

Abstract
Previously, using an inbred strain screen and QTL mapping strategies, we demonstrated the presence of loci in the mouse genome that significantly influenced the ability of a transgene-induced mammary tumor to metastasize to the lung. Here we present data supporting the signal transduction molecule, Sipa1, as a candidate for the metastasis efficiency modifier locus Mtes1. Sequence analysis of genes in a candidate haplotype block revealed a non-synonymous animo acid polymorphism in the Sipa1 PDZ protein-protein interaction domain. Biochemical analysis indicates that the missense substitution had a significant effect on the Sipa1 RapGAP function. Spontaneous metastasis assays using cells expressing ectopic Sipa1 or Sipa1 shRNA to modulate the expression of Sipa1demonstrate that the metastatic capacity of a highly aggressive mouse mammary tumor cell line is correlated with cellular Sipa1 levels. Examination of human gene expression data is consistent with the role of Sipa1 concentration in metastatic progression. Together these data suggest that the PDZ domain polymorphism is likely to be at least one of the underlying genetic polymorphisms responsible for the Mtes1 locus. This is also, to the best of our knowledge, the first demonstration of a constitutional genetic polymorphism having a significant impact on tumor metastasis.
Introduction
The process of metastasis is of great importance to the clinical management of cancer since the majority of cancer mortality is attributable to metastatic disease rather than the primary tumor 1. In most cases cancer patients with localized tumors have significantly better prognoses than those with disseminated tumors. The hypothesis that the first stages of metastasis can be an early event 2 has been reinforced by recent evidence that 60–70% of patients have initiated the metastatic process by the time of diagnosis 3, implying that it is critical to understand the factors leading to tumor dissemination. In addition, even patients who have no evidence of tumor dissemination at primary diagnosis are at risk for metastatic disease. Approximately one-third of women who are sentinel lymph node negative at the time of surgical resection of the primary breast tumor will subsequently develop clinically detectable secondary tumors 4. Early identification of these patients might alter their management and improve their prognosis.
To gain a better understanding of the many factors that can modulate metastatic progression, our laboratory initiated an investigation into the effects of constitutional genetic polymorphism on metastatic efficiency. Using the polyoma middle-T transgene-induced mouse mammary tumor model 5, we demonstrated that the genetic background upon which a tumor arose significantly influenced the ability of the tumor to form pulmonary metastases 6. Quantitative trait genetic mapping analysis revealed the probable presence of a metastasis efficiency locus, designated Mtes1, on proximal mouse chromosome 19 7 in a 10 megabase region orthologous to human chromosome 11q12-13, which is known to harbor the metastasis suppressor gene Brms1 8. However, extensive sequence analysis of mouse Brms1 did not reveal any polymorphisms associated with metastatic efficiency suppression 9, indicating that the causative polymorphism(s) was most likely associated with another linked gene (or genes).
To identify other potential candidates for the metastasis efficiency modifier locus Mtes1, a Multiple Cross Mapping (MCM) strategy was performed. The MCM strategy 10 takes advantage of shared haplotypes amongst different inbred strains of mice used in genetic mapping studies, to reduce the number of potential candidate genes 11,12 in a given candidate interval. Using this methodology, a medium resolution map of the 10 megabase region of mouse chromosome 19 was constructed 13 across four inbred strains used to map the Mtes1 locus. Two high (AKR/J, FVB/NJ) and two low metastatic (DBA/2J, NZB/B1NJ) genotype strains were included in the analysis. Identification of five haplotypes blocks that segregated appropriately across the inbred strains reduced the high priority candidate genes to be examined from approximately 500 to 23 13, a more tractable number for further characterization.
This study describes the further analysis of the potential Mtes1 candidate genes identified in the previous studies. Using a combination of bioinformatics, sequence analysis, and in vitro and in vivo experiments, we have identified the signal-induced proliferation-associated gene 1 (Sipa1 also know as Spa1; 14) as a strong candidate for the Mtes1 locus.
Materials and Methods
Sequence Analysis
Sequencing primers were designed using the Primer 3 software package 15. Primers were designed in intronic sequences to flank exons of interest where possible. The sequences of the primers are available on request. PCR products were generated under standard amplification conditions and purified with Qiagen PCR purification kits. Double strand sequencing was performed with a Perkin Elmer BigDye Dye Terminator sequence kit. Analysis was performed on a Perkin Elmer 3100 Automated Fluorescent Sequencer. Sequences were compiled and analyzed with the computer software packages PHRED and PHRAP 16 to identify polymorphisms.
Quantitative RT-PCR
mRNAs were transcribed into cDNA using ThermoScript™ RT-PCR System (Invitrogen, Carlsbad, CA) by following its protocol. SYBR Green Quantitative PCR was performed to detect the Sipa1 mRNA levels using an ABI PRISM 7900HT Sequence Detection System. The sense primer for Sipa1 was 5'-CCAGCTGGATACCAAAACGG-3', and the anti-sense primer, 5'-CCTCAGGAGCTGTTGCTGGT-3'. The sense primer for Sipa1L1 was 5'-CGAAGGGTTTGGGGTGAG-3', the antisense 5'-ACGTCGGCTCCATCTGGT-3'. The sense primer for Rap1ga was 5'- CCAAGAACAGAGTGGAGTCG -3', the antisense 5'- TGTGCAGGCCTGTATCATCC-3'. mRNA levels were normalized to glyceraldehyde-3-phosphate dehydrogenase (Gapdh) mRNA levels using primers purchased from Applied Biosystems.
Construction of Plasmids
The full length FVB Sipa1 construct (IMAGE Clone ID 5324326) was obtained from Invitrogen (Invitrogen, Carlsbad, CA). pcDNA-fl-sipa1-FVB-neo was constructed by inserting the full length IMAGE cDNA into the vector pcDNA™3.1/V5-His TOPO (Invitrogen, Carlsbad, CA). pcDNA-fl-sipa1-DBA-neo was constructed by replacing the FVB XbaI-to-BsrGI fragment encompassing the PDZ domain with the corresponding fragment from the DBA cDNA. Ligation reactions were transformed into DH5α competent cells and plasmids were isolated and screened by restriction enzyme digestion. The sequences were confirmed by sequencing with BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA).
pcDNA-3.1-V5/His-puro and pcDNA-sf-sipa1-FVB-puro plasmids were constructed by digesting pcDNA-3.1-V5/His (Invitrogen, Carlsbad, CA), pcDNA-fl-sipa1-DBA-neo, and pcDNA-fl-sipa1-FVB-neo plasmids with NsiI and BstZ17I to remove the neomycin resistant gene and replace it with the analogous SspI and NsiI fragment from the puromycin resistant gene from pSuper.retro (Oligoengine, Seattle, WA).
Complex formation between AQP2 and Sipa1
COS7 cells were transiently co-transfected with pcDNA3 vector alone or with pcDNA3-AQP2 (from ATCC) and pSRα-Sipa1 expressing human Sipa1, pcDNA-fl-sipa1-DBA-neo expressing DBA Sipa1, or pcDNA-fl-sipa1-FVB-neo expressing FVB Sipa1. Each dish received the same total amount of DNA. The transfection used lipofectamine (Invitrogen) according to the manufacturer's instructions. Two days after transfection, cells were lysed with Golden Lysis Buffer (GLB) containing 20 mM Tris, [pH 7.9], 137 mM NaCl, 5 mM EDTA, 1 mM EGTA, 10 mM NaF, 10% Glycerol, 1 mM sodium pyrophosphate, 1 mM Leupeptin, 1 mM PMSF and, aprotinin (10 ug/ml). Cell extracts were immunoprecipitated with anti-Sipa1 mAb, and protein A/G (PIERCE) was added with overnight rotation at 4°C. The immune complexes were washed once with GLB, once with high salt HNTG (20 mM Hepes, 500 mM NaCl, 0.1% of Triton-X 100, 10% of Glycerol), and twice with low salt of HNTG (20 mM Hepes, 150 mM NaCl, 0.1% of Triton-X 100, 10% of Glycerol). The immune complexes were then analyzed by immunoblotting with anti-AQP2 antibody (Santa Cruz). Cell extracts from transfectants were also analyzed for protein expression by immunoblotting with the anti-AQP2 antibody and an anti-Sipa1 mAb (BD BioSciences). For each blot, horseradish peroxisidase-conjugated anti-rabbit, anti-mouse or anti-goat immunogobulin G was used for the second reaction at a 1:10,000 dilution. Immune complexes were visualized by enhanced chemiluminescences with an ECL Kit from Amersham.
RalGDS pull-down assay
COS7 cells were transiently transfected as above, except that a plasmid encoding Epac-HA (a guanine nucleotide exchange factor for Rap) was also added, to elevate the level of GTP•Rap1. Alternatively, an astrocytoma cell line, U373MG, which in preliminary experiments was found to have high levels of endogenous GTP•Rap1, was transfected with the Sipa1 or Sipa1 alleles to establish stable clones expressing the respective allele. These stable clones were then transiently transfected with the AQP2 expressing plasmid or the vector control. Two days after transfection, cells were processed using a Rap1 activation Kit (Upstate Biotech. Inc.) according to the manufacturer's instructions. GTP•Rap1 protein was pulled-down by RalGDS beads, washed three times, and subjected to gel analysis and immunoblotting with an anti-Rap1 antibody (Santa Cruz). Cell extracts from transfectants were also analyzed as above for protein expression by immunoblotting with the anti-AQP2 antibody, anti-Sipa1 mAb, or anti –HA antibody (Convance).
Establishment of Stable Cell Lines Expressing Allelic Variants of Sipa1
The Mvt1 cell line 17, derived from a primary mammary tumor in an MMTV-VEGF/myc bi-trangenic mouse, was used to generate the stable cell lines expressing the FVB form of Sipa1. The plasmids pcDNA-3.1-puro, pcDNA-fl-sipa1-DBA-puro, or pcDNA-fl-sipa1-FVB-puro were linearized by PmeI digestion, and transfected into Mvt1 using Polyfect Transfection Reagent (Qiagen, Valencia, CA). Twenty four hours after transfection, the cells were selected in medium containing 10 μg/ml puromycin and transferred to 96 well plates and individual clones selected by limiting dilution. Colonies were screened by western blotting against V5 antibody to identify clones expressing full length Sipa1.
Sipa1 siRNA Assays
Short hairpin (sh) interfering RNA constructs against Sipa1 were generated by cloning oligos into the pSuper.Retro.puro vector (Oligoengine, Seattle, WA). Two different constructs were made. The oligos used to generate the constructs are as follows: construct 1 forward: 5'-AGCTTAAAAATACCTTTGAGCCGAGGCCATCTCTTGAATGGCCTCGGCTCAAAGGTAGGG-3'; construct 1 reverse: 5'-GATCCCCTACCTTTGAGCCGAGGCCATTCAAGAGATGGCCTCGGCTCAAAGGTATTTTTA; construct 2 forward: 5'- AGCTTAAAAATGCGGCTGTGTCCGTCCTGTCTCTTGAACAGGACGGACACAGCCGCAGGG-3'; construct 2 reverse: 5'- GATCCCCTGCGGCTGTGTCCGTCCTGTTCAAGAGACAGGACGGACACAGCCGCATTTTTA-3'. Plasmids were transfected with FuGene (Roche, Basel, Switzerland) following the manufacturers suggested protocol and selected by addition of puromycin to the media to a final concentration of 10 μg/ml.
Pulmonary Metastasis Assays
Cell lines were cultured in Dulbecco's modification of Eagle's Medium (Cellgro, VA) containing 5 μg/ml puromycin, 50 μg/ml gentamycin and supplemented with 10% FBS. Two days before injection, cells were passaged and permitted to grow to 80-90% confluence. The cells were then washed with PBS and trypsinized, collected, washed twice with cold PBS, counted in hemocytometer and resuspended at a concentration of 2.×106 cells/ml. 100 μl of cells were injected subcutaneously in the vicinity of the fourth mammary gland of 6 week old virgin FVB/NJ female mice. The mice were aged for 4 weeks before euthanizing them by anesthetic overdose. Tumors at the injection site were dissected and weighed. Lungs were isolated and the number of surface metastases enumerated using a dissecting microscope.
Electrophoresis and Western Blotting
Cells were trypsinized and collected, and cell pellets lysed in cell lysis buffer [M-PER® Mammalian Protein Extraction Reagent (PIERCE, Rockford, IL) with Halt™ Protease Inhibitor cocktail kit (PIERCE, Rockford, IL)]. SDS-PAGE was performed for 60-90 min at 100 V using the XCell SureLock™ Mini-Cell (Invitrogen, CA) with NuPAGE Novex Bis-Tris Gels (Invitrogen, CA). Proteins were transferred to Immobilon™-P membranes (Millipore, Bedford, MA) and immunoblotted against V5 to detect the exogenous Sipa1 protein or against anti-Sipa1 antibody to detect both endogenous and exogenous protein.
Quantitative Western Blot
Protein was extracted from tumor samples using the T-PER Tissue Protein Extraction Reagent (Pierce Biotechnology, Inc.) with Halt Protease Inhibitor Cocktail (Pierce Biotechnology, Inc.). Protein concentrations were determined by the BCA assay (Pierce Biotechnology, Inc.). Protein samples of each group were separated on SDS-PAGE pre-cast gels (Invitrogen Corp., Carlsbad, CA) then transferred to Immobilon-P membranes (Millipore). Western blotting against rabbit anti-Spa-1 (1:1000, a generous gift from Dr. Masakazu Hattori of Kyoto University) was performed as previously described18. The presence of the antigen was detected by using an ECL-Plus Kit (Amersham) as per the manufacturer's protocol. Quantification of protein signals was performed by using 1D quantification function of ImageQuant QL software (Amersham). The signal of the protein band was calculated by integration of local intensity subtracting local background intensity. GAPDH was used as a loading control.
RNA Extraction and Processing for Affymetrix GeneChip® Analysis
Total RNA was extracted as described above by using TRIzol® Reagent (Life Technologies, Inc.) according to the standard protocol. The quantity and quality of the RNA were determined by an Agilent Technologies 2100 Bioanalyzer (Bio Sizing Software version A.02.01., Agilent Technologies) and/or a GeneQuant Pro, (Amersham Biosciences Corp.). Samples containing high-quality total RNA with ratios between 1.8 and 2.1 were purified with the RNeasy Mini Kit (Qiagen). An on column digest was performed as part of this purification step using RNase-Free DNase Set (Qiagen). Purified total RNA for each strain was then pooled to produce a uniform sample containing 8 ug.
Double stranded cDNA was synthesized from this preparation using the SuperScript™ Choice System for cDNA Synthesis (Invitrogen) according to the protocol for Affymetrix GeneChip® Eukaryotic Target Preparation. The double stranded cDNA was purified using the GeneChip® Sample Cleanup Module (Qiagen, Valencia, CA). Synthesis of Biotin-Labeled cRNA was obtained by in vitro transcription of the purified template cDNA using the Enzo BioArray High Yield RNA Transcript Labeling Kit (T7) (Enzo Life Sciences, Inc.). The cRNA target was purified by using the GeneChip® Sample Cleanup Module (Qiagen, Valencia, CA). Hybridization cocktails from each fragmentation reaction were prepared according to the Affymetrix GeneChip® protocol. The hybridization cocktail was applied to Affymetrix Murine Genome Moe430A GeneChip® Arrays, processed on the Affymetrix® Fluidics Station 400, and then analyzed using the RMA function of the BRB-Array Tools software v. 3.6 19.
Results
Sequence analysis of candidate genes
Previously we utilized a multiple cross and haplotypes mapping strategy 10,20 to identify regions of mouse chromosome 19 that were most likely to contain the metastasis efficiency modifier gene Mtes1 13. Using this strategy, 5 regions containing ∼23 genes out of the approximately 500 genes present in the original candidate interval were prioritized for further characterization. The prioritized genes were further categorized based on their molecular function, analyzing genes of known function before attempting to analyze the more numerous genes of unknown function. Six genes in four of the five haplotype blocks were selected for the first round of analysis. The fifth haplotype block did not contain any gene of interest, based on a priori knowledge. Three genes in the intervals (RhoD, Map3k11[also known as Mlk3 or Kcnk7], Sipa1) were members of a signal transduction pathway that had been previously associated with metastatic progression21. However, none of the prioritized genes had been previously associated with metastasis. Three additional genes were also considered high priority candidates because of their role in global gene expression (Htatip, Rela) or DNA damage repair function (Ddb1). Three of the six genes considered for the preliminary analysis map within the same haplotypes block (Rela, Map3k11, Sipa1; figure 1a). Two additional genes also lie within this haplotype block (Pcnxl3, Kcnk8). These genes, however, were not in pathways previously associated with metastatic progression or were transcribed exclusively in neuronal tissues, and therefore were excluded from the initial analysis. Complete sequencing of the exons, intron-exon boundaries and the promoters as well as 1-2 kilobases of the regions immediately upstream of the promoters was performed in the two high metastatic (AKR/J, FVB/NJ) and two low metastatic (DBA/2J, NZB/B1NJ) strains of mice used to construct the haplotypes map 13. Although intronic and silent polymorphisms were found in the Rela, Ddb1, Htatip and RhoD genes, no obvious functionally relevant polymorphisms in the protein, promoter, or upstream regions was observed. These genes were therefore excluded from further consideration.
Figure 1.
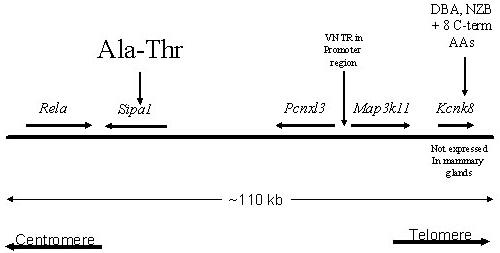
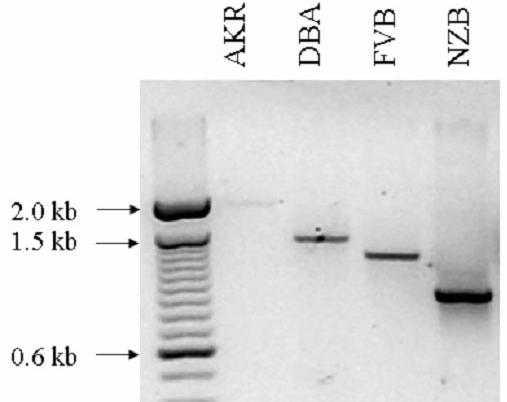
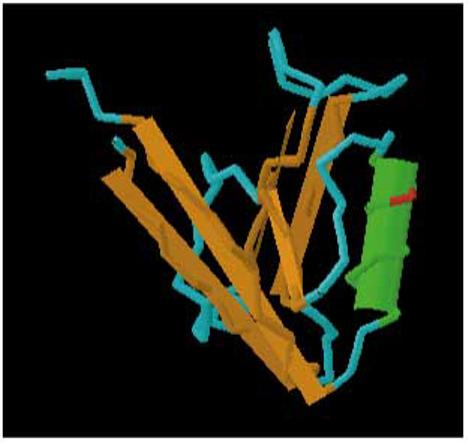
A) Schematic representation of the Mtes1/Sipa1 candidate haplotype interval. The chromosome is represented by the solid horizontal bar, with the centromere to the left and telomere to the right. The genes and their orientation are indicated by the arrow above the bar. Potentially significant polymorphisms are depicted above the genes. B) Size of the Map3k11 VNTR in four strains of mice as determined by PCR amplification. Size of the VNTR does not correlate with metastatic potential since the low metastatic strain, DBA/2J, is intermediate between the two highly metastatic strains, AKR/J and FVB/NJ. C) Three dimensional modeling of the PDZ domain to identify the location of the polymorphism. The A739T polymorphism is represented by the red section of the wire diagram on the open face of the alpha-helical region.
Sequence analysis of the four inbred strains reveal the presence of a variable tandem repeat (VTR) in the upstream region of the Map3k11 gene, containing a variable number of the MMS41 minisatellite tandem repeats 22. Since VTRs have been associated with alterations in transcriptional activity 23,24, the structure of the VTR in the four inbred strains and its effect on the transcriptional and translational activity were determined. PCR amplification across the VTR revealed that the number of repeats did not segregate with metastatic efficiency in these four strains (figure 1b). In addition, quantitative RT-PCR and western blot analysis did not reveal significant differences in either the mRNA levels of protein levels between the strains of interest (data not shown). Map3k11 was therefore excluded from further analysis.
Analysis of the Sipa1 gene revealed the presence of a polymorphism that results in an alanine (NZB, DBA*)-to-threonine (FVB, AKR) substitution in a PDZ domain, a motif frequently implicated in protein-protein interactions 25,. Modeling the polymorphism using the software package Cn3D 26, (available at http://www.ncbi.nlm.nih.gov/Structure/CN3D/cn3d.shtml) indicated that the polymorphism occurs in the open face of an α-helix (figure 1c). BLAST analysis 27 demonstrated that the alanine was conserved between mouse and human, suggesting the threonine substitution might have a functional consequence for protein-protein interactions. Because of the potential functional consequence of this polymorphism, Sipa1 was considered a potential candidate and subjected to further characterization.
Functional analysis of Sipa1 polymorphism
Sipa1 is a mitogen-inducible gene that encodes a GTPase activating protein (GAP) specific for Rap1 and Rap2 GTPases, which are members of the superfamily of Ras-related proteins 28. Sipa1 therefore negatively regulates Rap via its Rap-GAP activity, which catalyzes the hydrolysis of active GTP•Rap to inactive GDP•Rap. An interaction between the products of human SIPA1 and AQP2, a gene implicated in genetically determined nephrogenic diabetes insipidus, has recently been identified 29. AQP2 represents the first protein known to bind the PDZ domain of SIPA1, although the possible influence of this binding on the Rap-GAP activity of SIPA1 has not been examined.
Using AQP2 as a marker protein to monitor binding via the PDZ domain of SIPA1 or Sipa1, we sought to determine whether the efficiency of the interaction might be influenced by the polymorphism identified in the PDZ domain of Sipa1. COS7 cells, which express very low levels of endogenous AQP2 and Sipa1, were transiently co-transfected with AQP2 and either the DBA allele of Sipa1, the FVB allele, or the human SIPA1 allele (figure 2A). As with the DBA allele, the human allele encodes Ala at the codon corresponding to the polymorphic site in the PDZ domain of Sipa1. When extracts of the transfected cells were immunoprecipitated with an anti-SIPA1 antibody followed by immunoblotting with an anti-AQP2 antibody, the extracts containing the human allele or the Sipa1 DBA allele led to a stronger signal than the extracts containing the FVB allele (figure 2A). Therefore, AQP2 binds the protein encoded by the DBA allele more efficiently than that encoded by the FVB allele.
Figure 2.

The A739T polymorphism affects complex formation with AQP2 and the RapGAP function of Sipa1in an AQP2-dependent manner. A). Co-immunoprecipitation of AQP2 with Sipa1. AQP2 was transiently co-expressed with Human Sipa1 or mouse Sipa1 from DBA or FVB in COS7 cells. In the upper panel, anti-Sipa1 immunocomplexes were immunoblotted with anti-AQP2 antibodies. The middle and lower panels show, respectively, the expression of AQP2 and Sipa1 or Sipa1 in cell extracts. B) and C). Influence of AQP2 on the RapGAP activity of Sipa1. B: COS7 cells. C: U373MG cells. Both lines have very low levels of endogenous Sipa1 and AQP2. Human SIPA1 or mouse Sipa1 from DBA or FVB was expressed transiently in COS 7 cells (B) or as stable clones in U373MG (C). EPAC-HA, a Rap-specific guanine nucleotide exchange factor, was added transiently to the COS7 cells, to raise their level of GTP•Rap1. AQP2 was also transiently expressed in the COS7 and U373MG cells. A preliminary experiment (not shown) indicated that transfection of AQP2 alone into parental COS7 cells did not alter the endogenous level of GTP•Rap1. The upper panel of B and C shows the in vivo level of GTP•Rap1, which was assayed by RalGDS pull-down, as an indicator of the relative Rap GAP activity in the presence or absence of AQP2. The relative signal densities of GTP•Rap1 were quantified by Scion Image. The numbers under the upper panel refer to the “fold increase” in GTP•Rap1 for a particular Sipa1 allele when the GTP•Rap1 level with AQP2 was compared to the level without AQP2. The “fold increase” is 1 less than the fold difference. Thus, a 2-fold difference is designated a 1-fold increase. The remaining panels in B and C are immunoblots of cell extracts that document the relative level of the indicated protein. The Rap1 immunoblots also serve as loading controls.
We then examined whether the Sipa1 polymorphism might influence its in vivo Rap-GAP activity, in the presence or absence of AQP2, in transiently transfected COS7 cells (figure 2B). Mock-transfected cells had low levels of GTP•Rap1 (figure 2B, lane 1). To be able to assess the Rap-GAP activity of Sipa1 in COS7, we therefore co-transfected the cells with Sipa1 and Epac, a Rap-specific guanine nucleotide exchange factor, which should increase the level of GTP•Rap (figure 2, lanes 2-8). As expected, when Epac was added without Sipa1, it increased the level of GTP•Rap1 (Figure 2B, lane 2), and this level of GTP•Rap1 was reduced when Sipa1 was added together with Epac. Under these conditions, the human Sipa1 allele was more active than either of the mouse alleles in reducing the level of GTP•Rap1, and the DBA allele was somewhat more active than the FVB allele. The influence of AQP2 on in vivo Rap-GAP activity was assessed by co-tranfecting AQP2 along with Epac and a Sipa1 allele. In cells expressing the human or DBA allele, the presence of AQP2 was associated, respectively, with a ∼9-fold or ∼2-3-fold increase in the level of GTP•Rap1 compared with the corresponding control cells lacking AQP2 (figure 2B). By contrast, in cells expressing the FVB allele, AQP2 induced less than a 0.5-fold increase.
To confirm that these results could be extended to another cell line, we made stable transfectants of the Sipa1 alleles in U373MG, a human astrocytoma line (figure 2C). This cell line was chosen because in preliminary experiments it was found to contain high levels of endogenous GTP•Rap1 (figure 2C, lane 1) and low levels of endogenous Sipa1 (data not shown). In cells stably transfected with Sipa1, each of the mouse alleles induced a modest reduction in GTP•Rap1 (compare lane 1 with lanes 5 and 7), while the human allele (lane 3) induced a more substantial reduction. Transient transfection of AQP2 into these cells resulted in levels of GTP•Rap1 that were increased ∼2-fold in the human allele transfectants and ∼1-fold in the DBA allele transfectants. By contrast, AQP2 resulted in levels that were only marginally increased, by only 0.25, in the cells expressing the FVB allele.
Taken together, the above experiments lead us to conclude that expression of AQP2 induces a reduction in the Rap-GAP activity of Sipa1. The increased binding of AQP2 to the DBA allele of Sipa1 compared to the FVB allele results in a greater inhibition of Sipa1 activity, leading to higher concentrations activated Rap1 (Rap-GTP) in the DBA sample compared to the FVB sample (figure 2B, lanes 6 & 8; figure 2C, lanes 6 &8).
RNAi knockdown of Sipa1 reduces metastatic capacity
To test whether reduction of Sipa1 concentrations reduced metastatic capacity, as predicted by the dominant suppressor hypothesis, RNAi knock downs were therefore performed. Attempts were unsuccessful to establish cell lines that stably expressed shRNA with multiple independent single constructs that would knock down mRNA and protein levels for more than ∼10 days (data not shown). Therefore, the Mvt1 cell line, a highly metastatic mammary tumor cell line derived from a MMTV-myc/MMTV-vegf double transgenic animal on an FVB/N genetic background 17, was co-transfected with two shRNA constructs and individual colonies isolated resulting in a cell line with a stable ∼4-fold reduction in Sipa1 mRNA and protein levels (figure 3). The shRNA knockdown and empty vector control cell lines were implanted subcutaneously into virgin FVB/NJ female animals, tumors were permitted to grow for four weeks, and then the lungs were examined for metastases. As can be observed in figure 4a, macroscopic pulmonary surface metastases were significantly (p< 0.002) reduced in the shRNA knockdown cell line, suggesting a metastasis promoting role for Sipa1. The reduction in pulmonary metastases could not be explained by a non-specific reduction of the proliferative ability of the shRNA knockdown cells, since the primary tumors from the shRNA knockdown line were approximately 2-fold larger than those from the control cell line (figure 4B). The increased growth potential of the Sipa1 RNAi cell line was unexpected, as no significant difference in growth potential between the FVB and (DBA × FVB)F1 hybrids was observed in the original breeding study6,7, suggesting the presence of other modifiers masking this phenotype in the in vivo experiment6,7.
Figure 3.
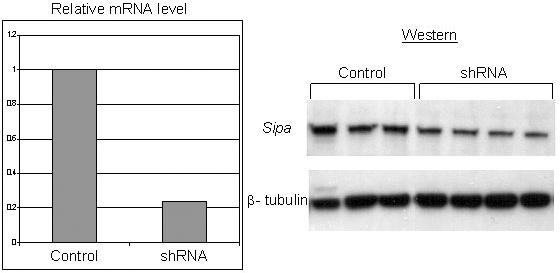
shRNA knockdown of Sipa1. The relative abundance of Sipa1 mRNA in the empty vector control cell line and the shRNA cell line is shown in the left panel. The right panel shows western blot analysis demonstrating the reduction of Sipa1 protein levels in the shRNA cells compared to the control cell line with β-tubulin as a loading control.
Figure 4.
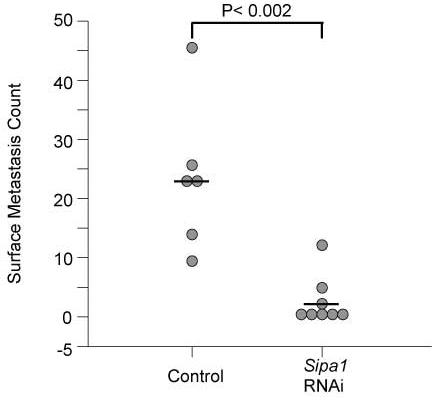
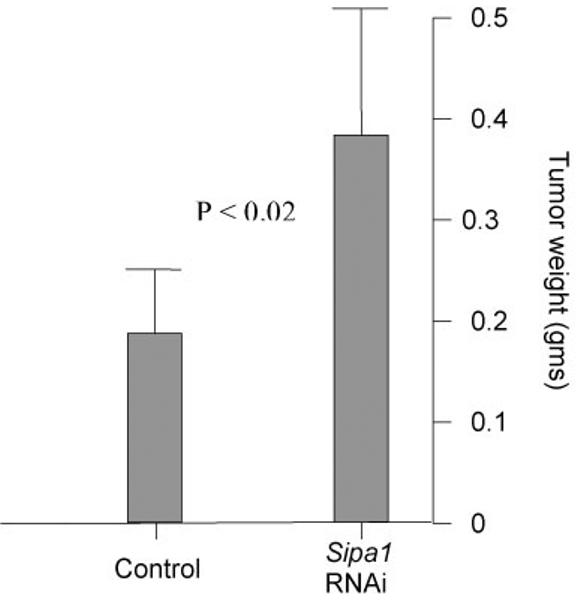
A) Scatterplot of the results of the shRNA experimental metastasis assay. The empty vector cell line is displayed on the left side of the graph, the cell line expressing the shRNA Sipa1 construct is shown on the right. B) Graphical representation of the tumor weights of the control and shRNA cell lines in the experimental metastasis assay. Standard deviation is shown by the error bars. P value for the difference between the two groups is also displayed.
Relative Sipa1 gene expression compared to other RapGAP genes
At present there are three known Rap1 GTPase activating proteins, Sipa1, Sipa1L130 [E6TP1], and Rap1ga131. If high concentrations of Sipa1L1 or Rap1ga were present in the Mvt1 cells, reduction of Sipa1 levels might not significantly change intracellular activated Rap1 levels, suggesting an alternative mechanism of metastasis suppression. The relative intracellular concentrations of the three Rap1GTPases mRNAs were therefore determined in the Mvt1 cell line by quantitative RT-PCR. Equivalent amounts of Sipa1 and Rap1ga mRNAs were observed, while a 6-8-fold higher level of Sipa1L1 was found (data not shown). These results, while not conclusive, suggest that either subtle changes in activated Rap1 concentrations may modulate metastatic efficiency or that Sipa1 may have other as of yet unknown functions that are effected by the polymorphism. Further studies will be required to address these possibilities.
shRNA alters cell morphology and adhesive properties
Previous studies demonstrated that overexpression of Sipa1 induces detachment of cells from the matrix 14. The RNAi cells were therefore examined to determine whether suppression of Sipa1 levels resulted in increased cell adhesion. Empty vector Mvt1 control cells grown to confluence consist of loose sheets of cells with round cells above the monolayer (figure 5a). Sipa1 knockdown cells, in contrast, form a solid monolayer without the presence of rounded or loosely attached cells (figure 5b), suggesting strong cellular adherence. High power magnification of the cells also revealed a significant change in cellular morphology between the clones. Mvt1 control cells display an epithelial morphology, while the Sipa1 knockdown cells appear more spindle shaped (figure 5c, d).
Figure 5.
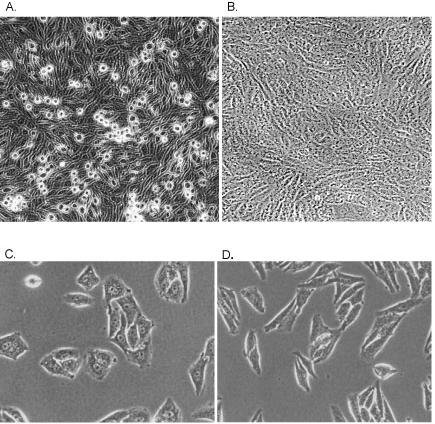
Photomicrographs of the grow properties and morphology of the empty vector (A, C) versus the shRNA (B, D) cell lines. The Mvt1 cell line produces many rounded cells when grown to confluence compared to the flat sheet observed in the RNAi knockdown cell line. The RNAi knockdown cell line also displays a more spindle shaped morphology than the more metastatic empty vector control.
Sipa1 modulates the expression of the metastasis-associated genes Kai1 and Tfpi
Examination of the gene expression data for the Sipa1 shRNA knockdown and vector control cell lines revealed that two genes previously associated with metastasis, Tfpi 32 and Kai1 33, were differentially regulated. Kai1 is a metastasis suppressor gene whose expression is lost during the progression of prostate cancer. Reintroduction of Kai1 into metastatic prostate cancer cell lines inhibits metastasis without changing primary tumor growth33. Similarly, ectopic expression of Tfpi has also been associated with reduction of metastasis in in vivo experiments32. The above data therefore suggest that Sipa1 may reduce metastatic potential in part by modulating the expression of the Kai1 and Tfpi genes. To validate this, quantitative RT-PCR was performed and demonstrated a ∼4-fold and ∼2-fold up-regulation, respectively, of these genes in the shRNA knockdown cells (data not shown). Quantitative RT-PCR of the Sipa1 ectopically expressing cell line demonstrated the opposite effect, with Tfpi and Kai1 both down regulated approximately 6-fold compared to the empty vector control cell line. The inverse correlation between the level of Sipa1 and that of both Kai1 and Tfpi is therefore consistent with the hypothesis that Sipa1may be an upstream regulator of these two metastasis inhibiting genes.
Recent evidence has demonstrated that short hairpin RNA (shRNA) has the potential to induce interferon responses, which might confound RNAi results 34. To determine whether the metastasis suppression observed in the Sipa1 RNAi experiments might be attributable to an interferon response, Affymetrix array analysis was performed. Duplicate arrays of each sample were analyzed to find genes consistently expressed differentially more than 1.5-fold, with a p value of less than 0.01. 101 probe sets were found to be differentially regulated in the shRNA cell line compared to the control line (see supplementary table 1). When the gene annotations and gene ontology terms available from Affymetrix for these probe sets were searched for associations with the interferon pathway, only two probe sets from genes that were related to interferon [interferon-related developmental regulator 1 (Ifrd1) gene and the interleukin 2 receptor gamma chain (Il2rg)] were observed to be differentially expressed (∼2-fold and ∼2.3-fold, respectively). A search of literature databases did not reveal additional interferon inducible genes from this list. While these analyses are not exhaustive, the paucity of interferon inducible genes observed, compared with previously reported results 34, increases the probability that the metastasis suppression resulted from specific inhibition of Sipa1 rather than from effects of shRNA interferon induction.
Sipa1 over-expression increases metastatic capacity
If reduction of Sipa1 activity, by either a dominant suppressor allele or a reduction in intracellular level, suppresses metastasis, it would be predicted that increasing the level of Sipa1 should enhance metastatic potential. To test this hypothesis, Sipa1 over-expressing cell lines were generated. If the reduced rate of metastasis reduction observed in the RNAi knockdown experiments were attributable to specific effects on Sipa1, rather than to non-specific shRNA-induced interferon induction 35 or off target effects 36, it would predict that over-expression of Sipa1 would enhance the metastatic potential of the Mvt1 mammary tumor cell line. Therefore, the Mvt1 cell line was transfected with a construct that encoded the Sipa1 FBV allele with an epitope tag, a clone expressing this construct was isolated, and it was implanted subcutaneously into FVB female mice. Lungs were harvested after 4 weeks and surface lesions counted. The ectopically expressing Sipa1 cell line developed ∼2-fold more surface pulmonary metastases compared to the empty vector cell line (36.6 vs. 16.6; p = 0.0004; see figure 6).
Figure 6.
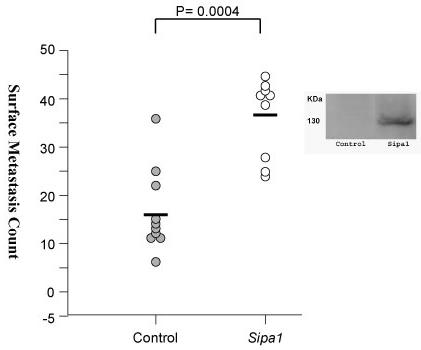
Scatterplot of the lung surface metastasis counts of mice implanted with the Sipa1 ectopically overexpressing cell line. The empty vector cell line is displayed on the left side of the graph, the cell line overexpressing the epitope tagged Sipa1 construct is shown on the right side of the graph. To the right of the graph is a western blot showing expression of the V5-His6 epitope tagged Sipa1 protein.
Sipa1 in human metastatic progression
To determine whether Sipa1 may play a role in human metastatic progression, a meta-analysis of human gene expression data was performed. The relative expression of Sipa1 in metastatic versus non metastatic tumors was performed via the Oncomine website (http://141.214.6.50/oncomine/main/index.jsp). Data were available from prostate37,38, lung39,40, and a meta-analysis of a variety of solid tumors41. Consistent with the data in the mouse breast cancer model, over-expression of Sipa1 was associated with metastatic progression in human prostate cancer (p = 0.001; see figure 7).
Figure 7.
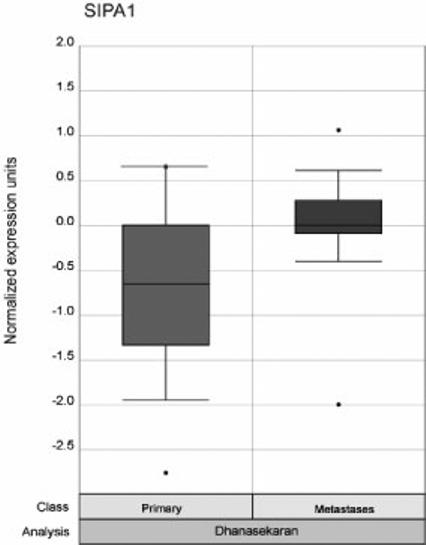
Graphical results of the Oncomine meta-analysis. The Oncomine website was analyzed for whether Sipa1 was significantly differentially expressed in metastatic versus non-metastatic tumors. Expression in the non-metastatic tumors is displayed as the box plot on the left of the figure and the metastatic tumors on the right. The p value for the meta-analysis is displayed at the top of the figure.
Discussion
Over the years a large number of genes have been associated with metastatic progression. Most of these genes have been identified by differential expression between the primary tumors and the disseminated lesions, mediated by loss of heterozygosity 42, epigenetic modulation 43 or other somatic events. These events are thought to influence metastatic progression by increasing the activity of metastasis-promoting genes or down-regulating or abrogating the inhibitory effects of metastasis suppressing factors. The mouse has been an integral component in the identification of these genes. Introduction of candidate metastasis-associated genes into cell culture followed by introduction of the cells into the lateral tail vein, ectopic or orthotopic implantation is used routinely to assess the effects of particular genes in the establishment of pulmonary metastases. It is believed that unlike human, the lungs are the primary site of metastasis in the mouse due to inability of tumor cells to pass through the pulmonary capillary beds due to the narrow diameter of the pulmonary capillaries. Metastasis to other sites, replicating the diverse metastatic target organs in the human population, can be achieved by introduction of cells into the vasculature in post-pulmonary sites, for example intracardiac, intrasplenic or portal vein injection44. These strategies have yielded a wealth of information regarding loss or gain of gene functions and the cellular processes that modulate and accompany metastatic progression. Somatic genomic changes resulting in increased or decreased expression of genes are not, however, the only ways to modulate metastasis-associated gene functions. Polymorphism may also influence gene function. The interaction of functional polymorphisms is likely to underlie the vast majority of the non-hereditary cancer susceptibility that exists in the human population. It should not be surprising therefore that similar susceptibility exists for all aspects of tumorigenesis, including metastatic progression.
The study presented here represents an ongoing effort in our laboratory to apply the QTL cloning strategy known as Multiple Cross Mapping (MCM) in parallel with conventional QTL precision mapping efforts, to identify and characterize metastasis efficiency modifier genes. The conventional strategy is based on generating and testing recombinant haplotypes between a “susceptible” and “resistant” chromosomal region by the construction of congenic and subcongenic animals, till the locus is mapped to a very short genomic region (1 Mb or less). Although potential epistatic effects can confound this strategy, it has a good probability of success and has been successfully used for the identification of cancer QTLs45. We are pursuing the congenic-based strategy in our ongoing efforts46.
A disadvantage of the conventional strategy is that it is based on the generation of congenic and subcongenic animals, by breeding. Such strains require substantial time (years) with the associated expense of genotyping and maintenance of breeding colonies and little intellectual return during the initial phases. To accelerate the process of QTL identification, Hitzemann et al.47 proposed the MCM strategy. This strategy relies on the co-localization of QTLs in multiple independent experimental crosses using different pairs of inbred strains. Since the majority of common laboratory of inbred strains are derived from common ancestors48, the co-localization of QTLs has a significant likelihood of being due to identity-by-decent. Examination of the haplotype structure across the candidate region might reveal regions that segregate appropriately with the phenotype and therefore may harbor the causative polymorphism. If the conserved haplotype blocks are sufficiently small, this method has the potential of significantly reducing the number of candidate genes in the 10-20 cM regions initially defined in preliminary complex trait analysis10,13,20,47,49. As greater amounts of mouse inbred strain sequence and SNPs12,50 become available, this method has the potential to dramatically accelerate the mapping-to-candidate gene analysis phase of complex trait analysis.
However, important caveats remain for the MCM strategy. First, MCM mapping requires the ability to rapidly identify haplotype blocks in the inbred strains of interest. At present, limited numbers of inbred strains have been sequenced, reducing the general applicability of this technique. Fortunately, efforts are currently underway to increase the number of sequenced inbred strains, which will greatly expand our knowledge of the haplotype structure for 15 additional commonly used inbred strains (www.niehs.nih.gov/oc/factsheets/mouse.htm). In addition, efforts are being made to type the known SNPs on large panels of inbred strains12,50, to expand the haplotype structure information into less commonly used strains. However, to be maximally effective, MCM requires a SNP map dense enough to capture the vast majority of the haplotypes present. The haplotype map generated in our previous studies had a resolution of approximately 110 kb13. Recent work, however, has indicated that the mouse haplotype structure is much more complex and fragmented than previously believed51,52, with larger haplotype blocks interspersed with regions of very short haplotypes. The average haplotype block observed in these studies was significantly shorter the ∼110 kb resolution used in our previous study, suggesting that additional appropriately segregating haplotype blocks may be present in the chromosome 19 interval. Further analysis of the region will be necessary to identify other potential candidate genes that might contribute to the metastasis suppressive phenotype of the Mtes1 locus.
In this study we identify a polymorphism in the signal transduction gene Sipa1 as a candidate for the metastasis efficiency modifier locus Mtes1. Biochemical analysis demonstrated that the polymorphism, which is located in the PDZ domain of Sipa1, can influence the known Rap1GAP function of this gene, while experimental manipulation of cellular Sipa1 mRNA levels significantly affected the ability of a highly metastatic mammary tumor cell line to colonize the lung. These data suggest that the relative functional concentration of Sipa1, as determined by its overall protein concentration and/or its availability to inactivate Rap1, modulate metastatic progression. These data also predict that further reduction of functional Sipa1 activity in cells homozygous for the DBA allele, compared to the [DBA × FVB]F1's used in these studies, would further reduce metastatic capacity. However, it is not possible to unambiguously perform these experiments. The limitations of meiotic recombination preclude the specific isolation of the Sipa1 DBA allele free of surrounding chromosomal DNA in an otherwise homozygous FVB background. Since there is evidence for at least one other potential candidate gene near Sipa1 contributing to the Mtes1 locus the results of a congenic experiment could not unambiguously be ascribed to the Sipa1 polymorphism. The ideal experiment would be a knockin, but the lack of FVB derived ES cells preclude performing this experiment.
The observation that Sipa1 modulation was effective in a cell line derived from a transgenic animal other than the PyMT transgenic used in the previous genetic modifier mapping studies 6,7suggests that the postulated role of Sipa1 in metastasis is not limited to the particular oncogenic events induced by the polyoma middle-T antigen. It remains to be determined how broadly this gene may function in metastatic progression. Our data and the meta-analysis from the Oncomine website suggest that Sipa1 may be important in more than a single cancer type.
The role of Sipa1 in human populations will also require additional studies to resolve. A number of polymorphisms in SIPA1 are present in the public databases, including two non-synonymous missense mutations near the RAPGAP domain. Association studies in the Anglian Breast cancer cohort 53,54 reveal a possible weak, but not statistically significant, risk of distant metastases in this cohort (data not shown). However, the SIPA1 gene has not been entirely re-sequenced at this point so there may be additional haplotypes that need to be explored to determine whether polymorphisms in SIPA1 play a role in human breast cancer. The Oncomine data also suggest a possible role in prostate cancer and studies are underway to try to validate this result, as well as to examine other solid tumor types.
The precise mechanism by which Sipa1 modulates metastasis efficiency is currently unclear. The results presented here do, however, provide some clues to how the polymorphism may be functioning. Sipa1 was originally cloned as a mitogen-inducible protein 55 that was subsequently shown to be a negative regulator of Rap1 by serving as a GTPase activating protein (GAP) for Rap1 28. Sipa1 has been shown to have significant effects on cellular adhesion 14, primarily related to its effects on Rap1, which has been implicated in maintaining the integrity of polarized epithelial 56 and intercellular adherens junctions 57. Consistent with these activities, we found that knock downs of Sipa1 appeared to increase the adhesive properties of the Mvt1 cells and eliminated the rounded cells observed in the untransfected cells and empty vector controls (figure 5). Interestingly, the RNAi knockdown cells also appeared to acquire a more spindle shaped morphology, as compared to the more cuboidal morphology of the control cells, similar to morphological changes observed in other metastasis suppressed cell systems 58. Rap1 has also been implicated in maintaining the integrity of polarized epithelial 56 and maintenance of intercellular adherens junctions 57. Part of Sipa1's role in metastasis may therefore be potentiating tumor cells escaping from the primary tumor by modulating cell morphology and the strength of intercellular contacts. In this scenario, the FVB Sipa1 allele would be more active than the DBA allele in promoting lung metastases in the PyMT model, presumably via regulation of Rap1. One might anticipate that modulating cellular adhesion could result in different histologies between tumors of the high and low metastatic genotypes. However, examination of the tumors by H & E analysis revealed no obvious histological differences (R. Cardiff, personal communication), suggesting either changes in adhesion or cell morphology are not the primary mechanism of Sipa1 metastatic suppression or that the alterations in adhesion or cell morphology do not significantly influence structures at this level of resolution.
In addition to activities that may promote metastases, Sipa1 may have other effects that curtail cell growth and tumor formation, just as Rap1 has activities that may stimulate or inhibit growth 59. has been demonstrated to have effects on cell cycle progression 55,60. Sipa1 has recently been shown to interact with a bromodomain protein, Brd4, and alterations in the relative ratio of these two proteins disrupted normal cell cycle proliferation 60. In some contexts, the negative growth functions of Sipa1 may predominate. For example, Sipa1 homozygous knockout animals are viable, but eventually develop a myeloproliferative stem cell disorder 61. The different allelic variants may therefore differentially affect the ability of disseminated cells to enter the cell cycle and proliferate into macroscopic lesions. We have not yet explored this possible effect of the Sipa1 polymorphism. Further studies will be required to address this possibility.
We also observed that Sipa1 levels appear to be inversely correlated to two genes that have previously been implicated in metastasis. Tfpi, tissue factor pathway inhibitor, is a natural inhibitor of tissue factor mediated coagulation. Ectopic expression of Tfpi in B16 melanoma cells has been demonstrated to reduce experimental metastasis in this model 32. Kai1 is a metastasis suppressor gene, whose loss has been correlated with increased metastatic potential of prostate 33 and other tumors 62-64. The inverse relationship between Sipa1 mRNA levels and these genes suggest that Sipa1 may lie upstream in the signal transduction networks that regulate Kai1 and Tfpi, and mutations or polymorphisms that alter Sipa1 function would be expected to have a similar effect. Review of our microarray data 65 is consistent with this possibility, with inbred strains carrying the low metastatic allele (DBA/2J and NZB/B1NJ; data not shown) exhibiting increased Affymetrix signal for both Tfpi and Kai1 compared with the high metastatic FVB/NJ background.
The strategy used in this study coupled the use of MCM and a priori knowledge of gene function to prioritize genes for analysis. While this approach appears to have been successful in identifying Sipa1 as either the underlying cause of the Mtes1 locus or a component of this locus, there is no reason to believe that genes of unknown function or known genes in pathways not previously associated with metastasis would not also be potential candidates. The method used here was utilized to try to eliminate the most obvious and easily testable genes before tackling the more difficult collection of genes of unknown function or functions not thought to be cancer related. Using other methods to prioritize genes, for example gene expression20, would likely result in additional candidate genes. Recent efforts in our laboratory to further investigate the Mtes1 locus, by this and other methods, has revealed the presence of a second candidate gene of unknown function whose expression correlates with metastatic potential that may contribute to the Mtes1 locus. Further investigations into the role of this uncharacterized gene in metastatic efficiency are currently in progress.
In conclusion, we have presented data implicating the Sipa1 gene as a strong candidate for the Mtes1 metastasis efficiency modifier locus. To the best of our knowledge, this is the first demonstration that a constitutional genetic polymorphism can significantly influence the metastatic process and strongly supports genetic background as an important determinant in metastatic progression 66-68. Identification of Sipa1 as a component of the metastatic cascade provides novel insight into the molecular mechanisms that function during tumor progression and provides an additional target to perturb in our efforts to control or eradicate disseminated disease, or that might eventually serve as a marker for primary tumors at high risk of metastasis.
Acknowledgments
The authors gratefully acknowledge Dr. Bruce Ponder and the members of the Laboratory of Population Genetics for useful discussions.
Footnotes
Reference
- 1.Liotta LA, Stetler-Stevenson WG. Principles of molecular cell biology of cancer: Cancer metastasis. J.B. Lippincott Co.; Philadelphia, PA: 1993. pp. 134–149. [Google Scholar]
- 2.Fisher B. From Halsted to prevention and beyond: advances in the management of breast cancer during the twentieth century. Eur J Cancer. 1999;35:1963–73. doi: 10.1016/s0959-8049(99)00217-8. [DOI] [PubMed] [Google Scholar]
- 3.Schmidt-Kittler O, et al. From latent disseminated cells to overt metastasis: genetic analysis of systemic breast cancer progression. Proc Natl Acad Sci U S A. 2003;100:7737–42. doi: 10.1073/pnas.1331931100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heimann R, Lan F, McBride R, Hellman S. Separating favorable from unfavorable prognostic markers in breast cancer: the role of E-cadherin. Cancer Res. 2000;60:298–304. [PubMed] [Google Scholar]
- 5.Guy CT, Cardiff RD, Muller WJ. Induction of mammary tumors by expression of polyomavirus middle T oncogene: A transgenic mouse model for metastatic disease. MCB. 1992;12:954–961. doi: 10.1128/mcb.12.3.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lifsted T, et al. Identification of inbred mouse strains harboring genetic modifiers of mammary tumor age of onset and metastatic progression. Int J Cancer. 1998;77:640–4. doi: 10.1002/(sici)1097-0215(19980812)77:4<640::aid-ijc26>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 7.Hunter KW, et al. Predisposition to efficient mammary tumor metastatic progression is linked to the breast cancer metastasis suppressor gene Brms1. Cancer Res. 2001;61:8866–72. [PubMed] [Google Scholar]
- 8.Seraj MJ, Samant RS, Verderame MF, Welch DR. Functional evidence for a novel human breast carcinoma metastasis suppressor, BRMS1, encoded at chromosome 11q13. Cancer Res. 2000;60:2764–9. [PubMed] [Google Scholar]
- 9.Park YG, et al. Comparative sequence analysis in eight inbred strains of the metastasis modifier QTL candidate gene Brms1. Mamm Genome. 2002;13:289–92. doi: 10.1007/s00335-001-2151-6. [DOI] [PubMed] [Google Scholar]
- 10.Hitzemann R, et al. Multiple cross mapping (MCM) markedly improves the localization of a QTL for ethanol-induced activation. Genes Brain Behav. 2002;1:214–22. doi: 10.1034/j.1601-183x.2002.10403.x. [DOI] [PubMed] [Google Scholar]
- 11.Wade CM, et al. The mosaic structure of variation in the laboratory mouse genome. Nature. 2002;420:574–8. doi: 10.1038/nature01252. [DOI] [PubMed] [Google Scholar]
- 12.Wiltshire T, et al. Genome-wide single-nucleotide polymorphism analysis defines haplotype patterns in mouse. Proc Natl Acad Sci U S A. 2003;100:3380–5. doi: 10.1073/pnas.0130101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park YG, Clifford R, Buetow KH, Hunter KW. Multiple cross and inbred strain haplotype mapping of complex-trait candidate genes. Genome Res. 2003;13:118–21. doi: 10.1101/gr.786403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsukamoto N, Hattori M, Yang H, Bos JL, Minato N. Rap1 GTPase-activating protein SPA-1 negatively regulates cell adhesion. J Biol Chem. 1999;274:18463–9. doi: 10.1074/jbc.274.26.18463. [DOI] [PubMed] [Google Scholar]
- 15.Rozen S, Skaletsky H. Primer 3. 1998 [Google Scholar]
- 16.Gordon D, Abajian C, Green P. Consed: a graphical tool for sequence finishing. Genome Res. 1998;8:195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- 17.Pei XF, et al. Explant-cell culture of primary mammary tumors from MMTV-c-Myc transgenic mice. In Vitro Cell Dev Biol Anim. 2004;40:14–21. doi: 10.1290/1543-706X(2004)40<14:ECOPMT>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 18.Le Voyer T, et al. An epistatic interaction controls the latency of a transgene-induced mammary tumor. Mamm Genome. 2000;11:883–9. doi: 10.1007/s003350010163. [DOI] [PubMed] [Google Scholar]
- 19.Hedenfalk I, et al. Gene-expression profiles in hereditary breast cancer. N Engl J Med. 2001;344:539–48. doi: 10.1056/NEJM200102223440801. [DOI] [PubMed] [Google Scholar]
- 20.Hitzemann R, et al. A strategy for the integration of QTL, gene expression, and sequence analyses. Mamm Genome. 2003;14:733–47. doi: 10.1007/s00335-003-2277-9. [DOI] [PubMed] [Google Scholar]
- 21.Steeg PS. Metastasis suppressors alter the signal transduction of cancer cells. Nat Rev Cancer. 2003;3:55–63. doi: 10.1038/nrc967. [DOI] [PubMed] [Google Scholar]
- 22.Bois P, et al. Isolation and characterization of mouse minisatellites. Genomics. 1998;50:317–30. doi: 10.1006/geno.1998.5329. [DOI] [PubMed] [Google Scholar]
- 23.Nakamura Y, Koyama K, Matsushima M. VNTR (variable number of tandem repeat) sequences as transcriptional, translational, or functional regulators. J Hum Genet. 1998;43:149–52. doi: 10.1007/s100380050059. [DOI] [PubMed] [Google Scholar]
- 24.Bailly S, Israel N, Fay M, Gougerot-Pocidalo MA, Duff GW. An intronic polymorphic repeat sequence modulates interleukin-1 alpha gene regulation. Mol Immunol. 1996;33:999–1006. doi: 10.1016/s0161-5890(96)00042-9. [DOI] [PubMed] [Google Scholar]
- 25.van Ham M, Hendriks W. PDZ domains-glue and guide. Mol Biol Rep. 2003;30:69–82. doi: 10.1023/a:1023941703493. [DOI] [PubMed] [Google Scholar]
- 26.Hogue CW. Cn3D: a new generation of three-dimensional molecular structure viewer. Trends Biochem Sci. 1997;22:314–6. doi: 10.1016/s0968-0004(97)01093-1. [DOI] [PubMed] [Google Scholar]
- 27.Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kurachi H, et al. Human SPA-1 gene product selectively expressed in lymphoid tissues is a specific GTPase-activating protein for Rap1 and Rap2. Segregate expression profiles from a rap1GAP gene product. J Biol Chem. 1997;272:28081–8. doi: 10.1074/jbc.272.44.28081. [DOI] [PubMed] [Google Scholar]
- 29.Noda Y, et al. Aquaporin-2 trafficking is regulated by PDZ-domain containing protein SPA-1. FEBS Lett. 2004;568:139–45. doi: 10.1016/j.febslet.2004.05.021. [DOI] [PubMed] [Google Scholar]
- 30.Gao Q, Srinivasan S, Boyer SN, Wazer DE, Band V. The E6 oncoproteins of high-risk papillomaviruses bind to a novel putative GAP protein, E6TP1, and target it for degradation. Mol Cell Biol. 1999;19:733–44. doi: 10.1128/mcb.19.1.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gould KA, et al. Genetic evaluation of candidate genes for the Mom1 modifier of intestinal neoplasia in mice. Genetics. 1996;144:1777–85. doi: 10.1093/genetics/144.4.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Amirkhosravi A, et al. Tissue factor pathway inhibitor reduces experimental lung metastasis of B16 melanoma. Thromb Haemost. 2002;87:930–6. [PubMed] [Google Scholar]
- 33.Dong J-T, Lamb PW, Rinkler-Schaeffer CW, Vukanovic J, Ichikawa T, Isaacs JT, Barrett JC. KAI1, a metastasis suppressor gene for prostate cancer on human chromosome 11p11.2. Science. 1995;268 doi: 10.1126/science.7754374. [DOI] [PubMed] [Google Scholar]
- 34.Bridge AJ, Pebernard S, Ducraux A, Nicoulaz AL, Iggo R. Induction of an interferon response by RNAi vectors in mammalian cells. Nat Genet. 2003;34:263–4. doi: 10.1038/ng1173. [DOI] [PubMed] [Google Scholar]
- 35.Sledz CA, Holko M, de Veer MJ, Silverman RH, Williams BR. Activation of the interferon system by short-interfering RNAs. Nat Cell Biol. 2003;5:834–9. doi: 10.1038/ncb1038. [DOI] [PubMed] [Google Scholar]
- 36.Snove O, Jr., Holen T. Many commonly used siRNAs risk off-target activity. Biochem Biophys Res Commun. 2004;319:256–63. doi: 10.1016/j.bbrc.2004.04.175. [DOI] [PubMed] [Google Scholar]
- 37.Dhanasekaran SM, et al. Delineation of prognostic biomarkers in prostate cancer. Nature. 2001;412:822–6. doi: 10.1038/35090585. [DOI] [PubMed] [Google Scholar]
- 38.LaTulippe E, et al. Comprehensive gene expression analysis of prostate cancer reveals distinct transcriptional programs associated with metastatic disease. Cancer Res. 2002;62:4499–506. [PubMed] [Google Scholar]
- 39.Bhattacharjee A, et al. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci U S A. 2001;98:13790–5. doi: 10.1073/pnas.191502998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garber ME, et al. Diversity of gene expression in adenocarcinoma of the lung. Proc Natl Acad Sci U S A. 2001;98:13784–9. doi: 10.1073/pnas.241500798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003;33:49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- 42.Miele ME, et al. Metastasis suppressed, but tumorigenicity and local invasiveness unaffected, in the human melanoma cell line MelJuSo after introduction of human chromosomes 1 or 6. Mol. Carcinog. 1996;15:284–299. doi: 10.1002/(SICI)1098-2744(199604)15:4<284::AID-MC6>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 43.Sekita N, et al. Epigenetic regulation of the KAI1 metastasis suppressor gene in human prostate cancer cell lines. Jpn J Cancer Res. 2001;92:947–51. doi: 10.1111/j.1349-7006.2001.tb01185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khanna C, Hunter K. Modeling metastasis in vivo. Carcinogenesis. 2005;26:513–23. doi: 10.1093/carcin/bgh261. [DOI] [PubMed] [Google Scholar]
- 45.Zhang S, Ramsay ES, Mock BA. Cdkn2a, the cyclin-dependent kinase inhibitor encoding p16INK4a and p19ARF, is a candidate for the plasmacytoma susceptibility locus, Pctr1. Proc Natl Acad Sci U S A. 1998;95:2429–34. doi: 10.1073/pnas.95.5.2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lancaster M, Rouse J, Hunter K. Modifiers for mammary tumor latency, progression and metastasis are present on mouse chromosomes 7, 9 and 17. Mamm Genome. 2005;16:120–126. doi: 10.1007/s00335-004-2432-y. [DOI] [PubMed] [Google Scholar]
- 47.Hitzemann R, et al. Effect of genetic cross on the detection of quantitative trait loci and a novel approach to mapping QTLs. Pharmacol Biochem Behav. 2000;67:767–72. doi: 10.1016/s0091-3057(00)00421-4. [DOI] [PubMed] [Google Scholar]
- 48.Beck JA, et al. Genealogies of mouse inbred strains. Nat Genet. 2000;24:23–5. doi: 10.1038/71641. [DOI] [PubMed] [Google Scholar]
- 49.Lindblad-Toh K, et al. Large-scale discovery and genotyping of single-nucleotide polymorphisms in the mouse. Nat Genet. 2000;24:381–6. doi: 10.1038/74215. [DOI] [PubMed] [Google Scholar]
- 50.Pletcher MT, et al. Use of a dense single nucleotide polymorphism map for in silico mapping in the mouse. PLoS Biol. 2004;2:e393. doi: 10.1371/journal.pbio.0020393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yalcin B, et al. Unexpected complexity in the haplotypes of commonly used inbred strains of laboratory mice. Proc Natl Acad Sci U S A. 2004;101:9734–9. doi: 10.1073/pnas.0401189101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang J, et al. A high-resolution multistrain haplotype analysis of laboratory mouse genome reveals three distinctive genetic variation patterns. Genome Res. 2005;15:241–9. doi: 10.1101/gr.2901705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pharoah PD, et al. Polygenic susceptibility to breast cancer and implications for prevention. Nat Genet. 2002;31:33–6. doi: 10.1038/ng853. [DOI] [PubMed] [Google Scholar]
- 54.Day N, et al. EPIC-Norfolk: study design and characteristics of the cohort. European Prospective Investigation of Cancer. Br J Cancer. 1999;80(Suppl 1):95–103. [PubMed] [Google Scholar]
- 55.Hattori M, et al. Molecular cloning of a novel mitogen-inducible nuclear protein with a Ran GTPase-activating domain that affects cell cycle progression. Mol Cell Biol. 1995;15:552–60. doi: 10.1128/mcb.15.1.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ohba Y, et al. Requirement for C3G-dependent Rap1 activation for cell adhesion and embryogenesis. Embo J. 2001;20:3333–41. doi: 10.1093/emboj/20.13.3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yajnik V, et al. DOCK4, a GTPase activator, is disrupted during tumorigenesis. Cell. 2003;112:673–84. doi: 10.1016/s0092-8674(03)00155-7. [DOI] [PubMed] [Google Scholar]
- 58.Seftor EA, et al. Molecular determinants of human uveal melanoma invasion and metastasis. Clin Exp Metastasis. 2002;19:233–46. doi: 10.1023/a:1015591624171. [DOI] [PubMed] [Google Scholar]
- 59.Stork PJ. Does Rap1 deserve a bad Rap? Trends Biochem Sci. 2003;28:267–75. doi: 10.1016/S0968-0004(03)00087-2. [DOI] [PubMed] [Google Scholar]
- 60.Farina A, et al. Bromodomain Protein Brd4 Binds to GTPase-Activating SPA-1, Modulating Its Activity and Subcellular Localization. Mol Cell Biol. 2004;24:9059–69. doi: 10.1128/MCB.24.20.9059-9069.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ishida D, et al. Myeloproliferative stem cell disorders by deregulated Rap1 activation in SPA-1-deficient mice. Cancer Cell. 2003;4:55–65. doi: 10.1016/s1535-6108(03)00163-6. [DOI] [PubMed] [Google Scholar]
- 62.Goncharuk VN, et al. Co-downregulation of PTEN, KAI-1, and nm23-H1 tumor/metastasis suppressor proteins in non-small cell lung cancer. Ann Diagn Pathol. 2004;8:6–16. doi: 10.1016/j.anndiagpath.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 63.Farhadieh RD, et al. Down-regulation of KAI1/CD82 protein expression in oral cancer correlates with reduced disease free survival and overall patient survival. Cancer Lett. 2004;213:91–8. doi: 10.1016/j.canlet.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 64.Zheng HC, et al. Expression of maspin and kai1 and their clinicopathological significance in carcinogenesis and progression of gastric cancer. Chin Med Sci J. 2004;19:193–8. [PubMed] [Google Scholar]
- 65.Qiu TH, et al. Global expression profiling identifies signatures of tumor virulence in MMTV-PyMT-transgenic mice: correlation to human disease. Cancer Res. 2004;64:5973–81. doi: 10.1158/0008-5472.CAN-04-0242. [DOI] [PubMed] [Google Scholar]
- 66.Hunter KW, Welch DR, Liu ET. Genetic background is an important determinant of metastatic potential. Nat Genet. 2003;34:23–24. doi: 10.1038/ng0503-23b. [DOI] [PubMed] [Google Scholar]
- 67.Hunter KW. Allelic diversity in the host genetic background may be an important determinant in tumor metastatic dissemination. Cancer Lett. 2003;200:97–105. doi: 10.1016/s0304-3835(03)00420-8. [DOI] [PubMed] [Google Scholar]
- 68.Hunter KW. Host genetics and tumour metastasis. Br J Cancer. 2004;90:752–5. doi: 10.1038/sj.bjc.6601590. [DOI] [PMC free article] [PubMed] [Google Scholar]