

Abstract
The syntrophins are a multigene family of intracellular dystrophin-associated proteins comprising three isoforms, α1, β1, and β2. Based on their domain organization and association with neuronal nitric oxide synthase, syntrophins are thought to function as modular adapters that recruit signaling proteins to the membrane via association with the dystrophin complex. Using sequences derived from a new mouse β1-syntrophin cDNA, and previously isolated cDNAs for α1- and β2-syntrophins, we prepared isoform-specific antibodies to study the expression, skeletal muscle localization, and dystrophin family association of all three syntrophins. Most tissues express multiple syntrophin isoforms. In mouse gastrocnemius skeletal muscle, α1- and β1-syntrophin are concentrated at the neuromuscular junction but are also present on the extrasynaptic sarcolemma. β1-syntrophin is restricted to fast-twitch muscle fibers, the first fibers to degenerate in Duchenne muscular dystrophy. β2-syntrophin is largely restricted to the neuromuscular junction.
The sarcolemmal distribution of α1- and β1-syntrophins suggests association with dystrophin and dystrobrevin, whereas all three syntrophins could potentially associate with utrophin at the neuromuscular junction. Utrophin complexes immunoisolated from skeletal muscle are highly enriched in β1- and β2-syntrophins, while dystrophin complexes contain mostly α1- and β1-syntrophins. Dystrobrevin complexes contain dystrophin and α1- and β1-syntrophins. From these results, we propose a model in which a dystrophin–dystrobrevin complex is associated with two syntrophins. Since individual syntrophins do not have intrinsic binding specificity for dystrophin, dystrobrevin, or utrophin, the observed preferential pairing of syntrophins must depend on extrinsic regulatory mechanisms.
Syntrophins are intracellular peripheral membrane proteins of 58–60 kD originally identified as proteins enriched at the postsynaptic apparatus in Torpedo electric organ (17). More recently, syntrophins in mammalian skeletal muscle have been shown to be part of a complex of proteins that associate with dystrophin, the product of the Duchenne/Becker muscular dystrophy gene (4, 28, 50, 54). Many of the dystrophin-associated proteins (DAPs)1 are transmembrane proteins. Thus, the dystrophin complex as a whole is thought to link cortical actin to the extracellular matrix, thereby stabilizing the sarcolemma during repeated cycles of contraction and relaxation (3). At the neuromuscular junction (NMJ), the DAPs have been implicated in agrin-stimulated clustering of nicotinic acetylcholine receptors (for review see reference 46). Dystrophin and DAPs are also found at synapses in the brain and retina (29, 33, 45). Thus, the syntrophins and other DAPs may participate in synaptogenesis as well as in sarcolemmal stabilization.
The three syntrophin isoforms, α1, β1, and β2, are encoded by different genes but have similar domain organizations. All known syntrophins contain two pleckstrin homology (PH) domains (2, 19), which are modules of ∼100 amino acids found in a wide array of signaling proteins. PH domains in other proteins bind phosphatidylinositol lipids and proteins, such as the βγ-subunits of trimeric G proteins (for review see reference 47). Thus, PH domains may mediate signal-dependent membrane association. Inserted within the first syntrophin PH domain is a PDZ domain (originally identified in postsynaptic density-95, discs large, ZO-1), a 90–amino acid domain found in more than 40 proteins, many of which are restricted to membrane specializations such as tight junctions or synapses (48). A trend emerging from study of other PDZ-containing proteins suggests that PDZ domains bind the cytoplasmic carboxy-terminal tails of transmembrane proteins (examples of which include NMDA receptors, K+ channels, Fas [42], and EGF receptors (for review see reference 48]). Finally, the COOH-terminal 57 amino acids of syntrophins are highly conserved among the three isoforms but are otherwise unique. This region, termed the syntrophin-unique (SU) domain, may contain the binding site for dystrophin family members (2, 6). Thus, the syntrophins are a family of multidomain proteins that likely function as modular adapters in recruiting signaling proteins to dystrophin complexes and the membrane.
Differential association of dystrophin with certain syntrophin isoforms and/or DAPs may play a role in tailoring the complex for a particular membrane specialization. Indeed, the protein complexes assembled by muscle dystrophin should be functionally distinct from those organized by retinal dystrophin. Likewise, each of the dystrophin-related proteins, utrophin, dystrophin-related protein 2 (DRP-2), and dystrobrevin, may differentially associate with particular DAPs in different cell types. All of these dystrophin family members contain amino acid sequences homologous to the dystrophin carboxy terminus, the region in dystrophin shown to bind syntrophins and the DAPs. Dystrophin, utrophin, and dystrobrevin have been shown to be capable of binding all three syntrophin isoforms in vitro (4, 6). With the exception of dystrobrevin, each dystrophin family member also has a WW domain postulated to bind the transmembrane DAP complex. Since each of the dystrophin family members is expressed in a wide range of cell types, their association with specific subsets of syntrophins/DAPs may be critical for cell-specific function.
Among all the DAPs, the syntrophins appear particularly well suited for differentially associating with the dystrophin family members. Each of the three syntrophins is postulated to function as a modular adapter recruiting signaling proteins to dystrophin membrane complexes. Yet, syntrophin isoforms share only ∼50% amino acid identity, suggesting that each may recruit a distinct set of proteins. Like members of the dystrophin family, the syntrophins are expressed in a wide range of tissues (1, 5). We have shown previously that in rat skeletal muscle, α1-syntrophin is localized on the sarcolemma with dystrophin, whereas β2-syntrophin is restricted to the NMJ, similar to utrophin (38). In contrast, the transmembrane dystroglycans are expressed in many tissues and are the products of a single gene (for review see reference 21). The sarcoglycans are comprised of multiple forms, but these are highly restricted to muscle (35, 53). In addition, most sarcoglycans are unrelated in amino acid sequence (32, 35), suggesting that they have distinct functions. Thus, among the known DAPs, syntrophins are leading candidates for associating with different dystrophin family members and could be involved in targeting of dystrophin family members to distinct membrane sites, or in conferring different functions to DAP complexes.
Here, we examine the associations of all three syntrophin isoforms with dystrophin family members expressed in skeletal muscle. We have previously isolated mouse cDNAs encoding α1- and β2-syntrophins (1) and examined the corresponding protein localization (38). However, these studies did not consider the recently described mammalian dystrobrevins (8, 41) or β1-syntrophin (5). We began this study by isolating the mouse β1-syntrophin cDNA and generating and characterizing isoform-specific antibodies. These isoform-specific antibodies were then used to define the distribution of each syntrophin and the association of syntrophins with dystrophin, utrophin, and dystrobrevin isoforms from normal, dystrophin-deficient, and Δ71–74 dystrophin transgenic mouse skeletal muscle. We find that pairs of syntrophin isoforms selectively copurify with dystrophin and utrophin. Based on these results, we propose a model in which particular pairs of syntrophin isoforms associate with dystrophin/dystrobrevin or utrophin/dystrobrevin complexes.
Materials and Methods
Mouse β1-Syntrophin cDNA Isolation
A λgt11 mouse liver cDNA library (Clontech Labs, Palo Alto, CA) was screened by hybridization with 32P-labeled human β1-syntrophin cDNA (a generous gift of Dr. Louis Kunkel, Howard Hughes Medical Institute, Boston, MA) by methods described previously (1). Five clones were isolated, but none of them contained the extreme 5′ coding region. Therefore, this sequence was obtained by two consecutive rounds of 5′ rapid amplification of cDNA ends using a kit purchased from GIBCO BRL (Gaithersburg, MD) and RNA isolated from C57Bl6 mouse liver (13). PCR products were amplified with Vent polymerase (NEB) using the sequence-specific primers 5′-CCTAATCTTGGAGACTCAGGTGG (round 1) and 5′-TCCCGCAGGTCTGCTCCGTTC (round 2). Resulting DNA from each round was cloned into Bluescript II (Stratagene, La Jolla, CA), and multiple clones were sequenced by the University of North Carolina Automated DNA Sequencing Facility (Chapel Hill, NC) on a DNA sequencer (model 373A; Applied Biosystems, Inc., Foster City, CA). Sequence was analyzed with the aid of a DNAStar Lasergene computer software package (Madison, WI).
Antibodies
Antisyntrophin.
mAb SYN1351 raised against Torpedo syntrophin has been described previously (17). Polyclonal antibodies (Abs) specific for each syntrophin isoform were prepared by immunizing rabbits with peptides according to standard methods (36). The Ab SYN37 was prepared against the peptide C-RLGGGSAEPLSSQSFSFHRDR, corresponding to amino acids 220–240 of mouse β1-syntrophin, plus an NH2-terminal cysteine (see boxed region in Fig. 1 B). The β2-syntrophin antibody, SYN28, was prepared against C-SGSEDSGSPKHQNTTKDR as an alternative to the weaker Ab SYN24 previously prepared against the same peptide (38). The α1-syntrophin antibody, SYN17, was described previously (38). Each antipeptide antibody used in this study was affinity purified with peptide coupled to Affi-Gel-10 or -15 (BioRad Labs, Hercules, CA) as described previously (28) and was used directly or after biotinylation with sulfo-NHS-biotin (Pierce, Rockford, IL) according to the manufacturer's protocol.
Figure 1.

Cloning, sequence, and domain structure of murine β1-syntrophin sequence. (A) Strategy for cloning mouse β1-syntrophin and structure of the combined cDNAs, showing the coding region bounded by start and stop codons. (B) The deduced amino acid sequence of mouse β1-syntrophin is aligned with mouse α1- and β2-syntrophins (1, 2). Identical amino acids are shaded. The boundaries of PH (6, 19), PDZ (2), and SU (2) domains are indicated by arrows. The boxed region denotes sequences used to generate synthetic peptides for production of isoform-specific antibodies. (C) Schematic diagram showing the relative organization of PH, PDZ, and SU domains in syntrophins. Mouse β1-syntrophin cDNA sequence data are available from GenBank/EMBL/DBJ under the accession number U89997.
Antidystrobrevin.
mAb 13H1 (a gift of J.B. Cohen, Harvard Medical School) was raised against Torpedo dystrobrevin (12). Abs DB308 and DB433 were prepared against peptides corresponding to mouse dystrobrevin, as previously described (8).
Antidystrophin.
Ab DYS3669 was prepared against COOH-terminal 10 amino acids of mouse dystrophin (22) plus a terminal cysteine (C-PGKPMREDTM) according to standard methods (36). mAb Mandys-8 (34) was purchased from Sigma Chemical Co. (St. Louis, MO).
Antiutrophin.
Ab UTR3165 was previously described (28). mAb DRP-1 was purchased from Novacastra Laboratories (Newcastle upon Tyne, UK).
Anti–myosin heavy chain (MHC).
mAbs BF-F3 (diluted 1:30) (44) and NCL-MHCf (Novacastra Laboratories, diluted 1:30) are specific for MHC expressed in type IIB fibers and all type II fibers, respectively. mAb A4.74 (diluted 1:30) (52) strongly labels rat MHC in type IIA and labels type IID fibers to a lesser extent.
Immunoaffinity Purification of Protein Complexes
Tissues from control (C57BL10 SNJ; Jackson Laboratory, Bar Harbor, ME), mdx (Jackson Laboratory), and Δ71–74/mdx mice (J.S. Chamberlain, University of Michigan, Ann Arbor, MI) (39) were dissected and frozen in liquid nitrogen. Protein complexes were partially purified as described previously (28). Briefly, tissues (5 g) were homogenized in 10 vol (wt/vol) of ice-cold homogenization buffer (HB; 10 mM Na phosphate, 0.4 M NaCl, 5 mM EDTA, pH 7.8, plus the protease inhibitors aprotinin, leupeptin, and antipain at 0.5 μg/ml each, pepstatin A, 0.05 μg/ml, and 2 mM PMSF) (28). The particulate fraction was pelleted (10 min at 12,000 g, model JA-20 rotor; Beckman Instruments, Fullerton, CA), resuspended in HB (10 vol), and recentrifuged. Washed pellets were solubilized in 1% Triton X-100/HB (3 vol) and incubated on ice for 15 min. For preparation of individual syntrophin isoforms (see Fig. 2), the particulate fractions were dissolved in 1% SDS/HB (3 vol) to disrupt possible complexes of multiple syntrophins. After incubation for 15 min, excess 1% Triton/HB (30 vol) was added. In all cases, the detergent-solubilized extracts were clarified by centrifugation (20 min at 53,000 g, model Ti 60 rotor; Beckman Instruments, Fullerton, CA) and incubated with antibody resins prepared by coupling 2 mg of affinity-purified antibody to 2 ml of Affi-gel 10 (BioRad Labs) according to manufacturer's specifications. After agitation for 2 h at 4°C, resins were collected in a column and washed sequentially with 1% Triton/HB (50 ml), 0.1% Triton/HB (10 ml), and HB (5 ml). Bound proteins were eluted with 0.1 M triethylamine, pH 11.5, precipitated with TCA, washed with 95% ethanol, and resuspended in SDS-PAGE sample buffer.
Figure 2.

Isoform specificity of syntrophin antibodies. Antibodies were characterized by immunoblotting samples enriched in individual syntrophin isoforms (as indicated above lanes). α1-, β1-, and β2-syntrophins were solubilized from crude membrane preparations from skeletal muscle, liver, and kidney, respectively, in 1% SDS to disrupt potential multi-syntrophin complexes. After addition of excess Triton, syntrophins were immunoaffinity purified with the appropriate antibody (Abs SYN17, SYN37, and SYN28). Blots were probed with mAb SYN1351 or biotinylated polyclonal antibodies as indicated. Labeling with each polyclonal antibody was eliminated by preincubation with the appropriate antigenic peptide (not shown). Positions of two molecular mass markers (52 and 59 kD) are shown on the right in the top panel.
Immunoblotting
Samples were resolved by electrophoresis on 8% acrylamide/0.8% bis-acrylamide gels buffered with tris-tricine (43). Proteins were transferred to Immobilon-P (Millipore Corp., Bedford, MA) in 192 mM glycine, 25 mM Tris, 20% methanol using a Trans-Blot apparatus (BioRad Labs). After blocking in 5% milk/TBS, 0.1% tween-20 (TBST), membranes were incubated with primary antibodies (30 nM) in 1% milk/TBST. After three 5-min washes in TBST, bound primary antibodies were detected with appropriate secondary antibodies conjugated to horseradish peroxidase (Jackson Immunoresearch, West Grove, PA). Because the heavy chain of polyclonal antibodies that leaked from resins comigrated with syntrophins, selected blots were probed with biotinylated antisyntrophin polyclonal antibodies and detected with peroxidase-avidin detection reagent (Vectastain ABC elite; Vector Laboratories, Burlingame, CA). Bands were visualized by enhanced chemiluminescence (Pierce) and exposed to film (Dupont/ NEN, Wilmington, DE). In some cases, blots were stripped for 1 h at 50°C in 2% SDS, 0.1 M β-mercaptoethanol, 62.5 mM Tris, pH 6.7, and reprobed as above. Prestained standards (Sigma Chemical Co.) were used after calibration with unstained molecular weight markers (BioRad Labs).
Immunohistochemistry
The gastrocnemius, plantaris, and soleus were removed intact from mice, flash-frozen in liquid nitrogen–cooled isopentane, and cryosectioned (7 μm). Before labeling, sections were fixed with 0.5% paraformaldehyde, permeabilized with 0.5% Triton X-100/PBS/0.2 M NH4Cl, and blocked with PBS containing 2% fish gelatin/0.08% BSA. Sections were incubated with primary antibodies (30 nM). After several PBS washes, sections were incubated with a cocktail containing either goat anti–rabbit IgG or anti–mouse IgM conjugated to Texas red (Jackson Immunoresearch; 1:200) mixed with either donkey anti–mouse IgG conjugated FITC or α-bungarotoxin (Tx) conjugated to BODIPY-fluorescence (Molecular Probes, Eugene, OR; 1:300). Washed sections were fixed with 4% paraformaldehyde and mounted in glycerol containing n-propyl gallate to reduce photobleaching (20). Antibody specificity was tested by preincubating antibodies with their antigenic peptide (100 μM) for 30 min before labeling. Adjacent sections were stained with Mayers haematoxylin-eosin (30). Sections were viewed on a fluorescence microscope (model Axioskop; Carl Zeiss, Inc., Thornwood, NY) and photographed (TMax 400 film; Kodak, Rochester, NY).
Results
Isolation of Mouse β1-syntrophin cDNA
The cDNA encoding the mouse form of β1-syntrophin was obtained by screening a mouse λgt11 cDNA library with the human β1-syntrophin cDNA. Five clones were isolated containing overlapping sequence that included >70% of the protein coding sequence and 820 base pairs of 3′ untranslated region. The remaining 5′ coding sequence was obtained by rapid amplification of cDNA ends (see Materials and Methods). The resulting cDNA (Fig. 1 A) contains 354 nucleotides 5′ of a 1,611-nucleotide open reading frame. The first methionine codon is in a context favoring translation initiation (27) and aligns with that described for the human cDNA (5). The deduced protein contains 537 amino acids and has a predicted mass of 58,088 D and a pI of 8.3. Its amino acid sequence is 90% identical to human β1-syntrophin while sharing only 48 and 55% identity with human α1- and β2-syntrophins, respectively, indicating that it is the mouse ortholog of human β1-syntrophin (5). After the initial methionine, the amino termini of both mouse and human β1-syntrophin have a stretch of nine hydrophobic amino acids (alanine and valine). A similar sequence is present in the human β2-syntrophin (6) sequence but is absent in mouse β2-syntrophin (2) and the α1-syntrophins (1, 6, 54). Comparison of the amino acid sequences of the three mouse syntrophins shows marked conservation, particularly within the PDZ and SU domains (Fig. 1 B).
Specificity of Syntrophin Antibodies
To investigate the expression of syntrophins and their association with dystrophin and dystrophin-related proteins, we prepared and characterized a series of antisyntrophin antibodies. Polyclonal antibodies were generated against synthetic peptides corresponding to syntrophin sequences from a region poorly conserved among the three isoforms (see boxed region in Fig. 1 B). To test the specificity of these antibodies, syntrophin isoforms were partially purified with the appropriate syntrophin peptide antibody from mouse tissues rich in either α1-, β1-, or β2-syntrophin (skeletal muscle, liver, and kidney, respectively). To ensure that each preparation contained only a single syntrophin, samples were first treated with SDS to disrupt possible multimeric complexes and were then diluted with Triton X-100 before immunopurification. As expected from previous results (38), immunoblotting with the anti–α1-syntrophin antibody, SYN17, labeled a single ∼60-kD protein in the α1-syntrophin preparation but did not recognize proteins in either the β1- or β2-syntrophin preparations (Fig. 2). Likewise, the anti–β1-syntrophin antibody, SYN37, strongly labeled proteins in the β1-syntrophin preparation without recognizing proteins in either the α1- or β2-syntrophin preparations. Finally, the β2-syntrophin antibodies, SYN24 and SYN28, recognized only β2-syntrophin. In contrast, mAb SYN1351, which recognizes an epitope in the PDZ domain (M.E. Adams, S.H. Gee, and S.C. Froehner, unpublished results), strongly labeled each syntrophin isoform (Fig. 2). Thus, we conclude that mAb SYN1351 is pan specific, while each of the syntrophin peptide antibodies is isoform-specific in immunoblotting and in immunoaffinity purification.
In addition to proteins of the size expected for full-length syntrophins, smaller proteins were recognized by the anti–β1- and anti–β2-syntrophin antibodies (Fig. 2). These proteins were also recognized by mAb SYN1351, thus confirming their identities as syntrophins. The tissue distribution patterns for these smaller syntrophin-related proteins are distinct from each other and from the corresponding full-length isoforms (Fig. 3). Although it is possible that the smaller proteins are proteolytic fragments, a more likely possibility is that they are generated by posttranslational modification or by alternative splicing of the β1- and β2-syntrophin mRNAs. Northern blot analysis identified multiple transcripts for β1- and β2-syntrophins, while only a single transcript for α1-syntrophin was found (1, 5, 6). Although the basis of these multiple transcripts is unknown, it is certainly possible that they encode modified forms of β1- and β2-syntrophins.
Figure 3.

Tissues express multiple syntrophin isoforms. Syntrophins were isolated from Triton X-100–solubilized tissue extracts with the pan-specific syntrophin antibody SYN1351. Sample loadings were adjusted to contain approximately equal amounts of total syntrophin, as judged by immunoblotting with SYN1351 (pan anti-syn). Individual syntrophin isoforms were identified by blotting with Abs SYN17 (anti–α1 syn), SYN37 (anti–β1 syn), or SYN24 (anti–β2 syn). Positions of two molecular mass markers (52 and 59 kD) are shown on the right in the top panel.
Differential Expression of Syntrophin Isoforms
To examine the relative amounts of the three syntrophin isoforms expressed in different tissues, syntrophins were partially purified from detergent-solubilized tissue extracts using the pan-specific mAb SYN1351. Equal amounts of total syntrophins from each tissue (as judged by immunoblotting with SYN1351) were subjected to immunoblotting with the isoform-specific antibodies (Fig. 3). The relative tissue distribution of each syntrophin isoform is consistent with previous Northern blot analysis (1, 5, 6). Thus, skeletal muscle contains the highest levels of α1-syntrophin, while liver expresses the highest levels of β1-syntrophin, and testis expresses predominantly β2-syntrophin. Almost every tissue expresses two or three syntrophin isoforms, but there did not appear to be any bias for particular pairs of syntrophins to be expressed together.
Previously, we proposed that dystrophin and related proteins might each be associated with a particular syntrophin isoform (1). This proposal was based largely on the common expression patterns between dystrophin family members and individual syntrophin isoforms. In vitro binding studies have demonstrated, however, that all three syntrophins are able to bind to a region in the COOH-terminal domain of dystrophin encoded by exon 74, and to the analogous regions in utrophin and dystrobrevin (6, 15), with no intrinsic binding specificity. However, the associations that occur in vivo might be regulated by additional factors. To examine this possibility, we have used a combination of immunofluorescence microscopy and biochemical analysis to determine which syntrophin isoforms are associated with dystrophin, utrophin, and dystrobrevin complexes.
Skeletal muscle was chosen for these studies for several reasons. First, skeletal muscle expresses all three syntrophins, although α1-syntrophin is the predominant isoform (see Fig. 3 and results in references 1, 5, 6). Since the distributions of dystrophin, utrophin, and dystrobrevin are known in this tissue, a comparison of their localizations with the syntrophin isoforms can be used to corroborate the biochemical studies on isolated complexes. Furthermore, previous studies have shown that two syntrophins, α1 and β2, have distinct distributions in muscle, an observation that supports the idea of differential association of syntrophins and dystrophin family members (38). Finally, Duchenne muscular dystrophy has its most profound effects on skeletal muscle. Since the absence of dystrophin results in a loss of dystrophin-associated proteins, including syntrophins, understanding syntrophin's interactions in this tissue may be especially important in deciphering the molecular causes of this disease.
We performed immunofluorescence microscopy on sections of adult mouse muscle with the syntrophin isoform-specific antibodies. As reported previously (38, 55), labeling for α1-syntrophin was strong on sarcolemma with particular enrichment at NMJs (Fig. 4). A similar labeling pattern was identified for β1-syntrophin, although the staining intensity of individual fibers varied. Slow-twitch fibers (type I) displayed little or no β1-syntrophin labeling, while a subset of fast-twitch fibers (type II) showed intense labeling (Fig. 5 A). Among fast fibers, the most glycolytic ones (type IIB) showed stronger labeling than oxidative fibers (type IIA and D) (Fig. 5 B). Although this was the typical staining pattern, exceptions could be found. For example, in predominantly fast muscles, staining for β1-syntrophin in slow fibers was weak but clearly detectable (not shown). In contrast, differential staining of fiber types was not seen for α1-syntrophin (Fig. 5 A) or dystrophin (22).
Figure 4.

Localization of syntrophin isoforms in skeletal muscle. Regions of mouse gastrocnemius muscle containing NMJs were identified by α-bungarotoxin (α-BgTx). Distributions of α1-, β1-, and β2-syntrophins were determined with Abs SYN17, SYN37, and SYN28, respectively. In each case, the specificity of immunolabeling was confirmed in adjacent serial sections by preincubating antibodies with the appropriate antigenic peptide. Bar, 50 μm.
Figure 5.

Fiber-type specificity of β1-syntrophin. (A) Syntrophins were examined in sections of mouse hind limb muscle containing the mixed slow/fast-twitch plantaris (left of arrows) with the adjacent fast-twitch gastrocnemius (right of arrows). Labeling for α1- and β1-syntrophins (SYN17 and SYN37) is compared to that of fast fiber (type II) MHC staining (mAb NCL-MHCf). Note that β1-syntrophin labeling is highly restricted to a subset of type II myofibers, while α1-syntrophin labeling shows uniform fiber-type staining. (B) β1-syntrophin labeling was examined in subtypes of fast fibers identified by mAb BF-F3 specific for type IIB MHC (IIB) and mAb A4.74 specific for IIA, and to a lesser extent type IID MHC (IIA, D). β1 and IIA, D show a single cryosection double labeled as indicated, while IIB shows an adjacent serial section. Note the correspondence between strong labeling for β1-syntrophin (β1) and that of type IIB myosin (IIB). Bars: (A) 50 μm; (B) 80 μm.
We have previously shown that β2-syntrophin is concentrated at NMJs (38). This labeling pattern was established on rat skeletal muscle with Ab SYN24, which reacts only weakly with mouse β2-syntrophin. We have now prepared an additional antibody, SYN28, which gives much stronger labeling of β2-syntrophin at the neuromuscular junction in mouse and rat skeletal muscle. In both rat and mouse, faint but specific labeling for β2-syntrophin was occasionally detected on the sarcolemma with Ab SYN28 (Fig. 4). Thus, although it is likely that small amounts of this isoform are also present on the sarcolemma, these results are in general agreement with our original finding that β2-syntrophin is concentrated at the NMJ. Thus, each of the three syntrophins exhibits a differential distribution in skeletal muscle, either within a single muscle fiber or across muscle fiber types.
Multiple Syntrophin Isoforms in Dystrophin and Utrophin Complexes
Previous biochemical studies showed that purified dystrophin complexes contain a triplet of syntrophin bands (55). However, the isoform identity of these syntrophin bands was not determined. Likewise, the syntrophin isoforms associated with utrophin in skeletal muscle have not been established. To determine which syntrophin isoforms are present in dystrophin and utrophin complexes, we partially purified dystrophin and utrophin from skeletal muscle extracts using immunoaffinity purification. When isolated in this way, dystrophin preparations contain no detectable utrophin, and utrophin preparations are free of detectable dystrophin (Fig. 6 A). Given the relative paucity of NMJ membrane (the site of highest concentration of utrophin) in skeletal muscle fibers, it is likely that much of the utrophin complex originated from nerve and blood vessels. In addition to the full-length utrophin (predicted to be 395 kD), a smaller utrophin-immunoreactive protein was identified. This ∼140-kD protein appears to be larger than G-utrophin and thus may either result from proteolysis of full-length utrophin or represent a previously undescribed utrophin homologue of Dp140. Aliquots of dystrophin and utrophin complexes containing approximately equal amounts of total syntrophin were analyzed by immunoblotting, thereby allowing comparison of the relative amounts of individual syntrophin isoforms. As shown in Fig. 6 A, dystrophin complexes were highly enriched in α1- and β1-syntrophins, while utrophin complexes contained mostly β1- and β2-syntrophins.
Figure 6.

Dystrophin and utrophin complexes contain distinct pairs of syntrophin isoforms. (A) Dystrophin and utrophin complexes were immunoaffinity purified from Triton-solubilized extracts of mouse skeletal muscle with antibodies DYS3669 and UTR3165, respectively. Sample loadings were adjusted to contain approximately equal amounts of syntrophin, as judged by immunoblotting (pan-Syn, mAb SYN1351). Duplicate blots were probed with mAbs Mandys-8 (Dys), and DRP-1 (Utr) or biotinylated polyclonal antibodies SYN17 (α1-syn), SYN37 (β1-syn), and SYN28 (β2-syn). (B) Syntrophins were immunoaffinity purified from skeletal muscle extracts with Abs SYN17, SYN37, and SYN28. Sample loadings were adjusted to contain similar amounts of total syntrophin, as judged by immunoblotting (pan-Syn, mAb SYN1351). A duplicate blot was probed with mAb Mandys-8 (Dys), stripped, and reprobed with mAb DRP-1 (Utr). Positions of molecular mass markers (in kD) are shown in some panels. These results were replicated twice, and representative blots from a single experiment are shown.
To corroborate these results, syntrophin isoform complexes were isolated by immunopurification. Samples containing approximately equal amounts of total syntrophin were then tested for relative levels of dystrophin and utrophin. As shown in Fig. 6 B, α1-syntrophin preparations were particularly enriched in dystrophin, while β1- and β2-syntrophin preparations contained smaller amounts of dystrophin. The same blot was stripped and reprobed for utrophin. Highest levels of utrophin were found in the β2-syntrophin preparations, while lower levels were detected in the β1-syntrophin preparations (Fig. 6 B). Only with much longer exposures was utrophin detected in α1-syntrophin complexes. Together, these results indicate that, in skeletal muscle, dystrophin associates preferentially with α1- and β1-syntrophin. In contrast, utrophin complexes contain predominantly β1- and β2-syntrophin.
Syntrophin Distribution in mdx Skeletal Muscle
Previous studies with the pan-specific syntrophin mAb SYN1351 showed that the loss of dystrophin from the sarcolemma causes a dramatic decrease of syntrophin staining (11). This decrease occurred despite the fact that total syntrophin, as measured by immunoblot analysis, was essentially unchanged in mdx muscle. To determine which isoforms are affected, we compared syntrophin isoform staining in normal and mdx gastrocnemius muscle. In wild-type mice, fibers of the gastrocnemius muscle, which are predominantly fast fiber type, have high levels of α1- and β1-syntrophins on the sarcolemma. However, in mdx muscle, the absence of dystrophin results in a dramatic reduction of both α1- and β1-syntrophin sarcolemmal staining (compare Fig. 7, A and B, to Fig. 4). This result provides further support for the association of α1- and β1-syntrophins with dystrophin.
Figure 7.

Localization of syntrophins in mdx mouse skeletal muscle. (A) The distribution of syntrophin isoforms was examined in regions containing NMJs (identified with α-bungarotoxin; Tx). Labeling in the perisynaptic regions with antibodies SYN17 (α1), SYN37 (β1), SYN28 (β2), and UTR3165 (Utr) was characterized. (B) In 8-wk-old mdx mice, regenerating fibers with central nuclei were identified by haematoxylin-eosin (H&E) staining. Serial sections were labeled with antibodies UTR3165 (Utr), DYS3669 (Dys), SYN17 (α1), SYN37 (β1), and SYN28 (β2). Bars: (A; α1, β1, and Utr) 50 μm; (A; B2) 30 μm; (B) 50 μm.
At some sites in mdx muscle, intense syntrophin staining was retained, and in each case, its distribution paralleled that of utrophin. As previously shown (38, 55), labeling for α1-syntrophin in mdx muscle was particularly strong at the NMJ, a site of high utrophin concentration (Fig. 7 A). Staining for β1- and β2-syntrophin was also retained at the NMJ (Fig. 7 A). Utrophin labeling frequently extended beyond its normal highly restricted distribution on the postsynaptic membrane, spilling over onto the perisynaptic sarcolemma and diminishing in intensity with distance from the NMJ (Fig. 7 A). α1- and β1-syntrophins, and to a lesser extent β2-syntrophin, were also found perisynaptically in mdx muscle (Fig. 7 A).
In nonsynaptic regions, utrophin labeling was frequently seen on small caliber myofibers with centrally located nuclei, the hallmark of regenerated fibers (Fig. 7 B). In adjacent serial sections, staining for α1-syntrophin and especially β1-syntrophin mirrored that of utrophin. β2-syntrophin was not detectable under standard labeling conditions. These fibers are not revertants since antidystrophin labeling was negative (Fig. 7 B). Thus, although syntrophin immunoreactivity is dramatically reduced on the sarcolemma of mdx skeletal muscle, staining for at least two isoforms is retained in regions that express utrophin. When considered with the results of immunoaffinity purification, these data suggest that utrophin is capable of associating with each syntrophin isoform.
Two Syntrophin-binding Sites in the Dystrophin Complex
Results from other laboratories suggest that the syntrophin content in purified dystrophin complexes is approximately twice that of other dystrophin-associated proteins (16, 56). While only a single syntrophin-binding region has been clearly identified on dystrophin, a second syntrophin-binding protein, dystrobrevin, is known to be associated with the dystrophin complex in skeletal muscle (50, 51). Originally identified as a Torpedo phosphoprotein of 87 kD (51), dystrobrevin is homologous to the COOH-terminal region of dystrophin and contains a syntrophin-binding site (4, 15). This site is followed by two tandem heptad repeats of leucines predicted to form coiled-coils (7). Ozawa and colleagues have shown that dystrophin binds in vitro to a protein called A0, and they have mapped its binding site to the first heptad repeat of dystrophin (50). Subsequently, A0 was shown to be equivalent to dystrobrevin (58). A weaker binding site was found in the region of dystrophin that contains the second heptad repeat (50). Thus, we propose a model in which dystrophin and dystrobrevin associate via coiled-coil interactions (7) and could thus recruit two syntrophins to the dystrophin complex.
In complexes that contain two syntrophin-binding proteins (dystrophin and dystrobrevin), it is difficult to determine the type of syntrophin associated with either protein independently. In this regard, the Δ71–74 transgenic mdx mouse is particularly useful since it expresses a dystrophin transgene in skeletal muscle that lacks exons 71–74, which includes the syntrophin-binding site (for review see reference 18). In mdx muscle, Δ71–74 dystrophin produced by the transgene is targeted to the sarcolemma, such that normal levels of membrane-bound dystrophin-associated proteins are restored (39, 40). Surprisingly, the Δ71–74 dystrophin transgene restores syntrophin immunoreactivity to the sarcolemma, despite lacking the syntrophin-binding site (39, 40). Since the Δ71–74 dystrophin transgene product retains most of the coiled-coil region encoded by exon 75, it might still associate with dystrobrevin, which could in turn bind syntrophin and target it to the membrane. Indeed, we find dystrobrevin localized on the sarcolemma along with both α1- and β1-syntrophins (Fig. 8 B). Thus, α1- and β1-syntrophins may be restored to the membrane in these transgenic mice via association with dystrobrevin bound to dystrophin.
Figure 8.


Dystrobrevin associates with α1- and β1-syntrophins in Δ71–74/mdx skeletal muscle. (A) Schematic comparing the structure of the cysteine-rich COOH-terminal region of normal dystrophin, of the dystrophin transgene lacking exons 71–74, and the corresponding structure of dystrobrevin. The locations of the sequences used for preparation of dystrobrevin antipeptide antibodies DB308 and DB433 are indicated by bars. H1, H2, and WW, position of helix 1 and helix 2 and the WW domain, respectively. (B) Immunofluorescence labeling for α1-syntrophin (α1, Ab SYN17), β1-syntrophin (β1, Ab SYN37), and dystrobrevin labeling (Db, mAb 13H1), with the corresponding labeling with α-bungarotoxin (Tx) shown to the right of each image. The labeling for β2-syntrophin (β2, Ab SYN28) and α-bungarotoxin (Tx) was reduced in size and intensity compared to control mice. (C) Dystrobrevin complexes were immunoaffinity purified from Triton extracts of Δ71–74/mdx skeletal muscle with antipeptide dystrobrevin antibodies that recognize either a central region (Ab DB308, left lane) or the linker between the two coiled-coils (Ab DB433, right lane). Sample loadings were adjusted for approximately equal amounts of dystrobrevin immunoreactivity in each lane (Db, mAb 13H1). Complexes purified with Ab DB433 contained dramatically lower amounts of dystrophin, as expected if this antibody inhibits a coiled-coil interaction between dystrophin and dystrobrevin; see text (Dys, Mandys-8). The total amount of syntrophin (pan-Syn, mAb SYN1351) purified with dystrobrevins was approximately the same with either Db antibody. Both α1-syntrophin (α1-syn, Ab SYN17) and β1-syntrophin (β1-syn, Ab SYN37) copurified with dystrobrevin, independent of the presence of dystrophin. Utrophin and β2-syntrophin were detectable only with much longer exposure times.
As an additional test of association of α1- and β1-syntrophins with dystrobrevin, we immunoaffinity purified dystrobrevin from Δ71–74 transgenic mdx mouse skeletal muscle. Using an antibody (DB308) directed against a central region of dystrobrevin, we purified complexes and found that both α1- and β1-syntrophins and Δ71–74 dystrophin copurified with dystrobrevin (Fig. 8 C, lanes DB308). Complexes isolated with a second dystrobrevin antibody, Ab DB433 made against the short linker region between dystrobrevin's coiled-coils (see Fig. 8 A), also contained α1- and β1-syntrophins. These results suggest that dystrobrevin can associate with either syntrophin isoform in vivo.
We did note one important difference in the dystrobrevin complexes isolated with Abs DB308 and DB433. The dystrophin content in Ab DB433 preparations was dramatically reduced in comparison with the amounts obtained when Ab DB308 was used (Fig. 8 C, lanes DB433). It appears that Ab DB433, which recognizes the linker sequence between the coiled-coils of dystrobrevin, disrupts the interaction between dystrophin and dystrobrevin. Alternatively, it is possible that the epitope for Ab DB433 is more accessible in dystrobrevins that are not bound to dystrophin. Both of these possibilities are consistent with the idea that interaction between dystrobrevin and dystrophin is mediated by their coiled-coil regions.
Discussion
The syntrophins are a multigene family of modular adapter proteins thought to recruit signaling proteins to the membrane via association with dystrophin and other members of the dystrophin family. The existence of three isoforms derived from distinct genes makes the syntrophins unique among dystrophin-associated proteins. In skeletal muscle, we find that the syntrophins have distinct but overlapping distributions. These differential localizations imply that each syntrophin has a unique function, probably derived in part from association with either dystrophin or utrophin, in combination with dystrobrevin.
Our results suggest that pairs of syntrophin isoforms associate with dystrophin and utrophin complexes. Previous stoichiometric analyses of purified dystrophin complexes is in good agreement with two syntrophins per complex (16, 56). Several features of the dystrophin complex could account for the presence of two syntrophins. In addition to the known syntrophin-binding site in dystrophin encoded by the first half of exon 74 (4, 50), a second syntrophin-binding site has been proposed. A peptide corresponding to the latter half of 74 and exon 75 of dystrophin binds an ∼60-kD DAP (50). Although this protein was thought to be β1-syntrophin, it may instead be the 60-kD form of dystrobrevin. Indeed, the peptide also bound a larger protein (A0) (50), which has since been identified as full-length dystrobrevin (58). Thus, the putative second syntrophin-binding site, which encompasses the first coiled-coil of dystrophin, may instead be a site of interaction between dystrophin and dystrobrevin. A second possibility is that two syntrophins in a dystrophin complex could result from syntrophin dimerization. Syntrophins have been shown to bind an ∼60-kD DAP, leading to the suggestion that syntrophins form dimers (55). However, this binding may also represent syntrophin association with the ∼60-kD dystrobrevin. In fact, syntrophin also bound a larger protein of the approximate molecular weight of full-length dystrobrevin (55). Finally, multiple syntrophins may also be recruited to dystrophin complexes by association with other DAPs (31).
The model we favor incorporates a previous proposal (7) that dystrophin and dystrobrevin interact via their coiled-coils. Considerable evidence, including new results that we present here, supports an association of dystrophin with dystrobrevin. Dystrophin and dystrobrevin colocalize in skeletal muscle (8, 12), copurify biochemically (51), and associate directly in vitro via the coiled-coil region of dystrophin (50). We now find that dystrobrevin complexes isolated with an antibody to the coiled-coil region of dystrobrevin contain only small amounts of dystrophin when compared to complexes isolated with an antibody directed to another site. Thus, antibody binding to the coiled-coil region is incompatible with dystrophin–dystrobrevin association. Finally, purified dystrophin complexes can be partially dissociated into three groups, a dystroglycan subcomplex, a sarcoglycan subcomplex, and a dystrophin–dystrobrevin– syntrophin subcomplex by treatment with n-octyl β-d-glucoside (57). Detailed characterization of this association will be needed to determine the precise stoichiometry and binding orientation of the coiled-coil interaction. Despite this reservation, the evidence for an interaction between dystrophin and dystrobrevin is quite strong and leads us to suggest a new model for the dystrophin COOH-terminal region in which dystrophin and dystrobrevin combine to recruit two syntrophins per complex (Fig. 9 A).
Figure 9.
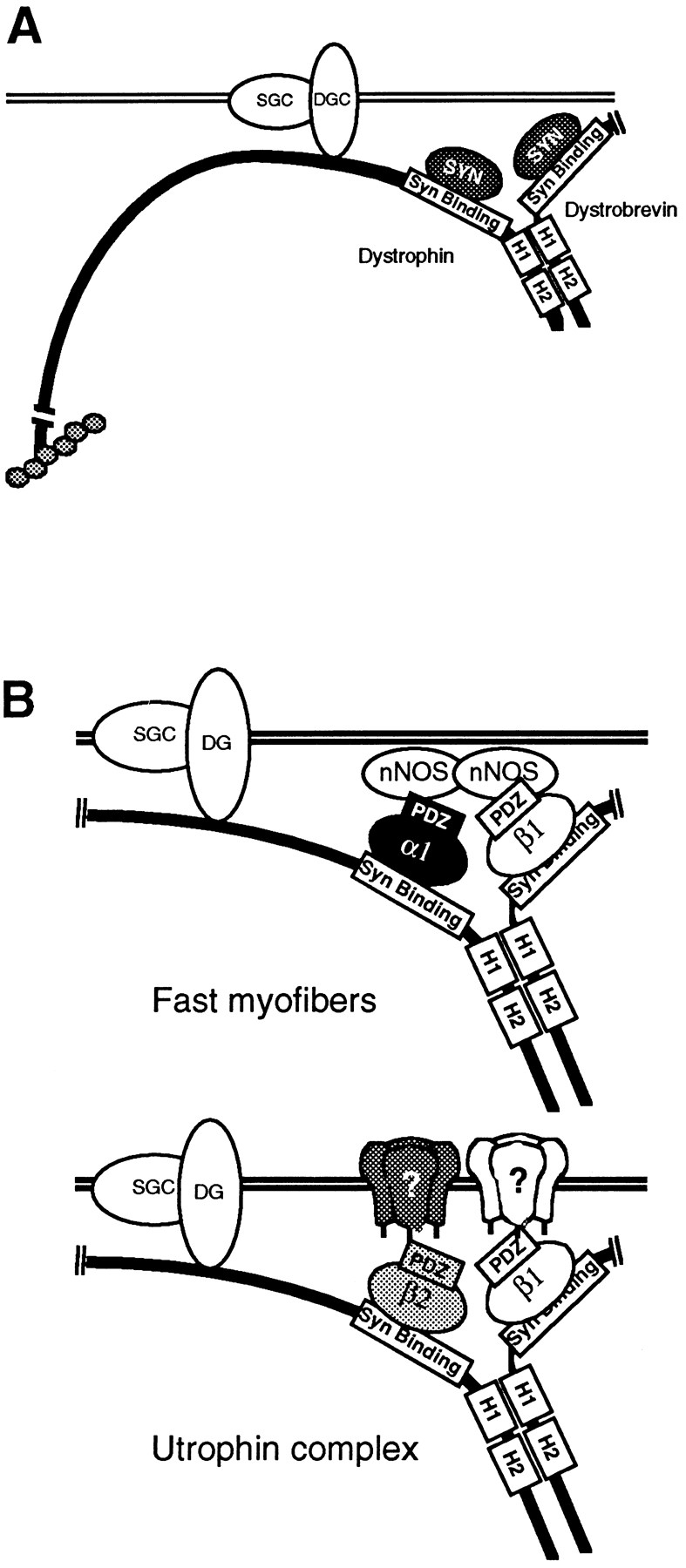
(A) Hypothetical model of the dystrophin complex containing two syntrophin-binding proteins. Syntrophins bind the exon 74–encoded region of dystrophin and the homologous region of dystrobrevin (6, 15). In both proteins, the syntrophin-binding site is followed by two heptad repeats of leucines (H1 and H2), separated by a short linker region (7). (B) Hypothetical examples of syntrophin isoform combinations as adapters linking membrane proteins or effector enzymes to dystrophin and utrophin. In fast-twitch myofibers, α1- and/or β1-syntrophin bind nNOS (10). In slow-twitch myofibers, α1-syntrophin is the predominant isoform present on the sarcolemma. Since slow-twitch fibers lack nNOS, the α1-syntrophin binding partners are unknown. Utrophin complexes contain β1- and β2-syntrophins. The binding partners for both syntrophins may be of particular relevance to NMJ function. DGC, dystroglycan complex; SGC, sarcoglycan complex.
In skeletal muscle, which expresses all three syntrophin isoforms, particular pairs of syntrophins preferentially associate with dystrophin or utrophin complexes. These preferential associations are not likely to result from intrinsic selectivity of either dystrophin or utrophin for particular syntrophins. Previous studies have shown that dystrophin, utrophin, and dystrobrevin are each able to bind any of the three syntrophin isoforms in vitro (5, 6). From our studies of native complexes from control, mdx, and mdx transgenic mice, we conclude that these associations are selective but not absolutely exclusive. Nevertheless, the pairings of syntrophin isoforms with dystrophin family members appear to be more highly regulated than suggested by in vitro studies. Potential mechanisms that could account for this selectivity include cell-specific expression of particular syntrophin isoforms and posttranslational modifications, such as phosphorylation, that alter the binding affinity of syntrophins for dystrophin and related proteins.
Since the pairing of syntrophin isoforms is thus unlikely to be determined by intrinsic selectivity for dystrophin family members, several possible pairings within an individual dystrophin–dystrobrevin complex can be envisioned. For example, the observed pairing of α1- and β1-syntrophins with dystrophin could represent either α1/β1 heterodimers or similar amounts of α1/α1 and β1/β1 homodimers with dystrophin. Indeed, these two possibilities are not mutually exclusive and may both occur in a single membrane region or may be differentially regulated at particular membrane regions. Distinguishing between these two possibilities in native complexes may prove technically difficult. Perhaps identification of the mechanisms that determine the syntrophin pairs may help to address these possibilities.
Syntrophins in Duchenne Muscular Dystrophy
A role for the syntrophins in muscular dystrophies or other myopathies has not received much attention, despite the fact that sarcolemmal expression of syntrophins, like that of the other proteins of the dystrophin complex, is dramatically reduced in Duchenne muscular dystrophy (DMD). Defects in other proteins of the dystrophin complex are the primary causes of other muscular dystrophies in which dystrophin is normal. For example, mutations in the genes encoding the sarcoglycans have been implicated in severe childhood autosomal recessive muscular dystrophy and in several forms of limb-girdle muscular dystrophy (for review see reference 37). Although no myopathies or other diseases have been linked to primary defects in syntrophins, our finding that β1-syntrophin is enriched in fast-twitch type IIB muscle fibers raises new issues with regard to the importance of syntrophins in DMD. Normally, dystrophin is expressed in all myofiber types (23), yet DMD preferentially affects human fast type IIB fibers (52). Although the mechanisms underlying this selective pathology remain to be determined, an intriguing possibility is that among the syntrophins, the function of β1-syntrophin is particularly important in maintaining sarcolemmal integrity.
Identifying other proteins associated with β1-syntrophin may be particularly relevant to understanding the molecular origin of the DMD myopathy. One possibility is that β1-syntrophin mediates the targeting of the neuronal form of nitric oxide synthase (nNOS) to the membrane. Like β1-syntrophin, nNOS is expressed preferentially in fast-twitch myofibers and is the only other protein known to correlate so closely with the fiber-type specific onset of DMD (26, 52). In normal muscle, nNOS is found on the sarcolemma in association with the dystrophin complex (9). This association is mediated, at least in part, by PDZ– PDZ heterodimerization between nNOS and syntrophin (10). Thus far, this nNOS interaction has been studied only in vitro with α1-syntrophin. It is possible that in vivo β1-syntrophin alone or paired with α1-syntrophin may be essential for targeting of nNOS to the sarcolemma, and could thus explain the fiber-type differences seen for this enzyme.
The physiological role of nNOS in normal skeletal muscle remains unknown, although some results suggest that it promotes muscle relaxation through a cGMP signaling pathway (26). The mechanism by which nNOS is activated in skeletal muscle is also poorly understood, although it is known that activation requires calcium/calmodulin and nNOS homodimerization (25). Neither the source of activating calcium nor the requirements for dimerization has been determined. Syntrophin association may play a role in both aspects of nNOS regulation. For example, close pairing of two syntrophins (discussed below) may facilitate nNOS dimerization, while syntrophin-mediated membrane localization may target nNOS to an appropriate Ca2+ source. Furthermore, loss of the syntrophin–nNOS complex from the membrane in dystrophic muscle may disrupt the nNOS-mediated relaxation pathway in favor of inappropriate cytoplasmic nNOS activation. This combination of loss of a potentially protective activity and acquisition of a potentially cytotoxic function may be central to DMD pathogenesis.
Syntrophins at the Neuromuscular Synapse
All three syntrophins are concentrated at the NMJ, but only β2-syntrophin is highly restricted to the synapse. α1- and β1-syntrophins are found on the entire sarcolemma with particularly high concentrations at synapses, a distribution similar to dystrophin. In contrast, the distribution of β2-syntrophin more closely resembles that of utrophin. Using immunoaffinity purification from whole skeletal muscle, we find that dystrophin preparations are enriched in α1- and β1-syntrophins, while utrophin complexes contain β1- and β2-syntrophin. Thus, while it is tempting to speculate that these preferential syntrophin–dystrophin/ utrophin pairings hold within the postsynaptic apparatus, two considerations make this conclusion premature. First, we find that all three syntrophins remain concentrated at the NMJ in mdx mice, possibly in association with utrophin. Second, the utrophin complexes isolated from skeletal muscle by biochemical means are not derived exclusively from the postsynaptic membrane, but come in large part from nonmuscle cells. Thus, it remains to be seen which syntrophin or combination of syntrophins colocalize with utrophin at the AChR-rich crests of the postsynaptic folds, or with dystrophin in the troughs, the site of high sodium channel density. Future studies will address this issue and the identification of synapse-specific binding partners for β2-syntrophin since it is unique among all the DAPs in its restriction to the postsynaptic apparatus.
Paired Syntrophins: Implications for Function
The discovery of two syntrophin isoforms in dystrophin/ utrophin complexes has clear implications for the mechanism of syntrophin function within membrane specializations. Specifically, the pairing of two PDZ-containing proteins in a submembrane complex resembles the membrane-associated guanylate kinase (MAGUK) complex. Like the syntrophins, the MAGUKs are a multigene family of PDZ-containing proteins that form submembranous protein scaffolds. The best characterized MAGUKs are the neuronal forms: PSD-95, Chapsyn-110, SAP97, and SAP102, which when expressed in COS cells combine to form homotypic and heterotypic multimers (24). MAGUKs have recently been shown to cluster membrane proteins such as K+ channels and NMDA-type glutamate receptors (for review see reference 48). The PDZ domains of certain MAGUKs mediate binding to the extreme COOH terminus of the cytoplasmic tail of membrane proteins (14, 49). Because this binding is dependent on the sequence of the COOH-terminal tail of the membrane protein, different MAGUK isoforms appear to bind different sets of membrane proteins. Thus, the combination of MAGUK isoforms at a particular membrane site may determine the composition of membrane proteins clustered there.
In contrast to the MAGUKs, which have three PDZ domains, syntrophins have only a single PDZ motif. This would appear to limit the ability of syntrophins to act as adapters in membrane organization by binding simultaneously multiple types of membrane proteins (such as ion channels) or a single membrane protein and an effector enzyme (such as nNOS). However, an attractive feature of the model in which two syntrophins are present in a single dystrophin complex is that each simultaneously could bind different proteins. Furthermore, a single complex might bind different combinations of proteins, depending on the specificity of the syntrophin PDZ domains. Thus, by regulating the syntrophin isoforms associated with dystrophin and dystrobrevin, or utrophin and dystrobrevin, the associated membrane or signaling protein might be different. The models shown in Fig. 9 show hypothetical combinations that might confer distinct functions on utrophin and dystrophin complexes, depending on the binding partners for the syntrophin PDZ domains.
Further studies will be required to determine if syntrophins play a role similar to that of the MAGUKs in membrane organization. Identifying the syntrophin PDZ domain binding partners and understanding the mechanisms that regulate the combinations of syntrophin isoforms in dystrophin complexes will be important for understanding the role of syntrophins in muscular dystrophy and in synapse formation.
Acknowledgments
We thank our colleagues in the Froehner and Sealock laboratories for helpful discussions and comments on the manuscript. We are indebted to A.H. Ahn and L.M. Kunkel for providing human β1-syntrophin cDNA and results before publication, to J.S. Chamberlain for supplying the Δ71– 74/mdx transgenic mice, to Katherine North for assistance with skeletal muscle fiber-typing, to J.B. Cohen for providing mAB 13H1, to R. Sealock and N.R. Kramarcy for providing Ab DYS3669, and to Kirk McNaughton and Curtis Conner for extensive assistance with cryosectioning.
This work was supported by National Institutes of Health Grants NS33145 (to S.C. Froehner and R. Sealock) and NS14871 (to S.C. Froehner) and a Muscular Dystrophy Association (MDA) Grant (to S.C. Froehner). M.E. Adams was an MDA postdoctoral fellow.
Footnotes
1. Abbreviations used in this paper: DAPs, dystrophin-associated proteins; DMD, Duchenne muscular dystrophy; DRP, dystrophin-related protein; HB, homogenization buffer; MAGUK, membrane-associated guanylate kinase; MHC, myosin heavy chain; NMJ, neuromuscular junction; nNOS, neuronal nitric oxide synthase; PDZ, protein domain originally identified in postsynaptic density-95, discs large, ZO-1; PH, pleckstrin homology; SU, syntrophin-unique.
Address all correspondence to S.C. Froehner, Department of Physiology, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599-7545. Tel.: (919) 966-1239. Fax: (919) 966-6413. E-mail: froehner@med.unc.edu
References
- 1.Adams ME, Butler MH, Dwyer TM, Peters MF, Murnane AA, Froehner SC. Two forms of mouse syntrophin, a 58 kd dystrophin-associated protein, differ in primary structure and tissue distribution. Neuron. 1993;11:531–540. doi: 10.1016/0896-6273(93)90157-m. [DOI] [PubMed] [Google Scholar]
- 2.Adams ME, Dwyer TM, Dowler LL, White RA, Froehner SC. Mouse α1- and β2-syntrophin gene structure, chromosome localization, and homology with a discs large domain. J Biol Chem. 1995;270:25859–25865. doi: 10.1074/jbc.270.43.25859. [DOI] [PubMed] [Google Scholar]
- 3.Ahn AH, Kunkel LM. The structural and functional diversity of dystrophin. Nat Genet. 1993;3:283–291. doi: 10.1038/ng0493-283. [DOI] [PubMed] [Google Scholar]
- 4.Ahn AH, Kunkel LM. Syntrophin binds to an alternatively spliced exon of dystrophin. J Cell Biol. 1995;128:363–371. doi: 10.1083/jcb.128.3.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahn AH, Yoshida M, Anderson MS, Feener CA, Selig S, Hagiwara Y, Ozawa E, Kunkel LM. Cloning of human basic A1, a distinct 59-kDa dystrophin-associated protein encoded on chromosome 8q23-24. Proc Natl Acad Sci USA. 1994;91:4446–4450. doi: 10.1073/pnas.91.10.4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahn AH, Feener CA, Gussoni E, Yoshida M, Ozawa E, Kunkel LM. The three human syntrophin genes are expressed in diverse tissues, have distinct chromosomal locations, and each bind to dystrophin and its relatives. J Biol Chem. 1996;271:2724–2730. doi: 10.1074/jbc.271.5.2724. [DOI] [PubMed] [Google Scholar]
- 7.Blake DJ, Tinsley JM, Davies KE, Knight AE, Winder SJ, Kendrick-Jones J. Coiled-coil regions in the carboxy-terminal domains of dystrophin and related proteins: potentials for protein-protein interactions. Trends Biochem Sci. 1995;20:133–135. doi: 10.1016/s0968-0004(00)88986-0. [DOI] [PubMed] [Google Scholar]
- 8.Blake DJ, Nawrotzki R, Peters MF, Froehner SC, Davies KE. Isoform diversity of dystrobrevin, the murine 87-kDa postsynaptic protein. J Biol Chem. 1996;271:7802–7810. doi: 10.1074/jbc.271.13.7802. [DOI] [PubMed] [Google Scholar]
- 9.Brenman JE, Chao DS, Xia H, Aldape K, Bredt DS. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell. 1995;82:743–752. doi: 10.1016/0092-8674(95)90471-9. [DOI] [PubMed] [Google Scholar]
- 10.Brenman JE, Chao DS, Gee SH, McGee AW, Craven SE, Santillano DR, Wu Z, Huang F, Xia H, Peters MF, et al. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and α1-syntrophin mediated by PDZ domains. Cell. 1996;84:757–767. doi: 10.1016/s0092-8674(00)81053-3. [DOI] [PubMed] [Google Scholar]
- 11.Butler MH, Douville K, Murnane AA, Kramarcy NR, Cohen JB, Sealock R, Froehner SC. Association of the Mr 58,000 postsynaptic protein of electric tissue with Torpedodystrophin and the Mr 87,000 postsynaptic protein. J Biol Chem. 1992;267:6213–6218. [PubMed] [Google Scholar]
- 12.Carr C, Fischbach GD, Cohen JB. A novel 87,000-Mr protein associated with acetylcholine receptors in Torpedoelectric organ and vertebrate skeletal muscle. J Cell Biol. 1989;109:1753–1764. doi: 10.1083/jcb.109.4.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chomczynski P, Sacchi N, Harrison SC. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 14.Cohen NA, Brenman JE, Synder SH, Bredt DS. Binding of the inward rectifier K+ channel Kir 2.3 to PSD-95 is regulated by protein kinase A phosphorylation. Neuron. 1996;17:759–767. doi: 10.1016/s0896-6273(00)80207-x. [DOI] [PubMed] [Google Scholar]
- 15.Dwyer TM, Froehner SC. Direct binding of Torpedosyntrophin to dystrophin and the 87 kDa dystrophin homologue. FEBS (Fed Eur Biochem Sci) Lett. 1995;375:91–94. doi: 10.1016/0014-5793(95)01176-f. [DOI] [PubMed] [Google Scholar]
- 16.Ervasti JM, Campbell KP. Membrane organization of the dystrophin-glycoprotein complex. Cell. 1991;66:1121–1131. doi: 10.1016/0092-8674(91)90035-w. [DOI] [PubMed] [Google Scholar]
- 17.Froehner SC, Murnane AA, Tobler M, Peng HB, Sealock R. A postsynaptic Mr 58,000 (58K) protein concentrated at acetylcholine receptor–rich sites in Torpedoelectroplaques and skeletal muscle. J Cell Biol. 1987;104:1633–1646. doi: 10.1083/jcb.104.6.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Froehner, S.C., M.E. Adams, M.F. Peters, and S.G. Gee. 1997. Syntrophins—modular adapter proteins at the neuromuscular junction and the sarcolemma. In Cytoskeletal Regulation of Membrane Function. S.C. Froehner and V. Bennett, editors. The Rockefeller University Press, New York. 197–207. [PubMed]
- 19.Gibson TJ, Hyvonen M, Musacchio A, Saraste M, Birney E. PH domain: the first anniversary. Trends Biochem Sci. 1994;19:349–353. doi: 10.1016/0968-0004(94)90108-2. [DOI] [PubMed] [Google Scholar]
- 20.Giloh H, Sedat JW. Fluorescence microscopy: reduced photobleaching of rhodamine and fluorescein protein conjugates by n-propyl gallate. Science (Wash DC) 1982;217:1252–1255. doi: 10.1126/science.7112126. [DOI] [PubMed] [Google Scholar]
- 21.Henry MD, Campbell KP. Dystroglycan: an extracellular matrix receptor linked to the cytoskeleton. Curr Opin Cell Biol. 1996;8:625–631. doi: 10.1016/s0955-0674(96)80103-7. [DOI] [PubMed] [Google Scholar]
- 22.Hoffman EP, Monaco AP, Feener CC, Kunkel LM. Conservation of the Duchenne muscular dystrophy gene in mice and humans. Science (Wash DC) 1987;238:347–350. doi: 10.1126/science.3659917. [DOI] [PubMed] [Google Scholar]
- 23.Hoffman EP, Hudecki MS, Rosenberg PA, Pollina CM, Kunkel LM. Cell and fiber-type distribution of dystrophin. Neuron. 1988;1:411–420. doi: 10.1016/0896-6273(88)90191-2. [DOI] [PubMed] [Google Scholar]
- 24.Kim E, Cho K, Rothschild A, Sheng M. Heteromultimerization and NMDA receptor-clustering activity of chapsyn-110, a member of the PSD-95 family of proteins. Neuron. 1996;17:103–113. doi: 10.1016/s0896-6273(00)80284-6. [DOI] [PubMed] [Google Scholar]
- 25.Klatt P, Schmid M, Leopold E, Schmidt K, Werner ER, Mayer B. The pteridine binding site of brain nitric oxide synthase. Tetrahydrobiopterin binding kinetics, specificity, and allosteric interaction with the substrate domain. J Biol Chem. 1994;269:13861–13866. [PubMed] [Google Scholar]
- 26.Kobzik L, Reid MB, Bredt DS, Stamler JS. Nitric oxide in skeletal muscle. Nature (Lond) 1994;372:546–548. doi: 10.1038/372546a0. [DOI] [PubMed] [Google Scholar]
- 27.Kozak M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell. 1986;44:283–292. doi: 10.1016/0092-8674(86)90762-2. [DOI] [PubMed] [Google Scholar]
- 28.Kramarcy NR, Vidal A, Froehner SC, Sealock R. Association of utrophin and multiple dystrophin short forms with the mammalian M(r) 58,000 dystrophin-associated protein (syntrophin) J Biol Chem. 1994;269:2870–2876. [PubMed] [Google Scholar]
- 29.Lidov HG, Byers TJ, Watkins SC, Kunkel LM. Localization of dystrophin to postsynaptic regions of central nervous system cortical neurons. Nature (Lond) 1990;348:725–728. doi: 10.1038/348725a0. [DOI] [PubMed] [Google Scholar]
- 30.Luna, L.G. 1968. Manual of Histologic Staining Methods of the Armed Forces Institute of Pathology. McGraw-Hill, New York. 36–40.
- 31.Madhavan R, Jarrett HW. Interactions between dystrophin glycoprotein complex proteins. Biochemistry. 1995;34:12204–12209. doi: 10.1021/bi00038a014. [DOI] [PubMed] [Google Scholar]
- 32.McNally MN, Duggan D, Gorospe RJ, Bonnemann CG, Fanin M, Pegoraro E, Lidov HGW, Noguchi S, Ozawa E, Finkel RS, et al. Mutations that disrupt the carboxyl-terminus of delta-sarcoglycan cause muscular dystrophy. Hum Mol Genet. 1996;5:1841–1847. doi: 10.1093/hmg/5.11.1841. [DOI] [PubMed] [Google Scholar]
- 33.Montanaro F, Carbonetto S, Campbell KP, Lindenbaum M. Dystroglycan expression in the wild type and mdx mouse neural retina: synaptic colocalization with dystrophin, dystrophin-related protein but not laminin. J Neurosci Res. 1995;42:528–538. doi: 10.1002/jnr.490420411. [DOI] [PubMed] [Google Scholar]
- 34.Nguyen thi M, Cartwright AJ, Morris GE, Love DR, Bloomfield JF, Davies KE. Monoclonal antibodies against defined regions of the muscular dystrophy protein, dystrophin. FEBS (Fed Eur Biochem Sci) Lett. 1990;262:237–240. doi: 10.1016/0014-5793(90)80199-s. [DOI] [PubMed] [Google Scholar]
- 35.Nigro V, Piluso P, Belsito A, Politano L, Puca AA, Papparella S, Rossi E, Viglietto G, Esposito MG, Abbondanza C, et al. Identification of a novel sarcoglycan gene at 5q33 encoding a sarcolemmal 35 kDa glycoprotein. Hum Mol Genet. 1996;5:1179–1186. doi: 10.1093/hmg/5.8.1179. [DOI] [PubMed] [Google Scholar]
- 36.Ousley AH, Froehner SC. An anti-peptide antibody specific for the class A calcium channel α1 subunit labels mammalian neuromuscular junction. Proc Natl Acad Sci USA. 1994;91:12263–12267. doi: 10.1073/pnas.91.25.12263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ozawa E, Yoshida M, Suzuki A, Mizuno Y, Hagiwara Y, Noguchi S. Dystrophin-associated proteins in muscular dystrophy. Hum Mol Genet. 1995;4:1711–1716. doi: 10.1093/hmg/4.suppl_1.1711. [DOI] [PubMed] [Google Scholar]
- 38.Peters MF, Kramarcy NR, Sealock R, Froehner SC. β2-Syntrophin: localization at the neuromuscular junction in skeletal muscle. Neuroreport. 1994;5:1577–1580. [PubMed] [Google Scholar]
- 39.Rafael JA, Sunada Y, Cole NM, Campbell KP, Faulkner JA, Chamberlain JS. Prevention of dystrophic pathology in mdxmice by a truncated dystrophin isoform. Hum Mol Genet. 1994;3:1725–1733. doi: 10.1093/hmg/3.10.1725. [DOI] [PubMed] [Google Scholar]
- 40.Rafael JA, Cox GA, Corrado K, Jung D, Campbell KP, Chamberlain JS. Forced expression of dystrophin deletion constructs reveals structure-function correlations. J Cell Biol. 1996;134:93–102. doi: 10.1083/jcb.134.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sadoulet-Puccio HM, Khurana TS, Cohen JB, Kunkel LM. Cloning and characterization of the human homologue of a dystrophin related phosphoprotein found at the Torpedoelectric organ post-synaptic membrane. Hum Mol Genet. 1996;5:489–496. doi: 10.1093/hmg/5.4.489. [DOI] [PubMed] [Google Scholar]
- 42.Sato T, Irie S, Kitada S, Reed J. FAP-1: a protein tyrosine phosphatase that associates with Fas. Science (Wash DC) 1995;268:411–415. doi: 10.1126/science.7536343. [DOI] [PubMed] [Google Scholar]
- 43.Schagger H, von Jagow G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- 44.Schiaffino S, Gorza L, Sartore S, Saggin L, Ausoni S, Vianello M, Gundersen K, Lomo T. Three myosin heavy chain isoforms in type 2 skeletal muscle fibres. J Musc Res Cell Motil. 1989;10:197–205. doi: 10.1007/BF01739810. [DOI] [PubMed] [Google Scholar]
- 45.Schmitz F, Holbach M, Drenckhahn D. Colocalization of retinal dystrophin and actin in postsynaptic dendrites of rod and cone photoreceptor synapses. Histochemistry. 1993;100:473–479. doi: 10.1007/BF00267828. [DOI] [PubMed] [Google Scholar]
- 46.Sealock R, Froehner SC. Dystrophin-associated proteins and synapse formation: is α-dystroglycan the agrin receptor? . Cell. 1994;77:617–619. doi: 10.1016/0092-8674(94)90045-0. [DOI] [PubMed] [Google Scholar]
- 47.Shaw G. The pleckstrin homology domain: an intriguing multifunctional protein module. Bioessays. 1996;18:35–46. doi: 10.1002/bies.950180109. [DOI] [PubMed] [Google Scholar]
- 48.Sheng M. PDZs and receptor/channel clustering: rounding up the latest suspects. Neuron. 1996;17:575–578. doi: 10.1016/s0896-6273(00)80190-7. [DOI] [PubMed] [Google Scholar]
- 49.Songyang Z, Fanning AS, Fu C, Xu J, Marfatia SM, Chishti AH, Crompton A, Chan AC, Anderson JM, Cantley LC. Recognition of unique carboxyl-terminal motifs by distinct PDZ domains. Science (Wash DC) 1997;275:73–77. doi: 10.1126/science.275.5296.73. [DOI] [PubMed] [Google Scholar]
- 50.Suzuki A, Yoshida M, Ozawa E. Mammalian α1- and β1-syntrophin bind to the alternative splice-prone region of the dystrophin COOH terminus. J Cell Biol. 1995;128:373–381. doi: 10.1083/jcb.128.3.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wagner KR, Cohen JB, Huganir RL. The 87K postsynaptic membrane protein from Torpedois a protein-tyrosine kinase substrate homologous to dystrophin. Neuron. 1993;10:511–522. doi: 10.1016/0896-6273(93)90338-r. [DOI] [PubMed] [Google Scholar]
- 52.Webster C, Silberstein L, Hays AP, Blau HM. Fast muscle fibers are preferentially affected in Duchenne muscular dystrophy. Cell. 1988;52:503–513. doi: 10.1016/0092-8674(88)90463-1. [DOI] [PubMed] [Google Scholar]
- 53.Yamamoto H, Mizuno Y, Hayashi K, Nonaka I, Yoshida M, Ozawa E. Expression of dystrophin-associated protein 35DAG (A4) and 50DAG (A2) is confined to striated muscles. J Biochem. 1994;115:162–167. doi: 10.1093/oxfordjournals.jbchem.a124294. [DOI] [PubMed] [Google Scholar]
- 54.Yang B, Ibraghimov-Beskrovnaya O, Moomaw CR, Slaughter CA, Campbell KP. Heterogeneity of the 59-kDa dystrophin-associated protein revealed by cDNA cloning and expression. J Biol Chem. 1994;269:6040–6044. [PubMed] [Google Scholar]
- 55.Yang B, Jung D, Rafael JA, Chamberlain JS, Campbell KP. Identification of α-syntrophin binding to syntrophin triplet, dystrophin, and utrophin. J Biol Chem. 1995;270:4975–4978. doi: 10.1074/jbc.270.10.4975. [DOI] [PubMed] [Google Scholar]
- 56.Yoshida M, Ozawa E. Glycoprotein complex anchoring dystrophin to sarcolemma. J Biochem. 1990;108:748–752. doi: 10.1093/oxfordjournals.jbchem.a123276. [DOI] [PubMed] [Google Scholar]
- 57.Yoshida M, Suzuki A, Yamamoto H, Noguchi S, Mizuno Y, Ozawa E. Dissociation of the complex of dystrophin and its associated proteins into several unique groups by n-octyl β-D-glucoside. Eur J Biochem. 1994;222:1055–1061. doi: 10.1111/j.1432-1033.1994.tb18958.x. [DOI] [PubMed] [Google Scholar]
- 58.Yoshida M, Yamamoto H, Noguchi S, Mizuno Y, Hagiwara Y, Ozawa E. Dystrophin-associated protein A0 is a homologue of the Torpedo87K protein. FEBS (Fed Eur Biochem Sci) Lett. 1995;367:311–314. doi: 10.1016/0014-5793(95)00574-s. [DOI] [PubMed] [Google Scholar]