

Abstract
Small GTPases of the Ypt/rab family are involved in the regulation of vesicular transport. These GTPases apparently function during the targeting of vesicles to the acceptor compartment. Two members of the Ypt/rab family, Ypt1p and Sec4p, have been shown to regulate early and late steps of the yeast exocytic pathway, respectively. Here we tested the role of two newly identified GTPases, Ypt31p and Ypt32p. These two proteins share 81% identity and 90% similarity, and belong to the same protein subfamily as Ypt1p and Sec4p. Yeast cells can tolerate deletion of either the YPT31 or the YPT32 gene, but not both. These observations suggest that Ypt31p and Ypt32p perform identical or overlapping functions. Cells deleted for the YPT31 gene and carrying a conditional ypt32 mutation exhibit protein transport defects in the late exocytic pathway, but not in vacuolar protein sorting. The ypt31/ 32 mutant secretory defect is clearly downstream from that displayed by a ypt1 mutant and is similar to that of sec4 mutant cells. However, electron microscopy revealed that while sec4 mutant cells accumulate secretory vesicles, ypt31/32 mutant cells accumulate aberrant Golgi structures. The ypt31/32 phenotype is epistatic to that of a sec1 mutant, which accumulates secretory vesicles. Together, these results indicate that the Ypt31/32p GTPases are required for a step that occurs in the transGolgi compartment, between the reactions regulated by Ypt1p and Sec4p. This step might involve budding of vesicles from the trans-Golgi. Alternatively, Ypt31/ 32p might promote secretion indirectly, by allowing fusion of recycling vesicles with the trans-Golgi compartment.
The passage of proteins through the secretory pathway involves their orderly progression through a series of membranous compartments (Palade, 1975) and is mediated by vesicles that bud from one compartment and fuse with the next (Novick and Brennwald, 1993; Rothman, 1994). Intracellular transport has been extensively studied using yeast genetics, cell-free assays, and neuronal synapses. The combined information indicates that most of the components of the machinery used in constitutive vesicular trafficking in yeast are related to the components that mediate neurotransmitter release (Bennett and Scheller, 1993; Ferro-Novick and Jahn, 1994). Models are beginning to emerge for the action of these components in vesicle budding and in vesicle targeting and fusion. Budding of vesicles is driven by cytosolic proteins that associate transiently with the membrane and form a coat complex. Different budding events in the secretory pathway are mediated by at least three types of coat complexes: clathrin, COPI, and COPII. Another class of coat proteins may be involved in budding of secretory vesicles from the TGN (Jones et al., 1993; Ladinsky et al., 1994; Narula and Stow, 1995). GTP-binding proteins of the ARF/Sar1 family were shown to be part of the COPI and COPII coat complexes and/or to regulate the assembly and disassembly of these complexes through guanine nucleotide cycling (Rothman, 1994, Schekman and Orci, 1996). After coat disassembly, a vesicle is targeted to the appropriate acceptor compartment. A current model for the molecular basis of this targeting reaction proposes the association of a specific vesicle-associated soluble N-ethylmaleimide–sensitive factor attachment protein (SNAP) receptor (v-SNARE)1 with a target-associated SNAP receptor (t-SNARE) (Rothman, 1994; Rothman and Wieland, 1996).
The Ypt/rab/Sec4 family of small GTPases plays an important role in vesicular trafficking both in yeast and in mammalian cells. These GTPases are involved in the regulation of protein transport through the different steps of the exocytic, endocytic, and transcytotic pathways (Pfeffer, 1992; Ferro-Novick and Novick, 1993; Novick and Brennwald, 1993; Zerial and Stenmark, 1993). An important question concerning the mode of action of Ypt/rab proteins is whether they function in vesicle budding, targeting, or fusion. Convincing evidence already exists for an important role for rab proteins in regulating vesicle targeting (Brennwald et al., 1994; Lian et al., 1994; Søgarrd et al., 1994). In yeast, Ypt1p was shown to act after vesicle formation, in the targeting of vesicles to the acceptor compartment in the ER-to-Golgi step (Rexach and Schekman, 1991; Segev, 1991), and Sec4p was shown to act in the targeting of trans-Golgi–derived vesicles to the plasma membrane (Novick and Brennwald, 1993). However, whether Ypt/rab proteins also have a role in vesicle budding still needs to be resolved. Another question concerning Ypt/ rab GTPases is whether they regulate every step of intracellular protein transport. In mammalian cells several rab proteins, including rab1, rab2, rab3, rab6, rab8, and rab11, have been assigned to the different exocytic steps, based on intracellular localization studies and analysis of dominant mutants (Goud et al., 1990). In yeast, a number of Ypt proteins, including Ypt51-53p, Ypt6p, and Ypt7p, have been implicated in the endocytic pathway and in the sorting of proteins to the vacuole (Wichmann et al., 1992; Horazdovski et al., 1994). Interestingly, only two Ypt GTPases have been shown to function in the yeast exocytic pathway: Ypt1p in the first two steps (Segev et al., 1988; Bacon et al., 1989; Baker et al., 1990; Jedd et al., 1995) and Sec4p in the last step (Novick et al., 1981; Goud et al., 1988; Walworth et al., 1989). In this paper, we wished to determine whether two newly identified Ypt proteins have a role in exocytic steps between those regulated by Ypt1p and Sec4p.
Two steps in the yeast exocytic pathway have been studied extensively: ER to cis- and trans-Golgi–derived vesicles to the plasma membrane. A large number of proteins that function in these secretory steps have been identified using genetic screens, but only a few proteins were identified as components of the machinery that functions at intermediate steps of the pathway, during protein transit through the Golgi complex (Novick et al., 1980; Achstetter et al., 1988; Bankaitis et al., 1989). The compartmental organization of the yeast Golgi complex has been characterized primarily by using two mutants, sec18 and sec7, and by cell fractionation studies (Cunningham and Wickner, 1989; Franzusoff and Schekman, 1989; Graham and Emr, 1991). Three functionally distinct Golgi compartments have been defined that contain, from cis to trans: α-1,6mannosyltransferase, α-1,3-mannosyltransferase, and the Kex2 protease. However, very little is known about proteins that mediate and regulate transport between the Golgi compartments. To date, only Sec7p and Sec14p have been suggested to function exclusively within the yeast Golgi (Achstetter et al., 1988; Bankaitis et al., 1989).
In this paper we report the identification of two new Ypt GTPases that belong to the exocytic Ypt/rab subfamily. Using inactivating mutations and intracellular localization experiments, we show that these two proteins have a role in secretory steps that lie between those regulated by Ypt1p and Sec4p and that involve transport through the trans-Golgi compartment. Our results suggest that the Ypt31/32 GTPases have a role in budding of vesicles from the trans-Golgi compartment. These findings provide the first in vivo indication that Ypt/rab GTPases may function not only in vesicle targeting, but also in vesicle budding. A possible implication of our data is that similar molecular processes underlie vesicle fusion and vesicle budding. Alternatively, Ypt31/32 might have a role in fusion of recycling vesicles with the trans-Golgi compartment, a process that is presumably necessary for exit from this compartment.
Materials and Methods
Yeast Strains, Media, and Reagents
Yeast strains used in this study are listed in Table I. NSY313 was derived from NSY290 (ypt31::HIS3) in which the ypt32A141D mutation was targeted to the chromosome (see Site-directed Mutagenesis and Plasmid Construction). NSY 348 was derived from a backcross of NSY313 to NSY125. NSY 368 was derived from a cross of PNY504 to NSY348.
Table I.
Yeast Strains
| Strain | Genotype | Source | ||
|---|---|---|---|---|
| NSY128: | MATα, ade2, his3Δ200, leu2-3,112, lys2-801, ura3-52 | DBY4975 David Botstein | ||
| NSY125: | MATa, his4-539, lys2-801, ura3-52 | DBY1034 David Botstein | ||
| NSY290: | NSY128, Δypt31::HIS3 | This study | ||
| NSY296: | NSY128, Δypt32::HIS3 | This study | ||
| NSY301: | NSY290 transformed with pRS316-YPT31 | This study | ||
| NSY302: | NSY301, Δypt32::KANr | This study | ||
| NSY306: | NSY302, transformed with pRS315-YPT31 | This study | ||
| NSY313: | MATα, ade2, his3Δ200, leu2-3,112, lys2-801, ura3-52, Δypt31::HIS3, ypt32-A141D | This study | ||
| NSY348: | MATa, lys2, his4, ura3-52, Δypt31::HIS3, ypt32-A141D | This study | ||
| NSY368: | MATα, leu2, ura3-52, suc2Δ9, Δypt31::HIS3, ypt32-A141D, sec1-1 | This study | ||
| PNY404: | MATα, his4-619, sec4-8 | Peter Novick, Yale University, New Haven, CT | ||
| PNY504: | MATα, leu2-3,112, ura3-52, suc2Δ9, sec1-1 | Peter Novick | ||
| NSY222: | MATα, his4, ura3-52, ypt1-A136D | Jedd et al., 1995 | ||
| JK9-3d: | MATa/α, leu2/leu2, his4/his4, trp1/trp1, ura3-52/ura3-52 | Mike Hall, Basel, Switzerland |
Yeast cultures were grown in synthetic medium (0.67% nitrogen base without amino acids) supplemented with the appropriate nutritional requirements or 5-fluoro-orotic acid (5-FOA) (Rose et al., 1990), and with glucose added to 2% (wt/vol).
Reagents were obtained from the following sources: Recombinant yeast lytic enzyme, Zymolyase 100T, and 35S-Translabel used for in vivo labeling were from ICN Pharmaceuticals Inc. (Irvine, CA). Secondary antibodies used for immunofluorescence were from Jackson ImmunoResearch (West Grove, PA). Rhodamine-phalloidin was from Molecular Probes (Eugene, OR). Horseradish peroxidase–conjugated goat anti–rabbit IgG used for Western blotting and the enhanced chemiluminescence detection kit were from Amersham International (Buckinghamshire, England). Immobilon-P membrane was from Millipore Corp. (Bedford, MA). Chemical reagents were purchased from Sigma Chemical Co. (St. Louis, MO). The following antibodies were kindly donated: antiinvertase by Chris Kaiser (Massachusetts Institute of Technology, Boston, MA); anti–carboxypeptidase Y (CPY) by Tom Stevens (University of Oregon, Eugene, OR); anti–α-factor by Tod Graham (Vanderbilt University, Nashville, TN); anti-α1,3 mannose by Alex Franzusoff (University of Colorado, Denver, CO).
Gene Deletion
Gene disruption was performed using PCR for replacement of the entire coding region with selectable markers, either HIS3 (Baudin et al., 1993) or the kanr gene (Wach et al., 1994). Oligonucleotides contained 35 bp of target homology at their ends and in all cases oligos were designed such that the structural gene was precisely deleted. For the deletion of YPT31 using HIS3 as a selectable marker, the following oligonucleotides were used: YPT31-HIS3-pro (GCA AAG GGA TTC TGA CGG CGT CTG GGG ATT TCA ACA CTC TTG GCC TCC TCT AG) and YPT31-HIS3-term (TCA CAT GCA AGT GCG CAA CTG CTG CAA AAT ATC TCT CGT TCA GAA TGA CAC G). The deletion of YPT32 was executed using oligonucleotides YPT32-HIS3-pro (TGA TAA AGA GCG AAC CAA GCA TAT TGT TTT CCA AGA CTC TTG GCC TCC TCT AG) and YPT32-HIS3-term (GAC AGA TAT GCA TGA AAC CAT GCG ACC TTA GTA GGG TCG TTC AGA ATG ACA CG) for disruption by HIS3. The kanr gene was used to disrupt YPT32 using the oligos YPT32-kanr-pro (TGA TAA AGA GCG AAC CAA GCA TAT TGT TTT CCA AGA GCA TAG GCC ACT AGT GGA TCT G) and YPT32kanr-term (GAC AGA TAT GCA TGA AAC CAT GCG ACC TTA GTA GGG CAG CTG AAG CTT CGT ACG C). PCR and transformation of yeast strains were performed as described by Baudin et al. (1993). Selection of G418 resistance was performed as described (Wach et al., 1994). Deletions were confirmed using Southern blotting (Rose et al., 1990) and/ or PCR. For the confirmation of YPT31 disruption by HIS3, the following oligos were used: YPT31dv (GCC GGA TCC GAT CTA TCC CAG CGC AAT C) and HIS3-1 (CAC TTG CGA TTG TGT GGC CTG). YPT32 deletion was confirmed using the oligos YPT32dv (GCA TGG TTC GCG CTG GAA AGA G) and HIS3-1, or kanr-1 (GTG AGA ACT GTA TCC TAG CA) for deletion by HIS3 and kanr, respectively.
Site-directed Mutagenesis and Plasmid Construction
The ypt32-A141D mutant was made by site-directed mutagenesis using the method of Kunkel (1985) in the Escherichia coli phagemid vector pBluescript II SK +. Mutagenesis was performed on a partial YPT32 gene generated by PCR using the oligonucleotides YPT32-b4 (CTC CGG TGT GGG TAA ATC TA) and YPT32-XbaI (GCG TCT AGA GGT TAG TAA TAA ATA ACT TG). The PCR fragment was cut with EcoRI and XbaI, and cloned EcoRI to XbaI in BSIISK. The resulting YPT32 gene is truncated by 144 bp at its 5′ end. The mutagenic oligonucleotide was YPT32A-D (CCA ACC GAT GAA GAC AAA AAT TTC GCA ATG). The mutation was confirmed by sequencing, and the gene was transferred to the integrating vector pRS306 (Sikorski and Hieter, 1989) using EcoRI and XbaI. This construct was cut with BglII and targeted to the chromosome in the NSY290 strain (Δypt31::HIS3) as previously described (Segev and Botstein, 1987).
Centromeric YPT31 expression plasmids were constructed as follows: A 1.26-kb HindIII-PstI genomic DNA fragment containing the YPT31 promoter and structural gene was cloned into the multiple cloning site of pBluescript II SK +. The insert was then excised as a BamHI-HindIII fragment and cloned into either pRS316 (URA3) or pRS315 (LEU2) (Sikorski and Hieter, 1989) to yield pRS316::YPT31 (pNS220) and pRS315::YPT31 (pNS221).
Protein Purification
The yeast proteins Ypt31, Ypt32, and Sec4 were expressed in bacteria as glutathione S–transferase (GST) fusions in the bacterial pGEX-KG expression plasmid (reference numbers YPT31, pNS210; YPT32, pNS211; SEC4, pNS212) (Guan and Dixon, 1991). This was done by PCR amplification of plasmid DNA using oligonucleotides that engineer BamHI and XbaI sites upstream and downstream of the YPT genes. In all three cases, the BamHI site was engineered directly upstream of the start codon such that the junction between GST and Ypt protein was identical in all cases. The following oligonucleotides were used (restriction sites are bold): YPT31-1 (GCC GAA TCC ATG AGC AGC GAG GAC TAC GGG) and YPT31-2 (CGC TCT AGA GTC ATT CAC ATG CAA GTG CGC), YPT 32-1 (GCC GGA TCC ATG AGC AAC GAA GAT TAC GGA TAC G) and YPT32-XbaI (GCG TCT AGA GGT TAG TAA TAA ATA ACT TG), SEC4-1 (GCC GGA TCC ATG TCA GGC TTG AGA ACT GTT TC), and SEC4-2 (CGC TCT AGA CCT GAT GAA AAT ACC TTC CAG A). The PCR product was digested with BamHI and XbaI, and cloned into the pGEX-KG vector, which was similarly cut. The construction of the GST–Ypt1 fusion protein has been previously described (Jones et al., 1995). GST-Ypt fusion proteins were purified as previously described (Jones et al., 1995).
Preparation of Antiserum Directed Against Ypt31 and Affinity Purification
Purified fusion protein was used to immunize two New Zealand white rabbits as previously described (Horazdovsky and Emr, 1993). The resulting antisera were tested by immunoblotting of whole cell yeast extracts. Anti-Ypt31p polyclonal antibodies were affinity purified using recombinant Ypt31p immobilized on cyanogen bromide–activated Sepharose (3 mg Ypt31p/ml packed bead volume) as described by the manufacturer (Pharmacia Biotechnology Inc., Piscataway, NJ). For affinity purification, crude rabbit serum was diluted 1:2 with buffer A (20 mM Tris, pH 7.5, 0.1 M NaCl) and bound to immobilized Ypt31p in batch for 3 h at room temperature. Beads were then washed with 20 column volumes of buffer A followed by 10 column volumes of buffer B (20 mM Tris, pH 7.5, 0.5 M NaCl). Immobilized antibody was then eluted with 0.1 M glycine, pH 2.5, elluate was collected in 0.9-ml fractions, which were immediately neutralized with 0.1 ml 1 M Tris, pH 8. Fractions were extensively dialyzed against TBS (10 mM Tris, pH 7.4, 0.1 M NaCl) and then against TBS-40% (vol/vol) glycerol and stored at −20°C. All experiments shown used affinity-purified antibodies.
Immunoblot Analysis
Cell extracts or purified Ypt proteins were resolved on 12% SDS–polyacrylamide gels and transferred onto Immobilon-P in transfer buffer (25 mM Tris, 380 mM glycine, 20% methanol) at 80 V for 1 h at 4°C. Membranes were treated with Ponceau S for 5 min, destained with water, and blocked for 1 h in TBST–5% milk (10 mM Tris, pH 7.4, 100 mM NaCl, 0.05% [vol/vol] Tween-20). Membranes were then probed with anti-Ypt31 (1:2,000) or anti-Ypt1 (1:2,000) in TBST–1% milk for 1 h at room temperature. The membrane was then washed 5 times with TBST–1% milk over the course of 30 min and probed with HRP-conjugated anti–rabbit IgG secondary at 1:5,000 for 1 h. Membranes were washed in TBST–1% milk six times over the course of 30 min and developed for chemiluminescence as described (Amersham International). Chemiluminescence was quantified by densitometry using a Howtek Scanmaster3+ densitometer.
Immunofluorescence and Electron Microscopy
Immunofluorescence microscopy was done as previously described (Pringle et al., 1989), with some modifications. Cells were grown in synthetic minimal medium to an OD600 between 0.5 and 1. Cells were fixed by directly adding 37% formaldehyde to the growth medium to a final concentration of 3.7%. After 15 min at 26°C, cells were pelleted at 2,500 rpm in a low-speed centrifuge and then resuspended in an equal volume of 3.7% formaldehyde in 0.1 M potassium phosphate, pH 6.5, and fixed for an additional 2 h at 26°C with rotation. Cells were washed twice with an equal volume of 0.1 M phosphate buffer and transferred to 1.5-ml Eppendorf tubes. Cells were pelleted at ∼5,000 rpm in a tabletop centrifuge and washed a final time in 1 ml of spheroplasting buffer (1.4 M sorbitol, 0.1 M potassium phosphate, pH 7.5). After this final wash, cells were resuspended in spheroplasting buffer to a final concentration of 1 OD600/200 μl buffer, and 1 M DTT was added to a final concentration of 5 mM. Spheroplasting was initiated by the addition of 10 μl of yeast lytic enzyme (20,000 U/ml) and carried out at 37°C for 1–2 h. Spheroplasts were harvested by centrifugation at 3,000 rpm in a tabletop centrifuge and washed twice in 200 μl spheroplasting buffer. After the final wash, cells were resuspended in 200 μl spheroplasting buffer. 20 μl of cells were seeded onto either multiwell slides or coverslips that had been coated with poly-l-lysine (0.1% wt/vol). Cells were allowed to settle for 5 min, buffer was aspirated, and the slides were allowed to air dry for 20 min.
Slides were blocked for 5 min with TBS-BSA (10 mM Tris, pH 7.4, 100 mM NaCl, 2.5 mg/ml BSA), after which primary affinity-purified antibodies in TBS-BSA were applied at the following dilutions: anti-Ypt1p, 1:500; anti-Ypt31p, 1:200. Incubation was carried out for 1 h. Slides were washed 12 times by rapid aspiration and addition of TBS-BSA. Secondary antibody fluoresceinated anti-rabbit FAB was then applied at 1:50 dilutions. Rhodamine-phalloidin was used at a dilution of 1:20 as described (Pringle et al., 1989). Coverslips or slides were mounted as described (Pringle et al., 1989) and viewed using a microscope (model Axioskop; Carl Zeiss, Inc., Thornwood, NY) equipped with a 100-W power source for epifluorescence (Carl Zeiss, Inc.).
Electron microscopy was performed on cells grown in synthetic minimal medium, where indicated cells were shifted to 37°C for 2 hours. Processing of cells for EM and quantification of micrographs was carried out as previously described (Kaiser and Schekman, 1990). The following rules were used to distinguish small vesicles, large vesicles, and cisternae/Berkeley bodies, all of which were manifested as electron-dense profiles of varying size and shape. Profiles were counted as small vesicles when they were 50–80 nm in diameter and as large vesicles if they were 100–150 nm in diameter. Linear membranous profiles >200 nm and <1 μm in length were scored as cisternae. Multilamellar circular membranous structures >200 nm in diameter were scored as Berkeley bodies (Novick et al., 1981). Thirty cells were counted for each strain. Immunoelectron microscopy was conducted as previously described (Mulholland et al., 1994). The use of anti-Ypt1p antibodies for immunoelectron microscopy has also been described (Preuss et al., 1992).
Cell Labeling and Immunoprecipitation
Cell labeling and immunoprecipitation were carried out as previously described (Jedd et al., 1995) with the following modifications: For the induction of invertase, cells were grown to mid-logarithmic phase in SD plus 2% dextrose (wt/vol) minus methionine and cysteine at 26°C and washed twice in SD plus 0.1% dextrose (wt/vol) minus methionine and cysteine. Cells were resuspended in the same medium at a density of 1 OD600 U/ml and incubated at 26°C for 30 min to induce invertase. For spheroplasting, Zymolyase 100T was used at a concentration of 1.0 μg/OD600 U of cells. Pulse-chase experiments were quantified using a phosphorimaging system (DuPont/New England Nuclear, Wilmington, DE). Quantification in the text refers to results presented in corresponding figures.
Results
Two New Functionally Homologous Exocytic Ypt GTPases
We have found two new YPT genes as high-copy suppressors of the dominant YPT1D124N mutation (Jones et al., 1995; Jones, S., and N. Segev, manuscript in preparation). The sequences of the two genes revealed that they belong to the Ypt/rab family of GTPases and that their protein products share 81% identity and 90% similarity (Fig. 1 A). These genes were submitted to the yeast database by other researchers as YPT31 or YPT8 (Lai et al., 1994) and YPT32. (These sequence data are available from GenBank/EMBL/ DDBJ under accession numbers U18778 and X72834, respectively.) The high degree of sequence similarity between the two genes suggested that they might be functionally homologous. The protein products are most similar to the mammalian rab11 GTPase and to Schizosaccharomyces pombe Ypt3 (62 and 63% identity, respectively), and their closest Saccharomyces cerevisiae homologues are Ypt1 and Sec4 (42–46% identity; Ypt1 and Sec4 share 48% identity). Phylogenetic analysis of all Ypt proteins present in S. cerevisiae, using the completed genome sequence available for this organism, and of their human homologues reveals that Ypt/rab GTPases fall into two subfamilies: exocytic and endocytic/vacuolar. The two new Ypt proteins, Ypt31 and Ypt32, seem to belong to the subfamily of exocytic Ypt/rab GTPases (Fig. 1 B). We wished to study the possible role of Ypt31 and Ypt32 proteins in the yeast exocytic pathway since very little is known about components that regulate the secretory steps between those regulated by the Ypt1 and Sec4 GTPases.
Figure 1.



The YPT31 and YPT32 genes encode two functionally homologous exocytic GTPases. (A) Comparison of the amino acid sequences of Ypt31 and Ypt32 proteins. The predicted protein sequence of Ypt32 was compared to Ypt31 using the MegAlign program (DNAStar Inc., Madison, WI, Clustal method with the PAM250 residue weight table). Identities are shaded with solid black, and residues conserved to within two distance units are shaded. Overall, the two proteins are 81.1% identical and 89.6% similar when compared using the bestfit program (Genetics Computer Group, Madison, WI). (B) Ypt31 and Ypt32 proteins belong to a subfamily of exocytic Ypt GTPases by phylogenetic analysis of the Ypt/rab family of small GTPases. The predicted amino acid sequence of Ypt31 and Ypt32 were compared to all other Ypt proteins, using the completed S. cerevisiae genome sequence and their human homologues. The analysis shows that Ypt/rab proteins fall into two functional subfamilies: those involved in endocytosis and vacuolar protein sorting (Endocytosis-Vac.) and those involved in exocytosis. Sequences were aligned as above. The scale at the bottom indicates the number of substitutions between sequences. hum., Homo sapiens; H-ras, Harvey murine sarcoma virus ras protein; all other sequences, Saccharomyces cerevisiae. These sequence data are available from GenBank/EMBL/DDBJ under accession numbers: Ypt31, U18778; Ypt32, X72834; rab11, X56740; Ypt7, X68144; rab7, U44104; Ypt1, X00209; rab1a, M28209; Sec4, M16507; Ypt6, U17244; rab6a, M28212; Ypt51, X76173; Ypt52, X76174; Ypt53, X76175; rab5b, X54871; and H-ras, X00740. (C) Requirement of YPT31 or YPT32 genes for cell viability. The YPT31 gene was precisely deleted using the HIS3 gene, and this strain was transformed with a URA3-marked CEN vector containing the YPT31 gene under control of its own promoter (NSY301, first row). This strain (NSY301) was subsequently deleted for the YPT32 gene using the KANr gene as a dominant delectable marker (NSY302, middle row). Finally, this strain (NSY302) was transformed with a second plasmid marked with LEU2 and carrying the YPT31 gene (NSY306, bottom row). The three strains were grown in synthetic media maintaining selection for plasmids. Serial dilutions of cells were then spotted onto either SD or SD-FOA and grown at 26°C. Cells deleted for both genes do not grow on SD-FOA plates, indicating that they cannot lose the URA-marked YPT31 plasmid unless they carry also the LEU-marked YPT31 plasmid.
To determine whether the YPT31 and YPT32 genes are essential for viability, we studied the effect of their deletion on cell growth. YPT31 was deleted by precise replacement of its entire coding region with the HIS3 gene, and YPT32 by its precise replacement with either the kanr or the HIS3 gene. The replacement of both genes was confirmed by PCR analysis (not shown). Cells deleted for either YPT31 or YPT32 alone are viable (see Fig. 3 B). Thus, neither gene is essential for viability. The effect of deletion of YPT31 and/or YPT32 genes on cell growth was determined also by analyzing the ability of the cells to lose the YPT31 gene carried on a plasmid marked with URA3. Cells deleted for YPT31 and carrying the YPT31 gene on a plasmid can grow on 5-FOA plates, indicating that the cells can lose the plasmid (Fig. 1 C, top line). However, cells deleted for both YPT31 and YPT32 genes and carrying the YPT31 gene on a plasmid do not grow on 5-FOA plates, indicating that they cannot lose the YPT31 gene carried by the plasmid (Fig. 1 C, middle line). These cells can grow on 5-FOA plates if they contain in addition another plasmid (marked with LEU2) that carries the YPT31 gene (Fig. 1 C, bottom line). Together, these results show that while neither gene is required for cell growth, at least one of them is necessary for viability. These data and the high degree of homology between the two genes suggest that they are functional homologues or perform overlapping functions.
Figure 3.


The YPT32-A141D mutation is in a conserved residue and confers a tight conditional growth phenotype. (A) Comparison of the amino acid sequence of the α-helix 5 region in Ypt/rab and ras proteins. Arrow indicates conserved residue, which when mutated to Asp results in temperature-sensitive function in Ypt1 (ypt1-A136D), Ypt32 (ypt32-A141D), and Sec4 (sec4-8, sec4-G147D). (B) The ypt32-A141D mutation confers a temperaturesensitive growth phenotype in cells deleted for the YPT31 gene. Serial dilutions of wild-type (NSY128), Δypt31 (NSY290), Δypt32 (NSY296), and Δypt31,ypt32-A141D (NSY313) cells were spotted on YPD plates and incubated at 26° or 37°C.
Ypt31/32 GTPases Exhibit a Novel Intracellular Localization Pattern
To study the intracellular expression and localization of Ypt31/32 GTPases, we purified recombinant Ypt31 and Ypt32 proteins and raised polyclonal antibodies against Ypt31p. The bacterially purified proteins both migrate on a denaturing gel with apparent molecular mass of ∼23 kD, with Ypt31p migrating somewhat more slowly than Ypt32p (Fig. 2 A, top). The antibody recognizes both bacterially produced proteins by immunoblot analysis, but its level of detection of Ypt31p is about threefold higher than that for Ypt32p. The antibodies did not react with the closest Ypt31/32p homologues, Ypt1p and Sec4p (Fig. 2 A, bottom). The affinity-purified anti-Ypt31p antibodies reveal one protein band in wild-type yeast cells. This band corresponds to Ypt31p since it is absent in cells deleted for the YPT31 gene and is more abundant in cells expressing the YPT31 gene on a 2μ high copy number plasmid. However, the antibodies also recognize Ypt32p when it is expressed from a 2μ plasmid in a YPT31-null strain (Fig. 2 B). Comparing the levels of Ypt31 and Ypt32 proteins in cells expressing them from a 2μ plasmid, in a strain deleted for YPT31 gene, suggests that Ypt31 is considerably more abundant than Ypt32 in yeast cells (about 5- to 10-fold more abundant, taking into account a threefold better detection of Ypt31p by the antibody). Fractionation of yeast cells followed by immunoblot analysis revealed that ∼90% of the Ypt31p molecules are associated with the membranous fraction (100,000 g pellet; data not shown).
Figure 2.



Ypt31 protein is found at sites of polarized cell growth. (A) Characterization of anti-Ypt31 antibodies. Antibodies raised against Ypt31p react with Ypt31p, and to a lesser degree with Ypt32p, but not Ypt1 and Sec4 proteins. Bacterially purified Ypt1, Ypt31, Ypt32, and Sec4 proteins were analyzed by SDS-PAGE followed by either Coomassie staining (1 μg protein in each lane) or Western blotting (5 ng protein in each lane) using anti-Ypt1 or anti-Ypt31 antibodies. (B) Detection of Ypt31 and Ypt32 proteins in yeast cell extracts: Ypt31p is present in excess of Ypt32p in wild-type yeast cells. Wild-type (NSY128) or deletion strains (Δypt31, NSY 290 and Δypt32, NSY296) transformed with the indicated plasmids were grown in synthetic medium maintaining selection for deletions and plasmids present. Cells were lysed by the addition of 2× Laemmli buffer and boiling for 5 min. 0.5 OD600 U of lysate from each strain was then subjected to immunoblot analysis using affinity-purified anti-Ypt31 antibodies. (C) Localization of the Ypt31 protein in yeast cells using immunofluorescence microscopy. Diploid yeast cells (JK9-3d) were double stained with rhodamine-phalloidin (to visualize filamentous actin) and with anti-Ypt31 antibodies. Cells were also photographed using Nomarski optics to distinguish budded from unbudded cells (top). Arrowheads indicate polarized Ypt31 staining, which localizes to regions of cell growth in (from left to right) a cell in late G1, a cell in early S phase, and a cell undergoing cytokinesis. Bar, 10 μm.
We examined the localization of Ypt31/32 proteins in growing wild-type yeast cells using immunofluorescence microscopy with the affinity-purified anti-Ypt31/32 antibodies. Filamentous actin (F-actin) was also visualized to label the presumptive bud site and identify cells undergoing cytokinesis. Two prominent staining features were observed; punctate staining, which is typical of the yeast Golgi (Segev et al., 1988; Franzusoff et al., 1989; Redding et al., 1991), and localization to the presumptive bud site and small buds. Sec4p, which resides on late secretory vesicles, also localizes to small buds (Novick and Brennwald, 1993). The asymmetric distribution of Ypt31p seen in some unbudded cells is due to the localization of this protein to the bud emergence site, which is marked by F-actin. In addition, late mitotic cells frequently showed Ypt31p localization at the site of cytokinesis (Fig. 2 C). Thus, periods in the cell cycle which are characterized by highly polarized growth, late G1/early S phase and cytokinesis (Lew and Reed, 1993), show polarized Ypt31p staining. In contrast, cells in early G1 (unbudded cells with random actin staining) and late G2 (large-budded cells) show punctate Ypt31p staining. These patterns of staining primarily reflect the distribution of Ypt31p since there is almost no signal in cells deleted for the YPT31 gene. However, it seems that the localization of Ypt32p is similar to that observed for Ypt31p since the very faint staining seen in ypt31-null cells is similar to that seen in wild-type cells (data not shown). The fact that the Ypt31p localization resembles both that of Golgi and late secretory vesicles suggests that Ypt31/32 GTPases localize to a late secretory compartment. Since to the best of our knowledge, no Golgi marker, including the trans-Golgi Kex2 protease, has been shown by immunofluorescence microscopy to be dramatically concentrated in sites of cell growth, we suggest that the polarized staining of Ypt31p reflects its localization to post-Golgi secretory vesicles. These vesicles were shown by electron microscopy to localize to sites of cell growth (Matile et al., 1969; Byers, 1981).
The ypt32-A141D Mutation Confers a Block in the trans-Golgi Compartment
To study the role of Ypt31/32 GTPases in the yeast secretory pathway, we constructed a conditional lethal ypt32A141D recessive mutation that confers temperature sensitivity for growth in cells deleted for the YPT31 gene. This mutation was constructed by analogy to the ypt1-A136D mutation, which was characterized previously in our lab (Jedd et al., 1995). The mutation changes an amino acid that is conserved in structure (a small, uncharged residue, A, S, or G) among Ypt/rab proteins (Fig. 3 A). By analogy to the tertiary structure of ras, this amino acid is predicted to be on the hydrophobic face of a conserved amphipathic α-helix (Tong et al., 1989). This mutation conferred a tight and rapid block of Ypt1p function (Jedd et al., 1995). Since the level of Ypt1-A136D mutant protein does not change after incubation at the restrictive temperature (Jedd et al., 1995), it is likely that this mutation encodes a protein that undergoes a temperature-sensitive change in function. The analogous mutation in SEC4-G147D (sec4-8; Moya et al., 1993) also confers a tight temperature-sensitive block in secretion of invertase (data not shown). We predicted that introducing an analogous mutation in other Ypt/rab proteins might result in a temperature-sensitive loss-of-function phenotype. As shown below, the ypt32-A141D mutation confers conditional blocks in growth and secretion. Therefore, this substitution has proven to be a useful tool for obtaining a conditional mutation in three different Ypt proteins and might be useful for determining the functions of other Ypt/rab proteins or of ras-like proteins in general.
Cells carrying the ypt32-A141D mutation and deleted for the YPT31 gene exhibit a tight temperature sensitivity for growth at 37°C (Fig. 3 B). We tested the effect of this mutation on the transport of two secreted proteins, invertase and α-factor, and a vacuolar protein, CPY. To determine the effect of the ypt31Δ/32-A141D (ypt31/32) mutation on protein transport at the permissive temperature, we compared the processing of invertase at 26°C in wildtype and mutant cells. Cells were labeled at 26°C for 7 min, and then chased for 10 or 30 min. Spheroplasts were separated from periplasmic contents, and invertase was immunoprecipitated from both fractions and analyzed by gel electrophoresis. It is possible to distinguish between invertase that resides in early secretory compartments (ER; core glycosylated and cis-Golgi; α-1,6-mannosylated), invertase in medial- or trans-Golgi (α-1,3-mannosylated), and secreted invertase (periplasmic). After 10 min of chase, wildtype cells secrete most (∼90%) of the invertase labeled during the pulse. ypt31/32 mutant cells also secrete most of the invertase (81%), which is fully glycosylated, but only after 30 min of chase (Fig. 4 A). Thus, the ypt31/32 mutant cells display a kinetic defect in secretion at the permissive temperature.
Figure 4.

ypt31-Δ/ypt32-A141D mutant cells exhibit a tight conditional secretory block of invertase transport downstream of the medial-Golgi. Wild-type (NSY349, left) and mutant (NSY348, right) cells were labeled for 7 min at 26°C. Cells were then chased at 26°C (A) or shifted to 37°C (B) with prewarmed media and chased for the indicated times. At each time point, cells were separated from the periplasmic fraction by spheroplasting, lysates were prepared, and samples were immunoprecipitated with antiinvertase antibodies. Equal portions of precipitate from each time point were then separated on 8% SDS–polyacrylamide gels. (C) Intracellular invertase from the indicated time point was divided into three equal portions and precipitated with antisera against invertase, α1,6-mannose, or α1,3-mannose residues as indicated. ER (core), Golgi (outerchain), and cytoplasmic (cyto.) forms of invertase are indicated in the left margin.
To determine the effect of the ypt31/32 mutations on protein transport at the nonpermissive temperature, the different secretory compartments were marked with radiolabeled transport intermediates of the marker proteins using a brief pulse under permissive conditions, and the cells were then chased at the nonpermissive temperature. First, to examine the effect of the mutation on the ability of cells to secrete a marker protein, we followed the processing of invertase at the restrictive temperature. Wild-type and mutant cells were labeled at the permissive temperature (26°C) for 7 min and then shifted to the nonpermissive temperature (37°C) and chased for 10 or 30 min. In wildtype and mutant cells, the various secretory compartments were loaded with the different forms of invertase during the pulse, as seen by the presence of core- and fully glycosylated invertase (Fig. 4 B, chase time 0′). After a 10-min chase of wild-type cells, all the forms that were present during the pulse were converted to the mature fully glycosylated form and were secreted. (87% was secreted.) Invertase in ypt31/32-mutant cells shifted to the nonpermissive temperature was also converted to the fully glycosylated form, as shown by reprecipitation of invertase from mutant cells with antisera specific for cis- and medialGolgi modifications (α-1,6- and α-1,3-mannose linkages) (Fig. 4 C). However, mutant cells at the nonpermissive temperature exhibited a complete block of secretion of invertase even after 30 min of chase (93% remained inside the cells; Fig. 4 B). This secretory defect is much more dramatic than the decrease in the rate of secretion observed at the permissive temperature. These results suggest that the ypt31/32 mutations confer a primary block in the secretion of invertase in a step between the medial-Golgi and the plasma membrane.
To distinguish between a block in the medial- or transGolgi compartments, which contain α-1,3-mannosyltransferase and Kex2 protease, respectively, we followed the processing of α-factor. This protein contains recognition sites for the Kex2 protease. Wild-type, ypt31-Δ/ypt32-A141D, and sec4-G147D mutant cells were labeled at the permissive temperature (26°C) for 7 min, shifted to the nonpermissive temperature (37°C), and chased for 10 or 30 min. Cells were separated from the medium, and α-factor was immunoprecipitated and analyzed by gel electrophoresis. In wild-type and mutant cells, the various secretory compartments were loaded with the different forms of α-factor during the pulse, as seen by the presence of core, α-1,6modified, α-1,3-modified, and mature forms (Fig. 5, chase time 0′). In wild-type cells, even after a short chase, all of these forms were converted to the mature form. The limitation of using α-factor as a marker is that the mature form cannot be quantitatively detected, probably because of degradation, and the analysis of the processing is thus restricted to determining the disappearance of the intermediate forms. sec4-G147D mutant cells exhibited a defect in secretion of mature α-factor consistent with a defect in the fusion of post-Golgi vesicles with the plasma membrane (Fig. 5 A, mαf). Like sec4-G147D mutant cells, ypt31-Δ/ ypt32-A141D mutant cells were defective in the secretion of mature α-factor peptide but were also partially defective at earlier stages of the secretory pathway. In addition, both mutants secreted some α-factor that had not received proteolytic processing, indicating an additional defect in the function of the trans-Golgi (Fig. 5 A, arrows). However, this trans-Golgi processing defect is probably minor and secondary, since the immature forms accumulated after the chase in both mutant strains represent only a minor fraction of the α-factor labeled during the pulse (time 0). Because a significant amount of α-factor is secreted under the conditions of this experiment, we examined the secretion of α-factor after a 2-min preshift to the nonpermissive temperature. Under these conditions, less α-factor is secreted to the medium in both the sec4 and ypt31/32 mutants (Fig. 5 B), although the ypt31/32 mutant is still slightly leaky. Together, the results suggest that ypt31/32 mutant cells, like sec4 mutant cells, are defective in processes that occur in the exit from the trans-Golgi, or within this compartment.
Figure 5.

ypt31-Δ/ypt32-A141D (NSY313) and sec4-G147D (PNY404) mutant cells both show defects in the trans-Golgi processing and transport of α-factor at the nonpermissive temperature. (A) The indicated strains (wild-type is NSY128) were labeled for 7 min at 26°C, shifted to 37°C with prewarmed media, and chased for the indicated times. Cells were separated from medium by centrifugation and processed for immunoprecipitation with anti–α-factor antibodies. (B) Alternatively, cells were preshifted to 37°C for 2 min with prewarmed medium, labeled for 7 min and chased for the indicated times. Only mature peptide is shown, as higher molecular weight forms of α-factor behave similarly to those shown in A, except that both mutants secreted proportionally less high molecular weight α-factor after the preshift, and all strains (including wild-type) show translocation and processing defects. Positions of ER (core), cis-Golgi (α16), medial-Golgi (α1-3), and trans-Golgi (mαf) forms are noted in the left margin. Arrows in the right margin indicate secreted α-factor that was not fully processed in the trans-Golgi.
We wished to determine whether transport to the vacuole is affected by the ypt31/32 mutations. Proteins destined for the vacuole are sorted from secreted proteins at the trans-Golgi compartment (Graham and Emr, 1991). Thus, CPY processing is dependent on the function of SEC genes that function at all steps between ER and the transGolgi, and on VPS genes that regulate sorting at the transGolgi, but not on SEC genes that function at the post- trans-Golgi step of the exocytic pathway (Stevens et al., 1982). To examine CPY processing, the secretory compartments were loaded during a pulse at the permissive temperature with the various labeled forms of CPY (the p1 form in the ER, p2 form in the Golgi, and mature form in the vacuole) and then shifted to 37°C for the chase. In wild-type cells, all of the CPY was converted to the mature form after 30 min of chase. ypt31/32 mutant cells sorted most of their CPY to the vacuole as seen by the accumulation of the mature form (80% when compared to wildtype). However, CPY processing was somewhat slower in mutant cells than in wild-type cells: The ER form (p1) was still present after 10 min chase, and some CPY remained as the p2 form after 30 min of chase at the nonpermissive temperature in mutant, but not in wild-type cells (Fig. 6). Mutations in VPS genes result in secretion of the p2 form of CPY (Banta et al., 1988). Since no CPY was secreted by ypt31/32 mutant cells at the nonpermissive temperature, Ypt31/32 GTPases do not seem to function in the sorting of vacuolar proteins (Fig. 6). These results suggest that there is no major defect in the processing of vacuolar CPY in ypt31/32 mutant cells. The defect revealed by the accumulation of the p2 form might reflect a secondary defect at the trans-Golgi compartment after a 30-min incubation at the nonpermissive temperature, or a decrease in the rate of CPY processing through the whole pathway in mutant cells. Alternatively, Ypt31/32 GTPases might have a role in more than one step between the medial-Golgi and the plasma membrane.
Figure 6.

ypt31-Δ/ypt32-A141D mutant cells do not exhibit a major defect in targeting of CPY to the vacuole at the nonpermissive temperature. Extracts from the 37°C samples shown in Fig. 4 were subjected to precipitation with anti-CPY antibodies and resolved on 8% SDS–polyacrylamide gels. Positions of the ER (p1), Golgi (p2), and vacuolar (m) forms of CPY are indicated in the left margin.
We have established that the Ypt31/32 GTPases function in the yeast exocytic pathway. Our results suggest that these GTPases function at the level of the trans-Golgi compartment, in trans-Golgi to plasma membrane transport, and perhaps also in medial- to trans-Golgi transport. The secretory phenotype conferred by the ypt31/32 mutation resembles that exhibited by the analogous sec4 mutation. However, the electron microscopic analysis of the ypt31/32 mutant cells described below indicates that Ypt31/32 GTPases function at a step distinct from that regulated by Sec4p.
Accumulation of Aberrant Golgi Structures in ypt31/32 Mutant Cells
The fact that the secretory defect of ypt31/32 mutant cells is similar to that of sec4 cells could indicate that Ypt31/32 and Sec4 GTPases function in the same process, i.e., fusion of secretory vesicles with the plasma membrane. Alternatively, they might have roles in two different secretory steps or substeps, e.g., Ypt31/32 might function in vesicle budding or fusion at the trans-Golgi compartment, and Sec4 in fusion of trans-Golgi–derived vesicles with the plasma membrane. In general, sec mutants have been shown to accumulate aberrant membranes of secretory compartments that precede the step in which they first function (Novick et al., 1981; Kaiser and Schekman, 1990). Thus, sec4 mutant cells have been shown to accumulate secretory vesicles (Novick et al., 1980; Walworth et al., 1989). To determine whether ypt31/32 mutant cells accumulate such vesicles or whether they accumulate other membranous structures, cells were shifted from the permissive temperature (26°C) to the nonpermissive temperature (37°C) for 2 h and examined by electron microscopy. We quantified the accumulation of various aberrant membrane structures: small vesicles (50–80 nm), aberrant Golgi (cisternae and Berkeley bodies), and large vesicles (100–150 nm) (Fig. 7 E). We compared the effect of the ypt1-A136D mutation and the analogous mutations in YPT31/32 and SEC4 on the type of membrane accumulated. Unlike sec4-G147D mutant cells, which accumulate post-Golgi vesicles almost exclusively (Fig. 7, D and E), ypt31Δ/32-A141D mutant cells exhibited an abundance of aberrant Golgi-like structures (Fig. 7, C and E) and some ER (Fig. 7 C, arrowhead). This phenotype is also distinct from that observed for ypt1A136D mutant cells, which accumulate ER membranes and small secretory vesicles (Fig. 7 B), but not Golgi.
Figure 7.
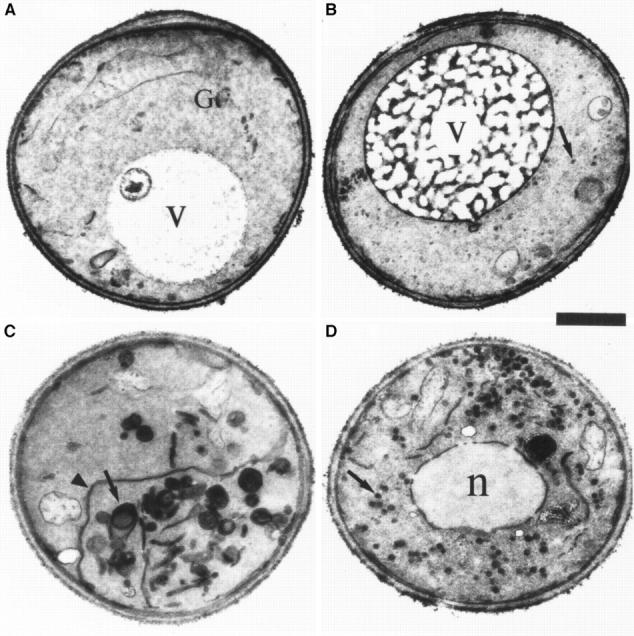

ypt31-Δ/ypt32A141D mutant cells accumulate aberrant Golgi-like structures at the nonpermissive temperature. Wild-type and three strains mutated at the analogous conserved residue in YPT1, YPT32, and SEC4 were analyzed by electron microscopy. Electron micrographs of representative cells are shown for the following strains: (A) wildtype (NSY128), (B) ypt1A136D (NSY222), (C) ypt31Δ/ypt32-A141D (NSY348), and (D) sec4-G147D (PNY 404). Cells were grown at 26°C, shifted to 37°C for 2 h, and then processed for thin section electron microscopy. Arrows point to membranous structures unique to each strain. Arrowheads indicate ER. G, wild-type Golgi cisterna (shown in detail in Fig. 9 A, second panel from left); V, vacuole; n, nucleus. (E) Quantification of the distinct membranous structures that accumulate in the different mutant strains: small vesicles (50–80 nm), Golgi (cisternae or Berkeley bodies), and large vesicles (100–150 nm). Bars represent the mean number of structures in 30 cell sections. Data is normalized to density per cubic micrometer for vesicle populations and density per 10 μm2 for cisternae and Berkeley bodies. Error bars represent one standard deviation (see Materials and Methods for details of the quantification procedure). The relatively high standard deviations are probably due to the aggregation of the aberrant membranes to one side of the cell, resulting in cell sections that are either rich in or devoid of the corresponding membranous structure. Bar, 1 μm.
ypt31-Δ/32-A141D mutant cells display a steady state increase in the frequency of Golgi cisternae even at the permissive temperature (26°C) when compared to wild-type cells (Fig. 8 A, B, and D). This result indicates that the kinetic defect in protein transport seen in the mutant at the permissive temperature (Fig. 4 A) is accompanied by some accumulation of Golgi cisternae. However, a more dramatic change was observed in mutant cells shifted to the nonpermissive temperature (37°C). Under this growth condition, the total number of aberrant Golgi profiles roughly doubled, and the population became dominated by Berkeley bodies (60% of total at 37°C vs. 30% of total at 26°C; Fig. 8 D). These multilamellar structures, in which one cisterna appears to engulf another, were first observed in sec7 and sec14 mutant cells, which are defective in Golgi function (Novick et al., 1981). Mutant Golgi structures are larger than those seen in wild-type cells (385 ± 107 nm, and 298 ± 81 nm, respectively), are frequently observed to form stacks, and are occasionally swollen to ∼100 nm at their periphery (Fig. 9 B), indicating a possible defect in vesicle formation.
Figure 8.
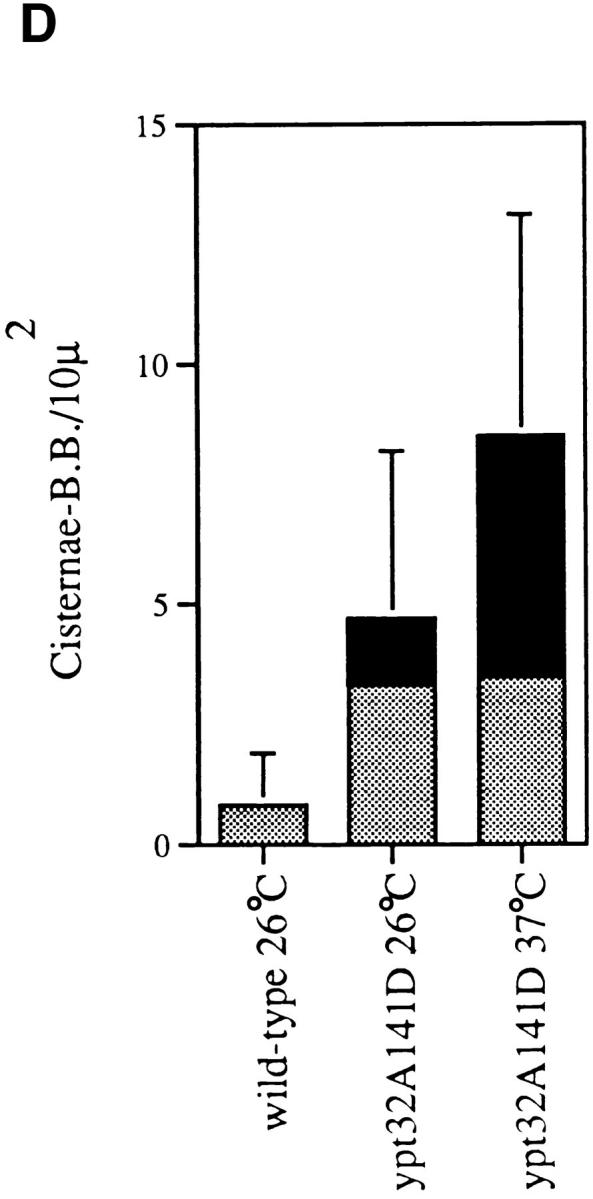


ypt31-Δ/ypt32A141D mutant cells accumulate Golgi membranes at the permissive temperature and Berkeley bodies at the nonpermissive temperature. Electron microscopy of (A) wild-type (NSY128), (B) ypt31-Δ/ypt32-A141D mutant strain (NSY348) at 26°C, and (C) ypt31-Δ/ypt32A141D mutant strain (NSY348) at 37°C. Arrows indicate a single Golgi cisterna in a wild-type cell and regions of Golgi accumulation in mutant cells. v, vacuole; n, nucleus. (D) Quantification of cisternal profiles and Berkeley bodies in wildtype cells at 26°C and ypt31Δ/ypt32-A141D mutant (NSY348) cells at 26° and 37°C. The percentage of the total structures counted that are cisternal is indicated by the gray bar, and the percentage of Berkeley bodies is represented by the black bar. At the permissive temperature, the mutant shows a fivefold increase in the frequency of Golgi profiles, the majority of which (70% of total) are cisternal. After 2 h at 37°C, the number of Golgi profiles has doubled and the population is dominated by Berkeley bodies (60% of total). (E) Immunofluorescence microscopy using anti-Ypt1p antibodies (Segev et al., 1988). (1) wild-type cells (NSY128) grown at 26°C; (2) the ypt31-Δ/ypt32-A141D mutant (NSY348) grown at 26°C; or (3) shifted to 37°C, for 90 min; (4) the same cells shown in 3 photographed with Nomarski optics to show the contours of the cells. Bars: (A–C) 1 μm; (E) 10 μm.
Figure 9.
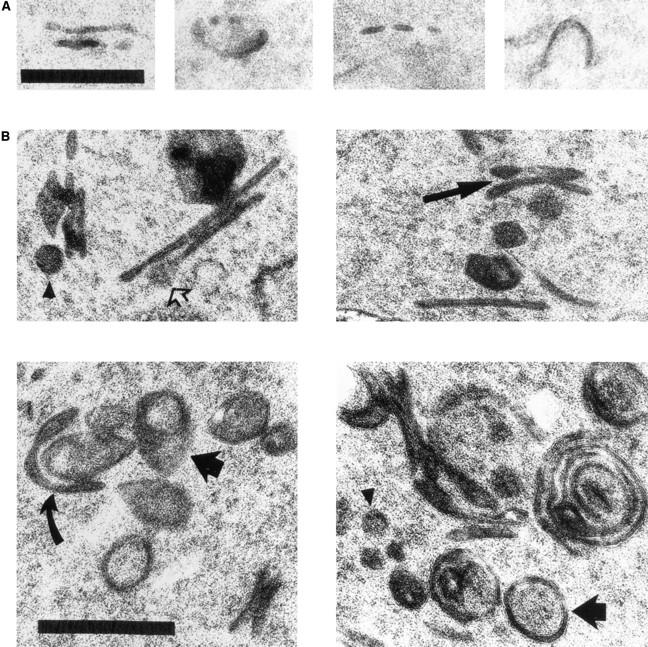
Details of Golgi structures in wild-type and ypt31-Δ/ypt32-A141D mutant cells. (A) Electron microscopy of wild-type (NSY128) cells. Wild-type Golgi is usually observed as an elongated or cup-shaped single cisternae (Preuss et al., 1992), which are not always continuous (third panel from left) because of fenestration (Rambourg et al., 1993). (B) Enlarged ypt31-Δ/ypt32-A141D mutant (NSY348) Golgi structures. Long arrows indicate an example of stacked cisternae in the mutant strain. Short arrows indicate spherical and tear drop–shaped Berkeley bodies. The tear-shaped structure suggests that Berkeley bodies are derived from individual cup-shaped cisternae that fuse at the rim (see curved arrow for a possible intermediate in this process). Open arrow indicates swelling to 100 nm at the cisternal rim, which may represent an intermediate preceding vesicle formation by membrane fission. Arrowheads indicate rare 100nm vesicles seen in the vicinity of cisternae, for size comparison with structures that look like budding vesicles. Note also the increased length of cisternal profiles in the mutant strain compared to wild-type. Regions of four different cells are shown for each strain. Bars, 0.5 μm.
A combination of immunofluorescence and immunoelectron microscopy was used to identify the origin of the aberrant membrane structures accumulated in ypt31Δ/32A141D mutant cells. In electron micrographs, these structures usually seem to cluster within one area of the cell. A similar clustering was visualized by immunofluorescence microscopy as a change in the Ypt1p staining pattern, from the wild-type punctate to a mutant asymmetric pattern (Fig. 8 E). To show that the structures visualized by immunofluorescence correspond to those seen by electron microscopy, we performed immunoelectron microscopy using affinity-purified antibodies against Ypt1p. After a 60-min incubation of the ypt31/32 mutant cells at 37°C, the cisternal and spherical structures that accumulate in these cells both contain Ypt1p on their membranes (Fig. 10). These membranes do not label with antibodies against the vacuolar H+-ATPase subunit Vph1p (data not shown), indicating that they differ from the multilamellar structures that accumulate in the class E vps28 mutant (Rieder et al., 1996). Because the aberrant membrane structures present in ypt31Δ/32-A141D mutant cells appear to label with the established Golgi marker Ypt1p (Preuss et al., 1992), and because similar structures are seen in sec mutants defective in Golgi function (Novick et al., 1981), we conclude that these structures are Golgi cisternae.
Figure 10.

Ypt1p is present on aberrant Golgi membranes accumulated in the ypt31-Δ/ypt32-A141D mutant cells. ypt31-Δ/ypt32A141D mutant cells were grown at 26°C and shifted to 37°C for 1 h before fixation and processing for immunoelectron microscopy using anti-Ypt1p antisera. Gold particles are seen specifically labeling stacked cisternae (long arrow) and spherical structures (short arrow). Note a strand of ER that is unlabeled (arrowhead) and the lack of gold particles over mitochondria (m) and cytosol. Bar, 250 nm.
To further test this interpretation, we constructed a ypt31Δ/32-A141D, sec1-1 triple mutant and determined the type of aberrant membranes that accumulated. The sec1-1 mutant has been shown to accumulate post-Golgi vesicles similar to those observed in the sec4-8 mutant (Harsay and Bretscher, 1995). If Ypt31/32p function in exit from the Golgi, the triple mutant should display the ypt31/32 mutant phenotype at the nonpermissive temperature. As controls we examined the ypt31/32 mutant and a triple mutant strain that had been transformed with YPT31 on a single copy vector. (This strain is hereafter referred to as the sec1-1 mutant.) While the sec1-1 mutant accumulated post-Golgi vesicles (Fig. 11 B), the triple mutant accumulated aberrant Golgi structures similar to those observed in the ypt31/32 mutant (Fig. 11, C and A, respectively). Thus, the ypt31/32 double mutation is epistatic to the sec1-1 mutation. However, the sec1-1 mutation appears to exacerbate the ypt31/32 phenotype. First, while the ypt31/32 mutant accumulated some ER (see Fig. 7 C, arrowhead), the triple mutant accumulated significant levels of fenestrated ER membranes that were continuous with the nuclear envelope (Fig. 11 C, arrowheads). Second, the triple mutant was inviable at 30°C, while both ypt31/32 and sec1-1 mutant cells were viable at this temperature (data not shown). sec1-1, like all late sec mutants, is defective in the uptake of the lipophilic dye FM4-64 from the plasma membrane, indicating a possible function for Sec1p in membrane recycling (Vida and Emr, 1995). One explanation for the synergistic effect in the triple mutant is that membrane accumulation due to the ypt31/32 mutations is enhanced by the defect in membrane recycling imposed by the sec1-1 mutation. Together, the ensemble of results presented show that Ypt31/32 GTPases function in a secretory step distinct from that regulated by Sec4p, probably in the exit of secretory proteins from the trans-Golgi compartment.
Figure 11.

The ypt31-Δ/ ypt32-A141D, sec1-1 triple mutant accumulates Golgi structures. Electron microscopy of (A) ypt31-Δ/ypt32A141D (NSY348) mutant, (B) the ypt31-Δ/ypt32A141D, sec1-1 triple mutant transformed with YPT31 on a single copy vector (phenotypically sec1-1), and (C) the ypt31-Δ/ypt32-A141D, sec1-1 (NSY368) triple mutant. Arrows point to structures characteristic of each strain; arrowheads indicate ER that can be seen in continuity with the nuclear envelope in the triple mutant, but are also present in ypt31-Δ/ ypt32-A141D mutant cells. n, nucleus. Bar, 1 μm.
Discussion
In this study, we show that two novel GTPases, Ypt31p and Ypt32p, have a role in the yeast exocytic pathway, in a step between those regulated by two other Ypt/rab GTPases, Ypt1p and Sec4p. The sequence similarity between Ypt31p and Ypt32p, and the fact that the presence of only one of them is required for cell viability, strongly suggests they are functional homologues. We propose that Ypt31p and Ypt32p function in exit from the trans-Golgi compartment (Fig. 12). This conclusion is based on phenotypic analysis of inactivating mutations in these genes, a comparison of ypt1, ypt31/32, and sec4 mutant strains by electron microscopy and epistasis analysis using the ypt31/32 mutant and a late secretory sec1 mutant.
Figure 12.
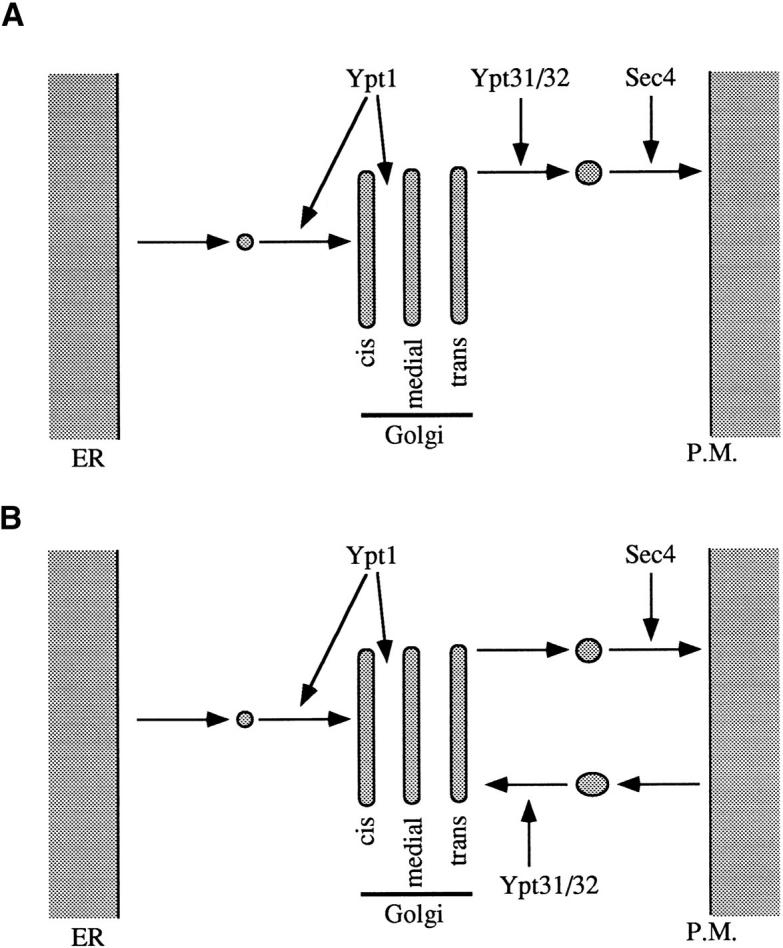
Model for the action of Ypt GTPases in the yeast exocytic secretory pathway. Ypt1p and Sec4p are essential for three secretory steps: ER to cis-Golgi, cis- to medial-Golgi, and transGolgi to the plasma membrane (Novick et al., 1981; Jedd et al., 1995). For two of these steps, it was shown that Ypt1 and Sec4 proteins function in targeting of ER- and trans-Golgi–derived vesicles, respectively (Novick et al., 1981; Rexach and Schekman, 1991; Segev, 1991). (A) Ypt31/32 proteins are suggested to have a role in promoting vesicle budding from the trans-Golgi. (B) Alternatively, Ypt31/32 may regulate a fusion step between the trans-Golgi and retrograde vesicles derived from a recycling compartment (see Discussion).
The inference that Ypt31/32 proteins are required for execution of a step(s) in the exocytic pathway is based on the tight, late-Golgi block in the secretion of invertase exhibited by ypt31/32 mutant cells. Thus, fully glycosylated invertase (α-1,6- and α-1,3- mannosylated) accumulates in these cells, indicating that the block must be after invertase arrives at the medial-Golgi compartment. As described below, our results using two other markers are most consistent with a role of Ypt31/32 GTPases in transport of proteins from the trans-Golgi compartment. First, the resemblance of the defect of α-factor processing in ypt31/32 mutant cells to that of sec4 mutant cells suggests that the primary secretory defect is at or beyond the trans-Golgi compartment. Second, there is no major block in transport of CPY to the vacuole, nor is CPY secreted to the medium. Finally, the ypt31/32 mutant phenotype is epistatic to that of sec1-1. Thus, Ypt31/32 proteins function upstream of the post-Golgi vesicle targeting step, and by inference upstream of Sec4p. These data suggest that the defect in the ypt31/32 mutant cells occurs after the arrival of secretory proteins at the trans-Golgi compartment and during their transport to the plasma membrane, but not to the vacuole. ypt31/32 mutant cells do show slight defects of CPY and α-factor transport during earlier steps of the secretory pathway and some accumulation of ER membranes, but these effects are probably indirect. A defect in a late secretory step might affect an earlier step indirectly, by blocking the recycling of transport factors back to an early compartment.
One possible mechanism that would involve a role for Ypt31/32p in exit from the trans-Golgi is that these GTPases function in the budding of secretory vesicles from the trans-Golgi compartment (Fig. 12 A). This suggestion is based on the accumulation of aberrant Golgi structures and the lack of accumulation of transport vesicles in ypt31/ 32 mutant cells, both at the permissive temperature (when the secretory phenotype is very mild) and after a long incubation at the nonpermissive temperature. In contrast, defects in Ypt1 and Sec4 proteins result in accumulation of 50- and 100-nm vesicles, respectively. Two observations support a role for Ypt31/32p in budding from the transGolgi. First, ypt31/32 mutant Golgi profiles are elongated relative to wild-type Golgi profiles. This result is consistent with a block in export of membranous material. Second, swollen membranous lobes as large as 100 nm, occasionally seen at the periphery of the mutant Golgi structures (see Fig. 9 B, open arrow), are consistent with a defect in membrane fission required for vesicle budding. An analogous electron microscopic approach was used to classify the early ER-to-Golgi Sec proteins into two distinct sets that function either in vesicle formation or in vesicle fusion (Kaiser and Schekman, 1990); this study was supported by subsequent in vitro analyses (Rexach and Schekman, 1991; Lupashin et al., 1996). Similarly, in vitro analysis is needed to confirm a role for Ypt/rab proteins in vesicle budding. For example, mammalian cell-free systems for post-TGN secretory vesicle formation (Tooze and Huttner, 1990; Jones et al., 1993; Deretic et al., 1996) could be used to analyze the role of Ypt31/32 proteins, or their mammalian homologues, in this process. Ypt/rab proteins have previously been implicated in vesicle budding, although the evidence was not conclusive. First, rab1 was suggested to act at the vesicle budding stage during in vitro ER-to-Golgi transport (Plutner et al., 1991; Peter et al., 1994); however, its yeast homologue, Ypt1p, has a somewhat different function (Rexach and Schekman, 1991; Segev, 1991). We have suggested that Ypt1p has a single site of action at the cis-Golgi compartment but is required for two successive transport steps (ER to cis-Golgi and cis- to medial-Golgi), regulating both fusion with and budding from the cis-Golgi (Jedd et al., 1995). Second, rab6 is associated with a protein complex, and using immunodepletion, this complex was shown to be needed for TGNderived vesicle budding in vitro. However, rab6 itself was not shown to be essential for this process (Jones et al., 1993). Third, GDP dissociation inhibitor abolishes vesicle formation in a cell-free system, indicating a possible role for rabs in this process (Deretic et al., 1996).
If Ypt/rab GTPases function both in vesicle budding and in vesicle targeting/fusion, how might they be acting? It has been suggested that Ypt/rab proteins regulate the formation of a SNARE complex that mediates vesicle fusion (Ferro-Novick and Jahn, 1994; Sollner, 1995; Rothman and Wieland, 1996). Another GTPase subfamily, Arf/Sar1, was shown to regulate coat assembly and disassembly during vesicle budding (Schekman and Orci, 1996). A common theme is that GTPases regulate protein complex assembly and/or disassembly. We hypothesize that Ypt/rab GTPases regulate such events during both vesicle budding and vesicle fusion, perhaps by interacting with the same set of partner proteins in each case. For example, disassembly of the SNARE complex may be required to reactivate the SNAREs for a new reaction cycle (Mayer et al., 1996). Ypt/rab proteins might regulate this disassembly during vesicle budding, while regulating assembly of the SNARE complex during vesicle fusion.
Previous studies have conclusively demonstrated a role for Ypt/rab proteins in regulating vesicle targeting (see introduction). Thus, an alternative interpretation of our data is that Ypt31/32, like Ypt1 and Sec4 GTPases, promote a targeting/fusion event in the Golgi. Two such mechanisms can be envisioned: (a) Ypt31/32 proteins may regulate an event in which cisternae fuse directly. This model implies that the fusion event does not involve vesicles because vesicles do not accumulate in ypt31/32 mutant cells. In addition, this alternative model implies the existence of an additional compartment beyond the Kex2p-containing transGolgi compartment in yeast because our results indicate that Ypt31/32 proteins mediate a step beyond the Kex2pcontaining compartment. Since there is no evidence for nonvesicular fusion within the yeast Golgi, nor for the existence of an additional secretory compartment beyond the Kex2p-containing compartment, this explanation currently seems unlikely. However, below we discuss a modification of this idea that involves a possible late-Golgi/ early endosome compartment. (b) Ypt31/32 proteins may regulate the fusion of retrograde vesicles that recycle components essential for the trans-Golgi budding machinery (Fig. 12 B). If the level of these components is limiting at the trans-Golgi, a tight and rapid exocytic block will occur when Ypt31/32 proteins are inactivated. Moreover, if the formation of such vesicles requires continuous input of membrane from exocytosis, they would not accumulate appreciably upon inactivation of Ypt31/32p. Although uncoupling of recycling to the trans-Golgi from exocytosis was shown for the ypt6 null mutant (Tsukada and Gallwitz, 1996), we cannot rule out the possibility that these processes are coupled in our experimental system.
Immunofluorescence microscopy revealed a novel staining pattern for Ypt31/32p: a polarized staining in sites of cell growth, plus a random punctate staining characteristic of the Golgi. This localization pattern shares similarities with the Ypt1p and Sec4p localization patterns, which are distinct from each other (Segev et al., 1988; Novick and Brennwald, 1993). The Ypt31/32p staining pattern is consistent with the possibility that these proteins act in secretory steps between those regulated by the Ypt1 and Sec4 GTPases. Ypt31/32p staining does not overlap significantly with that of the trans-Golgi marker Kex2p, nor with early Golgi markers (Ypt1p, Mnn1p, Pmr1p; data not shown). The reason for the lack of overlap with Kex2p staining is not clear. One possibility is that Ypt31/32 proteins associate with the Golgi transiently to promote vesicle budding and then remain bound to transport vesicles that are translocated to sites of exocytosis. This possibility is supported by the localization of Ypt31/32p to sites of cell growth. Localization to sites of cell growth has been shown for postGolgi secretory vesicles in yeast (Matile et al., 1969; Byers, 1981). The electron microscopic analysis of ypt31/32 mutant cells suggests that Ypt31/32 proteins function to promote the formation of vesicles from the trans-Golgi, but if Ypt31/32 proteins are present on exocytic transport vesicles, it is possible that these proteins also function after vesicle budding.
The closest homologues of Ypt31/32 GTPases in other species are mammalian rab11 and S. pombe Ypt3p. S. pombe Ypt3p was shown to be essential for cell viability (Miyake and Yamamoto, 1990). The rab11 amino acid sequence is 62% identical to that of Ypt31/32 proteins, but it is not clear that this level of sequence homology implies functional homology. Rab11 was shown to be associated with the TGN and TGN-derived vesicles of both the constitutive and regulated secretory pathways (Urbe et al., 1993; Deretic et al., 1996; Hori et al., 1996). The localization of rab11 is thus in agreement with our suggestion concerning the localization of Ypt31/32 GTPases and might indicate that rab11 and Ypt31/32 are functional homologues. Recently, rab11 protein was implicated in controlling traffic through the recycling endosome, which recycles plasma membrane proteins (Ulrich et al., 1996). If recycling to the trans-Golgi is coupled with exit from this compartment, it is not clear which of these reactions Ypt31/32p or rab11 regulates. One possibility is that exocytosis and endocytosis converge in a late-Golgi/early-endosomal compartment and that transport through this compartment is regulated by these GTPases. In mammalian cells there is evidence for the existence of an endosomal intermediate for a subset of post-Golgi exocytic transport events (Futter et al., 1995; Leitinger et al., 1995). In such a case, some of the membranous structures that we observe in the ypt31/32 mutant cells may represent yeast analogs of this compartment. In the future, the use of newly defined protein markers of the yeast endocytic pathway (Chuang and Schekman, 1996; Hicke and Riezman, 1996) will allow us to test this idea using the ypt31/32 mutant.
We present a model for the role of four Ypt GTPases in the yeast secretory pathway (Fig. 12). In this model, Ypt1p functions in ER to cis-Golgi and cis- to medial-Golgi transport, mediating vesicle targeting in the ER to cis-Golgi step; Ypt31/32 proteins function in vesicle budding from, or targeting to, the trans-Golgi; and Sec4p functions in targeting of secretory vesicles to the plasma membrane. There is one step, medial- to trans-Golgi, for which no Ypt GTPase has been assigned. Although it is possible that Ypt31/ 32p also have a role in this step (see partial early secretory defect, Figs. 5 and 6), they do not seem to be essential for this step, based on the α-factor secretory defect (Fig. 5). We searched the complete S. cerevisiae genome sequence for the presence of additional exocytic Ypt proteins and did not find any. However, Ypt6/rab6 proteins have similar homology to both the endocytic and exocytic Ypt/rab proteins, and the function of these Ypt/rab family members is uncertain. While Ypt6p was implicated in vacuolar protein sorting (Tsukada and Gallwitz, 1996), rab6 was suggested to be involved in intra-Golgi transport or in budding of vesicles from the TGN (Jones et al., 1993; Martinez et al., 1994). One possibility is that Ypt6p functions in the medial- to trans-Golgi step in yeast.
Studying Ypt31/32 in conjunction with the two other known exocytic Ypt GTPases, Ypt1p and Sec4p, should allow us to analyze possible interactions between these regulators. For example, we found YPT31 and YPT32 as suppressors of dominant YPT1 mutations (Jones, S., and N. Segev, manuscript in preparation). In addition, questions can now be asked regarding the specificity of accessory factors that regulate the nucleotide cycling of Ypt/rab proteins. Moreover, since very little is known about protein transport out of the yeast Golgi compartment, Ypt31/32 proteins will serve as a handle for the further characterization of this process.
Acknowledgments
We are grateful to B. Glick, S. Jones, R. Dubreuil, A. Turkewitz, and A. Adams for helpful discussions and critical reading of the manuscript. We thank C. Kaiser, T. Stevens, T. Graham, and A. Franzusoff for generous gifts of antibodies and the Electron Microscopy Lab at the University of Chicago and Yi-mei Chen for excellent help with electron microscopy.
Footnotes
1. Abbreviations used in this paper: 5-FOA, 5-fluoro-orotic acid; CYP, carboxypeptidase Y; F-actin, filamentous actin; GST, glutathione S-transferase; t- and v-SNARE, target- and vesicle-associated soluble N-ethylmaleimide-sensitive factor attachment protein receptors.
G. Jedd was supported by the National Institutes of Health (NIH) predoctoral training grant No. 5T32 GM 07151-20, and J. Mullholland was supported by grant GM46406 to David Botstein (Stanford University, Stanford, CA). This research was supported by grant GM45444 from NIH to N. Segev.
Address all correspondence to Nava Segev, Department of Pharmacological and Physiological Sciences, The University of Chicago, 947 East 58th Street, Box 271, Chicago, IL 60637. Tel.: (773) 702-3526. Fax: (773) 7023774.
References
- Achstetter T, Franzusoff A, Field C, Schekman R. SEC7encodes an unusual, high molecular weight protein required for membrane traffic from the yeast Golgi apparatus. J Biol Chem. 1988;263:11711–11717. [PubMed] [Google Scholar]
- Bacon RA, Salminen A, Ruohola H, Novick P, Ferro-Novick S. The GTP-binding protein Ypt1 is required for transport in vitro: the Golgi apparatus is defective in ypt1mutants. J Cell Biol. 1989;109:1015–1022. doi: 10.1083/jcb.109.3.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker D, Wuestehube L, Schekman R, Botstein D, Segev N. GTP-binding Ypt1 protein and Ca2+ function independently in a cell-free transport reaction. Proc Natl Acad Sci USA. 1990;87:355–359. doi: 10.1073/pnas.87.1.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankaitis VA, Malehorn DE, Emr SD, Greene R. The Saccharomyces cerevisiae SEC14gene encodes a cytosolic factor that is required for transport of secretory proteins from the yeast Golgi complex. J Cell Biol. 1989;108:1271–1281. doi: 10.1083/jcb.108.4.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banta LM, Robinson JS, Klionsky DJ, Emr SD. Organelle assembly in yeast: Characterization of yeast mutants defective in vacuolar biogenesis and protein sorting. J Cell Biol. 1988;107:1369–1383. doi: 10.1083/jcb.107.4.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudin A, Ozier-Kalogeropoulos O, Denouel A, Lacroute F, Cullin C. A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae. . Nucleic Acids Res. 1993;21:3329–3330. doi: 10.1093/nar/21.14.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MK, Scheller RH. The molecular machinery for secretion is conserved from yeast to neurons. Proc Natl Acad Sci USA. 1993;90:2559–2563. doi: 10.1073/pnas.90.7.2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennwald P, Kearns B, Champion K, Keranen S, Bankaitis V, Novick P. Sec9 is a SNAP-25-like component of a yeast SNARE complex that may be the effector of Sec4 function in exocytosis. Cell. 1994;79:245–258. doi: 10.1016/0092-8674(94)90194-5. [DOI] [PubMed] [Google Scholar]
- Byers, B. 1981. Cytology of the yeast life cycle. In The Molecular Biology of the Yeast Saccharomyces: Life Cycle and Inheritance. J.N. Strathern, E.W. Jones, and R.J. Broach, editors. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 59–96.
- Chuang JS, Schekman RW. Differential trafficking and timed localization of two chitin synthase proteins, Chs2p and Chs3p. J Cell Biol. 1996;135:597–610. doi: 10.1083/jcb.135.3.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham KW, Wickner WT. Yeast KEX2 protease and mannosyltransferase I are located to distinct compartments of the secretory pathway. Yeast. 1989;5:25–33. doi: 10.1002/yea.320050105. [DOI] [PubMed] [Google Scholar]
- Deretic D, Puleo-Scheppke B, Trippe C. Cytoplasmic domain of rhodopsin is essential for post-Golgi vesicle formation in a retinal cell free system. J Biol Chem. 1996;271:2279–2286. doi: 10.1074/jbc.271.4.2279. [DOI] [PubMed] [Google Scholar]
- Ferro-Novick S, Jahn R. Vesicle fusion from yeast to man. Nature (Lond) 1994;370:191–193. doi: 10.1038/370191a0. [DOI] [PubMed] [Google Scholar]
- Ferro-Novick S, Novick P. The role of GTP-binding proteins in transport along the exocytic pathway. Annu Rev Cell Biol. 1993;9:575–599. doi: 10.1146/annurev.cb.09.110193.003043. [DOI] [PubMed] [Google Scholar]
- Franzusoff A, Schekman R. Functional compartments of the yeast Golgi apparatus are defined by the sec7mutation. EMBO (Eur Mol Biol Organ) J. 1989;8:2695–2702. doi: 10.1002/j.1460-2075.1989.tb08410.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzusoff A, Redding K, Crosby J, Fuller SR, Schekman R. Localization of components involved in protein transport and processing through the yeast Golgi apparatus. J Cell Biol. 1989;112:27–37. doi: 10.1083/jcb.112.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futter CE, Connolly CN, Cutler DF, Hopkins CR. Newly synthesized transferrin receptors can be detected in the endosome before they appear on the cell surface. J Biol Chem. 1995;270:10999–11003. doi: 10.1074/jbc.270.18.10999. [DOI] [PubMed] [Google Scholar]
- Goud B, Salminen A, Walworth NC, Novick P. A GTP-binding protein required for secretion rapidly associates with secretory vesicles and the plasma membrane in yeast. Cell. 1988;53:753–768. doi: 10.1016/0092-8674(88)90093-1. [DOI] [PubMed] [Google Scholar]
- Goud B, Zahraoui A, Tavitian A, Saraste J. Small GTP-binding protein associated with Golgi cisternae. Nature (Lond) 1990;345:553–556. doi: 10.1038/345553a0. [DOI] [PubMed] [Google Scholar]
- Graham TR, Emr SD. Compartmental organization of Golgi-specific protein modification and vacuolar protein sorting events defined in a yeast sec18 (NSF) mutant. J Cell Biol. 1991;114:207–218. doi: 10.1083/jcb.114.2.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan KL, Dixon JE. Eukaryotic proteins expressed in Escherichia coli: an improved thrombin cleavage and purification procedure of fusion proteins with glutathione S-transferase. Anal Biochem. 1991;192:262–267. doi: 10.1016/0003-2697(91)90534-z. [DOI] [PubMed] [Google Scholar]
- Harsay E, Bretscher A. Parallel secretory pathways to the cell surface in yeast. J Cell Biol. 1995;131:297–310. doi: 10.1083/jcb.131.2.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicke L, Riezman H. Ubiquitination of a yeast plasma membrane receptor signals its ligand stimulated endocytosis. Cell. 1996;84:277–287. doi: 10.1016/s0092-8674(00)80982-4. [DOI] [PubMed] [Google Scholar]
- Horazdovsky B, Emr S. The VPS16gene product associates with a sedimentable protein complex and is essential for vacuolar protein sorting in yeast. J Biol Chem. 1993;268:4953–4962. [PubMed] [Google Scholar]
- Horazdovsky FB, Busch RG, Emr SD. VPS21encodes a rab5like GTP binding protein that is required for the sorting of yeast vacuolar proteins. EMBO (Eur Mol Biol Organ) J. 1994;13:1297–1309. doi: 10.1002/j.1460-2075.1994.tb06382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori Y, Takeyama Y, Hiroyoshi M, Ueda T, Maeda A, Ohyanagi H, Saitoh Y, Kaibuchi K, Takai Y. Possible involvement of Rab11 p24, a ras-like small GTP-binding protein, in intracellular vesicular transport of isolated pancreatic acini. Dig Dis Sci. 1996;41:133–138. doi: 10.1007/BF02208595. [DOI] [PubMed] [Google Scholar]
- Jedd G, Richardson CJ, Litt RJ, Segev N. The Ypt1 GTPase is essential for the first two steps of the yeast secretory pathway. J Cell Biol. 1995;131:583–590. doi: 10.1083/jcb.131.3.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Litt RJ, Richardson CJ, Segev N. Requirement of nucleotide exchange for Ypt1 GTPase mediated protein transport. J Cell Biol. 1995;130:1051–1061. doi: 10.1083/jcb.130.5.1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SM, Crosby JR, Salamero J, Howell KE. A cytosolic complex of p62 and rab6 associates with TGN38/41 and is involved in budding of exocytic vesicles from the trans-Golgi network. J Cell Biol. 1993;122:775–788. doi: 10.1083/jcb.122.4.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser CA, Schekman R. Distinct sets of SEC genes govern transport vesicle formation and fusion early in the secretory pathway. Cell. 1990;61:723–733. doi: 10.1016/0092-8674(90)90483-u. [DOI] [PubMed] [Google Scholar]
- Kunkel TA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci USA. 1985;82:488–492. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladinsky MS, Kremer JR, Furcinitti PS, McIntosh JR, Howell KE. HVEM tomography of the trans-Golgi network: structural insights and identification of a lace-like vesicle coat. J Cell Biol. 1994;127:29–38. doi: 10.1083/jcb.127.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai HM, Bard M, Kirsch RD. Identification of a gene encoding a new Ypt/Rab-like monomeric G-protein in Saccharomyces cerevisiae. . Yeast. 1994;10:399–402. doi: 10.1002/yea.320100313. [DOI] [PubMed] [Google Scholar]
- Leitinger B, Hille-Rehfeld A, Spiess M. Biosynthetic transport of the asialoglycoprotein receptor H1 to the cell surface occurs via endosomes. Proc Natl Acad Sci USA. 1995;92:10109–10113. doi: 10.1073/pnas.92.22.10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew DJ, Reed SI. Morphogenesis in the yeast cell cycle, regulation by Cdc28 and cyclins. J Cell Biol. 1993;120:1305–1320. doi: 10.1083/jcb.120.6.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian JP, Stone S, Jiang Y, Lyons P, Ferro-Novick S. Ypt1p implicated in v-SNARE activation. Nature (Lond) 1994;372:698–701. doi: 10.1038/372698a0. [DOI] [PubMed] [Google Scholar]
- Lupashin VV, Hamamoto S, Schekman RW. Biochemical requirements for the targeting and fusion of ER-derived transport vesicles with purified yeast Golgi membranes. J Cell Biol. 1996;132:277–289. doi: 10.1083/jcb.132.3.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez O, Schmidt A, Salamero J, Hoflack B, Roa M, Goud B. The small GTP-binding protein rab6 functions in intra-Golgi transport. J Cell Biol. 1994;127:1575–1588. doi: 10.1083/jcb.127.6.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matile, P., H. Moor, and C.F. Robinow. 1969. Yeast cytology. In The Yeasts. A.H. Rose, J.S. Harrison, editors. Academic Press, New York. 1:219–302.
- Mayer A, Wickner W, Haas A. Sec18 (NSF)-driven resease of Sec17p (α-SNAP) can precede docking and fusion of yeast vacuoles. Cell. 1996;85:83–94. doi: 10.1016/s0092-8674(00)81084-3. [DOI] [PubMed] [Google Scholar]
- Miyake S, Yamamoto M. Identification of ras-related, YPT family genes in Schizosaccharomyces pombe. . EMBO (Eur Mol Biol Organ) J. 1990;9:1417–1422. doi: 10.1002/j.1460-2075.1990.tb08257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moya M, Roberts D, Novick P. DSS4-1 is a dominant suppressor of sec4-8that encodes a nucleotide exchange protein that aids Sec4p function. Nature (Lond) 1993;361:460–463. doi: 10.1038/361460a0. [DOI] [PubMed] [Google Scholar]
- Mulholland J, Preuss D, Moon A, Wong D, Drubin D, Botstein D. Ultrastructure of the yeast cytoskeleton and its association with the plasma membrane. J Cell Biol. 1994;125:381–391. doi: 10.1083/jcb.125.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narula N, Stow J. Distinct coated vesicles labeled for p200 bud from trans-Golgi network membranes. Proc Natl Acad Sci USA. 1995;92:2874–2878. doi: 10.1073/pnas.92.7.2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick P, Brennwald P. Friends and family: the role of the Rab GTPases in vesicular traffic. Cell. 1993;75:597–601. doi: 10.1016/0092-8674(93)90478-9. [DOI] [PubMed] [Google Scholar]
- Novick P, Field C, Schekman R. Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway. Cell. 1980;21:205–215. doi: 10.1016/0092-8674(80)90128-2. [DOI] [PubMed] [Google Scholar]
- Novick P, Ferro S, Schekman R. Order of events in the yeast secretory pathway. Cell. 1981;25:461–469. doi: 10.1016/0092-8674(81)90064-7. [DOI] [PubMed] [Google Scholar]
- Palade G. Intracellular aspects of the process of protein synthesis. Science (Wash DC) 1975;189:347–358. doi: 10.1126/science.1096303. [DOI] [PubMed] [Google Scholar]
- Peter F, Huoffer C, Pind SN, Balch WE. Guanine nucleotide dissociation inhibitor is essential for Rab1 function in budding from the endoplasmic reticulum and transport through the Golgi stack. J Cell Biol. 1994;126:1393–1406. doi: 10.1083/jcb.126.6.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer S. GTP-binding proteins in intracellular transport. Trends Cell Biol. 1992;2:41–46. doi: 10.1016/0962-8924(92)90161-f. [DOI] [PubMed] [Google Scholar]
- Plutner H, Cox AD, Pind S, Khosravi-Far R, Bourne JR, Schvaninger R, Der CJ, Balch B. Rab1b regulates vesicular transport between the endoplasmic reticulum and successive Golgi compartments. J Cell Biol. 1991;115:31–43. doi: 10.1083/jcb.115.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preuss D, Mulholland J, Franzusoff A, Segev N, Botstein D. Characterization of the SaccharomycesGolgi complex through the cell cycle by immunoelectron microscopy. Mol Biol Cell. 1992;3:789–803. doi: 10.1091/mbc.3.7.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pringle JR, Preston RA, Adams AEM, Stearns T, Drubin D, Haarer BK, Jones E. Fluorescence microscopy methods for yeast. Methods Cell Biol. 1989;31:357–435. doi: 10.1016/s0091-679x(08)61620-9. [DOI] [PubMed] [Google Scholar]
- Rambourg A, Clermont Y, Kepes F. Modulation of the Golgi apparatus in Saccharomyces cerevisiaesec7 mutants as seen by three-dimensional electron microscopy. Anat Rec. 1993;237:441–452. doi: 10.1002/ar.1092370402. [DOI] [PubMed] [Google Scholar]
- Redding K, Holcomb C, Fuller SR. Immunolocalization of Kex2 protease identifies a putative late Golgi compartment in the yeast Saccharomyces cerevisiae. . J Cell Biol. 1991;113:527–538. doi: 10.1083/jcb.113.3.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rexach MF, Schekman RW. Distinct biochemical requirements for the budding, targeting, and fusion of ER-derived transport vesicles. J Cell Biol. 1991;114:219–229. doi: 10.1083/jcb.114.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder SE, Banta LM, Kohrer K, McCaffery MJ, Emr SD. Multilamellar endosome-like compartment accumulates in the yeast vps28vacuolar protein sorting mutant. Mol Biol Cell. 1996;7:985–999. doi: 10.1091/mbc.7.6.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose, M., F. Winston, and P. Heiter. 1990. Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 198 pp.
- Rothman JE. Mechanisms of intracellular protein transport. Nature (Lond) 1994;372:55–63. doi: 10.1038/372055a0. [DOI] [PubMed] [Google Scholar]
- Rothman JE, Wieland FT. Protein sorting by transport vesicles. Science (Wash DC) 1996;272:227–234. doi: 10.1126/science.272.5259.227. [DOI] [PubMed] [Google Scholar]
- Schekman R, Orci L. Coat proteins and vesicle budding. Science (Wash DC) 1996;271:1526–1538. doi: 10.1126/science.271.5255.1526. [DOI] [PubMed] [Google Scholar]
- Segev N. Mediation of the attachment or fusion step in vesicular transport by the GTP-binding Ypt1 protein. Science (Wash DC) 1991;252:1553–1556. doi: 10.1126/science.1904626. [DOI] [PubMed] [Google Scholar]
- Segev N, Botstein D. The ras-like yeast YPT1 gene is itself essential for growth, sporulation, and starvation response. Mol Cell Biol. 1987;7:2367–2377. doi: 10.1128/mcb.7.7.2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segev N, Mulholland J, Botstein D. The yeast GTP-binding YPT1 protein and a mammalian counterpart are associated with the secretion machinery. Cell. 1988;52:915–924. doi: 10.1016/0092-8674(88)90433-3. [DOI] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. . Genetics. 1989;19:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Søgarrd M, Tani K, Ye RR, Geromanos S, Tempst P, Kirchhausen T, Rothman JE, Söllner T. A rab protein is required for the assembly of SNARE complexes in the docking of transport vesicles. Cell. 1994;78:937–948. doi: 10.1016/0092-8674(94)90270-4. [DOI] [PubMed] [Google Scholar]
- Sollner T. SNAREs and targeted membrane fusion. FEBS (Fed Exp Biol Organ) Lett. 1995;369:80–83. doi: 10.1016/0014-5793(95)00594-y. [DOI] [PubMed] [Google Scholar]
- Stevens T, Esmon B, Schekman R. Early stages in the yeast secretory pathway are required for transport of carboxypeptidase Y to the vacuole. Cell. 1982;30:439–448. doi: 10.1016/0092-8674(82)90241-0. [DOI] [PubMed] [Google Scholar]
- Tong L, de Vos AM, Milburn MV, Jancarik J, Noguchi S, Nishimura S, Miura K, Ohisuka E, Kim S-H. Structural differences between a ras oncogene protein and the normal protein. Nature (Lond) 1989;337:90–93. doi: 10.1038/337090a0. [DOI] [PubMed] [Google Scholar]
- Tooze SA, Huttner WB. Cell-free protein sorting to the regulated and constitutive secretory pathways. Cell. 1990;60:837–847. doi: 10.1016/0092-8674(90)90097-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada M, Gallwitz D. Isolation and characterization of SYSgenes from yeast, multicopy suppressors of the functional loss of the transport GTPase Ypt6p. J Cell Sci. 1996;109:2471–2481. doi: 10.1242/jcs.109.10.2471. [DOI] [PubMed] [Google Scholar]
- Ulrich O, Reinsch S, Urbe S, Zerial M, Parton RG. Rab11 regulates recycling through the pericentriolar recycling endosome. J Cell Biol. 1996;135:913–924. doi: 10.1083/jcb.135.4.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbe S, Buber LA, Zerial M, Tooze SA, Parton RG. Rab11, a small GTPase associated with both constitutive and regulated secretory pathways in PC12 cells. FEBS (Fed Exp Biol Soc) Lett. 1993;334:175–182. doi: 10.1016/0014-5793(93)81707-7. [DOI] [PubMed] [Google Scholar]
- Vida TA, Emr SD. A new vital stain for visualizing vacuolar membrane dynamics and endocytosis in yeast. J Cell Biol. 1995;128:779–792. doi: 10.1083/jcb.128.5.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wach A, Brachat A, Pohlmann R, Philippsen P. New heterologous modules for classical or PCR–based gene disruptions in Saccharomyces cerevisiae. . Yeast. 1994;10:1793–1808. doi: 10.1002/yea.320101310. [DOI] [PubMed] [Google Scholar]
- Walworth NC, Goud B, Kabcenell AK, Novick PJ. Mutational analysis of SEC4suggests a cyclical mechanism for the regulation of vesicular traffic. EMBO (Eur Mol Biol Organ) J. 1989;8:1685–1693. doi: 10.1002/j.1460-2075.1989.tb03560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichmann H, Hengst I, Gallwitz D. Endocytosis in yeast: evidence for the involvement of a small GTP binding protein (Ypt7) Cell. 1992;71:1131–1142. doi: 10.1016/s0092-8674(05)80062-5. [DOI] [PubMed] [Google Scholar]
- Zerial M, Stenmark H. Rab GTPases in vesicular transport. Curr Opin Cell Biol. 1993;5:613–620. doi: 10.1016/0955-0674(93)90130-i. [DOI] [PubMed] [Google Scholar]