

Abstract
We hypothesized that the ambient air pollution particles (PM) induce cell cycle arrest in alveolar epithelial cells (AEC). Exposure of PM (25μg/cm2) to AEC induced cells cycle arrest in G1 phase, inhibited DNA synthesis, blocked cell proliferation and caused decrease in cyclin E, A, D1 and Cyclin E- cyclin-dependent kinase(CDK)-2 kinase activity after 4h. PM induced upregulation of CDK inhibitor, p21 protein and p21 activity in AEC. SiRNAp21 blocked PM–induced downregulation of cyclins and AEC G1 arrest. Accordingly, we provide the evidence that PM induces AEC G1 arrest by altered regulation of G1 cyclins and CDKs.
Keywords: Ambient air pollution particle, Cell cycle arrest, CDKs, G1 Cyclins, Particulate Matter
1. Introduction
Airborne particulate matter (PM 2.5μm) increases morbidity and mortality from cardiopulmonary diseases resulting in an estimated 500,000 deaths each year worldwide [1, 2]. PM is genotoxic to alveolar epithelial cell (AEC) by causing DNA damage and apoptosis [3–7]. The biochemical and molecular mechanisms underlying particle-induced cytotoxicity are poorly understood. However, the generation of reactive oxygen species (ROS) is known to mediate PM-induced toxicity to various cell components [3–5]. PM contains transition metals such as Fe, Cu, Ni, V, Co, and Cr, which may induce oxidative damage by generation of ROS [4, 6]. While, ROS-mediated activation of transcription factors, such as nuclear factor kappa B (NF-κB) and release of inflammatory mediators such as interleukin (IL)-6, IL-8 and tumor necrosis factor-alpha (TNF-α) may lead to lung injury [7]. Finally, we previously showed that activation of a mitochondria-regulated death pathway by augmented oxidative stress caused PM-induced apoptosis in AEC [3–5].
Oxidants generate signals that converge to cause wide range of cellular responses ranging from growth arrest; apoptosis and ultimately necrosis depending on the level of oxidative stress experienced [8–10]. H2O2, in particular, is known to induce multiphase cell cycle arrest [9]. However, the cellular responses after PM-induced oxidative stress on cell cycle regulation are not known. Control of cell cycle progression in response to oxidative stress is linked to activation of a checkpoint mechanism operating before entry into the S phase [10]. Progression through the G1 phase and the G1–S transition involves sequential assembly and activation of G1 cyclins and CDKs [10–12]. After oxidant injury, the rapidity of initiation of type II cell proliferation is crucial for a proper healing, as delay in the reepithelialization process has been implicated in the development of pulmonary fibrosis [3, 9]. Therefore, characterization of the mechanisms involved in the block of type II cell replication by oxidants; and the internal and external stimuli that regulate the repair mechanisms appear to be critical for the understanding and management of many lung diseases that are associated with oxidative stress. In this study, we sought to determine whether PM-induces AEC G1 arrest by altered regulation of G1 cyclins and CDKs.
2. Material and Methods
2.1. Particulate Matter
The ambient particle (2.5μm) is a well characterized Dusseldorf PM provided by the US EPA with known elemental composition comparable to US pollutant [3]. Elemental analyses of the PM were accomplished by infrared or thermal conductivity assays. Particles contain carbon (19.70±2.34%), hydrogen (1.4±0.3%), nitrogen (<.05%), oxygen (14.12±1.56), sulfur (2.09±0.55%) and ash (63.24±4.19%). Ionizable concentrations of metals include cobalt (103±13 ppm), copper (48±10 ppm), chromium (104±23 ppm), iron (14,521±572 ppm), manganese (21.3±37 ppm), nickel (1519±158 ppm), titanium (131±45 ppm) and vanadium (2767±190 ppm) [3].
2.2. Cell culture
A549 cells were obtained from the American Type Culture Collection and maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing L-glutamine (0.3μg/ml), nonessential amino acids, penicillin (100U/ml), streptomycin (200μg/ml), and 10% fetal bovine serum (FBS; GIBCO) in a humidified 95% air-5%CO2 incubator at 37°C. Targeting p21siRNA was done by cell transfection using commercially available p21siRNA duplexes (Santa Cruz Lab) exactly as per the manufactures protocol. After transfection, the cells were synchronized at G0/G1 phase by serum starvation exactly as given below.
2.3. Cell synchronization by serum starvation
Cells were synchronized at G0/G1 phase by serum starvation in DMEM with 0.5% bovine calf serum for 48h, then 10% serum was added to induce the cells to re-enter the cell cycle [5, 9].
2.4. Cell Cycle Analysis
Cells were synchronized as above, exposed to PM(25μg/cm2) with or without 10%FBS, incubated for variable period (0–24h) and then trypsinized, harvested, washed, resuspended gently in 5ml of 90% ethanol and fixed at 25°C for 1h. Then, cells were incubated with DNase-free RNase A(200μg/ml) at 37°C for 1h, followed by Propidium iodide (10μg/ml) at 37°C for 5min. Cells were separated by sonicating at 20% output level for 15s using a VirSonic 50 sonicator (Vitis), sorted by fluorescence-activated cell sorter and analyzed using FlowJo (Tree Start). For all of the experiments given below the cells were first synchronized by serum starvation exactly as described above and then treated with PM.
2.5. [3-H]-Thymidine incorporation assay
The cells were treated with PM (25μg/cm2), incubated with [3-H]-Thymidine (1μCi/ml [3-H]-TdR) for 6h. Then washed and reincubated with 10% trichloroacetic acid (TCA) solution twice for 5min each at 4°C followed by 10% SDS for 2min at room temperature. The radioactivity was quantified in a scintillation counter.
2.6. MTT cell viability assay
The cells were synchronized by serum starvation for 48h, then 10% serum was added to induce the cells to re-enter the cell cycle. The cells were then treated with PM (25μg/cm2) for 24h and incubated with 10% serum for 24h. Further, the cells were incubated with MTT (2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide, disodium salt) (20μl) for 3h. An absorbance at 490nm was measured to quantify the amount of formazan product.
2.7. TUNEL assays
AEC apoptosis was assessed by TUNEL stained nuclear morphology (Roche Diagnostics) as previously described using the manufacturer’s protocol [3].
2.8. Western blot analysis
Proteins were size fractionated by 10% gel electrophoresis and transferred to nitrocellulose membranes using a semi-dry transfer (Bio-Rad). The phospho-Rb (Ser780) and phospho-Rb (Ser795) antibodies were purchased from cell signaling technology while all of the following antibodies cyclin E (M-20), phosphorylated-cyclin E, cyclin A, CDK2(M-2), CDK4(C-22), cyclin D1(HD11), cyclin D1(H-295), p21(M-19), p27, c-Myc, Rb(M-15, C-15) and pRb were purchased from Santa Cruz Biotechnology. Blots were incubated with specific antibodies overnight at 4°C and developed with an enhanced chemiluninescence detection kit (Amersham). Immunoprecipitation was performed with anti-Cyclin E antibody using 200μg of total protein. Immunocomplexes were resolved by 10% SDS-PAGE and immunoblotted; the blot was probed with anti-Cyclin E or anti-p21 antibodies.
2.9. Kinase Assay
200μg of protein lysate was immunoprecipitated with 1–2μg of anti-cyclin E or anti-CDK2 antibody and incubated at 4°C overnight. Cyclin-CDKs were isolated by incubation at 4°C for 1h with 50μl of GSH-agarose beads (Pharmacia) and beads were washed. 500ng of histone H1, 20μM ATP and 0.1μCi of [γ-32P]ATP (3000Ci/mmol; Amersham) were added in 20μl of assay buffer and incubated for 30min at 37°C. The reaction was terminated by sample buffer (50μl) and boiling for 3min. The samples were resolved on 12% gel, fixed, Coomassie-stained, dried and then exposed to a PhosphorImager screen.
2.10. ELISA
p21 ELISA was performed by using TiterZyme ELISA Kits from Assay Designs as per the manufactures protocol.
2.11. Statistics
Data is reported as mean ± S.E.M. Statistical analysis was done by one-way ANOVA and Tukey tests. Results were considered significant when p<0.05.
3. Result and Discussion
In this study, we provide the evidence that PM induces AEC G1 arrest by altered regulation of G1 cyclins and CDKs in AEC. PM causes inhibition of the forward cell cycle regulatory cyclins (E, A), CDKs (2, 4), cyclin E-CDK2 kinase activity and induction of CDK inhibitor, p21 resulting in AEC arrest. PM is a complex mixture of organic and inorganic components [6, 9]. The biological/molecular and cellular effects of PM change significantly depending upon the elemental composition of PM. Therefore, in order to avoid the variability in examining the effects of air pollutants, we used the PM provided by US EPA with known elemental composition which is comparable to US pollutants [3]. Genotoxic effects of PM are mediated by ROS; however, the exact downstream targets of ROS are not well defined [3–7]. Activation of multiple signaling pathways such as Mitogen Activated Protein Kinases, tyrosine kinases, mitochondrial signaling, Bax, Bad, p53, NFkB, IL-6, IL-8, and TNF-α have been implicated in mediating genotoxic effects of PM [3–7]. However, the role of G1 cyclins has not been studied before. Given the important role of oxidants in the regulation of cell cycle, we examined the effects of PM on the regulation of G1 cyclins and AEC arrest.
Recent studies demonstrate that a sublethal dose of H2O2 induces multi-phase cell cycle arrest [5, 9]. Using this study model, we examined the effect of PM in AEC, with a focus on G1 phase. Cell cycle analysis after serum starvation of AEC for 48 hours revealed 92.3% cells were in G0/G1, with 5.47% in S and 2.33% in G2/M (Fig 1a). Addition of PM (25μg/cm2) alone did not change this distribution of serum starved AEC but prevented serum-induced G1 to S progression. Following addition of 10%FBS to AEC for 24h, 38.4±6.3% cells moved to S/G2/M phase while, 61.6±2.6% cells remained in G0/G1 phase. However, higher number of cells remained in G0/G1 when AEC were exposed to PM (25μg/cm2) for 24h in the presence of 10%FBS (G0/G1=87.2%, S=8.69%, G2M=4.11%), suggesting that PM-induces G1 arrest in AEC. The effect of PM on AEC arrest was seen as early as 4h. Further, we found that PM blocked serum induced (10%FBS) [3-H] Thymidine uptake and decreased cell survival as assessed by MTT assay at 24h in AEC (Fig 1b, 1c). However, as shown in Fig 1d, the cells exposed to PM (25μg/cm2) in presence of 10% FBS did not show significant apoptosis as assessed by the TUNEL assay suggesting that the reduction in MTT assay as compared to control in cells exposed to PM occurred because of growth arrest. These data suggest that an induction of G0/G1 arrest by PM does not constitute a decreased survival, but a decrease in proliferation. Collectively, we show that PM prevents progression of cell cycle by causing G0/G1 growth arrest in AEC.
Figure 1. a. PM induces alveolar epithelial cell cycle arrest in G1.
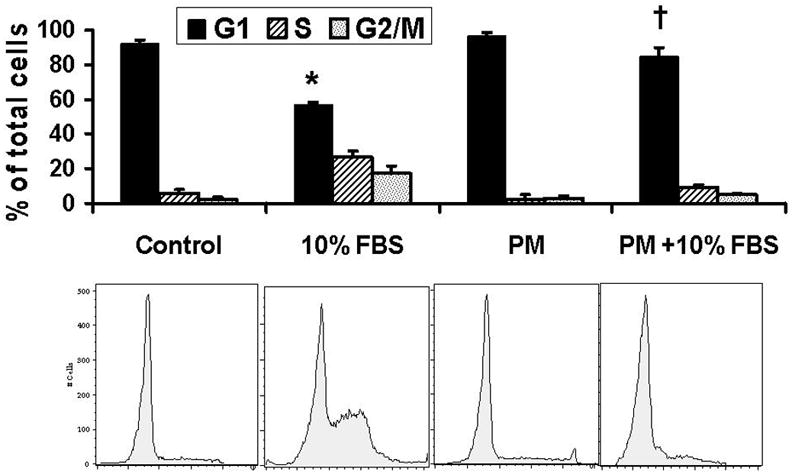
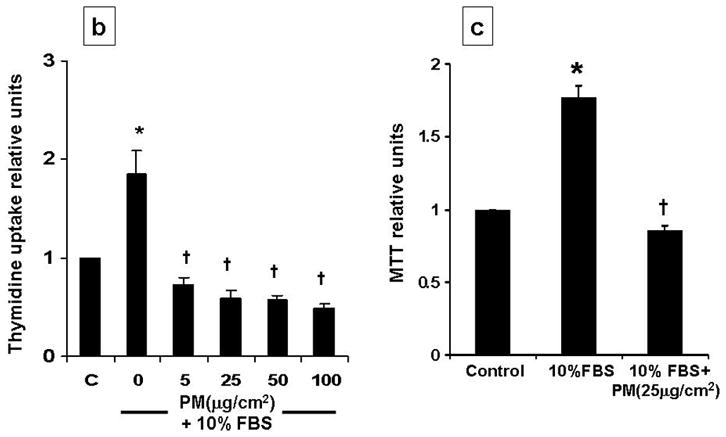
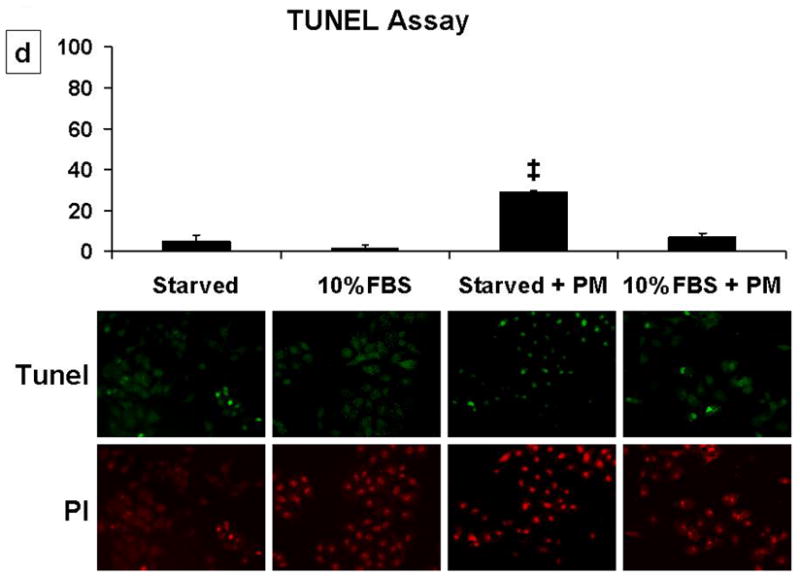
Cell cycle analysis shows that addition of PM (25μg/cm2) induced G1 arrest and prevented serum-induced G1 to S progression in AEC; this effect was seen as early as 4h. 1b, 1c: PM induced about 2-fold reduction in DNA synthesis and decreases cell survival in cells exposed to PM with 10%FBS as compared to 10%FBS alone. 1d: PM in presence of 10% FBS did not induce significant apoptosis as assessed by the TUNEL assay. Mean ± SEM, n=3. *p<0.05 control vs. 10% FBS, †p<0.05 10% FBS vs. 10% FBS + PM, ‡p<0.05 serum starved vs serum starved + PM.
The extent of AEC injury and repair are critical determinants of the toxic potential of PM. The cell may exhibits a wide range of adaptive cellular responses ranging from growth arrest, apoptosis to necrosis, depending on the level of oxidative stress experienced. The decisions of cells to arrest in G1 or progress through cell cycle after oxidative stress are determined by complex integration of extracellular and intracellular signals, such as, G1 cyclins and CDKs (Fig 4). Therefore, we examined the effect of PM on G1 cyclins such as cyclins (E, A, D1), CDKs (2, 4), c-Myc, pRb and CDK inhibitor, p21 in AEC. The cells were serum starved for 48h and then treated with PM (25μg/cm2) for variable times (0, 1, 2, 4, 8, 12, 24h). As shown in Fig 2a, the levels of p-cyclin E and cyclin A were progressively reduced after 4h in AEC exposed to PM while CDK-2 and 4 remained unchanged. The cyclins have specificity for different CDK subunits. G1-S transition is regulated by cyclin E in conjunction with its catalytic partner CDK-2 and their activity, which is the rate-limiting step for entry into the S phase of the cell cycle [8, 13–16]. Nuclear localization is crucial to the function of cyclin E/Cdk2 [13]. Cyclins are imported into the nucleus in G1 phase, possibly by piggy-backing on the p21Cip1/p27Kip1 Cdk-inhibitors [13, 14]. Cyclin E phosphorylation is necessary for both its activation and degradation [8, 13]. As shown in Fig 2b, although, total and p-cyclin E were reduced in the cell lysates, analysis of nuclear and cytoplasmic fractions showed that PM increased phosphorylated cyclin E at the nucleus at 4h which correlates with increase in p21 in AEC. Increased cyclin E phosphorylation, in the presence of PM-induced cell cycle arrest, suggest that phosphorylated cyclin E may be targeted for degradation which may in turn block the activation of transcription factors and DNA synthesis in AEC.
Figure 4.
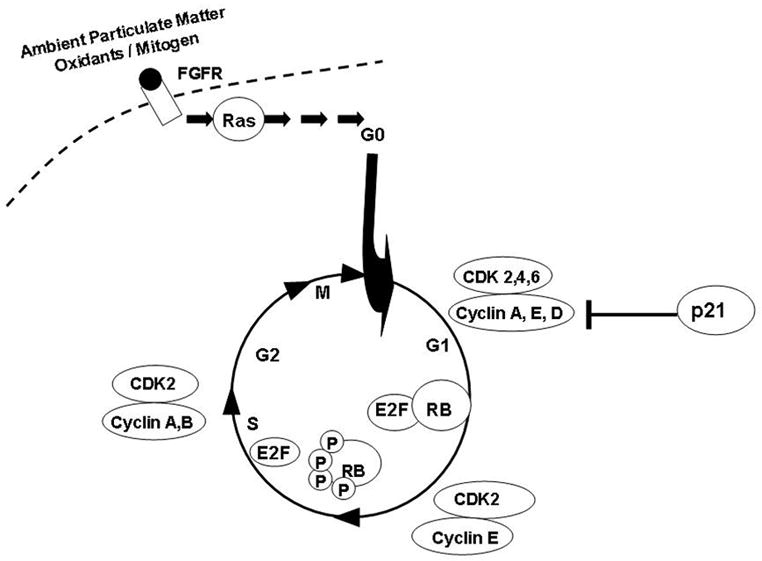
Schematic diagram of PM mediated signaling pathways involved in the regulation of cell cycle, G1 cyclins and CDKs.
Figure 2. PM regulates cyclins, CDKs, transcription factors c-Myc and pRb and cyclin E-CDK-2 kinase activity in AEC: 2a.
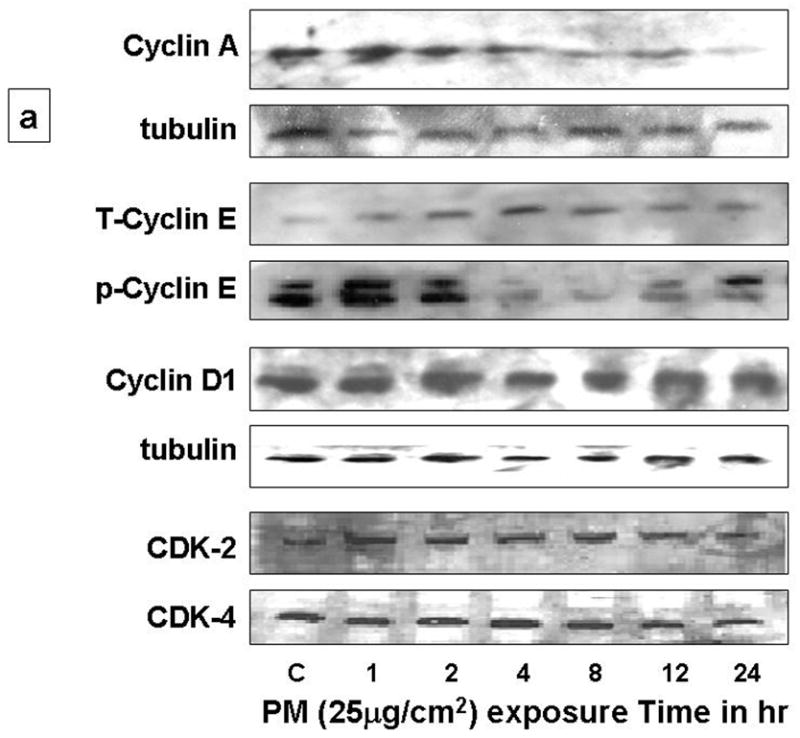
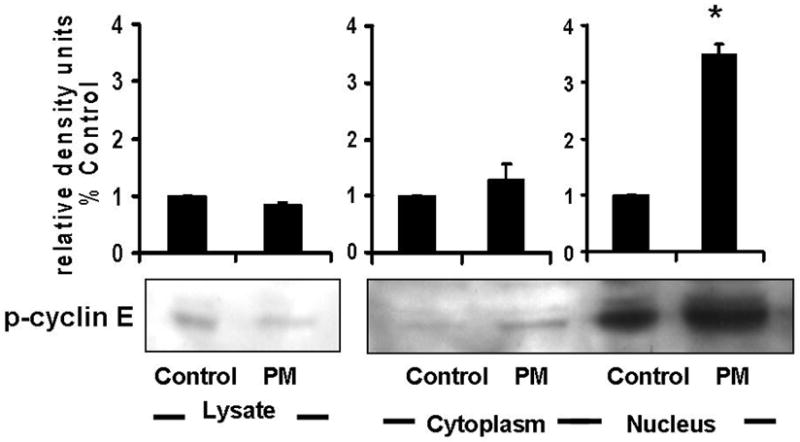
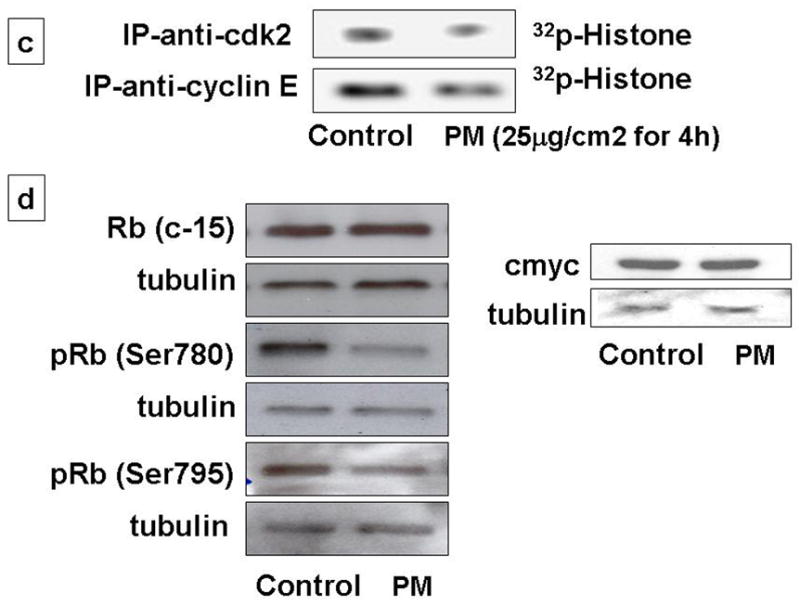
Exposure of AEC to PM (25μg/cm2) for variable times (0, 1, 2, 4, 8, 12, 24h) caused progressive reduction of total-cyclin-E (T-cyclin-E), phosphorylated-cyclin-E (p-cyclin-E), cyclin A and D1 after 4h, while CDK-2 and CDK-4 remained unchanged. 2b: Analysis of nuclear and cytoplasmic fractions of AEC after PM exposure show increased phosphorylated-cyclin-E at the nucleus. 2c: Exposure of cells to PM induced 2-fold reduction in cyclin E-CDK-2 kinase activity at 4h. 2d: c-Myc and Rb were unchanged, however phosphorylated Rb (Ser780 and Ser795) were reduced. n=3.
Further, we determined the cyclin E-CDK2 kinase activity in AEC exposed to PM [10]. As shown in Fig 2c, PM induced 2-fold reduction in cyclin E-CDK-2 kinase activity at 4h, suggesting that the PM-induced AEC G1 arrest may occur, in part, due to the reduction in cyclin E-CDK-2 kinase activity in AEC. The principal cellular substrates of the cyclin-CDKs are retinoblastoma protein (pRB). In addition to cyclin D1, G1/S transition is also mediated transcription factors pRb and c-Myc. Exposure of AEC to PM did not alter transcription factors c-Myc and Rb, however, PM caused reduction in phosphorylated Rb in our study (Fig 2d). These data show that PM-induced inhibition of the forward cell cycle regulatory cyclins (E, A), CDKs (2, 4) and cyclin E-CDK2 kinase activity causing AEC G1 arrest. Our findings are in agreement with previous reports by our group and others seen in H2O2 and hyperoxia-induced growth arrest in AEC [5, 9, 17]. It is likely that the events before the entry into the S phase may play a key role in growth inhibition after oxidative stress.
CDK inhibitor, p21 functions as a regulator of the G1/S phase checkpoint. The Cyclin-CDK complexes are negatively regulated by p21 [15–18]. Increased expression of p21 in lung epithelial cells has been reported after oxidative stress, such as hyperoxia [5, 17]. p21 is known to inhibit the cell cycle progression by causing inhibition of cyclin-CDK-2 and -CDK-4 complexes and by inhibition of Rb activation [17–18]. As shown in Fig 3a, we found that PM caused progressive increase in p21 levels after 4h. The levels of p27 were modestly increased after 4h. While, p21 activity was increased by 2-fold as assessed by ELISA in AEC exposed to PM (Fig 3b). Abrogation of p21 expression by small interfering RNA (siRNA) targeting in AEC prevented PM-induced downregulation of cyclin A and E; and prevented PM-induced AEC arrest (Fig 3c, 3d). Further as shown in Figure 3e, we found that immunoprecipitation of cyclin E caused increase in p21 binding in cells exposed to PM, while SiRNA to p21 blocked these effects suggesting that upregulation of p21 inhibits cyclinE-CDK2 causing reduction in phosphorylated Rb which results in AEC G1 arrest. Collectively, these data suggest that CDK inhibitor, p21 plays a critical role in PM-induced regulation of cyclins and PM-induced G1 arrest in AEC.
Figure 3. PM-induced upregulation of CDK inhibitor, p21 mediates PM-induced G1 arrest in AEC.
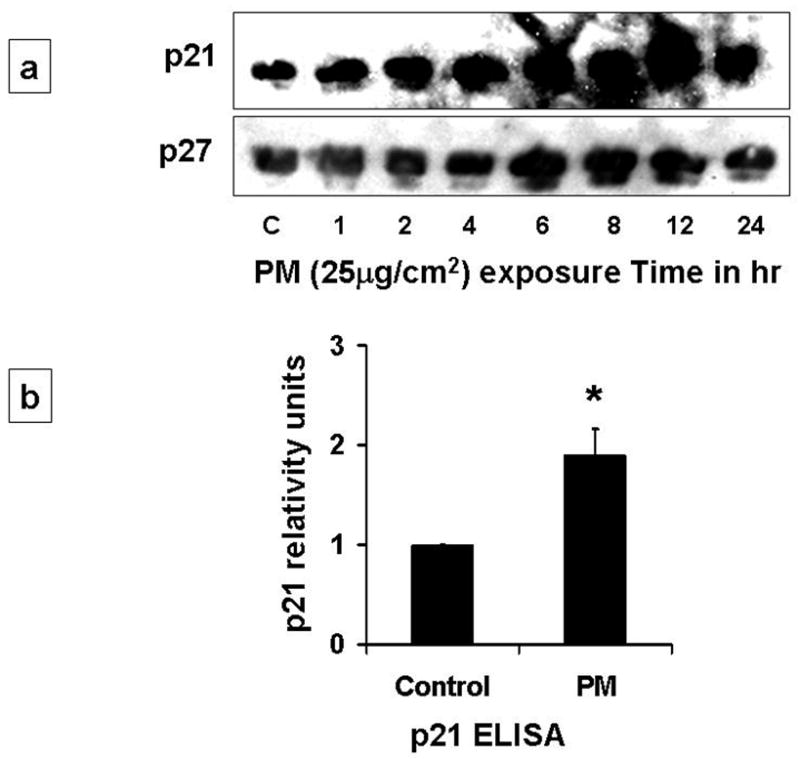
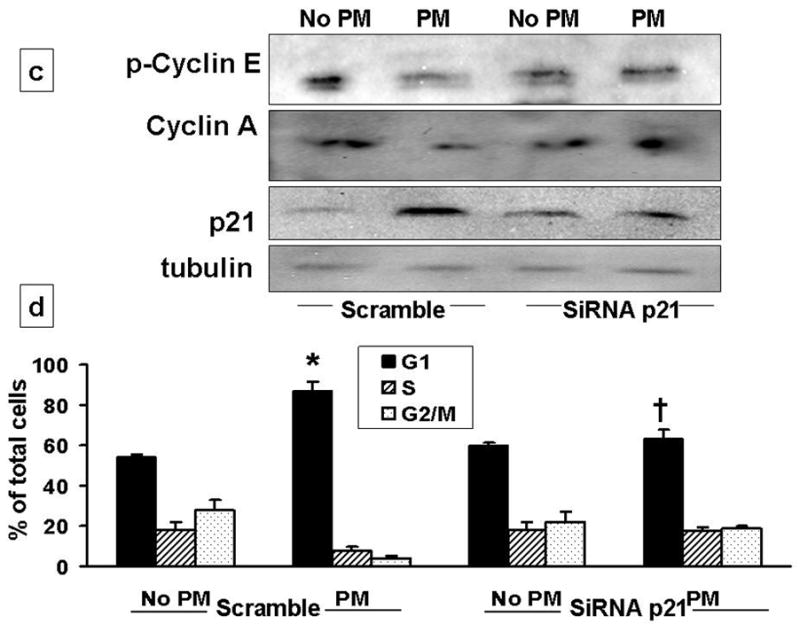
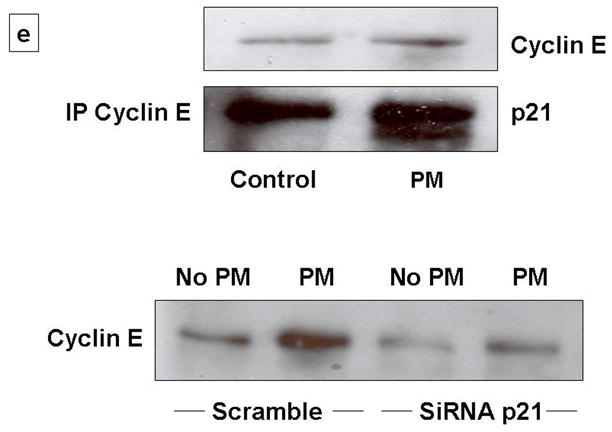
3a: PM caused progressive increase in p21 levels after 4h. The levels of p27 were modestly increased after 4h. 3b: p21 activity was increased by 2-fold as assessed by ELISA in AEC exposed to PM. 3c, 3d: siRNAp21 prevented PM-induced downregulation of cyclin A and phosphorylated-cyclin E; blocked the upregulation of p21; and prevented PM-induced AEC arrest. *p<0.05 control vs. PM (Scramble); †p<0.05 PM (Scramble) vs. PM (SiRNAp21) G1 Phase; n=3. 3e: Western blot with immunoprecipitation of cyclin E shows increase in p21 on exposure to PM, while siRNA to p21 caused reduction in cyclin E.
In summary, we provide the evidence that inhibition of forward cell cycle regulatory cyclins and CDKs; and induction of CDK inhibitor, p21 mediate PM-induced G1 arrest in AEC. These data provide insight into the mechanisms underlying the toxic effects of air pollution in the alveolar epithelium. An improved understanding of the cellular consequences induced by air-borne particulates may help us to design strategies armed at reducing the toxic effects of air pollution to the lung.
Acknowledgments
Supported in part by HL010487 and ALA-Research Grant (DU)
Abbreviations
- AEC
alveolar epithelial cells
- CDK
cyclin dependent kinases
- CKI
CDK inhibitory protein
- PM
Particulate Matter
- ROS
reactive oxygen species
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.U.N. Environment Program and WHO Report. Air pollution in the world’s mega cities. A Report from the U.N. Environment Program and WHO. Environment. 1994;36:5–37. [Google Scholar]
- 2.Pope CA, Burnett RT, Thun MJ, Calle EE, Krewski D, Kazuhiko I, Thurston GD. Lung cancer, cardiopulmonary mortality, and long-term exposure to fine particulate air pollution. JAMA. 2002;287:1132–1141. doi: 10.1001/jama.287.9.1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Upadhyay D, Panduri V, Ghio A, Kamp DW. Particulate Matter induces Alveolar Epithelial Cell DNA Damage and Apoptosis: Role of Free Radicals and Mitochondria. Am J Respir Cell Mol Biol. 2003;29(2):180–187. doi: 10.1165/rcmb.2002-0269OC. [DOI] [PubMed] [Google Scholar]
- 4.Anseth JW, Goffin AJ, Fuller GG, Ghio AJ, Kao PN, Upadhyay D. Lung surfactant gelation induced by epithelial cells exposed to air pollution or oxidative stress. Am J Respir Cell Mol Biol. 2005;33(2):161–168. doi: 10.1165/rcmb.2004-0365OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Upadhyay D, Chang W, Wei K, Gao M, Rosen GD. Fibroblast growth factor-10 prevents H2O2-induced cell cycle arrest by regulation of G1 cyclins and cyclin dependent kinases. FEBS Lett. 2007;581(2):248–52. doi: 10.1016/j.febslet.2006.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ghio AJ, Silbajoris R, Carson JL, Samet JM. Biological effects of oil fly ash. Environ Health Perspect. 2002;110(1):89–94. doi: 10.1289/ehp.02110s1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Monn C, Becker S. Cytotoxicity and induction of proinflammatory cytokines from human monocytes exposed to fine (PM2.5) and coarse particles (PM10-2.5) in outdoor and indoor air. Toxicol Appl Pharmacol. 1999;155(3):245–52. doi: 10.1006/taap.1998.8591. [DOI] [PubMed] [Google Scholar]
- 8.Lees E. Cyclin dependent kinase regulation. Curr Opin Cell Biol. 1995;7:773–780. doi: 10.1016/0955-0674(95)80060-3. [DOI] [PubMed] [Google Scholar]
- 9.Barnouin K, Dubuisson ML, Child ES, Fernandez de Mattos S, Glassford J, Medema RH, Mann DJ, Lam EWF. H2O2 Induces a Transient Multi-phase Cell Cycle Arrest in Mouse Fibroblasts through Modulating Cyclin D and p21Cip1 Expression. J Biol Chem. 2002;277:13761–13770. doi: 10.1074/jbc.M111123200. [DOI] [PubMed] [Google Scholar]
- 10.Clement A, Henrion-Caude A, Besnard V, Corroyer S. Role of Cyclins in Epithelial Response to Oxidants. Am J Respir Crit Care Med. 2001;164:S81–S84. doi: 10.1164/ajrccm.164.supplement_2.2106069. [DOI] [PubMed] [Google Scholar]
- 11.Bartek J, Lukas J. Pathways governing G1/S transition and their response to DNA damage. FEBS Lett. 2001;490:117–122. doi: 10.1016/s0014-5793(01)02114-7. [DOI] [PubMed] [Google Scholar]
- 12.Corroyer S, Maitre B, Cazals V, Clement A. Oxidant-induced growth arrest of lung alveolar epithelial cells involves inactivation of cyclin E-CDK2 complex. J Biol Chem. 1996;271:25117–25125. doi: 10.1074/jbc.271.41.25117. [DOI] [PubMed] [Google Scholar]
- 13.Jackman M, Kubota Y, den Elzen N, Hagting A, Pines J. Cyclin A- and cyclin E-Cdk complexes shuttle between the nucleus and the cytoplasm. Mol Biol Cell. 2002;13(3):1030–1045. doi: 10.1091/mbc.01-07-0361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diehl JA, Sherr CJ. A dominant-negative cyclin D1 mutant prevents nuclear import of cyclin-dependent kinase 4 (CDK4) and its phosphorylation by CDK-activating kinase. Mol Cell Biol. 1997;17:7362–7374. doi: 10.1128/mcb.17.12.7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shackelford RE, Kaufmann WK, Paules RS. Oxidative stress and cell cycle checkpoint function. Free Radic Biol Med. 2000;28:1387–1404. doi: 10.1016/s0891-5849(00)00224-0. [DOI] [PubMed] [Google Scholar]
- 16.Jackman MR, Pines JN. Cyclins and the G2/M transition. Cancer Surv. 1997;29:47–73. [PubMed] [Google Scholar]
- 17.O’Reilly MA, Staversky RJ, Watkins RH, Reed CK, de Mesy Jensen KL, Finkelstein JN, Keng PC. The cyclin-dependent kinase inhibitor p21 protects the lung from oxidative stress. Am J Respir Cell Mol Biol. 2001;24:703–710. doi: 10.1165/ajrcmb.24.6.4355. [DOI] [PubMed] [Google Scholar]
- 18.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]