

Abstract
SPA2 encodes a yeast protein that is one of the first proteins to localize to sites of polarized growth, such as the shmoo tip and the incipient bud. The dynamics and requirements for Spa2p localization in living cells are examined using Spa2p green fluorescent protein fusions. Spa2p localizes to one edge of unbudded cells and subsequently is observable in the bud tip. Finally, during cytokinesis Spa2p is present as a ring at the mother–daughter bud neck. The bud emergence mutants bem1 and bem2 and mutants defective in the septins do not affect Spa2p localization to the bud tip. Strikingly, a small domain of Spa2p comprised of 150 amino acids is necessary and sufficient for localization to sites of polarized growth. This localization domain and the amino terminus of Spa2p are essential for its function in mating. Searching the yeast genome database revealed a previously uncharacterized protein which we name, Sph1p (Spa2p homolog), with significant homology to the localization domain and amino terminus of Spa2p. This protein also localizes to sites of polarized growth in budding and mating cells. SPH1, which is similar to SPA2, is required for bipolar budding and plays a role in shmoo formation. Overexpression of either Spa2p or Sph1p can block the localization of either protein fused to green fluorescent protein, suggesting that both Spa2p and Sph1p bind to and are localized by the same component. The identification of a 150–amino acid domain necessary and sufficient for localization of Spa2p to sites of polarized growth and the existence of this domain in another yeast protein Sph1p suggest that the early localization of these proteins may be mediated by a receptor that recognizes this small domain.
Polarized cell growth and division are essential cellular processes that play a crucial role in the development of eukaryotic organisms. Cell fate can be determined by cell asymmetry during cell division (Horvitz and Herskowitz, 1992; Cohen and Hyman, 1994; Rhyu and Knoblich, 1995). Consequently, the molecules involved in the generation and maintenance of cell asymmetry are important in the process of cell fate determination. Polarized growth can occur in response to external signals such as growth towards a nutrient (Rodriguez-Boulan and Nelson, 1989; Eaton and Simons, 1995) or hormone (Jackson and Hartwell, 1990a , b ; Segall, 1993; Keynes and Cook, 1995) and in response to internal signals as in Caenorhabditis elegans (Goldstein et al., 1993; Kimble, 1994; Priess, 1994) and Drosophila melanogaster (St Johnston and Nusslein-Volhard, 1992; Anderson, 1995) early development.
Saccharomyces cerevisiae undergo polarized growth towards an external cue during mating and to an internal cue during budding. Polarization towards a mating partner (shmoo formation) and towards a new bud site requires a number of proteins (Chenevert, 1994; Chant, 1996; Drubin and Nelson, 1996). Many of these proteins are necessary for both processes and are localized to sites of polarized growth, identified by the insertion of new cell wall material (Tkacz and Lampen, 1972; Farkas et al., 1974; Lew and Reed, 1993) to the shmoo tip, bud tip, and mother–daughter bud neck. In yeast, proteins localized to growth sites include cytoskeletal proteins (Adams and Pringle, 1984; Kilmartin and Adams, 1984; Ford, S.K., and J.R. Pringle. 1986. Yeast. 2:S114; Drubin et al., 1988; Snyder, 1989; Snyder et al., 1991; Amatruda and Cooper, 1992; Lew and Reed, 1993; Waddle et al., 1996), neck filament components (septins) (Byers and Goetsch, 1976; Kim et al., 1991; Ford and Pringle, 1991; Haarer and Pringle, 1987; Longtine et al., 1996), motor proteins (Lillie and Brown, 1994), G-proteins (Ziman, 1993; Yamochi et al., 1994; Qadota et al., 1996), and two membrane proteins (Halme et al., 1996; Roemer et al., 1996; Qadota et al., 1996). Septins, actin, and actin-associated proteins localize early in the cell cycle, before a bud or shmoo tip is recognizable. How this group of proteins is localized to and maintained at sites of cell growth remains unclear.
Spa2p is one of the first proteins involved in bud formation to localize to the incipient bud site before a bud is recognizable (Snyder, 1989; Snyder et al., 1991; Chant, 1996). Spa2p has been localized to where a new bud will form at approximately the same time as actin patches concentrate at this region (Snyder et al., 1991). An understanding of how Spa2p localizes to incipient bud sites will shed light on the very early stages of cell polarization. Later in the cell cycle, Spa2p is also found at the mother–daughter bud neck in cells undergoing cytokinesis. Spa2p, a nonessential protein, has been shown to be involved in bud site selection (Snyder, 1989; Zahner et al., 1996), shmoo formation (Gehrung and Snyder, 1990), and mating (Gehrung and Snyder, 1990; Chenevert et al., 1994; Yorihuzi and Ohsumi, 1994; Dorer et al., 1995). Genetic studies also suggest that Spa2p has a role in cytokinesis (Flescher et al., 1993), yet little is known about how this protein is localized to sites of polarized growth.
We have used Spa2p green fluorescent protein (GFP)1 fusions to investigate the early localization of Spa2p to sites of polarized growth in living cells. Our results demonstrate that a small domain of ∼150 amino acids of this large 1,466-residue protein is sufficient for targeting to sites of polarized growth and is necessary for Spa2p function. Furthermore, we have identified and characterized a novel yeast protein, Sph1p, which has homology to both the Spa2p amino terminus and the Spa2p localization domain. Sph1p localizes to similar regions of polarized growth and sph1 mutants have similar phenotypes as spa2 mutants.
Materials and Methods
Strains, Media, and Microbiological Techniques
Yeast strains used in this study are listed in Table I. Strains were constructed by standard genetic techniques and grown at 30°C (except temperature-sensitive [ts] strains) in rich media-yeast extract/peptone/dextrose) or synthetic complete media lacking appropriate supplements for selection (Rose et al., 1991). RAY532 (cdc3-1), RAY520 (cdc10-1), and RAY525 (cdc11-1) strains were constructed by crossing mutants with SEY6210 and selecting for appropriate markers. Calcofluor was from Sigma Chemical Co. (St. Louis, MO) and α-factor was from Calbiochem-Novabiochem Corp. (La Jolla, CA).
Table I.
Yeast Strains Used in This Study
| Strain | Genotype | Source | ||
|---|---|---|---|---|
| 104 | Mata ade1 ade2 ura1 his7 lys2 tyr1 gal1 cdc3-1 | R. Mortimer (University of California, Berkeley) | ||
| 332 | Mata ade1 ade2 ura1 his7 lys2 tyr1 gal1 cdc11-1 | R. Mortimer (University of California, Berkeley) | ||
| 17012 | Mata ade1 ade2 ura1 his7 lys2 tyr1 gal1 cdc10-1 | R. Mortimer (University of California, Berkeley) | ||
| JC-F5 | Mata ura3-52 ade2-101 met1 bar1-1 bem1-s2 | Chenevert et al., 1994 | ||
| JC-Gll | Mata ura3-52 ade2-101 met1 bar1-1 bem1-s1 | Chenevert et al., 1994 | ||
| JC-J9 | Mata ura3-52 ade2-101 met1 bar1-1 pea1 (allelic to SPA2) | Chenevert et al., 1994 | ||
| JY426 | Mata his4-34 leu2-3112 ura3-52 fus1-Δ1 fus2-Δ3 | Cold Spring Harbor Yeast Genetics Course | ||
| JY429 | Matα trp1Δ1 ura3-52 cyh2 fus1-Δ1 fus2-Δ3 | Cold Spring Harbor Yeast Genetics Course | ||
| RAY416 | same SEY6210 plus URA3::SPA2GFP | This study | ||
| RAY520 | Matα ura3-52‡ his3-Δ200 cdc10-1 | This study | ||
| RAY525 | Matα leu2-3,112 ura3-52‡ his3-Δ200 trp1-Δ901 cdc11-1 | This study | ||
| RAY532 | Matα leu2-3,112 ura3-52‡ his3-D200 cdc3-1 | This study | ||
| RAY563 | same as SEY6210 plus sph1-Δ1::HIS3 | This study | ||
| RAY567 | same as SEY6211 plus sph1-Δ1::HIS3 | This study | ||
| RAY574 | same as SEY6210 plus spa2-Δ1::TRP | This study | ||
| RAY578 | same as SEY6211 plus spa2-Δ1::TRP | This study | ||
| RAY586 | same as SEY6210 plus spa2-Δ1::TRP sph1-Δ1::HIS3 | This study | ||
| RAY590 | same as SEY6211 plus spa2-Δ1::TRP sph1-Δ1::HIS3 | This study | ||
| RAY616 | same as SEY6210/11 plus spa2-Δ1::TRP | This study | ||
| RAY618 | same as SEY6210/11 plus sph1-Δ1::HIS3 | This study | ||
| RAY620 | same as SEY6210/11 plus spa2-Δ1::TRP sph1-Δ1::HIS3 | This study | ||
| RAY651 | same as RAY574 plus URA3::SPA1GFP | This study | ||
| RAY674 | same as SEY6211 plus URA3::SPA2GFP | This study | ||
| RAY685 | same as RAY520 plus URA3::SPA2GFP | This study | ||
| RAY691 | same as RAY525 plus URA3::SPA2GFP | This study | ||
| RAY696 | same as RAY532 plus URA3::SPA2GFP | This study | ||
| RAY697 | same as SEY6210 plus URA3 | This study | ||
| RAY698 | same as RAY574 plus URA3 | This study | ||
| RAY699 | same as SEY6210 plus URA3::S2pSPH1GFP | This study | ||
| RAY703 | same as RAY574 plus URA3::S2pSPH1GFP | This study | ||
| RAY705 | same as JC-F5 plus URA3::SPA2GFP | This study | ||
| RAY706 | same as JC-G11 plus URA3::SPA2GFP | This study | ||
| RAY709 | same as RAY563 plus URA3* | This study | ||
| RAY711 | same as RAY586 plus URA3 | This study | ||
| RAY712 | same as RAY719 plus URA3::SPA2GFP | This study | ||
| RAY719 | same as SEY6210 plus bem1-Δ1::HIS5 | This study | ||
| RAY765 | same as RAY578 plus URA3::SPA2GFP | This study | ||
| RAY773 | same as RAY578 plus URA3 | This study | ||
| RAY774 | same as RAY578 plus URA3::S2pSH1GFP | This study | ||
| RAY776 | same as RAY616 plus URA3 | This study | ||
| RAY778 | same as RAY618 plus URA3 | This study | ||
| RAY780 | same as RAY620 plus URA3 | This study | ||
| RAY775 | same as RAY616 plus URA3::SPA2GFP | This study | ||
| RAY777 | same as RAY618 plus URA3::SPA2GFP | This study | ||
| RAY779 | same as RAY620 plus URA3::SPA2GFP | This study | ||
| RAY786 | same as RAY574 plus URA3::SPA2 | This study | ||
| RAY787 | same as RAY578 plus URA3::SPA2 | This study | ||
| RAY811 | same as RAY590 plus URA3::SPA2 | This study | ||
| RAY836 | same as SEY6210 plus bem2-Δ1::HIS5 | This study | ||
| RAY854 | same as RAY618 plus URA3::S2pPH1GFP | This study | ||
| RAY867 | same as RAY836 plus URA3::SPA2GFP | This study | ||
| RAY871 | same as RAY616 plus URA3::S2pPH1GFP | This study | ||
| RAY872 | same as RAY620 plus URA3::S2pPH1GFP | This study | ||
| RAY873 | same as RAY567 plus URA3 | This study | ||
| RAY874 | same as RAY567 plus URA3::SPA2GFP | This study | ||
| RAY875 | same as RAY567 plus URA3::S2pSPH1GFP | This study | ||
| RAY876 | same as SEY6211 plus URA3 | This study | ||
| RAY877 | same as SEY6211 plus URA3::S2pSPH1GFP | This study | ||
| SEY6210 | Matα leu2-3,112 ura3-52 his3-Δ200 trp1-Δ901 lys2-801 suc2-Δ9 | S. Emr (University of California, San Diego) | ||
| SEY6211 | Mata leu2-3,112 ura3-52 his3-Δ200 trp1-Δ901 ade2 suc2-Δ9 | S. Emr (University of California, San Diego) | ||
| SEY6210/11 | Mata/α leu2-3,112/leu2-3,112 ura3-52/ura3-52 his3-Δ200/his3-Δ200 | S. Emr (University of California, San Diego) | ||
| trp1-Δ901/trp1-Δ901 LYS/lys2-801 ADE2/ade2 suc2-Δ9/suc2-Δ9 |
Strains were transformed with a Bg1II URA3 fragment from pFL34 to make URA3+.
Progeny of crosses were transformed with pRS416 to select for ura3-52 mutants.
Plasmid Construction and Mutagenesis
A 6.7-kb SalI–HindIII fragment of SPA2 from p203 (Gehrung and Snyder, 1990) was cloned into pRS406 (p406S2), pRS416 (p416S2), and pRS426 (p426S2). GFP was fused to the carboxyl terminus of SPA2 using PCR-amplified GFP coding region with an EcoRI site present at the 5′ end of the coding region and HindIII–XbaI sites at the 3′ end. GFP was fused to the SPA2 open reading frame (ORF) using the EcoRI site at the end of the SPA2 ORF, resulting in the last three amino acids of SPA2 being replaced by Asn Ile followed by GFP with an additional Leu-Val at its carboxyl terminus. We tested different versions of GFP to optimize sensitivity and minimize photobleaching. GFPS65T (Heim et al., 1995), made by oligonucleotide-directed mutagenesis (Kunkel, 1985), and mGFP5 (Siemering et al., 1996) were fused to SPA2 similarly. Both GFP mutants were significantly more sensitive than wild-type GFP and enabled observation at the fluorescein excitation wavelength. For all experiments SPA2GFPS65T was used and cloned into pRS405 (p405S2G), pRS406 (p406S2G), pRS416 (p416S2G), and pRS426 (p426S2G). Deletions were constructed using p406S2G in which the EcoRI site within the SPA2 ORF was removed by oligonucleotide-directed mutagenesis (Kunkel, 1985), and subsequently the EcoRI site upstream of the coding region was removed by partial digestion and subsequent filling in, leaving a single EcoRI site between SPA2 and GFP. In addition, oligonucleotide-directed mutagenesis (Kunkel, 1985) was used to replace the six nucleotides immediately 5′ of the SPA2 initiation codon with a BamHI restriction site, resulting in p406S2G1. All carboxyl-terminal deletions were generated by insertion of an EcoRI site by oligonucleotide-directed mutagenesis (Kunkel, 1985), which resulted in Asn-Ser after amino acid 1,074 (ΔZ), 655 (ΔY), 549 (ΔX), 511 (ΔW), and an Arg-Asn-Ser after amino acid 396 (ΔV). Amino-terminal deletions were constructed by inserting a BamHI site by oligonucleotide-directed mutagenesis (Kunkel, 1985) 5′ of amino acid 88 (ΔA), 288 (ΔB), 397 (ΔC), 511 (ΔD), and 625 (ΔE), which resulted in Gly-Ser immediately preceding a methionine residue (in ΔC a methionine was also inserted after Gly-Ser). Deletions were made by restriction digestion and followed by religation. All deletions were confirmed by dye terminator cycle sequencing (DNA Sequencing Kit; Perkin-Elmer Corp., Norwalk, CT) using an ABI377 automated sequencer. Integration vectors containing SPA2GFP and derivatives were linearized with either StuI for pRS406 or XcmI for pRS405. Typically, integration yielded two to four copies per cell by Southern analyses. For each integration, four independent transformants were examined by fluorescence microscope for GFP expression and confocal microscopy for localization.
A myc epitope tag was fused to the carboxyl terminus of SPA2 by PCR using p406S2G1 as a template, resulting in p406S2myc, which has the myc epitope (MEQKLISEEDL) followed by Lys-Leu-Val. Spa2myc from p406S2myc was liberated by BamHI–SacII digestion and cloned into BamHI–SacII-digested pRS425TPI, which is a derivative of pRS425 with the triose phosphate isomerase (TPI) promoter cloned into XhoI–HindIII sites, resulting in pRS425TPIS2myc.
Fluorescence and Confocal Microscopy
Transformants were initially screened using an Axiophot microscope (Zeiss, Oberkochen, Germany). Living cells were imaged at 22°C using a Biorad-MRC-600 confocal microscope (Bio Rad Laboratories, Hercules, CA) with a NA 1.4, ×60 objective and FITC (488-nm) excitation filter. For analyses of localization of Spa2GFP deletions, temperature-sensitive and mating mutants, 1–2 μl of an exponentially growing culture (grown in the appropriate synthetic deficient media) was spotted on a glass slide, and the coverslip was applied with gentle pressure. This resulted in cells sticking sufficiently to the slide to allow confocal image acquisition. Photobleaching was minimized by decreasing illumination intensity with neutral density filters.
For time course analyses, slides were prepared by cutting a square piece of 0.5-mm-thick silicon rubber with a hole cut in the center using a cork borer. This piece of silicon rubber was pressed onto a glass slide heated to 65°C, a drop of molten synthetic complete media lacking uracil with 2% agarose was spotted in the hole of the silicon rubber, and a No. 1 glass coverslip was placed on top to flatten the pad. The slide was cooled to room temperature for ∼10 min, and then the coverslip was slid off, leaving a flattened −ura agarose pad. 1–2 μl of an exponentially growing culture (grown in the appropriate synthetic deficient media) was spotted on this pad, allowed to partially dry for about 5 min, and covered with a coverslip, applying slight pressure to make a seal. Vaseline was coated along the edges of the coverslip to prevent the pad from drying out. Cells on this slide were viable and formed microcolonies upon extended incubation. For long confocal experiments, photobleaching and excessive irradiation were minimized by single scans with maximal neutral density filters at three different focal planes. Cell growth appeared to slow during long confocal experiments, yet cells continued to divide and Spa2GFP localization was identical to that in long experiments with infrequent scans. For examination of mating cells, a and α cells were both spotted on the same agarose pad and mating was followed by light microscopy and confocal microscopy.
Disruption of BEM1 and BEM2
BEM1 was cloned by PCR using Taq Polymerase (Perkin-Elmer Corp.) from yeast genomic DNA. A 2,136-bp fragment including 426 nucleotides 5′ of the initiation codon was cloned into the pGem-T vector (Promega, Madison, WI), resulting in pGBEM1. A BEM1 disruption plasmid was constructed by replacing SmaI–XcmI from pGBEM1 with S. pombe HIS5 (a homologue of S. cerevisiae HIS3) from pFA6a-HIS3MX6 (A. Wach, University of Basel, Switzerland), resulting in removal of all of BEM1 ORF except for the last 81 codons. BEM2 was disrupted using PCR-based gene disruption (Baudin et al., 1993). Oligonucleotides were used each with homology to 60 nucleotides 5′ and 3′ of the BEM2 ORF and 18 nucleotides to pFA6a-HIS3MX6, respectively, resulting in replacement of the entire BEM2 ORF with S. pombe HIS5 (a homologue of S. cerevisiae HIS3) (Wach et al., 1994). Linearized disruption cassettes or the PCR disruption cassette were used to create disruption strains using the one-step knockout procedure of Rothstein (1983). Disruption strains were confirmed by PCR analyses and both were temperature sensitive as previously reported (Bender and Pringle, 1991).
Quantitative Mating and Shmoo Formation
For mating function analyses of SPA2GFP deletion mutants, strain JC-J9 was transformed with StuI linearized p406S2G1 deletion mutants and transformants were used that had observable fluorescence signals (by fluorescence microscopy). Quantitative mating assays were carried out as described in Chenevert et al. (1994) using 1 × 106 cells from exponentially growing cultures of the enfeebled mating partners JY429 or JY426. Mating efficiency was calculated as the ratio of diploids to total cells.
Shmoo formation was determined by the addition of α-factor (final concentration 12.9 μM) to 1 × 106 log growing cells. After 2 h of growth, cells were fixed with 3.7% formaldehyde. Cells were sonicated and the number of shmoos was quantitated using a phase microscope. This concentration of α-factor and incubation time typically resulted in ∼80% shmooed cells in wild-type strains with very few cells having two shmoos. Both peanut- and pear-shaped shmoos were counted as shmoos. For observation of GFP fusions in shmooed cells, log cultures in selective media were grown for 3 h in rich media before α-factor addition.
Budding Pattern Assays
Calcofluor staining of bud scars was carried out as described in Pringle (1991). Budding patterns were quantitated and cells were photographed using a Zeiss Axiophot epifluorescence microscope with a NA 1.3, Plan-Neofluor ×100 objective and a UV-H 365 excitation filter.
Cloning and Sequencing of SPH1
SPH1 was cloned by PCR using Taq Polymerase (Perkin-Elmer Corp.) from yeast genomic DNA. Initially a 2-kb fragment including 346 nucleotides 5′ of the SPH1 initiation codon was cloned into the Promega pGem-T vector (pGSH1), and two independent clones were sequenced. Subsequently, an additional 2-kb fragment starting at 1,018 nucleotides into the SPH1 ORF was isolated by PCR from genomic DNA and cloned into pGem-T (pGSH2). Three independent clones of this PCR product were sequenced, and the full-length SPH1 including 1,068 nucleotides 3′ of the end of the coding region was constructed by replacement of a XhoI–NcoI restriction fragment from pGSH1 with an XhoI–NcoI fragment from pGSH2, yielding pGSH3. CEN and 2-μm vectors carrying SPH1 were constructed by subcloning a SalI–SacII fragment from pGSH3 into SalI– SacII pRS415 (p415SH3) and pRS425 (p425SH3) and XhoI–SacII pRS406 (p406SH3). The sequence data for SPH1 are available from EMBL/GenBank/DDBJ under accession number AF008236.
Disruption of SPA2 and SPH1
A SPA2 disruption plasmid was constructed by replacing the GFP sequence in p406S2G1 with the 3′ SPA2 noncoding sequence from p416S2 (resulting in p406S2.1), and subsequently all of the SPA2 ORF was replaced by TRP1 using the uniquely engineered BamHI site and the EcoRI site. SPH1 disruption plasmid was constructed by replacing StuI–XhoI from pGSH1 with HIS3. Linearized disruption cassettes were used to create disruption strains using the one-step knockout procedure of Rothstein (1983). Disruption strains were confirmed by PCR analyses.
Localization of Sph1p
An EcoRI site at nucleotide 1,157 in the SPH1 coding region (p406SH3) was removed by oligonucleotide-directed mutagenesis (Kunkel, 1985). In addition, oligonucleotide-directed mutagenesis (Kunkel, 1985) was used to replace the six nucleotides immediately preceding the initiation codon with a BamHI site, resulting in p406SH4. An EcoRI site was placed at the end of the SPH1 coding region by PCR with Pfu polymerase (Stratagene, La Jolla, CA) using p406SH4 as a template, yielding p406SH5. This resulted in the addition of the amino acids Ala-Asn-Ser to the carboxyl terminus of Sph1p. The sequence of p406SH5 was confirmed by dye terminator cycle sequencing using an ABI377 automated sequencer. The coding region of SPH1 from p406SH5 was liberated by BamHI–EcoRI digestion and cloned into BamHI–EcoRI-digested p406S2G1, resulting in p406SH5G in which SPH1GFP is driven by the SPA2 promoter. p406SH5G was linearized with either BsmI or XcmI and used to transform various yeast strains. Two p406SH5G transformants were examined by confocal microscopy.
A myc epitope tag was fused to the carboxyl terminus of Sph1p by PCR using p406SH4 as a template, resulting in p406SHmyc, which had two additional amino acids, Leu-Val, at the carboxyl terminus of Sph followed by the myc epitope (MEQKLISEEDLV). Sph1myc from p406SHmyc was liberated by BamHI–SacII digestion and cloned into BamHI–SacII-digested pRS425TPI, which is a derivative of pRS425 with the TPI promoter cloned into XhoI–HindIII sites, resulting in pRS425TPISHmyc.
Results
Spa2GFP Is Functional and Localizes Correctly
To investigate the function and dynamics of Spa2p localization, we fused GFP to its carboxyl terminus. For these studies, it was necessary that the expression level of the fusion protein reflect the normal level and be essentially constant throughout a population of cells. Hence we chose to use the endogenous SPA2 promoter and to integrate all constructs at the URA3 locus. We first examined the function of this fusion protein by testing its ability to complement the various phenotypes of a Δspa2 strain, such as a defect in shmoo formation (Gehrung and Snyder, 1990; Chenevert et al., 1994; Yorihuzi and Ohsumi, 1994; Valtz and Herskowitz, 1996), a mating defect (Gehrung and Snyder, 1990; Chenevert et al., 1994; Yorihuzi and Ohsumi, 1994; Dorer et al., 1995), and a random bud site selection pattern in a homozygous Δspa2 diploid (Snyder, 1989; Zahner et al., 1996). Fig. 1 A shows the results of a quantitative mating assay in which a Δspa2 strain carrying SPA2, SPA2GFP, or an empty plasmid is mated with an enfeebled mating partner and diploids are subsequently selected. Deletion of SPA2 results in ∼90-fold decrease in mating efficiency, which is fully complemented by SPA2GFP. Deletion of SPA2 results in a defect in shmoo formation (Gehrung and Snyder, 1990), which has subsequently been shown to be dependent on pheromone concentration and incubation time (Valtz and Herskowitz, 1996). A Δspa2 strain in the presence of saturating pheromone concentrations showed a lower percentage of shmooed cells than wild-type cells. Consistent with previous observations (Valtz and Herskowitz, 1996), this strain exhibited primarily peanut-shaped shmoos in contrast with the pear-shaped shmoos of the wild-type strain (data not shown). SPA2GFP fully complements the defect in shmoo formation observed at high pheromone concentration in a Δspa2 strain (Table II) and shmoo morphology (data not shown). Deletion of SPA2 has been shown to have no effect on bud site selection in haploids, yet homozygous diploid deletion mutants are defective in bipolar budding (Snyder, 1989; Zahner et al., 1996; Valtz and Herskowitz, 1996). Haploids (a or α cells) generally bud in an axial pattern, adjacent to the previous division site; however, diploids (a/α cells) bud in a bipolar pattern, distal or proximal to the division site (Freifelder, 1960; Hicks et al., 1977; Sloat et al., 1981). Fig. 1 B demonstrates that SPA2GFP is able to completely complement the bud site selection defect in homozygous Δspa2 diploids. Furthermore, deletion of SPA2 results in round cells and SPA2GFP also complements this morphological defect (data not shown). Together, these assays demonstrate that the Spa2GFP fusion protein is fully functional.
Figure 1.

(A) Complementation of spa2 mating defect by SPA2GFP. Δspa2 mutants with SPA2 (RAY786), SPA2GFP (RAY651), or an empty plasmid (RAY698) integrated at the URA3 locus were mated with the enfeebled mating tester strain (JY426). Diploids were selected on −ura, −lys plates. Mating efficiency with SPA2 (30.9%) was set to 100% efficiency. (B) Complementation of spa2 bipolar budding defect by SPA2GFP. Wild-type diploids (SEY6210/11) and homozygous Δspa2 diploids with either SPA2GFP (RAY775) or an empty plasmid (RAY776) integrated at the URA3 locus growing exponentially were stained with Calcofluor as described in Materials and Methods, and budding pattern was analyzed. For each strain, the position of the bud relative to the birth scar (see inset) was scored for ∼100 cells with two or more bud scars. (C) Localization of Spa2GFP at sites of polarized growth. Confocal microscopy of living cells with SPA2GFP integrated at URA3 (RAY416) at different stages in cell cycle. Note unbudded cells with patch of Spa2GFP, small- and medium-sized buds with Spa2GFP fluorescence on the periphery of the tip, and a cell undergoing cytokinesis with a “bar” of Spa2GFP at the mother–daughter bud neck. Bar, 5 μm.
Table II.
SPA2GFP Complements Shmoo Formation Defect of Δspa2 Mutant
| SPA2 construct | Percentage of shmoos* | |
|---|---|---|
| % | ||
| SPA2 | 77 | |
| − | 58 | |
| SPA2GFP | 83 |
Cells were treated with α-factor and quantitated as described in Materials and Methods. SPA2 denotes WT cells with pRS406 integrated at URA3 (RAY876), − are Δspa2 cells with pRS406 integrated at URA3 (RAY578), and SPA2GFP are Δspa2 cells with pRS406SPA2GFP integrated at URA3 (RAY765). For each strain 250 cells were counted. SPA2GFP had no effect on shmoo formation or morphology of wild-type cells (see Table V).
Peanut- and pear-shaped shmoos were designated as shmoos.
Cells containing the Spa2GFP were examined by fluorescence confocal microscopy. All experiments used GFP- (S65T) (Heim et al., 1995) mutant, which optimized sensitivity and minimized photobleaching. Fig. 1 C shows the localization of Spa2GFP by confocal microscopy. Consistent with previous indirect immunofluorescence studies, Spa2p localizes to a crescent in unbudded cells, which marks the new bud site, the bud tip of small buds, and the neck between the mother and daughter cell just before cytokinesis (Snyder, 1989; Snyder et al., 1991). Similar Spa2GFP localization was observed in a Δspa2 strain (data not shown), indicating that SPA2 is not required for the localization of the fusion protein. These results confirm previous immunolocalizations and show that they reflect the distribution of this protein in vivo. We also examined the effect of expression level on Spa2GFP localization to determine if localization was saturable. Expression level was varied by increasing the copy number of SPA2GFP from a single copy (replacement), SPA2GFP integrated at URA3, a centromere plasmid carrying SPA2GFP, and a multicopy 2-μm plasmid carrying SPA2GFP. The localization of Spa2p to the bud tip and mother–daughter bud neck was identical under all expression conditions; however, with expression from a multicopy 2-μm plasmid, cytoplasmic Spa2GFP was observed (data not shown). Furthermore, we did not observe a substantial increase in the intensity of the localized Spa2GFP fluorescence during overexpression, suggesting that Spa2p localization is saturable.
Spa2GFP Dynamics: Localization to Two Spatially and Temporally Distinct Structures
To examine the relationship between the two localizations of Spa2GFP (bud tip and mother–daughter bud neck), we investigated the dynamics of Spa2GFP in haploid cells (Fig. 2 A). Initially we examined Spa2GFP in living cells using confocal microscopy, because we observed substantial phototoxicity with conventional fluorescence microscopy, which was incompatible with such time course experiments. Fig. 2 A shows a time course of Spa2GFP dynamics at 22°C in which Spa2GFP within two cells was observed approximately every 5 min at minimally three different focal planes. This experiment involved a total of ∼100 confocal scans over ∼3 h, and illustrated in Fig. 2 A are the focal planes through the center of the cells. It should be noted that, although two cells are shown in this figure, Spa2GFP is found in a very specific location in the cell, making it difficult to observe such a structure in more than one cell at a time. The upper cell is in focus throughout this time course. During the first hour, Spa2GFP can be seen on the periphery of the bud and appears to spread out during this time. After >1 h, weak fluorescence becomes apparent at the neck between the mother and daughter cell. At 66 and 71 min, Spa2GFP can be seen at the bud periphery and mother–daughter bud neck, suggesting that these two structures can exist simultaneously. In panels (71–83 min), a ring structure is evident which appears as two dots (the cross-section of a ring) at the narrowest point in the neck between the mother and daughter cells. Movement of one focal plane (1 μm) in either direction in the z-axis was consistent with such a ring structure, revealing a solid bar perpendicular to the mother–daughter cell axis. At ∼1 h and 30 min, this Spa2p ring appears to have closed, now appearing as a “bar” in all focal planes. Subsequently, the Spa2p “bar” appears to get narrower in the axis perpendicular to the mother–daughter cell and thicker in the mother–daughter cell axis, indicative of cytokinesis. By ∼100 min, two Spa2p “bars” are apparent, one in the daughter and one in the mother cell. In the next 40 min (103–145-min panels), Spa2GFP fluorescence becomes concentrated in an intense spot axial to the first bud/birth site marking the next bud site. During this time and up to 160 min, the cells lose their round shape and initiate the next round of polarized growth. From these time courses Spa2p is observed to alternate between localized discrete structures (G1 phase unbudded cells, S phase bud emergence, and M phase during cytokinesis) and more diffuse structures as the daughter cells increase in size. In addition, Spa2p structures appear to change from cup-like crescents to donut-like rings during the cell cycle. Despite the slow apparent movement of these structures, these studies highlight the dynamic nature of Spa2p in living cells.
Figure 2.
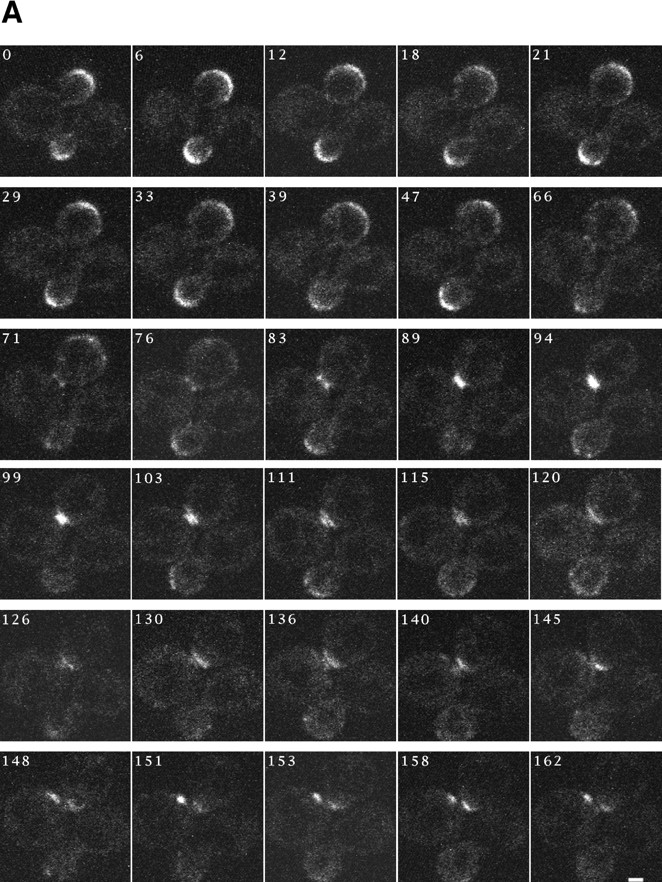

(A) Dynamics of Spa2GFP localization in living cells. Spa2GFP distribution in two cells (RAY416) at 6-min intervals. The Spa2GFP fluorescence in two cells is in different focal planes and the upper cell is in focus throughout the time course. Note the appearance at 76 min of a ring at the mother bud neck. Cells divided during and after confocal time course. (B) Localization of Spa2GFP in mating cells. Confocal microscopy of five representative mating pairs (RAY416 and RAY674) at different stages of mating/zygote formation. The diffuse background is due to the high gain setting to allow visualization of the entire cell. (C) Movement of Spa2GFP in budding zygotes. Time course of zygotes from RAY416 mated to RAY674. In the center of each panel is a dumbbell-shaped zygote with a separate round cell to its right and left. Note the ring of Spa2GFP fluorescence at the a/α shmoo neck at 0 min. Bars: (A and B) 1 μm; (C) 5 μm.
We also examined the localization and dynamics of Spa2GFP in mating cells. Previous immunofluorescence studies have revealed that Spa2p localizes to shmoo tips (Gehrung and Snyder, 1990) and Spa2GFP localization in mating cells showed similar localization (Fig. 2 B). Furthermore, Spa2GFP localized to the tips of mating projections (shmoos) after mating pheromone addition and this localization was also observed in the absence of wild-type SPA2 (data not shown). Upon zygote formation, Spa2GFP- labeled structures from each haploid appear to fuse into one structure at the site of cell fusion, which subsequently localizes to growing bud tip of the zygote. Fig. 2 C shows a time course of Spa2GFP localization in haploid cells that have just fused to form a zygote. The ring of Spa2GFP fluorescence at the neck of the a/α zygote persists for ∼20 min and then relocalizes to one side of the neck where the new bud will form. Subsequently, Spa2GFP can be seen on the tip of the growing bud. These experiments demonstrate that within a time frame of 10 min the Spa2GFP relocalizes from a ring structure at the site of cell fusion to the site of growth on the new bud.
Spa2GFP Localization in Mutants with Defects in Bud Emergence and Septation
We examined the effect of several cell cycle and bud emergence mutants on Spa2GFP localization to determine if it was possible to separate the two observed Spa2p structures during the cell cycle. Indirect immunofluorescence studies have revealed that Spa2p localizes very early in the cell cycle, marking the site of the incipient bud. Furthermore, neither actin mutants nor tubulin mutants, both of which result in a delocalized cytoskeleton, mislocalize Spa2p (Snyder et al., 1991). These results suggest that Spa2p localization is independent of the actin and microtubule cytoskeleton. Recently, additional cytoskeletal components termed septins have been shown to be involved in cytokinesis (Haarer and Pringle, 1987; Kim et al., 1991; Ford and Pringle, 1991; Cooper and Kiehart, 1996; Field et al., 1996; Longtine et al., 1996). The septins are encoded by CDC3, CDC10, CDC11, and CDC12, and these proteins are thought to form 10-nm neck filaments that have been observed by EM at the mother–daughter bud neck (Byers and Goetsch, 1976). Previous studies have shown that ts mutants in any of the septins result in a loss of localization of all the septins (Kim et al., 1991; Ford and Pringle, 1991; Haarer and Pringle, 1987) and 10-nm neck filaments (Byers and Goetsch, 1976). The role of the septins, Cdc3p, Cdc10p, and Cdc11p, in Spa2p localization was investigated. Flescher et al. (1993) have previously demonstrated a genetic interaction between CDC10 and SPA2, by showing that spa2 and an ochre truncation mutant of CDC10, cdc10-10, are synthetically lethal. These experiments implicated a connection between SPA2 and septin formation, although it is unclear whether such an interaction is CDC10 specific. SPA2GFP was integrated at the URA3 locus in the temperature-sensitive septin mutants cdc3-1, cdc10-1, and cdc11-1, and several transformants of each were examined by confocal microscopy after growth in liquid culture for 3 h at 25°C, 3 h at 37°C, and 6 h at 37°C (Fig. 3). At the permissive temperature, despite a small percentage of cells displaying the characteristic septin mutant phenotype, misshapen long cell, Spa2GFP localization appeared normal, with bud tip and mother–daughter bud neck fluorescence observable. At the restrictive temperature, the septin mutant phenotype was readily observable in most cells, resulting in the inability of cells to septate and thus an abundance of misshapen long cells with several long extended buds. In cdc3-1, cdc10-1, and cdc11-1 cells at 37°C, Spa2GFP was localized primarily to bud tips. Spa2GFP was not observed at the bud neck between mother and daughter cells (Fig. 3), consistent with Spa2p localization in a cdc10-10 mutant (Flescher et al., 1993). These results suggest that the septins are either required for Spa2p localization at the mother–daughter neck or, more likely, that these cdc mutants act at a point in the cell cycle before the Spa2p localization at the mother–daughter neck. Perhaps, the mother–daughter neck needs to be sufficiently constricted in order for the Spa2p localization to occur and therefore is not possible in such septin mutants. In contrast, the persistent localization of Spa2GFP to bud tips in the septin mutants at the nonpermissive temperature indicates that septins are not required to maintain Spa2p at this location.
Figure 3.
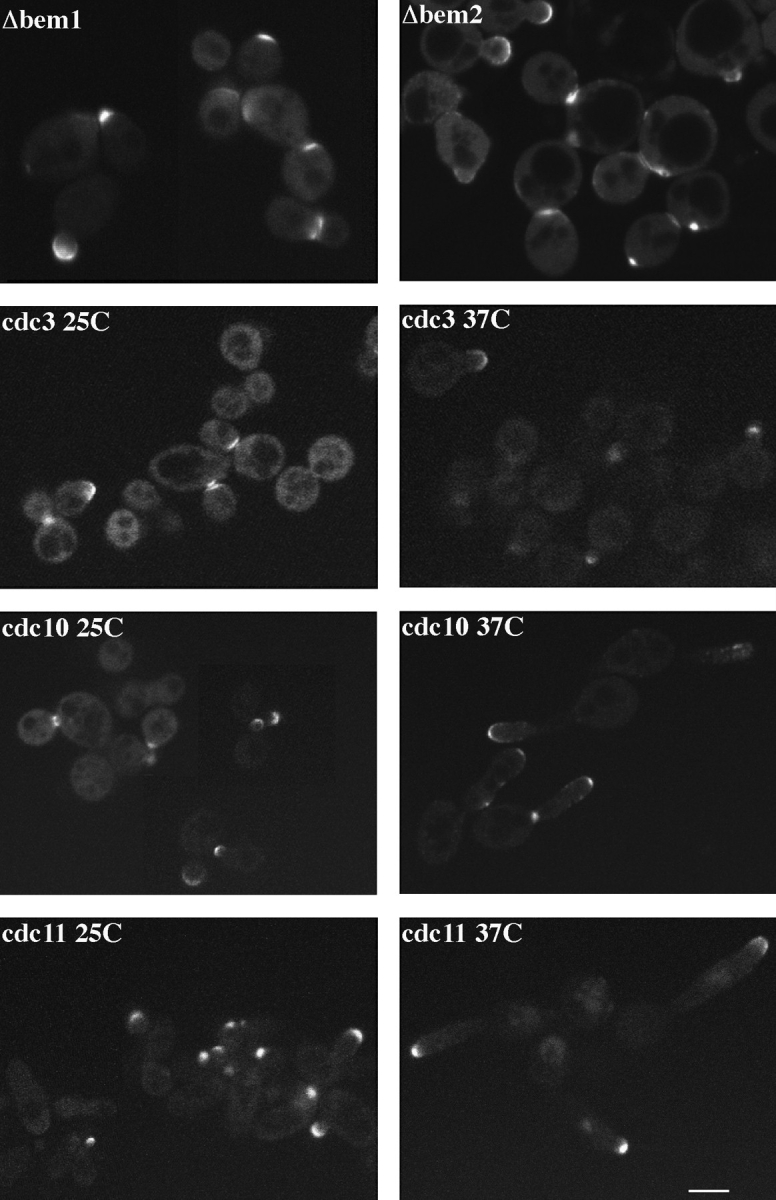
Spa2GFP localization in bem and septin mutants. Δbem1 (RAY712), Δbem2 (RAY836), cdc3-1 (RAY696), cdc10-1 (RAY685), and cdc11-1 (RAY691) strains with SPA2GFP integrated at the URA3 locus were grown at 25°C. Exponentially growing cultures of the septin mutants were shifted to 37°C for 3 h, and cells were examined by confocal microscopy. Note large cells in the bem mutants and the Spa2GFP fluorescence at both the bud tip and mother–daughter bud neck. Bar, 5 μm.
In addition, we examined the localization of Spa2GFP in bem1 and bem2 mutants that result in bud emergence defects (Bender and Pringle, 1991). BEM1 encodes a protein containing two SH3 domains (Chenevert et al., 1992) and BEM2 encodes a rho-GAP for Rho1p (Peterson et al., 1994; Kim et al., 1994). Bem1p has been shown to interact with proteins required for bud formation (Peterson et al., 1994; Zheng et al., 1995), bud site selection (Chant et al., 1991), and components of the pheromone-responsive mitogen-activated protein kinase cascade (Leeuw et al., 1995). In addition, BEM1 has been shown to be involved in cell mating and polarized growth during shmoo formation (Chenevert et al., 1992, 1994; Yorihuzi and Ohsumi, 1994). Bem2 mutants show genetic interactions with cytoskeletal components (Wang and Bretscher, 1995). BEM1 or BEM2 was deleted from haploid cells by one-step gene replacement (Rothstein, 1983); SPA2GFP was integrated at the URA3 locus; and several transformants of each were examined by confocal microscopy after growth in liquid culture (Fig. 3). Both bem1 and bem2 mutants show the bud emergence defects and are temperature sensitive as previously described (Bender and Pringle, 1991); however, Spa2GFP localization appears normal, with bud tip and mother–daughter bud neck fluorescence observable. These results suggest that BEM1 and BEM2 are not required for Spa2p localization. Furthermore, specific bem1 alleles, bem1-s1 and bem1-s2, affect cell mating (Chenevert et al., 1994) and both resultant truncated Bem1ps fail to interact with Ste20p (Leeuw et al., 1995). Ste20 transduces the signal from a membrane receptor, when it binds pheromone, to the mitogen-activated protein kinase cascade (Chenevert, 1994). Despite the inability of bem1-s cells to shmoo, a distinct patch of Spa2GFP was observed on the side of the cells adjacent to their mating partner (data not shown), suggesting that shmoo formation is not necessary for Spa2p localization and BEM1 acts after Spa2p localization in mating cells.
Delineation and Characterization of a Small Conserved Domain in Spa2p Both Necessary and Sufficient for its Localization and Function
To understand the early localization of Spa2p, we attempted to identify a region that was responsible for its striking localization in vivo. Fig. 4 shows a schematic drawing of Spa2GFP with recognizable secondary structural features indicated. As previously mentioned by Gehrung and Snyder (1990), the amino terminus of Spa2p contains a region (amino acid residues 286–378) with a very high probability, 0.99, of forming a coiled-coil structure using the Lupas program (Lupas et al., 1991). In addition, the carboxyl-terminal half of Spa2p contains 25 imperfectly repeated nonameric amino acid repeats between residues 816 and 1,087 (Gehrung and Snyder, 1990). Analysis of the yeast genome sequence using the BLAST homology search program (Altschul et al., 1990) revealed a significant homology between the amino-terminal third of Spa2p (the first 536 residues) and an ORF, YSCL8543.8. This homology existed primarily in two regions, the first 120 amino acid residues (32% identity) and amino acid residues 421– 536 of Spa2p (42% identity), with the last 63 residues of this second region displaying even greater identity, 54%. Consequently, we have denoted this second region the “Spa2 box.” Further BLAST searches (Altschul et al., 1990) using the Spa2 box from Spa2p or YSCL8543.8 did not reveal any other non-yeast proteins with significant homology.
Figure 4.

Diagram of Spa2GFP deletion constructs. Deletions ΔA, ΔB, ΔC, ΔD, and ΔE contain Spa2p residues 88, 288, 397, 511, and 625, respectively, to residue 1,463. Deletions ΔZ, ΔY, ΔX, ΔW, and ΔV contain Spa2p residue 1 to residue 1,074, 655, 549, 511, and 396, respectively. Constructs ΔBX, ΔBV, and ΔCX contain Spa2p residues 288–549, 288–396, and 397–549, respectively. All constructs contain Spa2p fused to GFP and were integrated at the URA3 locus in either SEY6210 for localization studies or JC-J9 for mating function analysis. See Fig. 5 A for localization data and Fig. 5 B for results from mating assays.
We generated the series of truncation and deletion mutants shown in Fig. 4 to assess the contributions of each of the defined structural domains in Spa2p to its in vivo localization and function. These mutants were made by inserting unique restriction sites that further allowed us to construct double mutants either lacking specific regions (not shown) or consisting only of specific regions fused directly to GFP, such as ΔBX, ΔBV, and ΔCX (Fig. 4). Each construct was integrated at the URA3 locus and at least four transformants of each mutant were examined microscopically. Fig. 4 illustrates the summary of the results of mutant localization and function, and Fig. 5 shows representative confocal micrographs of localization and mating function, respectively.
Figure 5.
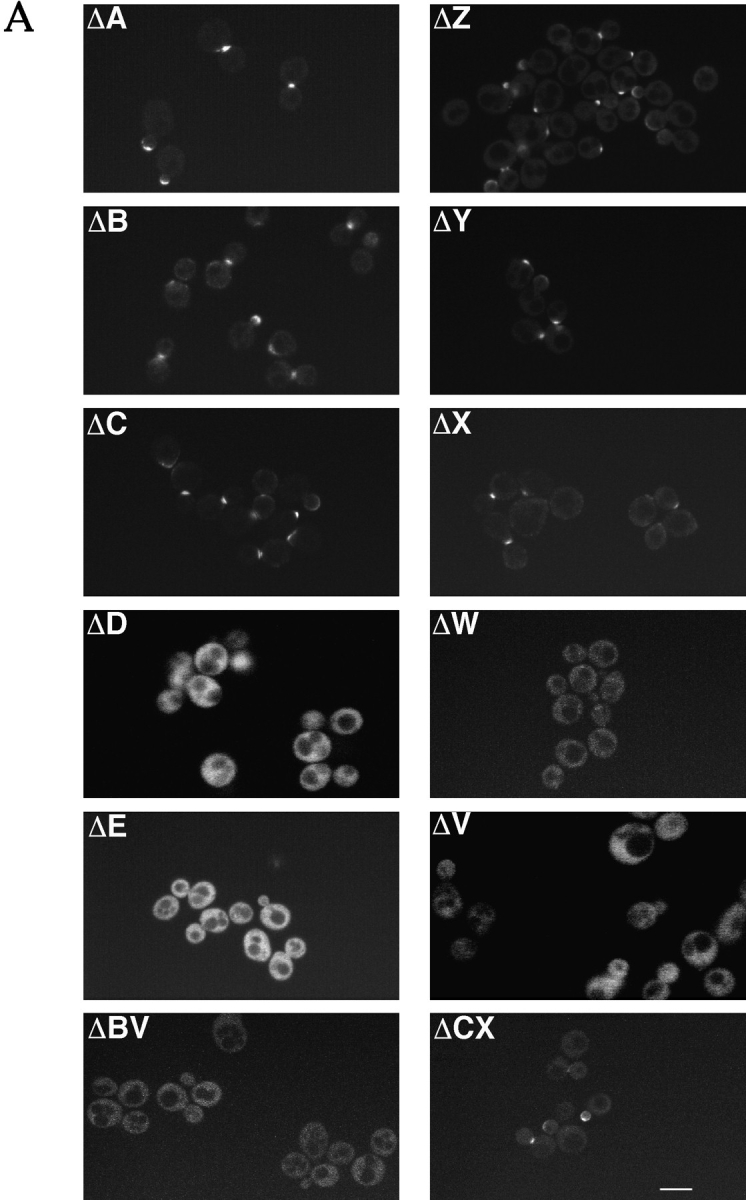

(A) Localization of Spa2GFP deletion mutants. Exponentially growing cultures of SEY6210 with deletion constructs integrated at the URA3 locus were examined by confocal microscopy. In ΔA, ΔB, ΔC, ΔZ, ΔY, ΔX, and ΔCX, note both bud tip and mother–daughter bud neck localization of Spa2GFP deletions. Panels ΔD, ΔE, ΔC, ΔW, ΔV, and ΔCX show general Spa2GFP fluorescence. (B) Mating function of Spa2GFP deletion mutants. The spa2 mutant (JC-J9) with SPA2GFP deletion constructs integrated at the URA3 locus was mated with an enfeebled mating partner (JY429), and diploids were selected on −met, −ura plates. In each experiment, mating of JC-J9 carrying full-length Spa2GFP was taken as 100% efficiency (absolute efficiency of 11.2% [left] and 7.9% [right]). Bar, 5 μm.
Amino-terminal truncations (ΔA–ΔC) and carboxyl-terminal truncations (ΔZ–ΔX) demonstrate that the amino terminus, the coiled-coil region (residues 286–378), and the nonameric amino acid repeat region (residues 816– 1,087) are not required for Spa2GFP localization in vivo, indicating that the Spa2 box is necessary for correct localization. Strikingly, removal of the last 38 amino acids from ΔX, resulting in ΔW which deletes the carboxyl-terminal 25 amino acids of the Spa2 box, results in a fusion that does not localize (Fig. 5 A), defining the carboxyl-terminal boundary of the Spa2p localization domain to amino acid residues 511–549. Furthermore, Spa2GFP lacking the B–E region (containing the coiled-coil and Spa2 box) or lacking the C–E region (largely the Spa2 box) did not localize (data not shown). To address whether the 152-residue Spa2 box, defined maximally by residues 397–549, was sufficient for localization, we made three constructs consisting of the coiled-coil region and Spa2 box (ΔBX), the coiled-coil region alone (ΔBV), and the Spa2 box alone (ΔCX) fused to GFP. Both constructs containing the Spa2 box, BX and CX, localized correctly, whereas the coiled-coil region alone (BV) did not localize to regions of growth. We were unable to detect expression of any Spa2GFP fusions in yeast by immunoblot analyses (using anti-GFP); however, all of the constructs that did not localize correctly were nonetheless expressed as seen by the substantial cytoplasmic fluorescence (Fig. 5 A). Altogether, these results show that the Spa2 box alone is necessary and sufficient for Spa2p bud tip and mother–daughter neck localization. Similar patterns of localization were observed in the spa2 mutant (pea1, strain JC-J9) isolated by Chenevert et al. (1994) as well as a Δspa2 strain (RAY574), which is deleted for the entire SPA2 coding region, demonstrating that the Spa2 box does not localize to sites of polarized growth via its interactions with Spa2p, i.e., oligomerization. Furthermore, we have not detected any interactions between Spa2p and itself using these various constructs by two-hybrid assays (Orger, M., and R. Arkowitz, unpublished data).
We also analyzed this collection of Spa2GFP truncation and deletion mutants for their function in cell mating. We examined strains carrying these Spa2GFP fusions to determine localization in cells exposed to high pheromone concentration, their ability to complement shmoo formation and morphology defects of a Δspa2 mutant, and their ability to complement the mating defect of a Δspa2 mutant. Spa2GFP localization in pheromone-treated cells and the ability of various constructs to complement shmoo formation defects were both carried out in a Mata Δspa2 strain (RAY578), and SPA2 mating function was assesed in Mata spa2 strains (JC-J9) by quantitative mating assays (Chenevert et al., 1994). All experiments were carried out with integrated copies of truncation or deletion constructs.
Localization and shmoo formation assays were carried out by treating cells with high pheromone concentration (12.9 μM) for 2 h followed by fixation. These pheromone concentrations have been shown by Dorer et al. (1995) to saturate the pheromone response pathway and shmoo formation. It was further shown that these saturating levels of pheromone resulted in the execution of a default mating pathway. We chose such high concentrations of pheromone both to increase shmoo formation and because SPA2 has been shown to be required for this default mating pathway (Dorer et al., 1995). Spa2GFP localization was assessed in Δspa2 cells by fluorescence and confocal microscopy. Table III shows that all constructs that localized to sites of polarized growth in budding cells localize to the shmoo tip. While none of the Spa2GFP truncations examined fully complemented the shmoo formation defect, ΔC, ΔD, and ΔY partially complemented this defect. These results suggest that a region substantially larger than the Spa2 box is required for Spa2p-mediated shmoo formation during the default mating pathway. Furthermore, the ΔX truncation localizes to the shmoo tip, yet it does not complement the shmoo formation defect, consistent with different requirements for these two processes.
Table III.
Shmoo Formation and Localization of Spa2GFP Deletion Mutants
| SPA2 construct | Percentage of shmoos* | Shmoo tip localization | ||
|---|---|---|---|---|
| % | ||||
| SPA2GFP | 83 | + | ||
| ΔC | 63 | + | ||
| ΔD | 66 | − | ||
| ΔY | 66 | + | ||
| ΔX | 54 | + | ||
| ΔW | 52 | − | ||
| − | 58 | − |
Δspa2 cells (RAY578) with the respective SPA2GFP deletion constructs integrated at URA3 were treated with α-factor and quantitated as described in Materials and Methods. For the percentage of shmoos, 250 cells from each strain were counted. Shmoo tip localization was determined on fixed cells using confocal microscopy.
Peanut- and pear-shaped shmoos were designated as shmoos.
The mating assays involved mixing equal amounts of an enfeebled tester strain with spa2 mutant JC-J9 carrying SPA2GFP constructs, allowing them to mate for 4 h at 30°C, and then determining mating efficiency by plating on nonselective and selective media. This functional assay was not affected by SPA2 copy number (data not shown). The results of mating assays are presented in Fig. 5 B and summarized in Fig. 4. The carboxyl-terminal two-thirds of Spa2p, including the nonameric amino acid repeat domain, is not necessary for mating function. Deletion ΔX, which removes amino acids 655–549, results in a fivefold decrease in mating efficiency, and further truncations drastically reduce mating efficiency to background levels, implicating this region in SPA2 mating function. The amino terminus of Spa2p has a greater role in mating function with a truncation of as little as 88 amino acid residues (ΔA), resulting in a reduction in mating efficiency to background levels, despite ΔA being expressed and localizing correctly (ΔA; Fig. 5 A). Together these results indicate that the Spa2 box also plays a role in mating function being necessary but not sufficient for mating. In addition, the first 90 amino acid residues are essential for mating function. The Spa2p requirements for shmoo formation during the default mating pathway appear to overlap but are not identical to the requirements for cell mating. For example, ΔC is partially functional in the shmoo formation assay, yet it is not functional for cell mating. These differences most likely reflect the roles of SPA2 in the different mating processes: normal mating along a pheromone gradient, and the default mating pathway in high pheromone concentration and the absence of a pheromone gradient. Our results suggest that Spa2p must be correctly localized (via the Spa2 box) for mating function and, in addition, the Spa2p amino terminus is necessary for mating function. Consistent with this interpretation, constructs consisting of the coiled-coil region and Spa2 box (ΔBX) alone or the Spa2 box alone (ΔCX), both of which localize correctly, were not functional in the mating assay. Conversely, Spa2GFP lacking either the B–D region (containing the coiled-coil and Spa2 box) or the C–D region (largely the Spa2 box), neither of which localized correctly, also were nonfunctional in mating (data not shown). The amino terminus of Spa2p, which is essential for mating, and the Spa2 box, which is required for localization, are both conserved in ORF YSCL8543.8 (32% and 42% identity, respectively), suggesting common functions and localizations for these two proteins.
SPH1, a Yeast Gene Containing the Spa2p Localization and Function Domains
Based upon the conserved Spa2p localization and functional domains in YSCL8543.8 (Fig. 6), we have named this ORF SPH1 for SPA2 homolog. We isolated SPH1 by PCR from genomic DNA. The sequence of several clones revealed a number of amino acid differences from the yeast genome project sequence (none within the conserved sequences), a frame shift, and a subsequent stop codon, resulting in a 648–amino acid protein (EMBL/GenBank/DDBJ accession No. AF008236). The frameshift in the SPH1 sequence reveals that, in addition to the two regions of homology discussed in the previous section, the carboxyl termini of Spa2p and Sph1p are also homologous with 28% identity in the terminal 84 amino acids of Spa2p (Fig. 6). These three regions of homology between Spa2p and Sph1p implicate common functions.
Figure 6.

Alignment of Spa2p and Sph1p sequences. A schematic representation of regions of identity as identified by the BLAST algorithm (Altschul et al., 1990) is shown. Aligned sequences are shown below. Sph1p is 15% identical to Spa2p over the entire protein. ∣, identical amino acids; ·, similar amino acids. A, B, and C, three regions of similarity between Spa2p and Sph1p. Numbers correspond to amino acid residue numbers of Spa2p and Sph1p.
The cloned SPH1 was used to construct a knockout strain using the one-step method of Rothstein (1983) in which SPH1 residues 26–408 were replaced with the HIS3 gene. Haploid (both a and α strains) and diploid homozygous Δsph1 strains were constructed, confirmed by PCR analyses, and examined for growth defects. All Δsph1 strains grew normally, did not exhibit ts or cold-sensitive (cs) growth defects, and appeared normal morphologically. Since SPA2 and SPH1 are both nonessential genes that display significant homology, we tested whether a double knockout strain displayed any synthetic phenotypes. Haploid Δspa2 Δsph1 strains were constructed both by sequential one-step deletion methods and also by mating Δsph1 haploids with Δspa2 haploids, selecting diploids, sporulation, and picking tetrads. Double knockout strains were confirmed by markers and PCR analyses, and in all cases cells grew normally, were not ts or cs, and appeared morphologically similar to Δspa2 cells. These results suggest that either SPA2 or SPH1 functions are nonessential or that there are other genes with similar functions. Synthetic lethal screens using the double knockout strain should allow us to address the latter possibility.
SPA2 has been shown to be involved in cell mating (Gehrung and Snyder, 1990; Chenevert et al., 1994; Yorihuzi and Ohsumi, 1994; Dorer et al., 1995) and we therefore examined the role of SPH1 in this process. The effect of Δsph1 alone and in combination with Δspa2 on the ability of cells to shmoo and mate was determined. Table IV shows quantitation of shmoo formation after treatment of cells with saturating concentrations of pheromone. Deletion of SPA2 results in a defect in shmoo formation (Gehrung and Snyder, 1990; Valtz and Herskowitz, 1996). Deletion of SPH1 resulted in a clear decrease in the percentage of shmoos, approximately halfway between wild-type and Δspa2 cells. However, Δsph1 shmoos were not peanut-shaped like the Δspa2 cells, but rather pear-shaped similar to wild-type cells (data not shown). Cells lacking both SPA2 and SPH1 show a shmoo formation defect similar to Δspa2 cells, and furthermore their shmoos were peanut-shaped, suggesting that SPA2 is epistatic to SPH1. To determine whether expression of SPH1 or SPA2 could suppress the shmoo formation defect of Δspa2 and Δsph1 strains, either SPA2GFP or SPH1GFP was expressed using the SPA2 promoter integrated at the URA3 locus. Table V shows that SPA2GFP is able to suppress the shmoo formation defect in both Δspa2 and Δsph1 cells, whereas SPH1GFP only suppresses the shmoo formation defect in Δsph1 cells.
Table IV.
Shmoo Formation of Δspa2 and Δsph1 Mutants
| Strain | Percentage of shmoos* | |
|---|---|---|
| % | ||
| WT | 84 | |
| WT | 80 | |
| Δspa2 | 58 | |
| Δspa2 | 58 | |
| Δsph1 | 74 | |
| Δsph1 | 70 | |
| Δspa2 Δsph1 | 64 | |
| Δspa2 Δsph1 | 63 |
Cells (SEY6211, RAY578, RAY567, and RAY590) were treated with α-factor and quantitated as described in Materials and Methods. Two independent isolates of each strain were used and for each isolate 250 cells were counted.
Peanut- and pear-shaped shmoos were designated as shmoos.
Table V.
SPA2GFP Complements Shmoo Formation Defect and Bud Site Selection Defect of Δsph1 Mutant
| Strain | Integrated gene | Percentage of shmoos* | Budding pattern | |||
|---|---|---|---|---|---|---|
| % | ||||||
| WT | − | 77 | Bipolar | |||
| WT | SPA2GFP | 78 | ND | |||
| WT | SPA1GFP | 76 | ND | |||
| Δspa2 | – | 58 | Random | |||
| Δspa2 | SPA2GFP | 83 | Bipolar | |||
| Δspa2 | SPH1GFP | 55 | Random | |||
| Δsph1 | − | 66 | Random | |||
| Δsph1 | SPA2GFP | 75 | Bipolar | |||
| Δsph1 | SPH1GFP | 79 | Bipolar |
Cells were treated with α-factor and quantitated as described in Materials and Methods. Integrants were selected that showed localized fluorescence. For percentage of shmoos, 250 Mata haploid cells from each strain were counted. Bud site selection was determined using Calcofluor staining on homozygous diploid cells as described in Materials and Methods.
Peanut- and pear-shaped shmoos were designated as shmoos.
The role of SPH1 in cell mating was examined by quantitative matings of deletion strains with an enfeebled mating partner. Fig. 7 A shows that Δsph1 either alone or in combination with Δspa2 has no observable effect on mating with an enfeebled mating partner. Furthermore, we examined the effect of Δsph1 on quantitative mating with a wild-type strain and were unable to detect any differences (data not shown). Because of the sequence conservation between Spa2p and Sph1p in two separate regions necessary for Spa2p mating function, we determined if SPH1 could suppress the mating defect in Δspa2 mutants. Fig. 7 B shows that expression of SPH1GFP using the SPA2 promoter integrated at the URA3 locus in a Δspa2 strain resulted in a substantial increase in mating efficiency with an enfeebled mating partner. In addition, overexpression of SPH1 from a multicopy (2 μm) plasmid using the SPH1 promoter also resulted in an increase in mating efficiency with an enfeebled mating partner (data not shown). These results suggest that Sph1p is involved in mating because it results in a defect in shmoo formation and can partially substitute for Spa2p mating function, implying that the sequence conservation between these two proteins is functionally significant.
Figure 7.
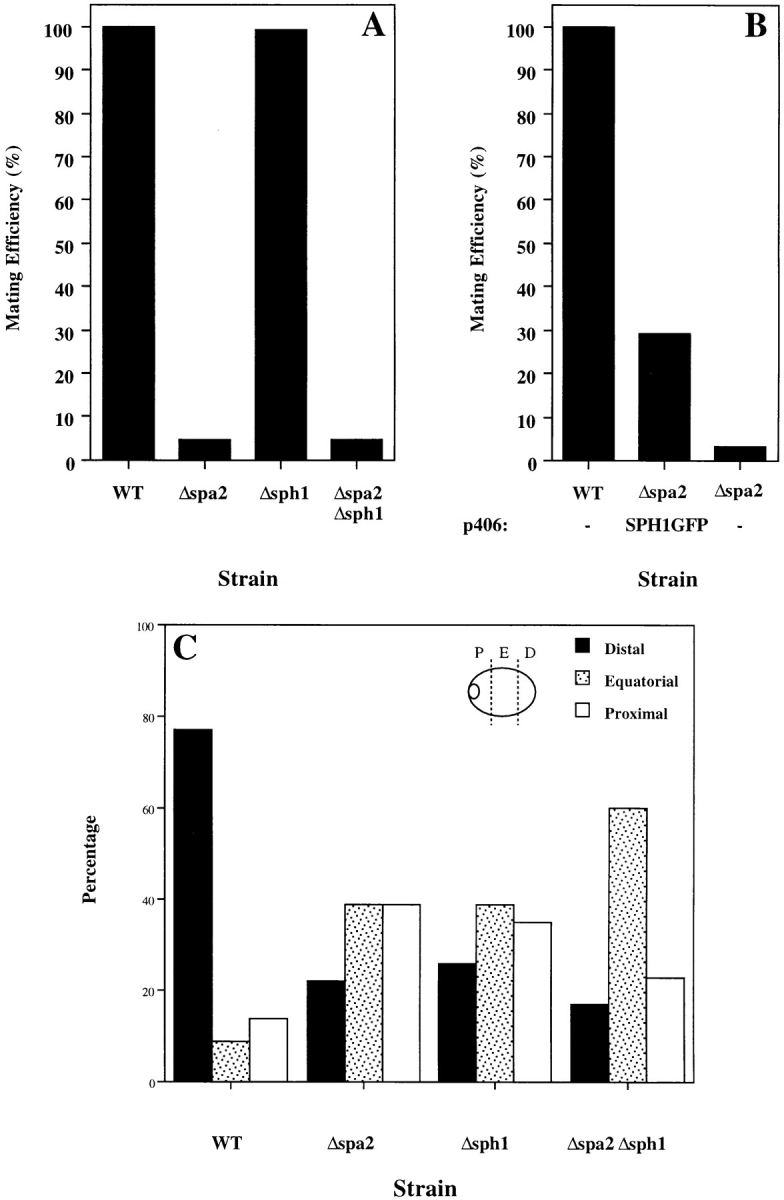

(A) Deletion of sph1 has no effect on mating efficiency. Exponential cultures of WT (RAY697), Δspa2 (RAY698), Δsph1 (RAY709), and Δspa2 Δsph1 (RAY711) were mated with an enfeebled mating tester strain (JY426) as described in Materials and Methods, and diploids were selected on −lys, −ura plates. Wild-type (WT) mating efficiency (4.4%) was set to 100% efficiency. (B) SPH1 can partially complement for mating deficiency of Δspa2 (RAY574). WT (RAY697), Δspa2 with SPH1GFP (RAY703), and Δspa2 (RAY698) were mated with an enfeebled mating tester strain (JY426) as described in Materials and Methods, and diploids were selected on −lys, −ura plates. WT mating efficiency (6.5%) was normalized to 100% efficiency. (C) SPH1 is required for bipolar bud site selection. Wild-type (SEY6210/11), homozygous Δspa2 (RAY616), homozygous Δsph1 (RAY618), and homozygous Δspa2 Δsph1 (RAY620) exponentially growing diploids were stained with Calcofluor as described in Materials and Methods, and budding pattern was analyzed. For each strain, the position of the bud relative to the birth scar (see inset) was scored for ∼150 cells with two or more bud scars. (D) Budding pattern of homozygous diploids sph1 and spa2 mutants. Representative fluorescence microscopy pictures of cells quantitated in C. Bar, 2.5 μm.
The effect of SPH1 deletion on bud site selection was investigated. Deletion of SPA2 has been shown to have no effect on bud site selection in haploids; however, homozygous diploid deletion mutants are defective in bipolar budding (Snyder, 1989; Zahner et al., 1996; Valtz and Herskowitz, 1996). Specifically, spa2 mutants are defective in bud site selection after the correct positioning of the first bud. Δsph1 haploid cells budded in an axial pattern identical to that of wild-type cells (data not shown). The budding pattern of homozygous diploids was determined by counting cells with two or more bud scars and determining the position of the bud relative to the birth scar (Fig. 7, inset). Fig. 7 C shows that SPH1 is required for bipolar bud site selection in diploids similar to SPA2. Homozygous Δsph1 diploids were able to correctly position the first bud, similar to spa2 mutants. The double homozygous diploid mutant Δsph1 Δspa2 showed a similar random bud site selection defect. Fig. 7 D shows representative pictures of Calcofluor staining of bud scars of the individual and double homozygous diploid deletions. It appears that the Δsph1 bud site selection defect is less random than that of Δspa2; however, further analyses are required. Similar to the ability of SPA2GFP to suppress the Δsph1 shmoo formation defect, SPA2GFP suppresses the bud site selection defect of homozygous Δsph1 diploids. Together these results demonstrate that SPH1 is required for bipolar budding after the positioning of the first bud and, together with shmoo formation and mating experiments, show that SPH1 and SPA2 have overlapping functions.
The conservation of the Spa2 box that is necessary and sufficient for localization to sites of polarized growth in Sph1p and the similar functions of SPH1 and SPA2 prompted us to examine the localization of Sph1p. To determine the localization of Sph1p, we used an SPH1GFP fusion that was driven by the SPA2 promoter, and this construct was integrated at the URA3 locus. Fig. 8 A shows the in vivo localization of Sph1GFP, which is strikingly similar to Spa2p localization. These experiments demonstrate that Sph1p also localizes to sites of polarized growth: at the site where the bud will form, the bud tip, and the mother–daughter bud neck. Similar localization was observed in haploids of the opposite mating type (Mata) and diploids, indicating that Sph1p localization is not cell type specific. Furthermore, Sph1GFP localized to sites of polarized growth in Δsph1, Δspa2, and Δsph1 Δspa2 cells, demonstrating that neither SPH1 nor SPA2 is required for Sph1GFP localization (data not shown). In addition, Sph1GFP localized to shmoo tips in Mata Δsph1 cells that had been treated with saturating concentrations of mating pheromone (Fig. 8 B). Together these results suggest that the Spa2 box in Sph1p is sufficient for localization to polarized growth during both budding and mating.
Figure 8.

(A) Sph1p localizes to sites of polarized growth in budding cells. Exponentially grown cells with SPH1GFP integrated at URA3 (RAY699) were analyzed by confocal microscopy. Immunoblot analyses of RAY699 revealed that a GFP fusion protein of the correct molecular mass (∼100 kD) was expressed. (B) Sph1p localizes to sites of polarized growth in shmoos. Exponentially grown Mata Δsph1 cells with SPH1GFP integrated at URA3 (RAY875) were treated with α-factor, as described in Materials and Methods, fixed, and viewed by confocal microscopy. Note that background fluorescence in cells is due to Ade fluorophore from ade2 mutation. Bar, 5 μm.
We also examined whether the localization of Spa2GFP or Sph1GFP could be blocked or competed out by overexpression of either protein. Cross competition of one protein for the other would suggest that these two proteins bind to the same site in vivo. Epitope-tagged Spa2p and Sph1p were overexpressed using a TPI promoter on a 2-μm multicopy plasmid in strains with either integrated SPA2GFP or SPH1GFP (Fig. 9 A). Overexpression of either Spa2p or Sph1p blocks localization of Spa2GFP. Conversely, overexpression of either Sph1p or Spa2p blocks localization of Sph1GFP. Immunoblot analyses demonstrated that both Spa2myc and Sph1myc were expressed (data not shown). Quantitation of the number of cells with localized Spa2GFP or Sph1GFP revealed that overexpressed Spa2myc blocked Spa2GFP or Sph1GFP localization more effectively than overexpressed Sph1myc (Fig. 9 B). Most cells (89%) showed localized Spa2GFP, whereas the number of cells with localized Spa2GFP was reduced twofold in the presence of overexpressed Sph1myc and reduced fourfold in the presence of overexpressed Spa2myc. Conversely, 88% of the cells had localized Sph1GFP that was reduced threefold in the presence of overexpressed Sph1myc and reduced fourfold in the presence of overexpressed Spa2myc. These results are consistent with ability of SPA2GFP to functionally replace Δsph1 in shmoo formation and bipolar bud site selection, whereas SPH1GFP is unable to replace Δspa2 function in shmoo formation and bipolar bud site selection. This cross competition of localization is consistent with the notion that these two proteins may be localized by the same cellular component and this interaction can be competed out by either protein.
Figure 9.


Localization of Spa2GFP or Sph1GFP is blocked by overexpression of either protein. A strain with SPA2GFP integrated at the URA3 locus (RAY416) was transformed with either pRS425 (−), pRS425TPISPH1myc (Sph1), or pRS425TPISPA2myc (Spa2). A strain with SPH1GFP integrated at the URA3 locus (RAY699) was transformed with either pRS425 (−), pRS425TPISPH1myc (Sph1), or pRS425TPISPA2myc (Spa2). Exponentially grown cultures were analyzed by confocal microscopy for GFP fluorescence. Each panel shows a representative field of view. (B) Quantitation of cells with localized Spa2GFP or Sph1GFP. For each condition, 300 cells were scored for the presence of fluorescence localized to regions of polarized growth using either fluorescence or confocal microscopy. Bar, 10 μm.
Discussion
Our studies with Spa2GFP demonstrate that Spa2p localizes to the bud tip, forming a cup-like crescent, and the bud neck between mother and daughter cell, forming a donut-like ring. These two localizations are distinct events that follow one another during the cell cycle. The changes in shape and localization of these structures occur on the minute time scale. In septin mutants (cdc3, cdc10, and cdc11) at the nonpermissive temperature, Spa2GFP is localized only to the bud tip, while the bud emergence mutants bem1 and bem2 have no effect on Spa2GFP localization. We have identified a small domain in Spa2p that is both necessary and sufficient for localization to sites of polarized growth in vivo. This 150–amino acid residue domain can target a heterologous protein to the sites of polarized cell growth in yeast. In addition to the amino-terminal 100 amino acids, the Spa2 box is also essential for Spa2p mating function, suggesting that Spa2p must be correctly localized to be functional. We have identified a novel protein, Sph1p, in which both the Spa2p localization domain and amino terminus are conserved. Deletion of SPH1 results in a defect in shmoo formation and a random bud site selection pattern in diploids. In addition, SPH1 can partially suppress the mating defect of Δspa2 cells and localizes to the same regions of polarized growth as Spa2p. Together these two proteins with similar functions constitute a novel protein family involved in polarized cell growth and appear to localize to regions of polarized growth via an interaction with the same cellular component.
Spa2p Dynamics and Localization
The biological and spectral characteristics of GFP have enabled the analyses of Spa2p localization in vivo during normal cell growth and specifically have shed light on Spa2p dynamics. In contrast with actin cortical patch movements (Doyle and Botstein, 1996; Waddle et al., 1996), Spa2p-containing structures change shape and localization on a much slower time scale. These movements could be due to subcellular reorganizations or association/dissociation reactions. While the ring of Spa2p at the mother–daughter bud neck is reminiscent of the septin ring (Kim et al., 1991; Ford and Pringle, 1991; Haarer and Pringle, 1987), it differs in that it does not appear as two rings, like the septin rings, until the cells have divided. The septins appear as two separate rings flanking the mother–daughter constriction in cells with large buds that are dividing, whereas Spa2GFP appears as a single ring until cell division and thereafter stays at the site of division in both cells.
What component localizes Spa2p to such specific regions and maintains this localization? Spa2p is one of the first proteins to localize to the site of the new bud (Snyder, 199l; Snyder et al., 1991) at approximately the same time as actin patches concentrate at the region of the incipient bud (Snyder et al., 1991). However, in budding cells when actin patches are delocalized, Spa2p localization is normal (Snyder et al., 1991), indicating that Spa2p localization is not dependent on a polarized cytoskeleton. Experiments using septin mutants, in which the neck filaments do not form at the nonpermissive temperature (Byers and Goetsch, 1976; Kim et al., 1991; Ford and Pringle, 1991; Haarer and Pringle, 1987), show that Spa2p is still localized to the bud tip. These results suggest that the septins are not required for maintaining this polarized localization. Our demonstration that a small domain of Spa2p is necessary and sufficient for this localization raises the attractive possibility that this region is binding to a receptor that marks the new bud. Consistent with the notion of a receptor is the result that Spa2p localization appears to be saturable. Furthermore, Spa2GFP or Sph1GFP localization can be competed out by either Spa2p or Sph1p overexpression. While a number of proteins localize to very similar regions as Spa2p and Sph1p, possible candidates for such a receptor include the polytopic membrane protein Fks1p, which encodes a β(1→ 3)glucan synthase (Qadota et al., 1996), and Axl2p/Bud10p (Roemer et al., 1996; Halme et al., 1996), which encodes a type I single transmembrane protein, both of which localize to similar regions as Spa2p. We have begun to examine proteins involved in localizing Spa2p by screening for mislocalization mutants. Preliminary results with ethylmethane sulfonate mutagenized cells indicate that it should be possible to identify and analyze such mutants using Spa2GFP and a visual screen (Lowe, N., and R. Arkowitz, unpublished data). Identification of proteins interacting with Spa2p and involved in its early localization will be greatly facilitated by the definition of a minimal Spa2p domain that localizes correctly.
SPA2 and SPH1 Function
By sequence comparisons we have identified a homologue of SPA2 that contains both a region required for mating function and a localization domain necessary and sufficient for polarized growth localization. Furthermore, at positions −234 to −239 in the promoter of SPA2 is the match to a MluI cell cycle box (MCB), which also includes an imperfect MluI site typically found adjacent to the MluI site. The position of this cell cycle box is in the range −100 to −250 upstream of the start ATG where MCBs are typically found (Johnston et al., 1991; McIntosh et al., 1991). SPH1 has an identical MCB (including adjacent imperfect MluI site) −155 to −160 in its potential promoter (White et al., 1986; Kilmartin et al., 1993; Yamamoto et al., 1996). SPA2 and SPH1 have similar functions, with deletion of either gene resulting in a defect in shmoo formation at saturating pheromone concentration and random bud site selection in diploids. Furthermore, overexpression of SPH1 can partially complement the mating defect of a Δspa2 strain, and overexpression of SPA2 can complement the shmoo formation and bipolar bud site selection defect of Δsph1 cells. Deletion of SPA2 results in a stronger defect in shmoo formation and morphology at saturating pheromone concentration than in Δsph1, and the double mutant appears phenotypically similar to the Δspa2 mutant, suggesting that SPA2 is epistatic to SPH1.
Recently, Valtz and Herskowitz (1996) cloned and characterized PEA2, which was originally identified as a mutant that formed a peanut-shaped shmoo in the presence of mating pheromone. This protein contains a predicted coiled-coil domain, yet it does not show any sequence homology to either Spa2p or Sph1p. PEA2 is required for efficient mating to an enfeebled mating partner and necessary for bipolar budding. In addition, Pea2p localizes to sites of polarized growth in budding and mating cells similar to the localization of Spa2p and Sph1p. The immunolocalization of Spa2p is dependent on PEA2. The functional similarity between SPA2, SPH1, and PEA2 and their localization to similar sites of polarized growth suggest that these proteins constitute a protein family. Interestingly, all three of these genes function in bipolar bud site selection, shmoo formation, and cell mating. Both Pea2p and Spa2p have potential coiled-coil domains, whereas both Sph1p and Spa2p have the Spa2 box necessary for polarized localization. An additional member of this family may be BUD6 (Zahner et al., 1996), which results in a similar bipolar bud site selection defect. Analysis of Bud6p sequence using the Lupas program (Lupas et al., 1991) also reveals a region with high probability of forming a coiled-coil domain. The localization as well as shmooing and mating defects of bud6 mutants should help determine whether it is a member of this family.
What is the function of this protein family? We propose these proteins may be involved in initiating and maintaining the organization of the large number of proteins present at sites of polarized growth sites (in a sense, chaperones for polarized growth), and that there must be a number of additional components with such functions. It is possible that CDC10 links such scaffolds to the septin ring initially (Flescher et al., 1993); however, additional proteins are required to maintain the localization of these proteins at the bud site. Further genetic and biochemical experiments will certainly shed light on the functions of Spa2p and Sph1p. We have identified a saturable site that localizes both Spa2p and Sph1p very early in the process of bud formation and a minimal domain of Spa2p that is able to localize to these sites of polarized growth. This minimal domain will be crucial for the identification of additional components involved in the early stages of cell polarization.
Acknowledgments
We thank I. Herskowitz, M. Snyder, R. Mortimer, S. Emr, J. Haseloff, K. Siemering, and A. Wach for yeast strains and plasmids; H. Pelham, S. Munro, J. Kilmartin, and M. Bassilana for helpful discussions and critical reading of the manuscript; and A. Nern for cloning BEM1 and assistance with ABI sequencing.
Note Added in Proof
Bud6p has recently been shown to localize to sites of polarized growth (Evangelista, M., K. Blundell, M.S. Longtine, C.J. Chow, N. Adames, J.R. Pringle, M. Peter, and C. Boone. 1997. Bni1p, a yeast formin linking cdc42p and the actin cytoskeleton during polarized morphogenesis. Science (Wash. DC). 276:118–122).
Footnotes
1. Abbreviations used in this paper: cs, cold sensitive; GFP, green fluorescent protein; ORF, open reading frame; TPI, triose phosphate isomerase; ts, temperature sensitive; WT, wild type.
Please address all correspondence to Robert A. Arkowitz, Division of Cell Biology, MRC Laboratory of Molecular Biology, Hills Road, Cambridge, CD2 2QH, United Kingdom. Tel.: (44) 1223-402229. Fax: (44) 1223-412142. e-mail: ra2@mrc-lmb.cam.ac.uk
References
- Adams AE, Pringle JR. Relationship of actin and tubulin distribution to bud growth in wild-type and morphogenetic-mutant Saccharomyces cerevisiae. . J Cell Biol. 1984;98:934–945. doi: 10.1083/jcb.98.3.934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Amatruda JF, Cooper JA. Purification, characterization, and immunofluorescence localization of Saccharomyces cerevisiaecapping protein. J Cell Biol. 1992;117:1067–1076. doi: 10.1083/jcb.117.5.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson K. One signal, two body axes. Science (Wash DC) 1995;269:489–490. doi: 10.1126/science.7542799. [DOI] [PubMed] [Google Scholar]
- Baudin A, Ozier-Kalogeropoulos O, Denouel A, Lacroute R, Cullin C. A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae. . Nucleic Acids Res. 1993;21:3329–3330. doi: 10.1093/nar/21.14.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender A, Pringle JR. Use of a screen for synthetic lethal and multicopy suppressor mutants to identify two new genes involved in morphogenesis in Saccharomyces cerevisiae. . Mol Cell Biol. 1991;11:1295–1305. doi: 10.1128/mcb.11.3.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers B, Goetsch L. A highly ordered ring of membrane-associated filaments in budding yeast. J Cell Biol. 1976;69:717–721. doi: 10.1083/jcb.69.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chant J. Generation of cell polarity in yeast. Curr Opin Cell Biol. 1996;8:557–565. doi: 10.1016/s0955-0674(96)80035-4. [DOI] [PubMed] [Google Scholar]
- Chant J, Corrado K, Pringle JR, Herskowitz I. Yeast BUD5, encoding a putative GDP-GTP exchange factor, is necessary for bud site selection and interacts with bud formation gene BEM1. Cell. 1991;65:1213–1224. doi: 10.1016/0092-8674(91)90016-r. [DOI] [PubMed] [Google Scholar]
- Chant J, Mischke M, Mitchell E, Herskowitz I, Pringle JR. Role of Bud3p in producing the axial budding pattern of yeast. J Cell Biol. 1995;129:767–778. doi: 10.1083/jcb.129.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenevert J. Cell polarization directed by extracellular cues in yeast. Mol Biol Cell. 1994;5:1169–1175. doi: 10.1091/mbc.5.11.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenevert J, Corrado K, Bender A, Pringle J, Herskowitz I. A yeast gene (BEM1) necessary for cell polarization whose product contains two SH3 domains. Nature (Lond) 1992;356:77–79. doi: 10.1038/356077a0. [DOI] [PubMed] [Google Scholar]
- Chenevert J, Valtz N, Herskowitz I. Identification of genes required for normal pheromone-induced cell polarization in Saccharomyces cerevisiae. . Genetics. 1994;136:1287–1296. doi: 10.1093/genetics/136.4.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Hyman AA. Cell fate determination. When is a determinant a determinant? . Curr Biol. 1994;4:420–422. doi: 10.1016/s0960-9822(00)00091-9. [DOI] [PubMed] [Google Scholar]
- Cooper JA, Kiehart DP. Septins may form a ubiquitous family of cytoskeletal filaments. J Cell Biol. 1996;134:1345–1348. doi: 10.1083/jcb.134.6.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorer R, Pryciak PM, Hartwell LH. Saccharomyces cerevisiaecells execute a default pathway to select a mate in the absence of pheromone gradients. J Cell Biol. 1995;131:845–861. doi: 10.1083/jcb.131.4.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle T, Botstein D. Movement of yeast cortical actin cytoskeleton visualized in vivo. Proc Natl Acad Sci USA. 1996;93:3886–3891. doi: 10.1073/pnas.93.9.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drubin DG, Nelson WJ. Origins of cell polarity. Cell. 1996;84:335–344. doi: 10.1016/s0092-8674(00)81278-7. [DOI] [PubMed] [Google Scholar]
- Drubin DG, Miller KG, Botstein D. Yeast actin-binding proteins: evidence for a role in morphogenesis. J Cell Biol. 1988;107:2551–2561. doi: 10.1083/jcb.107.6.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton S, Simons K. Apical, basal, and lateral cues for epithelial polarization. Cell. 1995;82:5–8. doi: 10.1016/0092-8674(95)90045-4. [DOI] [PubMed] [Google Scholar]
- Farkas V, Kovarik J, Kosinova A, Bauer S. Autoradiographic study of mannan incorporation into growing walls of Saccharomyces cerevisiae. . J Bacteriol. 1974;117:265–269. doi: 10.1128/jb.117.1.265-269.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field CM, Alawar O, Rosenblatt J, Wong ML, Alberts B, Mitchison TJ. A purified Drosophilaseptin complex forms filaments and exhibits GTPase activity. J Cell Biol. 1996;133:605–616. doi: 10.1083/jcb.133.3.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flescher EG, Madden K, Snyder M. Components required for cytokinesis are important for bud site selection in yeast. J Cell Biol. 1993;122:373–386. doi: 10.1083/jcb.122.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford SK, Pringle JR. Cellular morphogenesis in the Saccharomyces cerevisiaecell cycle: localization of the CDC11 gene product and the timing of events at the budding site. Dev Genet. 1991;12:281–292. doi: 10.1002/dvg.1020120405. [DOI] [PubMed] [Google Scholar]
- Freifelder D. Bud position in Saccharomyces cerevisiae. . J Bacteriol. 1960;80:567–568. doi: 10.1128/jb.80.4.567-568.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrung S, Snyder M. The SPA2 gene of Saccharomyces cerevisiaeis important for pheromone-induced morphogenesis and efficient mating. J Cell Biol. 1990;111:1451–1464. doi: 10.1083/jcb.111.4.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein, B., S.N. Hird, and J.G. White. 1993. Cell polarity in early C. elegans development. Dev. Suppl. 279–287. [PubMed]
- Haarer BK, Pringle JR. Immunofluorescence localization of the Saccharomyces cerevisiaeCDC12 gene product to the vicinity of the 10-nm filaments in the mother-bud neck. Mol Cell Biol. 1987;7:3678–3687. doi: 10.1128/mcb.7.10.3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halme A, Michelitch M, Mitchell EL, Chant J. Bud10p directs axial cell polarization in budding yeast and resembles a transmembrane receptor. Curr Biol. 1996;6:570–579. doi: 10.1016/s0960-9822(02)00543-2. [DOI] [PubMed] [Google Scholar]
- Heim R, Cubitt AB, Tsien RY. Improved green fluorescence [letter] Nature (Lond) 1995;373:663–664. doi: 10.1038/373663b0. [DOI] [PubMed] [Google Scholar]
- Hicks JB, Strathern JN, Herskowitz I. Interconversion of yeast mating types. III. Action of the homothallism (HO) gene in cells homozygous for the mating type locus. Genetics. 1977;85:395–405. doi: 10.1093/genetics/85.3.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvitz HR, Herskowitz I. Mechanisms of asymmetric cell division: two Bs or not two Bs, that is the question. Cell. 1992;68:237–255. doi: 10.1016/0092-8674(92)90468-r. [DOI] [PubMed] [Google Scholar]
- Jackson CL, Hartwell LH. Courtship in S. cerevisiae: both cell types choose mating partners by responding to the strongest pheromone signal. Cell. 1990a;63:1039–1051. doi: 10.1016/0092-8674(90)90507-b. [DOI] [PubMed] [Google Scholar]
- Jackson CL, Hartwell LH. Courtship in Saccharomyces cerevisiae: an early cell-cell interaction during mating. Mol Cell Biol. 1990b;10:2202–2213. doi: 10.1128/mcb.10.5.2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston LH, Lowndes NF, Johnson AL, Sugino A. A cell-cycle-regulated trans-factor, DSC1, controls expression of DNA synthesis genes in yeast. Cold Spring Harbor Symp Quant Biol. 1991;56:169–176. doi: 10.1101/sqb.1991.056.01.022. [DOI] [PubMed] [Google Scholar]
- Keynes R, Cook GM. Axon guidance molecules. Cell. 1995;83:161–169. doi: 10.1016/0092-8674(95)90157-4. [DOI] [PubMed] [Google Scholar]
- Kilmartin JV, Adams AE. Structural rearrangements of tubulin and actin during the cell cycle of the yeast Saccharomyces. . J Cell Biol. 1984;98:922–933. doi: 10.1083/jcb.98.3.922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilmartin JV, Dyos SL, Kershaw D, Finch JT. A spacer protein in the Saccharomyces cerevisiaespindle poly body whose transcript is cell cycle-regulated. J Cell Biol. 1993;123:1175–1184. doi: 10.1083/jcb.123.5.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HB, Haarer BK, Pringle JR. Cellular morphogenesis in the Saccharomyces cerevisiaecell cycle: localization of the CDC3 gene product and the timing of events at the budding site. J Cell Biol. 1991;112:535–544. doi: 10.1083/jcb.112.4.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y-J, Francisco L, Chen G-C, Marcotte E, Chan CSM. Control of cellular morphogenesis by the Ipl2/Bem2 GTPase-activating protein: possible role of protein phosphorylation. J Cell Biol. 1994;127:1381–1394. doi: 10.1083/jcb.127.5.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimble J. An ancient molecular mechanism for establishing embryonic polarity? . Science (Wash DC) 1994;266:577–578. doi: 10.1126/science.7939710. [DOI] [PubMed] [Google Scholar]
- Kunkel TA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci USA. 1985;82:488–492. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeuw T, Fourest-Lieuvin A, Wu C, Chenevert J, Clark K, Whiteway M, Thomas DY, Leberer E. Pheromone response in yeast: association of Bem1p with proteins of the MAP kinase cascade and actin. Science (Wash DC) 1995;270:1210–1213. doi: 10.1126/science.270.5239.1210. [DOI] [PubMed] [Google Scholar]
- Lew DJ, Reed SI. Morphogenesis in the yeast cell cycle: regulation by Cdc28 and cyclins. J Cell Biol. 1993;120:1305–1320. doi: 10.1083/jcb.120.6.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillie SH, Brown SS. Immunofluorescence localization of the unconventional myosin, Myo2p, and the putative kinesin-related protein, Smy1p, to the same regions of polarized growth in Saccharomyces cerevisiae. . J Cell Biol. 1994;125:825–842. doi: 10.1083/jcb.125.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, DeMarini DJ, Valencik ML, Al-Awar OS, Fares H, De Virgilio C, Pringle JR. The septins: roles in cytokinesis and other processes. Curr Opin Cell Biol. 1996;8:106–119. doi: 10.1016/s0955-0674(96)80054-8. [DOI] [PubMed] [Google Scholar]
- Lupas A, Van Dyke M, Stock J. Predicting coiled coils from protein sequences. Science (Wash DC) 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- McIntosh EM, Atkinson T, Storms RK, Smith M. Characterization of a short, cis-acting DNA sequence which conveys cell cycle stage- dependent transcription in Saccharomyces cerevisiae. . Mol Cell Biol. 1991;11:329–337. doi: 10.1128/mcb.11.1.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson J, Zheng Y, Bender L, Myers A, Cerione R, Bender A. Interactions between the bud emergence proteins Bem1p and Bem2p and Rho-type GTPases in yeast. J Cell Biol. 1994;127:1395–1406. doi: 10.1083/jcb.127.5.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priess JR. Establishment of initial asymmetry in early Caenorhabditis elegansembryos. Curr Opin Genet Dev. 1994;4:563–568. doi: 10.1016/0959-437x(94)90073-c. [DOI] [PubMed] [Google Scholar]
- Pringle JR. Staining of bud scars and other cell wall chitin with Calcofluor. Methods Enzymol. 1991;194:732–735. doi: 10.1016/0076-6879(91)94055-h. [DOI] [PubMed] [Google Scholar]
- Qadota H, Python CP, Inoue SB, Arisawa M, Anraku Y, Zheng Y, Watanabe T, Levin DE, Ohya Y. Identification of yeast Rho1p GTPase as a regulatory subunit of 1,3-beta-glucan synthase. Science (Wash DC) 1996;272:279–281. doi: 10.1126/science.272.5259.279. [DOI] [PubMed] [Google Scholar]
- Rhyu MS, Knoblich JA. Spindle orientation and asymmetric cell fate. Cell. 1995;82:523–526. doi: 10.1016/0092-8674(95)90022-5. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Boulan E, Nelson WJ. Morphogenesis of the polarized epithelial cell phenotype. Science (Wash DC) 1989;245:718–725. doi: 10.1126/science.2672330. [DOI] [PubMed] [Google Scholar]
- Roemer T, Madden K, Chang J, Snyder M. Selection of axial growth sites in yeast requres Axl2p, a novel plasma membrane glycoprotein. Genes & Dev. 1996;10:777–793. doi: 10.1101/gad.10.7.777. [DOI] [PubMed] [Google Scholar]
- Rose, M.D., F. Winston, and P. Hieter. 1991. Methods in Yeast Genetics: A Laboratory Course Manual. Cold Spring Harbor Press, Cold Spring Harbor, NY. 198 pp.
- Rothstein RJ. One-step gene disruption in yeast. Methods Enzymol. 1983;101:202–211. doi: 10.1016/0076-6879(83)01015-0. [DOI] [PubMed] [Google Scholar]
- Segall JE. Polarization of yeast cells in spatial gradients of alpha mating factor. Proc Natl Acad Sci USA. 1993;90:8332–8336. doi: 10.1073/pnas.90.18.8332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemering KR, Golbik R, Sever R, Haseloff J. Mutations that suppress the thermosensitivity of green fluorescent protein. Curr Biol. 1996;6:1653–1663. doi: 10.1016/s0960-9822(02)70789-6. [DOI] [PubMed] [Google Scholar]
- Sloat BG, Adams A, Pringle JR. Roles of CDC24 gene product in cellular morphogenesis during Saccharomyces cerevisiaecell cycle. J Cell Biol. 1981;89:395–405. doi: 10.1083/jcb.89.3.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder M. The SPA2 protein of yeast localizes to sites of cell growth. J Cell Biol. 1989;108:1419–1429. doi: 10.1083/jcb.108.4.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder M, Gehrung S, Page BD. Studies concerning the temporal and genetic control of cell polarity in Saccharomyces cerevisiae. . J Cell Biol. 1991;114:515–532. doi: 10.1083/jcb.114.3.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Johnston D, Nusslein-Volhard C. The origin of pattern and polarity in the Drosophilaembryo. Cell. 1992;68:201–219. doi: 10.1016/0092-8674(92)90466-p. [DOI] [PubMed] [Google Scholar]
- Tkacz JS, Lampen JO. Wall replication in Saccharomycesspecies: use of fluorescein-conjugated concanavalin A to reveal the site of mannan insertion. J Gen Microbiol. 1972;72:243–247. doi: 10.1099/00221287-72-2-243. [DOI] [PubMed] [Google Scholar]
- Valtz N, Herskowitz I. Pea2 protein of yeast is localized to sites of polarized growth and is required for efficient mating and bipolar budding. J Cell Biol. 1996;135:725–739. doi: 10.1083/jcb.135.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wach A, Brachat A, Poehlmann R, Philippsen P. New heterologous modules for classical or PRC-based gene disruptions in Saccharomyces cerevisiae. . Yeast. 1994;10:1793–1808. doi: 10.1002/yea.320101310. [DOI] [PubMed] [Google Scholar]
- Waddle JA, Karpova TS, Waterston RH, Cooper JA. Movement of cortical actin patches in yeast. J Cell Biol. 1996;132:861–870. doi: 10.1083/jcb.132.5.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Bretscher A. The rho-GAP encoded by BEM2 regulates cytoskeletal structure in budding yeast. Mol Biol Cell. 1995;6:1011–1024. doi: 10.1091/mbc.6.8.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JH, Barker DG, Nurse P, Johnston LH. Periodic transcription as a means of regulating gene expression during the cell cycle: contrasting modes of expression of DNA ligase genes in budding and fission yeast. EMBO (Eur Mol Biol Organ) J. 1986;5:1705–1709. doi: 10.1002/j.1460-2075.1986.tb04414.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, Guacci V, Koshland D. Pds1p is required for faithful execution of anaphase in the yeast, Saccharomyces cerevisiae. . J Cell Biol. 1996;133:85–97. doi: 10.1083/jcb.133.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamochi W, Tanaka K, Nonaka H, Maeda A, Musha T, Takai Y. Growth site localization of Rho1 small GTP-binding protein and its involvement in bud formation in Saccharomyces cerevisiae. . J Cell Biol. 1994;125:1077–1093. doi: 10.1083/jcb.125.5.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorihuzi T, Ohsumi Y. Saccharomyces cerevisiaeMATa mutant cells defective in pointed projection formation in response to alpha-factor at high concentrations. Yeast. 1994;10:579–594. doi: 10.1002/yea.320100503. [DOI] [PubMed] [Google Scholar]
- Zahner JE, Harkins HA, Pringle JR. Genetic analysis of the bipolar pattern of bud site selection in the yeast Saccharomyces cerevisiae. . Mol Cell Biol. 1996;16:1857–1870. doi: 10.1128/mcb.16.4.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Bender A, Cerione RA. Interactions among proteins involved in bud-site selection and bud-site assembly in Saccharomyces cerevisiae. . J Biol Chem. 1995;270:626–630. doi: 10.1074/jbc.270.2.626. [DOI] [PubMed] [Google Scholar]
- Ziman M, Preuss D, Mulholland J, O'Brien JM, Botstein D, Johnson DI. Subcellular localization of Cdc42p, a Saccharomyces cerevisiaeGTP-binding protein involved in the control of cell polarity. Mol Biol Cell. 1993;4:1307–1316. doi: 10.1091/mbc.4.12.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]