

Abstract
Macrophages are a key component of the innate immune system. In this study, we investigate how focal adhesion kinase (FAK) and the related kinase Pyk2 integrate adhesion signaling and growth factor receptor signaling to regulate diverse macrophage functions. Primary bone marrow macrophages isolated from mice in which FAK is conditionally deleted from cells of the myeloid lineage exhibited elevated protrusive activity, altered adhesion dynamics, impaired chemotaxis, elevated basal Rac1 activity, and a marked inability to form stable lamellipodia necessary for directional locomotion. The contribution of FAK to macrophage function in vitro was substantiated in vivo by the finding that recruitment of monocytes to sites of inflammation was impaired in the absence of FAK. Decreased Pyk2 expression in primary macrophages also resulted in a diminution of invasive capacity. However, the combined loss of FAK and Pyk2 had no greater effect than the loss of either molecule alone, indicating that both kinases function within the same pathway to promote invasion.
Introduction
The ability of macrophages to quickly respond to diverse extracellular cues allows these cells to function as important mediators of innate and adaptive immunity. In response to migratory stimuli, macrophages polarize and extend broad lamellipodia and spikelike filopodia in the direction of the chemotactic gradient (Calle et al., 2006). Formation of these protrusive structures is controlled by dynamic reorganization of the actin cytoskeleton and tubulin-based microtubules (Jones, 2000; Worthylake and Burridge, 2001). These structures are subsequently stabilized by integrin-mediated adhesions with the ECM. Members of the FAK family, which includes FAK and Pyk2, are critical integrators of these and other processes involved in cell motility. Through its function as a kinase and signaling scaffold, FAK has been shown to regulate focal adhesion turnover and migration in fibroblasts (Hanks et al., 2003; Parsons, 2003; Schlaepfer et al., 2004), whereas Pyk2 is a known regulator of macrophage motility (Okigaki et al., 2003). However, the biological function of FAK and the molecular interplay between FAK and Pyk2 in highly motile cell types such as macrophages has been largely unexplored. FAK was initially considered to be absent or expressed at low levels in monocyte/macrophages (Lin et al., 1994; De Nichilo and Yamada, 1996). It is now clear that FAK is indeed present in this cell lineage (Kume et al., 1997; Okigaki et al., 2003; Rovida et al., 2005), allowing for investigation into its role during macrophage migration and the immune response to inflammation.
During migration, FAK coordinates lamellipodial formation and the turnover/disassembly of focal adhesions (Webb et al., 2004; Tilghman et al., 2005). Focal adhesions are highly dynamic structures that form at sites of membrane contact with the ECM and are associated with a dense network of bundled actin stress fibers (Zaidel-Bar et al., 2004; Vicente-Manzanares et al., 2005). A critical role for FAK during cell migration is highlighted by the fact that fibroblasts derived from FAK-null mice migrate poorly in response to chemotactic and haptotactic factors and contain exceptionally large and stable focal adhesions (Ilic et al., 1995). In contrast to fibroblasts, macrophages form small focal complexes rather than large focal adhesions and produce fine actin cables rather than the stress fibers observed in cells of mesenchymal derivation (Pixley et al., 2005; for review see Pixley and Stanley, 2004). Macrophage-substrate contact also gives rise to podosomes, which are believed to play a role in adhesion, motility, matrix remodeling, and invasion (Calle et al., 2006). The formation of focal contacts and/or podosomes as well as the lack of actin stress fibers is consistent with the rapid motile response required of these cells.
In macrophages, integrin-dependent signaling can be enhanced by the presence of cytokines and growth factors (Schneller et al., 1997; Kiosses et al., 2001; Faccio et al., 2003). Colony-stimulating factor-1 (CSF-1) is a pleiotrophic myeloid lineage-specific growth factor that stimulates cell survival, proliferation, and monocyte–macrophage differentiation (for reviews see Stanley et al., 1997; Pixley and Stanley, 2004). It also functions as a potent macrophage chemoattractant (Boocock et al., 1989). Macrophages undergo significant morphological changes in response to CSF-1, including lamellipodia formation, dorsal ruffling, polarization, and CSF-1–directed chemotaxis (Boocock et al., 1989; Webb et al., 1996; Allen et al., 1997). CSF-1 receptor activation results in activation of the small GTPases Rac1 and Cdc42, which contribute to membrane ruffling and cell polarization (Cox et al., 1997; Kraynov et al., 2000). Recent data suggest that FAK is also important in establishing a proper leading edge and maintaining the polarity of moving cells (Tilghman et al., 2005).
To examine the role of FAK in primary macrophages, we have generated myeloid-specific conditional FAK knockout mice. We show that macrophages derived from these mice display significant motility defects coincident with elevated protrusive activity at the cell periphery, reduced adhesion turnover, and a marked inability to form the stable lamellipodia necessary for directional locomotion. Although the reduced expression of Pyk2 also resulted in motility defects, the combined loss of both FAK and Pyk2 had no additional consequence above what was observed in the absence of either molecule alone. The effects of FAK deletion on macrophage functions in vitro corresponded with decreased infiltration of FAK-null inflammatory monocytes into sites of inflammation in vivo. For the first time, these data provide genetic evidence that FAK is critically involved in the regulation of macrophage motility. These findings have profound implications for considering the physiological importance of macrophages for the control of infection and the maintenance of tissue homeostasis.
Results
Generation of myeloid-specific conditional FAK knockout mice
Mice harboring a floxed-FAK allele (Beggs et al., 2003) were crossed with mice expressing Cre recombinase under the control of the myeloid-specific lysozyme M (LysM) promoter (Clausen et al., 1999). Recombination catalyzed by Cre recombinase results in excision of the floxed target exon (Fig. 1 A). FAK sequences from bone marrow macrophages (BMMs) isolated from LysMwt/cre-FAKfl/fl mice were efficiently deleted during recombination (Fig. 1 B), and FAK protein expression levels were significantly reduced compared with FAK levels observed in BMMs isolated from LysMwt/wt-FAKfl/fl littermates (Fig. 1 C, top). Unlike FAK-null mouse embryo fibroblasts, which exhibit up-regulated levels of Pyk2 (Ilic et al., 1995), both wild-type (WT) and FAK-deficient (FAK−/−) BMMs expressed equivalent levels of Pyk2 (Fig. 1 C, bottom).
Figure 1.

Generation of myeloid lineage-specific conditional FAK knockout mice. (A) Schematic diagram of the floxed-FAK locus and the structure of the FAK locus after LysM-driven Cre-mediated recombination. The second kinase domain exon of FAK (black box) is flanked by loxP sites (black triangles). Primers (short arrows) and PCR products (lines) are shown above each allele. (B) PCR of DNA isolated from BMMs using the primers LoxP and GenoRV to distinguish the FAK-flox allele (1.6 kB; lane 1) and the recombined locus (327 bp; lane 2). (C) Immunoblot analysis of total FAK (top) and Pyk2 (bottom) expression in BMMs isolated from LysMwt/wt-FAKfl/fl and LysMwt/cre-FAKfl/fl mice (lane 1 and lane 2, respectively).
The loss of FAK promotes elevated protrusive activity at the cell periphery and altered adhesion dynamics
Adherent BMMs represent a highly heterogeneous population of cells that can vary significantly in their shape and size. Despite this variability, the mean adhesive area exhibited by FAK−/− macrophages plated onto fibronectin (FN)-coated coverslips was consistently reduced regardless of whether cells were cultured in the absence of CSF-1 overnight (Fig. 2 A), stimulated with CSF-1 for 20 min, or grown in the continual presence of CSF-1 (Fig. 2 A, second and third datasets, respectively). To specifically examine how the loss of FAK in macrophages affects cell spreading, WT and FAK−/−cells were examined by time-lapse video microscopy (Videos 1 and 2, available at http://www.jcb.org/cgi/content/full/jcb.200708093/DC1). Macrophages were starved of CSF-1 overnight. The following day, time-lapse microscopy commenced 5 min before the addition of exogenous CSF-1 and continued for an additional 10 min after stimulation. Before the addition of CSF-1, FAK−/− macrophages were observed to extend and retract numerous short-lived protrusions, resulting in a highly irregular peripheral edge. These structures were evident upon examination of individual FAK−/− BMMs taken at 1-min intervals (Fig. 2 B). Immediately after CSF-1 stimulation, both WT and FAK−/−cells exhibited circumferential spreading and fewer discrete protrusions. However, by 4 min after the addition of CSF-1, increased protrusive activity was again evident in FAK-deficient macrophages.
Figure 2.

FAK−/− macrophages exhibit elevated protrusive behavior in response to CSF-1. (A) WT (black bars) and FAK−/− (gray bars) macrophages were fixed and stained for filamentous actin, and the total area of adhesion was measured with ImageJ. Results shown are the mean ± SEM (error bars) and represent ∼150 cells from each population over five separate experiments. Asterisks indicate a statistically significant difference from the mean at ≥95% confidence level relative to CSF-1–starved WT cells. (B) The periphery of representative WT and FAK−/− macrophages taken from time-lapse video microscopy were outlined at time points before and after CSF-1 stimulation. Arrows indicate protrusions. (C) Sample kymographs from WT and FAK−/− cells are shown. The boxed area highlights protrusions extending from the cell periphery, which are enlarged in the inset. P, protrusion; R, retraction. (D) Protrusion persistence (Δx), distance (Δy), protrusion rate (Δy/Δx), and retraction rate (−Δy/Δx) were determined for each cell examined. The data represent the mean ± SEM of 10 cells (four parameters of membrane activity per cell) from each population over three separate videos. Asterisks indicate values that are significantly different from WT cells. *, P <0.05; **, P < 0.001.
To compare the protrusive activity of WT and FAK−/− BMMs, these time-lapse videos were analyzed by kymography, a technique that allows for the quantitation of protrusion persistence (Δx), protrusion distance (Δy), protrusion rate (Δx/Δy), and retraction rate (−Δx/Δy; Fig. 2 C). Protrusions formed by FAK−/− BMMs were shorter lived, lasting ∼1 min before retraction compared with the 1.7-min lifespan of WT protrusions (Fig. 2 D). FAK−/− protrusions also extended further than those produced by WT cells (4 ± 0.37 μm vs. 3 ± 0.71 μm, respectively). Thus, the protrusion and retraction rates calculated for FAK−/− macrophages (7.2 ± 1.2 μm/min and 3.7 ± 0.7 μm/min, respectively) were significantly faster than those observed for WT cells (2.7 ± 0.27 μm/min and 1.6 ± 0.18 μm/min, respectively). Collectively, these data show that macrophages deficient for FAK expression exhibit high levels of activity at the cell periphery that are characterized by the rapid formation and retraction of small protrusions. These data suggest that although FAK−/− BMMs are capable of forming lamellipodia, they may not be able to establish functional adhesions to stabilize the protrusions.
To determine whether FAK is involved in the regulation of adhesion formation and/or disassembly in macrophages, WT and FAK−/− BMMs were transfected with GFP-vinculin and examined by total internal reflective fluorescence (TIRF)–based video microscopy (Fig. 3 A and Videos 3 and 4, available at http://www.jcb.org/cgi/content/full/jcb.200708093/DC1). GFP-vinculin localized to prominent peripheral adhesion structures in both WT and FAK−/− cells. FAK was also observed to localize to these vinculin-containing structures (Fig. S1). Although adhesions in FAK−/− BMMs appear larger than those observed in WT cells, adhesion size did not differ significantly between the cell types. To quantify adhesion turnover, the kinetics of adhesion formation and disassembly was determined for vinculin by integrating the fluorescent intensity in individual adhesions over time. The rate of adhesion formation was reduced twofold in the absence of FAK (1.9 ± 0.17 min−1 and 0.91 ± 0.19 min−1 for WT and FAK−/− cells, respectively; Fig. 3 B). Similarly, adhesion disassembly in FAK−/− BMMs (0.85 ± 0.11 min−1) was also impaired compared with WT cells (1.48 ± 0.21 min−1). These results are consistent with the role of FAK as a mediator of adhesion turnover and provide support for the hypothesis that altered adhesion dynamics negatively impacts the ability of cells to establish stable protrusions.
Figure 3.

FAK regulates adhesion assembly and disassembly in macrophages. (A) TIRF-based video microscopy was used to examine adhesion formation and turnover in macrophages. WT and FAK−/− BMMs were starved of CSF-1 overnight and restimulated with 120 ng/ml CSF-1 just before filming. GFP-vinculin localizes in adhesion structures observed in both WT and FAK−/− macrophages. Bars, 10 μm. (B) Quantitative analysis of adhesion turnover. Adhesions from ∼8–10 cells were examined (three to five adhesions per cell). Asterisks indicate values that are significantly different from WT cells (*, P < 0.05). Error bars represent SEM.
FAK−/− BMMs exhibit a generalized defect in cell migration and invasion
Because FAK-deficient BMMs displayed poor lamellipodial stabilization in combination with altered adhesion dynamics, live cell imaging was used to examine chemokinesis, or random migration, in the presence of CSF-1 (Videos 5 and 6, available at http://www.jcb.org/cgi/content/full/jcb.200708093/DC1). WT macrophages stimulated with CSF-1 were observed to spread, extend lamellipodia, and move in a directed fashion throughout the course of the 2.5-h video analysis (Fig. 4 A). In contrast, FAK−/− BMMs rarely migrated >15 μm from their point of origin compared with 45 μm for WT cells (Fig. 4 B). To investigate whether FAK is required for directed cell migration in addition to random motility, WT and FAK-deficient macrophages were analyzed for their ability to migrate toward CSF-1 in a Boyden chamber assay. The loss of FAK inhibited CSF-1–induced motility by 53% (Fig. 4 C). The CSF-1–induced motility of BMMs extracted from LysMwt/wt-FAKwt/wt and LysMwt/cre-FAKwt/wt mice was identical to that observed for LysMwt/wt-FAKfl/fl BMMs (unpublished data), demonstrating that the presence of flox sites in the absence of Cre or the expression of Cre in the absence of a floxed allele had no effect on migration.
Figure 4.
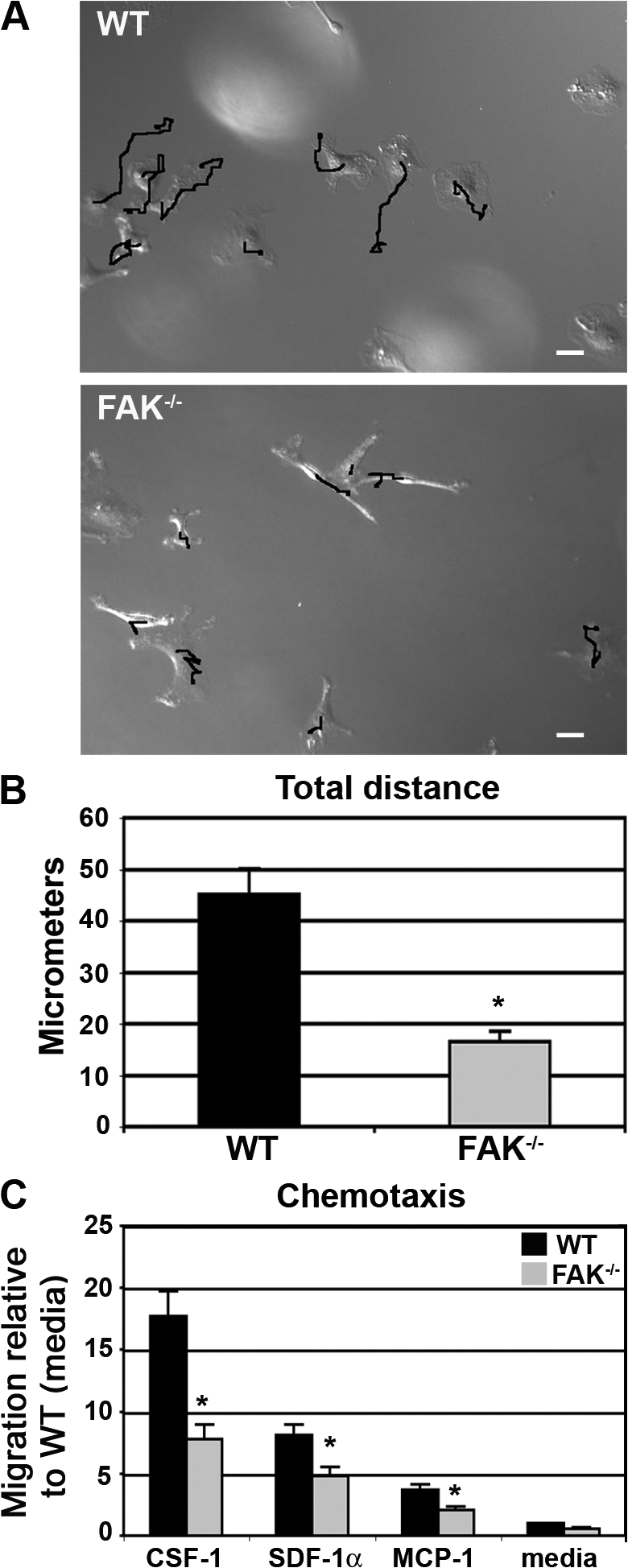
FAK−/− BMMs exhibit impaired CSF-1–dependent motility. (A) Time-lapse video microscopy was used to examine cell movement in response to CSF-1. WT and FAK−/− BMMs were starved of CSF-1 overnight before restimulation with 120 ng/ml CSF-1 for 2.5 h (Videos 5 and 6, available at http://www.jcb.org/cgi/content/full/jcb.200708093/DC1). Migration tracks overlay the final still image taken upon completion of the video. Bars, 10 μm. (B) The total distance traveled for WT (black bar) and FAK−/− (gray bar) BMMs was determined as described in Materials and methods. The data represent the mean ± SEM (error bars) of ∼30 cells from each population over three separate videos. Asterisks indicate values that are significantly different from WT cells (*, P < 0.05). (C) WT (black bars) and FAK−/− cells (gray bars) were starved of CSF-1 overnight before seeding onto Boyden chambers. Cells were then allowed to migrate toward 120 ng/ml CSF-1, 100 nM SDF-1α, or 100 nM MCP-1 for 4 h at 37°C. The number of migrated cells was determined and expressed relative to the migration of WT cells toward media alone. The data represent the mean ± SEM for four to six separate experiments. Asterisks indicate values that are significantly different from the migration of WT cells toward media (*, P < 0.05).
To determine whether the migration defect toward CSF-1 was specific for this cytokine, we next examined the ability of WT and FAK−/− BMMs to migrate toward stromal cell–derived factor-1α (SDF-1α) and macrophage chemoattractant protein-1 (MCP-1). These factors signal through chemokine receptors, which are structurally and mechanistically distinct from the receptor tyrosine kinase CSF-1 receptor. Migration of FAK−/− BMMs toward SDF-1α and MCP-1 was reduced 42% and 46%, respectively, compared with WT BMMs (Fig. 4 C), indicating that the defect in chemotaxis exhibited by FAK−/− macrophages is not restricted to CSF-1.
Invasion through 3D ECMs may require alternative signaling pathways to those regulating migration over 2D substrates (Wells et al., 2004). To determine the requirement for FAK during this process, WT and FAK−/− BMMs were seeded onto matrigel-coated Boyden chambers and allowed to invade toward CSF-1 for 24 h. The invasive capacity of FAK−/− BMMs was decreased by 60% compared with WT macrophages (Fig. 5 A). One possible explanation for the reduced levels of invasion exhibited by FAK−/− BMMs is that these cells may be less efficient in degrading matrix. To test this hypothesis, WT and FAK−/− BMMs were plated onto coverslips containing a layer of fluorescein-labeled FN. The pattern of fluorescein degradation was similar for both WT and FAK−/− cells (Fig. 5 B, b and e; arrowheads). These areas of matrix degradation colocalized with actin rosette structures characteristic of macrophage invadopodia (Fig. 5 B, c and f; arrowheads; Yamaguchi et al., 2006). Quantitative analysis revealed no difference in the area of proteolysis for either cell type (Fig. 5 C). Collectively, these data indicate that the absence of FAK in macrophages results in a generalized motility defect affecting chemotaxis, random migration, and invasion through a 3D matrix.
Figure 5.

FAK−/− BMMs are impaired in their ability to invade through 3D matrices. (A) WT and FAK−/− BMMs were starved of CSF-1 overnight before seeding onto matrigel-coated Boyden chambers. Cells were allowed to invade toward 120 ng/ml CSF-1 for 24 h at 37°C. Upon completion of the assay, the total number of cells that successfully invaded was determined. The data represent the mean ± SEM (error bars) for three separate experiments. Asterisks indicate values that are significantly different from WT cells (*, P < 0.05). (B) WT and FAK−/− BMMs were starved of CSF-1 overnight before plating onto collagen-coated coverslips containing an underlying layer of fluorescein-labeled FN (prepared as described in Materials and methods) in media containing 120 ng/ml CSF-1. After 4 h, cells were fixed and stained for filamentous actin (a and d). Degradation of ECM results in a cleared area of fluorescein (b and e; arrowheads). Overlayed images appear in panels c and f. (C) Areas of ECM degradation were measured in 10 randomly selected fields using ImageJ. The data represent the mean ± SEM for three separate experiments. Bar, 16 μm.
FAK−/− macrophages exhibit high basal levels of activated Rac1
We next examined whether the loss of FAK affected the activity of molecules involved in CSF-1–induced signaling. Specifically, the small GTPase Rac1 has been shown to regulate actin polymerization during lamellipodia formation in macrophages (Allen et al., 1997; Wells et al., 2004). Rac1 activity was measured in WT and FAK−/− BMMs that were starved of cytokine overnight and stimulated with CSF-1 for 0–30 min. Although the kinetics of activation were similar between the cell types, with peak Rac activity at ∼1 min of CSF-1 treatment, basal Rac1-GTP levels were ninefold higher in FAK-deficient macrophages than in WT cells (Fig. 6 A, top; compare lane 1 with lane 6). The overall CSF-1–dependent increase in Rac1 activity was not as great in FAK−/− BMMs as in WT BMMs (2.2-fold compared with 13.9-fold), largely as a result of the abnormally high basal activity in these cells. In contrast, activation of ERK1/2 occurred with nearly identical amplitude and kinetics in both cell types, peaking after 5 min of CSF-1 stimulation (Fig. 6 B).
Figure 6.

FAK−/− BMMs exhibit high basal levels of Rac1-GTP. Cells were plated onto tissue culture plastic and starved of CSF-1 overnight before stimulation with 120 ng/ml CSF-1 for 0–30 min. (A) GTP-bound Rac1 was isolated from lysates by incubation with Pak-1–binding domain agarose. Bound proteins (top) and total Rac1 (bottom) were detected by immunoblotting with Rac1 antibodies. (B) Cellular proteins immunoblotted with antibodies recognizing phospho-ERK1/2 (top) and total ERK1/2 (bottom). (A and B) Relative band intensities are displayed in graph form to the right. WT, squares; FAK−/−, triangles. Each immunoblot represents multiple independent experiments.
To determine whether the elevated basal Rac1 activity exhibited by FAK−/− BMMs contributed to the increased protrusive activity and/or decreased overall migratory potential of these cells, dominant-negative Rac1 (GFP-N17Rac1) or GFP alone was expressed in FAK−/− BMMs. No difference in invasion was detected between FAK−/− BMMs expressing GFP-N17Rac1 compared with FAK−/− BMMs expressing GFP alone (unpublished data). However, the expression of GFP-N17Rac1 in FAK-deficient macrophages resulted in a significant reduction in membrane protrusiveness (unpublished data), indicating that Rac1 activity contributes to membrane dynamics in these cells.
Pyk2 and FAK coordinately regulate macrophage invasion
The data presented thus far support a role for FAK in the regulation of macrophage motility. However, it was important to confirm that the defects exhibited by FAK−/− BMMs were directly caused by the loss of FAK. As an alternative to the genetic deletion of FAK, BMMs isolated from control mice were treated with siRNA duplexes targeting FAK. This resulted in a 90% reduction in FAK expression (Fig. 7 A, lane 8). Control and siRNA-treated BMMs were seeded onto matrigel-coated Boyden chambers and allowed to invade toward CSF-1 overnight. BMMs treated with siRNAs targeting FAK exhibited significantly reduced invasion relative to WT BMMs treated with vehicle or control siRNAs (Fig. 7 B, black bars). These data lend further support for an essential role of FAK in CSF-1–induced macrophage motility.
Figure 7.

Macrophage invasion requires Pyk2 and FAK expression. (A) WT and FAK−/− BMMs were treated with vehicle (H2O), siControl, siPyk2, and/or siFAK. 48 h after siRNA transfection, cells were lysed and immunoblotted for total FAK and Pyk2 (top) and ERK1/2 (bottom). (B) Vehicle and siRNA-treated cells were starved of CSF-1 overnight before seeding onto matrigel-coated Boyden chambers. WT (black bars) and FAK−/− (gray bars) BMMs were then allowed to invade toward 120 ng/ml CSF-1 for 24 h at 37°C. The data represent the mean ± SEM (error bars) for four to six separate experiments. Asterisks indicate values that are significantly different from vehicle-treated WT cells (*, P < 0.05). ^ indicates a value that is significantly different from WT siControl-treated cells (P < 0.05).
Although the loss of FAK from primary BMMs via both genetic deletion and RNAi resulted in a significant reduction in invasion, residual invasion was still evident under both conditions (Figs. 4 C and 7 B). This would suggest that a second, FAK-independent pathway is also involved in the regulation of macrophage motility. Macrophages derived from Pyk2−/− mice exhibit altered cell polarization and diminished contractility associated with reduced chemokine-induced motility (Okigaki et al., 2003). To more definitively assess the relationship between FAK and Pyk2 during macrophage invasion, siRNA oligonucleotides were used to reduce Pyk2 expression by ∼85–95% in both WT and FAK−/− BMMs (Fig. 7 A, middle; lanes 5 and 6). This treatment had no effect on FAK expression in WT cells (Fig. 7 A, top; lane 5). As before, the invasive capacity of FAK−/− BMMs treated with vehicle or siControl was reduced by 55% compared with WT macrophages (Fig. 7 B, gray bars). Invasion was similarly decreased by 55% in siPyk2-treated WT cells (Fig. 7 B, third dataset; black bar). Importantly, reduced expression of Pyk2 in FAK−/− BMMs did not cause an additional reduction in invasion over the effect of FAK deletion alone (Fig. 7 B, third dataset; gray bar). Combined knockdown of both FAK and Pyk2 from WT cells by siRNA also decreased invasion by a similar level. These data indicate that both FAK and Pyk2 contribute to the regulation of macrophage invasion in response to CSF-1 and that they appear to function within the same pathway.
FAK regulates the recruitment of monocyte/macrophages to sites of inflammation in vivo
To assess the role of FAK during leukocyte recruitment to sites of inflammation in vivo, mice were injected intraperitoneally with 4% thioglycollate (TG) to induce an inflammatory response. The peritoneal cavities of LysMwt/wt-FAKfl/fl (phenotypically WT) mice and myeloid lineage-specific conditional FAK knockout mice were lavaged 8, 16, and 72 h after injection, and the exudates were analyzed by flow cytometry using markers to distinguish between resident macrophages, infiltrating monocytes, and neutrophils (Melnicoff et al., 1989; Chan et al., 1998). Resident macrophages are characterized by high CD11b and F4/80 expression and low expression of GR-1 and Ly6G. In contrast, GR-1 is up-regulated in infiltrating inflammatory monocytes, whereas F4/80 expression is reduced in this model of inflammation (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200708093/DC1). Neutrophils were characterized by the high expression of GR-1 and Ly6G. In the resting peritoneum, equivalent numbers of resident macrophages were extracted from both mouse genotypes (Fig. 8 A). After 16 h, twice as many infiltrating CD11b-positive cells were recovered from the peritoneum of WT mice compared with conditional knockout mice. By 72 h after TG treatment, CD11b-positive cells continued to infiltrate the peritoneum of WT mice, whereas the accumulation of these cells was delayed in the knockout animals. This impaired recruitment was not true of all leukocyte populations, however, as both WT and conditional knockout animals exhibited similar recruitment of GR-1–positive cells in response to TG (Fig. 8 B).
Figure 8.

Recruitment of CD11b-positive cells is impaired in conditional FAK knockout mice during TG challenge. (A and B) WT and FAK−/− animals were injected intraperitoneally with 4% TG, and lavage fluid was collected at the indicated time points. To examine resident populations of cells, lavage fluid was also collected from the peritoneal cavities of unstimulated mice. Total cell numbers were obtained by hemocytometer, and the expression of CD11b and GR-1 was determined by flow cytometry. Data are expressed as cell numbers based on the percentage of cells designated as resident/inflammatory monocytes (A) or neutrophils (B; Fig. S1 and Table S1, available at http://www.jcb.org/cgi/content/full/jcb.200708093/DC1). WT, squares; FAK−/−, triangles. Data are the means ± SEM (error bars) from four mice per time point (two experiments). Asterisks indicate values that are significantly different from WT cells (*, P < 0.05). (C) PCR products of DNA isolated from cultured BMMs (lanes 1 and 2), CD11b positively selected cells from unstimulated (lanes 3 and 4) or TG-stimulated mice (lanes 5 and 6), and GR-1 positively selected cells from TG-stimulated mice (lanes 7 and 8) to distinguish the FAK-flox allele (1.6 kB) and the recombined locus (327 bp). (D) Cellular proteins from cultured BMMs, CD11b positively selected cells from unstimulated (lanes 3 and 4) or TG-stimulated mice (lanes 5 and 6), and GR-1 positively selected cells from TG-stimulated mice (lanes 7 and 8) were immunoblotted with antibodies recognizing FAK (top) and total ERK1/2 (bottom).
To confirm that FAK was genetically deleted from the CD11b-positive population of cells isolated from the peritoneum of TG-stimulated conditional FAK knockout mice, cells harvested from these mice were positively selected for CD11b surface expression, and the extent of Cre-mediated recombination was examined by PCR. CD11b-positive cells lavaged from FAK-deficient mice exhibited high levels of Cre-mediated recombination compared with cells obtained from TG-stimulated WT mice (Fig. 8 C, compare lane 5 with lane 6) or cultured WT BMMs (Fig. 8 C, lane 1). Resident cells harvested from unstimulated conditional FAK knockout animals also exhibited Cre-mediated recombination compared with cells taken from WT littermates (Fig. 8 C, compare lane 3 with lane 4). The high levels of recombination seen in FAK knockout mice corresponded to a concomitant loss of FAK expression from elicited and resident cells (Fig. 8 D, top; lane 6 and lane 4, respectively).
The infiltration kinetics of GR-1–positive neutrophils was similar between the two mouse genotypes (Fig. 8 B). This was somewhat surprising because LysM-Cre–mediated recombination is reported to occur with ∼99% efficiency in neutrophils (Clausen et al., 1999). To determine whether FAK was similarly deleted in GR-1–expressing cells, GR-1–positive cells were harvested from TG-stimulated WT and conditional FAK knockout animals and examined for Cre-mediated recombination by PCR. GR-1–positive cells from the conditional knockout mice exhibited efficient cre-mediated recombination (Fig. 8 C, compare lane 7 with lane 8). However, we were unable to detect FAK protein in GR-1–positive cells obtained from either WT or FAK-deficient mice, indicating that FAK is not normally expressed in neutrophils (Fig. 8 D, lanes 7 and 8). Thus, neutrophil infiltration was most likely not affected by the genetic deletion of FAK because FAK is not expressed in these cells. This suggests that the recruitment of neutrophils to sites of inflammation occurs via a FAK-independent mechanism.
Discussion
As critical effectors of the innate immune system, macrophages use adhesion signaling to accomplish many essential cellular functions, including adhesion to and extravasation from blood vessels, chemotaxis, and phagocytosis. In this study, we demonstrate for the first time that FAK plays a critical role in processes involving macrophage adhesion and motility in vitro. We also show that disruption of FAK-dependent adhesion pathways in macrophages in vivo results in an attenuated immune response marked by reduced monocyte/macrophage infiltration into sites of inflammation. Collectively, these results provide a framework for examining the signaling pathways controlling both FAK-dependent and -independent–based motility.
Regulation of macrophage motility by FAK family kinases
CSF-1 is a macrophage chemoattractant that interacts with its cognate receptor tyrosine kinase (CSF-1 receptor) and is produced endogenously by activated endothelial cells and tissue fibroblasts. FAK autophosphorylation increases in BMMs (unpublished data) and in the Bac1.2F5 macrophage cell line during CSF-1 stimulation, suggesting a functional relationship between CSF-1 signaling and FAK (Rovida et al., 2005). In this study, we demonstrate that FAK−/− BMMs were significantly impaired in their ability to migrate toward CSF-1. The chemotaxis of FAK-deficient macrophages toward SDF-1α and MCP-1, chemokines that signal through different subclasses of heptahelical G protein–coupled receptors (CXCR4 and CCR2, respectively), was similarly reduced. These results suggest that the deficiencies in macrophage chemotaxis observed in FAK−/− macrophages may be a consequence of a fundamental breakdown in the cell migration machinery rather than an inability to respond to specific migratory stimuli. This conclusion is supported by data showing that macrophages lacking FAK exhibited impaired invasion through matrigel despite the fact that they were still able to effectively degrade FN. The essential contribution of FAK to macrophage function was further confirmed by the finding that knockdown of FAK expression via siRNA treatment inhibited the invasive capacity of macrophages to the same extent as did genetic deletion of this molecule.
To delineate the mechanisms underlying the migratory defects observed in FAK-deficient macrophages, we first compared the morphology and actin dynamics of WT and FAK−/− BMMs. CSF-1 induces monocyte/macrophage spreading, polarization, and extension of lamellipodia (Boocock et al., 1989; Webb et al., 1996; Jones, 2000). FAK-deficient macrophages were observed to continuously extend and retract numerous short-lived protrusive structures. The inability of FAK−/− macrophages to form broad lamellipodia is consistent with a role for FAK in regulating and/or maintaining a leading edge during migration. This is in accordance with a previous study showing that FAK plays a central role in organizing and propagating signals required for directional migration in fibroblasts (Tilghman et al., 2005).
Although FAK−/− BMM migration and invasion were significantly impaired relative to control cells, the loss of FAK did not result in the complete abolishment of migratory activity, indicating that FAK-independent mechanisms also regulate macrophage motility. The genetic deletion of other signaling molecules such as Pyk2 and phosphoinositide 3-kinase–γ from macrophages induces a phenotype similar to FAK−/− BMMs, which is characterized by the formation of multidirectional lamellipodia, reduced polarization, and diminished migration (Jones et al., 2003; Okigaki et al., 2003). However, Pyk2 does not appear to be responsible for the residual invasive/migratory activity exhibited by FAK-deficient cells because the invasive/migratory capacity of FAK−/− macrophages was not further impaired by a loss of Pyk2. Nonetheless, depletion of Pyk2 from WT BMMs had an inhibitory effect on invasion/migration similar to that observed in the absence of FAK, suggesting that FAK and Pyk2 may regulate macrophage motility through a single pathway. We are currently investigating whether phosphoinositide 3-kinase–γ or other known regulators of cell motility contribute to the FAK-independent pathways controlling the migration of macrophages. We are also exploring the possibility that the method of migration used by macrophages in the absence of FAK may be intrinsically different from FAK-dependent motility. One attractive possibility is that in the absence of FAK, the cells may switch from a proteolysis-dependent mesenchymal mode of migration to a Rho kinase (ROCK)–dependent amoeboid mode of migration similar to that observed during the invasive migration of tumor cells (Torka et al., 2006).
Regulation of macrophage adhesions and membrane protrusions by FAK
In addition to defects in motility, FAK−/− BMMs exhibited low rates of adhesion turnover, increased membrane protrusiveness, and elevated basal Rac1-GTP levels. The relationship between these defects and impaired migration remains to be determined. In fibroblasts, FAK signaling promotes adhesion disassembly, leading to inhibition of adhesion maturation and promotion of adhesion turnover (Webb et al., 2004). Based on the highly stable adhesions observed in FAK−/− macrophages, FAK likely performs a similar function in these cells. However, the adhesions in BMMs are qualitatively different from those in fibroblasts. The large, mature focal adhesions present in fibroblasts are not evident in BMMs (for review see Pixley and Stanley, 2004). Moreover, the adhesion turnover rates observed in protruding regions of WT BMMs are considerably greater than the adhesion turnover rates reported for fibroblasts. Finally, BMM adhesion dynamics were measured in the presence of CSF-1. These differences may account for why both adhesion assembly and disassembly rates are decreased in FAK−/− BMMs, whereas only adhesion disassembly rates were reduced in FAK−/− fibroblasts.
The abnormally high protrusive activity of the FAK−/− BMMs may be related, in part, to defects in adhesion dynamics. In FAK−/− fibroblasts, slow adhesion turnover results in a reduced ability of cells to form new adhesions that stabilize membrane protrusions (Webb et al., 2004). Consistent with this finding, we were unable to detect adhesions in areas of hyperprotrusiveness in the FAK−/− BMMs, at least within the timescale of our measurements. A failure to form new adhesions at the leading edge after lamellipodia formation could also account for the decrease in directional motility exhibited by FAK−/− BMMs.
The contribution of high basal Rac1 activity to the migration defect exhibited by FAK−/− BMMs is less clear. High GTP-Rac1 in fibroblasts has been reported to correlate with an overall decrease in motility coincident with the adoption of either a rounded, unruffled appearance or a flattened, highly ruffled morphology (Pankov et al., 2005). The FAK−/− macrophages exhibit a similar ruffled and protrusive appearance, which could be reversed by the expression of dominant-negative Rac1-N17 (unpublished data). Thus, the hyperprotrusiveness exhibited in the absence of FAK may be the result of elevated Rac1-GTP. However, Rac1-N17 expression did not rescue the migration defect exhibited by FAK−/− BMMs (unpublished data). This is consistent with data from Rac1 knockout mice that show that Rac1 is not required for BMM migration (Wells et al., 2004; Wheeler et al., 2006).
Regulation of macrophage functions associated with the inflammatory response by FAK
Macrophages play a central role in the inflammatory process, releasing cytokines that control key events in the initiation, resolution, and repair processes of inflammation (Henderson et al., 2003). Neutrophils are rapidly recruited into sites of acute infection and are the principal cell type during the initial influx of infiltrating leukocytes (Issekutz and Movat, 1980). However, monocytes/macrophages replace neutrophils as the predominant infiltrating population within 16 h of an inflammatory reaction (Henderson et al., 2003). The induction of an inflammatory response in myeloid-specific conditional FAK knockout mice resulted in the delayed recruitment of inflammatory CD11b-positive monocytes/macrophages relative to control mice, whereas the recruitment of GR-1–positive neutrophils occurred with identical kinetics in both mouse genotypes. That the floxed-FAK allele underwent recombination in neutrophils from conditional FAK knockout mice is not surprising because efficient LysM-driven Cre-mediated excision of floxed target genes has been reported in this cell type (Clausen et al., 1999). However, because FAK protein is not endogenously expressed in this cell lineage, the genetic deletion of FAK had no effect on neutrophil recruitment. These findings underscore the importance of FAK during the recruitment of macrophages for which FAK-dependent mechanisms of migration are essential and suggest that other FAK- independent mechanisms of motility are used in cell lineages that do not typically express FAK.
Based on these data, we suggest that it may be possible to alter the macrophage-specific host response to inflammation by targeting FAK in the monocyte/macrophage lineage. This has significant consequences when considered within the context of diseases in which the accumulation of macrophages contributes to disease progression, such as chronic inflammatory diseases and cancer. Adhesion signaling in immune cells has already been established as a viable therapeutic target. For example, antibodies that block α4-integrin functions on the surface of T helper 1 cells or activated macrophages have been shown to be beneficial for treating both Crohn's disease and multiple sclerosis (Ghosh et al., 2003; Miller et al., 2003). Similar approaches might be useful for treating solid tumors, which are often found to contain macrophages, because high numbers of tumor-associated macrophages correlate with a poor prognosis (Pollard, 2004). Recent experiments using small molecular inhibitors of FAK, which specifically target FAK catalytic activity, have shown that cell migration and focal adhesion turnover are inhibited by the drug (Slack-Davis et al., 2007). Although further studies will be required to elucidate the FAK-dependent and -independent signaling pathways required for macrophage migration, the data presented herein highlight potential therapeutic applications involving the inhibition of FAK activity in macrophages.
Materials and methods
Generation of myeloid-specific conditional FAK knockout mice
Mice homozygous for cre recombinase under the control of the myeloid-specific LysM promoter were purchased from The Jackson Laboratory (stock # 004781) and have been described previously (Clausen et al., 1999). To generate myeloid-specific conditional FAK knockout mice and their control littermates, mice homozygous at the FAK locus (FAKfl/fl; Beggs et al., 2003) were crossed with mice homozygous for cre at the LysM locus (LysMcre/cre). Mice heterozygous at both loci were backcrossed to generate LysMwt/wt-FAKfl/fl (WT control) and LysMwt/cre-FAKfl/fl (FAK−/−) mice and the controls LysMwt/wt -FAKwt/wt and LysMwt/cre -FAKwt/wt.
Genotyping of mice and analysis of Cre-mediated recombination
Animals were routinely genotyped from tail DNA and subjected to PCR analysis. The following primers were used for PCR of the FAK locus: P1 (5′-GAGAATCCAGCTTTGGCTGTTG-3′) and GenoRV (5′-GAATGCTACAGGAACCAAATAAC-3′). This primer set generates 290- (WT) and 400-bp (FAK-flox) fragments. To determine the status of the LysM locus, the following primers were used: LysM1 (5′-CTTGGGCTGCCAGAATTTCTC-3′), LysM2 (5′-TTACAGTCGGCCAGGCTGAC-3′), and Cre8 (5′-CCCAGAAATGCCAGATTACG-3′). This primer set generates 350- (WT) and 700-bp (Cre) fragments. To check for Cre-mediated recombination in BMMs, DNA was isolated from macrophages and subjected to PCR with the primers LoxP (5′-GACCTTCAACTTCTCATTTCTCCC-3′) and GenoRV (listed above). The amplified PCR products consisted of a WT (1.4 kb), FAK-flox (1.6 kb), and Cre-mediated recombined fragment (327 bp). All PCR fragments were separated on 1.5% agarose gels.
Isolation of BMMs
BMMs were isolated from 6–8-wk-old mice by flushing femurs and tibias with PBS containing 0.5% BSA and 1% of 0.5 M EDTA. Cells were magnetically labeled using anti-CD11b microbeads (Miltenyi Biotec) and were positively selected on an MS MACS column (Miltenyi Biotec) according to the manufacturer's instructions. Cells were seeded onto bacterial plates and cultured in α-MEM supplemented with 10% heat-inactivated FBS, 10% CMG 14-12 cell-conditioned medium as a source of CSF-1 (provided by G. Longmore, Washington University, St. Louis, MO), 100 U/ml penicillin, and 100 U/ml streptomycin (Invitrogen). Media was replaced every 3–4 d until confluent.
Antibodies and reagents
Polyclonal FAK C-20 was purchased from Santa Cruz Biotechnology, Inc. A mAb recognizing phospho-ERK1/2, FN, and collagen I were all obtained from Sigma-Aldrich. mAbs recognizing Pyk2 and Rac1 were purchased from BD Biosciences. A polyclonal anti-ERK1/2 antibody was purchased from Cell Signaling Technology. Phycoerythrin (PE)-Cy5–conjugated rat anti–mouse Ly6G and 7AAD were purchased from eBioscience. FITC-conjugated rat anti–mouse CD45, PE-conjugated rat anti–mouse GR-1, allophycocyanin-conjugated CD11b, PE-Cy7–conjugated rat anti–mouse CD11b, PE-conjugated anti–mouse F4/80, and rat anti–mouse CD16/32 antibodies were purchased from Caltag Laboratories. Texas red–conjugated phalloidin was purchased from Invitrogen. HRP-conjugated sheep anti–mouse Ig and HRP-conjugated donkey anti–rabbit antibodies were purchased from GE Healthcare. CSF-1 was purchased from PeproTech. SDF-1α and MCP-1 were purchased from R&D Systems.
Transfection procedures
siGenome SmartPool siRNAs targeting murine FAK, Pyk2, and nonspecific siControl were purchased from Dharmacon. Transfection of 100 nmol of siRNA or 1 μg of plasmids encoding GFP-vinculin, GFP-FAK, GFP-N17Rac1, monomeric Kusabria orange–vinculin, and siRNAs into BMMs was achieved by nucleofection according to the manufacturer's instructions using the Mouse Macrophage Nucleofector kit (Amaxa Corp.).
Time-lapse video microscopy and analysis
To study cell spreading in response to CSF-1, 1 × 105 cells were seeded on 35-mm bacterial dishes and starved of CSF-1 overnight. The following day, cell images were collected for 5 min before the addition of 120 ng/ml exogenous CSF-1 using a microscope (Diaphot; Nikon) with a video camera (KY-F55B; Victor Company of Japan) every 15 s for a total of 45 min. To quantitate protrusive behavior, kymographs of cells from the videos were generated using ImageJ (National Institutes of Health) to create time space plots. In brief, a minimal intensity projection was performed for each video. Four lines of interest were drawn at 90° angles along the perimeter of the cell. Kymographs obtained from each region of interest allow for the visualization of individual protrusive and retracting areas. Protrusion distance and persistence were measured using the segmented line tool in ImageJ, and the data were exported to Excel (Microsoft) for statistical analysis.
To study random cell migration, 105 cells were seeded onto FN-coated (10 μg/ml) 35-mm Delta T dishes (Bioptechs) designed for live cell imaging and were starved of CSF-1 overnight. The following day, the media was changed to Leibovitz L-15 media (Invitrogen) containing 10% FBS and 120 ng/ml CSF-1. Time-lapse videos were made by capturing images every 5 min for 2.5 h using an inverted microscope (TE200; Nikon) with a 20× differential interference contrast objective and a heated stage (Bioptechs). Images were captured with a camera (ORCA; Hamamatsu) and were compiled using OpenLab software (Improvision). For analysis, each cell in the first frame was tracked for the entire time-lapse sequence, and the distance traveled was measured in OpenLab.
To study adhesion dynamics by TIRF microscopy, 105 cells expressing GFP-vinculin were seeded onto FN-coated (2 μg/ml) 35-mm glass-bottomed dishes and starved of CSF-1 overnight. Before filming, media was replaced with CCM1 medium (Hyclone) supplemented with 120 ng/ml exogenous CSF-1. Images were captured using an inverted microscope (1X70; Olympus) with a 60× objective. TIRF images were captured with a cooled charged-coupled device (Retiga Exi; Qimaging).
Quantification of adhesion dynamics
The fluorescent intensity of individual adhesions from cells expressing GFP-vinculin was measured over time as follows: images were acquired every 5 s using MetaMorph software (MDS Analytical Technologies). Adhesions located at the cell periphery and/or protruding edge were selected for analysis. ImageJ software was then applied to the entire image stack to subtract the background fluorescent intensity and to correct for overall photobleaching. The incorporation of vinculin into adhesions was linear on a semilogarithmic plot of the fluorescent intensity as a function of time. The apparent rate constants for the formation of vinculin-containing adhesions were determined from the slope of these graphs. Similarly, semilogarithmic graphs of the decrease in fluorescent intensity plotted as a function of time were also linear. From these plots, rate constants for the disassembly of vinculin from adhesions could be determined from the slope. For each rate constant determination, measurements were obtained for three to five individual adhesions on 8–10 cells.
Quantification of cell-adhesive area
For analysis of cell-adhesive area, ∼2 × 105 cells were seeded onto FN-coated (10 μg/ml) coverslips and incubated for 24 h at 37°C. Cells were then starved of CSF-1 overnight or left in complete (CSF-1 containing) media. Where indicated, cells were restimulated with 120 ng/ml CSF-1 for 20 min and were fixed and stained for F-actin. Cells were visualized through a fluorescence microscope (TE2000-E Eclipse; Nikon) and photographed with an ORCA CCD camera controlled by OpenLab software. To quantify cell area, digitized images acquired by immunofluorescence microscopy were analyzed with ImageJ.
Migration and invasion assays
For chemotaxis assays, the lower chamber of a modified Boyden chamber (6.5 mm and 8.0-μm Transwell Costar membrane; Corning International) was preincubated for 2 h with α-MEM and one of the following chemoattractants: 120 ng/ml CSF-1, 100 nM SDF-1α, or 100 nM MCP-1. 5 × 104 WT and FAK−/− BMMs previously starved of CSF-1 and serum overnight were loaded into the top chamber in CSF-1–free media and were allowed to migrate toward each chemoattractant for 4 h at 37°C. For invasion, the top and bottom of Biocoat invasion chambers (24-well 8.0-μm growth factor–reduced matrigel matrix; BD Biosciences) were preincubated in CSF-1–free media for 2 h. The media in the bottom chamber was then changed to include 120 ng/ml CSF-1, and cells were loaded into the top chamber in CSF-1–free media and allowed to invade through the matrigel toward CSF-1 for 24 h at 37°C. After migration or invasion, nonmigratory cells were removed from the top of the membrane using cotton swabs. The underside of each membrane was fixed, stained using the Diff-Quik staining set (Dade Behring), and mounted onto coverslips using Cytoseal 60 (Richard Allen Scientific). For migration assays, the number of cells migrated in 10 random fields was determined using light microscopy. For invasion assays, the total number of cells invading after 24 h was determined.
Matrix degradation assay
Glass coverslips were first coated with 100 μg/ml of type I collagen (Sigma-Aldrich) overnight at 4°C. The collagen layer was over-coated with N-hydroxysuccinimide–fluorescein (Thermo Fisher Scientific) initially dissolved in 50 μl N,N-dimethyl formamide (Sigma-Aldrich) and was brought to a final concentration of 33 μg/ml in sodium carbonate, pH 9, for 15 min at room temperature. Coverslips were washed twice with sodium carbonate and once with PBS before coating with a final layer of 20 μg/ml FN. 2 × 105 BMMs were plated onto the coverslips for 4 h at 37°C. Cells were fixed with 4% PFA in PBS for 20 min and permeabilized for 2–3 min with 0.4% Triton X-100 in PBS before staining with Texas red–phalloidin. Cells were visualized through a fluorescence microscope (TE2000-E Eclipse; Nikon) and photographed with an ORCA CCD camera controlled by OpenLab software (Improvision). To quantify degradation, areas cleared of N-hydroxysuccinimide–fluorescein in 10 randomly selected fields were measured with ImageJ.
TG-induced peritonitis
8–9-wk-old mice were administered 1 ml of 4% TG broth (Sigma-Aldrich) intraperitoneally. At various time points, mice were killed by carbon dioxide exposure, and peritoneal cavities were flushed with 5 ml PBS containing 0.5% BSA and 1% of 0.5 M EDTA. Cells recovered from peritoneal lavage were suspended in red blood cell lysis buffer (eBioscience) for 3 min on ice, counted with a hemocytometer (Bright-Line), and analyzed by flow cytometry.
Positive cell selection and flow cytometry
Cells flushed from the peritoneal cavities of mice were magnetically labeled using anti-CD11b or anti–GR-1 microbeads (Miltenyi Biotec) and were positively selected on an MS MACS column according to the manufacturer's instructions. After red blood cell lysis, 106 cells were incubated for 25 min on ice with fluorophore-conjugated antibodies recognizing the cellular antigens (CD45, GR-1, Ly6G, F4/80, and CD11b) diluted in PBS containing 0.5% BSA and 0.05% sodium azide. Cells were also stained with the viability dye 7AAD or DAPI. Before staining, Fc receptors were blocked with anti-CD16/32 (1 μg per 106 cells) antibodies. After staining, cells were washed twice and resuspended in PBS containing 0.5% BSA and 0.05% sodium azide, and gated cells (based on live cells) were analyzed for fluorescence staining on a FACSCalibur system (Becton Dickinson).
GTP-Rac1 pull-downs and immunoblotting
BMMs were starved of CSF-1 overnight before restimulation the following day with 120 ng/ml CSF-1. Cells were rinsed twice with PBS and lysed in modified radioimmunoprecipitation assay (50 mM Tris, 150 mM NaCl, 1% Igepal CA-630, and 0.5% deoxycholate) containing protease and phosphate inhibitors (100 μM leupeptin, 1 mM PMSF, 0.15 U/ml aprotinin, and 1 mM vanadate) as previously described (Kanner et al., 1989). Protein concentrations were determined with the BCA Assay kit (Thermo Fisher Scientific). Active GTP-bound Rac pull-down assays were performed with a Rac activation kit according to the manufacturer's instructions (Millipore). For immunoblotting, 6 μg of total cell lysate was resolved by 10% SDS-PAGE. Proteins were transferred to nitrocellulose membranes and immunoblotted as previously described (Burnham et al., 2000). Proteins were detected by HRP-conjugated anti–mouse or anti–rabbit Ig followed by enhanced chemiluminescence (Millipore). To quantify changes in the levels of protein phosphorylation, densitometry was performed (Molecular Dynamics). Band intensities were quantified by ImageQuant 5.0 (Molecular Dynamics), and values for phosphorylated proteins were divided by those for total protein and expressed relative to values obtained for unstimulated cells.
Statistical analysis
A two-sample t test assuming unequal variance was used to determine statistical significance between condition means with a significance level of ≤0.05.
Online supplemental material
Fig. S1 shows that FAK localizes in vinculin-containing adhesions. Fig. S2 shows the flow cytometry analysis of surface markers on resident and infiltrating cells isolated from the peritoneal cavity after TG treatment. Table S1 lists the markers that are expressed on resident and infiltrating cell subsets. Videos 1 and 2 show that FAK−/− BMMs exhibit elevated protrusive behavior in response to CSF-1. Videos 3 and 4 show that BMMs exhibit altered adhesion dynamics in the absence of FAK. Videos 5 and 6 show that FAK−/− BMMs exhibit impaired random migration compared with cells expressing FAK. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200708093/DC1.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants AI050733 to A.H. Bouton, CA26504 and PO1 CA100324 to E.R. Stanley, CA40042 and CA29243 to J.T. Parsons, EY0117379 and RPB444933-85773 to H.E. Beggs, and GM23244 to A.F. Horwitz.
Abbreviations used in this paper: BMM, bone marrow macrophage; CSF-1, colony-stimulating factor-1; FN, fibronectin; LysM, lysozyme M; MCP-1, macrophage chemoattractant protein-1; PE, phycoerythrin; SDF-1α, stromal cell–derived factor-1α; TG, thioglycollate; TIRF, total internal reflective fluorescence; WT, wild type.
References
- Allen, W.E., G.E. Jones, J.W. Pollard, and A.J. Ridley. 1997. Rho, Rac and Cdc42 regulate actin organization and cell adhesion in macrophages. J. Cell Sci. 110:707–720. [DOI] [PubMed] [Google Scholar]
- Beggs, H.E., D. Schahin-Reed, K. Zang, S. Goebbels, K.A. Nave, J. Gorski, K.R. Jones, D. Sretavan, and L.F. Reichardt. 2003. FAK deficiency in cells contributing to the basal lamina results in cortical abnormalities resembling congenital muscular dystrophies. Neuron. 40:501–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boocock, C.A., G.E. Jones, E.R. Stanley, and J.W. Pollard. 1989. Colony-stimulating factor-1 induces rapid behavioral responses in the mouse macrophage cell line, BAC1.2F5. J. Cell Sci. 93:447–456. [DOI] [PubMed] [Google Scholar]
- Burnham, M.R., P.J. Bruce-Staskal, M.T. Harte, C.L. Weidow, A. Ma, S.A. Weed, and A.H. Bouton. 2000. Regulation of c-SRC activity and function by the adapter protein CAS. Mol. Cell. Biol. 20:5865–5878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calle, Y., S. Burns, A.J. Thrasher, and G.E. Jones. 2006. The leukocyte podosome. Eur. J. Cell Biol. 85:151–157. [DOI] [PubMed] [Google Scholar]
- Chan, J., P.J. Leenen, I. Bertoncello, S.I. Nishikawa, and J.A. Hamilton. 1998. Macrophage lineage cells in inflammation: characterization by colony-stimulating factor-1 (CSF-1) receptor (c-Fms), ER-MP58, and ER-MP20 (Ly-6C) expression. Blood. 92:1423–1431. [PubMed] [Google Scholar]
- Clausen, B.E., C. Burkhardt, W. Reith, R. Renkawitz, and I. Forster. 1999. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 8:265–277. [DOI] [PubMed] [Google Scholar]
- Cox, D., P. Chang, Q. Zhang, P.G. Reddy, G.M. Bokoch, and S. Greenberg. 1997. Requirements for both Rac1 and Cdc42 in membrane ruffling and phagocytosis in leukocytes. J. Exp. Med. 186:1487–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Nichilo, M.O., and K.M. Yamada. 1996. Integrin alpha v beta 5-dependent serine phosphorylation of paxillin in cultured human macrophages adherent to vitronectin. J. Biol. Chem. 271:11016–11022. [DOI] [PubMed] [Google Scholar]
- Faccio, R., S. Takeshita, A. Zallone, F.P. Ross, and S.L. Teitelbaum. 2003. c-Fms and the alphavbeta3 integrin collaborate during osteoclast differentiation. J. Clin. Invest. 111:749–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh, S., E. Goldin, F.H. Gordon, H.A. Malchow, J. Rask-Madsen, P. Rutgeerts, P. Vyhnalek, Z. Zadorova, T. Palmer, and S. Donoghue. 2003. Natalizumab for active Crohn's disease. N. Engl. J. Med. 348:24–32. [DOI] [PubMed] [Google Scholar]
- Hanks, S.K., L. Ryzhova, N.Y. Shin, and J. Brabek. 2003. Focal adhesion kinase signaling activities and their implications in the control of cell survival and motility. Front. Biosci. 8:d982–d996. [DOI] [PubMed] [Google Scholar]
- Henderson, R.B., J.A. Hobbs, M. Mathies, and N. Hogg. 2003. Rapid recruitment of inflammatory monocytes is independent of neutrophil migration. Blood. 102:328–335. [DOI] [PubMed] [Google Scholar]
- Ilic, D., Y. Furuta, S. Kanazawa, N. Takeda, K. Sobue, N. Nakatsuji, S. Nomura, J. Fujimoto, M. Okada, and T. Yamamoto. 1995. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 377:539–544. [DOI] [PubMed] [Google Scholar]
- Issekutz, A.C., and H.Z. Movat. 1980. The in vivo quantitation and kinetics of rabbit neutrophil leukocyte accumulation in the skin in response to chemotactic agents and Escherichia coli. Lab. Invest. 42:310–317. [PubMed] [Google Scholar]
- Jones, G.E. 2000. Cellular signaling in macrophage migration and chemotaxis. J. Leukoc. Biol. 68:593–602. [PubMed] [Google Scholar]
- Jones, G.E., E. Prigmore, R. Calvez, C. Hogan, G.A. Dunn, E. Hirsch, M.P. Wymann, and A.J. Ridley. 2003. Requirement for PI 3-kinase gamma in macrophage migration to MCP-1 and CSF-1. Exp. Cell Res. 290:120–131. [DOI] [PubMed] [Google Scholar]
- Kanner, S.B., A.B. Reynolds, and J.T. Parsons. 1989. Immunoaffinity purification of tyrosine-phosphorylated cellular proteins. J. Immunol. Methods. 120:115–124. [DOI] [PubMed] [Google Scholar]
- Kiosses, W.B., S.J. Shattil, N. Pampori, and M.A. Schwartz. 2001. Rac recruits high-affinity integrin alphavbeta3 to lamellipodia in endothelial cell migration. Nat. Cell Biol. 3:316–320. [DOI] [PubMed] [Google Scholar]
- Kraynov, V.S., C. Chamberlain, G.M. Bokoch, M.A. Schwartz, S. Slabaugh, and K.M. Hahn. 2000. Localized Rac activation dynamics visualized in living cells. Science. 290:333–337. [DOI] [PubMed] [Google Scholar]
- Kume, A., H. Nishiura, J. Suda, and T. Suda. 1997. Focal adhesion kinase upregulated by granulocyte-macrophage colony-stimulating factor but not by interleukin-3 in differentiating myeloid cells. Blood. 89:3434–3442. [PubMed] [Google Scholar]
- Lin, T.H., A. Yurochko, L. Kornberg, J. Morris, J.J. Walker, S. Haskill, and R.L. Juliano. 1994. The role of protein tyrosine phosphorylation in integrin-mediated gene induction in monocytes. J. Cell Biol. 126:1585–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnicoff, M.J., P.K. Horan, and P.S. Morahan. 1989. Kinetics of changes in peritoneal cell populations following acute inflammation. Cell. Immunol. 118:178–191. [DOI] [PubMed] [Google Scholar]
- Miller, D.H., O.A. Khan, W.A. Sheremata, L.D. Blumhardt, G.P. Rice, M.A. Libonati, A.J. Willmer-Hulme, C.M. Dalton, K.A. Miszkiel, and P.W. O'Connor. 2003. A controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 348:15–23. [DOI] [PubMed] [Google Scholar]
- Okigaki, M., C. Davis, M. Falasca, S. Harroch, D.P. Felsenfeld, M.P. Sheetz, and J. Schlessinger. 2003. Pyk2 regulates multiple signaling events crucial for macrophage morphology and migration. Proc. Natl. Acad. Sci. USA. 100:10740–10745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankov, R., Y. Endo, S. Even-Ram, M. Araki, K. Clark, E. Cukierman, K. Matsumoto, and K.M. Yamada. 2005. A Rac switch regulates random versus directionally persistent cell migration. J. Cell Biol. 170:793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons, J.T. 2003. Focal adhesion kinase: the first ten years. J. Cell Sci. 116:1409–1416. [DOI] [PubMed] [Google Scholar]
- Pixley, F.J., and E.R. Stanley. 2004. CSF-1 regulation of the wandering macrophage: complexity in action. Trends Cell Biol. 14:628–638. [DOI] [PubMed] [Google Scholar]
- Pixley, F.J., Y. Xiong, R.Y. Yu, E.A. Sahai, E.R. Stanley, and B.H. Ye. 2005. BCL6 suppresses RhoA activity to alter macrophage morphology and motility. J. Cell Sci. 118:1873–1883. [DOI] [PubMed] [Google Scholar]
- Pollard, J.W. 2004. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer. 4:71–78. [DOI] [PubMed] [Google Scholar]
- Rovida, E., B. Lugli, V. Barbetti, S. Giuntoli, M. Olivotto, and P. Dello Sbarba. 2005. Focal adhesion kinase is redistributed to focal complexes and mediates cell spreading in macrophages in response to M-CSF. Biol. Chem. 386:919–929. [DOI] [PubMed] [Google Scholar]
- Schlaepfer, D.D., S.K. Mitra, and D. Ilic. 2004. Control of motile and invasive cell phenotypes by focal adhesion kinase. Biochim. Biophys. Acta. 1692:77–102. [DOI] [PubMed] [Google Scholar]
- Schneller, M., K. Vuori, and E. Ruoslahti. 1997. Alphavbeta3 integrin associates with activated insulin and PDGFbeta receptors and potentiates the biological activity of PDGF. EMBO J. 16:5600–5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack-Davis, J.K., K.H. Martin, R.W. Tilghman, M. Iwanicki, E.J. Ung, C. Autry, M.J. Luzzio, B. Cooper, J.C. Kath, W.G. Roberts, and J.T. Parsons. 2007. Cellular characterization of a novel focal adhesion kinase inhibitor. J. Biol. Chem. 282:14845–14852. [DOI] [PubMed] [Google Scholar]
- Stanley, E.R., K.L. Berg, D.B. Einstein, P.S. Lee, F.J. Pixley, Y. Wang, and Y.G. Yeung. 1997. Biology and action of colony–stimulating factor-1. Mol. Reprod. Dev. 46:4–10. [DOI] [PubMed] [Google Scholar]
- Tilghman, R.W., J.K. Slack-Davis, N. Sergina, K.H. Martin, M. Iwanicki, E.D. Hershey, H.E. Beggs, L.F. Reichardt, and J.T. Parsons. 2005. Focal adhesion kinase is required for the spatial organization of the leading edge in migrating cells. J. Cell Sci. 118:2613–2623. [DOI] [PubMed] [Google Scholar]
- Torka, R., F. Thuma, V. Herzog, and G. Kirfel. 2006. ROCK signaling mediates the adoption of different modes of migration and invasion in human mammary epithelial tumor cells. Exp. Cell Res. 312:3857–3871. [DOI] [PubMed] [Google Scholar]
- Vicente-Manzanares, M., D.J. Webb, and A.R. Horwitz. 2005. Cell migration at a glance. J. Cell Sci. 118:4917–4919. [DOI] [PubMed] [Google Scholar]
- Webb, D.J., K. Donais, L.A. Whitmore, S.M. Thomas, C.E. Turner, J.T. Parsons, and A.F. Horwitz. 2004. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat. Cell Biol. 6:154–161. [DOI] [PubMed] [Google Scholar]
- Webb, S.E., J.W. Pollard, and G.E. Jones. 1996. Direct observation and quantification of macrophage chemoattraction to the growth factor CSF-1. J. Cell Sci. 109:793–803. [DOI] [PubMed] [Google Scholar]
- Wells, C.M., M. Walmsley, S. Ooi, V. Tybulewicz, and A.J. Ridley. 2004. Rac1-deficient macrophages exhibit defects in cell spreading and membrane ruffling but not migration. J. Cell Sci. 117:1259–1268. [DOI] [PubMed] [Google Scholar]
- Wheeler, A.P., C.M. Wells, S.D. Smith, F.M. Vega, R.B. Henderson, V.L. Tybulewicz, and A.J. Ridley. 2006. Rac1 and Rac2 regulate macrophage morphology but are not essential for migration. J. Cell Sci. 119:2749–2757. [DOI] [PubMed] [Google Scholar]
- Worthylake, R.A., and K. Burridge. 2001. Leukocyte transendothelial migration: orchestrating the underlying molecular machinery. Curr. Opin. Cell Biol. 13:569–577. [DOI] [PubMed] [Google Scholar]
- Yamaguchi, H., F. Pixley, and J. Condeelis. 2006. Invadopodia and podosomes in tumor invasion. Eur. J. Cell Biol. 85:213–218. [DOI] [PubMed] [Google Scholar]
- Zaidel-Bar, R., M. Cohen, L. Addadi, and B. Geiger. 2004. Hierarchical assembly of cell-matrix adhesion complexes. Biochem. Soc. Trans. 32:416–420. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}