

Abstract
We purified from rat brain a novel actin filament (F-actin)–binding protein of ∼180 kD (p180), which was specifically expressed in neural tissue. We named p180 neurabin (neural tissue–specific F-actin– binding protein). We moreover cloned the cDNA of neurabin from a rat brain cDNA library and characterized native and recombinant proteins. Neurabin was a protein of 1,095 amino acids with a calculated molecular mass of 122,729. Neurabin had one F-actin–binding domain at the NH2-terminal region, one PSD-95, DlgA, ZO-1–like domain at the middle region, a domain known to interact with transmembrane proteins, and domains predicted to form coiled-coil structures at the COOH-terminal region. Neurabin bound along the sides of F-actin and showed F-actin–cross-linking activity. Immunofluorescence microscopic analysis revealed that neurabin was highly concentrated in the synapse of the developed neurons. Neurabin was also concentrated in the lamellipodia of the growth cone during the development of neurons. Moreover, a study on suppression of endogenous neurabin in primary cultured rat hippocampal neurons by treatment with an antisense oligonucleotide showed that neurabin was involved in the neurite formation. Neurabin is a candidate for key molecules in the synapse formation and function.
During the development of the nervous system, the distal tip of the elongating axon—the growth cone—actively migrates toward its target cell in response to the combined actions of attractive and repulsive guidance molecules in the extracellular environment (Garrity and Zipursky, 1995; Keynes and Cook, 1995; Chiba and Keshishian, 1996; Culotti and Kolodkin, 1996; Friedman and O'Leary, 1996; Tessier-Lavigne and Goodman, 1996). When the growth cone contacts with the target cell, it is transformed into the functional presynaptic terminal (Garrity and Zipursky, 1995; Chiba and Kishishian, 1996). The actin cytoskeleton has been shown to play crucial roles in these processes of the synapse formation (Mitchison and Kirschner, 1988; Smith, 1988; Bentley and O'Connor, 1994; Lin et al., 1994; Mackay et al., 1995; Tanaka and Sabry, 1995).
In the developing nervous system, the actin cytoskeleton is prominent in two structural domains of the growth cone, filopodia and lamellipodia (Mitchison and Kirschner, 1988; Smith, 1988; Bentley and O'Connor, 1994; Lin et al., 1994; Mackay et al., 1995; Tanaka and Sabry, 1995). In these domains, actin filament (F-actin)1 assembled at the leading edge are transported into the center of the growth cone and disassembled there. It has been suggested that this retrograde flow of F-actin is crucial for the growth cone motility. Drugs that disrupt F-actin have also been shown to cause the lamellipodial and filopodial collapse and block the ability of neurons to extend the growth cone in the correct direction (Marsh and Letourneau, 1984; Forscher and Smith, 1988; Bentley and Toroian-Raymond, 1986; Chien et al., 1993). These results suggest that the actin cytoskeleton regulates not only the growth cone motility but also the growth cone directionality. Recently, a variety of guidance molecules and their receptors have been identified (Garrity and Zipursky, 1995; Keynes and Cook, 1995; Chiba and Keshishian, 1996; Culotti and Kolodkin, 1996; Friedman and O'Leary, 1996; Tessier-Lavigne and Goodman, 1996). However, which molecules of the actin cytoskeleton are essential for the growth cone motility and directionality is not well understood.
When the growth cone contacts with the target cell, the target cell regulates the development of the presynaptic nerve terminal and the formation of the functional synapse (Bowe and Fallon, 1995; Chiba and Keshishian, 1996). In the established nervous system, the presynaptic and postsynaptic membranes get aligned in space and constitute the synaptic junction (Burns and Augustine, 1995; Garner and Kindler, 1996). Electron microscopic studies have revealed the ultrastructural features of the synaptic junction (Burns and Augustine, 1995; Garner and Kindler, 1996). The presynaptic cytoplasm is characterized by synaptic vesicles (SVs). SVs are not distributed uniformly; SVs cluster together in the vicinity of the presynaptic plasma membrane, where F-actin forms a network and is associated with the presynaptic plasma membrane (Hirokawa et al., 1989). Most SVs within the cluster are linked through thin strands to each other, to F-actin, or to both (Hirokawa et al., 1989). A subset of SVs within the cluster are attached by fine filamentous threads to neurotransmitter release zone at the presynaptic plasma membrane (Hirokawa et al., 1989). The presynaptic submembranous cytoskeleton is assumed to be involved in recruiting Ca2+ channels and the components of the SV fusion complex, delivering SVs to the neurotransmitter release zone, and keeping them in place (Burns and Augustine, 1995; Garner and Kindler, 1996). At the inner surface of the post-synaptic plasma membrane, there is an electron dense thickening, called postsynaptic density. The postsynaptic density is assumed to be involved in the selective targeting and accumulation of ion channels and receptors (Burns and Augustine, 1995; Garner and Kindler, 1996). It is also assumed that the presynaptic and postsynaptic submembranous cytoskeleton elements are linked to cell adhesion molecules to regulate the synaptic stabilization and plasticity (Fields and Itoh, 1996; Garner and Kindler, 1996). The presynaptic and postsynaptic submembranous cytoskeleton elements are thought to be composed of spectrin/fodrin, ankyrin, α-adducin, and protein 4.1 isoforms and to be linked to F-actin through these cytoskeleton proteins (Garner and Kindler, 1996). However, little is known about which molecules of the submembranous cytoskeleton are essential for the synaptic transmission and/or the synaptic stabilization.
To understand the regulation of the actin cytoskeleton during and after the development of the nervous system, it is of crucial importance to identify F-actin–binding proteins implicated in the synapse formation and function. Therefore, we attempted here to isolate neural tissue–specific F-actin–binding proteins. We isolated a novel neural tissue–specific F-actin–binding protein from rat brain, which may be involved in neurite formation, and named it neurabin (neural tissue–specific F-actin–binding protein).
Materials and Methods
125I-Labeled F-Actin Blot Overlay
125I-Labeled F-actin blot overlay was done as described (Chia et al., 1991; Pestonjamasp et al., 1995). Briefly, purified actin monomer (G-actin) was labeled with 125I–Bolton Hunter reagent. 125I-Labeled G-actin (1 mg/ml, average specific activity, 63.3 μCi/mg) was polymerized with 18 μg/ml of gelsolin (Sigma Chemical Co., St. Louis, MO) (molar ratio, 100:1) by incubation for 10 min at 4°C in a solution containing 20 mM Pipes, pH 7.0, 50 mM KCl, and 2 mM MgCl2. Phalloidin was then added to give a final concentration of 40 μM. The mixture was then incubated for another 15 min at room temperature and stored at 4°C as 125I-labeled F-actin. The sample to be tested was subjected to SDS-PAGE and transferred to a nitrocellulose membrane. The membrane was blocked in TBS containing 5% defatted powder milk. The membrane was then incubated for 1 h at room temperature with 10 μg/ml of 125I-labeled F-actin in TBS containing 5% defatted powder milk, and 4 μM phalloidin. After the incubation, the membrane was washed with TBS containing 0.5% Tween 20, followed by autoradiography using an image analyzer (Fujix BAS-2000II; Fuji Photo Film Co., Tokyo, Japan).
For competition experiments, 125I-labeled F-actin was prepared as described above except that the concentration of gelsolin was reduced to 7.2 μg/ml (molar ratio, 250:1). 20 μg/ml of 125I-labeled F-actin was incubated for 30 min at room temperature with 0.42 μg/ml of myosin subfragment 1 (S1) (Sigma Chemical Co.) in a solution containing 20 mM Pipes, pH 7.0, 58 mM KCl, 2 mM MgCl2, 130 μM CaCl2, and 0.8 μM phalloidin. Where indicated, 4 mM MgATP was added to the mixture. After the incubation, the mixture was diluted with an equal volume of TBS containing 10% defatted powder milk, and it was added to the blot membrane, followed by incubation for 1 h at room temperature.
Purification of Neurabin
All the purification procedures were carried out at 0–4°C. The synaptic soluble fraction was prepared from 240 adult rat brains as described previously (Mizoguchi et al., 1990) and stored at −80°C until use. One sixth of the fraction (420 ml, 360 mg of protein) was adjusted to 0.2 M NaCl with 4 M NaCl, and applied to a Q-Sepharose FF column (2.6 × 10 cm; Pharmacia Biotechnology Inc., Piscataway, NJ) equilibrated with buffer A (20 mM Tris/Cl, pH 7.5, and 1 mM DTT) containing 0.2 M NaCl. After the column was washed with 250 ml of buffer A containing 0.2 M NaCl, elution was performed with 350 ml of buffer A containing 0.5 M NaCl at a flow rate of 5 ml/min. Fractions of 10 ml each were collected. Neurabin appeared in fractions 5–19. These fractions (150 ml, 152 mg of protein) were collected, and NaCl was added to give a final concentration of 2 M. The sample was applied to a phenyl-Sepharose column (2.6 × 10 cm; Pharmacia) equilibrated with buffer A containing 2 M NaCl. After the column was washed with 250 ml of the same buffer, elution was performed with a 360-ml linear gradient of NaCl (2.0–0 M) in buffer A, followed by 180 ml of buffer A, at a flow rate of 3 ml/min. Fractions of 6 ml each were collected. Neurabin appeared in fractions 53–59. The active fractions (42 ml, 6.1 mg of protein) were collected, and CHAPS was added to give a final concentration of 0.6%. The sample was then applied to a HPLC hydroxyapatite column (0.78 × 10 cm; Koken Co. Ltd., Tokyo, Japan) equilibrated with buffer B (20 mM potassium phosphate, pH 7.8, 1 mM DTT, 0.6% CHAPS, and 10% glycerol). After the column was washed with 120 ml of the same buffer, elution was performed with a 75-ml linear gradient of potassium phosphate (20–100 mM) in buffer B, and a subsequent 75-ml linear gradient (100–300 mM) in buffer B, followed by a 50-ml linear gradient (300– 500 mM) in buffer B, at a flow rate of 0.5 ml/min. Fractions of 2.5 ml each were collected. Neurabin appeared in fractions 68–74. The active fractions (17.5 ml, 0.22 mg of protein) were collected. The rest of the synaptic soluble fraction was also subjected to the successive column chromatographies in the same manner as described above. The active fractions of the six hydroxyapatite column chromatographies were combined. One third of the combined sample was diluted with an equal volume of buffer C (20 mM Tris/Cl, pH 7.5, 0.5 mM EDTA, 1 mM DTT, and 0.6% CHAPS), and then applied to a Mono Q HR 5/5 column (Pharmacia Biotechnology, Inc.) equilibrated with buffer C. After the column was washed with 15 ml of buffer C, elution was performed with 5 ml of buffer C containing 0.2 M NaCl and a subsequent 15-ml linear gradient of NaCl (0.2–0.5 M) in buffer C, followed by 5 ml of buffer C containing 1.0 M NaCl, at a flow rate of 0.5 ml/min. Fractions of 0.5 ml each were collected. Neurabin appeared in fractions 31–34. The active fractions (2 ml, 0.11 mg of protein) were collected. The rest of the combined sample of the hydroxyapatite column chromatographies was also subjected to the Mono Q column chromatography in the same manner as described above. The active fractions of the three Mono Q column chromatographies were combined and diluted with an equal volume of buffer D (20 mM Hepes, pH 7.5., 0.5 mM EGTA, and 1 mM DTT). The sample was applied to a Mono S PC 1.6/5 column (Pharmacia Biotechnology, Inc.) equilibrated with buffer D. After the column was washed with 2.0 ml of buffer D, elution was performed with a 1.5-ml linear gradient of NaCl (0–1.0 M), followed by 0.5 ml of buffer D containing 1.0 M NaCl, at a flow rate of 50 μl/min. Fractions of 50 μl each were collected. Neurabin appeared in fractions 16–17 (Fig. 1 B). The active fractions (100 μl, 30 μg of protein) were collected and stored at −80°C until use.
Figure 1.
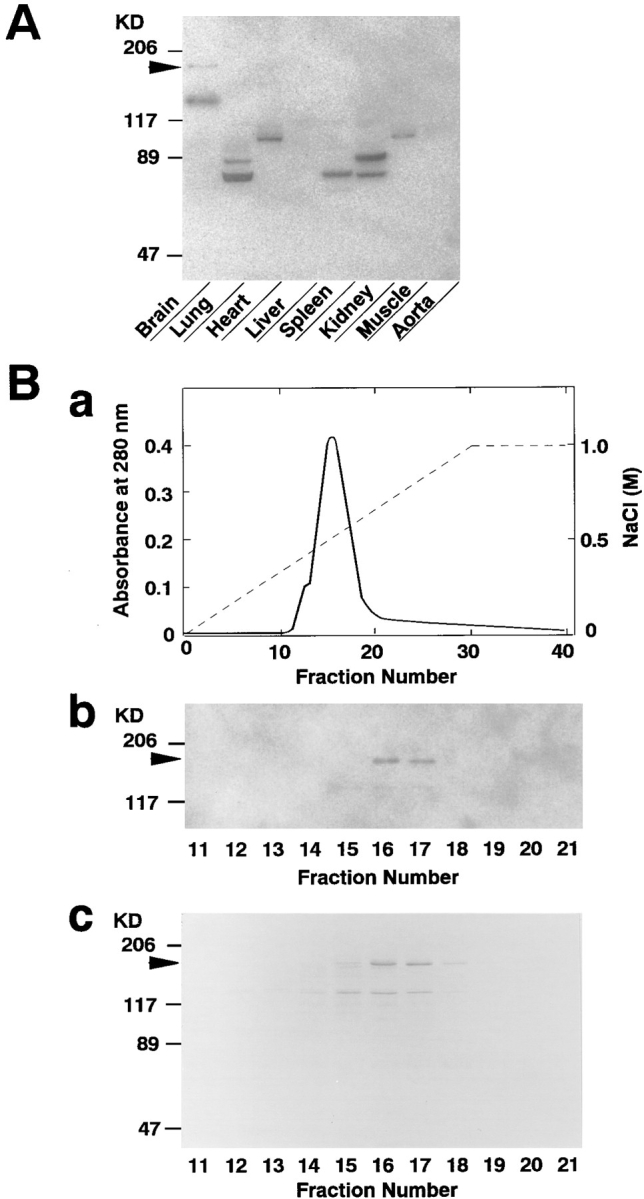
125I-Labeled F-actin–binding activities of various rat tissues and Mono S column chromatography of p180 (neurabin). (A) 125I-Labeled F-actin–binding activities of various rat tissues. The homogenates of various tissues (30 μg of protein each) were subjected to SDS-PAGE (8% polyacrylamide gel), followed by 125I-labeled F-actin blot overlay. (B) Mono S column chromatography. (a) Absorbance at 280 nm. (b) 125I-Labeled F-actin blot overlay. An aliquot 1-μl of each fraction was subjected to SDS-PAGE (8% polyacrylamide gel), followed by 125I-labeled F-actin blot overlay. (c) Protein staining with Coomassie brilliant blue. A 3-μl aliquot of each fraction was subjected to SDS-PAGE (8% polyacrylamide gel), followed by protein staining with Coomassie brilliant blue. Arrowheads indicate p180. Protein markers used are myosin (206), β-galactosidase (117), BSA (89), and ovalbumin (47).
Peptide Mapping of Neurabin and Molecular Cloning of Its cDNA
The purified Mono S sample (∼30 μg of protein) was subjected to SDS-PAGE (8% polyacrylamide gel). A protein band corresponding to a protein of ∼180 kD was cut out from the gel and digested with a lysyl endopeptidase, and the digested peptides were separated by TSKgel ODS-80Ts (4.6 × 150 mm; Tosoh, Tokyo, Japan) reverse phase high pressure liquid column chromatography as described (Imazumi et al., 1994). The amino acid (aa) sequences of the peptides were determined with a peptide sequencer. A rat brain cDNA library in λZAPII (Stratagene, La Jolla, CA) was screened using the oligonucleotide probes designed from the partial aa sequences. DNA sequencing was performed by the dideoxy nucleotide termination method using a DNA sequencer (ABI 373; Applied Biosystems, Inc., Foster City, CA).
For coiled-coil prediction, the MTK and MTIDK matrices of the COILS version 2.1 algorithm (Lupas et al., 1991; Lupas, 1996) were used with a 28-residue window. Weighting options were applied and a probability curve was generated.
Expression and Purification of Full-Length and Truncated Forms of Neurabin
Prokaryote and eukaryote expression vectors were constructed in pGexKG (Guan and Dixon, 1991), pCMV5, pCMV-myc (Takeuchi et al., 1997), and pAcYM1-His6 using standard molecular biology methods (Sambrook et al., 1989). Various glutathione-S-transferase (GST) fusion constructs of neurabin contained the following aa residues: pGex– neurabin-1, aa 1–144; pGex–neurabin-2, aa 1–210; pGex–neurabin-3, aa 145–293; pGex–neurabin-4, aa 286–615; pGex–neurabin-5, aa 505–791; pGex–neurabin-6, aa 665–1095; pGex–neurabin-7, aa 609–868; and pGex– neurabin-8, aa 890–1095. pCMV–neurabin contained full-length neurabin. Various myc-tagged constructs of neurabin contained the following aa residues to express the proteins with the NH2-terminal myc-epitope (MEQKLISEEDL); pCMV-myc–neurabin, full length; pCMV-myc–neurabin-1, aa 1–144; pCMV-myc–neurabin-2, aa 145–1095. The His6-tagged construct (pAcYM1-His6-neurabin) contained full-length neurabin to express the protein with the COOH-terminal six histidine residues.
For the GST fusion proteins, the GST fusion constructs were transformed into Escherichia coli. The GST fusion proteins were purified by use of glutathione-Sepharose beads. For the myc-tagged proteins, COS7 cells were transfected with the DEAE-dextran method (Hata and Südhof, 1995). For the His6-tagged protein, Spodoptera frugiperda (Sf9) cells were transfected with baculovirus carrying the His6-tagged construct (Kikuchi et al., 1995). The Sf9 cells were homogenized with buffer E (20 mM Tris/ Cl, pH 7.5, 1 mM DTT, 20 μg/ml leupeptin, 1 μg/ml pepstatin, and 20 μg/ ml aprotinin), and centrifuged at 100,000 g for 1 h. The supernatant was subjected to the phenyl-Sepharose column chromatography in the same manner as used for the purification of native neurabin. Each fraction was subjected to Western blot analysis using an antibody against neurabin (anti–neurabin-1) described below. The active fractions were collected, and 2 M imidazol/Cl, pH 7.2, was added to give a final concentration of 10 mM. The sample was subjected to Ni-agarose (Pharmacia Biotechnology, Inc.) column chromatography according to the manufacturer's protocol. The active fractions were collected and further subjected to the Mono S PC 1.6/5 column chromatography in the same manner as used for the purification of native neurabin. The active fractions were collected and used as His6-neurabin.
F-Actin–Cross-linking Activity
Low shear viscometry was performed as described (Pollard and Cooper, 1982; Kato et al., 1996). Briefly, the Mono S sample of native neurabin or His6-neurabin in an indicated amount was mixed with 0.28 mg/ml of G-actin in a solution containing 20 mM Tris/Cl, pH 7.2, 140 mM NaCl, 0.1 mM ATP, 0.5 mM DTT, and 1 mM EGTA, and the solution was sucked into a 0.1-ml micropipette. After the incubation at 25°C for various periods of time, the time for a stainless steel ball to fall a fixed distance in the pipette was measured.
Electron microscopy was performed as described (Endo and Masaki, 1982; Kato et al., 1996). Briefly, 0.28 mg/ml of G-actin was incubated at 25°C for 45 min with the Mono S sample of native neurabin (20 μg/ml of protein) in a solution containing 20 mM Tris/Cl, pH 7.2, 140 mM NaCl, 2 mM MgCl2, 0.1 mM ATP, 0.5 mM DTT, and 1 mM EGTA. The sample was negatively stained with 2% uranyl acetate and viewed with an electron microscope (model H-7100; Hitachi Ltd., Tokyo, Japan).
Activity of Cosedimentation with F-Actin
G-Actin was polymerized by incubation for 30 min at room temperature in a polymerization buffer (20 mM imidazol/Cl, pH 7.0, 2 mM MgCl2, 1 mM ATP, 0.5 mM DTT, and 90 mM KCl). GST–neurabin-1 (aa 1–144), GST–neurabin-2 (aa 1–210), or His6-neurabin in an indicated amount was incubated for 30 min at room temperature with 0.3 mg/ml of F-actin in a solution containing 20 mM imidazol/Cl, pH 7.0, 2 mM MgCl2, 1 mM ATP, 0.4 mM DTT, 27 mM KCl, 100 mM NaCl, and 0.2 mM EGTA, and the mixture (100 μl) was placed over a 50-μl cushion of 30% sucrose in the polymerization buffer. After the sample was centrifuged at 130,000 g for 20 min, the supernatant was removed from the cushion and the pellet was brought to the original volume in an SDS sample buffer. The comparable amounts of the supernatant and pellet fractions were subjected to SDS-PAGE, followed by protein staining with Coomassie brilliant blue to quantitate the fusion protein cosedimented with F-actin using a densitometer.
Gel Filtration and Sucrose Density Gradient Ultracentrifugation
In gel filtration analysis, His6-neurabin (2 μg of protein) was applied to a Superdex 200 PC 3.2/30 column (Pharmacia Biotechnology, Inc.) equilibrated with a buffer containing 20 mM Tris/Cl, pH 7.5, 150 mM NaCl, 1 mM EGTA, and 1 mM DTT. Elution was performed with the same buffer at a flow rate of 50 μl/min. Fractions of 50 μl each were collected. In sucrose density gradient ultracentrifugation analysis, His6-neurabin (0.2 ml, 5 μg of protein) was applied on a 4.8-ml, continuous sucrose density gradient (5–20% sucrose in 20 mM Tris/Cl, pH 7.5, 1 mM EGTA, and 1 mM DTT) and centrifuged at 235,000 g for 6 h at 4°C with a swing rotor (P55ST2; Hitachi Ltd.). After the ultracentrifugation, fractions of 150 μl each were collected. Each fraction was subjected to protein staining with silver and Western blot analysis using the anti–neurabin-1 antibody described below.
Primary Cultured Rat Hippocampal Neurons
Primary cultured rat hippocampal neurons were prepared as described (Takeuchi et al., 1997). Briefly, hippocampi were isolated from rat embryo (20-d gestation), dissociated, plated on poly-l-lysine–coated glass coverslips, and then cultured in MEM with 10% horse serum. After 4 d in culture, the medium was replaced with MEM supplemented with N2 supplement, 1 mg/ml of ovalbumin, 1 mM pyruvate, and 5 mM cytosine arabinoside, cultured for the indicated period, and then subjected to Western blot analysis and immunofluorescence microscopy.
Antibodies and Immunofluorescence Staining
A rabbit polyclonal antibody against neurabin was raised against GST– neurabin-7 (aa 609–868, anti–neurabin-1) or GST–neurabin-8 (aa 890– 1095, anti–neurabin-2). The antiserum was affinity purified with each GST fusion protein covalently coupled to NHS-activated Sepharose (Pharmacia Biotechnology, Inc.). The specificities of these antibodies were confirmed by Western blot analyses on the pCMV–neurabin-transfected COS7 cells versus mock cells. A monoclonal anti–α-actinin antibody was prepared as described (Kato et al., 1996). Monoclonal anti–myc-epitope (American Type Culture Collection, Rockville, MD), anti–synaptotagmin I (Wako Pure Chemical Industries, Ltd., Osaka, Japan), anti-synaptophysin (Boehringer Mannheim Corp., Indianapolis, IN), and anti–α-tubulin (Amersham Corp., Arlington Heights, IL) antibodies and a polyclonal anti–synapsin I (Chemicon International, Inc., Temecula, CA) were obtained from commercial sources.
For the immunofluorescence microscopy of cultured cells, the cells were washed with PBS, fixed with 4% paraformaldehyde in PBS for 1 h, washed with solution A (20 mM phosphate buffer, pH 7.2, and 0.45 M NaCl), and then treated for 30 min with a solution containing 20 mM Tris/ Cl, pH 7.5, 140 mM NaCl, 0.1% Triton X-100, and 20% defatted powder milk. The samples were incubated for 2 h with the rabbit anti–neurabin-1, mouse anti–α-tubulin, mouse anti–synaptotagmin I, and/or mouse anti-myc antibodies. After the samples were washed with solution A, they were incubated for 30 min with fluorescence-conjugated, anti–rabbit and/ or anti–mouse antibodies, including FITC-conjugated, rhodamine-conjugated, and/or Cy5-conjugated antibodies. For the double staining with F-actin, these second antibodies were mixed with rhodamine-phalloidin. After the incubation, the sample was washed with solution A, embedded, and then viewed with a confocal imaging system (MRC-1024; Bio-Rad Laboratories, Hercules, CA).
The immunofluorescence microscopy of adult rat neural tissue was done as described (Mizoguchi et al., 1990). Briefly, rats were perfused with 4% paraformaldehyde in PBS, and postfixed with 4% paraformaldehyde in PBS. Their tissues were sectioned in a cryostat, mounted on glass slides, and then air dried. The samples were incubated for 12 h with the rabbit anti–neurabin-1 or -2 antibody and the mouse anti-synaptophysin antibody, followed by incubation for 12 h with Texas red–conjugated anti– rabbit and FITC-conjugated anti–mouse antibodies.
Antisense Phosphorothioate Oligonucleotide
Six antisense phosphorothioate oligonucleotides (PONs) complementary to the neurabin sequence surrounding the initiation codon and the corresponding sense PONs were designed based on the neurabin sequence (see Fig. 9 A). Their sequences did not have significant homology to any other sequences in the database. They were synthesized on a synthesizer (Expedite; PerSeptive Biosystems, Cambridge, MA) and purified by two stepwise reverse phase high pressure liquid column chromatographies (Beaton et al., 1991).
Figure 9.


Inhibition of the neurite formation by an antisense phosphorothioate oligonucleotide complementary to the neurabin sequence in primary cultured rat hippocampal neurons. (A) Antisense PONs complementary to the neurabin sequence. (B) Western blot analysis. The samples (5 μg of protein each) were subjected to Western blot analysis using the anti–neurabin-1 or anti–α-actinin antibody. Protein markers used are myosin (206), β-galactosidase (117), and BSA (89). (C) Immunofluorescence microscopy. The neurons were treated with antisense-1 or sense-1, followed by the double staining using rhodamine-phalloidin and the anti–α-tubulin antibody. The anti–α-tubulin antibody was visualized with an FITC-conjugated, anti–mouse antibody. (a and d) with antisense-1; (b and e) with sense-1; and (c and f) control. (a–c) F-actin and (d–f) α-tubulin. These results are representative of three independent experiments. The neurite formation in thirty neurons was statistically analyzed in each experiment. Bars, 20 μm.
For antisense experiments, the primary cultured rat hippocampal neurons were prepared as described above and attached on poly-l-lysine– coated glass coverslips in MEM with 10% horse serum for 1–2 h (Torre et al., 1994). The coverslips with attached cells were then transferred to dishes containing glia-conditioned MEM with N2 supplements, 1 mg/ml of ovalbumin, and 1 mM pyruvate (Goslin and Banker, 1991) in the presence of 50 μM of each PON. Each PON was then added to the medium every 12 h, and half of the medium was renewed 24 h later. After 48 h, the neurons treated with each PON were subjected to Western blot analysis and immunofluorescence microscopy.
Other Procedures
G-Actin was purified from rabbit skeletal muscle as described (Pardee and Spudich, 1982). Protein concentrations were determined with BSA as a reference protein (Bradford, 1976).
Results
Purification of Neurabin and Molecular Cloning of Its cDNA
To identify novel F-actin–binding proteins from rat brain, we attempted to detect F-actin–binding proteins using a blot overlay method with 125I-labeled F-actin. The homogenates of various rat tissues were subjected to SDS-PAGE followed by the blot overlay. Several radioactive bands with various molecular masses were detected (Fig. 1 A). Of these 125I-labeled F-actin–binding proteins, two protein bands of ∼180 (p180) and 140 kD were detected only in brain. p180 was highly purified by column chromatographies, including Q-Sepharose, phenyl-Sepharose, hydroxyapatite, Mono Q, and Mono S column chromatographies. On the final Mono S column chromatography, the 125I-labeled F-actin–binding protein band well coincided with a protein of ∼180 kD, which was identified by protein staining (Fig. 1 B).
When the peptide mapping of the Mono S sample of p180 was performed, over 30 peptide peaks were observed. Of the peptide peaks, the aa sequences of the nine peptides were determined. Computer homology search revealed that they were not found in current protein database. On the basis of these aa sequences, we isolated a clone from a rat brain cDNA library and determined its nucleotide sequence. This clone contained a predicted initiation codon preceded by an in-frame stop codon, and a 3,285-bp coding region followed by a stop codon. The encoded protein consisted of 1,095 aa and showed a calculated molecular mass of 122,729 (these sequence data are available from GenBank/EMBL/DDBJ under accession number U72994) (Fig. 2). It included all the aa sequences of the peptides. The molecular mass calculated from the predicted aa sequence was far less than that estimated by SDS-PAGE. To confirm whether this clone contained a full-length cDNA of p180, we constructed the eukaryotic expression vector with this cDNA and expressed the protein in COS7 cells. The expressed protein showed the mobility similar to that of native p180 on SDS-PAGE and the 125I-labeled F-actin–binding activity (Fig. 3 A). Although the reason for the remarkable difference between the molecular mass value calculated from the predicted aa sequence and that estimated by SDS-PAGE is not known, we concluded that this clone encoded the full-length cDNA of p180.
Figure 2.

Deduced amino acid sequence of neurabin. The aa sequences of the nine peptide peaks derived from the purified sample of neurabin are indicated by underlines.
Figure 3.



Molecular characterization of neurabin. (A) 125I-Labeled F-actin–binding activity of recombinant p180 (neurabin). The pCMV-neurabin was transfected to COS7 cells, and the cell extract was subjected to SDS-PAGE (5–15% polyacrylamide gradient gel), followed by 125I-labeled F-actin blot overlay. The arrowhead indicates p180 (neurabin). The radioactive bands of 125I- labeled F-actin–binding activity shown by asterisks are endogenous proteins of COS7 cells. Protein markers used are myosin (206), β-galactosidase (117), BSA (89), and ovalbumin (47). (B) Schematic drawing of neurabin structure and its probabilities of forming coiled-coil structures. Probabilities were calculated for each residue with the weighted MTIDK matrix using a 28-residue window. (C) 125I-Labeled F-actin–binding activity of various truncated forms of neurabin. The purified proteins (0.2 μg of protein each) were subjected to SDS-PAGE (5–15% polyacrylamide gradient gel), followed by 125I-labeled F-actin blot overlay. GST–neurabin-1, aa 1–144; GST–neurabin-2, aa 1–210; GST–neurabin-3, aa 145–293; GST–neurabin-4, aa 286–615; GST–neurabin-5, aa 505–791; and GST–neurabin-6, aa 665–1095. Protein markers used are BSA (89) and ovalbumin (47). (D) Localizations of neurabin and F-actin in COS7 cells. Full-length and truncated forms of neurabin were expressed as myc-tagged proteins in COS7 cells. The cells were doubly stained using the anti-myc antibody and rhodamine-phalloidin. The anti-myc antibody was visualized with a Cy5-conjugated anti–mouse antibody. (a–c) myc-tagged protein; and (d–f) F-actin. (a and d) myc-neurabin (full-length); (b and e) myc–neurabin-1 (aa 1–144); and (c and f) myc–neurabin-2 (aa 145–1095). Bars, 20 μm.
We named p180 neurabin since p180 was specifically expressed in neural tissue and showed F-actin–binding activity as described below. Neurabin had one PSD-95, DlgA, ZO-1–like (PDZ) domain (aa 505–595), a domain known to interact with transmembrane proteins (Saras and Heldin, 1996). In the COOH-terminal region following the PDZ domain, several sequence stretches had high probabilities of forming coiled-coil structures (Lupas et al., 1991; Lupas, 1996) (Fig. 3 B). Especially, the coiled-coil probabilities of four sequence stretches (first one [Fig. 3 B, a], aa 675–736; second one [Fig. 3 B, b], aa 739–779; third one [Fig. 3 B, c], aa 787–834; and fourth one [Fig. 3 B, d], aa 1041–1080) were calculated to be 0.9–1.0. The probability of the sequence stretch (aa 598–628) was calculated to be 0.6–0.7. This stretch is unlikely to form a coiled-coil structure, because there was a difference of >0.2–0.3 in the probabilities between the MTK and MTIDK matrices (Lupas et al., 1991; Lupas, 1996). The NH2-terminal region of neurabin did not show any tendency to form a coiled-coil structure.
To determine the F-actin–binding domain of neurabin, we prepared fusion proteins of several truncated forms of neurabin with GST and examined the binding of 125I- labeled F-actin to these fusion proteins. GST–neurabin-1 (aa 1–144) and GST–neurabin-2 (aa 1–210) showed the 125I-labeled F-actin–binding activity, whereas the fusion proteins of other truncated forms of neurabin did not show the activity (Fig. 3 C). To confirm that neurabin interacted with F-actin through the F-actin–binding domain in intact cells, we compared the localization of truncated forms of neurabin with that of F-actin. When full-length neurabin was expressed in COS7 cells as a myc-tagged protein (myc-neurabin), it was colocalized with F-actin (Fig. 3 D, a and d). A similar result was obtained when a truncated form of neurabin (myc–neurabin-1, aa 1–144), which possessed the F-actin–binding domain alone, was expressed (Fig. 3 D, b and e). In contrast, when another truncated form of neurabin (myc–neurabin-2, aa 145–1095), which lacked the F-actin–binding domain, was expressed, the protein showed a diffuse distribution in the cytoplasm, but was partly concentrated at the F-actin–rich region of the cell periphery (Fig. 3 D, c and f). Taken together with the findings that GST–neurabin-1 (aa 1–144) showed the 125I-labeled F-actin–binding activity and was cosedimented with F-actin as described below, these results indicate that neurabin has one F-actin–binding domain at the NH2-terminal region (aa 1–144) (Fig. 3 C), and that neurabin interacts with F-actin through this domain in intact cells. This F-actin–binding domain showed no significant homology to any protein in current protein database.
Biochemical Properties of Neurabin
In addition to 125I-labeled F-actin blot overlay, the binding of neurabin to F-actin was examined by cosedimentation of neurabin with F-actin. When GST–neurabin-2 (aa 1–210) was incubated with F-actin followed by ultracentrifugation, the fusion protein was recovered with F-actin in the pellet (Fig. 4 A). A similar result was obtained with GST– neurabin-1 (aa 1–144) and His6-neurabin (data not shown). The stoichiometry of the binding of His6-neurabin to actin was calculated to be one His6-neurabin molecule per about eight actin molecules (Fig. 4 B). The K d value was calculated to be ∼8 × 10−7 M. It was examined by competition experiments whether neurabin bound along the sides of F-actin or at the ends. The binding of the Mono S sample of neurabin to 125I-labeled F-actin was completely inhibited by an excessive amount of myosin S1, a well-characterized protein that binds along the sides of F-actin (Rayment et al., 1993; Schröder et al., 1993) (Fig. 4 C). This inhibition was reversed by the addition of MgATP because MgATP dissociates the actin–myosin complex (Fraser et al., 1975). These results indicate that neurabin binds along the sides of F-actin.
Figure 4.

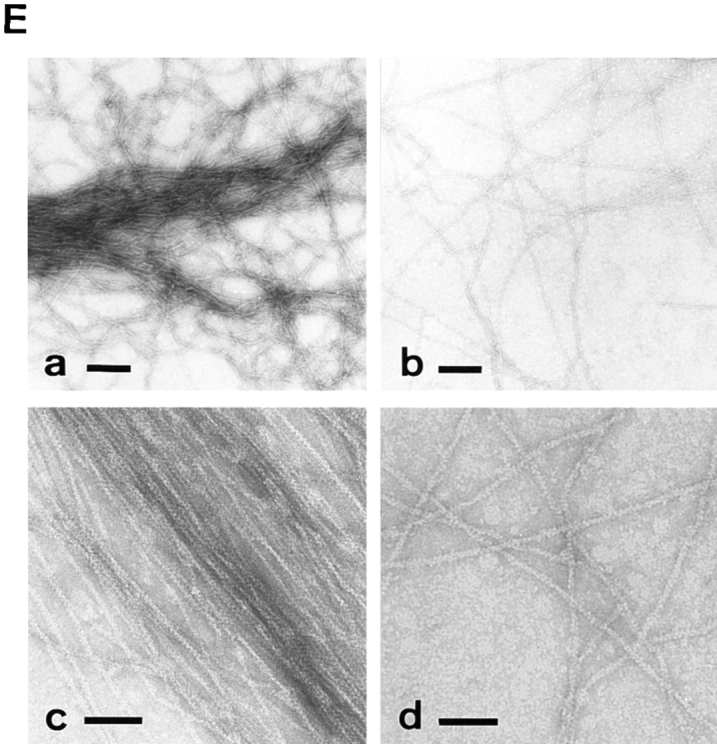
Biochemical properties of neurabin. (A) Cosedimentation of GST–neurabin-2 with F-actin. GST–neurabin-2 (aa 1–210) (10 μg of protein) was mixed with F-actin, followed by ultracentrifugation. S, supernatant; and P, pellet. Arrow and arrowhead indicate actin and GST–neurabin-2, respectively. Asterisks indicate the proteolytic products of GST–neurabin-2, which were not cosedimented with F-actin. Protein markers used are ovalbumin (45) and carbonic anhydrase (31). (B) Binding of His6-neurabin to F-actin. Various amounts of His6-neurabin were mixed with F-actin, followed by ultracentrifugation. Amounts of the free and bound His6-neurabin were calculated by determining the protein amounts from the supernatant and pellet fractions with a densitometer. (C) Inhibition by myosin S1 of the binding of neurabin to 125I-labeled F-actin. The Mono S sample of native neurabin (0.1 μg of protein) was subjected to SDS-PAGE (8% polyacrylamide gel), followed by the blot overlay with 125I-labeled F-actin pretreated with myosin S1 in the presence or absence of ATP. Protein markers used are myosin (206), β-galactosidase (117), and BSA (89). (D) F-actin–cross-linking activity of neurabin estimated by low shear viscometry. G-actin was mixed with the Mono S sample of native neurabin or His6-neurabin in an indicated amount and incubated for 45 min, followed by low shear viscometry (left). •, With native neurabin; and ○, with His6-neurabin. Protein staining of His6-neurabin with Coomassie brilliant blue (inset). Protein markers used are myosin (206), β-galactosidase (117), BSA (89), and ovalbumin (45). G-actin was mixed with 15 μg/ml of the Mono S sample of native neurabin and incubated for an indicate time, followed by low shear viscometry (right). •, With neurabin; and ○, without neurabin. (E) F-actin– cross-linking activity of neurabin estimated by electron microscopy. (a and c) with neurabin; and (b and d) without neurabin. Bars: (a and b) 200 nm; (c and d) 50 nm.
The F-actin–cross-linking activity of the Mono S sample of neurabin was examined by the falling ball method for low shear viscometry. Neurabin increased the viscosity in time- and dose-dependent manners, suggesting that neurabin showed the cross-linking activity (Fig. 4 D). A similar result was obtained with His6-neurabin purified from Sf9 cells. Whereas the Mono S sample of native neurabin was contaminated with a protein of ∼130 kD (Fig. 1 B), these results indicate that neurabin itself indeed shows the F-actin–cross-linking activity. This cross-linking activity of neurabin was confirmed by transmission electron microscopy of negatively stained specimens. Neurabin caused F-actin to associate into bundles (Fig. 4 E).
The molecular mass values of neurabin were estimated by gel filtration and sucrose density gradient ultracentrifugation in the presence of a reducing agent. When His6-neurabin was subjected to Superdex 200 column chromatography, it appeared at a position corresponding to ∼440 kD (Stokes' radius, about 66 Å) (Fig. 5 A). On sucrose density gradient ultracentrifugation, His6-neurabin appeared at a position corresponding to ∼380 kD (S value, ∼15.6) (Fig. 5 B). The molecular mass value of neurabin was calculated to be ∼440 kD from both the Stokes' radius and S value (Siegel and Monty, 1966). The frictional ratio of neurabin was calculated to be about 1.3. Because the molecular mass value of neurabin estimated by SDS-PAGE was ∼180 kD, and neurabin had predicted coiled-coil structures at the COOH-terminal region, these results suggest that neurabin forms a dimer. It is also possible that neurabin forms a trimer or tetramer because the molecular mass value of neurabin calculated from its predicted aa sequence was ∼120 kD.
Figure 5.
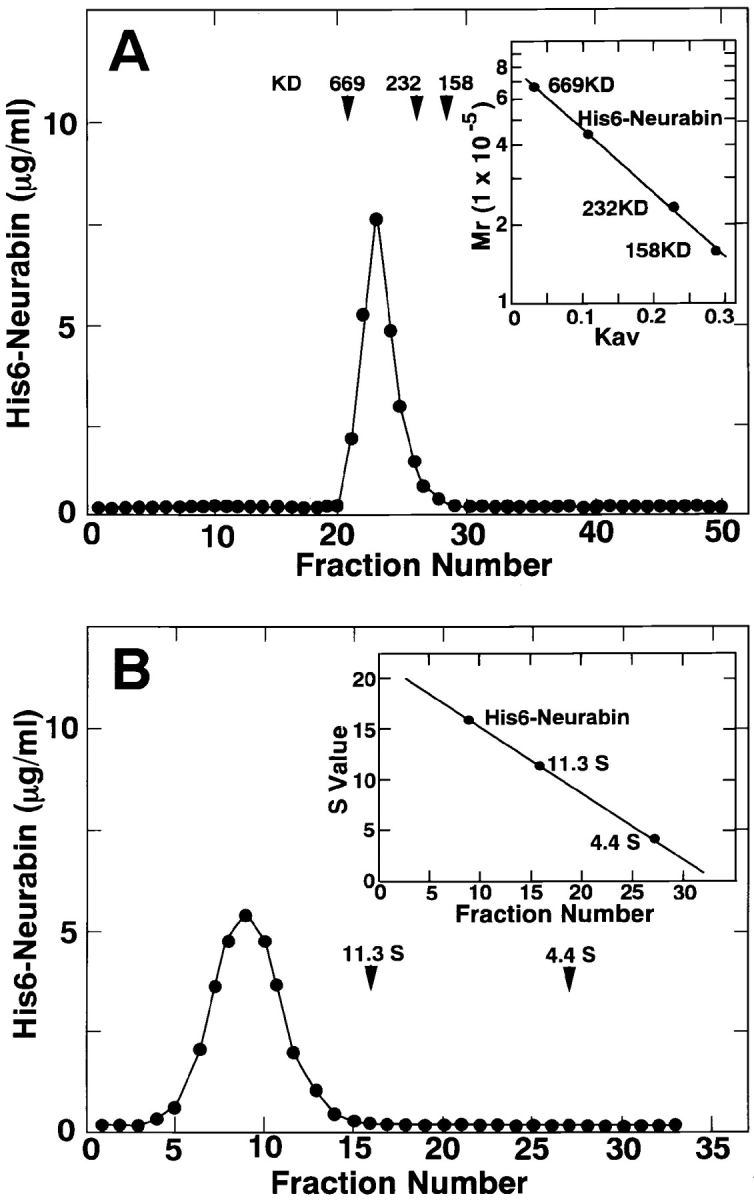
Oligomerization property of neurabin. (A) Superdex 200 column chromatography. Each fraction was subjected to protein staining with silver and Western blot analysis using the anti– neurabin-1 antibody. The amount of His6-neurabin of each fraction was determined with the densitometer. Protein markers used are thyroglobulin (669 kD, Stokes' radius 85.0 Å), catalase (232 kD, Stokes' radius 52.2 Å), and aldolase (158 kD, Stokes' radius 48.1 Å). Estimation of the molecular mass value of His6-neurabin (inset). (B) Sucrose density gradient ultracentrifugation. Each fraction was subjected to protein staining with silver and Western blot analysis using the anti– neurabin-1 antibody. The amount of His6-neurabin of each fraction was determined with the densitometer. Protein markers used are catalase (232 kD, 11.3 S), and BSA (67 kD, 4.4 S). Estimation of the S value of His6-neurabin (inset).
Tissue Distribution of Neurabin
Northern blot analysis showed that ∼9.5 kb mRNA was hybridized in rat brain (Fig. 6 A). In rat testis, mRNAs with smaller sizes were weakly hybridized, but the significance of these weak signals is unknown. Other rat tissues examined, including heart, spleen, lung, liver, skeletal muscle, and kidney, did not show any detectable signal.
Figure 6.
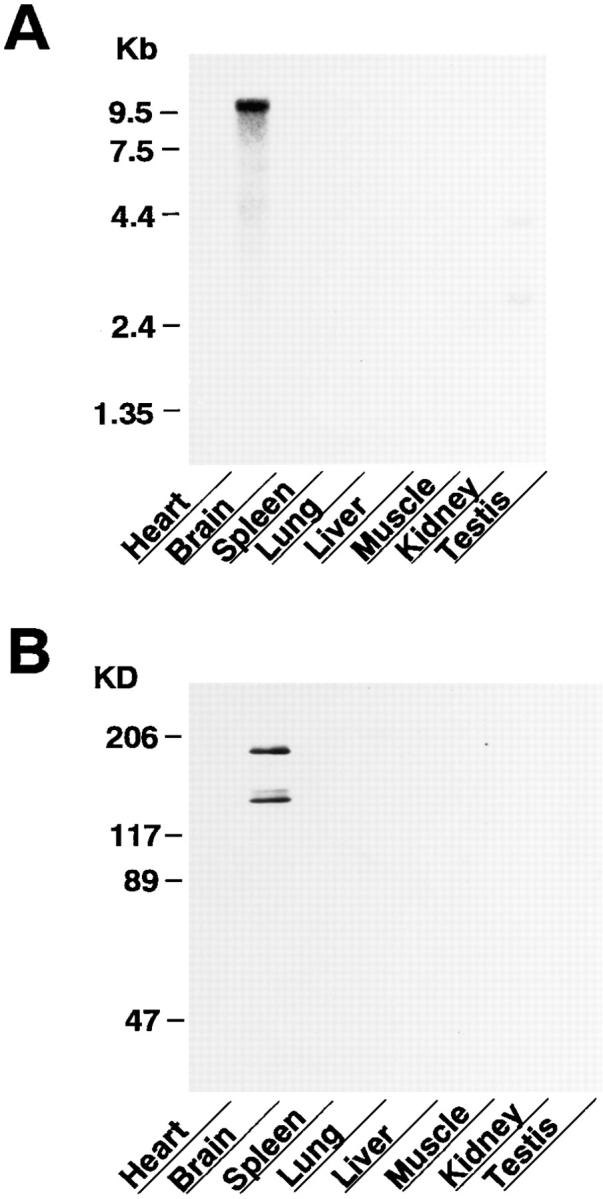
Tissue distribution of neurabin. (A) Northern blot analysis. A RNA blot membrane (CLONTECH, Palo Alto, CA) was hybridized with the 32P-labeled, 2.3-kbp KpnI-XhoI fragment of the neurabin cDNA according to the manufacturer's protocol. (B) Western blot analysis. The homogenates of various rat tissues (10 μg of protein each) were subjected to SDS-PAGE (8% polyacrylamide gel), followed by immunoblot using the anti– neurabin-1 antibody. Protein markers used are myosin (206), β-galactosidase (117), BSA (89), and ovalbumin (47).
Western blot analysis showed that a polyclonal antibody (anti–neurabin-1), which was raised against GST–neurabin-7 (aa 609–868), recognized a protein band of ∼180 kD and two proteins of ∼140 kD in rat brain (Fig. 6 B). No reactive band was observed in other rat tissues examined. The protein bands with the smaller molecular masses in rat brain appear to be proteolytic products of neurabin (see Discussion).
Localization of Neurabin in Cultured Hippocampal Neurons
It was first examined by Western blot analysis whether neurabin was expressed in primary cultured rat hippocampal neurons ranging in age 1–10 d. Neurabin was expressed in the neurons and the expression amounts were almost constant throughout the course of development in culture, whereas those of the SV proteins, including synapsin I and synaptophysin, increased (Fig. 7 A). Neurabin showed double immunoreactivity bands. This may be attributed to the posttranslational modifications, such as phosphorylation. The cultured hippocampal neurons were then used to assess the localization of neurabin by the immunofluorescence staining using the anti–neurabin-1 antibody. After 24 h in culture, almost all the neurons extend an axon and several minor processes that eventually become dendrites (Dotti et al., 1988). At this stage, neurabin was concentrated in the lamellipodia of the growth cone (Fig. 7 B), where F-actin is enriched (Goslin et al., 1989; Letourneau and Shattuck, 1989). Neurabin was not clearly concentrated in the filopodia. After 10 d in culture, both axons and dendrites develop and the synapses are observed (Basarsky et al., 1994). At this stage, neurabin showed dotty signals along the dendrites (Fig. 7 C, a). Synaptotagmin I, known to be highly concentrated at the presynaptic nerve terminal (Matthew et al., 1981), showed the similar staining pattern (Fig. 7 C, b). This result suggests that neurabin is highly concentrated in the synapse.
Figure 7.

Expression and localization of neurabin during the development of primary cultured rat hippocampal neurons. (A) Expression of neurabin during the development. The cultured cells at various stages were subjected to Western blot analysis using the anti–synapsin I, anti-synaptophysin, or anti–neurabin-1 antibodies. (B) Localization of neurabin in the hippocampal neurons at 24 h in culture. The sample was incubated with the anti– neurabin-1 antibody and visualized with an FITC-conjugated, anti–rabbit antibody. High magnification of the indicated box (inset). Arrows and arrowheads indicate the growth cone and cell body, respectively. (C) Localization of neurabin in the hippocampal neurons at 10 d in culture. The sample was doubly stained using the rabbit anti–neurabin-1 and mouse anti–synaptotagmin I antibodies. They were visualized with FITC-conjugated, anti– rabbit and rhodamine-conjugated, anti–mouse antibodies. (a) neurabin and (b) synaptotagmin I. Bars, 20 μm.
Localization of Neurabin in Rat Neural Tissue
The in situ distribution and localization of neurabin were examined by the immunofluorescence staining of cerebellum, hippocampus, retina, neuromuscular junction, and adrenal gland of adult rat using the anti–neurabin-1 antibody. The staining patterns of neurabin were compared with those of synaptophysin, known to be highly concentrated at the presynaptic nerve terminal (Navone et al., 1986). In the cerebellum, neurabin showed dotty signals in the molecular layer and discontinuous signals along the Purkinje cell body (Fig. 8 A). It also showed intense signals in the gromerulus of the granular layer, where the synapses are complexed. In the hippocampus, neurabin showed dotty signals in the stratum oriens, discontinuous signals along the cell body in the stratum pyramidale, and intense signals in the stratum radiatum (Fig. 8 C). These staining patterns of neurabin were similar to those of synaptophysin (Fig. 8, A–D). These results indicate that neurabin is highly concentrated in the synapse of these tissues. In the retina, neurabin was highly concentrated in the inner and outer plexiform layers, where the synapses are formed (Fig. 8 E). Neurabin was also expressed in the neuromuscular junction, where the synapses are formed between motor neurons and muscle fibers (Fig. 8 G), and in adrenal chromaffin cells (Fig. 8 I). In these tissues other than brain, the staining patterns of neurabin were also similar to those of synaptophysin (Fig. 8, E–J). Similar results were obtained with another antibody (anti–neurabin-2), which was raised against GST–neurabin-8 (aa 890–1095) (data not shown). These results demonstrate that neurabin is widely distributed in neural tissue and highly concentrated in the synapse of the developed neurons.
Figure 8.
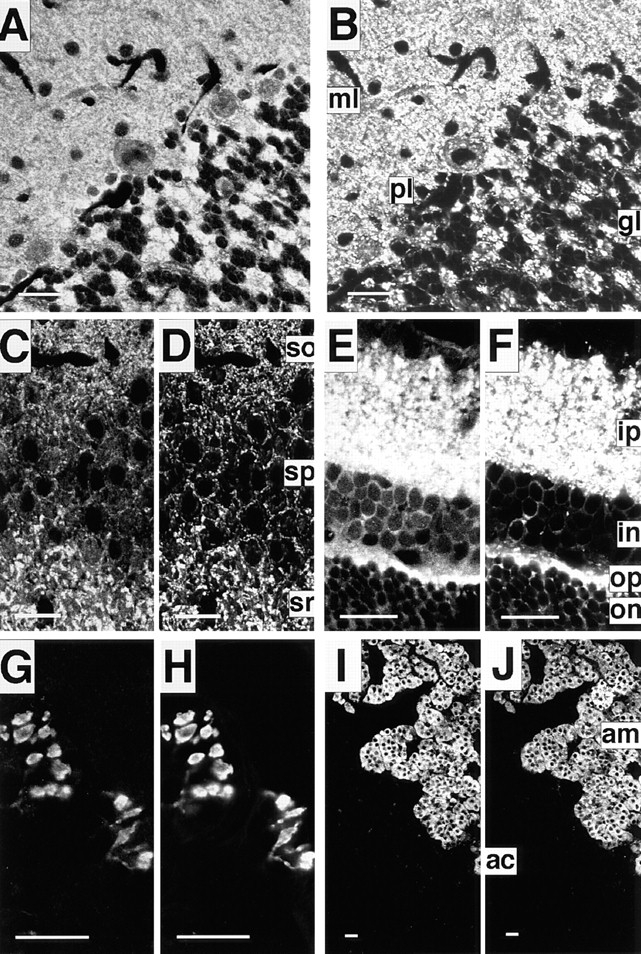
Localization of neurabin in rat neural tissue. The samples were doubly stained using the rabbit anti–neurabin-1 and mouse anti-synaptophysin antibodies. They were visualized with Texas red–conjugated, anti–rabbit and FITC-conjugated, anti– mouse antibodies. (A, C, E, G, and I) neurabin, and (B, D, F, H, and J) synaptophysin. (A and B) cerebellum. ml, molecular layer; pl, Purkinje cell layer; and gl, granular layer. (C and D) hippocampus (CA3). so, stratum oriens; sp, stratum pyramidale; and sr, stratum radiatum. (E and F) retina. ip, inner plexiform layer; in, inner nuclear layer; op, outer plexiform layer; and on, outer nuclear layer. (G and H), neuromuscular junction. (I and J) adrenal gland. am, adrenal medulla; and ac, adrenal cortex. Bars, 20 μm.
Involvement of Neurabin in Neurite Formation
To examine the physiological function of neurabin, we attempted to suppress the expression of endogenous neurabin in primary cultured rat hippocampal neurons by treatment with its antisense oligonucleotides. We designed six antisense PONs complementary to the neurabin sequence surrounding the initiation codon (Fig. 9 A). The corresponding sense PONs were also constructed. Each PON was added to the serum-free medium at 50 μM after plating, and again at 25 μM every 12 h. After 48 h, we examined by the immunofluorescence staining using the anti–neurabin-1 antibody that antisense PON was effective to suppress the neurabin expression. Of these antisense PONs, antisense-1 and -2 were effective, whereas other antisense PONs, including antisense-3, -4, -5, and -6, as well as all the sense PONs, were not (data not shown). Therefore, the effect of antisense-1 on the hippocampal neurons was precisely studied. When the hippocampal neurons were treated with neurabin antisense-1, the expression of neurabin was reduced as estimated by Western blot analysis using the anti–neurabin-1 antibody (Fig. 9 B). The treatment of the neurons with neurabin sense-1 did not affect the expression level of neurabin. In these neurons, the level of α-actinin, an F-actin–bundling protein, was not affected, suggesting that antisense-1 specifically reduced the expression of neurabin. The neurons treated with antisense-1 were morphologically compared with those treated with sense-1 by the immunofluorescence staining of F-actin and tubulin. In the neurons treated with sense-1, as well as in the control neurons, >90% of the cells were characterized by one long axon and several processes (Fig. 9 C). In contrast, in >90% of the neurons treated with antisense-1, any neurite formation was not observed. Trypan blue exclusion test showed that the neurons treated with antisense-1 were alive (data not shown). Moreover, this effect of antisense-1 was reversible. When antisense-1 was washed out after the last addition, the neurite formation of the hippocampal neurons began to be observed 48 h later (data not shown).
Discussion
We have purified here from rat brain a 125I-labeled F-actin– binding protein of ∼180 kD and characterized it. Because this protein is specifically expressed in neural tissue, including central and peripheral nerves, and adrenal chromaffin cells, we have named it neurabin. Structural analysis of neurabin has revealed that it consists of at least one F-actin–binding domain at the NH2-terminal region, one PDZ domain at the middle region, and domains for predicted coiled-coil structures at the COOH-terminal region.
Western blot analysis using the anti–neurabin-1 antibody in rat brain detected a protein band of ∼180 kD and two protein bands of ∼140 kD as shown in Fig. 6 B. The molecular mass value of neurabin calculated from the predicted aa was ∼120 kD. Therefore, there is a possibility that the 180- and 140-kD bands may be partially and fully denatured forms on the gel, respectively. However, this possibility is unlikely and the 140-kD proteins appear to be proteolytic products of neurabin because of the following observations. (a) Before the sample was subjected to SDS-PAGE, the sample was always boiled for 10 min in an SDS sample buffer containing 2-mercaptoethanol. Even when the sample was treated with 8 M urea, neurabin showed a molecular mass of ∼180 kD on SDS-PAGE (data not shown); (b) The 140-kD proteins were separated from the 180-kD protein by hydroxyapatite column chromatography (data not shown); (c) When the major 140-kD protein was highly purified and the peptide mapping was performed, its peptide map was almost identical to that of neurabin, but the 140-kD protein lacked some peptide peaks (data not shown). The aa sequences of the peptide peaks of the 140-kD protein were identical to those of neurabin (data not shown); (d) The amounts of the 140-kD proteins in rat brain varied from preparation to preparation; and (e) When the cell extract from the neurabin cDNA-transfected COS7 cells was subjected to Western blot analysis, the antibody recognized the expressed neurabin and its proteolytic products. The molecular mass values of these proteolytic products in the COS7 cells were ∼140 kD (data not shown).
We have concluded that neurabin functions as an F-actin– binding protein in intact cells and that the F-actin–binding domain is located at the NH2-terminal region (aa 1–144), on the basis of the following observations: (a) Full-length neurabin and its NH2-terminal region (aa 1–144) showed the 125I-labeled F-actin–binding activity; (b) full-length neurabin and its NH2-terminal region (aa 1–144) were cosedimented with F-actin; (c) full-length neurabin showed the F-actin–cross-linking activity; and (d) when full-length neurabin and its NH2-terminal region (aa 1–144) were expressed in COS7 cells, both proteins were colocalized with F-actin. There is no significant homology between the primary structures of F-actin–binding domains of neurabin and any known F-actin–binding protein, but three-dimensional structure of the F-actin–binding domain of neurabin may be similar to those of known F-actin–binding proteins.
We have moreover shown here that neurabin shows F-actin–cross-linking activity. Many actin-binding proteins have thus far been isolated, and of these proteins, the α-actinin/spectrin family members have most extensively been studied as F-actin–binding proteins having F-actin– cross-linking activity (Hartwig and Kwiatkowski, 1991; Matsudaira, 1991). They usually form oligomers by association at their rod domains and thereby show the F-actin– cross-linking activity. Neurabin has the domains for predicted coiled-coil structures. Coiled-coil structures have been identified in a variety of cytoskeleton proteins (Lupas et al., 1991; Lupas, 1996). They typically form rodlike, homodimeric, or higher order structures. The molecular mass of neurabin estimated by SDS-PAGE and that calculated by its predicted aa sequence are ∼180 and 120 kD, respectively, whereas those estimated by gel filtration and sucrose density gradient ultracentrifugation are ∼440 and 380 kD, respectively. The molecular masses of neurabin was calculated to be ∼440 from both the Stokes' radius and S value (Siegel and Monty, 1966). These results together with the dimerization or oligomerization property of the coiled-coil structures suggest that neurabin forms an oligomer with multiple F-actin–binding heads via its predicted coiled-coil structures and thereby shows the F-actin– cross-linking activity.
The PDZ domain has originally been identified in PSD-95/SAP90, one of membrane-associated guanylate kinases (Cho et al., 1992; Kistner et al., 1993), and implicated in protein–protein interactions (Saras and Heldin, 1996). This domain has been found in a variety of proteins that are typically located at specific regions of cell–cell junctions, such as the tight junction, the synaptic junction, and the septate junction. Recent studies have revealed that the PDZ domain binds the unique COOH-terminal motifs of target proteins (Doyle et al., 1996; Songyang et al., 1997), which are found in a large number of transmembrane proteins, such as N-methyl-d-aspartate receptors and Shaker-type K+ channel (Saras and Heldin, 1996). Of many proteins having the PDZ domain(s) thus far reported, no protein having F-actin–binding activity has been reported. Conversely, of many F-actin–binding proteins thus far reported, no protein having the PDZ domain(s) has been reported. We have shown here that neurabin is highly concentrated in the synapse. These results, together with the unique structural and biochemical properties of neurabin, suggest that it is linked to a transmembrane protein(s) through its PDZ domain, providing a linkage between the synaptic junction and the actin cytoskeleton. We have recently identified another F-actin–binding protein having one PDZ domain, and named it afadin (Mandai et al., 1997). Afadin appears to provide a linkage between cadherin-based cell–cell adherens junction and the actin cytoskeleton in various epithelia. These F-actin–binding proteins having one PDZ domain may constitute a functional family, of which members provide a linkage between cell–cell junctions and the actin cytoskeleton.
We have found here that neurabin is expressed in both the developing and developed neurons; in the developing neurons it is highly concentrated in the lamellipodia of the growth cone, whereas in the developed neurons it is highly concentrated in the synapse. We have moreover shown that suppression of the neurabin expression in the hippocampal neurons by treatment with the antisense oligonucleotide inhibits the neurite formation. These results, together with the biochemical properties of neurabin, suggest two possible roles: (a) it is involved in the vesicle trafficking in both the developing and developed neurons; and (b) it is involved in the formation and the maintenance of the synaptic junction. To clarify the role of neurabin, it is essential to identify its interacting molecule(s) other than F-actin. We have shown that myc– neurabin-2 (aa 145–1095), which lacks the F-actin–binding domain, is partly concentrated at the F-actin–rich region of the cell periphery in the COS7 cells. This result suggests that there is a molecule(s) other than F-actin that directly interacts with neurabin through the PDZ domain and/or the coiled-coil domains, which is present at the F-actin– rich region at the cell periphery. Identification of such an interacting molecule(s) is now under investigation.
Acknowledgments
We thank Dr. D.W. Russell (University of Texas Southwestern Medical Center at Dallas, Dallas, TX) for providing us the pCMV vector, and Dr. M. Imamura (National Institute of Neuroscience, Kodaira, Japan) for providing us the anti–α-actinin antibody. We also thank Dr. Sh. Tsukita (Kyoto University, Kyoto, Japan), and Dr. Y. Hata (Takai Biotimer Project, ERATO, Kobe, Japan) for helpful discussions.
Abbreviations used in this paper
- aa
amino acid
- F-actin
actin filament
- G-actin
actin monomer
- GST
glutathione-S-transferase
- neurabin
neural tissue–specific F-actin–binding protein
- PDZ
PSD-95, DlgA, ZO-1-like
- PON
phosphorothioate oligonucleotide
- S1
subfragment 1
- SV
synaptic vesicle
Footnotes
Address all correspondence to Y. Takai, Department of Molecular Biology and Biochemistry, Osaka University Medical School, Suita 565, Japan. Tel.: 81-6-879-3410. Fax: 81-6-879-3419. E-mail: ytakai@molbio.med.osaka-u.ac.jp
Received for publication 7 April 1997 and in revised form 8 August 1997.
References
- Basarsky TA, Parpura V, Haydon PG. Hippocampal synaptogenesis in cell culture: developmental time course of synapse formation, calcium influx, and synaptic protein distribution. J Neurosci. 1994;14:6402–6411. doi: 10.1523/JNEUROSCI.14-11-06402.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaton, G., D.D. Dellinger, W.S. Marshall, and M.H. Caruthers. 1991. Synthesis of oligonucleotide phosphorodithioates. In Oligonucleotides and Analogues: a Practical Approach. F. Eckstein, editor. IRL Press, Oxford. 109–134.
- Bentley D, Toroian-Raymond A. Disoriented pathfinding by pioneer neurone growth cones deprived of filopodia by cytochalasin treatment. Nature. 1986;323:712–715. doi: 10.1038/323712a0. [DOI] [PubMed] [Google Scholar]
- Bentley D, O'Connor TP. Cytoskeletal events in growth cone steering. Curr Opin Neurobiol. 1994;4:43–48. doi: 10.1016/0959-4388(94)90030-2. [DOI] [PubMed] [Google Scholar]
- Bowe MA, Fallon JR. The role of agrin in synapse formation. Annu Rev Neurosci. 1995;18:443–462. doi: 10.1146/annurev.ne.18.030195.002303. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Burns ME, Augustine GJ. Synaptic structure and function: dynamic organization yields architectural precision. Cell. 1995;83:187–194. doi: 10.1016/0092-8674(95)90160-4. [DOI] [PubMed] [Google Scholar]
- Chia CP, Hitt AL, Luna EJ. Direct binding of F-actin to ponticulin, an integral plasma membrane glycoprotein. Cell Motil Cytoskeleton. 1991;18:164–179. doi: 10.1002/cm.970180303. [DOI] [PubMed] [Google Scholar]
- Chiba A, Keshishian H. Neuronal pathfinding and recognition: roles of cell adhesion molecules. Dev Biol. 1996;180:424–432. doi: 10.1006/dbio.1996.0316. [DOI] [PubMed] [Google Scholar]
- Chien C-B, Rosenthal DE, Harris WA, Holt CE. Navigational errors made by growth cones without filopodia in the embryonic Xenopus brain. Neuron. 1993;11:237–251. doi: 10.1016/0896-6273(93)90181-p. [DOI] [PubMed] [Google Scholar]
- Cho K-O, Hunt CA, Kennedy MB. The rat brain postsynaptic density fraction contains a homolog of the Drosophila disc-large tumor suppressor protein. Neuron. 1992;9:929–942. doi: 10.1016/0896-6273(92)90245-9. [DOI] [PubMed] [Google Scholar]
- Culotti JG, Kolodkin AL. Functions of netrins and semaphorins in axon guidance. Curr Opin Neurobiol. 1996;6:81–88. doi: 10.1016/s0959-4388(96)80012-2. [DOI] [PubMed] [Google Scholar]
- Dotti CG, Sullivan CA, Banker GA. The establishment of polarity by hippocampal neurons in culture. J Neurosci. 1988;8:1454–1468. doi: 10.1523/JNEUROSCI.08-04-01454.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle DA, Lee A, Lewis J, Kim E, Sheng M, MacKinnon R. Crystal structures of a complexed and peptide-free membrane protein-binding domain: molecular basis of peptide recognition by PDZ. Cell. 1996;85:1067–1076. doi: 10.1016/s0092-8674(00)81307-0. [DOI] [PubMed] [Google Scholar]
- Endo T, Masaki T. Molecular properties and functions in vitro of chicken smooth-muscle alpha-actinin in comparison with those of striated-muscle alpha-actinins. J Biochem (Tokyo) 1982;92:1457–1468. doi: 10.1093/oxfordjournals.jbchem.a134070. [DOI] [PubMed] [Google Scholar]
- Fields RD, Itoh K. Neural cell adhesion molecules in activity- dependent development and synaptic plasticity. Trends Neurosci. 1996;19:473–480. doi: 10.1016/S0166-2236(96)30013-1. [DOI] [PubMed] [Google Scholar]
- Forscher P, Smith SJ. Actions of cytochalasins on the organization of actin filaments and microtubles in a neuronal growth cone. J Cell Biol. 1988;107:1505–1516. doi: 10.1083/jcb.107.4.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser AB, Eisenberg E, Kielley WW, Carlson FD. The interaction of heavy meromyosin and subfragment 1 with actin. Physical measurements in the presence and absence of adenosine triphosphate. Biochemistry. 1975;14:2207–2214. doi: 10.1021/bi00681a025. [DOI] [PubMed] [Google Scholar]
- Friedman GC, O'Leary DDM. Eph receptor tyrosine kinases and their ligands in neural development. Curr Opin Neurobiol. 1996;6:127–133. doi: 10.1016/s0959-4388(96)80018-3. [DOI] [PubMed] [Google Scholar]
- Garner CC, Kindler S. Synaptic proteins and the assembly of synaptic junctions. Trends Cell Biol. 1996;6:429–433. doi: 10.1016/s0962-8924(96)10036-2. [DOI] [PubMed] [Google Scholar]
- Garrity PA, Zipursky SL. Neuronal target recognition. Cell. 1995;83:177–185. doi: 10.1016/0092-8674(95)90159-0. [DOI] [PubMed] [Google Scholar]
- Goslin K, Birgbauer E, Banker G, Solomon F. The role of cytoskeleton in organizing growth cones: a microfilament-associated growth cone component depends upon microtubules for its localization. J Cell Biol. 1989;109:1621–1631. doi: 10.1083/jcb.109.4.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goslin, K., and G. Banker. 1991. Rat hippocampal neurons in low-density culture. In Culturing nerve cells. G. Banker, and K. Goslin, editors. MIT Press, Cambridge, MA. 251–281.
- Guan KL, Dixon JE. Eukaryotic proteins expressed in Escherichia coli: an improved thrombin cleavage and purification procedure of fusion proteins with glutathione S-transferase. Anal Biochem. 1991;192:262–267. doi: 10.1016/0003-2697(91)90534-z. [DOI] [PubMed] [Google Scholar]
- Hartwig JH, Kwiatkowski DJ. Actin-binding proteins. Curr Opin Cell Biol. 1991;3:87–97. doi: 10.1016/0955-0674(91)90170-4. [DOI] [PubMed] [Google Scholar]
- Hata Y, Südhof TC. A novel ubiquitous form of Munc-18 interacts with multiple syntaxins. Use of the yeast two-hybrid system to study interactions between proteins involved in membrane traffic. J Biol Chem. 1995;270:13022–13028. doi: 10.1074/jbc.270.22.13022. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Sobue K, Kanda K, Harada A, Yorifuji H. The cytoskeletal architecture of the presynaptic terminal and molecular structure of synapsin 1. J Cell Biol. 1989;108:111–126. doi: 10.1083/jcb.108.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imazumi K, Sasaki T, Takahashi K, Takai Y. Identification of a rabphilin-3A-interacting protein as GTP cyclohydrolase I in PC12 cells. Biochem Biophys Res Commun. 1994;205:1409–1416. doi: 10.1006/bbrc.1994.2822. [DOI] [PubMed] [Google Scholar]
- Kato M, Sasaki T, Ohya T, Nakanishi H, Nishioka H, Imamura M, Takai Y. Physical and functional interaction of rabphilin-3A with α-actinin. J Biol Chem. 1996;271:31775–31778. doi: 10.1074/jbc.271.50.31775. [DOI] [PubMed] [Google Scholar]
- Keynes R, Cook GMW. Axon guidance molecules. Cell. 1995;83:161–169. doi: 10.1016/0092-8674(95)90157-4. [DOI] [PubMed] [Google Scholar]
- Kikuchi A, Nakanishi H, Takai Y. Purification and properties of Rab3A. Methods Enzymol. 1995;257:57–70. doi: 10.1016/s0076-6879(95)57010-1. [DOI] [PubMed] [Google Scholar]
- Kistner U, Wenzel BM, Veh RW, Cases-Langhoff C, Garner AM, Appeltauer U, Voss B, Gundelfinger ED, Garner CC. SAP90, a rat presynaptic protein related to the product of the Drosophila tumor suppressor gene dlg-A. . J Biol Chem. 1993;268:4580–4583. [PubMed] [Google Scholar]
- Letourneau PC, Shattuck TA. Distribution and possible interactions of actin-associated proteins and cell adhesion molecules of nerve growth cones. Development (Camb) 1989;105:505–519. doi: 10.1242/dev.105.3.505. [DOI] [PubMed] [Google Scholar]
- Lin C-H, Thompson CA, Forscher P. Cytoskeletal reorganization underlying growth cone motility. Curr Opin Neurobiol. 1994;4:640–647. doi: 10.1016/0959-4388(94)90004-3. [DOI] [PubMed] [Google Scholar]
- Lupas A. Coiled coils: new structures and new functions. Trends Biochem Sci. 1996;21:375–382. [PubMed] [Google Scholar]
- Lupas A, Van Dyke M, Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- Mackay DJG, Nobes CD, Hall A. The Rho's progress: a potential role during neuritogenesis for the Rho family of GTPases. Trends Neurosci. 1995;18:496–501. doi: 10.1016/0166-2236(95)92773-j. [DOI] [PubMed] [Google Scholar]
- Mandai K, Nakanishi H, Satoh A, Obaishi H, Wada M, Nishioka H, Itoh M, Mizoguchi A, Aoki T, Fujimoto T, et al. Afadin: A novel actin filament–binding protein with one PDZ domain localized at cadherin-based cell-to-cell adherens junction. J Cell Biol. 1997;139:517–528. doi: 10.1083/jcb.139.2.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh L, Letourneau PC. Growth of neurites without filopodial or lamellipodial activity in the presence of cytochalasin B. J Cell Biol. 1984;99:2041–2047. doi: 10.1083/jcb.99.6.2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsudaira P. Modular organization of actin crosslinking proteins. Trends Biochem Sci. 1991;16:87–92. doi: 10.1016/0968-0004(91)90039-x. [DOI] [PubMed] [Google Scholar]
- Matthew WD, Tsavaler L, Reichardt LF. Identification of a synaptic vesicle-specific membrane protein with a wide distribution in neuronal and neurosecretory tissue. J Cell Biol. 1981;91:257–269. doi: 10.1083/jcb.91.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison T, Kirschner M. Cytoskeletal dynamics and nerve growth. Neuron. 1988;1:761–772. doi: 10.1016/0896-6273(88)90124-9. [DOI] [PubMed] [Google Scholar]
- Mizoguchi A, Kim S, Ueda T, Kikuchi A, Yorifuji H, Hirokawa N, Takai Y. Localization and subcellular distribution of smg p25A, a ras p21-like GTP-binding protein, in rat brain. J Biol Chem. 1990;265:11872–11879. [PubMed] [Google Scholar]
- Navone F, Jahn R, Di Gioia G, Stukenbrok H, Greengard P, De Camilli P. Protein p38: An integral membrane protein specific for small vesicles of neurons and neuroendocrine cells. J Cell Biol. 1986;103:2511–2527. doi: 10.1083/jcb.103.6.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardee JD, Spudich JA. Purification of muscle actin. Methods Enzymol. 1982;85:164–181. doi: 10.1016/0076-6879(82)85020-9. [DOI] [PubMed] [Google Scholar]
- Pestonjamasp K, Amieva MR, Strassel CP, Nauseef WM, Furthmayr H, Luna EJ. Moesin, ezrin, and p205 are actin-binding proteins associated with neutrophil plasma membranes. Mol Biol Cell. 1995;6:247–259. doi: 10.1091/mbc.6.3.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD, Cooper JA. Methods to characterize actin filament networks. Methods Enzymol. 1982;85:211–233. doi: 10.1016/0076-6879(82)85022-2. [DOI] [PubMed] [Google Scholar]
- Rayment I, Holden HM, Whittaker M, Yohn CB, Lorenz M, Holmes KC, Milligan RA. Structure of the actin-myosin complex and its implications for muscle contraction. Science. 1993;261:58–65. doi: 10.1126/science.8316858. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., E.F Fritsch, and T. Maniatis. 1989. Molecular Cloning: A Laboratory Manual, 2nd Ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 1650 pp.
- Saras J, Heldin C-H. PDZ domains bind carboxy-terminal sequences of target proteins. Trends Biochem Sci. 1996;21:455–458. doi: 10.1016/s0968-0004(96)30044-3. [DOI] [PubMed] [Google Scholar]
- Schröder RR, Manstein DJ, Jahn W, Holden H, Rayment I, Holmes KC, Spudich JA. Three-dimensional atomic model of F-actin decorated with Dictyostelium myosin S1. Nature. 1993;364:171–174. doi: 10.1038/364171a0. [DOI] [PubMed] [Google Scholar]
- Siegel LM, Monty KJ. Determination of molecular weights and frictional ratios of proteins in impure systems by use of gel filtration and density gradient centrifugation. Application to crude preparations of sulfite and hydroxylamine reductases. Biochim Biophys Acta. 1966;112:346–362. doi: 10.1016/0926-6585(66)90333-5. [DOI] [PubMed] [Google Scholar]
- Smith SJ. Neuronal cytomechanics: the actin-based motility of growth cones. Science. 1988;242:708–715. doi: 10.1126/science.3055292. [DOI] [PubMed] [Google Scholar]
- Songyang Z, Fanning AS, Fu C, Xu J, Marfatia SM, Chishti AH, Crompton A, Chan AC, Anderson JM, Cantley LC. Recognition of unique carboxyl-terminal motifs by distinct PDZ domains. Science. 1997;275:73–77. doi: 10.1126/science.275.5296.73. [DOI] [PubMed] [Google Scholar]
- Takeuchi M, Hata Y, Hirao K, Toyoda A, Irie M, Takai Y. SAPAPs: a family of PSD-95/SAP90-associated proteins localized at postsynaptic density. J Biol Chem. 1997;272:11943–11951. doi: 10.1074/jbc.272.18.11943. [DOI] [PubMed] [Google Scholar]
- Tanaka E, Sabry J. Making the connection: cytoskeletal rearrangements during growth cone guidance. Cell. 1995;83:171–176. doi: 10.1016/0092-8674(95)90158-2. [DOI] [PubMed] [Google Scholar]
- Tessier-Lavigne M, Goodman CS. The molecular biology of axon guidance. Science. 1996;274:1123–1133. doi: 10.1126/science.274.5290.1123. [DOI] [PubMed] [Google Scholar]
- Torre E, Mcniven MA, Urrutia R. Dynamin 1 antisense oligonucleotide treatment prevents neurite formation in cultured hippocampal neurons. J Biol Chem. 1994;269:32411–32417. [PubMed] [Google Scholar]