

Abstract
Little is known about the pathways used by cyclins and cyclin-dependent kinases to induce the events of the cell cycle. In budding yeast, a protein called Nap1 binds to the mitotic cyclin Clb2, and Nap1 is required for the ability of Clb2 to induce specific mitotic events, but the role played by Nap1 is unclear. We have used genetic and biochemical approaches to identify additional proteins that function with Nap1 in the control of mitotic events. These approaches have both identified a protein kinase called Gin4 that is required for the ability of Clb2 and Nap1 to promote the switch from polar to isotropic bud growth that normally occurs during mitosis. Gin4 is also required for the ability of Clb2 and Nap1 to promote normal progression through mitosis. The Gin4 protein becomes phosphorylated as cells enter mitosis, resulting in the activation of Gin4 kinase activity, and the phosphorylation of Gin4 is dependent upon Nap1 and Clb2 in vivo. Affinity chromatography experiments demonstrate that Gin4 binds tightly to Nap1, indicating that the functions of these two proteins are closely tied within the cell. These results demonstrate that the activation of Gin4 is under the control of Clb2 and Nap1, and they provide an important step towards elucidating the molecular pathways that link cyclin-dependent kinases to the events they control.
Cell division requires the precise coordination of an extraordinary number of events. Recent work has demonstrated that these events are induced and coordinated by two large families of proteins called cyclins and cyclin-dependent kinases (for reviews see King et al., 1994; Murray and Hunt, 1993; Norbury and Nurse, 1992). The cyclins appear at specific times during the cell cycle to bind and activate cyclin-dependent kinases, thereby inducing cell cycle events. Although much has been learned about how cyclin-dependent kinases are turned on and off during the cell cycle, we still know virtually nothing about the mechanisms used by these kinases to induce cell cycle events (Nigg, 1993). This problem is of particular interest in simple organisms like budding yeast, which uses a single cyclin-dependent kinase called p34CDC28 to induce both interphase and mitosis. This demonstrates that the same kinase is somehow able to induce different events when activated at different times. The mechanisms that make such specificity possible are not understood, and in no case do we know the pathway leading from activation of a cyclin-dependent kinase to the execution of a specific cell cycle event.
Since cyclin-dependent kinases are bound to different cyclins at each stage of the cell cycle, it seems likely that the cyclins somehow function to provide cyclin-dependent kinase complexes with specificity. To learn more about how cyclins might function in this capacity, we used affinity chromatography to identify proteins that interact with one kind of cyclin, but not with others. We reasoned that such proteins would be likely to play a role in the specific cell cycle events induced by the cyclin to which they bind (Kellogg et al., 1995 a; Kellogg and Murray, 1995). Using this approach, we found that members of the Nap/Set family of proteins interact specifically with mitotic cyclins in organisms as evolutionarily divergent as budding yeast and Xenopus. Further experiments in budding yeast demonstrated that one member of this family, a protein called Nap1, is required for the ability of the mitotic cyclin Clb2 to execute a subset of its normal mitotic activities. Without Nap1, Clb2 is unable to properly induce mitotic events even though it activates cyclin-dependent kinase activity to normal levels. These results indicate that Nap1 plays a role in the induction of mitotic events by an activated cyclin-dependent kinase complex, and they provide a starting point for understanding how cyclin-dependent kinase complexes induce specific cell cycle events.
We are interested in learning more about how the cyclin Clb2 and Nap1 function to control mitotic events. The specific mitotic event that we have chosen to study is a switch in the pattern of bud growth that occurs as yeast cells enter mitosis. When a yeast cell begins to divide, a new bud emerges from the mother cell during G1 phase and undergoes polar growth at its tip (Lew and Reed, 1995). As the cell enters mitosis, the Clb cyclins appear and induce a switch from polar bud growth to isotropic growth, so that the bud begins to grow over its entire surface. (Lew and Reed, 1993, 1995). Cells that lack Clb function fail to make this switch and continue polar bud growth during mitosis, giving rise to highly elongated buds (Amon et al., 1993; Lew and Reed, 1993; Richardson et al., 1992). Nap1 is required for the ability of Clb2 to induce this switch (Kellogg and Murray, 1995), demonstrating that it is an essential part of a pathway initiated by Clb2 that controls bud growth during mitosis.
Several factors make the pathway used by Clb2 and Nap1 to control bud growth ideally suited for genetic analysis. In our previous work with Nap1, we found that the proper control of bud growth during mitosis is not necessary for viability, so that cells with severe defects in this pathway can be maintained and studied (Kellogg et al., 1995 a; Kellogg and Murray, 1995). We also found that loss of bud growth control during mitosis causes cells to have characteristic elongated buds and to form unusual colonies that have a rough surface and an irregular shape. Mutations that disrupt this pathway can therefore be easily identified simply by screening for this unusual colony morphology.
Analysis of mitotic control in budding yeast is complicated by the fact that there are four functionally redundant cyclins that appear during mitosis, Clb1, Clb2, Clb3, and Clb4 (Fitch et al., 1992; Richardson et al., 1992). However, analysis of cyclin function can be simplified by deleting the genes for CLB1, CLB3, and CLB4, thereby creating a strain that is completely dependent upon CLB2 for survival. In previous studies, we used this CLB2-dependent strain background to study the role played by Nap1 in the induction of mitotic events by Clb2 (Kellogg and Murray, 1995). We found that the control of bud growth during mitosis in CLB2-dependent cells occurs through Clb2 and Nap1, whereas in wild-type cells there appear to be additional pathways that work through the other redundant Clb cyclins (Kellogg et al., 1995 a; Lew and Reed, 1993).
In this report, we describe the use of both genetic and biochemical approaches to identify and characterize additional proteins that function in the Nap1-dependent mitotic control pathway. Our experiments have identified a kinase called Gin4 that is specifically activated during mitosis and functions with Nap1 in the control of mitotic events.
Materials and Methods
Strains and Culture Conditions
Except where noted, all cells were grown in yeast/peptone/dextrose (YPD)1 media. All strains are in the W303 strain background (leu2-3,112 ura3-52 can1-100 ade2-1 his3-11 trp1-1), with the exception of the JB811 protease-deficient strain. The additional features of the strains used in this study are listed below. Strains carrying deletions of the CLB genes were derived from crosses between strains K2652 and K3080 (Amon et al., 1993; Fitch et al., 1992). Strain RSN31-8d was a gift from Robert Nash (University of California, San Francisco, CA).
DK177: Mata
RA4: Mata Δgin4::LEU2
DK131: Mata, Δnap1::LEU2
DK186: Mata Δbar1
DK166: Mata Δclb1 Δclb3::TRP1 Δclb4::HIS3
DK212: Mata Δbar1 Δclb1 Δclb3::TRP1 Δclb4::HIS3
DK213: Mata Δnap1::LEU2 Δclb1 Δclb3::TRP1 Δclb4::HIS3 Δbar1
DK214: Mata Δgin4::LEU2 Δclb1 Δclb3::TRP1 Δclb4::HIS3 Δbar1
DK216: Mata gin4K48A Δclb1 Δclb3::TRP1 Δclb4::HIS3 Δbar1
DK247: Mata Δbar1 ura::gal-Clb2Δ176
RSN31-8d: Mata Δbar1 cdc28-4
RA16: Mata Δgin4::LEU2 Δnap1::LEU2 Δclb1::URA3 Δclb3::TRP1 Δclb4::HIS3
JB811: Mata prb1 pep4-3 trp1 leu2-3,112 ura3-52
Mutagenesis and Screening
Mutagenesis with ethylmethane sulfate was carried out as previously described using strain DK166 (Lawrence, 1991). After mutagenesis, cells were plated on YPD media at a density of 2,000 colonies per 100-mm plate and were grown at 30°C for 2 d before screening colonies with a dissecting microscope.
Cloning and Deletion of the GIN4 Gene
To demonstrate that the GIN4 gene alone can rescue the ecm1 mutant phenotype, we used PCR to amplify and clone the GIN4 gene into YCplac111 to create pRA1 (oligos: GCGCAAGCTTGGAGTTTATTCATTCCCGCT and CGCGGTACCCTGTTACATAATTTATGTTTA). To further confirm that GIN4 is the rescuing gene, we deleted all sequences upstream of the BamH1 site found 120 bases downstream of the starting ATG in the GIN4 coding region in pRA1.
To generate a deletion of the GIN4 gene in the yeast genome, the GIN4 gene was amplified by PCR and cloned into the KpnI and HindIII sites of the bluescript cloning vector (oligos: GCGCAAGCTTGGAGTTTATTCATTCCCGCT and CGCGGTACCCTGTTACATAATTTATGTTTA). This vector was then used as a template for a second PCR reaction that amplified the bluescript vector with regions of homology to the GIN4 gene on each end (oligos: GCGCGCGGGCCCCAATGAATTGCGTAAACAGAA and GCGCGCCCTAGGTGTATTGTGCTGCGAATATCT). A fragment carrying the LEU2 gene was then ligated into this PCR product to produce a vector carrying the LEU2 gene flanked by sequences homologous to the 5′ and 3′ ends of the GIN4 gene (pRA5). A linearized fragment carrying the disrupted GIN4 gene was transformed into yeast, and disruptions of the GIN4 gene were confirmed both by PCR and by Western blotting with antibodies that recognize the COOH terminus.
Generation of Antibodies that Recognize Gin4
Antibodies that recognize Gin4 were raised by immunizing rabbits with a COOH-terminal fragment of Gin4, purified from bacteria as a 6X-histidine fusion protein. The COOH-terminal fragment was amplified by PCR and cloned into the vector pQE10 (QIAGEN, Inc., Chatsworth, CA; PCR primers: GCGGGATCCCAGTTCGAACGATGAGAGATT and GCGCAAGCTTCTATTTTTGTAGAACGCCTT). Antibodies were affinity purified from the serum using the Gin4 fusion protein as previously described (Kellogg and Alberts, 1992). The antibodies do not recognize any proteins in extracts from strains carrying a deletion of the GIN4 gene.
Cell Cycle Arrests and Immunofluorescence Methods
For all experiments, strains that are BAR1 were arrested with 20 μg/ml α-factor, whereas bar1 − strains were arrested with 1 μg/ml α-factor. Arrest with α factor was generally carried out for 3–3.5 h at room temperature, with the exception of the experiment shown in Fig. 3, which was carried out for 5 h at 30°C. Cells were arrested in mitosis by incubation in 30 μg/ml benomyl for 3 h at room temperature, as previously described (Li and Murray, 1991). Staining of mitotic spindles was carried out as previously described (Pringle et al., 1991).
Figure 3.

Deletion of the GIN4 gene causes a prolonged mitotic arrest in cells that are dependent upon Clb2 for survival. Cells were grown overnight at 30°C until they reached an OD of 0.5 (control cells) or 0.8 (Δgin4, Δclb1,3,4, Δbar1 cells), and α-factor was then added to 2 μg/ml at t = 0. At each time point, 1.5 ml of culture was removed and analyzed for Clb2 levels or for the presence of mitotic spindles, as previously described (Kellogg and Murray, 1995; Pringle et al., 1991). The strains used in this experiment carry deletions of the BAR1 gene to prevent them from breaking through the α-factor arrest. (A) A plot of the percentage of cells with a mitotic spindle as a function of time after the addition of α-factor to log phase cultures of a Δgin4, Δclb1,3,4, Δbar1 strain and a Δclb1,3,4, Δbar1 control strain. The percentage of cells with mitotic spindles was determined by counting spindles in random fields of cells. Over 200 cells were counted for each data point. After 3 h, the spindles began to appear unusually thick and bent, and it became difficult to accurately count the number of cells with spindles. (B) A Western blot showing the amount of Clb2 present as a function of time after the addition of α-factor to log phase cultures of the same strains shown in A. (C) Examples of the short spindles observed in the Δgin4, Δclb1,3,4, Δbar1 strain 2 h after addition of α-factor to a log phase culture.
Affinity Purification of Nap1-binding Proteins
We affinity purify Nap1-binding proteins using essentially the same protocol that we used to purify cyclin-binding proteins (Kellogg et al., 1995 a). Briefly, Nap1 is expressed and purified from Escherichia coli as a glutathione-S-transferase fusion, and is then coupled to Affigel 10 beads (Bio Rad Laboratories, Hercules, CA) to make an affinity column matrix. An extract is made from 7.5 g of log phase JB811 protease-deficient cells in extract buffer (50 mM K+-Hepes, pH 7.6, 275 mM KCl, 1 mM MgCl2, 1 mM EGTA, 0.15% Tween-20, 1 mM PMSF, 1 mM leupeptin, 1 mM chymostatin, 1 mM pepstatin, 1 mM DTT). The cells are broken open by extensive grinding under liquid nitrogen using a mortar and pestle. The crude extract is centrifuged at 20,000 g for 10 min, followed by 100,000 g for 90 min. The supernatant from the first spin is labeled “LS Supernatant” in Fig. 5, and the supernatant from the second spin is labeled “HS Supernatant”. The final supernatant is first passed through a 5-ml precolumn filter made by coupling BSA to Affigel 10, and the flowthrough from the precolumn is loaded directly onto a 2-ml affinity column containing 6 mg of Nap1. After loading, the column is washed with 30 ml of extract buffer containing 10% glycerol, 0.05% Tween-20, and no PMSF. The column is eluted with a salt gradient going from 0.3 M to 1.0 M KCl. The gradient is made by pipetting 400-μl aliquots of elution buffer onto the top of the column, with each aliquot increasing in KCl concentration by 50 mM. During elution, 400-μl fractions are collected. The fractions are concentrated by TCA precipitation, resuspended in 1× protein gel sample buffer, and 1/10 of each fraction is loaded onto each lane of an SDS-polyacrylamide gel.
Figure 5.

Affinity purification of Nap1-binding proteins. (A) Crude extracts from log phase yeast cells were loaded onto a Nap1 affinity column. After washing with buffer, the column was eluted with a gradient of KCl, and samples from each fraction were precipitated with TCA and loaded onto a 12.5% SDS-polyacrylamide gel. The first few fractions before the start of the salt gradient show the final fractions of the wash. The gel is stained with Coomassie blue. (B) The same fractions shown in A were loaded onto a 10% SDS-polyacrylamide gel and transferred to nitrocellulose, which was then probed with an anti-Gin4 antibody. For the affinity column elution fractions we loaded 1/10 the amount of protein that was loaded onto the gel shown in A.
Coimmunoprecipitation of Gin4 and Nap1
Immunoaffinity beads for the precipitation of Gin4 are made by cross-linking anti-Gin4 antibody to protein A beads as previously described, using 1 μg of affinity-purified antibody for each microliter of protein A beads (Kellogg et al., 1995 b). 1.5-liter cultures of strains DK186 and RA4 are grown to an OD of 0.7 and the cells are pelleted and frozen on liquid nitrogen. The cells are then broken open by extensive grinding under liquid nitrogen as previously described (Kellogg et al., 1995 a), and approximately 0.3-g aliquots of the frozen cell powder are stored at −80°C. To make a cell extract, 400 μl of IP buffer (50 mM K+-Hepes, pH 7.6, 400 mM NaCl, 1 mM EGTA, 1 mM MgCl2, 0.1% Tween-20, 2 μg/ml leupeptin, 2 μg/ml pepstatin, 2 μg/ml chymostatin, 2 mM PMSF, 10% glycerol) is added to an aliquot of cells from each strain. After a 5-min incubation on ice, the extract is centrifuged for 10 min in a microfuge at 4°C, and 450 μl of the supernatant from each strain is added to a tube containing 40–50 μl of anti-Gin4 beads. The tubes are gently mixed end over end at 4°C for 1.5 h, and the beads are then washed three times with 500 μl of IP buffer. At the end of these washes, the beads from each strain are equally split into two new tubes, and one set of tubes from each strain is washed three more times with IP buffer, while the other set is washed in IP buffer containing 1.0 M NaCl. At the end of the washes, 100 μl of 1× protein gel sample buffer is added to each tube and the tubes are placed in a boiling water bath for 5 min to release proteins from the beads. For Western blots, 10 μl of each sample are loaded onto a 10% SDS-polyacrylamide gel.
Construction of gin4K48A Strains
A 4.5-kb fragment containing the GIN4 gene was amplified by PCR and cloned into the KpnI and HindIII sites of the integrating vector YIplac211 to create plasmid pDK46 (PCR primers: GCGCAAGCTTGGAGTTTATTCATTCCCGCT and CGCGGTACCCTGTTACATAATTTATGTTTA). A BamHI site lies 21 bases away from the codon for the conserved lysine in subdomain II of the kinase domain (Hanks and Quinn, 1991). A PCR primer was made that incorporates this BamHI site and changes the conserved lysine to an alanine (AATGGATCCACAGGACAAGAGGCGGCAGTTGCGGTAATATCAAAAGCAG). A second primer was made that includes a HpaI site that occurs 683 bases downstream of the BamHI site, and these two primers were used to amplify a fragment that carries the kinase domain mutation, which was then used to replace the wild-type fragment in pDK46 to create pDK49. This plasmid was cut with HpaI to target integration at GIN4 in strains DK186 or DK166, creating a duplication of the GIN4 gene, with one mutant copy and one wild-type copy. Recombination events between these two copies were selected by plating cells on 5-fluoroorotic acid, and events that leave the kinase domain mutation in the genome were identified by looking for the altered cellular morphology characteristic of mutant gin4 strains. The phenotype of the gin4K48A, Δclb1,3,4 mutation is not quite as severe as the Δgin4, Δclb1 ,3,4 phenotype, perhaps because of the presence of residual kinase activity.
Gin4 Kinase Assays
To measure Gin4 kinase activity during the cell cycle, 650 ml of strains DK212 and DK213 cells are grown overnight at room temperature to ODs of 0.8 and 0.85, respectively. α-Factor is added to 1 μg/ml, and the cells are incubated at room temperature for 4 h. The α-factor is removed by washing the cells once with 500 ml of YPD media prewarmed to 30°C, followed by two washes with 50 ml of prewarmed media. At each time point after release from arrest, 50 ml of culture is removed and the cells are pelleted at 3,000 rpm for 2 min. The cells are resuspended in 1 ml YPD, transferred to a 1.8-ml screw-top tube, and then pelleted by centrifugation for 1 min in a microfuge. After removal of the supernatant, the tube containing the cell pellet is frozen in liquid nitrogen. To each tube containing frozen cells, 500 μl of acid-washed glass beads (0.5 mm diam; Biospec Products, Inc., Bartlesville, OK) is added, followed by 300 μl of ice-cold Lysis Buffer (50 mM K+-Hepes, pH 7.6, 1M NaCl, 1 mM EGTA, 1 mM MgCl2, 0.1% Tween-20, 2 μg/ml leupeptin, 2 μg/ml pepstatin, 2 μg/ml chymotrypsin, 2 mM PMSF, 10% glycerol), and the tubes are immediately placed in a Biospec Multibeater-8 and beaten at top speed for 25 s. The tubes are removed and cooled in an ice water bath before a 5-min spin in a microfuge. 300 μl of supernatant is removed and replaced with 200 μl of fresh buffer and the tubes are beaten again for 25 s. The supernatants are pooled and centrifuged again for 5 min. 25 μl is removed from each sample and used for Clb2-associated kinase assays as previously described (Kellogg and Murray, 1995).
Anti-Gin4 beads are prepared by binding affinity-purified anti-Gin4 antibody to protein A beads (Bio Rad Laboratories) in PBS for several hours (5 μg antibody/20 μl beads). The beads are then washed several times with lysis buffer (without PMSF), and 400 μl of each lysate is added to 20 μl of protein A beads in 0.6-ml tubes. The suspension is mixed for 1 h on a rotator at 4°C. The beads are then washed three times in lysis buffer, and three times in kinase buffer (50 mM K+-Hepes, pH 7.6, 1 mM EGTA, 2 mM MgCl2, 0.1% Tween-20, 10% glycerol) with a transfer to fresh tubes after the fifth wash. After the final wash, the supernatant is completely removed and 20 μl of assay buffer is added (kinase buffer containing 1 mM DTT, 0.25 mM ATP, 0.1 mCi/ml [γ32P]ATP, 50 μg/ml histone H1, 2 μg/ml leupeptin, 2 μg/ml pepstatin, 2 μg/ml chymotrypsin). The tubes are vortexed gently and incubated at 30°C for 30 min, with gentle mixing every 10 min. The reaction is stopped by the addition of 10 μl of 4× protein gel sample buffer, and 10 μl is loaded onto each lane of a 15% SDS-polyacrylamide gel. For Western blotting, 10 μl of the lysate used for immunoprecipitation is diluted into 90 μl of 1× sample buffer and incubated in a boiling water bath for 3 min, and 10 μl of each sample is loaded onto an SDS-polyacrylamide gel. The assays shown in Figs. 7 and 9 were carried out the same as above, except that we used cells arrested either in mitosis with benomyl, or in interphase with α-factor.
Figure 7.
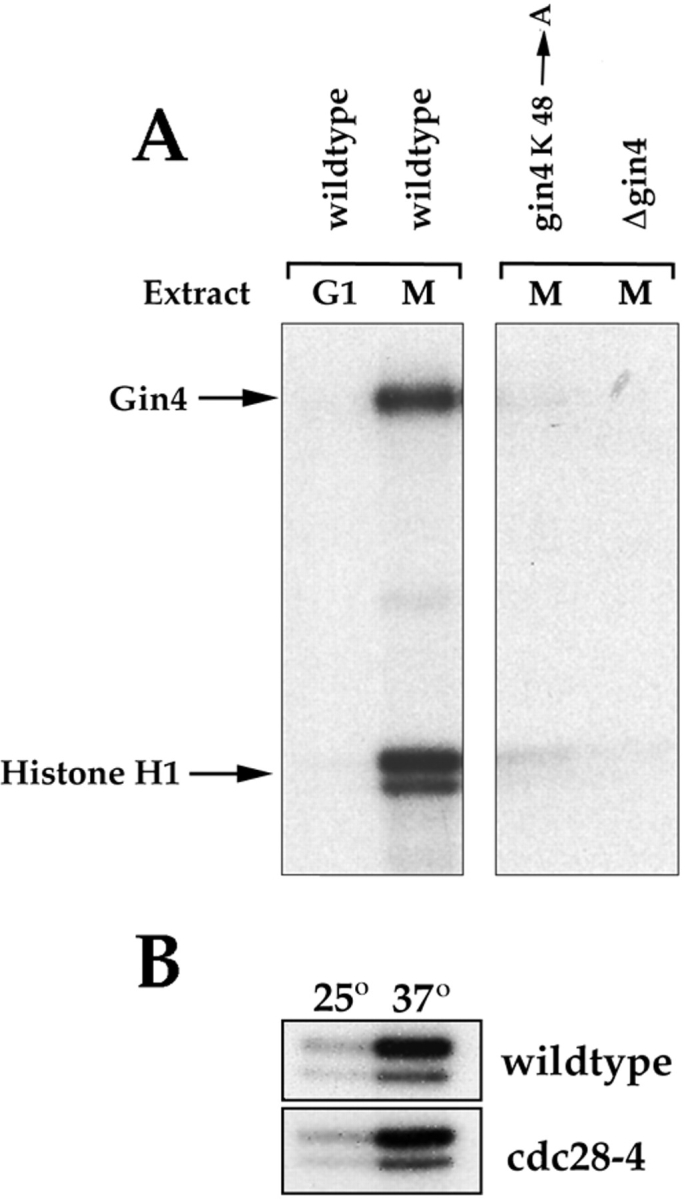
Gin4 from mitotic cells phosphorylates histone H1 and undergoes autophosphorylation in vitro. (A) Cells from the indicated strains were arrested in G1 with α-factor or in mitosis with benomyl. Extracts were then made from the arrested cells and Gin4 was immunoprecipitated and assayed for kinase activity using histone H1 as a substrate. (B) A control showing that the kinase activity present in Gin4 immunoprecipitates is not due to Cdc28. Wild-type or cdc28-4 cells were grown at 22°C and arrested in mitosis with benomyl. Gin4 was then immunoprecipitated from the arrested cells and kinase activity was measured either at 22°C or 37°C.
Figure 9.

The kinase activity of Gin4 is activated by phosphorylation. (A) The Gin4 protein was immunoprecipitated from mitotic cells and then split into two samples. One sample was treated with lambda phosphatase (New England Biolabs Inc.), while the other sample was treated identically, but with no added phosphatase. The precipitated protein was then resolved on a 9% SDS-polyacrylamide gel and detected by Western blotting. (B) The Gin4 protein was immunoprecipitated and treated with lambda phosphatase as in A. The immunoprecipitate was then washed with kinase buffer several times and assayed for kinase activity.
To measure Gin4-associated kinase activity in the cdc28-4 mutant background we carried out the same assay described above using log phase cultures of strains RSN31-8d and DK186. After the fifth wash, the immunoprecipitates from each strain are divided into two identical aliquots. After the sixth wash, the supernatant is removed and one aliquot of beads from each strain is preincubated at 37°C for 15 min. 40 μl of kinase assay buffer is warmed to 37°C before adding 20 μl to each sample of beads, and the tubes are vortexed gently and incubated at 37°C for 30 min, with gentle mixing every 10 min. The identical procedure is carried out at 25°C using the other aliquots of beads from each strain.
Treatment of Gin4 with Phosphatase
For experiments involved in treatment of Gin4 with phosphatase, Gin4 is immunoprecipitated from DK186 cells arrested in mitosis using the same procedure described above for coimmunoprecipitation of Gin4 and Nap1, except that the IP buffer contains 1.0 M NaCl. The anti-Gin4 beads are washed three times with IP buffer followed by two times with phosphatase buffer (50 mM Tris-HCl, pH 7.5, 5 mM DTT, 2 mM MnCl2, 100 μg/ml BSA). After the fourth wash, the beads are split into two identical aliquots, and after the final wash, the supernatant is removed leaving a total volume of 25 μl in each tube. 1.5 μl of lambda phosphatase (New England Biolabs Inc., Beverly, MA) is added to one tube and both tubes are incubated at 30°C for 30 min. The beads are then washed once in IP buffer containing 1.0 M NaCl and twice in kinase buffer before adding 15 μl of assay buffer. The tubes are mixed gently and incubated at 30°C for 30 min, with gentle mixing every 10 min. The reaction is stopped by the addition of 20 μl of 2× sample buffer. 15 μl is loaded onto a 15% SDS-polyacrylamide gel for the kinase assay, and 2 μl is loaded onto a 9% gel for a Gin4 Western blot. To ensure that all phosphatase was washed away before the kinase assay, we carried out controls in which we mixed phosphatase-treated Gin4 beads with untreated Gin4 beads, and observed no inhibition of Gin4 kinase activity (not shown).
Expression of Clb2Δ 176 in Interphase Cells
To induce expression of Clb2Δ176 in cells arrested in interphase, a 30-ml culture of strain DK247 is grown overnight to an OD of 0.65 in yeast extract peptone (YEP) media containing 2% raffinose. The cells are arrested in interphase by the addition of 1.5 μg/ml α-factor followed by incubation at 30°C for 3 h. The culture is divided in half, and the cells are pelleted and resuspended in the same volume of YEP media containing 1.5 μg/ml α-factor and either galactose or raffinose. At each time point after addition of galactose, 1.6-ml samples of each culture are taken and probed for the Gin4 protein by Western blotting, as described below.
PAGE and Western Blotting
PAGE and Western blotting are carried out as previously described (Anderson et al., 1973; Harlow and Lane, 1988). For the experiments shown in Figs. 3, 10, and 11, 1.6-ml samples of culture are taken at each of the indicated time points. The cells are then rapidly pelleted in a 1.8-ml screw-top tube, the supernatant is removed, and the tube is frozen on liquid nitrogen. After all of the samples have been collected, 300 μl of glass beads are added to each tube, followed by 125 μl of 1× protein gel sample buffer containing 2 mM PMSF, 2 μg/ml leupeptin, 2 μg/ml pepstatin, 2 μg/ml chymotrypsin. The tubes are immediately placed in a Biospec Multibeater-8 and beaten at top speed for 90 s, centrifuged briefly, and immediately incubated in a boiling water bath for 5 min. After centrifugation in a microfuge for 3 min, 10 μl of each sample is loaded onto gels for Western blotting. To see the phosphorylation-induced shift in the electrophoretic mobility of Gin4, samples are electrophoresed for 2.5 h at 170 V on 9% SDS-polyacrylamide gels.
Figure 10.

The Gin4 protein appears to undergo autophosphorylation in vivo. gin4K48A, Δclb1,3,4 cells and Δclb1,3,4 control cells were released from an α-factor arrest, and samples were taken at the indicated time points and probed for the Gin4 protein by Western blotting.
Figure 11.

Expression of Clb2 in cells arrested in interphase leads to phosphorylation of Gin4. A bar1 strain carrying an integrated copy of Clb2Δ176 under the control of the gal1 promoter was grown in raffinose and arrested in interphase by incubation in the presence of α-factor for 3 h. The culture was then divided in half, and Clb2Δ176 expression was induced in one half by transferring the cells to media containing galactose and α-factor. At the times indicated, samples were taken from each culture and used for an anti-Gin4 Western blot.
Figure 1.

An example of the cellular morphology observed for the ecm1 complementation group.
Results
Identification of Mutations That Disrupt the Mitotic Control of Bud Growth
In our previous work with Nap1, we found that cells with defects in the switch from polar to isotropic bud growth have a characteristic elongated bud morphology and form colonies that have a rough surface. To identify mutations in genes that are involved in the control of bud growth during mitosis, we mutagenized cells with ethylmethane sulfate to 80% lethality and screened through 80,000 colonies using a dissecting microscope to look for rough colonies. As a secondary screen, we examined the cells from these colonies under a microscope to determine which ones have the elongated bud morphology that is characteristic of a defect in the control of bud growth during mitosis. Since we wanted to identify mutations that affect the pathway used by Clb2 to control bud growth, we carried out our genetic screen in a strain carrying deletions of the genes for the redundant cyclins CLB1, CLB3, and CLB4. Throughout this paper we refer to this strain as a CLB2-dependent strain, or Δclb1,3,4.
We identified 40 mutations and subjected these to backcrossing and complementation analysis. We found 6 alleles of nap1, 12 alleles of a gene we named ecm1, for elongated cell morphology, and 5 alleles of a gene we named ecm2. Examples of the cellular and colony morphology observed for the ecm1 complementation group are shown in Fig. 1. 10 of the original mutations either lost their phenotype upon backcrossing or were judged to have a phenotype that is too weak for further characterization at this time. Analysis of the remaining seven mutations is in progress.
We cloned the gene corresponding to the ecm1 complementation group by transforming a mutant strain with a low copy genomic library to find plasmids that rescue the rough colony phenotype. We identified a plasmid carrying a genomic fragment that completely rescues the ecm1 phenotype, and found two open reading frames within the fragment. One open reading frame encoded a protein with homology to metabolic enzymes, whereas the other encoded a previously identified protein that causes morphological abnormalities and inhibits the growth of cells when its COOH-terminal end is overexpressed (growth inhibitory gene 4, GIN4; these sequence data are available from EMBL/GenBank/DDBJ under accession number D28142). To test whether GIN4 represents the rescuing gene, we used PCR to amplify the GIN4 gene by itself, and cloned it into a centromere-containing vector. The resulting plasmid gave complete rescue of the ecm1 mutant phenotype. As a further test, we showed that deletion of the 5′ end of the GIN4 gene completely eliminated the ability of this plasmid to rescue the ecm1 phenotype. To demonstrate that the GIN4 gene actually corresponds to ecm1, we integrated a marker near the GIN4 gene and showed that it segregated with the ecm1 mutation in 32 spores. Finally, we found that deletion of the GIN4 gene causes a phenotype that is indistinguishable from the phenotype of the original ecm1 mutant alleles (see below).
The GIN4 gene encodes a protein of 1,143 amino acids that has strong homology to serine/threonine protein kinases. The kinase domain of Gin4 is located within the NH2-terminal 300 amino acids and is most homologous to the kinase domains found in members of the SNF1 and nim1 protein kinase families.
Gin4 Is Required for the Proper Control of Bud Growth
As a first step towards learning more about the function of the Gin4 protein, we generated a deletion of the GIN4 gene. We were particularly interested in comparing the phenotype of Δgin4 cells with the phenotype of Δnap1 cells as a means of determining whether these two proteins are involved in similar functions within the cell. An important aspect of the Δnap1 phenotype is that it is most severe in cells that are dependent upon CLB2 for survival (Kellogg et al., 1995 a). To determine whether the same is true for GIN4, we deleted the GIN4 gene in a diploid strain heterozygous for deletions of the CLB1, CLB3, and CLB4 genes. Sporulation of this diploid generated all possible haploid combinations of the GIN4 deletion and the CLB deletions. We found that deletion of the GIN4 gene in a wild-type background causes a mild elongated bud phenotype and some cell clumping, although colonies form at a normal rate and have a smooth appearance (Fig. 2 B). This is similar to the phenotype seen for a NAP1 deletion in a wild-type background (Kellogg et al., 1995 a). Deletion of the GIN4 gene in a CLB2-dependent background (Δgin4, Δclb1,3,4) causes a severe phenotype that is indistinguishable from the phenotype of the original ecm1 mutant alleles that we identified in our genetic screen (Fig. 2 C). The cells grow as large interconnected clumps and have a highly elongated bud morphology that is identical to the morphology observed for a NAP1 deletion in the same genetic background (Fig. 2 D). The phenotype of the Δgin4, Δclb1,3,4 strain is also similar to the Δnap1, Δclb1,3,4 strain in that the cells grow very slowly at 30°C, and wild-type copies of CLB1 or CLB3 partially rescue the elongated bud phenotype, whereas wild-type CLB4 has no effect. The bud growth phenotypes of the strains described above are summarized in Table I.
Figure 2.

Deletion of the GIN4 gene causes cells to have an elongated bud morphology. Cells were grown to log phase in YPD liquid media at 30°C, and were photographed using a ×100 objective with Nomarski optics. The genotype of each strain is indicated above each picture.
Table I.
Summary of Elongated Bud Phenotypes
| Genotype | Elongated bud phenotype | |
|---|---|---|
| Wild type | − | |
| Δgin4 | + | |
| Δnap1 | + | |
| Δclb1, Δclb3, Δclb4 | −/+ | |
| Δclb1, Δclb3, Δclb4, Δgin4 | +++++ | |
| Δclb1, Δclb3, Δclb4, Δnap1 | +++++ | |
| Δclb1, Δclb3, Δgin4 | +++++ | |
| Δclb1, Δclb4, Δgin4 | ++ | |
| Δclb3, Δclb4, Δgin4 | ++ | |
| Δclb1, Δclb3, Δclb4, Δgin4, Δnap1 | +++++ | |
| Δgin4, Δnap1 | +++ |
The bud growth phenotype for each strain is judged by the relative length of the buds. The severity of the elongated bud phenotype is indicated by the number of plus signs.
These results demonstrate that both Gin4 and Nap1 are required for the repression of polar bud growth. Since the loss of either Gin4 or Nap1 in the Clb2-dependent background can be largely rescued by the presence of the other mitotic Clb cyclins, we can argue that Gin4 and Nap1 are mediating the functions of Clb2. This result also argues that the other Clbs are able to control bud growth through alternative pathways. We found that deletion of the genes for both NAP1 and GIN4 in a CLB2-dependent background causes a phenotype that is no different from either single deletion, further arguing that Gin4 and Nap1 work together. However, deletion of the genes for both NAP1 and GIN4 in a wild-type background causes an elongated bud phenotype that is more severe than either single deletion (see Table I). This result argues that Clb2 may not be the only mitotic cyclin that exerts its effects through Gin4 and Nap1. For example, the other Clb cyclins might control bud growth through alternative pathways that also work through Gin4 or Nap1. In this case, deletion of the genes for both GIN4 and NAP1 in a wild-type background could affect the function of more than one pathway.
Δgin4, Δclb1,3,4 Cells Undergo a Prolonged Mitotic Delay
To further test whether or not Gin4 and Nap1 are involved in similar functions, we determined whether Δgin4, Δclb1,3,4 cells undergo a mitotic delay in a manner similar to Δnap1, Δclb1,3,4 cells. In our previous studies we found that Δnap1, Δclb1,3,4 cells proceed through the cell cycle normally until they enter mitosis, at which point they undergo a prolonged delay at the short spindle stage with high Clb2 protein levels (Kellogg and Murray, 1995). To determine whether Δgin4, Δclb1,3,4 cells undergo a similar mitotic delay, we attempted to use α-factor to synchronize cells in G1. However, we found that even after 5 h in the presence of α-factor, many Δgin4, Δclb1,3,4 cells still have mitotic spindles and the Clb2 protein is still present, suggesting that cells are arrested in mitosis. This was true even in strains carrying a deletion of the BAR1 gene to prevent cells from breaking through the α-factor arrest. To study this mitotic arrest more carefully, we added α-factor to log phase cultures of Δgin4, Δclb1,3,4, Δbar1 cells, and to Δclb1,3,4, Δbar1 control cells, and then carried out assays every 30 min to measure Clb2 protein levels and the fraction of cells with a mitotic spindle (Figs. 3, A and B). After 90 min of exposure to α-factor, the control cells become synchronously arrested in G1 with no spindles and no Clb2 protein, as expected. In contrast, the Clb2 protein levels in Δgin4, Δclb1,3,4, Δbar1 cells do not start to fall until 30–60 min after the control strain, and then remain nearly constant for the remainder of the 5-h time course. Similarly, the fraction of cells with mitotic spindles does not start to fall until 30–60 min after the control strain, and a significant number of cells still have spindles after 3 h. These results suggest that some of the cells are able to exit mitosis after a delay of 30–60 min, while other cells are unable to exit mitosis even after 5 h. The majority of the cells appear to be arrested at the short spindle stage, which is the same stage at which Δnap1, Δclb1,3,4 cells arrest (Fig. 3 C; and Kellogg and Murray, 1995). Many of the cells in the Δgin4, Δclb1,3,4 strain have a dark and condensed appearance, suggesting that they are dead, which may be a consequence of the prolonged mitotic arrest. These observations are likely to explain the extremely slow growth rate of the Δgin4, Δclb1,3,4 cells.
Gin4 Is Not Required for the Activation of Cyclin-dependent Kinase Activity by Clb2
The previous experiments demonstrate that Gin4 is required for the ability of Clb2 to promote normal mitotic progression. A possible explanation for these results is that Gin4 is required for the formation of active Clb2/ p34CDC28 kinase complexes. In the case of Nap1, we were able to rule out this possibility by assaying the kinase activity of Clb2/p34CDC28 kinase complexes during the cell cycle in Δnap1 cells (Kellogg and Murray, 1995). To determine whether the same is true for Gin4, we assayed the activity of Clb2/p34CDC28 kinase complexes during the cell cycle in Δgin4 cells. We found that Clb2-associated kinase activity rises to normal levels during mitosis in Δgin4 cells, and that the kinase activity peaks 10 min later than in the control cells (Fig. 4). These results are similar to those obtained for Δnap1 cells (Kellogg and Murray, 1995). We were unable to obtain clean results when we carried out the same experiment in Δgin4, Δclb1,3,4 cells, due to the fact that this strain cannot be completely synchronized with α-factor (Fig. 3). However, the results that we were able to obtain using partially synchronized cultures agreed with the results obtained using the Δgin4 cells (not shown). Therefore, neither Nap1 nor Gin4 are required for activation of cyclin-dependent kinase activity by Clb2. The delay in the activation of kinase activity may be due to a defect in the pathway used by the cyclin-dependent kinase complex to stimulate its own activation (King et al., 1994).
Figure 4.

Gin4 is not required for the activation of cyclin-dependent kinase activity by Clb2. (A) A time course showing the appearance of Clb2-associated kinase activity during mitosis. Wild-type and Δgin4 cells were released from an α-factor arrest and then assayed for Clb2-associated kinase activity during the cell cycle as previously described (Kellogg and Murray, 1995). (B) The same samples used for the kinase assay in A were probed for Clb2 levels by Western blotting as previously described (Kellogg and Murray, 1995).
Gin4 Binds to Nap1
The experiments described above demonstrate that the GIN4 deletion and the NAP1 deletion have nearly identical phenotypes. These results provide strong support for the idea that Gin4 and Nap1 are involved in similar functions within the cell. Nap1 affinity chromatography experiments provide additional support for a close functional interaction between Gin4 and Nap1. For these experiments, we obtain purified Nap1 using an E. coli expression system, and then couple the Nap1 to a column matrix. Crude extracts from log phase yeast cells are loaded onto the affinity column, which is then washed with buffer and eluted with a gradient of 0.35–1 M KCl. We find that a number of different proteins bind to Nap1 (Fig. 5 A). These results are striking because the crude cell extract is made in a buffer that contains 0.275 M salt, and the column is washed with 15 column volumes of buffer containing the same salt concentration. The fact that many proteins remain bound to the column under these stringent wash conditions suggests that they bind to Nap1 with relatively high affinity, and are likely to represent specific interactions. For the present, however, we have focused our attention on the proteins that elute from Nap1 with 0.8–1.0 M KCl, since these are the most tightly bound and are therefore the most likely to be directly involved in Nap1 function. We noticed that one of these proteins is approximately the size that would be predicted for Gin4 (Fig. 5 A, arrow). The sequence of a tryptic peptide obtained from this protein identified it as Gin4, and Western blotting with anti-Gin4 antibodies provided further confirmation that it is the Gin4 protein (Fig. 5 B). Note that the Gin4 protein is quantitatively depleted from the extract as it passes over the Nap1 affinity column, suggesting a tight and specific interaction between Nap1 and Gin4. We also carried out immunoprecipitation experiments using anti-Gin4 antibodies to demonstrate that the endogenous Nap1 protein coprecipitates with the endogenous Gin4 protein in crude extracts (Fig. 6). We found that the interaction between Gin4 and Nap1 in crude extracts is stable to repeated washes with 0.4 M NaCl, and is disrupted by 1.0 M NaCl, as expected from the affinity column results. These results demonstrate that Gin4 binds to Nap1 with high affinity, and that these two proteins are likely to function together within the cell as a protein complex.
Figure 6.

Nap1 coprecipitates with Gin4 in crude cell extracts. Crude cell extracts were made from either wild-type cells or Δgin4 control cells, and the extracts were then used for Gin4 immunoprecipitations. The Gin4 immunoprecipitates were washed either with buffer containing 0.4 M NaCl or 1.0 M NaCl, and were then probed with anti-Nap1 antibody to detect coprecipitated Nap1.
In Western blotting experiments, we are unable to detect Clb2 in the fractions that elute from the Nap1 affinity column. This is not surprising, however, since in previous experiments we found that Nap1 can be quantitatively eluted from a Clb2 affinity column with 0.35 M salt (Kellogg et al., 1995 a). In these experiments, the Nap1 affinity column is washed with 15 column volumes of buffer containing 0.275 M salt before elution, which would be likely to wash off the Clb2 protein.
Nap1-dependent Activation of Gin4
The phenotype of Δgin4 cells and the fact that Gin4 binds tightly to Nap1 both suggest that Gin4 functions during mitosis, and that Nap1 and Gin4 are involved in similar functions. To learn more about when Gin4 functions, we developed an assay that would allow us to follow the kinase activity of Gin4 during the cell cycle. We immunoprecipitated Gin4 from cells arrested in interphase or mitosis, and then assayed the precipitated Gin4 for kinase activity using histone H1, myelin basic protein, or casein as test substrates (Fig. 7 A). We found that Gin4 from mitotic cells is able to phosphorylate histone H1, while Gin4 from interphase cells has little detectable kinase activity. Gin4 is also able to phosphorylate myelin basic protein (not shown). As controls, we carried out identical assays using mitotic extracts from cells that carry either a deletion of the GIN4 gene, or a point mutation in the Gin4 kinase domain (lysine 48 changed to alanine). Neither of these controls show any histone H1 kinase activity (Fig. 7 A). In addition to phosphorylation of histone H1 in these assays, we also detect phosphorylation of a protein that is the same size as Gin4. Phosphorylation of this protein is not detected in Δgin4 or in gin4K48A cells, suggesting that it is the Gin4 protein and that Gin4 is capable of undergoing autophosphorylation.
Since Nap1 is able to interact with Gin4 and Clb2, it is important to demonstrate that the kinase activity we observe in this assay is not due to Clb2/p34CDC28 kinase complexes that might coprecipitate with the Gin4 protein. The fact that no kinase activity is observed for the gin4K48A mutant argues strongly against this possibility. Control experiments demonstrate that the gin4K48A mutant protein is expressed at normal levels (Fig. 10, bottom) and is immunoprecipitated by the anti-Gin4 antibody (not shown). In addition, the immunoprecipitates used for these kinase assays are washed repeatedly with buffer containing detergent and 1M NaCl, which should wash off proteins that associate with Gin4. Indeed, neither Clb2 nor Nap1 can be detected in the Gin4 immunoprecipitates by Western blotting. (Note that the immunoprecipitation protocol used to detect an interaction between Nap1 and Gin4 shown in Fig. 6 uses a lower concentration of salt in the wash buffer.) Even when Gin4 immunoprecipitations are carried out at physiological salt concentrations we are unable to detect Clb2, and we suspect that Clb2 and Gin4 do not associate with Nap1 at the same time within the cell. In our previous work with Clb2 affinity columns, we found that Clb2 binds Nap1 but not Gin4 (Kellogg et al., 1995 a). This result supports the idea that Clb2 and Gin4 are not found in the same complex, and it suggests that Clb2 can only bind to Nap1 that is not complexed with Gin4. As an additional precaution, we immunoprecipitated Gin4 from a strain that carries the cdc28-4 temperature-sensitive allele of CDC28. Previous experiments have demonstrated that this allele produces a Cdc28 protein that is temperature sensitive in vitro (Ghiara et al., 1991). When we assayed the kinase activity of Gin4 immunoprecipitates at the restrictive temperature for cdc28-4, we observed no difference in kinase activity relative to a control strain, demonstrating that the kinase activity we observe is not due to Cdc28 (Fig. 7 B). Finally, in recent experiments we have found that fusion proteins purified from bacteria that contain the Gin4 kinase domain are able to phosphorylate histone H1 in vitro, demonstrating that Gin4 has the capacity to phosphorylate histone H1 (not shown).
We next used the phosphorylation of histone H1 as an assay to follow Gin4-associated kinase activity during the cell cycle. We arrested cells in G1 with α-factor, and then removed the α-factor and took samples every 10 min as the cells proceeded synchronously through the cell cycle. At each time point, we assayed Gin4-associated kinase activity, Clb2 protein levels, and Clb2-associated kinase activity (Fig. 8 A). We found that Gin4-associated kinase activity peaks during mitosis, and in multiple independent experiments we always observe that the peak of Gin4 kinase activity occurs slightly later than the peak of Clb2-associated kinase activity (Fig. 8 B). We also found that the putative autophosphorylation of Gin4 peaks at the same time as the kinase activity of Gin4.
Figure 8.

The Gin4 kinase is activated and phosphorylated during mitosis in a Nap1-dependent manner. (A) Δclb1,3,4, Δbar1 cells and Δnap1, Δclb1,3,4, Δbar1 cells were released from an α-factor arrest, and samples were taken at the indicated time points and assayed for Gin4-associated kinase activity using histone H1 as a substrate. (B) The same samples shown in A were assayed for Clb2-associated kinase activity. (C) The same samples shown in A were probed for the Clb2 protein by Western blotting. (D) The same samples shown in A were probed for the Gin4 protein by Western blotting.
Since Gin4 binds tightly to Nap1, and these two proteins appear to be involved in the same activities, we next sought to determine whether Nap1 is required for activation of the Gin4 kinase. To do this, we assayed Gin4-associated kinase activity during the cell cycle in Δnap1, Δclb1,3,4 cells. We found that there is a low level of Gin4-associated kinase activity in these cells that undergoes relatively little change during the cell cycle, demonstrating that Nap1 is required for the proper regulation of Gin4-associated kinase activity (Fig. 8 A). Interestingly, it appears that there is a low but significant level of Gin4 kinase activity during interphase in the Δnap1, Δclb1,3,4 cells (compare the kinase activity at the zero time points for the two different strains in Fig. 8 A). This observation suggests that Nap1 might be required both for the activation of Gin4 during mitosis, and for the efficient inactivation of Gin4 during interphase.
Nap1-dependent Phosphorylation of Gin4
We next addressed the mechanism by which Gin4 is activated during mitosis. Western blotting experiments reveal that the Gin4 protein in cells arrested in mitosis has a slower electrophoretic mobility than Gin4 from interphase cells, suggesting that Gin4 undergoes posttranslational modification during mitosis. Treatment of Gin4 immunoprecipitated from mitotic cells with phosphatase causes it to shift to the same electrophoretic mobility as Gin4 from interphase cells, demonstrating that Gin4 in mitotic cells is phosphorylated (Fig. 9 A). To determine the timing of Gin4 phosphorylation, we used Western blotting to follow the phosphorylation of Gin4 during the cell cycle in the same samples we used to follow the activation of Gin4 kinase activity in Fig. 8. We found that Gin4 phosphorylation occurs in parallel with the appearance of Gin4-associated kinase activity, and that phosphorylation of Gin4 is completely dependent upon the presence of Nap1 (Fig. 8 D).
In multiple experiments, we always observed that the time point that shows maximal phosphorylation of Gin4 occurs concurrently with the peak of Gin4 kinase activity (120-min time point in Fig. 8). This observation suggested that the kinase activity of Gin4 is activated during mitosis by phosphorylation. To confirm this, we immunoprecipitated Gin4 from cells arrested in mitosis, treated the Gin4 with phosphatase, and then assayed its kinase activity. We found that dephosphorylation of Gin4 from mitotic cells causes nearly a complete loss of Gin4 kinase activity (Fig. 9 B). The dephosphorylated Gin4 appears to be capable of undergoing autophosphorylation, but this result should be treated with caution. If a small fraction of the Gin4 protein is not completely dephosphorylated or inactivated by the phosphatase treatment, it would be likely to undergo autophosphorylation. This would give a deceptively strong signal relative to the phosphorylation of histone H1 because intramolecular phosphorylation will be more efficient than the intermolecular phosphorylation.
Gin4 Autophosphorylation
The Gin4 kinase assays shown in Figs. 7 and 8 suggest that Gin4 is capable of undergoing autophosphorylation. To test whether this is the case, we synchronized the gin4K48A strain with α-factor and then assayed phosphorylation of the mutant Gin4 protein during the cell cycle by Western blotting. We found that Gin4 with an inactive kinase domain completely fails to undergo mitotic phosphorylation, supporting the idea that autophosphorylation is occurring (Fig. 10). We obtain the same result in both a wild-type background and in the Clb2-dependent background.
Phosphorylation of Gin4 Is Induced by Clb2
Since Nap1 interacts with Clb2 and Gin4, and both Nap1 and Gin4 are required for the proper control of mitotic events by Clb2, one would predict that the phosphorylation and activation of Gin4 are also mediated by Clb2. To directly test whether this is true, we expressed the Clb2 protein in cells arrested in interphase and then used Western blotting to determine whether Gin4 is phosphorylated. For these experiments, we used a version of Clb2 that has a deletion of the first 176 amino acids (called Clb2Δ176), which removes the cyclin destruction box that normally prevents accumulation of mitotic cyclins during interphase. A bar1 strain carrying clb2Δ176 under the control of the gal1 promotor (gift of Adam Rudner, University of California, San Francisco, CA) was arrested in interphase by treatment with α-factor. The arrested cells were then divided into two equal cultures and galactose was added to one to induce expression of clb2Δ176 in the continued presence of α-factor. We found that expression of clb2Δ176 in cells arrested in interphase rapidly led to hyperphosphorylation of Gin4 (Fig. 11). Expression of clb2Δ176 during interphase did not, however, induce bud emergence, as reported previously (Amon et al., 1994).
Discussion
It has been known for a number of years that entry into mitosis is induced by the activation of a cyclin-dependent kinase by B-type cyclins. Yet we still know almost nothing about how B-type cyclins and cyclin-dependent kinases induce the actual events of mitosis. It remains unclear, for example, whether cyclin-dependent kinases initiate signaling pathways that lead to the activation of many other kinases during mitosis, or whether they function more directly to phosphorylate the proteins involved in the execution of mitotic events (Nigg, 1993). It is also unclear how the specificity of cyclin-dependent kinases is controlled, since simple organisms like budding yeast are somehow able to use the same cyclin-dependent kinase to induce completely different events during mitosis and interphase.
The pathway used by Clb2 to control bud growth during mitosis provides an excellent model system for understanding how cyclins and cyclin-dependent kinases control mitotic events. The function of this pathway is not required for viability, and mutations that disrupt the pathway can be easily identified because they give rise to both an unusual cell morphology and an unusual colony morphology. We have now identified two proteins that function in this pathway—Nap1 and Gin4. Nap1 was identified in a previous study in which we used affinity chromatography to identify proteins that bind specifically to Clb2 (Kellogg et al., 1995 a), while Gin4 was identified in the present study using a genetic screen for mutations that disrupt the control of bud growth during mitosis. We have also identified Gin4 biochemically as a protein that binds tightly to Nap1 in affinity chromatography experiments.
Gin4, Nap1, and Clb2 Function Together in the Control of Mitotic Events
A number of different experimental approaches demonstrate that Gin4, Nap1, and Clb2 work together in the control of mitotic events. First, the phenotypes caused by deletion of the GIN4 gene or the NAP1 gene are nearly indistinguishable, consistent with these two proteins being involved in carrying out similar functions. Second, loss of Gin4 or Nap1 function in cells that are dependent upon Clb2 causes a prolonged mitotic delay, consistent with these three proteins being required for the proper execution of mitotic events. Third, loss of Gin4, Nap1, or Clb2 function in cells that are dependent upon Clb2 causes the formation of highly elongated buds, demonstrating that all three of these proteins function in the switch from polar to isotropic bud growth that normally occurs during mitosis (Kellogg et al., 1995 a; Kellogg and Murray, 1995; Lew and Reed, 1995; Lew and Reed, 1993). Fourth, biochemical experiments demonstrate that Gin4 and Clb2 interact directly with Nap1, indicating that the functions of these three proteins must be closely tied (Kellogg et al., 1995 a). Fifth, the Gin4 kinase is phosphorylated and activated during mitosis, consistent with a mitotic role for Gin4. Sixth, Nap1 is required in vivo for the phosphorylation and activation of Gin4 during mitosis, demonstrating functional interaction between these two proteins in vivo. Finally, expression of Clb2 in cells arrested in interphase leads to the phosphorylation of Gin4, indicating that Gin4 phosphorylation occurs in response to Clb2 activity.
Loss of Gin4 Function in Clb2-dependent Cells Causes a Prolonged Mitotic Delay
In cells that are dependent upon CLB2 for survival, deletion of either the GIN4 gene or the NAP1 gene causes a prolonged mitotic delay at the short spindle stage. The mitotic delay observed for the GIN4 deletion is more severe, since many of the cells appear to be permanently arrested in mitosis and eventually die. The primary cause of this mitotic arrest is unknown. One possible explanation is that the cells fail to execute specific mitotic events, leading to the activation of checkpoint controls that delay the cells in mitosis until the completion of these events. Previous studies have demonstrated the existence of checkpoint controls that delay the cell cycle in response to spindle defects (Li and Nicklas, 1995; Murray, 1995; Rieder et al., 1994), and the fact that the mutant cells arrest with a short spindle suggests that they are defective in some aspect of spindle assembly or function. We have found that Δnap1 and Δgin4 cells are both resistant to conditions that destabilize microtubules, suggesting that Nap1 and Gin4 may play a role in regulating microtubule stability (Kellogg and Murray, 1995; and data not shown). A failure to properly control microtubule stability could cause a mitotic spindle defect and lead to activation of a spindle assembly checkpoint. A number of genes have been identified that are required for a spindle assembly checkpoint in yeast (Hoyt et al., 1991; Li and Murray, 1991), but we have not yet been able to test whether these genes are required for the mitotic delay seen in Δgin4, Δclb1,3,4 cells or in Δnap1, Δclb1,3,4 cells, since at least some of these checkpoint genes are synthetically lethal with a Δclb3, Δclb4 double deletion (Kellogg, D., unpublished data). Interestingly, we observe that cells arrested by benomyl at the mitotic spindle assembly checkpoint have fully phosphorylated and activated Gin4 (Figs. 7 and 9). This suggests that the spindle assembly checkpoint must be arresting mitotic events at a point after the activation of Gin4. Further analysis of this kind on other components of mitotic control pathways may help define where checkpoint controls are acting to arrest the cell cycle in mitosis.
Another possible explanation for the mitotic arrest is that Gin4 and Nap1 play a role in the pathway that triggers the destruction of Clb2 and the exit from mitosis. When cyclins induce entry into mitosis they initiate a series of events that culminates in the activation of the cyclin destruction machinery and the exit from mitosis (King et al., 1994, 1995). A number of experiments suggest that, in addition to destroying the cyclins, the cyclin destruction machinery may also function to induce chromosome separation by destroying proteins involved in holding sister chromosomes together (Holloway et al., 1993; Irniger et al., 1995). Interestingly, defects in the cyclin destruction machinery cause yeast cells to arrest with short spindles and unseparated chromosomes (Heichman and Roberts, 1996; Irniger et al., 1995). We know virtually nothing about the pathway that leads to activation of the cyclin destruction machinery, and it is possible that Gin4 and Nap1 function in this pathway. However, a role for Nap1 and Gin4 in the activation of cyclin destruction cannot explain the loss of bud growth control caused by deletion of these genes, indicating that Gin4 and Nap1 would have to be involved in several mitotic control pathways.
Mitosis-specific Phosphorylation and Activation of the Gin4 Kinase
Our results demonstrate that the Gin4 kinase is activated and phosphorylated during mitosis in a manner that is dependent upon Clb2 and Nap1. In addition, we found that a mutant version of Gin4 with an inactive kinase domain is not phosphorylated during mitosis, suggesting that the phosphorylation of Gin4 is due to autophosphorylation.
How is the Gin4 kinase phosphorylated and activated during mitosis? The results that we have obtained thus far suggest that Clb2 and Nap1 somehow work to activate Gin4 autophosphorylation. There are several models that might explain the requirement for Nap1 in the phosphorylation of Gin4. In previous studies, we found that purified Xenopus Nap1 can be phosphorylated by cyclin-dependent kinase complexes that contain cyclin B, but not by complexes that contain cyclin A (Kellogg et al., 1995 a). This result suggests that Nap1 is only phosphorylated during mitosis when B-type cyclins are present. Once phosphorylated, Nap1 could then stimulate Gin4 autophosphorylation. In this model, the specific induction of certain mitotic events by Clb2 would be due entirely to the ability of Clb2 to form a cyclin-dependent kinase complex that can phosphorylate Nap1. A problem with this model is that we have not yet been able to reconstitute phosphorylation of yeast Nap1 in vitro to recapitulate the results that we obtained using Xenopus proteins, but this may be due to technical differences between the two systems.
Another possible model for activation of Gin4 is that Nap1 functions as part of a mechanism that targets Gin4 for phosphorylation by the Clb2 cyclin-dependent kinase complex. The initial phosphorylation of Gin4 might then stimulate Gin4 autophosphorylation and lead to full phosphorylation and activation of the Gin4 kinase. A third model postulates that Gin4 autophosphorylation is activated simply by binding to both Nap1 and the Clb2 cyclin-dependent kinase complex, in a manner analogous to the activation of cyclin-dependent kinases by binding of cyclins. A problem with this kind of model is that we have thus far been unable to detect Clb2 in Gin4 immunoprecipitates, suggesting that these two proteins may not exist in the same complex within the cell.
The Induction of Cell Cycle Events and the Kinase Specificity Problem
Gin4 is able to phosphorylate both histone H1 and myelin basic protein in vitro, which means that its substrate specificity overlaps with the specificity of cyclin-dependent kinases and MAP kinases (Nigg, 1993; Peter et al., 1992). Since all of these kinases are known to be activated during mitosis (Gotoh et al., 1991; Heider et al., 1994; Minshull et al., 1994), this result emphasizes the importance of exercising caution when interpreting the results of kinase assays carried out in vitro. Many proteins have been identified that can be phosphorylated by cyclin-dependent kinases in vitro, but the increasing number of kinases with substrate specificities that overlap with cyclin-dependent kinases makes it difficult to conclude whether these proteins are genuinely phosphorylated by cyclin-dependent kinases in vivo. It is therefore difficult to use in vitro phosphorylation assays to study the pathways used by cyclin-dependent kinases to induce cell cycle events unless one has independent means of verifying that proteins are phosphorylated by cyclin-dependent kinases in vivo.
Understanding the Specificity of Cyclin-dependent Kinase Complexes
We found that expression of Clb2 in cells arrested in interphase can induce the phosphorylation of Gin4 at an inappropriate time during the cell cycle. Although expression of Clb2 in interphase cells can induce the phosphorylation of Gin4, it cannot induce bud emergence, which is normally induced by the G1 cyclins during interphase (Amon et al., 1994; Lew and Reed, 1995). Conversely, the fact that Gin4 is normally phosphorylated only during mitosis demonstrates that the G1 cyclins do not induce phosphorylation of Gin4. These results make it clear that different cyclins are somehow able to induce different cell cycle events. A further understanding of the molecular mechanism of Gin4 activation should provide an important step towards understanding how cyclins are able to do this. The finding that phosphorylation and activation of Gin4 are dependent upon Nap1 in vivo provides an important criterion for reconstituting the activation of Gin4 in vitro.
Acknowledgments
This work was supported by National Institutes of Health grant GM53959-01.
Footnotes
1. Abbreviation used in this paper: YPD, yeast/peptone/dextrose.
The initial stages of the genetic screen for mutants in bud growth control were carried out by D. Kellogg while working as a postdoctoral researcher in the lab of A. Murray, who also provided helpful advice and comments on the manuscript. We thank Y. Jin and A. Chisholm for use of their microscope, and we thank C. Wilson for use of his DNA synthesizer. We also thank A. Straight, K. Hardwick, D. Morgan, B. Sullivan, L. Wang, A. Kashyap, Z. Zimmerman, and A. Sreenivasan for helpful advice regarding these experiments, and/or critical reading of the manuscript.
Please address all correspondence to Douglas Kellogg, Sinsheimer Laboratories, Department of Biology, University of California at Santa Cruz, Santa Cruz, CA 95064. Tel.: (408) 459-5659. Fax: (408) 459-3139. e-mail: kellogg@darwin.ucsc.edu
References
- Amon A, Tyers M, Futcher B, Nasmyth K. Mechanisms that help the yeast cell cycle clock tick: G2 cyclins transcriptionally activate G2 cyclins and repress G1 cyclins. Cell. 1993;74:993–1007. doi: 10.1016/0092-8674(93)90722-3. [DOI] [PubMed] [Google Scholar]
- Amon A, Irniger S, Nasmyth K. Closing the cell cycle circle in yeast: G2 cyclin proteolysis initiated at mitosis persists until the activation of G1 cyclins in the next cycle. Cell. 1994;77:1037–1050. doi: 10.1016/0092-8674(94)90443-x. [DOI] [PubMed] [Google Scholar]
- Anderson CW, Baum PR, Gesteland RF. Processing of adenovirus 2-induced proteins. J Virol. 1973;12:241–252. doi: 10.1128/jvi.12.2.241-252.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitch I, Dahmann C, Surana U, Amon A, Nasmyth K, Goetsch L, Byers B, Futcher B. Characterization of four B-type cyclin genes of the budding yeast Saccharomyces cerevisiae. . Mol Biol Cell. 1992;3:805–818. doi: 10.1091/mbc.3.7.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiara JB, Richardson HE, Sugimoto K, Henze M, Lew DJ, Witenberg C, Reed SI. A cyclin B homolog in S. cerevisiae: chronic activation of the Cdc28 protein kinase by cyclin prevents exit from mitosis. Cell. 1991;65:163–174. doi: 10.1016/0092-8674(91)90417-w. [DOI] [PubMed] [Google Scholar]
- Gotoh Y, Moriyama K, Matsuda S, Okumura E, Kishimoto T, Kawasaki H, Suzuki K, Yahara I, Sakai H, Nishida E. XenopusM phase MAP kinase: isolation of its cDNA and activation by MPF. EMBO (Eur Mol Biol Organ) J. 1991;10:2661–2668. doi: 10.1002/j.1460-2075.1991.tb07809.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanks SK, Quinn AM. Protein kinase catalytic domain sequence database: identification of conserved features of primary structure and classification of family members. Methods Enzymol. 1991;200:38–62. doi: 10.1016/0076-6879(91)00126-h. [DOI] [PubMed] [Google Scholar]
- Harlow, E., and D. Lane. 1988. Antibodies. A Laboratory Manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- Heichman KA, Roberts JM. The yeast CDC16 and CDC27 genes restrict DNA replication to once per cell cycle. Cell. 1996;85:39–48. doi: 10.1016/s0092-8674(00)81080-6. [DOI] [PubMed] [Google Scholar]
- Heider H, Hug C, Lucocq JM. A 40-kDa myelin basic protein kinase, distinct from erk1 and erk2, is activated in mitotic HeLa cells. Eur J Biochem. 1994;219:513–520. doi: 10.1111/j.1432-1033.1994.tb19966.x. [DOI] [PubMed] [Google Scholar]
- Holloway SL, Glotzer M, King RW, Murray AW. Anaphase is initiated by proteolysis rather than by the inactivation of MPF. Cell. 1993;73:1393–1402. doi: 10.1016/0092-8674(93)90364-v. [DOI] [PubMed] [Google Scholar]
- Hoyt MA, Trotis L, Roberts BT. S. cerevisiaegenes required for cell cycle arrest in response to loss of microtubule function. Cell. 1991;66:507–517. doi: 10.1016/0092-8674(81)90014-3. [DOI] [PubMed] [Google Scholar]
- Irniger S, Piatti S, Michaelis C, Nasmyth K. Genes involved in sister chromatid separation are needed for B-type cyclin proteolysis in budding yeast. Cell. 1995;81:269–277. doi: 10.1016/0092-8674(95)90337-2. [DOI] [PubMed] [Google Scholar]
- Kellogg DR, Alberts BM. Purification of a multiprotein complex containing centrosomal proteins from the Drosophila embryo by chromatography with low-affinity polyclonal antibodies. Mol Biol Cell. 1992;3:1–11. doi: 10.1091/mbc.3.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg DR, Murray AW. NAP1 acts with Clb2 to perform mitotic functions and suppress polar bud growth in budding yeast. J Cell Biol. 1995;130:675–685. doi: 10.1083/jcb.130.3.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg DR, Kikuchi A, Fujii-Nakata T, Turck CW, Murray A. Members of the NAP/SET family of proteins interact specifically with B-type cyclins. J Cell Biol. 1995;130:661–673. doi: 10.1083/jcb.130.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg DR, Oegema K, Raff J, Schneider K, Alberts BM. CP60: a microtubule-associated protein that is localized to the centrosome in a cell cycle-specific manner. Mol Biol Cell. 1995;6:1673–1684. doi: 10.1091/mbc.6.12.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King RW, Jackson PK, Kirschner MW. Mitosis in transition. Cell. 1994;79:563–572. doi: 10.1016/0092-8674(94)90542-8. [DOI] [PubMed] [Google Scholar]
- King RW, Peters J-M, Tugendreich S, Rolfe M, Hieter P, Kirschner MW. A 20S complex containing CDC27 and CDC16 catalyzes the mitosis specific conjugation of ubiquitin to cyclin B. Cell. 1995;81:279–288. doi: 10.1016/0092-8674(95)90338-0. [DOI] [PubMed] [Google Scholar]
- Lawrence CW. Classical mutagenesis techniques. Methods Enzymol. 1991;194:273–281. doi: 10.1016/0076-6879(91)94021-4. [DOI] [PubMed] [Google Scholar]
- Lew DJ, Reed SI. Morphogenesis in the yeast cell cycle: regulation by Cdc28 and cyclins. J Cell Biol. 1993;120:1305–1320. doi: 10.1083/jcb.120.6.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew DJ, Reed SI. Cell cycle control of morphogenesis in budding yeast. Curr Opin Gen Dev. 1995;5:17–23. doi: 10.1016/s0959-437x(95)90048-9. [DOI] [PubMed] [Google Scholar]
- Li R, Murray AW. Feedback control of mitosis in budding yeast. Cell. 1991;66:519–531. doi: 10.1016/0092-8674(81)90015-5. [DOI] [PubMed] [Google Scholar]
- Li X, Nicklas RB. Mitotic forces control a cell cycle checkpoint. Nature (Lond) 1995;373:630–632. doi: 10.1038/373630a0. [DOI] [PubMed] [Google Scholar]
- Minshull J, Sun H, Tonks NK, Murray AW. MAP-kinase dependent mitotic feedback arrest in Xenopusegg extracts. Cell. 1994;79:475–486. doi: 10.1016/0092-8674(94)90256-9. [DOI] [PubMed] [Google Scholar]
- Murray A. The genetics of cell cycle checkpoints. Curr Opin Gen Dev. 1995;5:5–11. doi: 10.1016/s0959-437x(95)90046-2. [DOI] [PubMed] [Google Scholar]
- Murray, A., and T. Hunt. 1993. The Cell Cycle: An Introduction. Oxford University Press, New York. 243 pp.
- Nigg EA. Cellular substrates of p34cdc2and its companion cyclin-dependent kinases. Trends Cell Biol. 1993;3:296–301. doi: 10.1016/0962-8924(93)90011-o. [DOI] [PubMed] [Google Scholar]
- Norbury C, Nurse P. Animal cell cycles and their control. Annu Rev Biochem. 1992;61:441–470. doi: 10.1146/annurev.bi.61.070192.002301. [DOI] [PubMed] [Google Scholar]
- Peter M, Sanghera JS, Pelech SL, Nigg EA. Mitogen-activated protein kinases phosphorylate nuclear lamins and display sequence specificity overlapping that of mitotic protein kinase p34cdc2 . Eur J Biochem. 1992;205:287–294. doi: 10.1111/j.1432-1033.1992.tb16779.x. [DOI] [PubMed] [Google Scholar]
- Pringle JR, Adams AEM, Drubin DG, Haarer BK. Immunofluorescence methods for yeast. Methods Enzymol. 1991;194:565–602. doi: 10.1016/0076-6879(91)94043-c. [DOI] [PubMed] [Google Scholar]
- Richardson H, Lew DJ, Henze M, Sugimoto K, Reed S. Cyclin B homologs in Saccharomyces cerevisiaefunction in S phase and in G2. Genes & Dev. 1992;6:2021–2034. doi: 10.1101/gad.6.11.2021. [DOI] [PubMed] [Google Scholar]
- Rieder CL, Schultz A, Cole R, Sluder G. Anaphase onset in vertebrate somatic cells is controlled by a checkpoint that monitors sister kinetochore attachment to the spindle. J Cell Biol. 1994;127:1301–1310. doi: 10.1083/jcb.127.5.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]