

Abstract
The vascular pathophysiology of sickle cell disease (SCD) is influenced by many factors including adhesiveness of red and white blood cells to endothelium, increased coagulation and homeostatic perturbation. The vascular endothelium is central to disease pathogenesis because it displays adhesion molecules for blood cells, balances procoagulant and anticoagulant properties of the vessel wall and regulates vascular homeostasis by synthesizing vasoconstricting and vasodilating substances. Occurrence of intermittent vascular occlusion in SCD leads to reperfusion injury associated with granulocyte accumulation and enhanced production of reactive oxygen species. The participation of nitric oxide (NO) in oxidative reactions causes a reduction in NO bioavailability and contributes to vascular dysfunction in SCD. Therapeutic strategies designed to counteract endothelial, inflammatory and oxidative abnormalities may reduce the frequency of hospitalization, blood transfusion, the incidence of pain and the occurrence of acute chest syndrome and pulmonary hypertension in patients with SCD.
Keywords: Sickle cell disease, endothelium, nitric oxide
Introduction
The presence of abnormal hemoglobin (Hb) in red cells of sickle cell disease (SCD) patients was demonstrated by Pauling and colleagues in 1949 (1). By 1957, Vernon Ingram identified the biochemical difference between normal and sickle hemoglobin (HbS) (2). Sickle hemoglobin results from a glutamic acid to valine substitution at the sixth amino acid position of the β globin chain, with minimal structural differences between hemoglobin A (HbA) and HbS when the Hb molecule is liganded with oxygen. When HbS is deoxygenated however, the mutant valine residue on the deoxy-HbS molecule participates in an intermolecular contact by fitting into a hydrophobic pocket on a nearby Hb molecule present in both HbA and HbS (3). This interaction leads to reversible polymer formation, with subsequent depolymerization occurring without delay upon oxygenation. Irreversible polymerization only occurs in a minority of severely dehydrated and oxidatively damaged cells (4).
Typically, a red cell transits the arterial circulation in 1–2 sec, the microcirculation in 1 sec and transits the the venous circulation in 15 sec (5). Thus, an erythrocyte passes through the lung an average of four times a minute, with HbS polymerization and depolymerization in individual red cells a chronic, recurring process for the life of the red cell. According to the kinetic theory of HbS polymerization, there is a delay period between the generation of deoxy-HbS and initiation of polymerization. This period is reported to be long enough for most erythrocytes to reach the lung and be reoxygenated (4). From the perspective of polymerization kinetics and oxygen binding, HbS remains unaggregated and behaves like HbA during this time. If the capillary transit time exceeds this delay period, red blood cells (RBCs) sickle inside the capillary, cause transient or permanent physical occlusion and initiate downstream signaling events that affect hemodynamics.
The pathobiology of SCD is instigated by episodic vascular occlusion, with multiple inciting events beyond frank HbS polymerization and mechanical obstruction induced by sickled RBCs. This includes adherence of circulating blood elements with endothelial cells, coagulopathy and endothelial dysfunction (6). These central pathophysiological mechanisms leading to sickle cell vaso-occlusion and therapeutic strategies aimed to alleviate disease progression will be discussed in the context of this review.
Increased red cell adhesion
Abnormal interactions between erythrocytes and endothelium are a primary initiating factor in the development of microvascular occlusions in SCD. This view is supported by the significant correlation between clinical severity of SCD and extents of RBC adhesiveness (7), where increased sickle red cell adhesion to vascular endothelium occurs both in static and dynamic flow conditions (8). Studies using static flow conditions reflect lower affinity binding mechanisms of RBC surface structures, controlled by time and equilibrium, while dynamic flow models largely reflect high affinity blood cell-endothelial interactions (9). Considering that sickle red cells interact with not only vascular endothelial cells, but also leukocytes, platelets, plasma proteins, and other erythrocytes (10), both experimental models are applicable to understanding processes of cellular adhesion in SCD.
A number of adhesion molecules expressed by both normal and sickle RBCs have been identified and play a key role in the pathogenesis of SCD (Table and Figure 1). Mature RBCs in SCD are shown to express increased levels of Lutheran blood group glycoprotein (Lu gp) (11) and LW glycoprotein (LW gp) (12). Sickle erythrocytes expressing increased Lu gp bind significant amounts of laminin, an extracellular matrix (ECM) protein distributed throughout most vascular beds (11). Endothelial damage observed in patients with SCD can bring the underlying matrix laminin into direct contact with flowing blood (13), allowing adherence of RBCs to exposed matrix elements. Furthermore, plasma levels of laminin are elevated in sickle cell disease (14), suggesting that laminin may be deposited on the surface of the endothelium where it could also serve as an adhesive substrate. Epinephrine elevates cAMP levels in sickle RBCs and increases adhesion to laminin in a protein kinase A (PKA)–dependent manner (15).
Table 1.
Molecules Involved in Sickle Red Blood Cell Adhesion
| Adhesion molecule and alternate name(s) | Ligand /major location |
|---|---|
| Lutheran glycoprotein (B-CAM/LU) | Laminin/ECM |
| LW glycoprotein (ICAM-4) | LFA-1 (αLβ2)/leukocytes
Mac-1 (αMβ2)/leukocytes αIIb, β 3/platelets αVβ3/endothelial cells |
| VLA-4 (α4β1) | VCAM-1/endothelial cells
Fibronectin/ECM |
| Glycoprotein IV (CD36) | Thrombospondin/plasma, ECM |
| CD47 (integrin-associated protein; IAP) | Thrombospondin/plasma, ECM |
| GP IIb/IIIa (CD41/CD61) | Von Willebrand Factor/plasma |
ECM; extracellular matrix.
Figure 1.
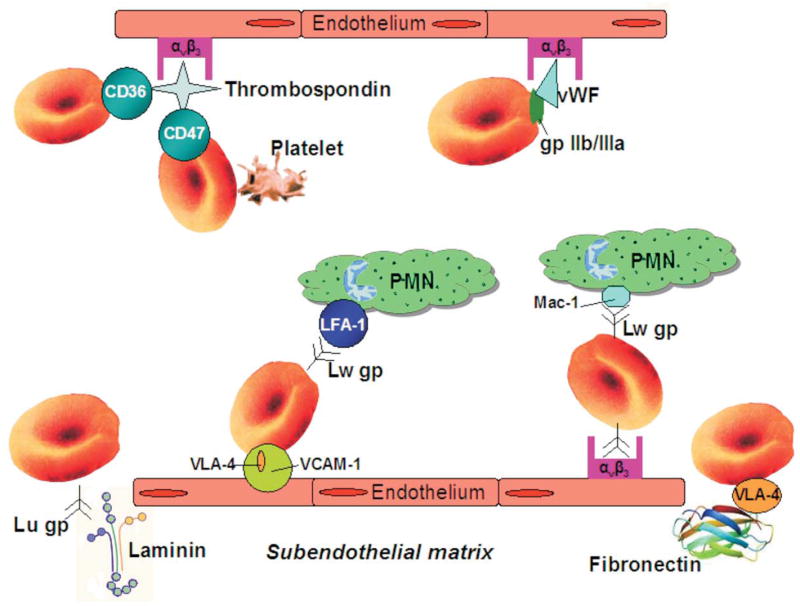
Increased red cell adhesion in sickle cell disease. Lu gp, Lutheran glycoprotein; PMN, polymorphonuclear leukocytes;VCAM-1, vascular cell adhesion molecule-1; vWF, von Willebrand factor.
The LW gp, increased in sickle red cells, is homologous to members of the intercellular adhesion molecule (ICAM) family and thus is also known as ICAM-4 (16). Studies have shown that LW gp associates with specific integrins expressed on leukocytes [LFA-1 (αLβ2), Mac-1 (αMβ2)] (17) and platelets (αIIb,β3) (18), suggesting a potential physiological significance of ICAM-4 mediated RBC–platelet interaction in hemostasis and thrombosis. A recent study has shown that LW gp on HbS, but not HbA RBCs, can be activated via the adrenergic signaling pathway to mediate adhesion to αVβ3 integrin on cultured endothelial cells (19). Epinephrine is a major mediator of vascular stress under these conditions and induces a three-fold increase in sickle cell cAMP via stimulation of β-adrenergic receptors. Increased levels of cAMP cause only minimal responses in normal RBCs, while up-regulating serine phosphorylation in sickle red cell LW gp (19). This epinephrine-induced mechanism that results in increased cell adhesiveness wherein Lu and LW gps bind to counter-receptors can contribute to the robust relationship between SCD pain crises and stress responses.
Reticulocytes also participate in adhesive interactions in SCD, mediated by increased levels of integrin VLA-4 (α4β1) and membrane glycoprotein IV (CD36) (20, 21). Both molecules have been shown to mediate sickle reticulocyte adhesion to different vascular ligands. VLA-4 mediates adhesion both to endothelial vascular cell adhesion molecule-1 (VCAM-1) and to fibronectin, an abundant ECM protein that may be bound to the surface of activated endothelium (21). As shown in figure 1, CD36 mediates adhesion through a thrombospondin (TSP) bridge to αVβ3 integrin on activated microvascular and large vessel endothelium (22, 23). Thrombospondin in turn is a protein present both in the circulation and subendothelial matrix (24). In the circulation, TSP is normally sequestered in platelet α-granules that, upon activation, release TSP to cause increased plasma concentrations (25). Consistent with this, patients with SCD have elevated plasma levels of soluble TSP (26). Immobilized subendothelial TSP is a primary adhesive substrate for endothelial cells (27) and may be exposed to circulating blood cells as a result of the vascular damage inherent in SCD patients (13). Although sickle reticulocyte adherence can occur through CD36, the frequency and characteristics of vaso-occlusive events do not significantly differ in CD36-deficient SCD patients (28), indicating that a single type of adhesion molecule is insufficient to counteract the promoting role of red cell adhesion in sickle cell vaso-occlusion.
Thrombospondin has also been shown to bind CD47 (integrin-associated protein; IAP) expressed on both normal and HbS RBCs (29). Although HbA and HbS red cells express approximately the same amount of CD47, adhesion to TSP differs significantly between the two groups. Under basal flow conditions, CD47 is reported to be the primary cell-surface adhesive molecule on HbS red cells for immobilized TSP, whereas on HbA red cells this binding is negligible (29). It now appears that soluble TSP can up regulate the activity of CD47 to adhere immobilized TSP (30). The ability of soluble TSP to promote red cell adhesion to immobilized TSP is relevant when considering that soluble TSP levels are elevated in SCD (26). The signaling activity mediating red cell adhesion to immobilized TSP is enhanced when red cells are exposed to shear stress, and can be mimicked using a specific CD47 agonist peptide (30). Recent evidence indicates that CD47 mediates reticulocyte adhesion to immobilized TSP through activation of integrin VLA-4 (α4β1) (31).
Von Willebrand factor (vWF), a multimeric plasma protein synthesized by megakaryocytes (32) and endothelial cells (33), also mediates adhesive interactions between sickle RBCs and the endothelium. High molecular weight forms of vWF secreted by cultured endothelial cells promote sickle red cell adhesion to endothelium cultured in flow chambers (34). Likewise, vWF also induces sickle erythrocyte adhesion to ex vivo mesocecum vasculature pretreated with desmopressin, an agent which causes the release of large multimeric forms of the protein from endothelial cells (35). GPIIb/IIIa-like receptors expressed on erythroid cells are thought to promote red cell adhesion to vWF, which interacts with endothelial αVβ3 integrin (36) (figure 1).
Generation of transgenic mouse models and the application of intravital imaging of microvascular networks has furthered understanding of in vivo microcirculatory flow characteristics of HbA and sickle RBCs. Four different fractions of sickle RBCs, reticulocytes, discocytes, dense discocytes, or irreversibly sickled cells (ISCs), have been identified in SCD blood separated on a percoll-renografin density gradient (37). The relative contribution of red cell density to adhesive and obstructive events was investigated in ex vivo mesoceum microvasculature of the rat. Microvascular adhesion of reticulocytes was greater than dense discocytes and ISCs, suggesting that dense cells more readily deform and better contact the endothelial cell surface (38). In vivo adhesion of sickle red cells was examined in the cremaster muscle preparation of transgenic Antilles mice expressing human α, human βS and residual mouse globins (39). In vivo flow experiments revealed that red cells adhered exclusively to postcapillary venules in sickle, but not wild type, mice, thus identifying sickle cell vaso-occlusion as a microcirculatory phenomenon (40). Data from ex vivo and in vivo experimental systems support the hypothesis that sickle vaso-occlusion is a two step process, with initiation due to endothelial adhesion of deformable red cells, followed by a propagation phase caused by physical obstruction induced by dense ISCs (41).
Increased leukocyte adhesion
The participation of leukocytes in the pathophysiology of sickle cell disease is supported by large epidemiologic studies that show high baseline leukocyte counts as a risk factor for disease severity (42), acute chest syndrome (43) and silent cerebral infarcts (44). Notably, the administration of granulocyte colony-stimulating factor causes fatal and severe sickle cell crisis in homozygous and heterozygous SCD patients, respectively (45, 46).
Experimental data demonstrating the significant role of leukocytes in sickle cell vasoocclusion comes from studies utilizing intravital microscopy. These studies show that sickle RBCs interact with adherent leukocytes in postcapillary venules, rather than directly with the endothelium (47). It is important to note that in vivo leukocyte-endothelial interactions are confined to the venular segments of the microcirculation and are rarely observed in arterioles (48). The preferential adhesion of leukocytes to venular endothelium is not explained by the existence of lower shear rates in venules, compared to arterioles (49), but rather suggests a heterogenous distribution of adhesion molecules between arterioles and venules. In fact, intravital confocal microscopy and immunofluorescent labeling has recently shown that the spatial distribution and expression levels of adhesion molecules in the microcirculation significantly impact on leukocyte adhesion in vivo (50).
Recurrent vaso-occlusive crisis and tissue ischemia induce a continuous inflammatory response in SCD (6). One of the important features of inflammation is the migration of leukocytes from the circulation, across the endothelium and into affected sites of tissue injury. Figure 2 shows a scheme of inflammatory events leading to increased leukocyte adhesion in SCD.
Figure 2.

Vascular inflammation in sickle cell disease. PMN, polymorphonuclear leukocytes; IL-1, interleukin-1; IL-6, interleukin-6; IL-8, interleukin-8; TNF-α, tumor necrosis factor-alpha; PGSL-1, P-selectin glycoprotein ligand-1; Fcγ, high-affinity Fc gamma receptor; ICAM-1, intercellular adhesion molecule-1.
The migration of leukocytes from the center of blood flow to the vascular endothelium is induced by pro-inflammatory mediators cytokines (51), including interleukin-1 (IL-1), IL-6, IL-8, and tumor necrosis factor-alpha (TNF-α) secreted by tissue macrophages (52) and found to be elevated in the plasma of SCD patients (53, 54, 55, 56). Aside from tissue macrophages, monocytes also serve as a source of increased serum levels of IL-1 and TNF-α in patients with SCD. Unstimulated circulating monocytes from SCD patients have been shown to produce more TNF-α and IL-1β compared to the control group (57). Interleukin-1 and TNF-α activate vascular endothelial cells leading to increased expression of endotheial markers, E-selectin, P-selectin, VCAM-1 and ICAM-1, which function as receptors for leukocyte adhesive proteins (53). Circulating endothelial cells in patients with sickle cell disease, regardless of clinical status, express elevated levels of all of these markers of endothelial-cell activation (13). Interleukin-1, TNF-α and IL-6 also activate hepatocytes to synthesize acute-phase proteins, some of which (alpha-1-antitrypsin, ceruloplasmin, alpha 2-macroglobulin, and C-reactive protein) are significantly elevated in SCD patients (58).
Electron microscopy has shown that IL-8 can be immobilized on the luminal surface of the endothelium after being secreted by tissue macrophages. Binding of IL-8 to the abluminal side of endothelial cells enables its internalization and transportation to the luminal surface (59). Interleukin-8 bound to glycosaminoglycans on the surface of endothelial cells activates leukocytes via transmembrane G protein-coupled receptors, allowing for rapid clustering of cell surface integrins necessary for leukocyte arrest (60). The activation state of neutrophils from SCD patients is determined by the expression of several surface antigens and levels of circulating proteins released by neutrophils. The high-affinity Fc receptor (FcγR1/CD64) expressed on immature neutrophils is considered a marker of PMN activation (61) and is significantly increased in SCD patients, especially in crisis (62, 63). Another sensitive marker of neutrophil activation is decreased surface expression of L-selectin (CD62L), which is shed upon PMN stimulation via proteolytic cleavage (64). CD62L is significantly decreased in non-symptomatic SCD patients, which is further reduced in vaso-occlusive crisis (63). Neutrophil activation also causes degranulation, leading to increased plasma levels of lactoferrin and elastase (65), both of which are reported to be significantly increased in sickle vaso-occlusive crisis (63).
Migration of leukocytes through vascular endothelium is a multistage process and involves multiple adhesion molecules, listed in Table 2. Leukocytes first roll on activated endothelial cells by an interaction mediated largely by P- and E-selectin on the endothelium and L-selectin on the leukocyte, which bind to counter glycoprotein structures containing O-linked oligosaccharide chains (66). Rolling leukocytes may become detached or alternatively arrest, flatten, and transmigrate. Chemokines, such as the IL-8 found on the endothelial surface, activate leukocytes and promote the rapid clustering of surface integrins necessary for leukocyte arrest (60). This arrest phase is mediated by leukocyte α4β1 (CD49d/CD29; VLA-4), αLβ2 (CD11a/CD18; LFA-1) and αMβ2 (CD11b/CD18; Mac-1) integrins binding to endothelial counter-receptors that include VCAM-1, ICAM-1, ICAM-2 and ICAM-3 (67). Flow cytometric analysis show that leukocyte adhesion molecules LFA-1 (CD11a/CD18), Mac-1 (CD11b/CD18) and L-selectin (CD62L) are normally expressed by sickle cell patient PMNs in or out of crisis (62), suggesting that the endothelium plays a very prominent role in the increased leukocyte adhesion observed in SCD. Indeed, sickle cell endothelial P- and E-selectin doubly deficient mice (SSP−/E−) are protected from the occurrence of hypoxia-induced vascular occlusion (47). Specifically, intravital microscopy studies employing a sickle mouse model expressing human α (αH) globins and βS globins on a homozygous mouse βmajor deletional background, αHβS[βMDD], revealed an attenuated leukocyte adhesion response in posthypoxic cremasteric venules following treatment with a blocking antibody against P-selectin (68). The dual radiolabeled mAb technique enabled the quantification of E- and P-selectin expression in different vascular beds in Paszty βS transgenic mice, which are heterozygous for murine β globin and human βS. Up regulation of P-selectin was demonstrated in the lung, heart, small bowel, large bowel and penis, while E-selectin expression only increased in the penis (69).
Table 2.
Leukocyte Adhesion Molecules
| Adhesion molecule and alternate name(s) | Endothelial Ligand |
|---|---|
| L-Selectin (CD62L) | L-Selectin Ligand |
| Leukocyte Function Associated Antigen (LFA-1; αLβ2; CD11a/CD18) | ICAM-1, 2, 3 |
| Mac-1 (αMβ2; CD11b/CD18) | ICAM-1, 2, 3 |
| VLA-4 (α4β1; CD49d/CD29) | VCAM-1 |
| P-Selectin Glycoprotein Ligand (PSGL-1; CD162) | P-Selectin (CD62P)
E-Selectin (CD62-L) |
| Platelet-Endothelial Cell Adhesion Molecule-1 (PECAM-1; CD31) | PECAM-1 (CD31) |
Increased coagulant activity
Whole blood tissue factor (TF), the physiologic initiator of coagulation, is significantly elevated in patients with SCD (70). Tissue factor is present on cell surfaces as an integral membrane protein. Under normal physiologic conditions no vascular cells display an active TF, although endothelial cells, monocytes, polymorphonuclear leukocytes can all be induced to express the protein (71). The inflammatory mediators reported to be increased in SCD plasma, such as IL-1, TNF-α and C-reactive protein (53, 58), can induce TF synthesis in monocytes and endothelial cells (66). Indeed, circulating endothelial cells express increased amounts of TF in SCD (72). Likewise, TF bearing microparticles, shed from activated monocytes and endothelial cells, are present in the blood of SCD patients (73). Immunostaining of tissue sections from transgenic sickle mice reveals that endothelial expression of TF is confined almost exclusively to the pulmonary veins and is augmented by hypoxia/reoxygenation (74).
The interaction of TF with factor VIIa results in hydrolysis of factors IX and X, which in turn trigger the coagulation cascade (71). The rate of factor X hydrolysis is shown to be reduced by approximately 50-fold when factor VIIa is assembled with solution-phase TF, indicating the important influence of membrane surface properties on TF activation (75). Indeed, the presence of phosphatidylserine (PS) in TF-bearing vesicles has been shown to significantly enhance the hydrolysis of factor X (75). Externalized PS on sickle red cell surfaces also provides a milieu for TF activation and increased coagulant activity associated with the disease (76). Phosphotidylserine is normally sequestered on the inner leaflet of membrane bilayers, but translocates to the outer surface following cell injury. There are energy-dependent enzyme systems that maintain membrane phospholipid asymmetry in red blood cells (77). However, membranes of sickle erythrocytes show substantial alteration of phospholipid asymmetry, findings observed in both reticulocytes and mature erythrocytes (78).
Once hydrolyzed via TF, factor Xa forms a prothrombinase complex with factor Va and participates in the cleavage of prothrombin into thrombin and fragment 1.2 (79). Increased plasma concentrations of prothrombin fragment 1.2 (80) and reduced plasma levels of factor V in sickle cell patients suggests increased thrombin generation in SCD (81). Thrombin cleaves fibrinopeptides A and B from fibrinogen to form fibrin monomers that spontaneously polymerize into a fibrin clot (82). High levels of fibrinopeptide A are also seen in SCD patients, implying the presence of enhanced fibrinogen proteolysis. The degradation of cross-linked fibrin by plasmin forms D-dimer, a product that has emerged as a potentially useful serological coagulation marker (82). Plasma markers of fibrinolysis, D-dimer, and plasmin:antiplasmin complex are reported to be significantly increased in asymptomatic subjects with SCD compared to controls (80).
Thrombin self-production is inhibited by a negative feedback mechanism via the thrombomodulin (TM)–protein C system (83). Thrombomodulin is an integral membrane protein expressed on the surface of vascular endothelial cells that binds thrombin, localizing thrombin to endothelial cell surfaces and inducing conformational changes such that the formed protein complex can activate protein C (PC). Activated PC, with the aid of its nonenzymatic cofactor protein S, inhibits multiple steps in the coagulation pathway (83). Patients with SCD have low circulating levels of protein C and S (84, 85). Protein C and protein S levels in children with sickle cell anemia and stroke were compared to those with sickle cell anemia who were neurologically normal. There were significantly decreased levels of both protein C and protein S in patients with SCD who have had thrombotic strokes, compared to patients who have not experienced cerebrovascular events (86). Reduced levels of protein C and S in SCD patients may reflect low protein synthesis due to hepatic dysfunction, or may be the result of chronic consumption due to increases in TF and thrombin generation.
Thrombin is a potent platelet activator that signals via at least two types of receptors, which include the G protein-linked, proteolytically activated receptors PAR-1 and PAR-4 (87). Platelet levels of both P-selectin (CD62P) and CD40 ligand, two markers of platelet activation (88), are higher in children with SCD than in healthy control subjects (80, 89). Activated platelets bind monocytes and neutrophils through an interaction between P-selectin on platelets and P-selectin glycoprotein ligand-1 (PSGL-1) on leukocytes (90). Indeed, platelet-WBC complexes have been detected by FACS analysis in the blood of sickle cell patients (91).
In summary, multiple studies indicate increased coagulant activity in SCD subjects that is induced by platelet activation, thrombin generation and fibrinolysis. Figure 3 illustrates the scheme of events leading to increased coagulant activity and fibrinolysis in SCD.
Figure 3.

Pathogenesis of increased coagulant activity in sickle cell disease.
Altered nitric oxide bioavailability and impaired vascular function
An central aspect of sickle cell vasculopathy is the impairment of endothelial regulation of vasomotor tone, thrombosis and inflammation (6). Reduced nitric oxide (NO) bioavailability in SCD is a critical component of these underlying abnormalities (92). Nitric oxide, a diffusible intercellular messenger, is produced by most mammalian cells including vascular endothelium, neurons, smooth muscle cells, macrophages, neutrophils, platelets and epithelium (93). Once formed by endothelial NO synthase (eNOS or NOS3), NO diffuses to nearby smooth muscle cells, where it reacts with the ferrous iron of the heme group of guanylate cyclase, resulting in enhanced synthesis of cyclic guanosine monophosphate (cGMP) from guanosine triphosphate (GTP) (94). Cyclic GMP then activates protein kinase G (PKG), which in turn stimulates Ca2+-ATPase-dependent refilling of intracellular calcium stores and lowering of intracellular calcium levels, thus mediating smooth muscle cell relaxation (95). This cGMP/PKG-dependent signaling mechanism also promotes a potent inhibition of platelet aggregation (96). Reduced intracellular calcium levels in platelets lead to decreased P-selectin expression and suppress expression of the active conformation of glycoprotein IIb/IIIa (GPIIb/IIIa) required for binding fibrinogen (97).
Nitric oxide is also a potent antagonist of inflammation (98). This effect of NO is in part a result of inhibition of nuclear factor-κB (NF-κB) activation through induction of expression of the NF-κB inhibitor IκBα, and stabilization of the NF-κB/IκBα complex (99). NF-κB controls the expression of a variety of genes involved in inflammatory and immune responses (100), with VCAM-1, ICAM-1 and E-selectin key examples (101). Both antibody blocking of integrins and inhibition of integrin gene expression profoundly inhibit inflammatory cell-induced vascular dysfunction (102). Considering that sickle cell vaso-occlusion predominantly occurs in the microcirculation (40, 47), it is relevant that compensatory NO signaling mechanisms are operative to inhibit the preferential adhesion of circulating blood cells to venular endothelium. Inhibition of NO synthesis has no apparent effect on experimentally-induced arteriolar thrombosis, but enhances thrombosis in venules (103). Furthermore, exogenous L-arginine and NO donors have been shown to reverse the pro-thrombotic actions of NOS inhibition in venules (103). Considering that cytokine stimulated leukocyte-endothelial adhesions occur in the venular segments of the microcirculation in vivo (104) and that adhesion molecules demonstrate a heterogenous distribution between arterioles and venules (45), the venular actions of NO are particularly relevant.
Reduced NO bioavailability in SCD results from consumption of NO by plasma cell-free hemoglobin (105) and reactive oxygen species (106) (Figure 4). Increased tissue rates of reactive oxygen species production in SCD may arise from several factors, including increased expression and activity of circulating and vessel bound xanthine oxidase (XO) (107, 108) and endothelial cell NADPH oxidase/Nox activities (109), as well as the uncoupling of eNOS electron transfer (110).
Figure 4.

Increased intravascular consumption of nitric oxide in sickle cell disease. Superoxide (O2•−) generated by uncoupled endothelial nitric oxide synthase (eNOS), xanthine oxidase (XO) and NADPH oxidase, reacts with nitric oxide (NO) to form peroxynitrite (ONOO −). Nitric oxide is also consumed by plasma free hemoglobin, released by intravascular hemolysis.
Intravascular hemolysis and decompartmentalization of hemoglobin
Nitric oxide binds very rapidly to deoxyhemoglobin, forming a stable Hb (Fe+2)-NO complex (111). Nitric oxide also reacts with and converts oxygenated Hb to methemoglobin and nitrate (NO3−) (112). Rates of intravascular NO scavenging are reduced significantly when hemoglobin is sequestered within red cell membranes (113). Polymerization of hemoglobin S in patients with SCD leads to destabilization of the red blood cell membranes and intravascular hemolysis, releasing cell free hemoglobin into plasma (114). SCD patient plasma contains approximately 0.001–0.033 g/dL cell-free ferrous hemoglobin, which stoichiometrically can consume micromolar quantities of nitric oxide and abrogate forearm blood flow responses to infusion of NO donors (105). Serum haptoglobin (hp), the hemoglobin scavenging protein, can bind approximately 0.05– 0.15 g/dL free Hb (115), however hp levels are significantly reduced in patients with SCD relative to HbA study subjects (116). Moreover, a significant inverse correlation exists between steady-state LDH and hp levels in SCD patients, indicative of enhanced clearance of the hp/Hb complex formed via intravascular hemolysis (117).
Apart from releasing red cell hemoglobin into plasma, hemolysis also leads to the release of erythrocyte arginase, which converts L-arginine, the substrate for NO synthesis, to ornithine (118). Arginase activity is elevated almost two-fold in SCD patients with pulmonary hypertension, resulting in a lower arginine/ornithine ratio and indicating the presence of intravascular hemolysis (119). Intravascular hemolysis and subsequent decrease in NO bioavailability is associated with clinical complications of SCD, including pulmonary arterial hypertension, priapism and stroke (120). These clinical complications are also shared by other hemolytic anemias (120). Indeed, mice expressing exclusively human sickle hemoglobin and mice with acute alloimmune hemolytic anemia develop pulmonary hypertension that is associated with blunted NO-induced vasodilation (121).
Chronic hemolysis in sickle cell disease requires the upregulation of enzyme systems to catabolize the hemoglobin released from hemolyzed erythrocytes. Plasma hp-Hb complex displays an epitope recognized by the hemoglobin-scavenger protein expressed on reticuloendothelial cells (122). Receptor-ligand endocytosis of the complex rapidly clears it from blood plasma and drives expression of heme-oxygenase 1 (HO-1) and biliverdin reductase. Heme-oxygenase, found predominantly in Kupffer cells of the liver, is the rate-limiting enzyme in the degradation of heme to biliverdin, also releasing carbon monoxide and iron (123). Patients with sickle cell disease have increased cellular HO-1 protein levels in their peripheral blood mononuclear cells compared with healthy volunteers (124). Increased HO-1 expression is also observed in renal tubular epithelial cells and circulating endothelial cells of patients with sickle cell disease and could represent an adaptive response to the inflammatory stress inherent in SCD (125).
Haptoglobin-related protein (Hpr) also binds to free plasma Hb. Circulating Hpr-Hb complex is directed into specialized high-density lipoprotein particles, where they are sequestered with apolipoproteins apoA-I and apoL-I (126). Interestingly, an apolipoprotein-A1 mimetic peptide, L-4F, ameliorates the impaired carotid arteriole relaxation of mice expressing exclusively human sickle hemoglobin (127). The effect of this apolipoprotein-A1 mimetic peptide in sickle cell cerebrovascular responses may be due to the sequestration of the Hpr-Hb complex. If so, it is plausible that apolipoprotein binding limits NO scavenging by the Hpr-hemoglobin complex (128).
It is noted that cerebrovascular disease in SCD appears to involve predominantly large vessels (129), in contrast to microvascular involvement and associated complications. Such clinical manifestations have led scientists to propose two partially overlapping subphenotypes in SCD. The first subphenotype is associated with increased white blood cell count, vaso-occlusive pain crisis, acute chest syndrome, high steady state hemoglobin and low fetal hemoglobin levels, while the second phenotype is linked with increased hemolysis, stroke and pulmonary hypertension (130). The apparent localization of decreased venular vs. arteriolar NO bioavailability may be relevant in determining the observed sub-phenotypes of SCD patients.
Increased vascular and tissue oxidant generation
Xanthine oxidase (XO), a source of vascular O2•− and H2O2 production, is elevated in SCD patient plasma and in the arteriolar and venular walls of SCD mice (107, 131). Increased circulating and vessel wall XO in SCD contributes to hemodynamic instability and contribute to the pathogenesis of a broader systemic vascular dysfunction. This is exemplified by the observation of impaired NO-dependent vasorelaxation in SCD mice (107) and affirms the blunted vascular relaxation that occurs in SCD mouse vessels in response to a calcium ionophore and the NO donor DEA-NONOate (132). This impaired vessel relaxation is restored by denudation of endothelium (132), supporting the concept of increased generation of NO-inactivating species by the endothelium of SCD vessels (107). Importantly, the inhibition of vessel relaxation observed in SCD mice is completely restored by an NO donor, sodium nitroprusside (SNP). Because SNP is metabolized by smooth muscle cells (133), this excludes the existence of an endorgan defect in NO signaling at the level of guanylyl cyclase and further confirms that NO is being consumed by endothelial-derived redox reactions in SCD vessels. Indeed, basal rates of O2• − production are significantly increased in the aorta of SCD mice vs. controls (107). The impairment of NO-dependent vascular relaxation by increased rates of reactive oxygen species generation is further underscored by the observation that the superoxide dismutase (SOD) mimetic MnTE-2-PyP restores ACh-dependent relaxation of SCD mouse vessels (107).
Superoxide derived from endothelial cell NADPH oxidase also mediates microvascular dysfunction in sickle cell transgenic mice (109). Vascular endothelial cells contain all requisite phox proteins and the low molecular weight GTP binding protein of the multicomponent phagocytic NADPH oxidase system (134). Leukocyte-endothelial and platelet-endothelial cell adhesion was observed in the cerebral microvasculature of wild-type (WT), SOD1 transgenic (SOD1-TgN), and gp91phox-deficient mice transplanted with bone marrow from sickle cell transgenic mice. NADPH oxidase inactivation was as effective as SOD overexpression in abolishing both the leukocyte and platelet adhesion responses typically observed in SCD mice (109).
An increase in the production of superoxide uncoupled NO synthases (NOS) has also been implicated in the pathogenesis of SCD (109, 121). Nitric oxide is synthesized by a family of NOS enzymes that have a C-terminal reductase and an N-terminal oxygenase domain (93). The oxidative deamination of L-arginine to L-citrulline during NO formation requires the cofactors NADPH, flavin adenine dinucleotide (FAD), flavin mononucleotide (FMN), iron protoporphyrin IX (heme) and tetrahydrobiopterin (BH4) (135). Functionally, NOS catalyses a multi-electron, rather than a two-electron, oxidation and thus performs two separate NADPH oxidation cycles. The flavin cofactors FAD and FMN support NADPH-dependent heme reduction during NOS turnover, forming stable semiquinone radical intermediates. For endothelial and neuronal NOS, electron transfer is limited beyond the flavin centers, until the enzymes are activated by Ca2+-calmodulin (CaM). Binding of CaM facilitates the transfer of an electron from FMNH2, resulting in the reduction of the iron to the ferrous state (Fe2+). Oxygen then binds, forming the Fe2+-dioxygen (Fe2+=O2) complex, which undergoes oxygen-oxygen bond scission to induce hydroxylation of L-arginine to Nω-hydroxy-L-arginine (NHA). Once NHA is formed, it is reduced to yield the final products NO and L-citrulline (136). NOS enzymes have an absolute requirement for BH4, which provide an electron for the reaction that hydroxylates L-arginine to NHA (137). In the absence of BH4 or L-arginine, the Fe2+=O2 complex decays under formation of superoxide and Fe3+ (138), a phenomenon that has been termed ‘uncoupling’ of NOS.
Leukocyte and platelet adhesion responses are exacerbated in sickle cell transgenic mice that show overexpression of eNOS in microvessels. Conversely, a significant attenuation of endothelial leukocyte and platelet adhesion occurs when the vascular wall is rendered eNOS deficient (109). The eNOS-mediated enhancement of blood cell adhesion is reversible by pretreatment with a BH4 generating agent or polyethyleneglycol–superoxide dismutase, implicating an adverse role for eNOS-dependent superoxide production (109).
The neutrophil-derived, heme-containing myeloperoxidase (MPO) catalytically consumes NO directly and via the generation of radical intermediates, thereby modulating NO bioactivity and adversely impacting vasomotor function (139). The NO metabolite nitrite is also a substrate for MPO and becomes oxidized to the reactive oxidizing and nitrating species nitrogen dioxide (·NO2), explaining in part the close spatial correlation between NO2Tyr and MPO (140). Importantly, the diffuse immunoreactivity of MPO along the subendothelial space in tissues from patients with diverse inflammatory diseases, including SCD, reinforces ex vivo observations describing discrete interactions between endothelial cells and MPO, in particular the avidity of MPO for endothelial and other cells that results in its transcytosis into the subcellular matrix (141). Of note, there is elevated MPO immunoreactivity throughout the alveolar epithelium in lung tissues from patients with SCD, possibly contributing to the pulmonary hypertension and acute chest syndrome prevalent in SCD (140).
Reperfusion injury and tissue damage
The initiation, progression and resolution of vaso-occlusive crisis in SCD presents features common with reperfusion injury. The resupply of oxygen to ischemic tissues can, paradoxically, support a more pro-inflammatory state and contribute to multiple organ damage observed in SCD. Indeed, exposure of sickle cell mice to transient hypoxia (from 8 to 11% O2) and reoxygenation induces NF-κB activation in kidney and liver (142) and a P-selectin-dependent increase in vessel wall leukocyte adhesion (68)
Xanthine oxidoreductase (XOR) is an important source of the reactive species that mediate much of the tissue damage in reperfusion injury (143). During experimental hypoxia, conversion of XOR to the oxidant-generating XO is significantly greater in the liver and kidneys of sickle cell mice expressing a mixture of human HbS and murine Hb (142). Extensive hepatocellular injury, accompanied by increased plasma ALT levels, decreased liver XO immunoreactivity and catalytic activity was observed in knockout-transgenic SCD mice, even under normoxic conditions (107).
Increased expression of inducible NOS (iNOS or NOS2) and elevated tissue nitrite (NO2 −) and nitrate (NO3 −) occurs in animal models of cardiac, liver, and kidney ischemia/reperfusion (I/R). In many cases, NOS2 inhibitors significantly improve organ function in tissue I/R injury. Similarly, I/R-induced tissue injury is attenuated in the liver and kidney of NOS2 −/ − mice (144, 145). Accelerated NO production during I/R injury is cytotoxic via direct reactions of NO and its reactions with other oxygen-derived species, generating secondary products capable of further oxidation and nitration reactions. The oxidation and nitration of lipids, amino acids, and proteins will alter biomolecular structure and function and at the same time transduce the pathogenic actions of reactive species in various disease processes. For example, enhanced iNOS immunoreactivity and nitration of liver and kidney actin is prevalent in both SCD mice and humans (146).
PATHOGENIC MECHANISM-TARGETED THERAPIES FOR SICKLE CELL DISEASE
Inhibition of HbS polymerization
In a multicenter, randomized, placebo-controlled trial, SCD patients treated with hydroxyurea had lower rates of vaso-occlusive crises (VOCs), increased time between crises, a lower incidence of acute chest syndrome and a reduced need for blood transfusions (147). The mechanism underlying these benefits is, in part, related to an increase in the proportion of cells containing fetal Hb and an increase in the erythrocyte mean corpuscular volume, thereby reducing the potential for Hb polymerization (147). Hydroxyurea has been shown to increase both γ-globin mRNA levels and HbF significantly in mixed cultures of peripheral-blood mononuclear and erythroid progenitor cells from normal donors (148). Of note, it has also been demonstrated that hydroxyurea can be oxidized by heme groups to produce NO in vitro (149) and in vivo (150). Studies have further shown that soluble guanylate cyclase activators or analogues increase fetal haemoglobin gene expression in erythroleukaemic cells and in primary human erythroblasts (151). Agents other than hydroxyurea that increase the level of HbF include 5-aza-2’-deoxycytidine (decitibine). The presence of a nitrogen in the 5th position of the pyridine ring of cytidine analogs causes resistance to methylation. Hypomethylation induces γ-globin gene transcription in primates and patients with sickle cell anemia (152). In preliminary trials, patients who failed to respond to hydroxyurea with an increase in HbF responded to decitibine with a sustained approximately 14% (153) increase of HbF.
Anti-adhesive and Anti-inflammatory therapy
Sulfasalazine is a potent inhibitor of NF-κB that controls the expression of a variety of genes involved in inflammatory and immune responses (154). Intraperitoneal (IP) and oral administration of this drug to SCD mice and humans, respectively, significantly reduced endothelial cell expression of VCAM-1, ICAM-1 and E-selectin (155). Anionic polysaccharides such as high molecular weight dextran sulfate (HDS), chondroitin sulfate A (CSA) and heparin inhibit TSP-mediated adhesion of sickle erythrocytes to rat endothelium ex vivo (156). Likewise, the blockade of αVβ3 integrin, a site of sickle cell-endothelium interaction mediated by TSP, has also been suggested as a therapeutic approach to prevent cell-endothelial interactions in SCD (157).
TBC772, a peptide antagonist of α4β1 integrin (VLA-4) on reticulocytes, significantly inhibits sickle RBC retention in rat retina (158). Similarly, blocking endothelial monolayers with P-selectin monoclonal antibody (9E1) reduced adherence of sickle red cells to untreated and thrombin-treated endothelium by 30 and 67%, respectively (159). Peptides containing arginine-glycine-aspartic acid (RGD) sequences have also been shown to inhibit plasma -induced adhesion of sickle red cells to endothelium (160). As noted previously, high molecular weight vWF and TSP both serve as plasma bridging proteins in sickle cell adherence to endothelium.
Intravenous (i.v.) administration of polynitroxyl albumin, a superoxide dismutase and catalase mimetic, in SCD mice inhibited hypoxia/reoxygenation-induced expression of NF-κB, VCAM-1, ICAM-1 in tissues of the lung, liver and skin (161). Similarly, dexamethasone treatment inhibited NF-kappaB, VCAM-1, and ICAM-1 expression in the liver, lungs, and skin of sickle cell mice. However, rebound vasoocclusive crises were observed 3 days after cessation of glucocorticosteroid treatment, an event also reflected by the responses of patients with SCD (162).
Anticoagulant therapy
Despite the evidence for increased coagulant activity in SCD, randomized trials reveal that chronic oral anticoagulation by high-dose warfarin was not beneficial, due to major bleeding complications (163). However, a lower dose of warfarin therapy successfully reduced prothrombin fragment 1.2 by 50% in seven patients with symptomatic SCD (164). Long term minidose heparin adminstration to four SCD patients with severe recurrent painful crises caused a 73% reduction in hospitalization days and 74% reduction in hours spent in emergency rooms per year (165). A randomized double-blinded trial using ticlopidine blocked platelet activation, but did not improve platelet survival or prevent sickle crisis in asymptomatic patients with SCD (166). A pilot study of low-dose acenocoumarol in 22 patients with SCD demonstrated significant decreases in prothrombin fragment 1.2 levels, thrombin–antithrombin complexes and D-dimer fragments, but displayed no impact on the occurrence of vasoocclusive crises (167). Finally, no clear clinical benefit was observed in small, nonrandomized clinical studies of low dose aspirin (168) and aspirin plus dipyrimadole (169).
Three month dietary supplementation with fish oil containing omega-3-fatty acids has been shown to reduced prothrombotic activity in 10 sickle cell patients, as assessed by significantly decreased plasma levels of prothrombin fragment 1.2 and D-dimer. The clinical improvement produced by dietary fish oil therapy was reflected as reduced frequency of pain episodes in patients enrolled in this study (170).
Low molecular weight antioxidants
Since oxidative stress has long been recognized as part of the pathophysiology of SCD, small prospective studies have examined the effect of vitamin E supplementation on clinical outcomes of the disease. In one study, Vitamin E supplementation in 6 sickle cell patients led to an increase in α-tocopherol concentration from 0.7 ± 0.2 to 2.3 ± 0.3 mg/g lipid and a significant reduction in irreversibly sickled cells (from 25 ± 3% to 11 ± 1%) (171). However, no clinical outcomes were measured in this study. In another study, 13 patients with sickle cell disease received α-tocopherol along with ascorbic acid, zinc and soybean oil. After 8 months, there was a 37.5% reduction in the number of irreversibly sickled cells, but there was no effect on hemoglobin concentrations or number of vaso-occlusive and aplastic crises (172). Continuation of new trials of therapeutic interventions to reduce in vivo oxidative stress seem relevant in patients with SCD. A possibly more effective mode of protective therapy would be to start the therapeutic interventions at an earlier stage of the disease and target specific sites of reactive species generation with more potent catalytic oxidant scavengers rather than nutritional anti-oxidants.
Therapeutic strategies aimed to increase nitric oxide bioavailability
Therapeutic interventions that enhance NO in SCD include inhaled NO, L-arginine supplementation and administration of sildenafil. Inhalation of 80 ppm NO gas for 1.5 hours by patients with sickle-cell disease significantly reduces the NO scavenging potential of hemoglobin within the circulatory system, producing a measurable decrease in arterial plasma NO consumption (105). However, to date, only isolated case reports have described the use of inhaled NO in the treatment of SCD patients. Inhaled NO was reported to be beneficial for SCD patients with acute chest syndrome because of its ability to ameliorate pulmonary hypertension and ventilation/perfusion mismatch (173, 174, 175). Inhalation of NO has also been reported to decrease pain scores and opioid use in children with acute vaso-occlusive crisis (176), providing support for future clinical investigation of this therapeutic modality.
The observation that patients with sickle cell anemia have significantly reduced concentrations of arginine in their plasma and urine suggests that this group of patients may have increased use or catabolism of this key substrate of NO biosynthesis, and therefore increased nutritional requirements (177, 178). Indeed, L-arginine levels are depressed in patients with sickle cell disease, particularly during vaso-occlusive pain crisis and during acute chest syndrome (179). The potential role of reduced arginine bioavailability in patients with pulmonary hypertension secondary to SCD, and the effects of oral arginine therapy on pulmonary artery pressures, were evaluated in sickle cell patients (180). Ten patients with sickle cell disease complicated by secondary pulmonary hypertension were treated for five days with oral L-arginine. Pulmonary arterial systolic pressure was estimated by echocardiographic measurement of the tricuspid regurgitant jet velocity before and after the 5 days of treatment, and decreased by 15.2%. Pulmonary pressure decreased in 9 of 10 patients, suggesting greater vascular responsiveness and less end-stage vasculopathy than commonly observed in patients with primary pulmonary hypertension. Furthermore, the study showed a trend toward a 20% improvement in oxygen saturation after L-arginine supplementation. Plasma L-arginine levels rose significantly after therapy and methemoglobin levels increased from 0.55 to 1.10%, support that there was an increase in NO production.
Another NO-based therapeutic strategy for SCD is the phosphodiesterase 5-inhibitor, sildenafil, which potentiates the effect of NO by inhibiting cGMP degradation. Sildenafil treatment of 12 SCD patients with pulmonary hypertension resulted in a 10 mmHg decrease in estimated pulmonary artery systolic pressure and a 78 m improvement in the 6-min walk distance (181).
Conclusions
The unifying theme of this review is that an altered vascular endothelial phenotype, induced by sickled red cells, is central to the pathogenesis of the vaso-occlusion, reperfusion injury and end-organ damage in SCD. The activation of endothelium results in increased adherence of red cells, leukocytes and enhanced coagulation. Decreased bioavailability of the endothelial-derived relaxing and anti-inflammatory activities of NO is an important component of this pathophysiological setting in SCD. Nitric oxide induces vasorelaxation, inhibits platelet aggregation, and decreases expression of pro-inflammatory adhesion molecules by the endothelium. The application of pharmacologic strategies designed to increase NO levels, suppress HbS polymerization and decrease endothelial adherence and coagulation show clinical promise in decreasing the morbidity and mortality of this devastating vascular disease.
Acknowledgments
Supported in part by The Scientific and Technological Research Council of Turkey (TUBITAK) grant SBAG-2797, the Akdeniz University Research Foundation and National Institutes of Health Grants HL58115 and HL64937. The authors appreciate the editorial and computers graphics skills of Georgeanna Williams.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pauling L, Itano HA, Singer SJ, Wells IC. Sickle cell anemia a molecular disease. Science. 1949;110:543–548. doi: 10.1126/science.110.2865.543. [DOI] [PubMed] [Google Scholar]
- 2.Ingram VM. Gene mutations in human haemoglobin: the chemical difference between normal and sickle cell haemoglobin. Nature. 1957;180:326–328. doi: 10.1038/180326a0. [DOI] [PubMed] [Google Scholar]
- 3.Dykes G, Crepeau RH, Edelstein SJ. Three-dimensional reconstruction of the fibres of sickle cell haemoglobin. Nature. 1978;272:506–510. doi: 10.1038/272506a0. [DOI] [PubMed] [Google Scholar]
- 4.Eaton WA, Hofrichter J. Sickle cell hemoglobin polymerization. Adv Protein Chem. 1990;40:63–279. doi: 10.1016/s0065-3233(08)60287-9. [DOI] [PubMed] [Google Scholar]
- 5.Burtis CA, Ashwood ER, editors. Teitz Fundamentals of Clinical Chemistry. 4. Saunders Company; Philadelphia, PA: 1996. [Google Scholar]
- 6.Hebbel RP, Osarogiagbon R, Kaul D. The endothelial biology of sickle cell disease: inflammation and a chronic vasculopathy. Microcirculation. 2004;11:129–151. [PubMed] [Google Scholar]
- 7.Hebbel RP, Boogaerts MA, Eaton JW, Steinberg MH. Erythrocyte adherence to endothelium in sickle-cell anemia. A possible determinant of disease severity. N Engl J Med. 1980;302:992–995. doi: 10.1056/NEJM198005013021803. [DOI] [PubMed] [Google Scholar]
- 8.Smith BD, La Celle PL. Erythrocyte-endothelial cell adherence in sickle cell disorders. Blood. 1986;68:1050–1054. [PubMed] [Google Scholar]
- 9.Hebbel RP. Adhesive interactions of sickle erythrocytes with endothelium. J Clin Invest. 1997;100:S83–86. [PubMed] [Google Scholar]
- 10.Chiang EY, Frenette PS. Sickle cell vaso-occlusion. Hematol Oncol Clin North Am. 2005;19:771–784. doi: 10.1016/j.hoc.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 11.Udani M, Zen Q, Cottman M, Leonard N, Jefferson S, Daymont C, Truskey G, Telen MJ. Basal cell adhesion molecule/lutheran protein. The receptor critical for sickle cell adhesion to laminin. J Clin Invest. 1998;101:2550–2558. doi: 10.1172/JCI1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parsons SF, Spring FA, Chasis JA, Anstee DJ. Erythroid cell adhesion molecules Lutheran and LW in health and disease. Baillieres Best Pract Res Clin Haematol. 1999;12:729, 45. doi: 10.1053/beha.1999.0050. [DOI] [PubMed] [Google Scholar]
- 13.Solovey A, Lin Y, Browne P, Choong S, Wayner E, Hebbel RP. Circulating activated endothelial cells in sickle cell anemia. N Engl J Med. 1997;337:1584–1590. doi: 10.1056/NEJM199711273372203. [DOI] [PubMed] [Google Scholar]
- 14.Bolarin DM, Swerdlow P, Wallace AM, Littsey L. Serum concentrations of laminin P1 and aminoterminal propeptide of type III procollagen in sickle cell disease. Haematologia (Budap) 1998;29:51–58. [PubMed] [Google Scholar]
- 15.Hines PC, Zen Q, Burney SN, Shea DA, Ataga KI, Orringer EP, Telen MJ, Parise LV. Novel epinephrine and cyclic AMP-mediated activation of BCAM/Lu-dependent sickle (SS) RBC adhesion. Blood. 2003;101:3281–3287. doi: 10.1182/blood-2001-12-0289. [DOI] [PubMed] [Google Scholar]
- 16.Delahunty M, Zennadi R, Telen MJ. LW protein: a promiscuous integrin receptor activated by adrenergic signaling. Transfus Clin Biol. 2006;13:44–49. doi: 10.1016/j.tracli.2006.02.022. [DOI] [PubMed] [Google Scholar]
- 17.Hermand P, Huet M, Callebaut I, Gane P, Ihanus E, Gahmberg CG, Cartron JP, Bailly P. Binding sites of leukocyte beta 2 integrins (LFA-1, Mac-1) on the human ICAM-4/LW blood group protein. J Biol Chem. 2000;275:26002–26010. doi: 10.1074/jbc.M002823200. [DOI] [PubMed] [Google Scholar]
- 18.Hermand P, Gane P, Huet M, Jallu V, Kaplan C, Sonneborn HH, Cartron JP, Bailly P. Red cell ICAM-4 is a novel ligand for platelet-activated alpha IIbbeta 3 Integrin. J Biol Chem. 2003;278:4892–4898. doi: 10.1074/jbc.M211282200. [DOI] [PubMed] [Google Scholar]
- 19.Zennadi R, Hines PC, De Castro LM, Cartron JP, Parise LV, Telen MJ. Epinephrine acts through erythroid signaling pathways to activate sickle cell adhesion to endothelium via LW-alphavbeta3 interactions. Blood. 2004;104:3774–3781. doi: 10.1182/blood-2004-01-0042. [DOI] [PubMed] [Google Scholar]
- 20.Joneckis CC, Ackley RL, Orringer EP, Wayner EA, Parise LV. Integrin alpha 4 beta 1 and glycoprotein IV (CD36) are expressed on circulating reticulocytes in sickle cell anemia. Blood. 1993;82:3548–3555. [PubMed] [Google Scholar]
- 21.Swerlick RA, Eckman JR, Kumar A, Jeitler M, Wick TM. Alpha 4 beta 1-integrin expression on sickle reticulocytes: vascular cell adhesion molecule-1-dependent binding to endothelium. Blood. 1993;82:1891–1899. [PubMed] [Google Scholar]
- 22.Sugihara K, Sugihara T, Mohandas N, Hebbel RP. Thrombospondin mediates adherence of CD36+ sickle reticulocytes to endothelial cells. Blood. 1992;88:2634–2642. [PubMed] [Google Scholar]
- 23.Brittain HA, Eckman JR, Wick TM. Sickle erythrocyte adherence to large vessel and microvascular endothelium under physiologic flow is quantitatively different. J Lab Clin Med. 1992;120:538–545. [PubMed] [Google Scholar]
- 24.Jaffe EA, Ruggiero JT, Leung LK, Doyle MJ, McKeown-Longo PJ, Mosher DF. Cultured human fibroblasts synthesize and secrete thrombospondin and incorporate it into extracellular matrix. Proc Natl Acad Sci U S A. 1983;80:998–1002. doi: 10.1073/pnas.80.4.998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brittain HA, Eckman JR, Swerlick RA, Howard RJ, Wick TM. Thrombospondin from activated platelets promotes sickle erythrocyte adherence to human microvascular endothelium under physiologic flow: a potential role for platelet activation in sickle cell vaso-occlusion. Blood. 1993;81:2137–2143. [PubMed] [Google Scholar]
- 26.Browne PV, Mosher DF, Steinberg MH, Hebbel RP. Disturbance of plasma and platelet thrombospondin levels in sickle cell disease. Am J Hematol. 1996;51:296–301. doi: 10.1002/(SICI)1096-8652(199604)51:4<296::AID-AJH8>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 27.Murphy-Ullrich JE, Mosher DF. Interactions of thrombospondin with endothelial cells: receptor-mediated binding and degradation. J Cell Biol. 1987;105:1603–1611. doi: 10.1083/jcb.105.4.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee K, Gane P, Roudot-Thoraval F, Godeau B, Bachir D, Bernaudin F, Cartron JP, Galacteros F, Bierling P. The nonexpression of CD36 on reticulocytes and mature red blood cells does not modify the clinical course of patients with sickle cell anemia. Blood. 2001;98:966–971. doi: 10.1182/blood.v98.4.966. [DOI] [PubMed] [Google Scholar]
- 29.Brittain JE, Mlinar KJ, Anderson CS, Orringer EP, Parise LV. Integrin-associated protein is an adhesion receptor on sickle red blood cells for immobilized thrombospondin. Blood. 2001;97:2159–2164. doi: 10.1182/blood.v97.7.2159. [DOI] [PubMed] [Google Scholar]
- 30.Brittain JE, Mlinar KJ, Anderson CS, Orringer EP, Parise LV. Activation of sickle red blood cell adhesion via integrin-associated protein/CD47-induced signal transduction. J Clin Invest. 2001;107:1555–1562. doi: 10.1172/JCI10817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brittain JE, Han J, Ataga KI, Orringer EP, Parise LV. Mechanism of CD47-induced alpha4beta1 integrin activation and adhesion in sickle reticulocytes. J Biol Chem. 2004;279:42393–42402. doi: 10.1074/jbc.M407631200. [DOI] [PubMed] [Google Scholar]
- 32.Nachman R, Levine R, Jaffe EA. Synthesis of factor VIII antigen by cultured guinea pig megakaryocytes. J Clin Invest. 1977;60:914–921. doi: 10.1172/JCI108846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jaffe EA, Hoyer LW, Nachman RL. Synthesis of antihemophilic factor antigen by cultured human endothelial cells. J Clin Invest. 1973;52:2757–2764. doi: 10.1172/JCI107471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wick TM, Moake JL, Udden MM, Eskin SG, Sears DA, McIntire LV. Unusually large von Willebrand factor multimers increase adhesion of sickle erythrocytes to human endothelial cells under controlled flow. J Clin Invest. 1987;80:905–910. doi: 10.1172/JCI113151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaul DK, Nagel RL, Chen D, Tsai HM. Sickle erythrocyte-endothelial interactions in microcirculation: the role of von Willebrand factor and implications for vasoocclusion. Blood. 1993;81:2429–2438. [PubMed] [Google Scholar]
- 36.Wick TM, Moake JL, Udden MM, McIntire LV. Unusually large von Willebrand factor multimers preferentially promote young sickle and nonsickle erythrocyte adhesion to endothelial cells. Am J Hematol. 1993;42:284–292. doi: 10.1002/ajh.2830420308. [DOI] [PubMed] [Google Scholar]
- 37.Kaul DK, Fabry ME, Windisch P, Baez S, Nagel RL. Erythrocytes in sickle cell anemia are heterogeneous in their rheological and hemodynamic characteristics. J Clin Invest. 1983;72:22–31. doi: 10.1172/JCI110960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaul DK, Fabry ME, Nagel RL. Microvascular sites and characteristics of sickle cell adhesion to vascular endothelium in shear flow conditions: pathophysiological implications. Proc Natl Acad Sci U S A. 1989;86:3356–3360. doi: 10.1073/pnas.86.9.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fabry ME, Sengupta A, Suzuka SM, Costantini F, Rubin EM, Hofrichter J, Christoph G, Manci E, Culberson D, Factor SM, Nagel RL. A second generation transgenic mouse model expressing both hemoglobin S (HbS) and HbS-Antilles results in increased phenotypic severity. Blood. 1995;86:2419–2428. [PubMed] [Google Scholar]
- 40.Kaul DK, Fabry ME, Costantini F, Rubin EM, Nagel RL. In vivo demonstration of red cell-endothelial interaction, sickling and altered microvascular response to oxygen in the sickle transgenic mouse. J Clin Invest. 1995;96:2845–2853. doi: 10.1172/JCI118355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaul DK, Fabry ME, Nagel RL. The pathophysiology of vascular obstruction in the sickle syndromes. Blood Rev. 1996;10:29–44. doi: 10.1016/s0268-960x(96)90018-1. [DOI] [PubMed] [Google Scholar]
- 42.Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, Klug PP. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330:1639–1644. doi: 10.1056/NEJM199406093302303. [DOI] [PubMed] [Google Scholar]
- 43.Castro O, Brambilla DJ, Thorington B, Reindorf CA, Scott RB, Gillette P, Vera JC, Levy PS. The acute chest syndrome in sickle cell disease: incidence and risk factors. The Cooperative Study of Sickle Cell Disease. Blood. 1994;84:643–649. [PubMed] [Google Scholar]
- 44.Kinney TR, Sleeper LA, Wang WC, Zimmerman RA, Pegelow CH, Ohene-Frempong K, Wethers DL, Bello JA, Vichinsky EP, Moser FG, Gallagher DM, DeBaun MR, Platt OS, Miller ST. Silent cerebral infarcts in sickle cell anemia: a risk factor analysis. The Cooperative Study of Sickle Cell Disease. Pediatrics. 1999;103:640–645. doi: 10.1542/peds.103.3.640. [DOI] [PubMed] [Google Scholar]
- 45.Adler BK, Salzman DE, Carabasi MH, Vaughan WP, Reddy VV, Prchal JT. Fatal sickle cell crisis after granulocyte colony-stimulating factor administration. Blood. 2001;97:3313–3314. doi: 10.1182/blood.v97.10.3313. [DOI] [PubMed] [Google Scholar]
- 46.Grigg AP. Granulocyte colony-stimulating factor-induced sickle cell crisis and multiorgan dysfunction in a patient with compound heterozygous sickle cell/beta+ thalassemia. Blood. 2001;97:3998–3999. doi: 10.1182/blood.v97.12.3998. [DOI] [PubMed] [Google Scholar]
- 47.Turhan A, Weiss LA, Mohandas N, Coller BS, Frenette PS. Primary role for adherent leukocytes in sickle cell vascular occlusion: a new paradigm. Proc Natl Acad Sci U S A. 2002;99:3047–3051. doi: 10.1073/pnas.052522799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.House SD, Lipowsky HH. Leukocyte-endothelium adhesion: microhemodynamics in mesentery of the cat. Microvasc Res. 1987;34:363–379. doi: 10.1016/0026-2862(87)90068-9. [DOI] [PubMed] [Google Scholar]
- 49.Perry MA, Granger DN. Role of CD11/CD18 in shear rate-dependent leukocyte-endothelial cell interactions in cat mesenteric venules. J Clin Invest. 1991;87:1798–1804. doi: 10.1172/JCI115200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sumagin R, Sarelius IH. TNF-alpha activation of arterioles and venules alters distribution and levels of ICAM-1 and affects leukocyte-endothelial cell interactions. Am J Physiol Heart Circ Physiol. 2006;291:H2116–H2125. doi: 10.1152/ajpheart.00248.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Middleton J, Patterson AM, Gardner L, Schmutz C, Ashton BA. Leukocyte extravasation: chemokine transport and presentation by the endothelium. Blood. 2002;100:3853–60. doi: 10.1182/blood.V100.12.3853. [DOI] [PubMed] [Google Scholar]
- 52.Tan P, Luscinskas FW, Homer-Vanniasinkam S. Cellular and molecular mechanisms of inflammation and thrombosis. Eur J Vasc Endovasc Surg. 1999;17:373–389. doi: 10.1053/ejvs.1998.0759. [DOI] [PubMed] [Google Scholar]
- 53.Francis RB, Jr, Haywood LJ. Elevated immunoreactive tumor necrosis factor and interleukin-1 in sickle cell disease. J Natl Med Assoc. 1992;84:611–615. [PMC free article] [PubMed] [Google Scholar]
- 54.Makis AC, Hatzimichael EC, Mavridis A, Bourantas KL. Alpha-2-macroglobulin and interleukin-6 levels in steady-state sickle cell disease patients. Acta Haematol. 2000;104:164–168. doi: 10.1159/000046509. [DOI] [PubMed] [Google Scholar]
- 55.Akohoue SA, Shankar S, Milne GL, Morrow J, Chen KY, Ajayi WU, Buchowski MS. Energy expenditure, inflammation, and oxidative stress in steady-state adolescents with sickle cell anemia. Pediatr Res. 2007;61:233–238. doi: 10.1203/pdr.0b013e31802d7754. [DOI] [PubMed] [Google Scholar]
- 56.Michaels LA, Ohene-Frempong K, Zhao H, Douglas SD. Serum levels of substance P are elevated in patients with sickle cell disease and increase further during vaso-occlusive crisis. Blood. 1998;92:3148–3151. [PubMed] [Google Scholar]
- 57.Belcher JD, Marker PH, Weber JP, Hebbel RP, Vercellotti GM. Activated monocytes in sickle cell disease: potential role in the activation of vascular endothelium and vaso-occlusion. Blood. 2000;96:2451–2459. [PubMed] [Google Scholar]
- 58.Hedo CC, Aken'ova YA, Okpala IE, Durojaiye AO, Salimonu LS. Acute phase reactants and severity of homozygous sickle cell disease. J Intern Med. 1993;233:467–470. doi: 10.1111/j.1365-2796.1993.tb01000.x. [DOI] [PubMed] [Google Scholar]
- 59.Middleton J, Neil S, Wintle J, Clark-Lewis I, Moore H, Lam C, Auer M, Hub E, Rot A. Transcytosis and surface presentation of IL-8 by venular endothelial cells. Cell. 1997;91:385–395. doi: 10.1016/s0092-8674(00)80422-5. [DOI] [PubMed] [Google Scholar]
- 60.Wang L, Fuster M, Sriramarao P, Esko JD. Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat Immunol. 2005;6:902–910. doi: 10.1038/ni1233. [DOI] [PubMed] [Google Scholar]
- 61.Akerley WL, 3rd, Guyre PM, Davis BH. Neutrophil activation through high-affinity Fc gamma receptor using a monomeric antibody with unique properties. Blood. 1991;77:607–615. [PubMed] [Google Scholar]
- 62.Fadlon E, Vordermeier S, Pearson TC, Mire-Sluis AR, Dumonde DC, Phillips J, Fishlock K, Brown KA. Blood polymorphonuclear leukocytes from the majority of sickle cell patients in the crisis phase of the disease show enhanced adhesion to vascular endothelium and increased expression of CD64. Blood. 1998;91:266–274. [PubMed] [Google Scholar]
- 63.Lard LR, Mul FP, de Haas M, Roos D, Duits AJ. Neutrophil activation in sickle cell disease. J Leukoc Biol. 1999;66:411–415. doi: 10.1002/jlb.66.3.411. [DOI] [PubMed] [Google Scholar]
- 64.Ley K, Gaehtgens P, Fennie C, Singer MS, Lasky LA, Rosen SD. Lectin-like cell adhesion molecule 1 mediates leukocyte rolling in mesenteric venules in vivo. Blood. 1991;77:2553–2555. [PubMed] [Google Scholar]
- 65.de Haas M, Kerst JM, van der Schoot CE, Calafat J, Hack CE, Nuijens JH, Roos D, van Oers RH, von dem Borne AE. Granulocyte colony-stimulating factor administration to healthy volunteers: analysis of the immediate activating effects on circulating neutrophils. Blood. 1994;84:3885–3894. [PubMed] [Google Scholar]
- 66.McEver RP, Cummings RD. Perspectives series: cell adhesion in vascular biology. Role of PSGL-1 binding to selectins in leukocyte recruitment. J Clin Invest. 1997;100:485–491. doi: 10.1172/JCI119556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bevilacqua MP. Endothelial-leukocyte adhesion molecules. Annu Rev Immunol. 1993;11:767–804. doi: 10.1146/annurev.iy.11.040193.004003. [DOI] [PubMed] [Google Scholar]
- 68.Kaul DK, Hebbel RP. Hypoxia/reoxygenation causes inflammatory response in transgenic sickle mice but not in normal mice. J Clin Invest. 2000;106:411–420. doi: 10.1172/JCI9225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wood K, Russell J, Hebbel RP, Granger DN. Differential expression of E- and P-selectin in the microvasculature of sickle cell transgenic mice. Microcirculation. 2004;11:377–385. doi: 10.1080/10739680490437559. [DOI] [PubMed] [Google Scholar]
- 70.Key NS, Slungaard A, Dandelet L, Nelson SC, Moertel C, Styles LA, Kuypers FA, Bach RR. Whole blood tissue factor procoagulant activity is elevated in patients with sickle cell disease. Blood. 1998;91:4216–4223. [PubMed] [Google Scholar]
- 71.Eilertsen KE, Osterud B. Tissue factor: (patho)physiology and cellular biology. Blood Coagul Fibrinolysis. 2004;7:521–538. doi: 10.1097/00001721-200410000-00001. [DOI] [PubMed] [Google Scholar]
- 72.Solovey A, Gui L, Key NS, Hebbel RP. Tissue factor expression by endothelial cells in sickle cell anemia. J Clin Invest. 1998;101:1899–1904. doi: 10.1172/JCI1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shet AS, Aras O, Gupta K, Hass MJ, Rausch DJ, Saba N, Koopmeiners L, Key NS, Hebbel RP. Sickle blood contains tissue factor-positive microparticles derived from endothelial cells and monocytes. Blood. 2003;102:2678–2683. doi: 10.1182/blood-2003-03-0693. [DOI] [PubMed] [Google Scholar]
- 74.Solovey A, Kollander R, Shet A, Milbauer LC, Choong S, Panoskaltsis-Mortari A, Blazar BR, Kelm RJ, Jr, Hebbel RP. Endothelial cell expression of tissue factor in sickle mice is augmented by hypoxia/reoxygenation and inhibited by lovastatin. Blood. 2004;104:840–846. doi: 10.1182/blood-2003-10-3719. [DOI] [PubMed] [Google Scholar]
- 75.Krishnaswamy S. The interaction of human factor VIIa with tissue factor. J Biol Chem. 1992;267:23696–23706. [PubMed] [Google Scholar]
- 76.Franck PF, Bevers EM, Lubin BH, Comfurius P, Chiu DT, Op den Kamp JA, Zwaal RF, van Deenen LL, Roelofsen B. Uncoupling of the membrane skeleton from the lipid bilayer. The cause of accelerated phospholipid flip-flop leading to an enhanced procoagulant activity of sickled cells. J Clin Invest. 1985;75:183–190. doi: 10.1172/JCI111672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Seigneuret M, Devaux PF. ATP-dependent asymmetric distribution of spin-labeled phospholipids in the erythrocyte membrane: relation to shape changes. Proc Natl Acad Sci USA. 1984;81:3751–3755. doi: 10.1073/pnas.81.12.3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.de Jong K, Larkin SK, Styles LA, Bookchin RM, Kuypers FA. Characterization of the phosphatidylserine-exposing subpopulation of sickle cells. Blood. 2001;98:860–867. doi: 10.1182/blood.v98.3.860. [DOI] [PubMed] [Google Scholar]
- 79.Monroe DM, Hoffman M, Roberts HR. Platelets and thrombin generation. Arterioscler Thromb Vasc Biol. 2002;22:1381–1389. doi: 10.1161/01.atv.0000031340.68494.34. [DOI] [PubMed] [Google Scholar]
- 80.Tomer A, Harker LA, Kasey S, Eckman JR. Thrombogenesis in sickle cell disease. J Lab Clin Med. 2001;137:398–407. doi: 10.1067/mlc.2001.115450. [DOI] [PubMed] [Google Scholar]
- 81.Leslie J, Langler D, Serjeant GR, Serjeant BE, Desai P, Gordon YB. Coagulation changes during the steady state in homozygous sickle-cell disease in Jamaica. Br J Haematol. 1975;30:159–166. doi: 10.1111/j.1365-2141.1975.tb00530.x. [DOI] [PubMed] [Google Scholar]
- 82.Nesheim M. Fibrinolysis and the plasma carboxypeptidase. Curr Opin Hematol. 1998;5:309–313. doi: 10.1097/00062752-199809000-00001. [DOI] [PubMed] [Google Scholar]
- 83.Esmon CT. Regulation of blood coagulation. Biochim Biophys Acta. 2000;1477:349–360. doi: 10.1016/s0167-4838(99)00266-6. [DOI] [PubMed] [Google Scholar]
- 84.Westerman MP, Green D, Gilman-Sachs A, Beaman K, Freels S, Boggio L, Allen S, Zuckerman L, Schlegel R, Williamson P. Antiphospholipid antibodies, proteins C and S, and coagulation changes in sickle cell disease. J Lab Clin Med. 1999;134:352–362. doi: 10.1016/s0022-2143(99)90149-x. [DOI] [PubMed] [Google Scholar]
- 85.Wright JG, Malia R, Cooper P, Thomas P, Preston FE, Serjeant GR. Protein C and protein S in homozygous sickle cell disease: does hepatic dysfunction contribute to low levels? Br J Haematol. 1997;98:627–631. doi: 10.1046/j.1365-2141.1997.2663083.x. [DOI] [PubMed] [Google Scholar]
- 86.Tam DA. Protein C and protein S activity in sickle cell disease and stroke. J Child Neurol. 1997;12:19–21. doi: 10.1177/088307389701200103. [DOI] [PubMed] [Google Scholar]
- 87.Ruggeri ZM. Platelets in atherothrombosis. Nat Med. 2002;8:1227–1234. doi: 10.1038/nm1102-1227. [DOI] [PubMed] [Google Scholar]
- 88.Furie B, Furie BC. Role of platelet P-selectin and microparticle PSGL-1 in thrombus formation. Trends Mol Med. 2004;10:171–178. doi: 10.1016/j.molmed.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 89.Inwald DP, Kirkham FJ, Peters MJ, Lane R, Wade A, Evans JP, Klein NJ. Platelet and leucocyte activation in childhood sickle cell disease: association with nocturnal hypoxaemia. Br J Haematol. 2000;111:474–481. doi: 10.1046/j.1365-2141.2000.02353.x. [DOI] [PubMed] [Google Scholar]
- 90.Vandendries ER, Furie BC, Furie B. Role of P-selectin and PSGL-1 in coagulation and thrombosis. Thromb Haemost. 2004;92:459–466. doi: 10.1160/TH04-05-0306. [DOI] [PubMed] [Google Scholar]
- 91.Wun T, Cordoba M, Rangaswami A, Cheung AW, Paglieroni T. Activated monocytes and platelet-monocyte aggregates in patients with sickle cell disease. Clin Lab Haematol. 2002;24:81–88. doi: 10.1046/j.1365-2257.2002.00433.x. [DOI] [PubMed] [Google Scholar]
- 92.Aslan M, Freeman BA. Oxidant-mediated impairment of nitric oxide signaling in sickle cell disease--mechanisms and consequences. Cell Mol Biol (Noisy-le-grand) 2004;50:95–105. [PubMed] [Google Scholar]
- 93.Mayer B, Hemmens B. Biosynthesis and action of nitric oxide in mammalian cells. Trends Biochem Sci. 1997;22:477–481. doi: 10.1016/s0968-0004(97)01147-x. [DOI] [PubMed] [Google Scholar]
- 94.Moncada S, Higgs A. The L-arginine-nitric oxide pathway. N Engl J Med. 1993;329:2002–2012. doi: 10.1056/NEJM199312303292706. [DOI] [PubMed] [Google Scholar]
- 95.Lincoln TM, Cornwell TL. Intracellular cyclic GMP receptor proteins. FASEB J. 1993;7:328–338. doi: 10.1096/fasebj.7.2.7680013. [DOI] [PubMed] [Google Scholar]
- 96.Radomski MW, Palmer RM, Moncada S. The role of nitric oxide and cGMP in platelet adhesion to vascular endothelium. Biochem Biophys Res Commun. 1987;148:1482. doi: 10.1016/s0006-291x(87)80299-1. [DOI] [PubMed] [Google Scholar]
- 97.Loscalzo J. Nitric oxide insufficiency, platelet activation, and arterial thrombosis. Circ Res. 2001;88:756–762. doi: 10.1161/hh0801.089861. [DOI] [PubMed] [Google Scholar]
- 98.De Caterina R, Libby P, Peng HB, Thannickal VJ, Rajavashisth TB, Gimbrone MA, Jr, Shin WS, Liao JK. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J Clin Invest. 1995;96:60–68. doi: 10.1172/JCI118074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Peng HB, Libby P, Liao JK. Induction and stabilization of I kappa B alpha by nitric oxide mediates inhibition of NF-kappa B. J Biol Chem. 1995;270:14214–14219. doi: 10.1074/jbc.270.23.14214. [DOI] [PubMed] [Google Scholar]
- 100.Abraham E. NF-kappaB activation. Crit Care Med. 2000;28:N100–N104. doi: 10.1097/00003246-200004001-00012. [DOI] [PubMed] [Google Scholar]
- 101.Khan BV, Harrison DG, Olbrych MT, Alexander RW, Medford RM. Nitric oxide regulates vascular cell adhesion molecule 1 gene expression and redox-sensitive transcriptional events in human vascular endothelial cells. Proc Natl Acad Sci U S A. 1996;93:9114–9119. doi: 10.1073/pnas.93.17.9114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fujita H, Morita I, Murota S. A possible mechanism for vascular endothelial cell injury elicited by activated leukocytes: a significant involvement of adhesion molecules, CD11/CD18, and ICAM-1. Arch Biochem Biophys. 1994;309:62–69. doi: 10.1006/abbi.1994.1085. [DOI] [PubMed] [Google Scholar]
- 103.Broeders MA, Tangelder GJ, Slaaf DW, Reneman RS, oude Egbrink MG. Endogenous nitric oxide protects against thromboembolism in venules but not in arterioles. Arterioscler Thromb Vasc Biol. 1998;18:139–145. doi: 10.1161/01.atv.18.1.139. [DOI] [PubMed] [Google Scholar]
- 104.Kalogeris TJ, Kevil CG, Laroux FS, Coe LL, Phifer TJ, Alexander JS. Differential monocyte adhesion and adhesion molecule expression in venous and arterial endothelial cells. Am, J, Physiol. 1999;276:L9–L19. doi: 10.1152/ajplung.1999.276.1.L9. [DOI] [PubMed] [Google Scholar]
- 105.Reiter CD, Wang X, Tanus-Santos JE, Hogg N, Cannon RO, 3rd, Schechter AN, Gladwin MT. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med. 2002;8:1383–1389. doi: 10.1038/nm1202-799. [DOI] [PubMed] [Google Scholar]
- 106.Aslan M, Thornley-Brown D, Freeman BA. Reactive species in sickle cell disease. Ann NY Acad Sci. 2000;899:375–91. doi: 10.1111/j.1749-6632.2000.tb06201.x. [DOI] [PubMed] [Google Scholar]
- 107.Aslan M, Ryan TM, Adler B, Townes TM, Parks DA, Thompson JA, Tousson A, Gladwin MT, Patel RP, Tarpey MM, Batinic-Haberle I, White CR, Freeman BA. Oxygen radical inhibition of nitric oxide-dependent vascular function in sickle cell disease. Proc Natl Acad Sci U S A. 2001;98:15215–15220. doi: 10.1073/pnas.221292098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Houston M, Estevez A, Chumley P, Aslan M, Marklund S, Parks DA, Freeman BA. Binding of xanthine oxidase to vascular endothelium. Kinetic characterization and oxidative impairment of nitric oxide-dependent signaling. J Biol Chem. 1999;274:4985–4994. doi: 10.1074/jbc.274.8.4985. [DOI] [PubMed] [Google Scholar]
- 109.Wood KC, Hebbel RP, Granger DN. Endothelial cell NADPH oxidase mediates the cerebral microvascular dysfunction in sickle cell transgenic mice. FASEB J. 2005;19:989–991. doi: 10.1096/fj.04-3218fje. [DOI] [PubMed] [Google Scholar]
- 110.Wood KC, Hebbel RP, Lefer DJ, Granger DN. Critical role of endothelial cell-derived nitric oxide synthase in sickle cell disease-induced microvascular dysfunction. Free Radic Biol Med. 2006;40:1443–1453. doi: 10.1016/j.freeradbiomed.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 111.Gibson Q, Roughton FJW. The kinetics and equilibria of the reactions of nitric oxide with sheep hemoglobin. J Physiol (Lond) 1957;136:507–526. doi: 10.1113/jphysiol.1957.sp005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Herold S, Exner M, Nauser T. Kinetic and mechanistic studies of the NO-mediated oxidation of oxymyoglobin and oxyhemoglobin. Biochemistry. 2001;40:3385–3395. doi: 10.1021/bi002407m. [DOI] [PubMed] [Google Scholar]
- 113.Liu X. Diffusion-limited reaction of free nitric oxide with erythrocytes. J BiolChem. 1998;273:18709–18713. doi: 10.1074/jbc.273.30.18709. [DOI] [PubMed] [Google Scholar]
- 114.Bensinger TA, Gillette PN. Hemolysis in sickle-cell disease. Arch Intern Med. 1974;133:624–631. [PubMed] [Google Scholar]
- 115.Langlois MR, Delanghe JR. Biological and clinical significance of haptoglobin polymorphism in humans. Clin Chem. 1996;42:1589–1600. [PubMed] [Google Scholar]
- 116.Bourantas KL, Dalekos GN, Makis A, Chaidos A, Tsiara S, Mavridis A. Acute phase proteins and interleukins in steady state sickle cell disease. Eur J Haematol. 1998;61:49–54. doi: 10.1111/j.1600-0609.1998.tb01060.x. [DOI] [PubMed] [Google Scholar]
- 117.Kato GJ, McGowan V, Machado RF, Little JA, Taylor J, 6th, Morris CR, Nichols JS, Wang X, Poljakovic M, Morris SM, Jr, Gladwin MT. Lactate dehydrogenase as a biomarker of hemolysis-associated nitric oxide resistance, priapism, leg ulceration, pulmonary hypertension, and death in patients with sickle cell disease. Blood. 2006;107:2279–2285. doi: 10.1182/blood-2005-06-2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Morris CR, Kato GJ, Poljakovic M, Wang X, Blackwelder WC, Sachdev V, Hazen SL, Vichinsky EP, Morris SM, Jr, Gladwin MT. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. JAMA. 2005;294:81–90. doi: 10.1001/jama.294.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Morris CR, Morris SM, Jr, Hagar W, van Warmerdam J, Claster S, Kepka-Lenhart D, Machado L, Kuypers FA, Vichinsky EP. Arginine therapy: a new treatment for pulmonary hypertension in sickle cell disease? Am J Respir Crit Care Med. 2003;168:63–69. doi: 10.1164/rccm.200208-967OC. [DOI] [PubMed] [Google Scholar]
- 120.Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev. 2007;21:37–47. doi: 10.1016/j.blre.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hsu LL, Champion HC, Campbell-Lee SA, Bivalacqua TJ, Manci EA, Diwan BA, Schimel DM, Cochard AE, Wang X, Schechter AN, Noguchi CT, Gladwin MT. Hemolysis in sickle cell mice causes pulmonary hypertension due to global impairment in nitric oxide bioavailability. Blood. 2007;109:3088–3098. doi: 10.1182/blood-2006-08-039438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Moestrup SK, Moller HJ. CD163: a regulated hemoglobin scavenger receptor with a role in the anti-inflammatory response. Ann Med. 2004;36:347–354. doi: 10.1080/07853890410033171. [DOI] [PubMed] [Google Scholar]
- 123.Choi AM, Alam J. Heme oxygenase-1: function, regulation, and implication of a novel stress-inducible protein in oxidant-induced lung injury. Am J Respir Cell Mol Biol. 1996;15:9–19. doi: 10.1165/ajrcmb.15.1.8679227. [DOI] [PubMed] [Google Scholar]
- 124.Jison ML, Munson PJ, Barb JJ, Suffredini AF, Talwar S, Logun C, Raghavachari N, Beigel JH, Shelhamer JH, Danner RL, Gladwin MT. Blood mononuclear cell gene expression profiles characterize the oxidant, hemolytic, and inflammatory stress of sickle cell disease. Blood. 2004;104:270–280. doi: 10.1182/blood-2003-08-2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Nath KA, Grande JP, Haggard JJ, Croatt AJ, Katusic ZS, Solovey A, Hebbel RP. Oxidative stress and induction of heme oxygenase-1 in the kidney in sickle cell disease. Am J Pathol. 2001;158:893–903. doi: 10.1016/S0002-9440(10)64037-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Nielsen MJ, Petersen SV, Jacobsen C, Oxvig C, Rees D, Moller HJ, Moestrup SK. Haptoglobin-related protein is a high-affinity hemoglobin-binding plasma protein. Blood. 2006;108:2846–2849. doi: 10.1182/blood-2006-05-022327. [DOI] [PubMed] [Google Scholar]
- 127.Ou J, Ou Z, Jones DW, Holzhauer S, Hatoum OA, Ackerman AW, Weihrauch DW, Gutterman DD, Guice K, Oldham KT, Hillery CA, Pritchard KA., Jr L-4F, an apolipoprotein A-1 mimetic, dramatically improves vasodilation in hypercholesterolemia and sickle cell disease. Circulation. 2003;107:2337–2341. doi: 10.1161/01.CIR.0000070589.61860.A9. [DOI] [PubMed] [Google Scholar]
- 128.Kato GJ, Hsieh M, Machado R, Taylor J, 6th, Little J, Butman JA, Lehky T, Tisdale J, Gladwin MT. Cerebrovascular disease associated with sickle cell pulmonary hypertension. Am J Hematol. 2006;81:503–510. doi: 10.1002/ajh.20642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hillery CA, Panepinto JA. Pathophysiology of stroke in sickle cell disease. Microcirculation. 2004;11:195–208. doi: 10.1080/10739680490278600. [DOI] [PubMed] [Google Scholar]
- 130.Gladwin MT, Kato GJ. Cardiopulmonary complications of sickle cell disease: role of nitric oxide and hemolytic anemia. Hematology Am Soc Hematol Educ Program. 2005:51–57. doi: 10.1182/asheducation-2005.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kaul DK, Liu XD, Choong S, Belcher JD, Vercellotti GM, Hebbel RP. Anti-inflammatory therapy ameliorates leukocyte adhesion and microvascular flow abnormalities in transgenic sickle mice. Am J Physiol Heart Circ Physiol. 2004;287:H293–H301. doi: 10.1152/ajpheart.01150.2003. [DOI] [PubMed] [Google Scholar]
- 132.Nath KA, Shah V, Haggard JJ, Croatt AJ, Smith LA, Hebbel RP, Katusic ZS. Mechanisms of vascular instability in a transgenic mouse model of sickle cell disease. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1949–R1955. doi: 10.1152/ajpregu.2000.279.6.R1949. [DOI] [PubMed] [Google Scholar]
- 133.Kowaluk EA, Seth P, Fung HL. Metabolic activation of sodium nitroprusside to nitric oxide in vascular smooth muscle. J Pharmacol Exp Ther. 1992;262:916–922. [PubMed] [Google Scholar]
- 134.Bayraktutan U, Draper N, Lang D, Shah AM. Expression of functional neutrophil-type NADPH oxidase in cultured rat coronary microvascular endothelial cells. Cardiovasc Res. 1998;38:256–262. doi: 10.1016/s0008-6363(98)00003-0. [DOI] [PubMed] [Google Scholar]
- 135.White KA, Marletta MA. Nitric oxide synthase is a cytochrome P-450 type hemoprotein. Biochemistry. 1992;31:6627–6631. doi: 10.1021/bi00144a001. [DOI] [PubMed] [Google Scholar]
- 136.Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem, J. 2001;57:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bec N, Gorren AC, Voelker C, Mayer B, Lange R. Reaction of neuronal nitric-oxide synthase with oxygen at low temperature. Evidence for reductive activation of the oxy-ferrous complex by tetrahydrobiopterin. J Biol Chem. 1998 May;273:13502–13508. doi: 10.1074/jbc.273.22.13502. [DOI] [PubMed] [Google Scholar]
- 138.Presta A, Siddhanta U, Wu C, Sennequier N, Huang L, Abu-Soud HM, Erzurum S, Stuehr DJ. Comparative functioning of dihydro- and tetrahydropterins in supporting electron transfer, catalysis, and subunit dimerization in inducible nitric oxide synthase. Biochemistry. 1998;37:298–310. doi: 10.1021/bi971944c. [DOI] [PubMed] [Google Scholar]
- 139.Eiserich JP, Baldus S, Brennan ML, Ma W, Zhang C, Tousson A, Castro L, Lusis AJ, Nauseef WM, White CR, Freeman BA. Myeloperoxidase, a leukocyte-derived vascular NO oxidase. Science. 2002;296:2391–2394. doi: 10.1126/science.1106830. [DOI] [PubMed] [Google Scholar]
- 140.Baldus S, Eiserich JP, Brennan ML, Jackson RM, Alexander CB, Freeman BA. Spatial mapping of pulmonary and vascular nitrotyrosine reveals the pivotal role of myeloperoxidase as a catalyst for tyrosine nitration in inflammatory diseases. Free Radic Biol Med. 2002;33:1010. doi: 10.1016/s0891-5849(02)00993-0. [DOI] [PubMed] [Google Scholar]
- 141.Baldus S, Eiserich JP, Mani A, Castro L, Figueroa M, Chumley P, Ma W, Tousson A, White CR, Bullard DC, Brennan ML, Lusis AJ, Moore KP, Freeman BA. Endothelial transcytosis of myeloperoxidase confers specificity to vascular ECM proteins as targets of tyrosine nitration. J Clin Invest. 2001;108:1759–1770. doi: 10.1172/JCI12617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Osarogiagbon UR, Choong S, Belcher JD, Vercellotti GM, Paller MS, Hebbel RP. Reperfusion injury pathophysiology in sickle transgenic mice. Blood. 2000;96:314–320. [PubMed] [Google Scholar]
- 143.Parks DA, Granger DN. Ischemia induced vascular changes: role of xanthine oxidase and hydroxyl radicals. Am J Physiol. 1983;245:G285–G289. doi: 10.1152/ajpgi.1983.245.2.G285. [DOI] [PubMed] [Google Scholar]
- 144.Lee VG, Johnson ML, Baust J, Laubach VE, Watkins SC, Billiar TR. The roles of iNOS in liver ischemia-reperfusion injury. Shock. 2001;16:355–360. doi: 10.1097/00024382-200116050-00006. [DOI] [PubMed] [Google Scholar]
- 145.Ling H, Gengaro PE, Edelstein CL, Martin PY, Wangsiripaisan A, Nemenoff R, Schrier RW. Effect of hypoxia on proximal tubules isolated from nitric oxide synthase knockout mice. Kidney Int. 1998;53:1642–1646. doi: 10.1046/j.1523-1755.1998.00913.x. [DOI] [PubMed] [Google Scholar]
- 146.Aslan M, Ryan TM, Townes TM, Coward L, Kirk MC, Barnes S, Alexander CB, Rosenfeld SS, Freeman BA. Nitric oxide-dependent generation of reactive species in sickle cell disease. Actin tyrosine nitration induces defective cytoskeletal polymerization. J Biol Chem. 2003;278:4194–4204. doi: 10.1074/jbc.M208916200. [DOI] [PubMed] [Google Scholar]
- 147.Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert SV, McMahon RP, Bonds DR. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med. 1995;332:1317–1322. doi: 10.1056/NEJM199505183322001. [DOI] [PubMed] [Google Scholar]
- 148.Smith RD, Li J, Noguchi CT, Schechter AN. Quantitative PCR analysis of HbF inducers in primary human adult erythroid cells. Blood. 2000;95:863–869. [PubMed] [Google Scholar]
- 149.Stolze K, Nohl H. EPR studies on the oxidation of hydroxyurea to paramagnetic compounds by oxyhemoglobin. Biochem Pharmacol. 1990;40:799–802. doi: 10.1016/0006-2952(90)90318-f. [DOI] [PubMed] [Google Scholar]
- 150.Glover RE, Ivy ED, Orringer EP, Maeda H, Mason RP. Detection of nitrosyl hemoglobin in venous blood in the treatment of sickle cell anemia with hydroxyurea. Mol Pharmacol. 1999;55:1006–1010. doi: 10.1124/mol.55.6.1006. [DOI] [PubMed] [Google Scholar]
- 151.Ikuta T, Ausenda S, Cappellini MD. Mechanism for fetal globin gene expression: role of the soluble guanylate cyclase-cGMP-dependent protein kinase pathway. Proc Natl Acad Sci U S A. 2001;98:1847–1852. doi: 10.1073/pnas.041599798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ley TJ, DeSimone J, Noguchi CT, Turner PH, Schechter AN, Heller P, Nienhuis AW. 5-Azacytidine increases gamma-globin synthesis and reduces the proportion of dense cells in patients with sickle cell anemia. Blood. 1983;62:370–380. [PubMed] [Google Scholar]
- 153.DeSimone J, Koshy M, Dorn L, Lavelle D, Bressler L, Molokie R, Talischy N. Maintenance of elevated fetal hemoglobin levels by decitabine during dose interval treatment of sickle cell anemia. Blood. 2002;99:3905–3908. doi: 10.1182/blood.v99.11.3905. [DOI] [PubMed] [Google Scholar]
- 154.Wahl C, Liptay S, Adler G, Schmid RM. Sulfasalazine: a potent and specific inhibitor of nuclear factor kappa B. J Clin Invest. 1998;101:1163–1174. doi: 10.1172/JCI992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Solovey AA, Solovey AN, Harkness J, Hebbel RP. Modulation of endothelial cell activation in sickle cell disease: a pilot study. Blood. 2001;97:1937–1934. doi: 10.1182/blood.v97.7.1937. [DOI] [PubMed] [Google Scholar]
- 156.Barabino GA, Liu XD, Ewenstein BM, Kaul DK. Anionic polysaccharides inhibit adhesion of sickle erythrocytes to the vascular endothelium and result in improved hemodynamic behavior. Blood. 1999;93:1422–1429. [PubMed] [Google Scholar]
- 157.Kaul DK, Tsai HM, Liu XD, Nakada MT, Nagel RL, Coller BS. Monoclonal antibodies to alphaVbeta3 (7E3 and LM609) inhibit sickle red blood cell-endothelium interactions induced by platelet-activating factor. Blood. 2000;95:368–74. [PubMed] [Google Scholar]
- 158.Lutty GA, Taomoto M, Cao J, McLeod DS, Vanderslice P, McIntyre BW, Fabry ME, Nagel RL. Inhibition of TNF-alpha-induced sickle RBC retention in retina by a VLA-4 antagonist. Invest Ophthalmol Vis Sci. 2001;42:1349–1355. [PubMed] [Google Scholar]
- 159.Matsui NM, Borsig L, Rosen SD, Yaghmai M, Varki A, Embury SH. P-selectin mediates the adhesion of sickle erythrocytes to the endothelium. Blood. 2001;98:1955–1962. doi: 10.1182/blood.v98.6.1955. [DOI] [PubMed] [Google Scholar]
- 160.Kumar A, Eckman JR, Wick TM. Inhibition of plasma-mediated adherence of sickle erythrocytes to microvascular endothelium by conformationally constrained RGD-containing peptides. Am J Hematol. 1996;53:92, 98. doi: 10.1002/(SICI)1096-8652(199610)53:2<92::AID-AJH6>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 161.Mahaseth H, Vercellotti GM, Welch TE, Bowlin PR, Sonbol KM, Hsia CJ, Li M, Bischof JC, Hebbel RP, Belcher JD. Polynitroxyl albumin inhibits inflammation and vasoocclusion in transgenic sickle mice. J Lab Clin Med. 2005;145:204–211. doi: 10.1016/j.lab.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 162.Belcher JD, Mahaseth H, Welch TE, Vilback AE, Sonbol KM, Kalambur VS, Bowlin PR, Bischof JC, Hebbel RP, Vercellotti GM. Critical role of endothelial cell activation in hypoxia-induced vasoocclusion in transgenic sickle mice. Am J Physiol Heart Circ Physiol. 2005;288:H2715–H2725. doi: 10.1152/ajpheart.00986.2004. [DOI] [PubMed] [Google Scholar]
- 163.Salvaggio JE, Arnold CA, Banov CH. Long-term anti-coagulation in sickle-cell disease. A clinical study. N Engl J Med. 1963;269:182–186. doi: 10.1056/NEJM196307252690403. [DOI] [PubMed] [Google Scholar]
- 164.Wolters HJ, ten Cate H, Thomas LL, Brandjes DP, van der Ende A, van der Heiden Y, Statius van Eps LW. Low-intensity oral anticoagulation in sickle-cell disease reverses the prethrombotic state: promises for treatment? Br J Haematol. 1995;90:715–717. doi: 10.1111/j.1365-2141.1995.tb05607.x. [DOI] [PubMed] [Google Scholar]
- 165.Chaplin H, Jr, Monroe MC, Malecek AC, Morgan LK, Michael J, Murphy WA. Preliminary trial of minidose heparin prophylaxis for painful sickle cell crises. East Afr Med J. 1989;66:574–584. [PubMed] [Google Scholar]
- 166.Semple MJ, Al-Hasani SF, Kioy P, Savidge GF. A double-blind trial of ticlopidine in sickle cell disease. Thromb Haemost. 1984;51:303–306. [PubMed] [Google Scholar]
- 167.Schnog JB, Kater AP, Mac Gillavry MR, Duits AJ, Lard LR, van Der Dijs FP, Brandjes DP, ten Cate H, van Eps LW, Rojer RA. Low adjusted-dose acenocoumarol therapy in sickle cell disease: a pilot study. Am J Hematol. 2001;68:179–183. doi: 10.1002/ajh.1175. [DOI] [PubMed] [Google Scholar]
- 168.Zago MA, Costa FF, Ismael SJ, Tone LG, Bottura C. Treatment of sickle cell diseases with aspirin. Acta Haematol. 1984;72:61–64. doi: 10.1159/000206360. [DOI] [PubMed] [Google Scholar]
- 169.Chaplin H, Jr, Alkjaersig N, Fletcher AP, Michael JM, Joist JH. Aspirin-dipyridamole prophylaxis of sickle cell disease pain crises. Thromb Haemost. 1980;43:218–221. [PubMed] [Google Scholar]
- 170.Tomer A, Kasey S, Connor WE, Clark S, Harker LA, Eckman JR. Reduction of pain episodes and prothrombotic activity in sickle cell disease by dietary n-3 fatty acids. Thromb Haemost. 2001;85:966–974. [PubMed] [Google Scholar]
- 171.Phillips G, Tangney CC. Relationship of plasma alpha tocopherol to index of clinical severity in individuals with sickle cell anemia. Am J Hematol. 1992;41:227–231. doi: 10.1002/ajh.2830410402. [DOI] [PubMed] [Google Scholar]
- 172.Natta CL, Machlin LJ, Brin M. A decrease in irreversibly sickled erythrocytes in sicle cell anemia patients given vitamin E. Am J Clin Nutr. 1980;33:968–971. doi: 10.1093/ajcn/33.5.968. [DOI] [PubMed] [Google Scholar]
- 173.Oppert M, Jorres A, Barckow D, Eckardt KU, Frei U, Kaisers U. Inhaled nitric oxide for ARDS due to sickle cell disease. Swiss Med Wkly. 2004;134:165–167. doi: 10.4414/smw.2004.10521. [DOI] [PubMed] [Google Scholar]
- 174.Sullivan KJ, Goodwin SR, Evangelist J, Moore RD, Mehta P. Nitric oxide successfully used to treat acute chest syndrome of sickle cell disease in a young adolescent. Crit Care Med. 1999;27:2563–2568. doi: 10.1097/00003246-199911000-00039. [DOI] [PubMed] [Google Scholar]
- 175.Atz AM, Wessel DL. Inhaled nitric oxide in sickle cell disease with acute chest syndrome. Anesthesiology. 1997;87:988–990. doi: 10.1097/00000542-199710000-00037. [DOI] [PubMed] [Google Scholar]
- 176.Weiner DL, Hibberd PL, Betit P, Cooper AB, Botelho CA, Brugnara C. Preliminary assessment of inhaled nitric oxide for acute vaso-occlusive crisis in pediatric patients with sickle cell disease. JAMA. 2003;289:1136–1142. doi: 10.1001/jama.289.9.1136. [DOI] [PubMed] [Google Scholar]
- 177.Reyes AA, Karl IE, Klahr S. Role of arginine in health and in renal disease. Am J Physiol. 1994;267:F331–F346. doi: 10.1152/ajprenal.1994.267.3.F331. [DOI] [PubMed] [Google Scholar]
- 178.Enwonwu CO, Xu XX, Turner E. Nitrogen metabolism in sickle cell anemia: free amino acids in plasma and urine. Am J Med Sci. 1990;300:366–371. doi: 10.1097/00000441-199012000-00005. [DOI] [PubMed] [Google Scholar]
- 179.Morris CR, Kuypers FA, Larkin S, Vichinsky EP, Styles LA. Patterns of arginine and nitric oxide in patients with sickle cell disease with vaso-occlusive crisis and acute chest syndrome. J Pediatr Hematol Oncol. 2000;22:515–520. doi: 10.1097/00043426-200011000-00009. [DOI] [PubMed] [Google Scholar]
- 180.Morris CR, Vichinsky EP, van Warmerdam J, Machado L, Kepka-Lenhart D, Morris SM, Jr, Kuypers FA. Hydroxyurea and arginine therapy: impact on nitric oxide production in sickle cell disease. J Pediatr Hematol Oncol. 2003;25:629–634. doi: 10.1097/00043426-200308000-00008. [DOI] [PubMed] [Google Scholar]
- 181.Machado RF, Martyr S, Kato GJ, Barst RJ, Anthi A, Robinson MR, Hunter L, Coles W, Nichols J, Hunter C, Sachdev V, Castro O, Gladwin MT. Sildenafil therapy in patients with sickle cell disease and pulmonary hypertension. Br J Haematol. 2005;130:445–453. doi: 10.1111/j.1365-2141.2005.05625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]