

Abstract
Lamellipodial protrusion and directional migration of carcinoma cells towards chemoattractants, such as epidermal growth factor (EGF), depend upon the spatial and temporal regulation of actin cytoskeleton by actin-binding proteins (ABPs). It is generally hypothesized that the activity of many ABPs are temporally and spatially regulated by PIP2; however, this is mainly based on in vitro–binding and structural studies, and generally in vivo evidence is lacking. Here, we provide the first in vivo data that directly visualize the spatial and temporal regulation of cofilin by PIP2 in living cells. We show that EGF induces a rapid loss of PIP2 through PLC activity, resulting in a release and activation of a membrane-bound pool of cofilin. Upon release, we find that cofilin binds to and severs F-actin, which is coincident with actin polymerization and lamellipod formation. Moreover, our data provide evidence for how PLC is involved in the formation of protrusions in breast carcinoma cells during chemotaxis and metastasis towards EGF.
Introduction
EGF is an important chemoattractant in hematogenous metastasis of mammary tumors (Wyckoff et al., 2004; Kedrin et al., 2007). Upon EGF stimulation, mammary carcinoma cells start to form cellular protrusions critical for chemotaxis, such as lamellipodia and invadopodia. In carcinoma cells these changes in morphology are driven by restructuring of the actin cytoskeletal network by several actin-binding proteins (ABPs; Condeelis et al., 2005). Cofilin, an actin-severing protein, is important for the initial regulation of actin polymerization during EGF stimulation (Mouneimne et al., 2004, 2006). Active cofilin binds to and severs F-actin, thereby increasing the number of free barbed ends (Mouneimne et al., 2004). In addition, barbed ends may also be increased by the intrinsic nucleation activity of cofilin (Andrianantoandro and Pollard, 2006). The actin polymerization activity of cofilin is consistent with observations that the local activation of cofilin causes local actin polymerization and cellular protrusions in vivo (Ghosh et al., 2004; Mouneimne et al., 2006), which places cofilin as a key protein in chemotaxis of metastatic cancer cells. Indeed, the invasion signature of breast carcinoma cells showed that cofilin is an important contributor to the metastatic phenotype (Wang et al., 2004), and recent findings show that the activity status of cofilin is directly related to invasion, intravasation, and metastasis of mammary tumors (Wang et al., 2006, 2007).
In EGF-stimulated mammary carcinoma cells, the increase in cofilin-dependent barbed ends occurs in two transients: one at 1 min, and a late one at 3 min (Mouneimne et al., 2004). The first barbed end transient depends on PLC (Mouneimne et al., 2004), but not on PI3K or cofilin-dephosphorylation activity (Mouneimne et al., 2004; Song et al., 2006), and is required for directional sensing during chemotaxis (Mouneimne et al., 2006). However, it is not clear how PLC regulates cofilin activity. PLC cleaves the plasma membrane (PM) phospholipid PIP2 into DAG and IP3. Structural and in vitro–binding studies suggest that PIP2 can regulate ABPs such as cofilin. For example, structural studies showed that the putative PIP2-binding sites of many ABPs are overlapping with the sites that are important for F-actin binding, and therefore lipid binding would prevent actin binding (Yonezawa et al., 1990; Ojala et al., 2001; Yin and Janmey, 2003; Gorbatyuk et al., 2006). In vitro–binding studies showed that the activity of many ABPs is inhibited by PIP2-containing lipid micelles. Therefore, it has been hypothesized that ABPs bind to and are inhibited by PIP2 in the PM, and upon PLC-mediated PIP2 reduction, the ABPs get released and activated (Goldschmidt-Clermont et al., 1991, 1992; Janmey and Lindberg, 2004; Di Paolo and De Camilli, 2006; Janetopoulos and Devreotes, 2006; Logan and Mandato, 2006). Indeed gelsolin, a G-actin sequestering and F-actin capping ABP, is released from membrane fractions when PLC is activated (Chen et al., 1996). However, the direct visualization in living cells of the PIP2 hydrolysis and the subsequent translocation from the PM and activation of ABPs is generally lacking. Here, we have tested this hypothesis in living cells for cofilin because the EGF-induced activation of cofilin is PLC dependent in breast carcinoma cells, and because cofilin has been proposed to bind to PIP2 in vitro.
In this study, we provide evidence that in mammary carcinoma cells, cofilin is locally activated by release of a membrane-associated pool of cofilin upon EGF-stimulated PIP2 reduction. By colocalization, FRET, and membrane fractionation studies, we show that a fraction of cofilin is membrane associated, and that this association is reduced upon decreases in the PIP2 level. Using FRET- and FLIP-based experiments and a cofilin-severing assay, we show that the released cofilin locally binds to F-actin and severs actin filaments. This is the first direct in vivo demonstration that cofilin is regulated by PIP2. Moreover, to our knowledge this is the first report that directly visualizes the spatial and temporal regulation of an ABP regulation by PIP2 in living cells.
Results
EGF stimulates PLC-dependent PIP2 reduction in MTLn3 cells
To test whether cofilin activity can be regulated by PIP2, we first studied whether changes in the PIP2 level can be induced upon EGF stimulation in the rat MTLn3 carcinoma cell line. We have chosen this cell type for study because cofilin-induced formation of barbed ends, cell protrusion, and chemotaxis is PLC dependent (Mouneimne et al., 2006), and the metastatic potential of mammary tumors resulting from the growth of these cells is dependent on the activity status of cofilin (Wang et al., 2006). To follow rapid changes in PIP2 levels in vivo, we used a FRET-based PIP2 assay that provides high temporal resolution (van der Wal et al., 2001; van Rheenen and Jalink, 2002; for details see Materials and methods and Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200706206/DC1). A fast and significant drop in PIP2-FRET was observed upon EGF stimulation in MTLn3 (14 ± 5%) and EGFR-overexpressing MTLn3 cells (22 ± 2.5%), accompanied by an increase in cell protrusion activity measured as an increase in cell area (Fig. 1 A). This observation was confirmed by immunofluorescence of fixed MTLn3 cells using PIP2-specific antibodies (Fig. S2) before and 1 min after EGF stimulation (Fig. 1 A, middle). In resting cells, the antibody staining of PIP2 at the PM was higher compared with stimulated cells, indicating a decrease of this lipid (Fig. 1 A, right). Analysis of the fluorescent labeling intensities suggests a drop in the PIP2 levels of 40 to 60%. Biochemical analysis of low-density membrane fractions showed similar decreases in PIP2 after EGF stimulation (see below). These data suggest that the PLC-induced increase in cell area may indeed be initiated by PIP2 hydrolysis. If so, then inhibition of PLC should block PIP2 hydrolysis and subsequently the cell area increase. Mouneimne et al. (2004) observed a block in the cell protrusion activity in the first 3 min after EGF exposure in cells treated with the PLC inhibitor U73122, but they observed normal protrusion in cells treated with U73343, an inactive form of this drug. In line with this, we found that EGF-stimulated PIP2 hydrolysis is inhibited by U73122, but not by U73343 (Fig. 1, B and C), suggesting that the EGF-induced increase in protrusion (cell area) is dependent on PLC through reduction in PIP2. We have also reduced the level of PIP2 by an EGF-independent mechanism. In a recent report, it was noted that PIP2 levels can be reduced by cholesterol extraction with MβCD (Kwik et al., 2003). We observed the same phenomenon in our cell type. Both FRET level (33 ± 5%) and antibody staining were lower after MβCD treatment, suggesting decreased PIP2 levels (Fig. 1 D). Moreover, MβCD treatment resulted in an increase in cell area (Fig. 2 A). Although MβCD may have many side effects and is therefore a blunt tool to reduce PIP2 levels, the observed decrease in PIP2 and the increase in cell area are in line with the idea that PIP2 reduction is inducing formation of cellular protrusions.
Figure 1.

EGF stimulation induces a rapid loss of PIP2 in carcinoma MTLn3 cells. (A) EGF-induced decrease in PIP2-FRET (black, average 23 experiments) and increase in cell area (red) were measured in MTLn3 cells overexpressing the EGF-receptor (Xue et al., 2006) as described in Materials and methods and Fig. S1 (available at http://www.jcb.org/cgi/content/full/jcb.200706206/DC1). The middle panel shows PIP2-antibodies staining and the quantification before and after EGF (n > 25). (B) MTLn3-EGFR cells were treated with the PLC inhibitor U73122 (n = 16) or the nonactive drug U73343 (n = 9), and PIP2 changes were measured by FRET. (C) WT MTLn3 cells were treated as described in B, and PIP2 levels were analyzed by antibody staining. (n > 25) (D) The cholesterol from MTLn3 cells was extracted by methyl-β-cyclodextrin (MβCD). The change in the PIP2 level was monitored by FRET (n =10) and antibody staining. Bars, 5 μm. Values were statistically tested by t test. *, P < 0.05; **, P < 0.01; error bars represent SEM.
Figure 2.

PM-associated cofilin translocates from the PM upon PLC-dependent PIP2 reduction. (A) MTLn3 cells were stimulated with EGF or treated with MβCD. Shown are images of fixed cells stained for cofilin before and after stimulation or treated as indicated (n > 25). The Leica confocal software glow lookup table was used for all images. Bars, 10 μm. (B) MTLn3 cells were fixed and labeled for PIP2 and for total cofilin or the phosphorylated form of cofilin. Images were collected before and 1 min after EGF stimulation. The spatial relationship between PIP2 and cofilin was analyzed with Pearson's correlation coefficient analysis (all conditions n > 25). (C) Shown are the merge and the scatterplot images of the cofilin and PIP2 channels. Scatterplots are from the first 1 μm from the cell edge and were obtained from images that were smoothed ones using ImageJ. The loss in PIP2 signal was compensated by longer exposure times. Note that the higher the spatial relationship between PIP2 and cofilin, the more pixels in the scatterplot align along the diagonal (n = 8). (D) The amount of cofilin in the low-density membrane fraction was analyzed by Western blot in cells stimulated with EGF or treated with MβCD. Note that in contrast to cellular cofilin (Cell Cof.), pure cofilin added to the isolation procedure does not localize in these gradient fractions (three independent experiments). The amount of PIP2 in low-density membrane fractions of cells treated with the PLC-inhibitor U73122 or the nonactive form U73343 was analyzed by dot blot (E) and the amount of cofilin in the same fraction was determined by Western blot (F) (three independent experiments). Values were statistically tested by t test. *, P < 0.05; **, P < 0.01; error bars represent SEM.
Co-localization between cofilin and PIP2 is diminished upon PLC-dependent PIP2 reduction
Our model predicts that a fraction of cofilin is associated with PIP2 in the PM and is inactive, and that upon PIP2 reduction by EGF stimulation, this fraction of cofilin leaves the PM and becomes active. To test this, we studied the spatial distribution of cofilin before and after PIP2 reduction. In unstimulated cells, a fraction of cofilin is localized at the periphery of the cell, and is closely associated with the PM (Fig. 2 A, left). We hypothesize that this is the nonphosphorylated fraction of cofilin that is kept inactive by PIP2. Upon either EGF stimulation or MβCD treatment, cells form cellular protrusions resulting in the PLC-dependent increase in cell area (Fig. 2 A; (Mouneimne et al., 2004). Cofilin is locally released from its peripheral enrichment and translocates to the leading edge of the newly formed lamellipodia (Fig. 2 A, right). We hypothesize that the cofilin in the actin-rich leading edge of lamellipodia is not bound to the membrane PIP2 and therefore active. To test our hypothesis, we analyzed the immunostaining of PIP2 and cofilin at these two regions of the cell. First we compared the colocalization of PIP2 with the phosphorylated-and nonphosphorylated form of cofilin. We predict that the spatial relationship between PIP2 and membrane-bound cofilin should be greater than PIP2 and nonmembrane-bound cofilin. The spatial relationship was analyzed with scatterplots and Pearson's correlation coefficient analysis of immunofluorescence images, and they indeed showed that PIP2 has a higher spatial relationship with the membrane-bound and nonphosphorylated form of cofilin than with the cytosolic and phosphorylated form of cofilin (Fig. 2, B and C). This suggests that the membrane-bound fraction of cofilin is a nonphosphorylated form of cofilin. To test whether this fraction gets released at lower PIP2 levels, cells were stimulated with EGF or treated with MβCD (Fig. 1). Changes in PIP2 staining intensity do not influence the outcome because Pearson's correlation coefficient is insensitive to relative changes in the intensities of the two images (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200706206/DC1). As predicted by our hypothesis that cofilin is associated with PIP2 and released as PIP2 levels fall, the Pearson's correlation coefficient indeed drops (Fig. 2 B) and the scatterplot shows less spatial correlation (Fig. 2 C), suggesting that PIP2 and cofilin become spatially segregated after stimulation with EGF or treatment with MβCD. Biochemical analysis of membrane fractions by dot blots to detect PIP2 and Western blot to detect cofilin confirmed that cofilin is lost from the PM in association with a drop in the amount of PIP2 after EGF-stimulation or MβCD treatment (Fig. 2, D–F).
If a PLC-dependent drop in the PIP2 level is responsible for the translocation of cofilin from the PM and the subsequent protrusion of the cell, then inhibition of PLC should prevent cofilin translocation from the PM. In cells treated with U73122, but not cells treated with the control drug U73343, EGF neither induced a drop in the membrane-associated PIP2 (Fig. 2 E) nor a decrease in the PIP2 labeling levels (Fig. 1 C). Interestingly, the inhibition of PIP2 hydrolysis was associated with retention of membrane-bound cofilin (Fig. 2 F) and the inhibition of the disruption of colocalization between cofilin and PIP2 (Fig. 2, B and C). These data strongly suggest that cofilin is membrane-associated and that upon EGF-mediated PIP2 hydrolysis cofilin gets released from the PM.
Cofilin leaves the PM upon PLC-dependent PIP2 reduction in living cells
To follow the translocation of cofilin in living cells, we expressed a chimera of GFP and cofilin in MTLn3 cells. GFP-cofilin is recognized by GFP and cofilin antibodies and the localization by GFP is similar to that found by antibody staining (Fig. 3, A and B). Translocation of GFP-cofilin from the PM was measured by FRET between GFP-cofilin and a membrane-targeted mCherry (mCherry-CAAX) (Fig. 3 C). FRET (as measured by sensitized emission [sens]) was only observed at the PM (Fig. 3 D; for control cells see Fig. S4 A, available at http://www.jcb.org/cgi/content/full/jcb.200706206/DC1) because mCherry-CAAX labeled exclusively the PM and FRET only occurs for molecules in close proximity (<10 nm) (Fig. 3 C). FRET (sens) values may vary between cells due to (1) variations in expression levels of the fluorescent proteins (van Rheenen et al., 2005), and (2) different excitation and detection settings (e.g., PMT settings) (van Rheenen et al., 2004). However, we compared FRET (sens) values in the same cells before and after stimulation, thus the concentration of the probes or the detection sensitivity did not change during the experiment and FRET changes can be observed as relative changes to the initial FRET (sens) value. Translocation of cofilin from the PM led to a loss of FRET, and indeed, a rapid decrease in cofilin FRET (sens) was observed upon EGF stimulation and MβCD treatment (Fig. 3 D and for quantifications, Fig. S4 B), which was accompanied by an area increase resulting from EGF-stimulated protrusion (Fig. 3 E). GFP-cofilin translocation is PLC dependent because FRET (sens) decreases were not observed in cells treated with U73122 (Fig. 3 D and Fig. S4 B). To control for photobleaching (<5%) or the apparent redistribution of the probes during EGF-induced changes in cell shape, we divided the sensitized emission by the total amount of GFP or mCherry to obtain apparent donor (Ed) and acceptor (Ea) FRET efficiency. This concentration-independent FRET analysis confirmed EGF-induced FRET changes (Fig. S4 B) excluding any effects of bleaching and redistribution of the probe. To test this in more detail, we measured FRET (sens) between mCherry-CAAX and a GFP that was continuously tagged to the PM (GFP-CAAX) (Fig. S4 C). As expected, EGF stimulation did not result in lower FRET values. These data, together with the FRET efficiency analysis, exclude that FRET (sens) changes observed in Fig. 3, D and E, are a result of (1) changes in mCherry-CAAX, (2) changes in cells shape, and (3) photobleaching.
Figure 3.

Translocation of cofilin from the PM upon EGF stimulation in living cells. (A) MTLn3 cells were transiently transfected with GFP-cofilin. GFP-cofilin was detected by Western blot using antibodies against GFP or cofilin. (B) GFP images were taken from MTLn3 cells expressing GFP-cofilin before and 1 min after EGF stimulation. The LCS glow lookup table was used for the images. (C) Schematic representation of FRET between GFP-cofilin and mCherry-CAAX. Cofilin bound to PIP2 is in close proximity to the membrane, and FRET occurs. The distance between the PM and cofilin will increase upon the translocation of cofilin to cortical F-actin and FRET will diminish. (D) FRET (sens) was measured in nontreated (n = 19) and U73122-treated (n = 16) MTLn3 cells expressing GFP-cofilin and mCherry-CAAX as detailed in the Materials and methods. Shown are FRET images before and after EGF stimulation and before and after MβCD treatment. For controls and the quantification of FRET decreases, see Fig. S4 (available at http://www.jcb.org/cgi/content/full/jcb.200706206/DC1). (E) FRET signal (black line) and area increase (red line) of MTLn3 cells were measured in time upon EGF stimulation on the confocal microscope at 25°C. Plotted is the average of seven experiments. Bars, 5 μm.
To test whether GFP-cofilin translocation from the PM represents the endogenous cofilin translocation, we measured FRET between immunostained endogenous cofilin (Alexa 488) and mCherry-CAAX by acceptor photobleaching experiments. As illustrated in Fig. S4, D and E, these experiments indeed suggest that the in vivo–observed translocation of GFP-cofilin upon EGF represents the translocation of endogenous cofilin from the PM, and that this translocation is dependent of PLC-mediated PIP2 hydrolysis. Collectively, we conclude that cofilin leaves the PM upon PLC-mediated PIP2 reduction in living cells.
Cofilin leaves the PM upon PLC-independent PIP2 reduction
Collectively, our data suggest that the translocation of cofilin from the PM is mediated by PLC-dependent reduction in PIP2. If so, then the reduction of PIP2 by other means should also cause cofilin to leave the PM. Rapid loss of PIP2-independent PLC can be initiated by a rapamycin-induced recruitment of a phosphoinositide 5-phosphatase (5-ptase) enzyme domain to the PM (Fig. 4 A; Fig. S5, available at http://www.jcb.org/cgi/content/full/jcb.200706206/DC1). This translocation is mediated by the heterodimerization of the FRB domain at the PM and the FKBP12-tagged 5-ptase domain upon rapamycin exposure (Heo et al., 2006; Varnai et al., 2006). The 5-ptase domain dephosphorylates PIP2 into PIP, leading to the loss of PIP2 at the PM (Fig. 4 A). In line with our PLC data, rapamycin also induced the translocation of cofilin from the PM (Fig. 4 B), confirming that this translocation is mediated by a loss in PIP2. Moreover, upon rapamycin exposure, we observed the formation of membrane protrusions, suggesting an activation of cofilin (Fig. 4 C; Video 1, available at http://www.jcb.org/cgi/content/full/jcb.200706206/DC1). As expected, rapamycin-induced membrane protrusions are not inhibited by the PLC inhibitor U73122 (Fig. 4 C).
Figure 4.

5-ptase induced reduction of PIP2 leads to cofilin translocation from the PM. MTLn3 cells were transfected with an mRFP-tagged 5-ptase and a PM-targeted CFP-FRB domain. Upon rapamycin addition, the 5-ptase translocated to the PM (for explanation see Fig S5 and Varnai et al. [2006]). PIP2 levels were followed by YFP-PH(PLCδ1) (A), and the cofilin membrane localization was followed by GFP-cofilin (B). Below the images, the intensity profiles of FRB, PH, and 5-ptase fluoresence through the cell are shown. The blue lines represent pre and the red lines represent post-rapamycin (n > 15). Bars, 5 μm. (C) The formation of lamellipodia upon rapamycin (t = 0) addition was followed by confocal microscopy (see also Video 1, available at http://www.jcb.org/cgi/content/full/jcb.200706206/DC1). Shown are the images of PM-targeted CFP-FRB domains, before (t = 0) or 21 s (t = 21) after rapamycin addition. The right image represent a merge of binary threshold images (ImageJ) of image t = 0 and t = 21. Yellow represents no change, red represents increase lamellipod formation. The bottom images show the kymograph of the formation of the lamellipod, and the area of the cell that was used for the kymograph. From the kymographs, the size of the lamellipod was measured and plotted against time (right panel) for nonstimulated cells (control, red dashed line), rapamycin-stimulated cells (rapa, red line), and for cells that were treated with U73122 and stimulated with rapamycin (U73122 + rapa, blue line) and for cells that were treated with U73122 only (U73122 control, blue dashed line). n = 12; error bars represent SEM.
From all this, we conclude that cofilin translocates from the PM upon rapamycin-induced PIP2 reduction, and that this leads to the formation of membrane protrusions.
Cofilin released from the PM is active
Our model predicts that the translocation of the spatially confined and membrane-associated cofilin (Fig. 2 A, the xz projection) will lead to a local increase in activity. Active cofilin binds to and severs F-actin (Wang et al., 2007). To test whether cofilin activity increases upon PLC-mediated PIP2 reduction, we measured cofilin binding to F-actin by FRET. MTLn3 cells were fixed using a method that retains F-actin but loses G-actin (Song et al., 2006) and immunostained for endogenous cofilin (donor: Alexa 488) and F-actin (acceptor: rhodamine), and FRET was measured by acceptor photobleaching experiments. If cofilin is membrane bound, the distance between cofilin and F-actin is large, resulting in low FRET values. If cofilin is bound to F-actin, these molecules are in close proximity and FRET will be high in a relatively non-diffusible location. Cofilin/actin FRET increased upon EGF stimulation or MβCD-treatment, indicating that cofilin binds to actin filaments within 60 s after EGF stimulation (Fig. 5 A). Interestingly, cofilin–actin filament binding is reduced if PLC is inhibited, showing that the cofilin that binds to F-actin originates from the PM (Fig. 5 A). Strikingly, localized application of EGF by a micropipette resulted in a localized increase in FRET, where FRET values were two times higher at the front of the cell close to the pipette than at the opposite side of the cell (Fig. 5 B). This strongly suggests that localized EGF application results in a local translocation of cofilin from the PM to F-actin.
Figure 5.

Cofilin binds actin upon PLC-mediated PIP2 reduction. (A) Control or EGF-stimulated MTLn3 cells were fixed and labeled for cofilin (donor; Alexa 488) and F-actin (acceptor; rhodamine). Using a high laser power, a region was bleached in the acceptor fluorescence (white square box). The increase in the donor fluorescence was measured and FRET was calculated for un-treated, U73122-treated, or MβCD-treated cells (right panel; for all n > 15). (B) FRET was measured in a cell that was stimulated by local application of EGF using a pipette (asterisk). In the graph we plotted the normalized FRET values at the side of the cell close to the pipette (front) and the opposite side of the cell (back) (n = 5). Values were statistically tested by t test. *, P < 0.05; **, P < 0.01; error bars represent SEM.
Cofilin switches from PM binding to F-actin binding
To investigate the switch between PM and actin filament binding by cofilin after EGF stimulation, we performed fluorescence loss in photobleaching (FLIP) experiments. GFP-cofilin stains the periphery and cytosol. With an intense laser, a region in the cytosol was bleached, and we measured the fluorescence intensity at the periphery (Per), and in the cytosol (Cyt) over time (Fig. 6 A). Because cofilin molecules localized at the periphery are retained at the PM or on actin filaments, those molecules will be bleached less efficiently compared with the free diffusible cytosolic cofilin molecules, leading to an increase in the ratio of Per/Cyt staining (Fig. 6 A, right graph). The residence time of cofilin on actin filaments or the PM determines how fast the Per/Cyt ratio returns to baseline. If the actin filament and PM residence times are different, a change in the ratio of PM/actin binding will lead to a change in the overall residence time of cofilin at the periphery, and thus also the recovery time. Photobleaching during image acquisition or fluctuations in the laser power will equally influence the fluorescence loss of the periphery and the fluorescence increase in the cytosol, and therefore do not influence the outcome. To test the dynamic range of our FLIP experiment, a freely diffusible GFP and a PM-tagged GFP (coexpressed with free diffusible GFP as cytosolic marker) were tested. As can be seen in Fig. 6 B, free diffusible GFP recovers very fast, whereas the recovery of PM-tagged GFP recovers very slowly. To test whether cell shape changes influence the recovery rate caused by a change in PM background bleaching, the cells were stimulated with EGF. As shown in Fig. 6 B, this does not influence the recovery of PM-tagged GFP, excluding cell shape effects on recovery rates. The same experiments were performed for GFP-cofilin. In unstimulated cells, GFP-cofilin recovers rapidly, whereas in EGF-stimulated cells, the recovery is much slower. Inhibition of PLC by U73122 generates recovery rates for GFP-cofilin equal to the unstimulated rate (Fig. 6 C). We fitted the cofilin data with two component exponential decays representing the PM and actin binding, with a fast component of 3.7 and a slow component of 26 s. Unstimulated cells contain predominately the fast fraction (85%), whereas the stimulated cells contain predominately the slow fraction (62%) (Fig. 6 D). Because FRET experiments of cofilin-PM and cofilin-actin filaments showed that cofilin in unstimulated cells is predominately bound to the PM, and in EGF-stimulated cells predominately bound to F-actin, it is very likely that the fast component is membrane-bound cofilin, while the slow component is the F-actin–bound cofilin. This means that in unstimulated cells, 85% of cofilin molecules located at the periphery is PM bound, which goes down to 38% upon EGF stimulation, except in PLC-inhibited cells (Fig. 6 D). The actin fraction is 15% in unstimulated cells, which increases to 62% after EGF stimulation, which is blocked when PLC is inhibited (Fig. 6 D). Thus, our FLIP data are consistent with the interpretation that upon EGF stimulation, cofilin translocates from the PM to underlying F-actin.
Figure 6.

Cofilin translocates from the PM to F-actin upon PLC-mediated PIP2 reduction. (A) The membrane and actin binding of GFP-cofilin was followed by FLIP experiments on the confocal microscope at 25°C. At time 0, the cytosolic GFP-cofilin was bleached briefly (bleach region indicated in yellow) and the fluorescence at the cell periphery (Per in blue) and the cytosol (Cyt in red) were followed over time (middle graph). Upon bleaching, the Per/Cyt ratio increases, and because of exchange of molecules in the Cyt and Per, the ratio decreases to baseline. To normalize for bleach efficiencies, the initial ratio was set to 0 and the first point after bleaching was set to 1. (B) Experiments as described in A for cells expressing GFP and GFP-CAAX, and cells expressing GFP. The dark green and pink lines represent the GFP-CAAX/GFP experiments in unstimulated and EGF stimulated cells, respectively. The light green line represents the GFP experiment. Note that EGF-induced cell shape changes did not influence the GFP-CAAX/GFP FLIP, and therefore did not influence the GFP-cofilin FLIP results (for all n > 15). (C) Experiments as described in B for cells expressing GFP-cofilin. The blue and red squares represent unstimulated and EGF-stimulated (1 min) cells, respectively. The pink squares represent EGF-stimulated (1 min) U73122-treated cells. Black lines are the two component exponential decays fits with taus of 3.7 and 26 s (for both n > 15). (D) The fractions of the fast and slow taus before and after EGF (for 1 min) stimulation in U3122-treated or untreated cells (all n > 15). (E) Cofilin activity in untreated, 5 min MβCD-treated, 1 min EGF-stimulated, or EGF-stimulated and U73122-treated cells. Cofilin activity was standardized over total protein content and normalized to untreated cells. (average of three independent experiments). Bars, 5 μm. Values were statistically tested by t test. *, P < 0.05; **, P < 0.01; error bars represent SEM.
To investigate the activation state of cofilin during the time it is being released from the PM, we measured activity of cofilin in cell lysates prepared from cells either before or after EGF stimulation, and then using a cofilin-severing assay to measure cofilin activity in the cell lysate. Indeed, PIP2 reduction either induced by EGF-stimulation or MβCD treatment (Fig. 1) leads to an increase in the severing activity of cofilin (Fig. 6 E). Importantly, the PLC inhibitor U13122 potently inhibits the EGF-induced increase of cofilin-severing activity (Fig. 6 E), showing that this increased severing activity is dependent on PIP2 reduction and cofilin translocation from the PM to F-actin.
Discussion
PLC activity is required for EGF-induced actin polymerization and cell motility of mammary carcinoma cells (Chen et al., 1994; Mouneimne et al., 2004, 2006); however, the molecular basis of how PLC regulates actin dynamics and motility is not well established. The data presented in this manuscript explain how PLC affects the activity of a PM-associated fraction of cofilin, resulting in changes in actin polymerization and protrusion. The invasion signature of breast carcinoma cells has shown that cofilin is an important contributor to the metastatic phenotype (Wang et al., 2004, 2007), and recent findings show that the activity status of cofilin is directly related to the invasion, intravasation, and metastasis of mammary tumors (Mouneimne et al., 2006; Wang et al., 2006, 2007). The initial cofilin activation after EGF stimulation in metastatic tumor cells is independent of PI3K and cofilin dephosphorylation (Mouneimne et al., 2004; Song et al., 2006). Here, we provide evidence for the requirement of PLC during the initial activation of cofilin and provide the mechanism by which this occurs during EGF-mediated protrusion formation and metastasis. It is important to note that the mechanism described in this paper applies to metastatic amoeboid tumor cells in which migration is PLC dependent and cofilin dependent (Wang et al., 2007). In this cell type, the initial activation of cofilin upon EGF stimulation is uncoupled from its phosphorylation status, and therefore the activation of cofilin is highly dependent on PLC. However, it has been shown recently in less metastatic mesenchymal-type tumor cells that the phenotype of cofilin suppression is different, suggesting that cofilin may be regulated by alternate pathways which may be cell-type dependent (Sidani et al., 2007; Wang et al., 2007). Therefore, in some cell types, the activation may be more dependent on the dephosphorylation of cofilin by phosphatase such as slingshot, while the PLC-mediated pathway plays a less prominent role (Nishita et al., 2005; Soosairajah et al., 2005). This supports the idea that both mechanisms are physiologically important, but the contribution of either of the signaling pathways may differ in different cell types and under various conditions.
In this manuscript, we tested the hypothesis that a pool of cofilin is associated with and inhibited by PIP2 in the PM, and that it can be released and activated upon PLC-mediated PIP2 hydrolysis. Using biochemical, FRET, and FLIP experiments, we identified three cofilin compartments in the cell—the cytosolic, PM-associated, and F-actin–bound compartments—the latter two at the periphery of the cell. FLIP experiments showed that cofilin can rapidly cycle between these compartments and that upon EGF stimulation, cofilin molecules at the PM compartment translocate to the F-actin compartment. These observations suggest the activity cycle shown in Fig. 7. In unstimulated cells, cofilin at the cell periphery is mainly localized in the PM compartment. Upon EGF-induced PIP2 reduction, this pool is released from the PM and translocates to the nearby F-actin compartment. The increase in the number of cofilin molecules that bind to actin filaments leads to actin severing, which in turn leads to an increase in free barbed ends and cofilin–G-actin products. The cofilin–G-actin product is not able to bind to F-actin or the PM, and therefore diffuses to the cytosol compartment. Cofilin is released from the cofilin–G-actin complex by phosphorylation in order to reenter the PM or F-actin compartments. The result of releasing cofilin from the PM is an increase in the number of barbed ends in the F-actin compartment followed by the elongation of filaments by polymerization from the barbed ends of the severed filaments. The newly polymerized actin filaments are preferred by the ARP2/3 complex for binding (Ichetovkin et al., 2002). Each ARP2/3 complex nucleates a new daughter branch from the side of the cofilin-generated mother filament, resulting in a large amplification of actin polymerization resulting from cofilin activity. This actin polymerization is the driving force for the formation of membrane protrusions.
Figure 7.

Compartments involved in the cofilin activity cycle. Cofilin molecules cycle through three compartments in the cell; the cytosol, the actin, and the PM compartments. Cofilin at the PM compartment initially translocates to the F-actin compartment upon EGF mediated PIP2 reduction. Cofilin binds and severs actin filaments resulting in filaments with free barbed ends, and cofilin–G-actin complex. The cofilin–G-actin complex cannot bind actin or PM, and therefore diffuses to the cytosol compartment. In the cytosol compartment, cofilin is phosphorylated by LIM-kinase, resulting in the release of cofilin from the cofilin–G-actin complex. Upon cofilin dephosphorylation by SSH, cofilin can reenter the PM or F-actin compartment, starting a new cycle. The cycling of cofilin through the three compartments increases free barbed ends, resulting in newly polymerized actin filaments. The ARP2/3 complex prefers to bind to these newly formed actin filaments, which amplifies the cofilin-induced actin polymerization, resulting in protrusion formation.
In line with our model, we observed that rapamycin-induced PIP2 reduction leads to translocation of cofilin from the membrane with the subsequent formation of membrane protrusions. In contrast to EGF stimulation, the protrusions caused by rapamycin-induced PIP2 reduction are small and do not persist. This is consistent with recent work showing (1) that EGF-induced lamellipod extension requires both cofilin and ARP2/3, which cooperate and act synergistically in driving dendritic nucleation and membrane extension (Ichetovkin et al., 2002; DesMarais et al., 2004); and (2) that acute loss of PIP2 induced by rapamycin leads to loss of ARP2/3 subunits from lamellipods (Zoncu et al., 2007). Contrastingly, EGF stimulation leads to cofilin activation without negatively affecting ARP2/3 localization (DesMarais et al., 2004). Therefore, in the case of EGF stimulation, cofilin and ARP2/3 are activated together, resulting in the formation of stable and large protrusions. In the case of rapamycin, the initial cofilin-induced actin polymerization is not amplified by ARP2/3, leading to the formation of small protrusions with a subsequent collapse. These results suggest that rapamycin-induced PIP2 reduction that leads to depletion of ARP2/3 can lead to lamellipod collapse (Zoncu et al., 2007), and our results suggest that the extent of collapse can be counteracted by the activation of cofilin.
As illustrated in Fig. 7, our data demonstrated that cofilin molecules at the PM compartment are released and activated by decreases in the PIP2 level induced by PLC or a 5-ptase. Whether cofilin at the PM compartment is inactive through direct PIP2 binding or indirectly through an adaptor molecule cannot be concluded from our data. For example, we cannot exclude the possibility that cofilin interacts with a protein at the PM, resulting in cooperative PM binding and cofilin inactivation. Cooperative binding may lead to a higher apparent affinity for the PM than expected from in vitro–binding affinities (McLaughlin et al., 2002) and the strong selectivity of PM association for the dephosphorylated form of cofilin, which is not predicted from structural studies (Gorbatyuk et al., 2006). A reduction of PIP2 in the PM may lead to a direct reduction of cofilin–PIP2 binding, or in the case of cooperative binding, it may lead to a reduced cofilin adaptor protein binding or adaptor protein PIP2 binding. Although we cannot exclude cooperative binding, existing biochemical data point toward direct binding of cofilin to PIP2 because cofilin binds specifically to PIP2 in PIP-strips (unpublished data), and cofilin activity in vitro is inhibited by micelles containing PIP2 (Yonezawa et al., 1990, 1991; Ojala et al., 2001). Although the binding mechanisms in both models may differ, the output is the same: PIP2 reduction leads to the release from the PM and a local activation of cofilin.
In polarized cells, the activation of cofilin is restricted toward the chemoattractant source achieved by a local excitation global inhibition (LEGI) mechanism (Mouneimne et al., 2006). This LEGI mechanism involves the global phosphorylation of cofilin in the cytosol compartment by LIMK in response to EGF stimulation in breast carcinoma cells in concert with cofilin's local activation by PLC at the PM and its translocation to the F-actin compartment (Fig. 7). The rapid phosphorylation of cofilin close to the actin compartment acts to sharpen the spatial location of cofilin activation. Therefore, the local activation of cofilin by PLC as shown in this study is the key to the initiation of local actin polymerization required for chemotaxis, as postulated previously (Mouneimne et al., 2006; Song et al., 2006).
It has been widely hypothesized that the activities of various ABPs such as gelsolin, profilin, capping protein, and cofilin are modulated by phospholipid binding (Janmey and Lindberg, 2004; Di Paolo and De Camilli, 2006; Janetopoulos and Devreotes, 2006; Logan and Mandato, 2006). However, this hypothesis is mainly based on structural and in vitro–binding studies. Here, we provide compelling in vivo evidence demonstrating the activation of an ABP, cofilin, by varying a phospholipid, PIP2. Furthermore, we have demonstrated that the activation of cofilin by PLC-induced changes in PIP2 levels causes a translocation of cofilin from the PM compartment to the F-actin compartment, suggesting a mechanism for how the activities of other PIP2-regulated ABPs may be localized in cells.
Materials and methods
Cell culture
MTLn3 (rat mammary adenocarcinoma cell line) cells were maintained, starved as described previously (Song et al., 2006). For light microscopy experiments, MTLn3 cells were plated on glass-bottom dishes (MatTek Corporation) that had been treated with HCl and ethanol and washed with PBS. For PIP2 FRET experiments, the dishes were coated with poly-l- lysine (Sigma-Aldrich) and collagen. Before the experiments, cells were starved in L15 medium (Invitrogen) supplemented with 0.35% BSA for 2.5 to 4 h. A bath application of 5 to 15 nM EGF (Invitrogen) for 1 min or 5 mM methyl-β-cyclodextrin (MβCD) for 2.5 min was used to stimulate the cells.
DNA constructs and transfections
The pNA3 expression vectors with inserts eCFP-PH(PLCδ1), and GFP-CAAX were a gift of K. Jalink (Netherlands Cancer Institute, Amsterdam, The Netherlands). PM-FRB-CFP and mRFP-FKBP-5-ptase domain were a gift of T. Balla (National Institute of Child Health and Human Development, NIH, Bethesda, MD). pEYFP-Mem was from Clonetech Laboratories, Inc. pcDNA3 with the insert of mCherry-CAAX was obtained by replacing GFP of pcDNA3-GFP-CAAX with mCherry using the BamHI and EcoRI restriction sites. GFP-cofilin was constructed by subcloning the rat cofilin from peGFP-N1-cofilin (described in DesMarais et al. [2004]) into pEGFP-C1 with HindIII. A frame shift was corrected by removing the overhang of SacI by T4 polymerase. Constructs were transfected using Lipofectamine 2000 (Invitrogen) at 1μg DNA per MatTek dish. After transfection, the cells were washed with fresh medium and incubated until use.
Immunofluorescence
MTLn3 cells were fixed and permeabilized as previously described (Song et al., 2006). In brief, cells were simultaneously fixed and extracted using 3.7% paraformaldehyde, 0.1% glutaraldehyde (Electron Microscopy Sciences), and 0.075% mg/ml saponin (Sigma-Aldrich) in PBS. Compared with sequential fixation and detergent extraction protocols, simultaneous fixation and extraction results in depletion of soluble G-actin (Song et al., 2006). Primary antibodies diluted in blocking buffer were incubated with the fixed cells for 1 h. Secondary antibodies (AlexaFluor 488 or 555; Molecular Probes) were diluted in blocking buffer and incubated with the cells for 1 h. Cells were washed and mounted in PBS.
FRET-based PIP2 hydrolysis assay
Kinetic analysis of PIP2 hydrolysis by FRET was assayed as described (van der Wal et al., 2001) with some modifications. Cells were placed on an inverted microscope (model 470; Olympus) equipped with Zeiss EC Plan-NeoFluar 40×/1.3 OIL PH3 objective, a computer-driven cooled CCD camera, humidified CO2 chamber, and were kept at 37°C. Excitation was done with a Lambda DG-4 using a D430/25 CFP excitation filter, and IPLab Spectrum software (VayTek) was used to collect images. A ND2 filter was used in the excitation path to minimize photo toxicity during long experiments. A dual-band dichroic mirror 86002v2bs was used (Chroma Technology Corp.), and emission was collected on a dual-view 505 dcxr D465/30m (CFP) HQ535/30m (YFP) (Optical Insights, LLC). Images were collected every 3 s and stored on the computer. Using a custom-made Visual Basic (vb6.0) program, the images were imported and analyzed. Following a threshold step, background regions were determined and the background was measured and subtracted from the images. Cells were traced with a fixed threshold and the mean fluorescence level of CFP and YFP of the whole cells was measured. CFP leak-through was subtracted from the YFP values (leak-through factor was 0.84 and was established in cells expressing only CFP) and the YFP value was multiplied by a fixed factor which was determined at the onset of the experiment to achieve a YFP/CFP ratio of 1. FRET changes were measured as an offset of the initial value. For more details see Fig. S1.
Antibody-based PIP2 assay
PIP2 antibody was purchased from Assay Designs, Inc. Pictures were taken using 60× NA1.4 infinity-corrected optics on a microscope (model IX170; Olympus) supplemented with a computer-driven cooled CCD camera. IPLab Spectrum software (VayTek) was used to collect images, and exposure times were kept constant. Digital images were linearly converted in NIH ImageJ and analyzed using a customized macro. The macro averages the fluorescence intensity in 29 annuli, ranging from 1.1 μm outside the cell periphery and extending approximate 6 μm inside the cell (consecutive annuli are 0.22 μm apart from each other). The averaged fluorescence intensity of the first 1 μm inside the cell was used to represent the membrane PIP2 changes. For every condition, at least 30 cells were analyzed.
Membrane fractionation
A 100-mm plate of MTLn3 cells at ∼90% confluence was lysed with 100 μl of ice-cold lysis buffer (50 mM Tris, pH 7.5, 300 mM NaCl, 5 mM EGTA, 20 mM DTT, 2% Triton X-100, 1× protease inhibitor mixture, and 50 mM NaF). Cell lysates were collected after incubating the lysates on ice for 30 min. The harvested cell lysate was mixed with 60% OptiPrep (AXIS-SHIELD PoC AS) to yield 40% OptiPrep final. The lysate containing 40% OptiPrep was transferred to the bottom of a centrifuge tube. 1 ml 30% OptiPrep was added carefully on top of 40% OptiPrep lysate to avoid disrupting the interface. 300 μl of 5% OptiPrep was added carefully on top of 30% OptiPrep. Tubes were centrifuged for 12–14 h at 4°C in a rotor at 100,000 g (TLS55; Beckman Coulter). Gradient fractions (150 μl) were collected and cofilin content was analyzed by immunoblotting. To analyze the amount of PIP2, 1 μl of each gradient fraction was dot blotted onto nitrocellulose and dried for 1 h. Dot blot was then probed with a monoclonal antibody raised against PI(4,5)P2 (Assay Designs). Densitometry analysis was done in ImageJ (W.S. Rasband, NIH, Bethesda, MD; http://rsb.info.nih.gov/ij/). The amount of cofilin or PIP2 in the low-density fraction was normalized against the total amount of cofilin in the lysate.
Cofilin activity assay
The relative cofilin-severing activity was quantified using a slightly modified version of the established microscopy severing assay (Chan et al., 2000; Ichetovkin et al., 2000). In brief, MTLn3 cell lysates were prepared by lysing cells (3 × 107 cells/ml) in lysis buffer at different times after EGF stimulation. Rhodamine- and biotin-labeled F-actin were prepared by incubating 0.4 μM rhodamine-labeled actin, 0.2 μM biotin-labeled actin, and 1.4 μM unlabeled actin in polymerization buffer and 0.2 μM phalloidin for no more than 1.5 h at room temperature. A flow chamber precoated with 0.5% nitrocellulose in amylacetate was perfused in biotin antibody for 5 min. Rhodamine/biotin-labeled F-actin was diluted 30 times to perfuse in the flow chamber. The severing of actin filaments by cell lysates prepared from MTLn3 cells was observed under a TIRF microscope. Due to the cross-linking of filaments to the solid surface, the filaments are stable indefinitely, and 20 min was used routinely as the control check time to see if any filament severing occurred in the absence of cofilin. Filament numbers were quantified in ImageJ. The relative cofilin activity was measured by dividing the number of filaments 5–10 min after adding the lysates by the number of filaments before adding cell lysates in the same field. Confocal experiments using function-blocking cofilin antibodies were done to confirm that all severing activity was due to cofilin.
Sensitized emission
Sensitized emission was calculated from confocal GFP and mCherry images using a highly corrected algorithm as recently described (van Rheenen et al., 2004), in a custom-made Visual Basic program. In short, three images were collected with a confocal microscope (TCS SP2 AOBS; Leica) with 60× oil immersion optics at 25°C; a GFP image excited at 488 nm (488nmGFP), a mCherry image excited at 488 nm (488nmmCherry), and a mCherry image excited at 560 nm (560nmmCherry). Upon background subtraction, sensitized emission (FRET(sens)) was calculated as follows:
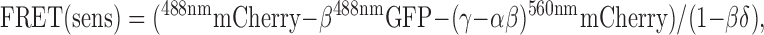 |
where α is the correction factor for mCherry fluorescence excitation at 488 nm and detected in the 488nmGFP channel, β is the leakthrough of GFP in the 488nmmCherry channel, δ that for leak-through of sensitized emission back into the 488nmGFP channel, and γ relates mCherry fluorescence detected in the 488nmGFP channel to the mCherry fluorescence detected in the 488nmmCherry channel. The α, β, γ, and δ correction factors were determined on-line from cells expressing either GFP or mCherry fluorescence proteins alone, present on the same coverslip (for more details see van Rheenen et al., 2004). For each image, the correction factors were redetermined to correct for laser fluctuations. Apparent FRET efficiencies (Ed and Ea) were obtained by relating FRET (sens) to the GFP or mCherry image, respectively. To compare independent measurements of Ea, Ea was divided by the γ factor (van Rheenen et al., 2004). Regions of interest such as the PM were determined by a threshold step in the mCherry image. FRET values were calculated by applying the same algorithms on the raw mean data of the region of interest.
Acceptor photobleaching experiments
Donor and acceptor confocal images were collected before and after photobleaching of the acceptor. In a custom-made Visual Basic program, the post-bleached donor image was corrected for imaging-bleaching and the gain in donor and the loss in acceptor fluorescence were determined at the cell periphery. FRET was calculated by relating the gain of donor fluorescence to the total amount post-bleach acceptor fluorescence. All FRET calculations were performed on the mean values of regions of interest of the periphery of the cell. No significant photo conversion of the acceptor was observed during photobleaching.
Colocalization analysis
Colocalization analyses were performed on the first 1 μm of a cell. Cells were precisely traced on phase-contrast images. Pixels along the perimeter were measured for intensity correlation. The perimeter was inset by one pixel and measured. This was repeated iteratively to provide Pearson's coefficient from the outer edge of the cell toward the center at 0.22-μm steps for the first 1 μm of the cell. Pearson's correlation coefficients (r) were calculated as previously described (Song et al., 2006) using the following equation:
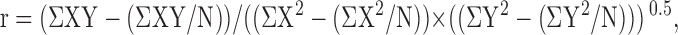 |
where element X was a pixel value of one probe, and Y the corresponding pixel value of the other probe at the same spatial location, and N the number of pixels.
For scatterplot analyses, the first 1 μm of the cell was precisely traced on the fluorescent images and the analysis was performed in a custom-made Visual Basic program, where the pixel intensity of the first image (image A) was plotted against the pixel intensity of the second image (image B). However, because of small misalignments in filter sets, the image of the second probe (image B) can have a pixel shift relative to the image of the first probe (image A). Before analysis, images were corrected for these pixel shifts as follows: The spatial correlation for pixels in which the gray value in both image A and B is >0 were analyzed with the Pearson's correlation coefficient. Then, image B was moved one pixel to the left, and the correlation was recalculated. If a misalignment in filter resulted in a pixel shift to the right, image B that is moved one pixel to the left will have a higher spatial correlation with image A then the non-moved image B. If this was true, image B was moved permanently. The correlation was calculated for a move of image B to the left, right, up, or down. This procedure was repeated iteratively until the coefficient no longer increased.
Online supplemental material
Figure S1 explains the PIP2 FRET assay. Figure S2 shows the specificity of PIP2 antibodies used in this study. Figure S3 explains why the Pearson's correlation coefficient is insensitive to relative changes in images. Figure S4 shows cofilin membrane FRET controls. Figure S5 explains the loss of PIP2 upon rapamycin addition. Video 1 shows the lamellipod increase upon rapamycin treatment. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200706206/DC1.
Supplementary Material
Acknowledgments
We thank all the members of our department for their helpful discussions. We especially thank Dr. K. Jalink and Dr. T. Balla for plasmids, and Dr. J. Segall of our department for the EGFR-overexpressing MTLn3 cell line.
J. van Rheenen was supported by a fellowship from the Dutch Cancer Society and the Cell Migration Consortium Grant No, U54GM064346; J.S. Condeelis by GM38511; and J.M. Backer by CA100324. This project was supported in part by the Cell Migration Consortium grant no. U54GM064346 from NIGMS: In particular, the Consortium supported the development of the cofilin FRET-biosensor.
Abbreviations used in this paper: ABP, actin-binding protein; FLIP, fluorescence loss in photobleaching; PM, plasma membrane.
References
- Andrianantoandro, E., and T.D. Pollard. 2006. Mechanism of actin filament turnover by severing and nucleation at different concentrations of ADF/cofilin. Mol. Cell. 24:13–23. [DOI] [PubMed] [Google Scholar]
- Chan, A.Y., M. Bailly, N. Zebda, J.E. Segall, and J.S. Condeelis. 2000. Role of cofilin in epidermal growth factor-stimulated actin polymerization and lamellipod protrusion. J. Cell Biol. 148:531–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, P., H. Xie, M.C. Sekar, K. Gupta, and A. Wells. 1994. Epidermal growth factor receptor-mediated cell motility: phospholipase C activity is required, but mitogen-activated protein kinase activity is not sufficient for induced cell movement. J. Cell Biol. 127:847–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, P., J.E. Murphy-Ullrich, and A. Wells. 1996. A role for gelsolin in actuating epidermal growth factor receptor-mediated cell motility. J. Cell Biol. 134:689–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condeelis, J., R.H. Singer, and J.E. Segall. 2005. The great escape: when cancer cells hijack the genes for chemotaxis and motility. Annu. Rev. Cell Dev. Biol. 21:695–718. [DOI] [PubMed] [Google Scholar]
- DesMarais, V., F. Macaluso, J. Condeelis, and M. Bailly. 2004. Synergistic interaction between the Arp2/3 complex and cofilin drives stimulated lamellipod extension. J. Cell Sci. 117:3499–3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo, G., and P. De Camilli. 2006. Phosphoinositides in cell regulation and membrane dynamics. Nature. 443:651–657. [DOI] [PubMed] [Google Scholar]
- Ghosh, M., X. Song, G. Mouneimne, M. Sidani, D.S. Lawrence, and J.S. Condeelis. 2004. Cofilin promotes actin polymerization and defines the direction of cell motility. Science. 304:743–746. [DOI] [PubMed] [Google Scholar]
- Goldschmidt-Clermont, P.J., J.W. Kim, L.M. Machesky, S.G. Rhee, and T.D. Pollard. 1991. Regulation of phospholipase C-gamma 1 by profilin and tyrosine phosphorylation. Science. 251:1231–1233. [DOI] [PubMed] [Google Scholar]
- Goldschmidt-Clermont, P.J., M.I. Furman, D. Wachsstock, D. Safer, V.T. Nachmias, and T.D. Pollard. 1992. The control of actin nucleotide exchange by thymosin beta 4 and profilin. A potential regulatory mechanism for actin polymerization in cells. Mol. Biol. Cell. 3:1015–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbatyuk, V.Y., N.J. Nosworthy, S.A. Robson, N.P.S. Bains, M.W. Maciejewski, C.G. dos Remedios, and G.F. King. 2006. Mapping the phosphoinositide-binding site on chick cofilin explains how PIP2 regulates the cofilin-actin interaction. Mol. Cell. 24:511–522. [DOI] [PubMed] [Google Scholar]
- Heo, W.D., T. Inoue, W.S. Park, M.L. Kim, B.O. Park, T.J. Wandless, and T. Meyer. 2006. PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science. 314:1458–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichetovkin, I., J. Han, K.M. Pang, D.A. Knecht, and J.S. Condeelis. 2000. Actin filaments are severed by both native and recombinant dictyostelium cofilin but to different extents. Cell Motil. Cytoskeleton. 45:293–306. [DOI] [PubMed] [Google Scholar]
- Ichetovkin, I., W. Grant, and J. Condeelis. 2002. Cofilin produces newly polymerized actin filaments that are preferred for dendritic nucleation by the Arp2/3 complex. Curr. Biol. 12:79–84. [DOI] [PubMed] [Google Scholar]
- Janetopoulos, C., and P. Devreotes. 2006. Phosphoinositide signaling plays a key role in cytokinesis. J. Cell Biol. 174:485–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janmey, P.A., and U. Lindberg. 2004. Cytoskeletal regulation: rich in lipids. Nat. Rev. Mol. Cell Biol. 5:658–666. [DOI] [PubMed] [Google Scholar]
- Kedrin, D., J. van Rheenen, L. Hernandez, J. Condeelis, and J. Segall. 2007. Cell motility and cytoskeletal regulation in invasion and metastasis. J. Mammary Gland Biol. Neoplasia. 12:143–152. [DOI] [PubMed] [Google Scholar]
- Kwik, J., S. Boyle, D. Fooksman, L. Margolis, M.P. Sheetz, and M. Edidin. 2003. Membrane cholesterol, lateral mobility, and the phosphatidylinositol 4,5-bisphosphate-dependent organization of cell actin. Proc. Natl. Acad. Sci. USA. 100:13964–13969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan, M.R., and C.A. Mandato. 2006. Regulation of the actin cytoskeleton by PIP2 in cytokinesis. Biol. Cell. 98:377–388. [DOI] [PubMed] [Google Scholar]
- McLaughlin, S., J. Wang, A. Gambhir, and D. Murray. 2002. PIP2 and proteins: interactions organization, and information flow. Annu. Rev. Biophys. Biomol. Struct. 31:151–175. [DOI] [PubMed] [Google Scholar]
- Mouneimne, G., L. Soon, V. DesMarais, M. Sidani, X. Song, S.C. Yip, M. Ghosh, R. Eddy, J.M. Backer, and J. Condeelis. 2004. Phospholipase C and cofilin are required for carcinoma cell directionality in response to EGF stimulation. J. Cell Biol. 166:697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouneimne, G., V. DesMarais, M. Sidani, E. Scemes, W. Wang, X. Song, R. Eddy, and J. Condeelis. 2006. Spatial and temporal control of cofilin activity is required for directional sensing during chemotaxis. Curr. Biol. 16:2193–2205. [DOI] [PubMed] [Google Scholar]
- Nishita, M., C. Tomizawa, M. Yamamoto, Y. Horita, K. Ohashi, and K. Mizuno. 2005. Spatial and temporal regulation of cofilin activity by LIM kinase and Slingshot is critical for directional cell migration. J. Cell Biol. 171:349–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojala, P.J., V. Paavilainen, and P. Lappalainen. 2001. Identification of yeast cofilin residues specific for actin monomer and PIP2 binding. Biochemistry. 40:15562–15569. [DOI] [PubMed] [Google Scholar]
- Sidani, M., D. Wessels, G. Mouneimne, M. Ghosh, S. Goswami, C. Sarmiento, W. Wang, M. El-Sibai, J.M. Backer, R. Eddy, et al. 2007. Cofilin determines the migration behavior and turning frequency of metastatic cancer cells. J. Cell Biol. 179:777–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, X., X. Chen, H. Yamaguchi, G. Mouneimne, J.S. Condeelis, and R.J. Eddy. 2006. Initiation of cofilin activity in response to EGF is uncoupled from cofilin phosphorylation and dephosphorylation in carcinoma cells. J. Cell Sci. 119:2871–2881. [DOI] [PubMed] [Google Scholar]
- Soosairajah, J., S. Maiti, O. Wiggan, P. Sarmiere, N. Moussi, B. Sarcevic, R. Sampath, J.R. Bamburg, and O. Bernard. 2005. Interplay between components of a novel LIM kinase-slingshot phosphatase complex regulates cofilin. EMBO J. 24:473–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Wal, J., R. Habets, P. Varnai, T. Balla, and K. Jalink. 2001. Monitoring agonist-induced phospholipase C activation in live cells by fluorescence resonance energy transfer. J. Biol. Chem. 276:15337–15344. [DOI] [PubMed] [Google Scholar]
- van Rheenen, J., and K. Jalink. 2002. Agonist-induced PIP(2) hydrolysis inhibits cortical actin dynamics: regulation at a global but not at a micrometer scale. Mol. Biol. Cell. 13:3257–3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rheenen, J., M. Langeslag, and K. Jalink. 2004. Correcting confocal acquisition to optimize imaging of fluorescence resonance energy transfer by sensitized emission. Biophys. J. 86:2517–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rheenen, J., E.M. Achame, H. Janssen, J. Calafat, and K. Jalink. 2005. PIP2 signaling in lipid domains: a critical re-evaluation. EMBO J. 24:1664–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnai, P., B. Thyagarajan, T. Rohacs, and T. Balla. 2006. Rapidly inducible changes in phosphatidylinositol 4,5-bisphosphate levels influence multiple regulatory functions of the lipid in intact living cells. J. Cell Biol. 175:377–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, W., S. Goswami, K. Lapidus, A.L. Wells, J.B. Wyckoff, E. Sahai, R.H. Singer, J.E. Segall, and J.S. Condeelis. 2004. Identification and testing of a gene expression signature of invasive carcinoma cells within primary mammary tumors. Cancer Res. 64:8585–8594. [DOI] [PubMed] [Google Scholar]
- Wang, W., G. Mouneimne, M. Sidani, J. Wyckoff, X. Chen, A. Makris, S. Goswami, A.R. Bresnick, and J.S. Condeelis. 2006. The activity status of cofilin is directly related to invasion, intravasation, and metastasis of mammary tumors. J. Cell Biol. 173:395–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, W., R. Eddy, and J. Condeelis. 2007. The cofilin pathway in breast cancer invasion and metastasis. Nat. Rev. Cancer. 7:429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyckoff, J., W. Wang, E. Lin, Y. Wang, F. Pixley, R. Stanley, T. Graf, J. Pollard, J. Segall, and J.S. Condeelis. 2004. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 64:7022–7029. [DOI] [PubMed] [Google Scholar]
- Xue, C., J. Wyckoff, F. Liang, M. Sidani, S. Violini, K.-L. Tsai, Z.-Y. Zhang, E. Sahai, J. Condeelis, and J.E. Segall. 2006. Epidermal growth factor receptor overexpression results in increased tumor cell motility in vivo coordinately with enhanced intravasation and metastasis. Cancer Res. 66:192–197. [DOI] [PubMed] [Google Scholar]
- Yin, H.L., and P.A. Janmey. 2003. Phosphoinositide regulation of the actin cytoskeleton. Annu. Rev. Physiol. 65:761–789. [DOI] [PubMed] [Google Scholar]
- Yonezawa, N., E. Nishida, K. Iida, I. Yahara, and H. Sakai. 1990. Inhibition of the interactions of cofilin, destrin, and deoxyribonuclease I with actin by phosphoinositides. J. Biol. Chem. 265:8382–8386. [PubMed] [Google Scholar]
- Yonezawa, N., Y. Homma, I. Yahara, H. Sakai, and E. Nishida. 1991. A short sequence responsible for both phosphoinositide binding and actin binding activities of cofilin. J. Biol. Chem. 266:17218–17221. [PubMed] [Google Scholar]
- Zoncu, R., R.M. Perera, R. Sebastian, F. Nakatsu, H. Chen, T. Balla, G. Ayala, D. Toomre, and P.V. De Camilli. 2007. Loss of endocytic clathrin-coated pits upon acute depletion of phosphatidylinositol 4,5-bisphosphate. Proc. Natl. Acad. Sci. USA. 104:3793–3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}