

Abstract
Secretory proteins enter the Golgi apparatus when transport vesicles fuse with the cis-side and exit in transport vesicles budding from the trans-side. Resident Golgi enzymes that have been transported in the cis-to-trans direction with the secretory flow must be recycled constantly by retrograde transport in the opposite direction. In this study, we describe the functional characterization of Golgi-derived transport vesicles that were isolated from tissue culture cells. We found that under the steady-state conditions of a living cell, a fraction of resident Golgi enzymes was found in vesicles that could be separated from cisternal membranes. These vesicles appeared to be depleted of secretory cargo. They were capable of binding to and fusion with isolated Golgi membranes, and after fusion their enzymatic contents most efficiently processed cargo that had just entered the Golgi apparatus. Those results indicate a possible role for these structures in recycling of Golgi enzymes in the Golgi stack.
How are proteins sorted in the Golgi apparatus? Secretory proteins enter on the cis-side and emerge on the trans-side (Palade, 1975). In between, they move through a sequence of subcompartments where they are processed by resident Golgi enzymes (Kornfeld and Kornfeld, 1985; Rothman, 1990). Models to explain how resident Golgi enzymes are separated from the secretory flow fall into three categories that are not mutually exclusive (Nilsson and Warren, 1994): First, a retention mechanism anchors resident Golgi enzymes in the cisternae in which they reside. Second, anterograde transport is selective and allows only secretory cargo to be transported to the next cisterna. Third, anterograde transport is nonselective, and resident Golgi enzymes are selectively retrieved by retrograde transport.
Vesicular transport between Golgi compartments is mediated by COP I (COat Protein)-coated vesicles. These vesicles were first identified when isolated Golgi membranes were incubated with cytosol and GTPγS (Orci et al., 1986; Malhotra et al., 1989; Serafini et al., 1991), and they are functional intermediates in the transport of vesicular stomatitis virus (VSV)1 glycoprotein (VSV-G) between different populations of Golgi membranes in vitro (Ostermann et al., 1993). Golgi-derived COP I–coated vesicles contain both secretory cargo and resident Golgi enzymes (Ostermann et al., 1993; Sonnichsen et al., 1996), which indicates that not only secretory cargo is transported between Golgi cisternae. Several other results also indicate that resident Golgi enzymes are mobile; enzymes in early Golgi compartments of yeast cells were processed by enzymes in the late Golgi (Graham and Krasnov, 1995; Harris and Waters, 1996), and a medial-Golgi enzyme of mammalian cells was modified by an enzyme in the cis-Golgi even after it had reached the medial-Golgi (Hoe et al., 1995). Golgi enzymes do not appear to be anchored in place but freely diffuse laterally in Golgi membranes (Cole et al., 1996).
Proteins enter the Golgi apparatus in transport vesicles that bud from the ER. Whereas the known Golgi-derived transport vesicles are COP I–coated, COP II–coated vesicles transport secretory cargo from the ER to the Golgi (Barlowe et al., 1994). Export from the ER is selective: COP II–coated vesicles are enriched in proteins that are destined for export from the ER and depleted of resident ER proteins (Rexach et al., 1994). These vesicles fuse with each other and with already fused vesicles to form tubular vesicular clusters (Aridor et al., 1995) that travel along microtubules to the cis-Golgi network on the cis-side of the Golgi apparatus (Presley et al., 1997). From there, vesicle components that are needed for another round of transport as well as ER proteins that were erroneously transported to the Golgi apparatus are returned to the ER by retrograde transport (Pelham, 1988).
Coatomer (CM), the cytosolic precursor of the COP I coat (Duden et al., 1991; Waters et al., 1991), specifically recognizes and binds to a retrieval signal on the cytoplasmic tail of ER membrane proteins (Cosson and Letourneur, 1994), which indicated a role for COP I–coated vesicles in the retrieval of ER proteins. This was confirmed in yeast mutant studies: A strain that failed to retrieve ER proteins was defective in the α-subunit of CM (Letourneur et al., 1994). Another CM mutant, sec21, was shown to be defective in the β′ subunit (Hosobuchi et al., 1992). Even though this mutant was originally identified as a mutant that failed to secrete proteins (Kaiser and Schekman, 1990), it was recently shown that the primary defect of this mutant is in the retrograde transport from the Golgi to the ER (Lewis and Pelham, 1996). The secretion defect can be explained as a secondary effect that results from the failure to retrieve vesicle components from the Golgi that are needed for another round of transport out of the ER (Gaynor and Emr, 1997).
Electron microscopic analysis of cells has demonstrated that transport vesicles are sufficiently long-lived to be present under steady-state conditions. In a recent analysis of insulin-producing cells, two populations of COP I– coated vesicles were identified. Whereas one contained a marker protein for Golgi-to-ER retrograde transport but not secretory cargo, the other population contained secretory cargo but excluded the retrograde transport marker (Orci et al., 1997). In this study, we address the transport of Golgi enzymes between Golgi subcompartments, and we provide functional evidence for the existence of retrograde transport carriers in cells that recycle resident Golgi enzymes between Golgi subcompartments.
Materials and Methods
Cell Growth, VSV Infection, and Labeling of VSV-G
CHO wild-type (WT) and 15B cells were grown on tissue culture plates in MEMα supplemented with 10% fetal calf serum plus penicillin and streptomycin in a 5% CO2 atmosphere at 37°C and split 1 to 10 every 2 d. For large scale preparations of CHO WT cell, cells were grown in spinner flasks until confluent. For infection, 15B cells were grown on plates until 90% confluent. Cells were incubated with VSV in 5 ml media without serum per 15-cm plate for 45 min. 15 ml of media with serum was added, and the infection was continued for an additional 3 h. Cells were harvested by trypsinization, resuspended in methionine-free media, and incubated with 0.5 mCi per ml of [35S]methionine/cysteine for 5 min at 37°C (“pulse”). 10 ml of methionine-containing media was added per 1 ml of labeling reaction and the mixture was incubated for an additional 0–20 min at 37°C (“chase”).
Permeabilization of Cells and Flotation of Vesicles
Cells were washed in PBS and ST (0.2 M sucrose, 10 mM Tris, pH 7.2) buffer, resuspended in 4 ml of ST per 1 ml of cell pellet, frozen in liquid nitrogen, and kept at −75°C until needed. Cells were thawed in a water bath at room temperature and transferred on ice immediately after thawing. Permeabilized cells were pelleted by 5 min of centrifugation at 1,000 g at 4°C. Vesicles remained in the supernatant. To separate vesicles from cytosol, vesicles were pelleted by ultracentrifugation and resuspended in 0.8 ml KHM buffer (150 mM KCl, 10 mM Hepes, pH 7.2, 2.5 mM MgOAc). 1.2 ml of 50% iodixanol (“optiprep”) in HM (10 mM Hepes, pH 7.2, 2.5 mM MgOAc) was added, and the mixture was overlaid in an SW55 tube (Beckman Instruments, Fullerton, CA) with 2 ml of 25% iodixanol in KHM and 1 ml of 10% iodixanol in KHM. Vesicles were floated to the 10%/25% interface by 3 h of centrifugation at 55,000 rpm at 4°C and harvested at the interface.
Fractionation by Velocity Sedimentation
1 ml each of 37.5, 35, 32.5, 30, 27.5, 25, 22.5, 20, 17.5, 15, and 12.5% (wt/wt) sucrose in KHM were overlaid in an SW41 tube (Beckman Instruments) and left at room temperature until linear. 1 ml sample was laid on top, and the gradient was centrifuged for 35 min at 40,000 rpm in an SW41 rotor at 4°C. 12 fractions of 1 ml were collected from the top. Galactosyl transferase (GalT) and N-acetylglucosaminyl transferase (NAGT) activity was measured as published (Brew et al., 1975; Vischer and Hughes, 1981). To fractionate budding reactions, a 100-μl budding reaction was loaded on a 600-μl 15–32.5% sucrose in KHM gradient in a 700-μl SW55 tube (5-mm diameter, 41-mm length). The gradient was centrifuged for 15 min at 50,000 rpm in an SW55 rotor (using the appropriate adaptors) at 4°C. 10 fractions of 70 μl each were collected from the top using a gel loading tip.
Binding and Fusion Assays
For binding experiments, vesicles (supernatant from permeabilized cells or after flotation) were mixed with CHO Golgi membranes and cytosol that were isolated as published (Balch et al., 1984). For example, 50 μl of supernatant was mixed with a 100-μl premix containing 10 μl WT Golgi membranes, 15 μl CHO cytosol, 75 μM ATP, 3 mM creatine phosphate, and 12 IU/ml creatine kinase in 10 mM Hepes, pH 7.2, and 2.5 mM MgOAc. KCl and sucrose were added to 50 and 200 mM final, respectively. Samples were incubated for 20 min on ice or at 37°C. Samples were either diluted in 7.5% sucrose in KHM and fractionated by velocity sedimentation, or Golgi membranes were pelleted by a sequence of two centrifugations at 17,000 g. Vesicles that remained in the supernatant were pelleted by ultracentrifugation.
For fusion assays, isolated Golgi membranes or aliquots of fractions from the velocity sedimentation gradients as indicated in the figure legends were mixed and incubated as described in the binding experiments, except that 1 mM UDP-GlcNAc was added, and samples were incubated for 1–2 h. When gradient fractions instead of Golgi membranes were mixed, 200 ng/ml NSF was added. After incubation, Golgi membranes were pelleted (see Fig. 2) or VSV-G was immunoprecipitated (see Fig. 4), dissolved in 50 mM citrate, pH 5.5, 1 mM DTT, and 0.4% SDS, and denatured for 3 min at 95°C. An equal volume of 50 mM citrate was added, and endoglycosidase H (Endo H) was added when indicated. After incubation for 1 h at 37°C, electrophoresis sample buffer was added, and the sample was loaded on a 10% acrylamide gel. After electrophoresis, gels were dried, and proteins were visualized by phosphorimaging or autoradiography. For phosphorimaging, exposures were typically chosen with a maximum intensity (100% black) between 102 and 103. The minimum intensity (0% black) was set to 1/100 of this value, and intensities in between were linearly assigned gray scale values from 0 to 100%.
Figure 2.

Fusion of Golgi-derived vesicles with cisternal membranes. (a) CHO 15B cells were infected with VSV, and Golgi- localized VSV-G was labeled for 5 min with [35S]methionine followed by a 10-min chase. Golgi membranes were isolated from those cells and were mixed with WT cell–derived vesicles in the presence of cytosol, ATP, and UDP-GlcNAc. After incubation on ice or at 37°C, membranes were pelleted by centrifugation, and proteins were digested with Endo H when indicated. Proteins were separated by gel electrophoresis, and VSV-G was visualized by phosphorimaging. (b) An aliquot of each fraction from the velocity sedimentation gradient shown in a was incubated with VSV-G–containing 15B Golgi membranes as described above. (c) Golgi membranes containing labeled VSV-G were prepared as described in a, except that the chase time was varied from 0–20 min. Golgi membranes from each preparation were allowed to fuse with WT cell–derived vesicles and analyzed as described above. (d) Comparison of Man I processing of VSV-G in Golgi membranes after different chase times with the percentage of Endo H resistance after membrane fusion. To determine Man I processing, Golgi membranes were incubated at 37°C without vesicles to allow for completion of Man I processing before fractionation by gel electrophoresis. VSV-G was visualized by phosphorimaging, and the amount of VSV-G in the more quickly migrating band representing Man I–processed VSV-G was quantified. Endo H resistance was quantified by phosphorimaging from the experiment shown in c.
Figure 4.

VSV-G is restricted to cisternal Golgi membranes. (a) Distribution of VSV-G in homogenate. 15B cells were infected with VSV, and Golgi-localized VSV-G was labeled. Homogenate from those cells was fractionated by velocity sedimentation, and VSV-G in each fraction was immunoprecipitated. (b) An aliquot from each fraction from the gradient shown in a was mixed with WT Golgi membranes to determine fusion competence. (c) Same as in b, except that vesicles derived from WT cells were added instead of WT Golgi membranes.
Isolation of Vesicles from AlF4 − Treated Cells
CHO WT cells grown in spinner culture to near confluency were incubated with 30 mM NaF and 50 μm AlCl3 for 20 min at 37°C. All subsequent steps were done at 4°C. Cells were pelleted by centrifugation, washed in PBS and ST, and homogenized in ST. NaF and AlCl3 were added to the PBS and ST buffers used in the wash and homogenization steps. To isolate vesicles from homogenate, a medium speed supernatant was prepared by 10 min of centrifugation at 17,000 g. Vesicles in the supernatant were pelleted by ultracentrifugation, resuspended in KHM buffer, and fractionated by velocity sedimentation on a 30-ml linear 15– 35% sucrose/KHM gradient in an SW28 tube (25 min at 28,000 rpm). Vesicle-containing fractions were identified by measuring GalT activity and were pooled and layered on top of a 0.75-ml cushion of 50% iodixanol in HM. Vesicles were pelleted on this cushion by 3 h of centrifugation at 41,000 rpm in an SW41 rotor. 1.5 ml was collected from the bottom and mixed with 0.5 ml of 50% iodixanol in HM. A step gradient of 2 ml of 25% and 1 ml of 10% iodixanol in KHM was layered on top of this sample, and the gradient was centrifuged for 3 h at 55,000 rpm in an SW55 rotor. Vesicles were harvested at the 10%/25% interface.
For immunoisolation, M450 magnetic beads coated with anti–mouse IgG (DYNAL, Inc., Great Neck, NY) were preincubated with saturating amounts of CM1A10 monoclonal antibody in binding buffer (KHM buffer plus 0.2 M sucrose and 0.5 mg/ml milk powder as a blocking agent). Beads were reisolated with a magnet and incubated with 5 μl of vesicles in binding buffer for 2 h with gentle agitation. Beads were collected with a magnet and washed three times in binding buffer plus one time in binding buffer without milk powder. The supernatant and the first wash buffer were combined, and vesicles that did not bind to beads or were released in the first wash were pelleted by ultracentrifugation. Half of each sample (beads or vesicles that were pelleted from the supernatant) was dissolved in electrophoresis sample buffer and NAGT or GalT assay buffer.
Formation of COP I–coated Vesicles In Vitro
COP I–coated vesicles were prepared as described (Ostermann et al., 1993), except that the salt wash step was omitted, and vesicle formation was done in a one-stage incubation with CM, myristylated ADP ribosylation factor (mARF), GTP, and PalCoA. At the end of the vesicle formation reaction, salt was added to 180 mM final, and Golgi membranes were pelleted by 10 min of centrifugation at 17,000 g. The supernatant was mixed with a transport reaction containing VSV-infected 15B Golgi, and fusion was measured using the Golgi transport assay as described previously (Balch et al., 1984).
Results
Two Populations of Golgi Enzyme–containing Membranes
We asked whether we could detect evidence for the existence of functional retrograde transport vesicles in cellular homogenate. If vesicle formation is faster than vesicle fusion, then Golgi-derived vesicles might be present in cells in sufficient amounts to allow their detection even without the use of inhibitors of vesicle fusion. Furthermore, if resident Golgi enzymes move forward with the secretory flow and are constantly retrieved, then even a large amount of those enzymes could be in vesicles at any given time.
To separate small Golgi-derived vesicles from large cisternal membranes in cellular homogenates, we used velocity sedimentation on sucrose density gradients. While Golgi and ER-derived transport vesicles form as coated vesicles, their coat proteins are rapidly removed from the vesicles after budding. Therefore, we expect that vesicles and cisternal membranes are of similar density and that their sedimentation speed only depends on their size and shape. Large membranes (for example, cisternal Golgi membranes) will sediment quickly, and small membranes (for example, transport vesicles) will sediment slowly. In each fraction we measured the activity of two Golgi-specific enzyme activities, GalT, and NAGT activity. GalT modifies N-linked glycans by attaching galactose residues and is mostly localized in late Golgi compartments. The bulk of NAGT activity is attributable to the enzyme NAGT I, which is mostly found in the medial-Golgi apparatus (Nilsson et al., 1993). We found that both GalT and NAGT activity in homogenate distributed in two peaks: Whereas most of the activity was found in quickly sedimenting membranes (Fig. 1 a, peak in fractions 8 to 10), ∼10% of NAGT and 25% of GalT activity fractionated differently and were found in more slowly sedimenting and therefore smaller membranes (Fig. 1 a, peak in fraction 3).
Figure 1.
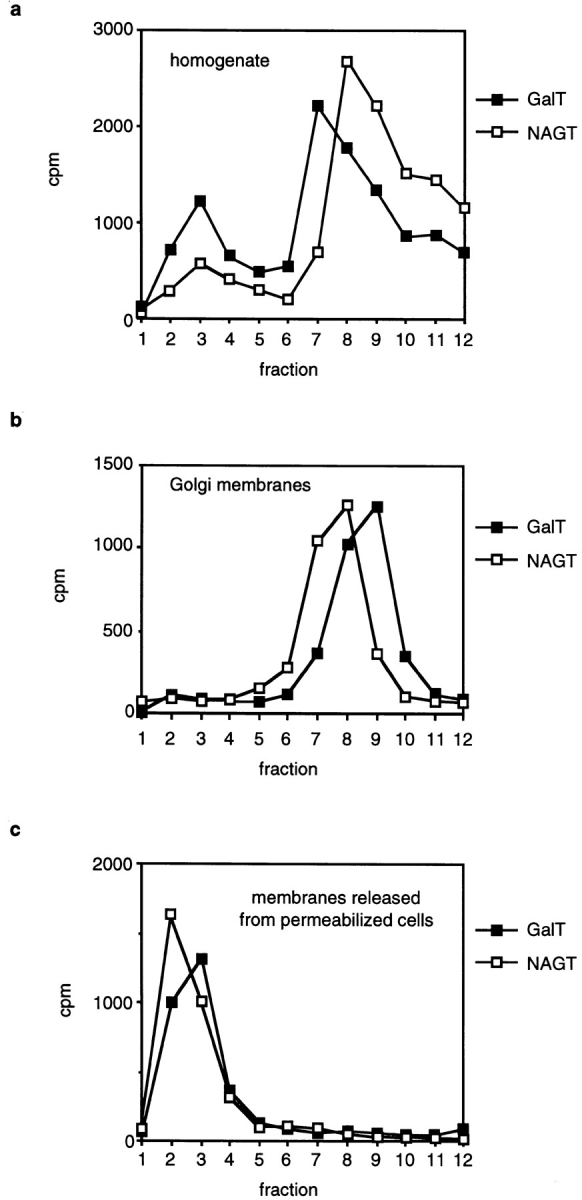
Fractionation of Golgi membranes by velocity sedimentation. (a) Homogenate of CHO cells was loaded on a sucrose gradient and centrifuged for 35 min at 40,000 rpm. Fractions were collected from the top, and GalT and NAGT activity in each fraction was measured. (b) Fractionation of isolated Golgi membranes by velocity sedimentation. (c) CHO cells were permeabilized and pelleted. The supernatant was loaded on a sucrose gradient and fractionated by velocity sedimentation. In b and c, measurements of NAGT and GalT activities were obtained from separate gradients. The differences of one fraction in the peak position are not thought to be significant.
To determine whether the quickly sedimenting membranes are cisternal Golgi membranes, we isolated Golgi membranes from homogenate according to the protocol that is used to prepare Golgi membranes for use in the in vitro transport assay (Balch et al., 1984). When those membranes were fractionated by velocity sedimentation, GalT and NAGT activity was found close to the bottom of the gradient in fractions 8–10 (Fig. 1 b). The smaller vesicles that were found in cellular homogenate were depleted from Golgi membrane preparations.
While the Golgi apparatus is tightly anchored in the cell, Golgi-derived transport vesicles are thought to be less tightly bound if not freely diffusible (Rothman et al., 1984). Therefore, we would expect that such vesicles are released when cells are permeabilized. To do so, we resuspended cells in an isotonic sucrose buffer and permeabilized them by rapid freezing in liquid nitrogen followed by quick thawing in a water bath at room temperature. Permeabilized cells were pelleted by low-speed centrifugation at 1,000 g. While most of the GalT- and NAGT-containing membranes remained inside the permeabilized cells and were pelleted, 5–10% of both GalT and NAGT activity remained in the supernatant. When those membranes were fractionated by velocity sedimentation, the fractionation profiles were similar to the fractionation profiles of the slowly sedimenting vesicles present in cellular homogenate (Fig. 1 c).
Golgi Enzymes in Vesicles Are Efficiently Transferred to Cisternal Membranes
If the vesicles identified by us were transport vesicles, we would expect them to fuse with isolated Golgi membranes. To determine their fusion competence, we used an assay that was modified from the well-known in vitro Golgi transport assay (Balch et al., 1984). Newly synthesized VSV-G was pulse labeled with [35S]methionine for 5 min, and cells were incubated for 10 min in the presence of unlabeled methionine before isolation of the Golgi membranes so that the majority of the labeled VSV-G would be in the Golgi apparatus of the 15B cells. The Golgi apparatus of 15B cells lacks functional NAGT I (Gottlieb et al., 1975), and, as a result of this defect, VSV-G remains sensitive to digestion by Endo H. If WT cell–derived vesicles would fuse with Golgi membranes derived from 15B mutant cells, then the glycosylation defect in the mutant Golgi should be corrected by NAGT I contained in the vesicles. If fusion occurred, VSV-G would be processed by NAGT I. This would allow subsequent processing by mannosidase II, which would render VSV-G Endo H resistant.
VSV-G in Golgi membrane preparations isolated from 15B cells can be separated into a doublet of bands by gel electrophoresis (Fig. 2 a, lane 1). The upper band is the only form seen when the chase is omitted and therefore must be the ER-localized high-mannose form of VSV-G. During the chase, this form is converted into the slightly faster migrating form of VSV-G, which was processed by mannosidase I (Man I) in the early Golgi. This lower band consists of Golgi-localized VSV-G, and it is this form of VSV-G that is processed by NAGT I. When Golgi membranes are isolated after a 10-min chase, on average two thirds of VSV-G is in the Golgi and one third in the ER form. The presence of the ER form is most likely due to an ER contamination in our Golgi membrane preparation, as well as Golgi localized VSV-G that has not yet been processed by Man I. After incubation of VSV-G–containing Golgi membranes with vesicles derived from WT cells, approximately half of the total VSV-G was converted to the Endo H–resistant form (Fig. 2 a, lane 4).
If the Golgi-derived vesicles are indeed transport intermediates, we would expect that NAGT I in vesicles is more efficiently transferred to Golgi membranes than NAGT I in cisternal membranes. Even though cisternal membranes can generate fusion competent NAGT I–containing vesicles, only part of the NAGT I in cisternae would be incorporated into vesicles and transferred to the other Golgi population. We fractionated homogenate from WT cells by velocity sedimentation and added an aliquot of each fraction to a fusion reaction (Fig. 2 b). Maximum conversion of VSV-G to the Endo H–resistant form was observed when membranes from fractions 2 and 3, which contained the vesicles, were added. Even though fractions 6–10 contained the bulk of the Golgi enzymes, much less of the total VSV-G was processed. The more efficient glycosylation of VSV-G after incubation with vesicles instead of cisternal membranes supports our view that these vesicles are indeed transport intermediates.
We determined next whether the NAGT I–containing vesicles fuse preferentially with early or late Golgi subcompartments. VSV-infected 15B cells were pulse labeled for 5 min, followed by a chase for 0, 5, 10, or 20 min at 37°C. Four different Golgi preparations were prepared from these cells, which now contained labeled VSV-G in either early or late compartments of the Golgi. We asked first how well VSV-G in those preparations was processed after incubation with WT cell–derived vesicles (Fig. 2 c) and found that maximal conversion to the Endo H–resistant form was observed after a chase of 5 min. Next, we determined which percentage of VSV-G could serve as a substrate for NAGT I processing. Mostly, this is VSV-G that was processed by Man I, but it also included VSV-G that had reached a Man I–containing compartment but was not yet processed by the time Golgi membranes were isolated, but will be processed by Man I during the later incubation. Man I catalyzes the first Golgi processing step, and the time of processing by Man I indicates when the wave of VSV-G labeled during the pulse enters the Golgi. To be certain that all VSV-G in a Man I–containing compartment was processed by Man I, Golgi membranes were incubated at 37°C to allow for completion of this reaction before a sample of these Golgi membranes was loaded on a gel. Processing of VSV-G by Man I could be detected by the increase in mobility caused by the removal of six mannose residues from VSV-G by Man I. The amount of Man I processing of VSV-G was quantified by phosphorimaging and compared to the amount of Endo H resistance that VSV-G could acquire after fusion of those Golgi membranes with WT cell–derived vesicles (Fig. 2 d).
We found that most of the Man I–processed VSV-G from the 0- and 5-min chase times could subsequently be processed by NAGT I supplied by vesicles and subsequently become Endo H resistant. After longer chase times, however, the percentage of Endo H–resistant VSV-G decreased, even though the amount of substrate for NAGT I processing (the Man I–processed VSV-G) increased. This result indicates that shortly after Man I processing, VSV-G moves into later Golgi compartments that no longer consume the NAGT I–containing vesicles. It appears that the vesicles fuse preferentially with the earliest Golgi compartment, which is where Man I processing occurs. If NAGT I–containing vesicles could fuse equally with all Golgi compartments, then we would expect Man I–processed VSV-G in all Golgi compartments to be processed by NAGT I with similar efficiency.
Processing by NAGT I also requires the presence of the UDP-GlcNAc/UMP antiporter, and proteins become Endo H resistant only after processing by mannosidase II, which cleaves mannose residues of glycans that were processed by NAGT I. Those enzymes would be expected to colocalize with NAGT I in the medial- and trans-Golgi. As VSV-G in the early Golgi is efficiently converted to the Endo H–resistant form after delivery of NAGT I from vesicles, it is possible that mannosidase II and the antiporter are present in these vesicles as well. On the other hand, the distributions of Golgi enzymes overlap in the Golgi stack (Nilsson et al., 1993), and it is therefore likely that the Man I–containing compartment of the 15B cells contains some mannosidase II and UDP-GlcNAc/UMP antiporter activity. Because of the long incubation time of the in vitro assay, these low activities may be sufficient so that processing of VSV-G to the Endo H–resistant form would require only the addition of WT NAGT I activity.
Vesicles Bind to Golgi Membranes in an Energy- and Cytosol-requiring Reaction
The results from the membrane fusion experiments demonstrated that a fraction of NAGT I activity is transferred to 15B Golgi membranes, but it did not allow us to determine how much of the vesicles fused with Golgi membranes. As an alternative assay, we studied the binding of vesicles to cisternal Golgi membranes by determining whether the vesicle-associated enzyme activity cofractionated with cisternal membranes after incubation. Such a cofractionation would at the least require stable binding of vesicles to cisternal membranes, but it would also include membrane fusion. We followed the vesicle-associated GalT activity in these experiments since a larger amount of this activity was found in the vesicle containing fractions.
We incubated vesicles with isolated Golgi membranes in the presence of cytosol and ATP for 20 min on ice or at 37°C. At the end of the incubation, the mixture was fractionated by velocity sedimentation (Fig. 3 a). While the GalT activity present in vesicles (fraction 2 and 3) and cisternal membranes (fraction 8 to 10) remained separated when the incubation was done on ice, after incubation at 37°C most of the vesicle-derived GalT was transferred to the fractions containing cisternal membranes. However, when vesicles were incubated in the absence of Golgi membranes, we observed no change in their rate of sedimentation (Fig. 3 b). We conclude that vesicles are capable of reassociating with cisternal Golgi membranes but will not reassemble with each other into larger structures. In addition to vesicles, the supernatant of permeabilized cells also contained other membrane fragments, possibly plasma membrane derived, that were generated by the freeze/thaw cycle. These fragments, which constituted the bulk of the membranes present in the supernatant of permeabilized cells, are larger than Golgi-derived vesicles and were therefore found in the bottom fractions of the gradient (as determined by protein staining, data not shown). We did not observe any association of the vesicles with these fragments after incubation, which indicates that Golgi-derived vesicles do not nonspecifically bind to all membranes.
Figure 3.

Binding of Golgi- derived vesicles to cisternal membranes. (a) Vesicles (supernatant of permeabilized cells) were mixed with isolated Golgi membranes, CHO cytosol, ATP, and an ATP-regenerating system. Samples were incubated for 20 min on ice or at 37°C before they were loaded on a sucrose gradient and fractionated by velocity sedimentation. (b) Same as in a, except that Golgi membranes were omitted and a larger amount of vesicles was loaded on the gradient. (c) Vesicles (supernatant of permeabilized cells) or Golgi membranes were incubated alone or together in the presence of cytosol and, unless indicated, ATP and an ATP-regenerating system. After incubation for 20 min on ice or at 37°C, large membranes were pelleted by a medium-speed centrifugation. Small vesicles remained in the supernatant. To determine the GalT activity in the supernatant, vesicles were pelleted by ultracentrifugation and dissolved in GalT assay buffer. (d) Vesicles were separated from cytosol by flotation on a density gradient. Cytosol-free vesicles were mixed with isolated Golgi membranes and the indicated amounts of cytosol and incubated on ice or at 37°C. At the end of the incubation, large membranes were pelleted, and the amount of GalT activity in the supernatant was determined. The amount of GalT activity in the supernatant was expressed as a percent of the corresponding control that was kept on ice.
To ascertain that the binding of vesicles to Golgi membranes is indeed the result of an enzymatic reaction and not simply nonspecific aggregation, we determined the energy and cytosol requirements of vesicle docking. We used a simplified assay that made use of the different sedimentation behavior of vesicles and cisternal Golgi membranes. Whereas GalT activity in vesicles remains in the supernatant after a medium-speed centrifugation, cisternal Golgi membranes are efficiently pelleted under those conditions. We incubated vesicles or Golgi membranes separately or together. When vesicles were incubated alone on ice or at 37°C, vesicle-derived GalT activity remained in the supernatant (Fig. 3 c, 1. pair). GalT activity of isolated Golgi membranes was efficiently pelleted under those conditions, and only a small amount remained in the supernatant (Fig. 3 c, 2. pair). When both were incubated together on ice, vesicle-derived GalT remained in the supernatant. However, after incubation at 37°C in the presence of cytosol and ATP, approximately two thirds of the vesicle- derived GalT was removed from the supernatant (Fig. 3 c, 3. pair). When ATP was omitted from the incubation, the vesicle-derived GalT activity remained in the supernatant, which demonstrates that vesicle docking requires energy (Fig. 3 c, 4. pair). To determine whether vesicle binding requires cytosol, we partially purified vesicles to remove cytosolic proteins that were also released when cells were permeabilized. Vesicles and Golgi membranes were incubated with the indicated concentrations of CHO cytosol on ice (control) or at 37°C. Golgi membranes and Golgi-bound vesicles were pelleted by centrifugation, and the amount of GalT that remained in the supernatant was determined and expressed as a percentage of the amount present in the control incubation that was kept on ice (Fig. 3 d). (Values greater than 100% are caused by partial fragmentation of cisternal Golgi membranes after incubation at 37°C in the absence of cytosol.) We found that vesicles efficiently bound to cisternal membranes only when cytosol was added.
Secretory Cargo Is Restricted to Cisternal Golgi Membranes
Is secretory cargo similarly found in both vesicles and cisternal membranes? To answer this question, we infected cells with VSV and asked which membranes contained VSV-G, the viral glycoprotein that is a marker for secretory protein transport. We chose VSV-G as our marker for anterograde transport for several reasons: VSV-G is expressed at high levels when cells are infected with VSV, which allows us to detect even small amounts of the protein. The steps in its glycosylation are well characterized (Kornfeld and Kornfeld, 1985), and by analyzing how VSV-G is glycosylated, we can determine whether it is derived from the ER, in transit through the Golgi, or emerging from the trans-side of the Golgi. But most importantly, much of the previous work on transport through the Golgi of mammalian cells studied transport of VSV-G, and there is no doubt that VSV-G moves through the Golgi subcompartments in the cis-to-trans direction.
15B cells were infected with VSV, and Golgi-localized VSV-G was labeled by a 5-min pulse followed by a 10-min chase at 37°C. We found that the GalT distribution between vesicles and cisternae was similar in 15B cells and WT cells and was not affected by the infection; with or without infection, 25–30% of GalT activity was found in the vesicle-containing fractions. Saturating amounts of VSV were used for the infection to assure that all cells were infected, and cells were harvested when they synthesized almost exclusively viral proteins. We homogenized those cells and fractionated the homogenate by velocity sedimentation (Fig. 4 a). The peak of VSV-G distribution was found in fraction 7, overlapping with the peak of Golgi enzyme activity in cisternal membranes. Contrary to our results with resident Golgi enzymes, no second peak of VSV-G–containing vesicles was observed. Only small amounts of VSV-G–containing membranes (∼5% of the total) were found in fractions 2–4. The lack of a peak in their distribution indicates that these membranes are of variable size, as would be expected if they were generated by mechanical fragmentation of larger structures.
We tested whether the VSV-G–containing membranes could fuse with WT Golgi membranes when they were incubated together. Glycosylation of VSV-G could occur as a result of vesicular transport (of VSV-G to WT Golgi membranes, or of the WT enzyme to the VSV-G–containing membranes) or by direct fusion (of WT Golgi with VSV-G–containing membranes). While our results in Fig. 2 indicate that cisternal Golgi membranes are much less efficient than Golgi-derived vesicles in transferring NAGT I to VSV-G–containing 15B Golgi membranes, we could still obtain an efficient glycosylation reaction by adding concentrated Golgi preparations. In the experiments shown in Fig. 2, homogenate was fractionated by velocity sedimentation, and since the cisternal membranes spread out over several fractions, they are more dilute in those fractions than they were in homogenate. The Golgi-enriched membranes that are routinely used in the in vitro Golgi transport assay are prepared by flotation from homogenate on a step gradient that yields a preparation that is much more concentrated.
While VSV-G in fraction 7 and neighboring fractions became Endo H–resistant after incubation with concentrated WT Golgi membranes, the small amount of VSV-G in fractions 2–4 could not be processed (Fig. 4 b). Similar results were obtained when WT Golgi–derived vesicles were added instead of WT Golgi membranes (Fig. 4 c). While VSV-G in fractions 6–8 was efficiently processed, VSV-G in fractions 2–5 was not. We have shown above that the Golgi enzyme containing vesicles could efficiently bind to cisternal Golgi membranes, and we also asked whether the VSV-G–containing small membranes could do the same. However, we did not find any specific association of VSV-G–containing membranes with cisternal Golgi membranes (data not shown).
In conclusion, fusion competent and functional VSV- G–containing membranes are only found in the fractions containing cisternal membranes. The majority of the small VSV-G–containing membranes are not functional and most likely are fragments. However, it is possible that at least some of those membranes are transport vesicles. We estimate that 5–10% of the total VSV-G in a gel lane must be processed to the Endo H–resistant form so that this band is readily detectable over the background. Therefore, at most, 0.25–0.5% of the total VSV-G (5–10% of the VSV-G in fractions 2–4, which is 5% of the total) could be in vesicles that escaped our detection. This is, however, a much smaller amount than the amount of resident Golgi enzymes contained in these fractions, which indicates that VSV-G is depleted from the Golgi enzyme–containing vesicles.
COP I Components Are Required for Vesicular Transport of Resident Golgi Enzymes between Golgi Membranes In Vitro
The data presented so far indicate that the Golgi enzyme– containing vesicles could be involved in retrograde transport. Golgi membranes are capable of producing COP I–coated vesicles, and these vesicles are intermediates in the transport between Golgi subcompartments in vitro (Ostermann et al., 1993). As COP I–coated vesicles are involved in recycling from the Golgi to the ER (Letourneur et al., 1994; Lewis and Pelham, 1996; Gaynor and Emr, 1997), it would seem reasonable to expect that they might also play a role in recycling between Golgi compartments. To determine the relationship between the putative retrograde transport carriers observed in this study and COP I–coated vesicles, and to provide supporting evidence that they might indeed be COP I-coated vesicles, we asked whether they cofractionate. Since the COP I coat is normally rapidly lost after budding, we made use of the inhibitor AlF 4 −. AlF 4 − treatment of cells results in an accumulation of coated vesicles, similar to the effect that is seen when isolated Golgi membranes are incubated with GTPγS and cytosol (Melancon et al., 1987).
After AlF 4 − treatment, cells were homogenized, and Golgi-derived membranes (both vesicles and cisternal membranes) in the homogenate were separated from soluble proteins by flotation followed by velocity sedimentation fractionation. We identified COP I–coated membranes with an antibody against the β-subunit of coatomer (βCOP) and measured GalT activity in each fraction to identify fractions containing Golgi-derived membranes (Fig. 5 a). We found that almost all of the bound βCOP cofractionated with small Golgi-derived vesicles (peak in fraction 3) and that only small amounts of βCOP were found on the cisternal Golgi membranes (peak in fraction 8).
Figure 5.
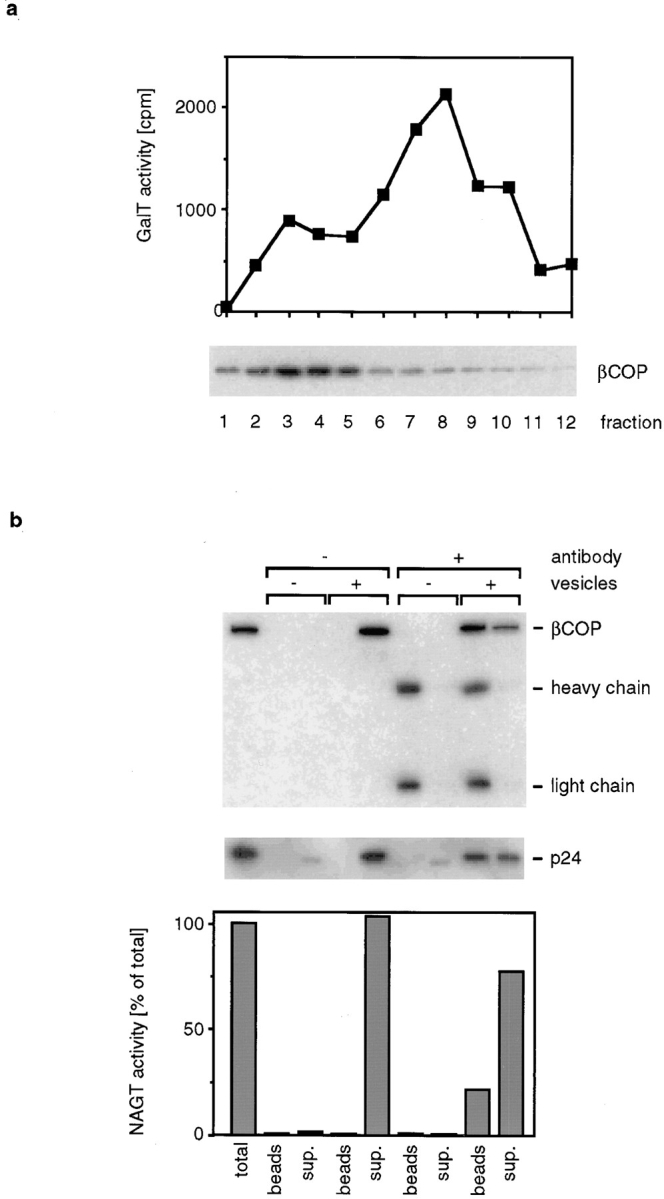
Vesicles cofractionate with COP I–coated vesicles. (a) CHO cells were treated with 50 μM AlF 4 − for 20 min before harvesting and homogenization. Membranes were separated from cytosolic proteins by flotation and further fractionated by velocity sedimentation. Coated membranes were detected by Western blotting with an antibody against the β-subunit of coatomer, and Golgi enzyme–containing membranes were detected by measuring GalT activity. (b) AlF 4 −-treated CHO cells were homogenized, and coated vesicles were isolated as described in Materials and Methods. An aliquot of these vesicles was incubated with M450 magnetic beads that had been preincubated with CM1A10 antibody. In controls, beads were not preincubated with antibody, or vesicles were omitted. After binding, beads were recovered with a magnet, and membranes that remained in the supernatant were pelleted by ultracentrifugation. Half of each sample was dissolved in electrophoresis sample buffer and in NAGT assay buffer. The amount of βCOP and p24 in each fraction was determined by Western blotting. The secondary antibody used to detect the monoclonal antibody bound to βCOP also binds to the heavy and light chain of the CM1A10 antibody. CM1A10 antibody is present in the electrophoresis samples since it is released when antibody-coated beads are extracted with electrophoresis sample buffer. The faint band visible in the p24 blot in the supernatant fractions of the lanes without vesicles is most likely a nonspecific cross reaction of the p24 antibody with milk powder proteins that were added as blocking agent.
To determine whether at least a subset of Golgi enzyme–containing vesicles remained COP I–coated under those conditions, we immunoisolated COP I–coated vesicles using the monoclonal antibody CM1A10 that recognizes the COP I coat (Palmer et al., 1993) (Fig. 5 b). We preincubated magnetic beads with saturating amounts of CM1A10, reisolated the beads to remove free antibody, and incubated the antibody-coated beads with vesicles that were isolated from AlF 4 −-treated cells. After allowing for vesicle binding, the beads were collected with a magnet, and the supernatant was removed. Beads were washed in buffer, and membranes that remained in the supernatant (plus the buffer of the first wash) were pelleted by ultracentrifugation. Half of each sample (vesicle pellet or vesicles bound to beads) was dissolved with electrophoresis sample buffer for electrophoresis and Western blotting, and the other half was used for a NAGT assay. To determine the yield of the immunoisolation, we determined how much of the COP I coat protein βCOP and how much of the COP I vesicle protein p24 (Stamnes et al., 1995) bound to antibody-coated beads. Binding of βCOP was efficient; more than 50% of the total βCOP bound to beads, and approximately half of the p24 present in the vesicle sample bound to the beads. When we determined the amount of NAGT activity that was bound to antibody-coated beads, we found that 15–25% of the NAGT activity bound to the beads when the antibody was added. No binding occurred to control beads that did not have antibody added. Similar results were obtained when GalT activity was measured instead of NAGT activity (data not shown). The efficient coprecipitation of p24 with COP I antibodies shows that the coat remains bound to the membranes during immunoisolation. Therefore, the lower yield of NAGT activity that is immunoprecipitated is most likely not caused by loss of coat proteins from the vesicles but rather indicates that only a subfraction of NAGT activity is in COP I–coated vesicles after AlF 4 − treatment. A possible explanation for this is that AlF 4 − treatment might accumulate mostly vesicles in the ER-to-Golgi pathway and that only a smaller fraction of the Golgi-derived vesicles remains COP I–coated under those conditions. Indeed, we found that under the AlF 4 − treatment conditions used by us, ER to Golgi transport of VSV-G was inhibited, and vesicles containing ER-derived VSV-G accumulated. Processing of VSV-G in the Golgi, however, was not inhibited under those conditions (data not shown).
We asked next whether resident Golgi enzymes can be transported between isolated Golgi stacks in COP I–coated vesicles. For this, we used the established Golgi transport assay (Balch et al., 1984) and asked whether a significant percentage of the signal measured in this assay was recovered when COP I–coated vesicles that budded from WT Golgi membranes were allowed to fuse with 15B Golgi membranes that had been isolated from VSV-infected cells. If COP I–coated vesicles transfer NAGT I between the Golgi stacks in this assay, then delivery of NAGT I to the 15B Golgi would allow glycosylation and incorporation of [3H]GlcNAc into VSV-G.
We followed the previously established protocol to assemble COP I–coated vesicles in vitro (Orci et al., 1993; Ostermann et al., 1993) and used WT Golgi membranes as donor membranes for vesicle formation. Membranes were incubated with CM and mARF in the presence of GTP and palmitoyl-CoA. At the end of the incubation, the salt concentration was increased to assure that no vesicles remained bound, and Golgi membranes were pelleted by centrifugation. The supernatant was mixed with Golgi membranes from VSV-infected 15B cells together with cytosol, ATP, and [3H]UDP GlcNAc. As controls, CM or mARF was omitted from the first incubation. We observed glycosylation of VSV-G only when the preincubation was done in the presence of both CM and mARF (Fig. 6 a), which demonstrates that glycosylation of VSV-G depended on the formation of COP I–coated vesicles in the first incubation. To see how completely the assay signal was reconstituted by the two-stage incubation as compared to an incubation that contained both types of Golgi membranes together, we measured the time course of the fusion reaction. Both the kinetics and the extent of glycosylation were similar when either vesicles were added to 15B Golgi membranes (Fig. 6 b, open squares) or when the same amount of WT Golgi membranes that had been used for the vesicle formation reaction was directly mixed with 15B Golgi membranes (Fig. 6 b, closed squares). We expected similar kinetics since the slow steps of the complete transport reaction are glycosylation of VSV-G and membrane fusion, but not vesicle formation (Taylor et al., 1994). We conclude that enough NAGT I is transported in a COP I–dependent manner between isolated Golgi cisternae to allow for efficient glycosylation of VSV-G.
Figure 6.

Transport of NAGT I between isolated Golgi membranes. (a) WT Golgi membranes were incubated at 37°C with or without CM and mARF as indicated. Golgi membranes were pelleted, and the supernatant was mixed with VSV-G–containing 15B Golgi membranes. Incorporation of [3H]GlcNAc into VSV-G was determined by scintillation counting. (b) WT Golgi membranes (closed squares) or WT Golgi–derived vesicles (supernatant from a budding reaction, open squares) were mixed with VSV- G–containing 15B Golgi. At the indicated times, aliquots were withdrawn, and the amount of VSV-G glycosylation was measured. (c) WT Golgi membranes were incubated for 10 min with budding components on ice or at 37°C. At the end of the incubation, the salt concentration was increased, and the mixture was fractionated by velocity sedimentation. An aliquot from each fraction was mixed with VSV-G–containing 15B Golgi under the conditions of the transport assay. VSV-G glycosylation was determined and expressed as a percent of the signal obtained in a control incubation that contained the amount of Golgi membranes that was used in the budding reaction. (d) NAGT activity in the gradient fractions of the experiment shown in c.
As an alternative to separating the budding reaction into a pellet and supernatant by medium-speed centrifugation, we fractionated the complete reaction by velocity sedimentation. This allowed us to directly compare the activity of slowly sedimenting vesicles with more quickly sedimenting cisternal membranes (Fig. 6 c), similarly to the experiments done with whole cellular homogenate. WT Golgi membranes were incubated with the budding components (CM, mARF, GTP, PalCoA) either on ice or at 37°C. At the end of the incubation, the salt concentration was increased to prevent vesicle binding to cisternal membranes, and the reaction mixture was fractionated by velocity sedimentation. After fractionation, we assayed in each fraction its activity in the transport assay (Fig. 6 c) as well as total NAGT activity (Fig. 6 d).
We found that both with or without incubation, most of the total NAGT activity was found in the bottom fractions of the gradient. However, when we measured the activity of each fraction in the transport assay, a different result was obtained. When the budding reaction was kept on ice, the highest activity in the transport assay was found in the membranes that contained the bulk of the NAGT activity. After incubation at 37°C, however, the peak of activity in the transport assay was found in fraction 4, which contained more slowly sedimenting membranes. It is important to note that the incubation did not result in drastic change in the sedimentation behavior of the bulk NAGT activity. Only a small amount of NAGT was redistributed from the cisternal membranes to the vesicle-containing fractions. This is consistent with previous findings that the bulk of the Golgi membranes stay intact and that only a fraction of the total membranes vesicularize under those conditions (Orci et al., 1993). However, just as we found with the vesicles isolated from cellular homogenate, and as we would expect for a functional transport intermediate, the small amount of NAGT I contained in vesicles was more efficiently transferred to the 15B Golgi membranes than the large amount remaining in cisternal membranes.
Discussion
In this study, we provide evidence that two different types of Golgi enzyme–containing membranes can be found in cellular homogenate. One population represents small vesicles that cofractionate with the known COP I–coated vesicles, whereas the other population fractionates as would be expected for Golgi cisternae. The most striking difference between these two populations was observed in membrane fusion experiments. When either vesicles or cisternal membranes from WT cells were mixed with VSV-G–containing Golgi membranes from the NAGT I–deficient 15B mutant, then the small amount of NAGT I contained in the vesicles glycosylated a larger percentage of VSV-G than the bulk of NAGT I that cofractionated with the cisternal membranes. The NAGT I–containing vesicles did not seem to fuse nonspecifically with all Golgi subcompartments. VSV-G that had just entered the Golgi apparatus, as determined by its processing by the first Golgi processing enzyme Man I, was most efficiently processed by NAGT I in vesicles. This indicates that the NAGT I–containing vesicles fuse preferentially with the cis-side of the Golgi apparatus.
Very different results were obtained when VSV-G in transit through the Golgi was analyzed. Whereas Golgi enzyme–containing vesicles were sufficiently abundant in homogenate to form a separate peak when homogenate was fractionated by velocity sedimentation, no such peak of VSV-G–containing vesicles was found. While some VSV-G was found in the vesicle-containing fractions, the VSV-G–containing membranes appeared to be incompetent to fuse under the conditions tested by us. While Golgi enzyme–containing vesicles efficiently bound to cisternal membranes, the VSV-G–containing membranes did not. These results indicate that the small amount of VSV-G–containing membranes in the vesicle fractions are indeed nonfunctional fragments of Golgi cisternae that are evenly distributed over most of the velocity sedimentation gradient fractions. The functional Golgi enzyme–containing vesicles appear to be depleted of secretory cargo.
We conclude that we have identified a novel type of Golgi-derived transport intermediates, and we propose that these are considered to be retrograde transport carriers. The fractionation behavior of these membranes indicates that they are vesicles, and in the light of our observation that vesicular transport of NAGT I between Golgi membranes in vitro depends on COP I components, it is reasonable to propose that these retrograde transport carriers are indeed COP I–coated vesicles. However, it is important to note that after accumulation of COP I–coated vesicles by AlF 4 − treatment of cells, only a fraction of the vesicle-associated NAGT activity could be immunoprecipitated with coatomer antibodies. This leaves open the possibility that other types of coat proteins might exist, or that our vesicle sample contains more than one type of Golgi-derived vesicles. While we favor the explanation that retrograde transport carriers are COP I–coated vesicles, other hypotheses must be considered. For example, retrograde transport might be mediated by tubular extensions from Golgi cisternae, or the fenestrated rims of Golgi cisternae could play a role in connecting neighboring cisternae (Mellman and Simon, 1992; Weidman et al., 1993). Such tubules or fenestrated membranes might easily fragment into vesicles upon homogenization, and their formation might require COP I components that might form and stabilize the tips of tubular extensions. A full understanding of retrograde transport will require a detailed morphological examination of retrograde transport carriers.
How do we reconcile our failure to identify intra-Golgi anterograde transport vesicles in this study with the in vitro and in vivo evidence that they exist? After all, a single round of vesicle formation and fusion had been reconstituted in vitro by following transport of VSV-G in COP I–coated vesicles (Ostermann et al., 1993). While this earlier study's principal goal was to demonstrate that COP I–coated vesicles are functional intermediates, some have concluded from it that the in vitro Golgi transport assay measures vesicular transport of VSV-G. However, only 5–10% of the VSV-G was incorporated into vesicles, and only one third of this became Endo H–resistant after fusion. The high efficiency of VSV-G processing observed in the experiments shown here cannot be explained by this relatively inefficient transport alone. The study presented here was unbiased towards detecting a specific subclass of transport intermediates but rather defined a pool of vesicles that should in principle contain all different types of vesicles. If Golgi membranes produced vesicles that travel either in the anterograde or retrograde direction, then this pool should contain equal numbers of each. At this time, we can not offer a convincing explanation why anterograde transport vesicles were not detected. They could differ from retrograde vesicles in their mobility or their stability of binding to the Golgi apparatus, or they might be present at different steady-state concentrations because of differences in the kinetics of their formation and consumption. On the other hand, it is appropriate to note that models have been proposed that explain both protein transport through the Golgi and the asymmetric distribution of Golgi enzymes over the Golgi stack and predict retrograde but not anterograde Golgi-derived transport vesicles (Becker et al., 1995; Bannykh and Balch, 1997; Glick et al., 1997).
Acknowledgments
The authors thank Peggy Weidman (St. Louis University, St. Louis, MO) for providing help and reagents to establish the transport assay, Mark Stamnes for p24 antiserum, and Jim Rothman and Thomas Söllner (all from Sloan Kettering Institute, New York) for CM1A10 antibody.
This work was supported by the American Heart Association, Tennessee Affiliate, the Vanderbilt University Research Council, the Diabetes Research and Training Center (Vanderbilt University, pilot and feasibility study), and the Vanderbilt Cancer Center (pilot and feasibility study).
Abbreviations used in this paper
- CM
coatomer
- Endo H
endoglycosidase H
- GalT
galactosyl transferase
- Man I
mannosidase I
- mARF
myristylated ADP ribosylation factor
- NAGT
N-acetylglucosaminyl transferase
- VSV
vesicular stomatitis virus
- VSV-G
VSV glycoprotein
- WT
wild-type
Footnotes
Address all correspondence to Joachim Ostermann, Department of Biochemistry, Vanderbilt University School of Medicine, Nashville, TN 37232-0146. Tel.: (615) 343-3803. Fax: (615) 343-0704. E-mail: ostermj@ctrvax.vanderbilt.edu
References
- Aridor M, Bannykh SI, Rowe T, Balch WE. Sequential coupling between COPII and COPI vesicle coats in endoplasmic reticulum to Golgi transport. J Cell Biol. 1995;131:875–893. doi: 10.1083/jcb.131.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch WE, Dunphy WG, Braell WA, Rothman JE. Reconstitution of the transport of protein between successive compartments of the Golgi measured by the coupled incorporation of N-acetylglucosamine. Cell. 1984;39:405–416. doi: 10.1016/0092-8674(84)90019-9. [DOI] [PubMed] [Google Scholar]
- Bannykh SI, Balch WE. Membrane dynamics at the endoplasmic reticulum Golgi interface. J Cell Biol. 1997;138:1–4. doi: 10.1083/jcb.138.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlowe C, Orci L, Yeung T, Hosobuchi M, Hamamoto S, Salama N, Rexach MF, Ravazzola M, Amherdt M, Schekman R. COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell. 1994;77:895–907. doi: 10.1016/0092-8674(94)90138-4. [DOI] [PubMed] [Google Scholar]
- Becker B, Bolinger B, Melkonian M. Anterograde transport of algal scales through the Golgi complex is not mediated by vesicles. Trends Cell Biol. 1995;5:305–307. doi: 10.1016/s0962-8924(00)89047-9. [DOI] [PubMed] [Google Scholar]
- Brew K, Shaper JH, Olsen KW, Trayer IP, Hill RL. Cross-linking of the components of lactose synthetase with dimethylpimelimidate. J Biol Chem. 1975;250:1434–1444. [PubMed] [Google Scholar]
- Cole NB, Smith CL, Sciaky N, Terasaki M, Edidin M, Lippincott-Schwartz J. Diffusional mobility of Golgi proteins in membranes of living cells. Science. 1996;273:797–801. doi: 10.1126/science.273.5276.797. [DOI] [PubMed] [Google Scholar]
- Cosson P, Letourneur F. Coatomer interaction with di-lysine endoplasmic reticulum retention motifs. Science. 1994;263:1629–1631. doi: 10.1126/science.8128252. [DOI] [PubMed] [Google Scholar]
- Duden R, Griffiths G, Frank R, Argos P, Kreis TE. β-COP, a 110 kd protein associated with non-clathrin-coated vesicles and the Golgi complex, shows homology to β-adaptin. Cell. 1991;64:649–665. doi: 10.1016/0092-8674(91)90248-w. [DOI] [PubMed] [Google Scholar]
- Gaynor EC, Emr SD. COP I–independent anterograde transport—cargo selective ER to Golgi protein transport in yeast COP I mutants. J Cell Biol. 1997;136:789–802. doi: 10.1083/jcb.136.4.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glick BS, Elston T, Oster G. A cisternal maturation mechanism can explain the asymmetry of the Golgi stack. FEBS Lett. 1997;414:177–181. doi: 10.1016/s0014-5793(97)00984-8. [DOI] [PubMed] [Google Scholar]
- Gottlieb C, Baenziger J, Kornfeld S. Deficient uridine diphosphate-N-acetylglucosamine:glycoprotein N-acetylglucosaminyltransferase activity in a clone of Chinese hamster ovary cells with altered surface glycoproteins. J Biol Chem. 1975;250:3303–3309. [PubMed] [Google Scholar]
- Graham TR, Krasnov VA. Sorting of yeast α1,3 mannosyltransferase is mediated by a lumenal domain interaction, and a transmembrane domain signal that can confer clathrin-dependent Golgi localization to a secreted protein. Mol Biol Cell. 1995;6:809–824. doi: 10.1091/mbc.6.7.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris SL, Waters MG. Localization of a yeast early Golgi mannosyltransferase, Och1p, involves retrograde transport. J Cell Biol. 1996;132:985–998. doi: 10.1083/jcb.132.6.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoe MH, Slusarewicz P, Misteli T, Watson R, Warren G. Evidence for recycling of the resident medial/trans-Golgi enzyme, N-acetylglucosaminyltransferase I, in ldlD cells. J Biol Chem. 1995;270:25057–25063. doi: 10.1074/jbc.270.42.25057. [DOI] [PubMed] [Google Scholar]
- Hosobuchi M, Kreis T, Schekman R. SEC21 is a gene required for ER to Golgi protein transport that encodes a subunit of a yeast coatomer. Nature. 1992;360:603–605. doi: 10.1038/360603a0. [DOI] [PubMed] [Google Scholar]
- Kaiser CA, Schekman R. Distinct sets of SEC genes govern transport vesicle formation and fusion early in the secretory pathway. Cell. 1990;61:723–733. doi: 10.1016/0092-8674(90)90483-u. [DOI] [PubMed] [Google Scholar]
- Kornfeld R, Kornfeld S. Assembly of asparagine-linked oligosaccharides. Annu Rev Biochem. 1985;54:631–664. doi: 10.1146/annurev.bi.54.070185.003215. [DOI] [PubMed] [Google Scholar]
- Letourneur F, Gaynor EC, Hennecke S, Demolliere C, Duden R, Emr SD. Coatomer is essential for retrieval of dilysine-tagged proteins to the endoplasmic reticulum. Cell. 1994;79:1199–1207. doi: 10.1016/0092-8674(94)90011-6. [DOI] [PubMed] [Google Scholar]
- Lewis MJ, Pelham HR. SNARE-mediated retrograde traffic from the Golgi complex to the endoplasmic reticulum. Cell. 1996;85:205–215. doi: 10.1016/s0092-8674(00)81097-1. [DOI] [PubMed] [Google Scholar]
- Malhotra V, Serafini T, Orci L, Shepherd JC, Rothman JE. Purification of a novel class of coated vesicles mediating biosynthetic protein transport through the Golgi stack. Cell. 1989;58:329–336. doi: 10.1016/0092-8674(89)90847-7. [DOI] [PubMed] [Google Scholar]
- Melancon P, Glick BS, Malhotra V, Weidman PJ, Serafini T, Gleason ML, Orci L, Rothman JE. Involvement of GTP-binding “G” proteins in transport through the Golgi stack. Cell. 1987;51:1053–1062. doi: 10.1016/0092-8674(87)90591-5. [DOI] [PubMed] [Google Scholar]
- Mellman I, Simons K. The Golgi complex: in vitro veritas? . Cell. 1992;68:829–840. doi: 10.1016/0092-8674(92)90027-A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson T, Warren G. Retention and retrieval in the endoplasmic reticulum and the Golgi apparatus. Curr Opin Cell Biol. 1994;6:517–521. doi: 10.1016/0955-0674(94)90070-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson T, Pypaert M, Hoe MH, Slusarewicz P, Berger EG, Warren G. Overlapping distribution of two glycosyltransferases in the Golgi apparatus of HeLa cells. J Cell Biol. 1993;120:5–13. doi: 10.1083/jcb.120.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orci L, Glick BS, Rothman JE. A new type of coated vesicular carrier that appears not to contain clathrin: its possible role in protein transport within the Golgi stack. Cell. 1986;46:171–184. doi: 10.1016/0092-8674(86)90734-8. [DOI] [PubMed] [Google Scholar]
- Orci L, Palmer DJ, Amherdt M, Rothman JE. Coated vesicle assembly in the Golgi requires only coatomer and ARF. Nature. 1993;364:732–734. doi: 10.1038/364732a0. [DOI] [PubMed] [Google Scholar]
- Orci L, Stamnes M, Ravazzola M, Amherdt M, Perrelet A, Söllner TH, Rothman JE. Bidirectional transport by distinct populations of COP I-coated vesicles. Cell. 1997;90:335–349. doi: 10.1016/s0092-8674(00)80341-4. [DOI] [PubMed] [Google Scholar]
- Ostermann J, Orci L, Tani K, Amherdt M, Ravazzola M, Elazar Z, Rothman JE. Stepwise assembly of functionally active transport vesicles. Cell. 1993;75:1015–1025. doi: 10.1016/0092-8674(93)90545-2. [DOI] [PubMed] [Google Scholar]
- Palade G. Intracellular aspects of the process of protein synthesis. Science. 1975;189:347–358. doi: 10.1126/science.1096303. [DOI] [PubMed] [Google Scholar]
- Palmer DJ, Helms JB, Beckers CJ, Orci L, Rothman JE. Binding of coatomer to Golgi membranes requires ADP-ribosylation factor. J Biol Chem. 1993;268:12083–12089. [PubMed] [Google Scholar]
- Pelham HR. Evidence that luminal ER proteins are sorted from secreted proteins in a post-ER compartment. EMBO (Eur Mol Biol Organ) J. 1988;7:913–918. doi: 10.1002/j.1460-2075.1988.tb02896.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJM, Lippincott-Schwartz J. ER-to-Golgi transport visualized in living cells. Nature. 1997;389:81–84. doi: 10.1038/38001. [DOI] [PubMed] [Google Scholar]
- Rexach MF, Latterich M, Schekman RW. Characteristics of endoplasmic reticulum–derived transport vesicles. J Cell Biol. 1994;126:1133–1148. doi: 10.1083/jcb.126.5.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman JE. The reconstitution of intracellular protein transport in cell-free systems. Harvey Lect. 1990;86:65–85. [PubMed] [Google Scholar]
- Rothman JE, Miller RL, Urbani LJ. Intercompartmental transport in the Golgi complex is a dissociative process: facile transfer of membrane protein between two Golgi populations. J Cell Biol. 1984;99:260–271. doi: 10.1083/jcb.99.1.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafini T, Stenbeck G, Brecht A, Lottspeich F, Orci L, Rothman JE. A coat subunit of Golgi-derived non-clathrin-coated vesicles with homology to the clathrin-coated vesicle coat protein β-adaptin. Nature. 1991;349:215–220. doi: 10.1038/349215a0. [DOI] [PubMed] [Google Scholar]
- Sonnichsen B, Watson R, Clausen H, Misteli T, Warren G. Sorting by COP I–coated vesicles under interphase and mitotic conditions. J Cell Biol. 1996;134:1411–1425. doi: 10.1083/jcb.134.6.1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamnes MA, Craighead MW, Hoe MH, Lampen N, Geromanos S, Tempst P, Rothman JE. An integral membrane component of coatomer-coated transport vesicles defines a family of proteins involved in budding. Proc Natl Acad Sci USA. 1995;92:8011–8015. doi: 10.1073/pnas.92.17.8011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor TC, Kanstein M, Weidman P, Melancon P. Cytosolic ARFs are required for vesicle formation but not for cell-free intra-Golgi transport: evidence for coated vesicle-independent transport. Mol Biol Cell. 1994;5:237–252. doi: 10.1091/mbc.5.2.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vischer P, Hughes CR. Glycosyl transferases of baby-hamster-kidney (BHK) cells and ricin-resistant mutants. Eur J Biochem. 1981;117:275–284. doi: 10.1111/j.1432-1033.1981.tb06334.x. [DOI] [PubMed] [Google Scholar]
- Waters MG, Serafini T, Rothman JE. “Coatomer”: a cytosolic protein complex containing subunits of non-clathrin-coated Golgi transport vesicles. Nature. 1991;349:248–251. doi: 10.1038/349248a0. [DOI] [PubMed] [Google Scholar]
- Weidman P, Roth R, Heuser J. Golgi membrane dynamics imaged by freeze-etch electron microscopy: views of different membrane coatings involved in tubulation versus vesiculation. Cell. 1993;75:123–133. [PubMed] [Google Scholar]