

Abstract
Detachment of epithelial cells from the extracellular matrix leads to induction of programmed cell death, a process that has been termed “anoikis.” It has been reported recently that detachment of MDCK cells from matrix results in activation of Jun–NH2-terminal kinases (JNKs) and speculated that these stress activated protein kinases play a causal role in the induction of anoikis (Frisch, S.M., K. Vuori, D. Kelaita, and S. Sicks. 1996. J. Cell Biol. 135:1377–1382). We report here that although JNK is activated by detachment of normal MDCK cells, study of cell lines expressing activated signaling proteins usually controlled by Ras shows that stimulation of JNK fails to correlate with induction of anoikis. Activated phosphoinositide 3-OH kinase and activated PKB/Akt protect MDCK cells from detachment-induced apoptosis without suppressing JNK activation. Conversely, activated Raf and dominant negative SEK1, a JNK kinase, attenuate detachment-induced JNK activation without protecting from apoptosis. zVAD-fmk, a peptide inhibitor of caspases, prevents MDCK cell anoikis without affecting JNK activation. p38, a related stress-activated kinase, is also stimulated by detachment from matrix, but inhibition of this kinase with SB 203580 does not protect from anoikis. It is therefore unlikely that either JNK or p38 play a direct role in detachment-induced programmed cell death in epithelial cells.
Integrin-mediated adhesion to the extracellular matrix provides an essential survival signal for many mammalian cell types (19). Upon detachment from the matrix, endothelial (20) and epithelial (5) cells enter into programmed cell death; this cell detachment–induced apoptosis has been referred to as “anoikis.” Normal adherent cells of epithelial and various other origins do not survive in the absence of correct cell surface integrin binding to extracellular matrix proteins and therefore are unable to proliferate in inappropriate sites or to survive in the absence of attachment. Transformation by v-ras and v-src oncogenes results in potent protection of MDCK cells from detachment-induced apoptosis (5); this is likely to underlie the ability of many tumor-derived cell lines to grow in suspension (26).
We have recently characterized part of the signal transduction pathway by which oncogenic Ras protein protects MDCK cells from detachment-induced apoptosis (12). In its activated, GTP-bound form, Ras is able to interact with and stimulate a number of families of target enzymes, including the Raf serine/threonine protein kinases, the heterodimeric phosphoinositide 3-OH kinase (PI 3-kinases)1, and the Ral/GDS family of guanine nucleotide exchange factors for the Ras-related protein Ral (16). Partial loss-of-function mutations in Ras have been identified, which distinguish between these various effector families, with T35S Ras still signaling through Raf, but not the other effectors, S37G Ras signaling through Ral/GDS only, and Y40C Ras signaling through PI 3-kinase only (10, 24, 28, 29). The ability of Ras to protect MDCK cells from detachment-induced apoptosis relies entirely on the PI 3-kinase effector pathway; activation of Raf or Ral/GDS provide no ability to survive in suspension (12). Adhesion to extracellular matrix normally provides a basal level of PI 3-kinase activity, which protects epithelial cells from apoptosis. In Ras-transformed cells, this signal can be provided in the absence of adhesion by the direct interaction of Ras/GTP with the catalytic p110 subunit of PI 3-kinase. Downstream of PI 3-kinase, the serine/threonine protein kinase PKB/Akt appears to play a critical role in protecting cells from apoptosis, both in this (12) and other (17) cell systems.
Many components of the machinery that regulates and executes programmed cell death have been identified (22). In addition to the central roles of the caspase (ICE) family of proteases and the Bcl-2 family of apoptosis regulators, recent reports have suggested that the stress activated protein kinases that phosphorylate the NH2-terminal region of Jun (SAPKs or Jun–NH2-terminal kinases [JNKs] [14]) may be involved in controlling apoptosis in certain systems. Activation of JNK in the absence of ERK activity induces apoptosis in neuronal cells (4, 7, 30), while ceramide-induced apoptosis in U937 leukemia cells and bovine aortic endothelial cells appears to involve induction of JNK activity (27). In certain fibroblastic cell lines, the JNK pathway has been reported to mediate cell death after injury induced by cisplatinum, UV irradiation, or heat shock (31). However, in other systems such as B cells, JNK activation appears to play a protective role with respect to apoptosis (25). In the case of detachment-induced apoptosis in MDCK cells, it has recently been reported that detachment from matrix causes activation of JNK and that this correlates with induction of apoptosis (6). Here we present evidence that although JNK, and additionally the related kinase p38, is activated by detachment of MDCK cells from extracellular matrix, neither of these kinases is necessary for detachment-induced apoptosis, nor is their activation sufficient to induce this form of programmed cell death. There is thus no causal correlation between activation of JNK and p38 and the induction of anoikis.
Materials and Methods
Materials
SB203580 and zVAD.fmk were purchased from Alexis (San Diego, CA).
Expression Vectors.
The Ras and Ras mutant expression vectors were generated as described in 24. Activated PI 3-kinase constructs were made by the addition of a COOH-terminal farnesylation signal from H-Ras to p110 to cause its membrane localization (myc-tagged p110-CAAX in pcDNA3). Raf-CAAX cDNA was provided by R. Marais and C.J. Marshall (Institute of Cancer Research, London, United Kingdom). Gag-PKB cDNA was provided by B. Burgering and P. Coffer (University of Utrecht, Utrecht, The Netherlands). HA-tagged Akt cDNA was provided by T. Franke (Harvard Medical School, Boston, MA). Competitive inhibitory SEK mutant SEK-AL cDNA was provided by J. Woodgett (Ontario Cancer Institute, Ontario, Canada). GST-jun (NH2-terminal amino acids 1–79) and GST-ATF2 (amino acids 51–53 MTL→ AAA) were provided by N. Jones (Imperial Cancer Research Fund). The ATF2-derived fusion protein GST-ATF2 (amino acids 51–53 MTL→ AAA) binds to p38, but not JNK.
Cell Lines.
Early passage MDCK cells were provided by J. Taylor-Papadimitriou (Imperial Cancer Research Fund) and were maintained in DME supplemented with 10% FBS. To generate stably transfected lines wild-type cells were transfected with the various constructs by lipofection (Lipofectamine; GIBCO BRL, Gaithersburg, MD). Cells were plated at 105 cells/well of a six-well plate and transfected the next day with 4 μg of DNA (plus 0.4 μg of a hygromycin resistance plasmid for those vectors not encoding a neomycin resistance gene) and 10 μl of Lipofectamine/ well. After 48 h, cells were replated into a 10-cm dish and selected with 250 μg/ml hygromycin or 500 μg/ml G418 as appropriate. Control cells were transfected with empty vector alone or were untransfected. After selection with antibiotic for 10 to 14 d, individual resistant colonies were picked and expanded. At least 24 clones were picked for each transfection and expression of protein assessed by Western bloting of cell lysates using appropriate antibodies. Expression of V12 H-Ras and the Ras mutant proteins (C40 and G37) varied between 5- and 10-fold excess over endogenous Ras levels. Confirmation of activation of the relevant signaling pathway in each cell type has been described previously (12).
Kinase Assays
The effect of detachment on JNK and p38 activation was measured in sub-confluent cells growing in DME/10% FBS. Cells were plated at 2 × 105 cells/well of a six-well plate and detached 24 h later with a brief incubation with 0.25% trypsin/2 mM EDTA. Trypsin was neutralized with one wash with DME/10% FBS; the cells were then resuspended in DME/BSA 2 mg/ml and incubated in six-well plates previously coated with polyHEMA for the period of time indicated. Cells were then rapidly pelleted and lysed in lysis buffer containing 150 mM NaCl, 50 mM Hepes (pH 7.5), 1% Triton X-100, 5 mM EDTA, 10 μg/ml aprotinin/pepstatin/leupeptin, 1 mM Na3VO4, and 1 mM PMSF. Lysates were centrifuged at 13,000 g for 10 min at 4°C, equalized for protein, and the supernatant incubated with 2 μg of GST-ATF2 (amino acids 51–53 MTL → AAA) for p38 assays, or GST-jun (NH2-terminal amino acids 1–79) for JNK assays for 1 h in the presence of glutathione-agarose. Pellets were washed twice in wash buffer (1% PBS/ Triton X-100/EDTA 1mM) and once in kinase buffer wash (25 mM Tris, pH 8, 10 mM MgCl2). The pellet was resuspended in kinase buffer and 5 μCi [γ-32P]ATP (Amersham Intl., Arlinton Heights, IL); after 30 min incubation at room temperature the reaction was stopped with sample buffer and boiling for 5 min. After SDS-PAGE the phosphorylation of glutathione-S-transferase (GST) fusion protein was quantified by phosphorimager (Molecular Dynamics, Sunnyvale, CA). In preliminary experiments, kinase assays carried out by immunoprecipitation of JNK using polyclonal antibody sc-571 (Santa Cruz Biotechnology, Santa Cruz, CA) or p38 using polyclonal antibody sc-535 (Santa Cruz Biotechnology), followed by phosphorylation of the same substrates used in the pull-out assays (GST-ATF2 [amino acids 51–53 MTL → AAA] for p38 assays, GST-jun [NH2-terminal amino acids 1–79] for JNK assays) gave essentially identical results to those obtained using the pull-out assay described above. Subsequent assays were therefore carried out using the GST fusion pull-out assay.
Anoikis Assays
Performed as described in 5. Cells were plated at 2 × 105 cells/well of a six-well plate and detached 24 h later with a brief incubation with 0.25% trypsin/2 mM EDTA. At the time of performing the assay the density of the cells was 4 × 104 cells/cm2, the same as that used by Frisch et al. (6).
Survival Assay.
Cells were detached and equal numbers either replated immediately or maintained in suspension as described above for 12 h and then replated into 96-well plates. 4 h after replating, cell survival was measured by a colorimetric method based on the conversion of MTS tetrazolium to formazan (CellTiter AQueous Kit; Promega, Madison, WI) used according to the manufacturer's instructions.
DNA Fragmentation ELISA.
Performed using the cell death detection ELISA kit (Boehringer Mannheim, Indianapolis, IN). Samples were assayed in duplicate according to the manufacturer's instructions. Lysates from 103 cells/point were assayed.
DNA Laddering.
5 × 106 Cells were maintained in suspension for 8 h. Cells were then washed once in PBS, and low molecular weight DNA was extracted. The cell pellet was lysed using 0.5 ml of extraction buffer (0.5% Triton X-100, 5 mM Tris, pH 7.5, 20 mM EDTA) for 20 min on ice. The low molecular weight DNA containing soluble fraction was isolated by centrifugation at 15,000 g for 10 min, phenol/chloroform extracted three times, ethanol precipitated, resuspended in Tris/EDTA, pH 8.0, containing 20 μg/ml RNase A, and incubated at 37°C for 2 h. DNA was run on a 1.5% agarose gel and visualized by ethidium bromide staining.
Results
JNK and p38 Are Activated after MDCK Detachment
After trypsinization and placing in suspension, JNK and p38 activity was assayed at various time points. This showed a rapid and sustained elevation in JNK (Fig. 1 A) and p38 activity (Fig. 1 B) with JNK activity still elevated up to 6 h after detachment. At longer time points, JNK activity remained elevated for at least 10 h after detachment, whereas p38 activity returned to near basal levels at that time (data not shown). Trypsinization itself was not responsible for these effects, as there was no significant difference in JNK activity in cells that were attached and those that had been detached and lysed immediately (Fig. 1 A); detachment by the use of EDTA had identical results (data not shown). A comparison between JNK activation by detachment and by ultraviolet irradiation showed that the effect of detachment was less marked than that of ultraviolet (Fig. 1 C). Reattachment of cells to collagen-coated plates after trypsinization prevented a significant rise in JNK activity (data not shown).
Figure 1.

Jun and p38 kinases are activated in response to MDCK detachment. JNK (A) or p38 (B) activity was assayed in attached cells or at varying time points after detachment. Data are expressed as fold increase in kinase activity over the basal level and represent the means of two separate experiments for each kinase. For comparison (C), the level of JNK activation in response to ultraviolet C irradiation is shown (assayed at 60 min after detachment or ultraviolet C exposure of 2,000 μJ/cm2). Representative autoradiographs of kinase reactions assayed as described in Materials and Methods are shown. Time point 0 represents cells that were detached and then immediately lysed.
Effect of Stable Expression of Activated Ras and Downstream Effectors on JNK Activation
MDCK cell lines expressing V12 H-Ras, activated Raf (Raf-CAAX), activated PI 3-kinase catalytic subunit (p110*), and activated Akt/PKB (gag-PKB) were established. In addition, MDCK lines expressing Ras effector mutants V12 C40 Ras and V12 G37 Ras were generated. The C40 mutant activates the PI 3-kinase but not the Raf or Ral-GDS pathways, and the G37 mutant interacts with the Ral-GDS but not PI 3-kinase or Raf pathways (24). In previously reported studies employing transient transfection assays, both activated Ras and PI 3-kinase have been reported to increase JNK activity (13). However, we could detect no significant elevation in basal JNK activity in any of the clones expressing V12 Ras or its effector molecules, including PI 3-kinase and Raf (Fig. 2 A). Others have reported Ras activation of JNK to be very weak compared with that of Rac/Cdc42 or physiological regulators of the pathway (3, 21). The effect of detachment from ECM on JNK activity was measured in the various lines. At least two independent clones for each construct were assayed in four separate experiments. Fig. 2 B shows that cells expressing V12 Ras or activated Raf have blunted JNK activation after suspension. In contrast, those clones expressing activated PI 3-kinase, the C40 or G37 mutants of V12 Ras, or gag-PKB showed detachment-induced JNK activity similar to wild-type cells.
Figure 2.

Effect of stable expression of activated Ras and downstream effectors on JNK activation. (A) Basal JNK activity in attached cells in MDCK clones is not significantly different from parental wild-type cells. Data are from four to six separate experiments with at least two individual clones for each mutant line and are normalized to the level of JNK activity in attached wild-type cells. (B) Detachment-induced JNK activation is suppressed in cells expressing activated Ras or Raf mutants. JNK activity was measured after 2 h in suspension. Data are from four to six separate experiments with at least two individual clones for each mutant line. The increment in kinase activity induced by detachment is derived by subtracting the basal activity in attached cells for each clone or wild-type cells and normalizing to the JNK activity in wild-type cells in suspension. Typical autoradiographs of JNK assays carried out with cells in suspension are shown.
JNK Activation in MDCK Clones Does Not Correlate with Apoptosis
Parental MDCK cells undergo apoptosis in suspension as measured by DNA fragmentation in agarose gels or by ELISA and by estimating cell survival (Figs. 3 and 12). The various MDCK clones were placed in suspension, and parallel measurement of JNK activity (see Fig. 2) and DNA fragmentation was carried out. Quantification of DNA fragmentation by ELISA showed that cells expressing V12 Ras, activated PI 3-kinase, V12 C40 Ras (the mutant that activates PI 3-kinase), or gag-PKB were all protected from detachment-induced apoptosis (Fig. 3). Cells expressing activated Raf or V12 G37 Ras were not protected from apoptosis. As we have recently shown, a PI 3-kinase/PKB pathway is critically involved in mediating the survival signal downstream from matrix adhesion and from oncogenic Ras. Cells expressing activated PI 3-kinase or Akt/PKB have JNK activity similar to wild-type cells (Fig. 2). This suggests that either PI 3-kinase and Akt/PKB exert their anti-apoptotic effect downstream of JNK activation or that the activation of JNK after detachment of wild-type MDCK cells is not a critical component of the apoptotic pathway. The latter explanation is favored by the finding that cells expressing activated Raf do not show increased JNK activity in response to detachment but still undergo apoptosis.
Figure 3.

JNK activation in MDCK clones does not correlate with detachment-induced apoptosis. After 8 h in suspension, DNA fragmentation was quantified by ELISA (A) or visualized by agarose gel electrophoresis. The ELISA data are from four to six separate experiments with at least two individual clones for each mutant line conducted in parallel with JNK assays shown in Fig. 2.
Inhibition of JNK or p38 Pathways Does Not Prevent Detachment-induced Apoptosis
Cell lines stably expressing the dominant-negative JNK kinase (SEK-AL) were examined for inhibition of JNK activity. After detachment, JNK was not activated in these clones (Fig. 4 A). However, although JNK activation after detachment was blocked by expression of SEK-AL there was no effect on the level of detectable apoptosis (Figs. 4 B and 3 B). Wild-type MDCK cells incubated with the p38 inhibitor SB203580 at 20 μM, a concentration that prevented p38 activation in suspended cells (data not shown), were also not protected from detachment-induced apoptosis (Fig. 4 B). In addition, use of a combination of SEK-AL and SB203580 to inhibit both JNK and p38 kinase activity failed to affect detachment induced apoptosis (Fig. 4 B). These results suggest that activation of the JNK and p38 pathways does not contribute significantly to apoptosis triggered by loss of adhesion.
Figure 4.
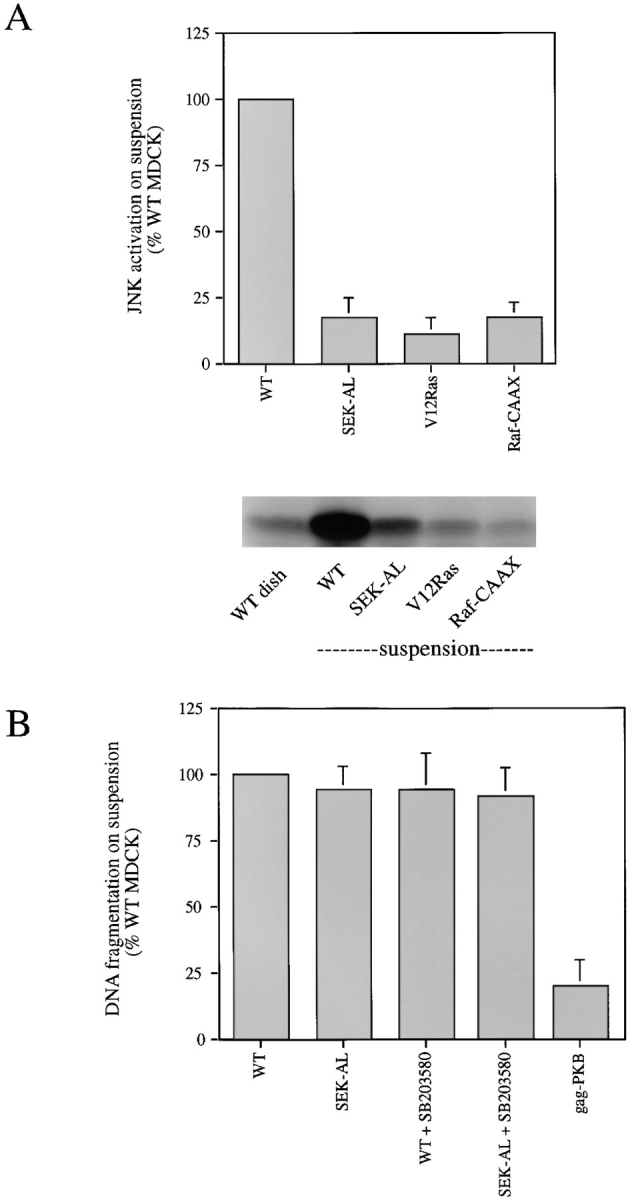
Inhibition of JNK or p38 pathways does not prevent detachment-induced apoptosis. (A) Expression of dominant inhibitory SEK-AL mutant inhibits detachment-induced JNK activation. Data for SEK-AL are from eight separate experiments with three individual clones and are compared with JNK activity levels in cells expressing activated Ras or Raf mutants. Representative autoradiographs of kinase reactions assayed as described in Materials and Methods are shown. The increment in kinase activity induced by detachment is derived by subtracting the basal activity in attached cells for each clone or wild-type cells and normalizing to the JNK activity in wild-type cells in suspension. (B) DNA fragmentation in response to detachment was quantified by ELISA in MDCK clones expressing SEK-AL (n = 6) or in wild-type cells treated with the p38 inhibitor SB203580 (n = 3). The level of DNA fragmentation in cells expressing the activated Akt/ PKB mutant is shown for comparison.
Inhibition of ICE-like Protease Activity with zVAD.fmk Protects MDCK from Apoptosis Without Affecting JNK Activity
The global inhibitor of ICE and related proteases (caspases), zVAD.fmk, was used to investigate the role of these proteases in JNK activation and apoptosis after cell detachment. Incubation with zVAD.fmk inhibited apoptosis as measured by assay of mitochondrial activity (Fig. 5 A) and also by DNA fragmentation (Fig. 5 B). A concentration of 100 μM zVAD.fmk strongly protected wild-type MDCK cells from detachment-induced apoptosis. In addition, zVAD.fmk enhanced MDCK survival in a dose-dependent manner after detachment, as measured by a clonogenic assay (data not shown). However, incubation with zVAD.fmk did not significantly affect JNK activation in suspended cells (Fig. 5 C), even at a 100 μM peptide concentration, which fully protects from apoptosis. It therefore appears unlikely that JNK activation is sufficient to cause apoptosis in the absence of caspase activity or that death-inducing levels of caspase activity are required for the detachment-induced activation of JNK.
Figure 5.
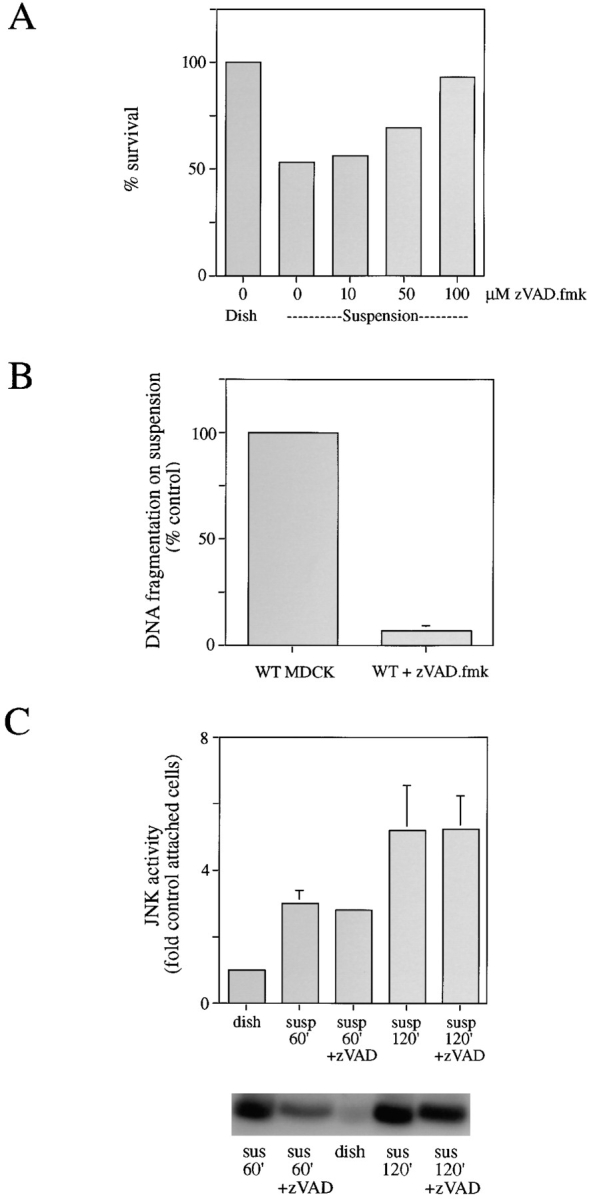
Inhibition of ICE-like protease activity with zVAD.fmk prevents apoptosis without affecting JNK activity. (A) Dose- response effect of treatment with zVAD.fmk on cell survival in response to detachment as assayed by mitochondrial integrity. Data are mean of two experiments. (B) zVAD.fmk at 100 μM prevents DNA fragmentation quantified by ELISA. Results are mean of three separate experiments. (C) zVAD.fmk at 100 μM does not affect JNK activation in response to detachment at 60 or 120 min. Representative autoradiographs of kinase reactions assayed as described in Materials and Methods are shown.
Discussion
In this paper we show that JNK and p38 are both rapidly activated upon detachment of MDCK epithelial cells from extracellular matrix. The activation of JNK on detachment of these cells has previously been reported by Frisch et al. (6); however, in contrast to that work, we find evidence that activation of JNK, and also p38, is not related to detachment-induced apoptosis. As also seen by Frisch et al., expression of activated Ras in MDCK cells both suppresses detachment-induced apoptosis (5) and JNK activation (6). By expressing downstream effectors of Ras and partial loss-of-function Ras mutants, we were able to show that the ability of Ras to prevent detachment-induced JNK activation is mediated by the Raf pathway (Fig. 2), whereas the ability of Ras to prevent detachment-induced apoptosis is mediated by the PI 3-kinase and PKB/Akt pathway. Cells expressing activated PI 3-kinase or PKB/Akt are highly protected from detachment-induced apoptosis, yet they show the same degree of detachment-induced JNK activity as do wild-type cells. The ability of Ras to protect from anoikis therefore is mediated by a different pathway from its ability to suppress JNK activation. A possible explanation for constitutive activation of Ras and Raf preventing JNK activation is that the resulting activation of ERK may upregulate the expression of phosphatases that have broad specificity for the entire family of JNKs and ERKs (2).
While cells that activate JNK normally upon suspension can be resistant to apoptosis, such as the PKB/Akt or PI 3-kinase–expressing cells, the converse is also true: cells that fail to activate JNK upon suspension can still be sensitive to detachment-induced cell death. This is found with activated Raf-expressing cells, as discussed above, and also with cells expressing SEK-AL, a dominant negative form of the JNKK, SEK1. Expression of SEK-AL reduces detachment-induced JNK activity to levels seen in activated Ras-expressing cells, yet has no protective effect. This is different from the effect of dominant negative JNKK reported by Frisch et al. (6). In that study, expression of dominant negative JNKK reduced JNK activation by detachment to a similar degree to what we find here, yet it completely protected MDCK cells from detachment- induced apoptosis. The reason for this discrepancy is not clear. The mutations used to create the dominant negative JNKK/SEK1 were slightly different: S257A and T261L here (31) and S257A and T261A in Frisch's study (6). However, since they had similar effects on JNK activation in both cases it is unlikely that this was significant. Perhaps a more likely explanation lies in clonal variation of the MDCK cells used. It should be noted that with the exception of the experiments using dominant negative JNKK, the MDCK cells used in our studies behave very similarly with respect to apoptosis and JNK as do those of Frisch et al.
It has been suggested recently that activation of the caspase family proteases may be responsible for activation of JNK and p38, possibly by proteolytic activation of upstream kinases such as MEKK (1, 11). It has been reported that transient expression of activated MEKK-1, an activator of SEK-1/JNKK, can induce apoptosis in fibroblasts, but that this may not be through the JNK or p38 pathways (8). If JNK is activated downstream of caspases, it would be expected that overexpression of Bcl-2 or expression of the cowpox anti-apoptotic protein CrmA, both of which inhibit activation of the proteases, would prevent JNK activation. Such an effect has indeed been reported (6). However, we find here that the broad specificity peptide-based caspase inhibitor zVAD.fmk fails to inhibit JNK activation by detachment at all at concentrations where it blocks apoptosis fully. It is possible that relatively small amounts of caspase activity are sufficient to activate JNKs, whereas a greater level of caspase activity is required to induce apoptosis. This indicates that although the JNK pathway may be one of the targets of caspases, its activation is not sufficient to induce programmed cell death, at least in this cell system. In the cases of cells constitutively expressing a high level of Bcl-2 or CrmA, caspase activity may be very strongly inhibited compared to the use of zVAD.fmk, to levels much lower than is required to inhibit detachment-induced apoptosis.
We have previously reported that levels of PI 3-kinase– produced phosphoinositide lipids drop rapidly on detachment of MDCK cells and are rapidly restored after replating on matrix (12). Since PI 3-kinase activation has been associated with stimulation of the small GTPase Rac, at least by their similar effects on the actin cytoskeleton (23), and activated Rac has been shown to stimulate JNK in cotransfection assays (3, 21), it might be expected that JNK activity in cells would mirror PI 3-kinase activity. However, clearly the opposite is true from the data reported here and previously (12): when cells are detached, PI 3-kinase activity goes down and JNK activity goes up, while the reverse happens on replating. We have also been unable to see an induction of JNK activity by constitutively activated PI 3-kinase in transient cotransfection experiments (18). A possible explanation for this apparent anomaly is that different pools of Rac protein may exist in the cell: one responds to PI 3-kinase and interacts with effectors that control the actin cytoskeleton, while another responds to other incoming signals and interacts with different effectors that regulate the JNK pathway. Recent reports have clearly shown that Rac controls the cytoskeleton and JNK through different effector systems (9, 15). At present the nature of the pathway whereby JNK is activated after detachment is unknown, and it is not clear if Rac or other small GTPases are involved. It is likely that JNK is regulated by several different pathways in addition to those that may involve Rac/Cdc42 or caspases.
The data reported here provide evidence that the JNK pathway is activated after the cellular stress of detachment from the extracellular matrix. However, JNK activation is neither necessary nor sufficient for the induction of programmed cell death by loss of adhesion. JNK appears to be regulated by several different pathways, one of which may lie downstream of the caspase family of ICE proteases. In this cell type, the JNK and p38 pathways appear not to be essential parts of the apoptosis signaling machinery. However, it is clear that the regulation of programmed cell death is complex and varies considerably between different cell types and also between different forms of death-inducing insult. The concept of JNK playing an essential role in the induction of apoptosis was derived largely from data obtained using neuronal cells; the experiments reported here suggest that considerable caution should be employed when extrapolating these ideas to other cellular systems.
Abbreviations used in this paper
- GST
glutathione-S-transferase
- JNK
Jun–NH2-terminal kinase
- PI 3-kinase
phosphoinositide 3-OH kinase
Footnotes
A. Khwaja was supported by a Medical Research Council clinician–scientist award.
Address all correspondence to Julian Downward, Imperial Cancer Research Fund, 44 Lincoln's Inn Fields, London WC2A 3PX, United Kingdom. Tel.: (44) 171 269 3533. Fax: (44) 171 269 3092. E-mail: downward@icrf.icnet.uk
Asim Khwaja's present address is Department of Haematology, University College London Medical School, London WC1E 6HX, United Kingdom.
References
- 1.Cahill MA, Peter ME, Kischkel FC, Chinnaiyan AM, Dixit VM, Krammer PH, Nordheim A. CD95 (APO-1/Fas) induces activation of SAP kinases downstream of ICE-like proteases. Oncogene. 1996;13:2087–2096. [PubMed] [Google Scholar]
- 2.Chu Y, Solski PA, Khosravi-Far R, Der CJ, Kelly K. The mitogen-activated protein kinase phosphatases PAC1, MKP-1, and MKP-2 have unique substrate specificities and reduced activity in vivo toward the ERK2 sevenmaker mutation. J Biol Chem. 1996;271:6497–6591. doi: 10.1074/jbc.271.11.6497. [DOI] [PubMed] [Google Scholar]
- 3.Coso OA, Chiariello M, Yu JC, Teramoto H, Crespo P, Xu N, Miki T, Gutkind JS. The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell. 1995;81:1137–1146. doi: 10.1016/s0092-8674(05)80018-2. [DOI] [PubMed] [Google Scholar]
- 4.Estus S, Zaks WJ, Freeman RS, Gruda M, Bravo R, Johnson EM. Altered gene expression in neurons during programmed cell death: identification of c-jun as necessary for neuronal apoptosis. J Cell Biol. 1994;127:1717–1727. doi: 10.1083/jcb.127.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frisch SM, Francis H. Disruption of epithelial cell–matrix interactions induces apoptosis. J Cell Biol. 1994;124:619–626. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frisch SM, Vuori K, Kelaita D, Sicks S. A role for Jun-N-terminal kinase in anoikis; suppression by bcl-2 and crmA. J Cell Biol. 1996;135:1377–1382. doi: 10.1083/jcb.135.5.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ham J, Babij C, Whitfield J, Pfarr CM, Lallemand D, Yaniv M, Rubin LL. A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 1995;14:927–939. doi: 10.1016/0896-6273(95)90331-3. [DOI] [PubMed] [Google Scholar]
- 8.Johnson NL, Gardner AM, Diener KM, Lange-Carter CA, Gleavy J, Jarpe MB, Minden A, Karin M, Zon LI, Johnson GL. Signal transduction pathways regulated by mitogen-activated/extracellular response kinase kinase kinase induce cell death. J Biol Chem. 1996;271:3229–3237. doi: 10.1074/jbc.271.6.3229. [DOI] [PubMed] [Google Scholar]
- 9.Joneson T, McDonough M, Bar-Sagi D, Van Aelst L. RAC regulation of actin polymerization and proliferation by a distinct pathway from Jun kinase. Science. 1996;274:1374–1376. doi: 10.1126/science.274.5291.1374. [DOI] [PubMed] [Google Scholar]
- 10.Joneson T, White MA, Wigler MH, Bar-Sagi D. Stimulation of membrane ruffling and MAP kinase activation by distinct effectors of Ras. Science. 1996;271:810–812. doi: 10.1126/science.271.5250.810. [DOI] [PubMed] [Google Scholar]
- 11.Juo P, Kuo CJ, Reynolds SE, Konz RF, Raingeaud J, Davis RJ, Blenis J. Fas activation of the p38 mitogen-activated protein kinase signalling pathway requires ICE/CED-3 family proteases. Mol Cell Biol. 1997;17:24–35. doi: 10.1128/mcb.17.1.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khwaja A, Rodriguez-Viciana P, Wennstrom S, Warne PH, Downward J. Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. EMBO (Eur Mol Biol Organ) J. 1997;16:2783–2793. doi: 10.1093/emboj/16.10.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klippel A, Reinhard C, Kavanaugh W M, Apell G, Escobedo MA, Williams LT. Membrane localization of phosphatidylinositol 3-kinase is sufficient to activate multiple signal transducing kinase pathways. Mol Cell Biol. 1996;16:4117–4127. doi: 10.1128/mcb.16.8.4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kyriakis JM, Avruch J. Protein kinase cascades activated by stress and inflammatory cytokines. Bioessays. 1996;18:567–577. doi: 10.1002/bies.950180708. [DOI] [PubMed] [Google Scholar]
- 15.Lamarche N, Tapon N, Stowers L, Burbelo PD, Aspenstrom P, Bridges T, Chant J, Hall A. Rac and Cdc42 induce actin polymerization and G1 cell cycle progression independently of p65PAK and the JNK/SAPK MAP kinase cascade. Cell. 1996;87:519–529. doi: 10.1016/s0092-8674(00)81371-9. [DOI] [PubMed] [Google Scholar]
- 16.Marshall CJ. Ras effectors. Curr Opin Cell Biol. 1996;8:197–204. doi: 10.1016/s0955-0674(96)80066-4. [DOI] [PubMed] [Google Scholar]
- 17.Marte BM, Downward J. PKB/Akt: connecting PI 3-kinase to cell survival and beyond. Trends Biochem Sci. 1997;22:355–358. doi: 10.1016/s0968-0004(97)01097-9. [DOI] [PubMed] [Google Scholar]
- 18.Marte BM, Rodriguez-Viciana P, Wennström S, Warne PH, Downward J. PI 3-kinase and PKB/Akt act as an effector pathway for R-Ras. Curr Biol. 1997;7:63–70. doi: 10.1016/s0960-9822(06)00028-5. [DOI] [PubMed] [Google Scholar]
- 19.Meredith JE, Schwartz MA. Integrins, adhesion and apoptosis. Trends Cell Biol. 1997;7:146–150. doi: 10.1016/S0962-8924(97)01002-7. [DOI] [PubMed] [Google Scholar]
- 20.Meredith JE, Fazeli B, Schwartz MA. The extracellular matrix as a cell survival factor. Mol Biol Cell. 1993;4:953–961. doi: 10.1091/mbc.4.9.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Minden A, Lin A, Claret FX, Abo A, Karin M. Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell. 1995;81:1147–1157. doi: 10.1016/s0092-8674(05)80019-4. [DOI] [PubMed] [Google Scholar]
- 22.Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- 23.Nobes CD, Hawkins P, Stephens L, Hall A. Activation of the small GTP-binding proteins rho and rac by growth factor receptors. J Cell Sci. 1995;108:225–233. doi: 10.1242/jcs.108.1.225. [DOI] [PubMed] [Google Scholar]
- 24.Rodriguez-Viciana P, Warne PH, Khwaja A, Marte BM, Pappin D, Das P, Waterfield MD, Ridley A, Downward J. Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell. 1997;89:457–467. doi: 10.1016/s0092-8674(00)80226-3. [DOI] [PubMed] [Google Scholar]
- 25.Sakata N, Patel HR, Terada N, Aruffo A, Johnson GL, Gelfand EW. Selective activation of c-jun kinase mitogen-activated protein kinase by CD40 on human B cells. J Biol Chem. 1995;270:30823–30828. doi: 10.1074/jbc.270.51.30823. [DOI] [PubMed] [Google Scholar]
- 26.Stoker M, O'Neill C, Berryman S, Waxman V. Anchorage and growth regulation in normal and virus-transformed cells. Int J Cancer. 1968;3:683–693. doi: 10.1002/ijc.2910030517. [DOI] [PubMed] [Google Scholar]
- 27.Verheij M, Bose R, Lin X-H, Yao B, Jarvis WD, Grabt S, Birrer MJ, Szabo E, Zon LI, Kyriakis JM, et al. Requirement for ceramide-initiated SAPK/JNK signalling in stress-induced apoptosis. Nature. 1996;380:75–79. doi: 10.1038/380075a0. [DOI] [PubMed] [Google Scholar]
- 28.White MA, Nicolette C, Minden A, Polverino A, Vanaelst L, Karin M, Wigler MH. Multiple Ras functions can contribute to mammalian-cell transformation. Cell. 1995;80:533–541. doi: 10.1016/0092-8674(95)90507-3. [DOI] [PubMed] [Google Scholar]
- 29.White MA, Vale T, Camonis JH, Schaefer E, Wigler MH. A role for the Ral guanine nucleotide dissociation stimulator in mediating Ras-induced transformation. J Biol Chem. 1996;271:16439–16442. doi: 10.1074/jbc.271.28.16439. [DOI] [PubMed] [Google Scholar]
- 30.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 31.Zanke BW, Boudreau K, Rubie E, Winnett E, Tibbles LA, Zon L, Kyriakis J, Liu FF, Woodgett JR. The stress-activated protein-kinase pathway mediates cell-death following injury induced by cisplatinum, UV irradiation or heat. Curr Biol. 1996;6:606–613. doi: 10.1016/s0960-9822(02)00547-x. [DOI] [PubMed] [Google Scholar]