous distribution over blisters (not shown). Distribution of laminin-5 to the epidermal sides of blisters was likely due to its binding to α6β4 on the basal keratinocytes, consistent with our adhesion studies using cultured keratinocytes, described below.
Type VII collagen and entactin, both components of the epidermal basement membrane (Martin, 1987; Timpl, 1989; Burgeson, 1993), were each detected in the basement membrane of wild-type epidermis (Fig. 4, C and D, respectively); entactin was also detected in the basement membranes of capillaries throughout the dermis (Fig. 4 D). Staining of adjacent sections from α3-null skin showed that type VII collagen and entactin each codistributed with laminin-5 to regions of disorganized basement membrane (compare Fig. 4, F–H, arrows). In addition, staining of adjacent sections through a blister showed that collagen VII and entactin, like laminin-5, each distributed to both the epidermal and dermal sides of the split (compare Fig. 4, J–L). Similar staining (not shown) was seen with an antiserum expected to recognize laminin isoforms containing the α1, β1, or γ1 chain (see Burgeson et al., 1994), including the basement membrane protein laminin-6 (α3, β1, and γ1), but not laminin-5 (α3, β3, and γ2).
An antiserum against fibronectin produced a fibrillar pattern of staining throughout the dermis (Fig. 5, A and B) and occasionally showed concentrated staining in the basement membrane zone (not shown), as reported previously during embryonic development (Peters and Hynes, 1996). An antiserum specific for the EIIIB segment of fibronectin produced similar staining patterns (Fig. 5, D and E). In α3-null skin, total fibronectin (Fig. 5 C) and EIIIB+ fibronectin (Fig. 5 F) were each detected at both the epidermal and dermal sides of blisters. Since levels of EIIIB+ fibronectin are normally very low in serum (Peters et al., 1995), these staining patterns suggest that fibronectin present in the blisters is derived from extracellular matrix rather than from infiltrating serum. However, we cannot rule out some contribution of serum fibronectin to the fibronectin detected in blisters. Although the role of fibronectin as a component of the epidermal basement membrane has been controversial (Hynes, 1990), our findings imply an association of fibronectin with the basal keratinocytes of the epidermis. Considered together, the distributions of the various matrix proteins indicate that blister formation in the α3-null epidermis is associated with disorganization and rupture of the basement membrane itself rather than with a simple defect in adhesion of the epidermis to the basement membrane.
Figure 5.

Fibronectin distributes to both epidermal and dermal sides of blisters in α3null skin. Frozen sections from wild-type (A, B, D, and E) or α3-null (C and F) skin were stained with an antiserum against fibronectin (B and C) or the preimmune serum (A), or with antiserum specific for the EIIIB segment of fibronectin (D–F). Recognition of fibronectin by anti-EIIIB requires treatment with N-glycanase (E and F), as described previously (Peters and Hynes, 1996); as a control, the section in D was not treated with N-glycanase. (A, B, D, and E) Arrowheads point to the dermal-epidermal junction. (C and F) Arrowheads and arrows point to the dermal and epidermal sides of blisters, respectively. Bar, 50 μm.
Distributions of Basement Membrane Proteins in Developing Skin
To compare skin development and basement membrane formation in normal and α3-null mice, frozen sections were prepared from embryos at days 11.5 (E11.5), 15.5 (E15.5), and 17.5 (E17.5) postcoitum. Phase contrast microscopy (not shown) revealed that skins from α3-null, wild-type, or heterozygous embryos were morphologically indistinguishable at each of these developmental stages, and appeared to stratify normally (for a review of skin development, see Watt and Hertle, 1994). The embryonic epidermis at E11.5 is composed of a basal cell layer and an outer cell layer (the periderm) and was distinguishable as a thin sheet of cells surrounding the embryo. At E15.5, the epidermis showed varying degrees of stratification. By E17.5, 2–3 d before birth, the epidermis was clearly stratified and resembled that of a newborn mouse.
The developmental expression and general distributions of several ECM proteins were identical in skin from wildtype, heterozygous, and α3-null embryos at each developmental stage examined; therefore, panels in Fig. 6 show representative data for entactin and type VII collagen. Entactin clearly defined a basement membrane zone at the dermal-epidermal junction at E11.5 (Fig. 6 A), E15.5 (Fig. 6 C), and E17.5 (Fig. 6 E). Type VII collagen was detected throughout the dermis at each of these stages, as it was at birth, but it became more concentrated in the basement membrane zone as development proceeded (compare Fig. 6 B, D, and F). The distribution of fibronectin at each stage was similar to that seen at birth; fibronectin was detected throughout the dermis up to the dermal-epidermal junction, and in some sections it was considerably more concentrated at this junction (data not shown), as reported previously (Peters and Hynes, 1996).
Figure 6.

Distributions of entactin and type VII collagen in the developing skin. Frozen sections from mouse embryonic skin at days E11.5 (A and B), E15.5 (C and D), or E17.5 (E and F) of development were stained with antisera against entactin (A, C, and E) or type VII collagen (B, D, and F). Staining patterns for entactin or type VII collagen were identical in wild-type (A, C, and D) and heterozygous (E and F) embryos; type VII collagen staining in B is from an α3-null, E11.5 embryo, but was identical to that of a wild-type embryo at this stage. e, epidermis; d, dermis. Bar, 50 μm.
Distributions of Laminin-5 and Its Integrin Receptors during Development of Normal and α3-Null Skin
To compare developmental expression of laminin-5 and its receptors α3β1 and α6β4, we first examined expression of these proteins in wild-type or heterozygous mice. α3β1 was expressed in the embryonic epidermis as early as E9.5 and remained restricted to basal keratinocytes, in a basolateral distribution, throughout development (Fig. 1 B and data not shown). The relative distributions of α6β4 and laminin-5 were determined by double-label immunofluorescence using a monoclonal antibody against the β4 subunit (346-11A; Kennel et al., 1989) and anti–laminin-5 serum (Fig. 7, A–D, G, and H). Laminin-5 was not detected in the unstratified skin of E11.5 embryos (data not shown). At E15.5, the degree of stratification varied throughout the embryo, and the appearance of laminin-5 in the basement membrane zone coincided roughly with increased epidermal stratification; anti–laminin-5 did not stain the basement membrane in less stratified regions (Fig. 7 B), but clearly stained the basement membrane in more stratified regions (Fig. 7 D). The distribution of β4 at E15.5 was similar; in less stratified regions it was present in a basolateral distribution within the basal keratinocyte layer (Fig. 7 A), similar to the distribution of α3β1 (not shown), and in more stratified regions it was concentrated at the basal surfaces of the basal keratinocytes, adjacent to laminin-5 in the basement membrane (compare Fig. 7, C and D). Therefore, recruitment of α6β4 to the basal aspects of basal keratinocytes appeared to coincide with incorporation of laminin-5 into the basement membrane, and may reflect the establishment of cell adhesion to laminin-5 mediated by α6β4. In E17.5 epidermis, α6β4 was always concentrated in the basal surfaces of basal keratinocytes adjacent to the laminin-5-rich basement membrane (Fig. 7, G and H), as in newborn skin (Fig. 4, A and B).
Figure 7.

Distributions of α6β4 and laminin-5 in the developing skin of normal and α3-null embryos. Frozen sections from mouse embryonic skin at days E15.5 (A–F) or E17.5 (G–L) of development were stained by double-label immunofluorescence with either monoclonal antibody 346-11A against the β4 integrin subunit (A, C, and G) or GoH3 monoclonal antibody against the α6 integrin subunit (E, I, and K), and anti–laminin-5 serum (B, D, H, and F, J, L, respectively). Control sections were from wild-type embryos (A–D) or heterozygous embryos (G and H). In wild-type E15.5 embryos, α6β4 and laminin-5 codistributed to the basement membrane zone in more stratified regions (C and D), but not in less stratified regions (A and B); the width of the epidermis in each panel is indicated by a double-headed arrow. In α3-null embryos at E15.5 (E and F) and E17.5 (I and J), arrowheads point to areas of laminin-5 staining in areas of disorganized basement membrane, below the α6-positive basal keratinocytes; the skin in E and F is folded back on itself. (K and L) Higher magnification of α3-null skin at E17.5 showing α6-negative, basal keratinocytes that have separated from the laminin-5 positive basement membrane, marked by arrowheads. e, epidermis; d, dermis. Bars: (shown in J for A–J) 50 μm; and (in L for K and L) 50 μm.
The temporal expression and distribution of α6β4 (i.e., α6 or β4 subunit) during development of α3-null embryos were identical to those in wild-type or heterozygous embryos. However, staining of E15.5 embryos with anti–laminin-5 revealed occasional regions of basement membrane disorganization in α3-null skin (Fig. 7, E and F). By E17.5 these regions were more common (Fig. 7, I and J), and became progressively more disorganized and more extensive by birth. Disorganized basement membrane was found on skin from both legs and torso in α3-null embryos, but was not detected in skin from wild-type or heterozygous embryos.
Although we cannot rule out prenatal blister formation in α3-null mice, we did not detect skin blisters greater than a few cells in diameter in α3-null embryos. In E17.5 embryos, we occasionally observed individual basal keratinocytes in which α6 staining was absent (Fig. 7 K). As expected for basal keratinocytes lacking α6β4 (van der Neut et al., 1996; Georges-Labouesse et al., 1996; Dowling et al., 1996), these cells were detached from the basement membrane, since laminin-5 distributed only to the dermal side of the separation (compare arrowheads in Fig. 7, K and L). However, we did not detect blisters at E17.5 that resembled the larger splits seen in neonatal skin. Embryos were collected by Cesarian section, thereby reducing the mechanical stress associated with birth. Otherwise, skin sections from E17.5 embryos and neonatal mice were prepared in the same manner, suggesting that blisters in α3-null neonates were caused by physical trauma during or after birth, rather than by mechanical stress during sample preparation.
Isolation of Primary Keratinocytes from α3-Null, Neonatal Mice
To address directly the importance of α3β1 as a laminin-5 receptor in the epidermis, we isolated primary keratinocytes from wild-type and α3-null, neonatal mice (Dlugosz et al., 1995; for details see Materials and Methods). To compare surface expression of integrins that bind to laminin-5 in wild-type and α3β1-deficient keratinocytes, 7-d, primary cultures were surface-iodinated with 125I, and detergent lysates were immunoprecipitated with antibodies against integrin subunits and analyzed by SDS-PAGE under nonreducing conditions (Fig. 8). As expected, antiserum against α3 immunoprecipitated an abundance of the α3 subunit (∼150 kD) associated with the β1 subunit (∼110 kD) from lysates of wild-type cells (lane 2), but did not detect α3β1 in lysates from α3-null cells (lane 5). An anti-β1 serum (lane 1) coimmunoprecipitated β1 and associated proteins of ∼150 kD, corresponding to α3 and other comigrating α subunits present in keratinocytes (Watt and Hertle, 1994). Surface levels of β1 integrins dropped significantly in α3null cells compared to wild-type cells (compare lanes 1 and 4), demonstrating that α3β1 constitutes the major proportion of total β1 integrins in mouse keratinocytes.
Figure 8.

Surface expression of integrins in primary keratinocytes isolated from wild-type (lanes 1–3) or α3-null (lanes 4–6), neonatal mice. Detergent lysates from 125I surface-labeled cells were immunoprecipitated with antisera against the cytoplasmic domains of the β1, α3, or α6 integrin subunits, as indicated at the top of each lane. Molecular weight markers are shown to the right of the autoradiograph. Migratory positions of certain integrin subunits are indicated to the left, including proteolytic fragments of β4, a, b, and c, described previously by Hemler et al. (1989). The β1-associated band in α3-null cells that comigrates with α3 (lane 4) represents other integrin α subunits that dimerize with β1 in keratinocytes, since α3 was not detected in α3-null cells (lane 5). ?, an unidentified band that may represent a proteolytic fragment of β4 (see text).
α6β4 was detected using a polyclonal antiserum against the α6 integrin subunit (lane 3), which coimmunoprecipitated a series of bands expected for the α6 subunit (∼140 kD) and various derivatives of the β4 subunit (Hemler et al., 1989; Kajiji et al., 1989; Adams and Watt, 1991). In nonreduced immunoprecipitates from human cells, the fulllength β4 subunit migrates at ∼210 kD (a fragment), and smaller fragments of 165 kD (b fragment), 125 kD (c fragment), and 85 kD are thought to result from proteolysis of β4 (Hemler et al., 1989). Indeed, this pattern of proteolysis has been useful as a diagnostic tool in studies of β4 integrins (Hemler et al., 1989). Bands corresponding to the a, b, and c fragments of β4 in mouse keratinocytes are indicated in Fig. 8; an unidentified band of ∼70 kD may correspond to the 85-kD fragment seen in human cells. Each of these fragments showed faster migration in the mouse keratinocytes than has been reported in human cells (Hemler et al., 1989), possibly due to species-specific differences in glycosylation of β4. Importantly, α6-associated bands were identical in lysates from wild-type cells and α3-null cells (compare lanes 3 and 6). A band corresponding to the α6 subunit was not detected in β1 immunoprecipitations (lanes 1 and 4), suggesting that primary mouse keratinocytes have little, if any, α6β1. Consistent with this notion, α6β1 was not detected in mouse keratinocytes either by immunoprecipitation of α6 integrins with GoH3 followed by Western blotting with anti-β1 serum, or by immunoprecipitation of β1 integrins followed by Western blotting with anti-α6 serum (data not shown).
α3β1 Is Required for Spreading of Keratinocytes on Laminin-5
We subcultured primary mouse keratinocytes on laminin5-rich ECM prepared from human keratinocyte cultures, as described in the Materials and Methods section; this ECM supports α3β1-mediated cell attachment (Carter et al., 1991; Weitzman et al., 1993). Both α3-positive cells (in this case from a mouse heterozygous for the α3-null mutation) and α3-null cells attached to laminin-5–rich ECM within 15 min. After attachment, α3-positive keratinocytes spread rapidly on the laminin-5–rich ECM (Fig. 9 A). In contrast, α3-null keratinocytes remained unspread for up to 2 h following attachment to this matrix (Fig. 9 B). The same results were obtained in spreading assays using purified laminin-5 (Fig. 9, C and D). α3-null keratinocytes were able to spread when fibronectin was present in the substrate with laminin-5 (Fig. 9, E and F), demonstrating that α3β1-deficient cells were not generally deficient in the ability to spread. The antibody GoH3, which blocks cell adhesion mediated by α6 integrins (Sonnenberg et al., 1987), only slightly inhibited attachment, and did not affect spreading, of wild-type keratinocytes on laminin-5 (Fig. 9 G). In contrast, the same concentration of GoH3 completely inhibited attachment of α3β1-deficient cells to laminin-5 (Fig. 9 H). Therefore, while α6β4 and α3β1 are each sufficient for attachment of mouse keratinocytes to laminin-5, α3β1 is essential for postattachment cell spreading on this ligand, consistent with previous, integrin-blocking studies using human foreskin keratinocytes (Xia et al., 1996).
Figure 9.
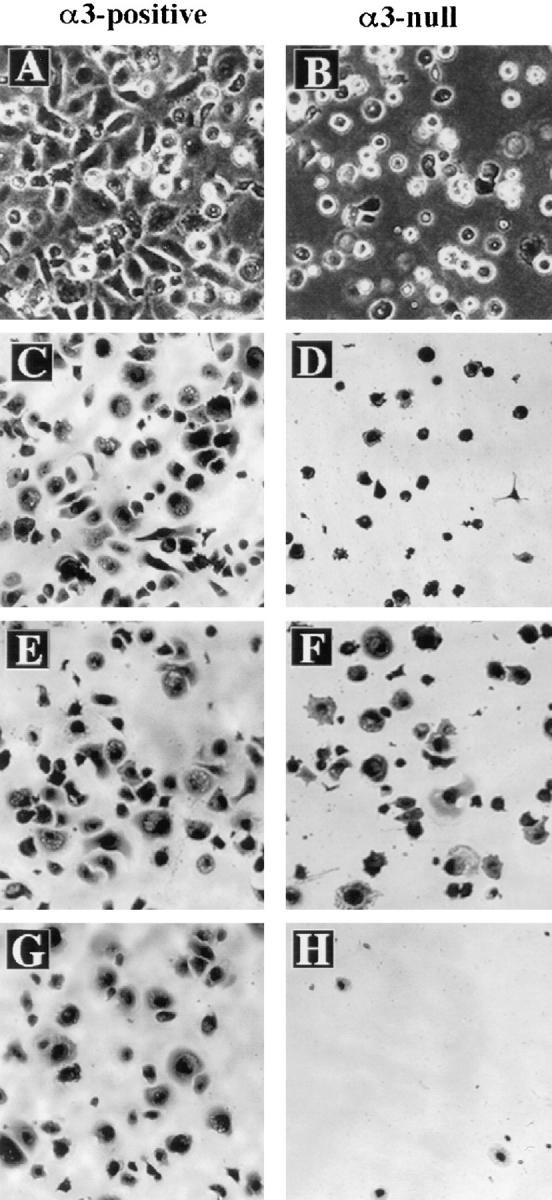
α3β1 is required for postadhesion spreading of mouse keratinocytes on laminin-5. (A and B) Primary keratinocytes from neonatal mice heterozygous (A) or homozygous (B) for the α3null mutation were subcultured on HEK-secreted, laminin-5–rich ECM (see Materials and Methods) and then photographed after 1.5 h. (C–H) Primary keratinocytes from wild-type (C, E, and G) or α3-null (D, F, and H) neonatal mice were subcultured on purified laminin-5 for 1 h and then fixed in 4% paraformaldehyde and stained with Giemsa. (E and F) Fibronectin was included with laminin-5 in the substrate. (G and H) Cells were treated with the monoclonal antibody GoH3, which blocks adhesion by α6 integrins.
Discussion
The importance of integrin α3β1 as an ECM receptor in the epidermis has been unclear. The generation of mutant mice lacking the α3 integrin subunit provides a useful animal model for the study of α3β1 functions in the epidermis. In addition, since α3-null mice develop fully until perinatal death, they provide a rich source of α3β1-deficient, epidermal keratinocytes for study in culture. We have examined both α3-null mice and α3-deficient keratinocytes isolated from these mice for defects in adhesionrelated functions.
α3β1 Is Required for Basement Membrane Integrity in the Skin
Although stratification of the skin from α3-deficient mice was comparable to that of wild-type mice, both immunofluorescence microscopy and electron microscopy of skin sections from α3-null mice revealed a basement membrane with extensive regions of disorganization. Furthermore, matrix disorganization was often associated with blistering at the dermal-epidermal junction in neonatal mice. A deficiency in α6β4 through null mutation of either the α6 subunit (Georges-Labouesse et al., 1996) or the β4 subunit (van der Neut et al., 1996; Dowling et al., 1996) also causes epidermal blistering, although the blistering phenotype in these mice is much more severe than in the α3-null mice. In addition, the α3-null phenotype differs from those of the α6-null and β4-null mice in another important way. The latter two mutations appear to cause detachment of basal epidermal keratinocytes from the basement membrane, since laminin-5 localized only to the dermal sides of blisters in both cases (Georges-Labouesse et al., 1996; van der Neut et al., 1996; Dowling et al., 1996). In contrast, blistering in α3-null epidermis appeared to result from defects within the basement membrane itself, since laminin-5 distributed to both the epidermal and the dermal sides of the split. Indeed, the α3-null epidermis appeared to retain its ability to bind to the basement membrane, probably via α6β4, since laminin-5 colocalized with α6β4 to the epidermal sides of blisters, and α3-null keratinocytes attached to laminin-5 in an α6-dependent manner. Thus, the epidermal phenotypes associated with α3β1-deficient and α6β4deficient mice appear distinct from one another, and suggest an involvement of α3β1 in establishing and/or maintaining integrity of the basement membrane.
The chemical composition and physical organization of the epidermal basement membrane are complex (for reviews see Borradori and Sonnenberg, 1996; Uitto et al., 1996). At the ultrastructural level, the dermal-epidermal junction is resolved into the lamina lucida, which abuts the plasma membranes of basal keratinocytes in the epidermis, and the underlying lamina densa. Entactin/nidogen localizes to the lamina densa along with type IV collagen, laminin-5, and other laminin isoforms, including laminin-6 and -7. Some basement membrane structures are specifically associated with hemidesmosomes. For example, anchoring fibrils, which consist of type VII collagen, extend from the dermis into the lamina densa of the basement membrane adjacent to hemidesmosomes. The anchoring filaments, which contain laminin-5, extend from the anchoring fibrils across the lamina lucida to the hemidesmosomes at the basal cell surface. Interestingly, type VII collagen, entactin, and laminin-5 each distributed to both sides of blisters in α3-null skin. These distributions suggest that the basement membrane does not rupture between distinct domains of the basement membrane but, instead, ruptures unevenly through different layers and structures. Alternatively, distributions of these proteins within blisters may reflect distinct fractions that are associated with different domains of the basement membrane. For example, type VII collagen present at the epidermal side of a blister may represent a fraction that has not been properly assembled into anchoring fibrils. In either case, the distributions of matrix proteins within blisters appear to reflect a general disorganization and weakening of the basement membrane in α3-null skin. Although the presence of fibronectin in the basement membrane has been controversial (discussed in Hynes, 1990), this matrix protein also distributed to both sides of blisters, suggesting either a direct or an indirect interaction between fibronectin and basal keratinocytes.
Disorganized Basement Membrane Appears during Development of α3-Null Skin
Our observations suggest that the temporal expression patterns and general distributions of α6β4, laminin-5, and several other ECM proteins in the developing skin of α3null mice are comparable to those in the skin of wild-type and heterozygous mice. However, examination of skin sections from α3-null embryos stained with anti–laminin-5 serum revealed defects in basement membrane organization by day E15.5 of development, which became progressively more evident as development proceeded. E15.5 was also the stage at which laminin-5 was first detected in the basement membrane beneath more stratified regions of the epidermis, where most of the α6β4 had relocated to the basal surface of the basal keratinocytes. Therefore, this stage of development appears to represent a time when laminin-5 is recruited into the basement membrane and, presumably, when maintenance of the dermal-epidermal junction begins to switch over to a laminin-5–dependent adhesion mechanism. α3β1 may be required at this time to help assemble and/or maintain this new laminin-5–dependent basement membrane (see below).
The causal relationships of the disorganized matrix and epidermal blistering seen in α3-null skin are unknown. However, as discussed above, it seems likely that matrix disorganization reflects a weak basement membrane that eventually ruptures and leads to blister formation. In an alternative but not exclusive model, it is possible that small blisters occur during skin development (i.e., see Fig. 7, K and L) and that, during fetal repair of the dermal-epidermal junction, remnants of old basement membrane are displaced below the plane of the newly deposited matrix, producing the disorganized appearance of the basement membrane. Indeed, dense concentrations of laminin-5 and other matrix proteins that resembled remnants of continuous basement membrane were often seen extending from the dermal-epidermal junction down into the dermal regions.
The Laminin-5 Receptors α3β1 and α6β4 Have Distinct Functions in Keratinocytes
Keratinocytes isolated from α3-null epidermis retained the ability to attach to laminin-5 in culture but, in contrast to wild-type or heterozygous keratinocytes, did not spread well under our experimental conditions. Attachment of α3null keratinocytes to laminin-5 was dependent on α6β4, since the monoclonal antibody GoH3 completely blocked attachment and α6β1 was not detected in these cells. On the other hand, the same concentrations of GoH3 had little effect on attachment of wild-type keratinocytes to laminin-5, suggesting that α3β1 was sufficient for attachment. These results show that α3β1 and α6β4 have distinct but overlapping functions in mouse keratinocytes; both can support initial cell attachment to laminin-5, but α3β1 is required for cell spreading. These roles for α3β1 and α6β4 have been demonstrated previously in function-blocking studies of human foreskin keratinocytes (Xia et al., 1996), and have been suggested for a murine mammary tumor cell line (Sonnenberg et al., 1993). Differences in function between these two integrins reflect their distinct roles in vivo; α6β4 is required for stable adhesion of the epidermis to the basement membrane, and α3β1 appears to have a post-adhesion role in basement membrane integrity. Indeed, although α3β1 may partially compensate for the absence of α6β4 in β4-null mice by relocalizing to the basal surfaces of basal keratinocytes (van der Neut et al., 1996), α3β1 was clearly not sufficient to maintain adhesion of the epidermis to the basement membrane in these mice.
Ultrastructure of the basement membrane in α3-null skin reflects these distinct functions for α6β4 and α3β1. In α3-null skin, the lamina densa was present directly beneath and adjacent to hemidesmosomes, but was not formed properly in regions between hemidesmosomes, suggesting that formation of the lamina densa in hemidesmosomefree regions is α3β1-dependent. Alternatively, hemidesmosomes may be associated with an electron-dense component of the basement membrane that is distinct from the rest of the lamina densa. Thus, α3β1 is necessary for formation of a continuous lamina densa, but an intact basement membrane is not required for hemidesmosome assembly. In contrast, α6β4 is necessary for assembly of hemidesmosomes but not for formation of a continuous lamina densa (Georges-Labouesse et al., 1996; van der Neut et al., 1996; Dowling et al., 1996).
α3β1 May Function as a Regulator of Basement Membrane Assembly/Remodeling
Analysis of kidneys from α3-null mice also revealed disorganization of the glomerular basement membrane (Kreidberg et al., 1996), suggesting that α3β1 may be generally important for organization of extracellular matrices. Consistent with this notion, α3β1 can interact weakly with a broad range of matrix proteins, including fibronectin (Wayner and Carter, 1987; Elices et al., 1991), laminin-1 (Wayner and Carter, 1987; Gehlsen et al., 1988; Elices et al., 1991), collagens (Wayner and Carter, 1987; Elices et al., 1991), and entactin/nidogen (Dedhar et al., 1992; Wu et al., 1995), and shows post-adhesion localization to focal contacts in cells cultured on most of these proteins (DiPersio et al., 1995).
There are several ways in which α3β1 might serve as a secondary receptor that functions in matrix organization. One possibility is that α3β1 is involved in initial assembly of the basement membrane by binding to laminin-5 or other matrix proteins during their processing or recruitment into sites of cell adhesion. Indeed, Wu et al. (1995) recently showed that α3β1 in transfected cells can mediate assembly of both fibronectin and entactin into the ECM. In the epidermal basement membrane, entactin binds to the type IV collagen network (Aumailley et al., 1989). Therefore, certain bridging interactions between matrix proteins that require α3β1 may be absent from α3-null skin, thereby compromising basement membrane integrity and weakening the dermal-epidermal junction. Thus, α6β4 may be a primary receptor for keratinocyte attachment to the basement membrane, while α3β1 functions as a relatively weak, postadhesion receptor (DiPersio et al., 1995) required for basement membrane assembly. A recent study demonstrated such distinct functions for fibronectin receptors in an embryonic stem cell line, where αvβ3 was dominant in focal contact formation, while α5β1 was primarily responsible for fibronectin matrix assembly (Wennerberg et al., 1996).
A second possibility is that α3β1 binds to laminin-5 or other matrix proteins in the pre-assembled basement membrane in order to stabilize primary interactions of cells with the ECM (i.e., via α6β4 in hemidesmosomes) and/or between ECM proteins themselves. In this way, α3β1 may be required to maintain strength and integrity of the basement membrane independently of its initial assembly.
Finally, interactions of α3β1 with basement membrane ligands may induce signal transduction events in keratinocytes that regulate matrix organization. α3β1-mediated adhesion can induce the tyrosine phosphorylation of focal adhesion kinase, or FAK (Kornberg et al., 1991; Jewell et al., 1995), as well as of other unidentified proteins in mouse keratinocytes (DiPersio, C.M., and R.O. Hynes, unpublished data). It is possible that α3β1-dependent signaling pathways regulate the production of matrix proteins or the activities of metalloproteinases or other enzymes that regulate matrix assembly and/or remodeling. Indeed, a balance between ECM structure/composition and the activities of matrix degrading enzymes has been demonstrated in other systems (Sympson et al., 1994; Tremble et al., 1994) and can be regulated through integrins (Huhtala et al., 1995; Brooks et al., 1996). Ligand binding by α3β1 may be required to signal the presence of basement membrane and provide positive or negative feedback to the cell that regulates basement membrane production.
A number of blistering skin diseases in humans are caused by defective basement membrane proteins or hemidesmosomal components that interfere with normal adhesion of the epidermis to the dermis. For example, some forms of JEB have been attributed to defects in the α3, β3, or γ2 chain of laminin-5 (Aberdam et al., 1994; Pulkkinen et al., 1994a ,b; Uitto et al., 1994; Kivirikko et al., 1995), and dystrophic forms of JEB are caused by mutations in the gene for type VII collagen (Uitto et al., 1994). Patients with cicatricial pemphigoid that develop anti–laminin-5 antibodies also form skin blisters (Kirtschig et al., 1995). Similarly, patients with bullous pemphigoid, an acquired blistering disease, develop antibodies against either BPAG-1e (bullous pemphigoid antigen-1e) or BPAG-2, both components of hemidesmosomes (Diaz et al., 1977; Mueller et al., 1989; Sawamura et al., 1991). At least two cases of JEB with pyloric atresia are associated with loss of α6β4 integrin through mutations in the β4 integrin subunit (Vidal et al., 1995; Niessen et al., 1996). The blistering phenotype associated with mice lacking integrin α3β1 suggests that this basement membrane receptor may also be a potential target of human blistering diseases. α3-null mice provide a useful system in which to study the roles of integrin receptors both in normal epidermal development and function, and in epidermal blistering diseases.
Acknowledgments
We are grateful to Robert Burgeson for providing us with purified laminin-5 and antisera to laminin-5 and type VII collagen, as well as for valuable advice and critical reading of the manuscript. We are also grateful to Ronald van der Neut, Arnoud Sonnenberg, and Elisabeth GeorgesLabouesse for sharing their research results before publication. We thank Albert Chung, Stephen Kennel, and Vito Quaranta for antibodies against entactin, the β4 integrin subunit, and the α6 integrin subunit, respectively. We thank Kim Mercer and Denise Crowley for histology, Pat Reilly for preparation of samples for electron microscopy, Stephen Cornwall for technical assistance, and Ed Clark, Laird Bloom, and Bernhard Bader for critical reading of the manuscript.
Footnotes
1. Abbreviations used in this paper: ECM, extracellular matrix; HEK, human epidermal keratinocytes; JEB, junctional epidermolysis bullosa.
This research was supported by a grant from the National Institutes of Health (RO1CA17007) to R.O. Hynes. K.M. Hodivala-Dilke was supported by the International Human Frontier Science Program. R.O. Hynes is an Investigator of the Howard Hughes Medical Institute.
The first two authors contributed equally to the work presented.
Please address all correspondence to R.O. Hynes, Howard Hughes Medical Institute, Center for Cancer Research, and Department of Biology, Massachusetts Institute of Technology, Cambridge, MA 02139. Tel.: (617) 253-6422. Fax: (617) 253-8357.
References
- Aberdam D, Galliano MF, Vailly J, Pulkkinen L, Bonifas J, Christiano K, Tryggvason K, Uitto KJ, Epstein EH, Ortonne J-P. Herlitz's junctional epidermolysis bullosa is genetically linked to mutations in the nicein/kalinin (laminin 5) LAMC2 gene. Nat Genet. 1994;6:299–304. doi: 10.1038/ng0394-299. [DOI] [PubMed] [Google Scholar]
- Adams JC, Watt FM. Changes in keratinocyte-extracellular matrix interactions during terminal differentiation: reduction in fibronectin binding precedes loss of α5β1 integrin from the cell surface. Cell. 1990;63:425–435. doi: 10.1016/0092-8674(90)90175-e. [DOI] [PubMed] [Google Scholar]
- Adams JC, Watt FM. Expression of β1, β3, β4, and β5 integrins by human epidermal keratinocytes and non-differentiating keratinocytes. J Cell Biol. 1991;115:829–841. doi: 10.1083/jcb.115.3.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aumailley M, Wiedemann H, Mann K, Timpl R. Binding of nidogen and the laminin-nidogen complex to basement membrane collagen type IV. Eur J Biochem. 1989;184:241–248. doi: 10.1111/j.1432-1033.1989.tb15013.x. [DOI] [PubMed] [Google Scholar]
- Baudoin C, Miquel C, Blanchet-Bardon C, Gambinin C, Meneguzzi G, Ortonne J-P. Herlitz junctional epidermolysis bullosa keratinocytes display heterogeneous defects of nicein/kalinin gene expression. J Clin Invest. 1994;93:862–869. doi: 10.1172/JCI117041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borradori L, Sonnenberg A. Hemidesmosomes: roles in adhesion, signaling and human diseases. Curr Opin Cell Biol. 1996;8:647–656. doi: 10.1016/s0955-0674(96)80106-2. [DOI] [PubMed] [Google Scholar]
- Brennen JK, Mansky J, Roberts G, Litchman MA. Improved methods for reducing calcium and magnesium concentrations in tissue culture medium. In Vitro. 1982;11:693–703. doi: 10.1007/BF02616371. [DOI] [PubMed] [Google Scholar]
- Brooks PC, Stromblad S, Sanders LC, von Schalscha TL, Aimes RT, Stetler-Stevenson WG, Quigley JP, Cheresh DA. Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin αvβ3. Cell. 1996;85:683–693. doi: 10.1016/s0092-8674(00)81235-0. [DOI] [PubMed] [Google Scholar]
- Burgeson RE. Type VII collagen, anchoring fibrils, and epidermolysis bullosa. J Invest Dermatol. 1993;101:252–255. doi: 10.1111/1523-1747.ep12365129. [DOI] [PubMed] [Google Scholar]
- Burgeson RE, Chiquet M, Deutzmann R, Ekblom P, Engel J, Kleinman H, Martin GR, Meneguzzi G, Paulsson M, Sanes J, et al. A new nomenclature for the laminins. Matrix Biol. 1994;14:209–211. doi: 10.1016/0945-053x(94)90184-8. [DOI] [PubMed] [Google Scholar]
- Carroll JM, Romero MR, Watt FM. Suprabasal integrin expression in the epidermis of transgenic mice results in developmental defects and a phenotype resembling psoriasis. Cell. 1995;83:957–968. doi: 10.1016/0092-8674(95)90211-2. [DOI] [PubMed] [Google Scholar]
- Carter WG, Wayner EA, Bouchard TS, Kaur P. The role of integrins α2β1 and α3β1 in cell-cell and cell-substrate adhesion of human epidermal cells. J Cell Biol. 1990a;110:1387–1404. doi: 10.1083/jcb.110.4.1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter WG, Kaur P, Gil SG, Gahr PJ, Wayner EA. Distinct functions for integrins α3β1 in focal adhesions and α6β4/bullous antigen in a new stable anchoring contact (SAC) of keratinocytes: relation to hemidesmosomes. J Cell Biol. 1990b;111:3141–3154. doi: 10.1083/jcb.111.6.3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter WG, Ryan MC, Gahr PJ. Epiligrin, a new cell adhesion ligand for integrin α3β1 in epithelial basement membranes. Cell. 1991;65:599–610. doi: 10.1016/0092-8674(91)90092-d. [DOI] [PubMed] [Google Scholar]
- Cavani A, Zambruno, Marconi A, Manca V, Marchetti M, Giannetti A. Distinctive integrin expression in the newly forming epidermis during wound healing in humans. J Invest Dermatol. 1993;101:600–604. doi: 10.1111/1523-1747.ep12366057. [DOI] [PubMed] [Google Scholar]
- Clark, R.A.F. 1990. Fibronectin matrix deposition and fibronectin receptor expression in healing and normal skin. J. Invest. Dermatol. 94 (suppl):128S– 134S. [DOI] [PubMed]
- Coulombe PA, Hutton ME, Vassar R, Fuchs E. A function for keratins and a common thread among different types of epidermolysis bullosa simplex diseases. J Cell Biol. 1991;115:1661–1674. doi: 10.1083/jcb.115.6.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedhar S, Jewell K, Rojiani M, Gray V. The receptor for the basement membrane glycoprotein entactin is the integrin α3β1. J Biol Chem. 1992;267:18908–18914. [PubMed] [Google Scholar]
- DeFreitas MF, Yoshida CK, Frazier WA, Mendrick DL, Kypta RM, Reichardt LF. Identification of integrin α3β1 as a neuronal thrombospondin receptor mediating neurite outgrowth. Neuron. 1995;15:333–343. doi: 10.1016/0896-6273(95)90038-1. [DOI] [PubMed] [Google Scholar]
- Delwel GO, de Melker AA, Hogervorst F, Jaspars LH, Fles DLA, Kuikman I, Lindblom A, Paulsson M, Timpl R, Sonnenberg A. Distinct and overlapping ligand specificities of the α3Aβ1 and α6Aβ1 integrins: recognition of laminin isoforms. Mol Biol Cell. 1994;5:203–215. doi: 10.1091/mbc.5.2.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz LA, Calvanico NJ, Tomasi TB, Jr, Jordon RE. Bullous pemphigoid antigen: isolation from normal human skin. J Immunol. 1977;118:455–460. [PubMed] [Google Scholar]
- DiPersio CM, Shah S, Hynes RO. α3Aβ1 integrin localizes to focal contacts in response to diverse extracellular matrix proteins. J Cell Sci. 1995;108:2321–2336. doi: 10.1242/jcs.108.6.2321. [DOI] [PubMed] [Google Scholar]
- Domloge-Hultsch N, Gammon WR, Briggaman RA, Gil SG, Carter WG, Yancey KB. Epiligrin, the major human keratinocyte integrin ligand, is a target in both an acquired autoimmune and an inherited subepidermal blistering skin disease. J Clin Invest. 1992;90:1628–1633. doi: 10.1172/JCI116033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling J, Yu Q-C, Fuchs E. β4 integrin is required for hemidesmosomal formation, cell adhesion and cell survival. J Cell Biol. 1996;134:559–572. doi: 10.1083/jcb.134.2.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dlugosz AA, Glick AB, Tennenbaum T. Isolation and utilization of epidermal keratinocytes for oncogene research. Methods Enzymol. 1995;254:3–20. doi: 10.1016/0076-6879(95)54003-2. [DOI] [PubMed] [Google Scholar]
- Elices MJ, Urry LA, Hemler ME. Receptor functions for the integrin VLA-3: fibronectin, collagen, and laminin binding are differentially influenced by ARG-GLY-ASP peptide and by divalent cations. J Cell Biol. 1991;112:169–181. doi: 10.1083/jcb.112.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehlsen KR, Dillner L, Engvall E, Ruoslahti E. The human laminin receptor is a member of the integrin family of cell adhesion receptors. Science (Wash DC) 1988;241:1228–1229. doi: 10.1126/science.2970671. [DOI] [PubMed] [Google Scholar]
- Georges-Labouesse E, Messaddeq N, Yehia G, Cadalbert L, Dierich A, Le Meur M. Absence of integrin α6 leads to epidermolysis bullosa and neonatal death in mice. Nat Genet. 1996;13:370–373. doi: 10.1038/ng0796-370. [DOI] [PubMed] [Google Scholar]
- Grenz H, Carbonetto S, Goodman SL. α3β1 integrin is moved into focal contacts in kidney mesangial cells. J Cell Sci. 1993;105:739–751. doi: 10.1242/jcs.105.3.739. [DOI] [PubMed] [Google Scholar]
- Guo L, Degenstein L, Dowling J, Yu Q-C, Wollmann R, Perman B, Fuchs E. Gene targeting of BPAG1: abnormalities in mechanical strength and cell migration in stratified epithelia and neurologic degeneration. Cell. 1995;81:233–243. doi: 10.1016/0092-8674(95)90333-x. [DOI] [PubMed] [Google Scholar]
- Hemler ME, Crouse C, Sonnenberg A. Association of the VLA α6 subunit with a novel protein. J Biol Chem. 1989;264:6529–6535. [PubMed] [Google Scholar]
- Hertle MD, Adams JC, Watt FM. Integrin expression during human epidermal development in vivo and in vitro. Development. 1991;112:193–206. doi: 10.1242/dev.112.1.193. [DOI] [PubMed] [Google Scholar]
- Hodivala KJ, Watt FM. Evidence that cadherins play a role in the downregulation of integrin expression that occurs during keratinocyte terminal differentiation. J Cell Biol. 1994;124:589–600. doi: 10.1083/jcb.124.4.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotchin NA, Watt FM. Transcriptional and post-translational regulation of β1 integrin expression during keratinocyte terminal differentiation. J Biol Chem. 1992;267:14852–14858. [PubMed] [Google Scholar]
- Huhtala P, Humphries MJ, McCarthy JB, Tremble P, Werb Z, Damsky CH. Cooperative signaling by α5β1 and α4β1 integrins regulates metalloproteinase gene expression in fibroblasts adhering to fibronectin. J Cell Biol. 1995;129:867–879. doi: 10.1083/jcb.129.3.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes RO. Alterations of cell surface proteins by viral transformation and by proteolysis. Proc Natl Acad Sci USA. 1973;70:3170–3174. doi: 10.1073/pnas.70.11.3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes, R.O. 1990. Fibronectins. Springer-Verlag, New York. 546 pp.
- Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- Hynes RO, Marcantonio EE, Stepp MA, Urry LA, Yee GH. Integrin heterodimer and receptor complexity in avian and mammalian cells. J Cell Biol. 1989;109:409–420. doi: 10.1083/jcb.109.1.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewell K, Kapron-Bras C, Jeevaratnam P, Dedhar S. Stimulation of tyrosine phosphorylation of distinct proteins in response to antibody-mediated ligation and clustering of α3 and α6 integrins. J Cell Sci. 1995;108:1165–1174. doi: 10.1242/jcs.108.3.1165. [DOI] [PubMed] [Google Scholar]
- Jones JCR, Kurpakus MA, Cooper HM, Quaranta V. A function for the integrin α6β4 in the hemidesmosome. Cell Regulation. 1991;2:427–438. doi: 10.1091/mbc.2.6.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajiji S, Tamura RN, Quaranta V. A novel integrin (αEβ4) from human epithelial cells suggest a fourth family of integrin adhesion receptors. EMBO (Eur Mol Biol Organ) J. 1989;8:673–680. doi: 10.1002/j.1460-2075.1989.tb03425.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennel SJ, Foote LJ, Falcioni R, Sonnenberg A, Stringer CD, Crouse C, Hemler ME. Analysis of the tumor-associated antigen TSP-180. Identity with α6-β4 in the integrin superfamily. J Biol Chem. 1989;264:15515–15521. [PubMed] [Google Scholar]
- Kirtschig G, Marinkovitch MP, Burgeson RE, Yancey KB. Antibasement membrane autoantibodies in patients with anti-epiligrin cicatricial pemphigoid bind the α subunit of laminin 5. J Invest Dermatol. 1995;105:543–548. doi: 10.1111/1523-1747.ep12323431. [DOI] [PubMed] [Google Scholar]
- Kivirikko S, McGrath JA, Baudoin C, Aberdam D, Ciatti S, Dunnill MGS, McMillan JR, Eady RAJ, Ortonne J-P, Meneguzzi G, et al. A homozygous nonsense mutation in the α3 chain gene of laminin 5 (LAMA3) in lethal (Herlitz) junctional epidermolysis bullosa. Hum Mol Genet. 1995;4:959–962. doi: 10.1093/hmg/4.5.959. [DOI] [PubMed] [Google Scholar]
- Kornberg LJ, Earp HS, Turner CE, Prockop C, Juliano RL. Signal transduction by integrins: increased protein tyrosine phosphorylation caused by clustering of β1 integrins. Proc Natl Acad Sci USA. 1991;88:8392–8396. doi: 10.1073/pnas.88.19.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreidberg JA, Donovan MJ, Goldstein SL, Rennke H, Shepherd K, Jones RC, Jaenisch R. α3 β1 integrin has a crucial role in kidney and lung organogenesis. Development. 1996;122:3537–3547. doi: 10.1242/dev.122.11.3537. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of the bacteriophage T4. Nature (Lond) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Mainiero F, Pepe A, Wary KK, Spinardi L, Mohammadi M, Schlessinger J, Giancotti F. Signal transduction by the α6β4 integrin: distinct β4 subunit sites mediate recruitment of Shc/Grb2 and associate with the cytoskeleton of hemidesmosomes. EMBO (Eur Mol Biol Organ) J. 1995;14:4470–4481. doi: 10.1002/j.1460-2075.1995.tb00126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcantonio EE, Hynes RO. Antibodies to the conserved cytoplasmic domain of the integrin β1 cytoplasmic domain by site directed mutagenesis. J Cell Biol. 1988;106:1765–1772. doi: 10.1083/jcb.106.5.1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinkovich MP, Lunstrum GP, Burgeson RE. The anchoring filament protein kalinin is synthesized and secreted as a high molecular weight precursor. J Biol Chem. 1992;267:17900–17906. [PubMed] [Google Scholar]
- Martin GR, Timpl R. Laminin and other basement membrane components. Annu Rev Cell Biol. 1987;3:57–85. doi: 10.1146/annurev.cb.03.110187.000421. [DOI] [PubMed] [Google Scholar]
- Mautner VM, Hynes RO. Surface distribution of LETS protein in relation to the cytoskeleton of normal and transformed fibroblasts. J Cell Biol. 1977;75:743–768. doi: 10.1083/jcb.75.3.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JA, Gatalica B, Christiano AM, Owaribe K, McMillan JR, Eady RA, Uitto J. Mutations in the 180-kD bullous pemphigoid antigen (BPAG2), a generalized atrophic benign epidermolysis bullosa. Nat Genet. 1995;11:83–86. doi: 10.1038/ng0995-83. [DOI] [PubMed] [Google Scholar]
- Mueller S, Klaus-Kovtun V, Stanley JR. A 230-kD basic protein is the major bullous pemphigoid antigen. J Invest Dermatol. 1989;92:33–38. doi: 10.1111/1523-1747.ep13070476. [DOI] [PubMed] [Google Scholar]
- Niessen CM, Hogervorst F, Jaspers LH, De Malker AA, Delwel GO, Hulsman EHM, Kuikman I, Sonnenberg A. The α6β4 integrin is a receptor for both laminin and kalinin. Exp Cell Res. 1994;211:360–367. doi: 10.1006/excr.1994.1099. [DOI] [PubMed] [Google Scholar]
- Niessen CM, van der Raaij-Helmer LMH, Hulsman EHM, van der Neut R, Jonkman MF, Sonnenberg A. Deficiency of the integrin β4 subunit in junctional epidermolysis bullosa with pyloric atresia: consequences for hemidesmosome formation and adhesion properties. J Cell Sci. 1996;109:1695–1706. doi: 10.1242/jcs.109.7.1695. [DOI] [PubMed] [Google Scholar]
- Pellegrini G, DeLuca M, Orecchia G, Balzac F, Cremona O, Savoia P, Cancedda R, Marchisio PC. Expression, topography, and function of integrin receptors are severely altered in keratinocytes from involved and uninvolved psoriatic skin. J Clin Invest. 1992;89:1783–1795. doi: 10.1172/JCI115782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltonen J, Larjava H, Jaakkola S, Gralnick H, Akiyama SK, Yamada SS, Yamada KM, Uitto J. Localization of integrin receptors for fibronectin, collagen, and laminin in human skin. J Clin Invest. 1989;84:1916–1923. doi: 10.1172/JCI114379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JH, Hynes RO. Fibronectin isoform distribution in the mouse. Cell Adhesion and Communication. 1996;4:103–125. doi: 10.3109/15419069609010766. [DOI] [PubMed] [Google Scholar]
- Peters JH, Trevithick JE, Johnson P, Hynes RO. Expression of the alternatively spliced EIIIB segment of fibronectin. Cell Adhesion and Communication. 1995;3:67–89. doi: 10.3109/15419069509081278. [DOI] [PubMed] [Google Scholar]
- Pulkkinen L, Christiano AM, Airenne T, Haakana H, Tryggvason K, Uitto J. Mutations in the γ2 chain gene (LAMC2) of kalinin/laminin 5 in the junctional forms of epidermolysis bullosa. Nat Genet. 1994a;6:293–297. doi: 10.1038/ng0394-293. [DOI] [PubMed] [Google Scholar]
- Pulkkinen L, Christiano AM, Gerecke DR, Wagman DW, Burgeson RE, Pittelkow MR, Uitto J. A homozygous nonsense mutation in the β3 chain of laminin 5 (LAMB3) in Herlitz junctional epidermolysis bullosa. Genomics. 1994b;24:357–360. doi: 10.1006/geno.1994.1627. [DOI] [PubMed] [Google Scholar]
- Raugi GJ, Olerud JE, Gown AM. Thrombospondin in early human wound tissue. J Invest Dermatol. 1987;89:551–554. doi: 10.1111/1523-1747.ep12461198. [DOI] [PubMed] [Google Scholar]
- Rheinwald JG, Green H. Serial cultivation of strains of human epidermal keratinocytes: the formation of keratinizing colonies from single cells. Cell. 1975;6:331–343. doi: 10.1016/s0092-8674(75)80001-8. [DOI] [PubMed] [Google Scholar]
- Rouselle P, Lunstrum GP, Keene DR, Burgeson RE. Kalinin: an epithelium-specific basement membrane adhesion molecule that is a component of anchoring filaments. J Cell Biol. 1991;114:567–576. doi: 10.1083/jcb.114.3.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawamura D, Ki K, Nomura K, Sugita Y, Christiano AM, Uitto J. Bullous pemphigoid antigen: cDNA cloning, cellular expression, and evidence for polymorphism of the human gene. J Invest Dermatol. 1991;96:908–915. doi: 10.1111/1523-1747.ep12475433. [DOI] [PubMed] [Google Scholar]
- Sonnenberg A, de Melker AA, Martinez de Velasco AM, Janssen H, Calafat J, Niessen CM. Formation of hemidesmosomes in cells of a transformed murine mammary tumor cell line and mechanisms involved in adherence of these cells to laminin and kalinin. J Cell Sci. 1993;106:1083–1102. doi: 10.1242/jcs.106.4.1083. [DOI] [PubMed] [Google Scholar]
- Sonnenberg A, Janssen H, Hogervorst F, Calafat J, Hilgers J. A complex of platelet glycoprotein Ic and Ia identified by a rat monoclonal antibody. J Biol Chem. 1987;262:10376–10383. [PubMed] [Google Scholar]
- Stepp MA, Spurr-Michaud S, Tisdale A, Elwell J, Gipson IK. α6β4 integrin heterodimer is a component of hemidesmosomes. Proc Natl Acad Sci USA. 1990;87:8970–8974. doi: 10.1073/pnas.87.22.8970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sympson CJ, Talhouk RS, Alexander CM, Chin JR, Clift SM, Bissell MJ, Werb Z. Targeted expression of stromelysin-1 in mammary gland provides evidence for a role of proteinases in branching morphogenesis and the requirement for an intact basement membrane for tissue-specific gene expression. J Cell Biol. 1994;125:681–693. doi: 10.1083/jcb.125.3.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennenbaum T, Yuspa SH, Brover A, Castronovo V, Sobel ME, Yamada Y, DeLuca LM. Extracellular matrix receptors in mouse skin carcinogenesis: altered expression linked to appearance of early markers of tumor progression. Cancer Res. 1992;52:2966–2976. [PubMed] [Google Scholar]
- Timpl R. Structure and biological activity of basement membrane proteins. Eur J Biochem. 1989;180:487–502. doi: 10.1111/j.1432-1033.1989.tb14673.x. [DOI] [PubMed] [Google Scholar]
- Tomaselli KJ, Hall DE, Reichardt LT, Flier LA, Gehlsen KR, Turner DC, Carbonetto S. A neuronal cell line (PC12) expresses two β1-class integrins-α1β1 and α3β1-that recognize different neurite outgrowth domains in laminin. Neuron. 1990;5:651–662. doi: 10.1016/0896-6273(90)90219-6. [DOI] [PubMed] [Google Scholar]
- Tremble P, Chiquet-Ehrismann R, Werb Z. The extracellular matrix ligands fibronectin and tenascin collaborate in regulating collagenase gene expression in fibroblasts. Mol Biol Cell. 1994;5:439–453. doi: 10.1091/mbc.5.4.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uitto, J., L. Pulkkinen, and A.M. Christiano. 1994. Molecular basis of the dystrophic and junctional forms of epidermolysis bullosa: mutations in the type VII collagen and kalinin (laminin 5) genes. J. Invest. Dermatol. 103(suppl): 39S–46S. [DOI] [PubMed]
- Uitto, J., A. Mauviel, and J. McGrath. 1996. The dermal-epidermal basement membrane zone in cutaneous wound healing. In The Molecular and Cellular Biology of Wound Repair. Richard A. F. Clark, editor. Plenum Press, NY. 513–560.
- van der Neut R, Krimpenfort P, Calafat J, Niessen CM, Sonnenberg A. Epithelial detachment due to absence of hemidesmosomes in integrin β4 null mice. Nat Genet. 1996;13:366–369. doi: 10.1038/ng0796-366. [DOI] [PubMed] [Google Scholar]
- Vidal F, Aberdam D, Miquel C, Christiano AM, Pulkkinen L, Uitto J, Ortonne J-P, Meneguzzi G. Integrin β4 mutations associated with junctional epidermolysis bullosa with pyloric atresia. Nat Genet. 1995;10:229–234. doi: 10.1038/ng0695-229. [DOI] [PubMed] [Google Scholar]
- Verrando P, Pisani A, Ortonne JP. The new basement membrane antigen recognized by the monoclonal antibody GB3 is a large size glycoprotein: modulation of its expression by retinoic acid. Biochim Biophys Acta. 1988;942:45–56. doi: 10.1016/0005-2736(88)90273-8. [DOI] [PubMed] [Google Scholar]
- Watt, F.M., and M.D. Hertle. 1994. Keratinocyte integrins. In The Keratinocyte Handbook. I.M. Leigh, E.B. Lane, and F.M. Watt, editors. Cambridge University Press, Cambridge, MA. 153–164.
- Wayner EA, Carter WG. Identification of multiple cell adhesion receptors for collagen and fibronectin in human fibrosarcoma cells possessing unique α and common β subunits. J Cell Biol. 1987;105:1873–1884. doi: 10.1083/jcb.105.4.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitzman JB, Pasqualini R, Takada Y, Hemler ME. The function and distinctive regulation of the integrin VLA-3 in cell adhesion, spreading, and homotypic cell aggregation. J Biol Chem. 1993;268:8651–8657. [PubMed] [Google Scholar]
- Wennerberg K, Lohikangas L, Gullberg D, Pfaff M, Johansson S, Fassler R. β1 integrin-dependent and -independent polymerization of fibronectin. J Cell Biol. 1996;132:227–238. doi: 10.1083/jcb.132.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Chung AE, McDonald JA. A novel role for α3β1 integrins in extracellular matrix assembly. J Cell Sci. 1995;108:2511–2523. doi: 10.1242/jcs.108.6.2511. [DOI] [PubMed] [Google Scholar]
- Xia Y, Gil SG, Carter WG. Anchorage mediated by integrin α6β4 to laminin 5 (epiligrin) regulates tyrosine phosphorylation of a membraneassociated 80-kD protein. J Cell Biol. 1996;132:727–740. doi: 10.1083/jcb.132.4.727. [DOI] [PMC free article] [PubMed] [Google Scholar]