

Abstract
Newly synthesized membrane proteins are transported by fast axonal flow to their targets such as the plasma membrane and synaptic vesicles. However, their transporting vesicles have not yet been identified. We have successfully visualized the transporting vesicles of plasma membrane proteins, synaptic vesicle proteins, and the trans-Golgi network residual proteins in living axons at high resolution using laser scan microscopy of green fluorescent protein-tagged proteins after photobleaching. We found that all of these proteins are transported by tubulovesicular organelles of various sizes and shapes that circulate within axons from branch to branch and switch the direction of movement. These organelles are distinct from the endosomal compartments and constitute a new entity of membrane organelles that mediate the transport of newly synthesized proteins from the trans-Golgi network to the plasma membrane.
Neurons synthesize membrane proteins in the cell body and convey them by fast axonal transport to their appropriate targets, such as the growth cone plasma membrane and synaptic vesicles. Accumulating evidence suggests that this transport is mediated by microtubule-based kinesin superfamily proteins (Hirokawa, 1996). However, the dynamics of these proteins during transport has not been revealed and the vesicles transporting these membrane proteins have not yet been identified. For example, until now we did not know whether synaptic vesicle proteins are transported in axons on spherical small clear vesicles just like the synaptic vesicles to the nerve terminals. More generally, except for the peptide-containing secretory vesicles that are stored in cells (Jung and Scheller, 1991), transporting vesicles from the TGN to the cell surface are largely unknown, not only in neurons, but in any kind of cells. There are several biochemical approaches for purification of the transporting vesicles using various types of cells (Walworth and Novick, 1987; Bennett et al., 1988; deCurtis and Simons, 1989), but lack of appropriate marker proteins makes it difficult to identify the vesicles in cells. Judging from the importance of the pathway from the TGN to the cell surface in cellular membrane trafficking, identification of the transporting vesicles from the TGN to the cell surface is necessary. Neurons are suitable for this purpose because the transporting vesicles have to travel a long distance in axons before they arrive at their destinations (Grafstein and Forman, 1980).
Previously axonal membrane transport was visualized by video-enhanced contrast differential interference contrast (VEC-DIC)1 video microscopy systems (Allen et al., 1982). This technique enabled the visualization of organelle transport in living axons and led to the discovery of the kinesin motor protein (Brady et al., 1985; Vale et al., 1985). However, this technique cannot be used to identify the molecular components of each vesicle. Moreover, this technique is limited with respect to visualizing small vesicles densely packed in the cytosol (Seiler et al., 1997), because adjacent structures interfere with the imaging of small vesicles. Fluorescence tracers have been applied to the visualization of endocytotic vesicles such as endosomes or lysosomes (Hollenbeck and Bray, 1987; Hollenbeck, 1993), but they cannot be applied to the visualization of anterogradely transported vesicles that transport newly synthesized membrane proteins. It is difficult to identify transporting vesicles by immunocytochemistry of fixed samples because the fraction of membrane proteins being transported is 10 or 100 times less than the fraction that has already reached targets such as the plasma membrane or synaptic vesicles, and accordingly, the level of staining of transporting vesicles is often below background levels. In addition, after fixation of the cells, we cannot tell whether the vesicles were actually moving or were stationary at the time of fixation. These technical limitations have prohibited the identification of the vesicles transporting newly synthesized proteins. We have overcome these difficulties by developing a new method combining adenovirus-mediated gene transfer, green fluorescent protein (GFP) technology, laser scan microscopy with photomultiplier detection, and laser photobleaching and have succeeded in visualizing the transporting vesicles of both synaptic vesicle and plasma membrane proteins at high resolution and revealing the dynamics of these proteins in real time in living axons.
Using this approach, we identified tubulovesicular organelles as comprising a general transport machinery for newly synthesized proteins in living axons. These vesicles transport plasma membrane proteins, synaptic vesicle proteins, and TGN residual proteins from the TGN to the plasma membrane. We revealed their dynamics in living axons, such as switching of the direction of movement and division of the vesicles. We also detected the transport of proteins between axon branches. These results indicate that the tubulovesicular organelles are not simply transported unidirectionally to targets, but are circulated within axons until they fuse with the plasma membrane. In addition, our findings contribute to the clarification of two fundamental issues in neuronal cell biology; (a) the elucidation of synaptic vesicle formation by directly showing that synaptic vesicle precursors are transported not by endosomal compartments but by tubulovesicular organelles, indicating that the synaptic vesicles are synthesized locally by recycling at the nerve terminal; and (b) the elucidation of the mechanisms of axonal growth by showing that the supply of plasma membrane precursors by the tubulovesicular organelles is sufficient for axonal growth, and that circulation of the tubulovesicular organelles enables efficient use of newly synthesized proteins at the growing tip of bifurcating axons.
Materials and Methods
Recombinant Adenovirus Construction
Rat GAP-43 (Karns et al., 1987), mouse SNAP-25 (Oyler et al., 1989), rat synaptophysin (Sudhof et al., 1987), and rat TGN-38 (Luzio et al., 1990) cDNAs were cloned by RT-PCR and the sequences were confirmed by sequencing. TrkA cDNA was kindly provided by Dr. E.M. Shooter (Stanford University, Stanford, CA) and Dr. T. Hunter (Salk Institute, LaJolla, CA; Meakin et al., 1992). Chimeric cDNAs were constructed to encode full-length GAP-43–S65T-mutated GFP-flag tag (cDNA of S65T-mutated GFP was kindly provided by Dr. R. Tsien, University of California San Diego, La Jolla, CA), full-length SNAP-25–S65T-mutated GFP-flag tag, full-length synaptophysin linker–EGFP (CLONTECH Laboratories, Inc., Palo Alto, CA) HA tag, full-length trkA linker–EGFP-flag tag, and full-length TGN-38–EGFP. All the constructs were made to fuse the GFP molecules with the COOH-terminal of the membrane proteins with/without the intervening linker sequence (see Fig. 1). The aminoacid sequence of the linker was KGVEPKTYCYYSS (Grote et al., 1995). In some constructs, flag-tag (DYKDDDDK) or HA-tag (CYPYDVPDYASL) was added to the COOH-terminal of the GFP. The cDNAs were subcloned into pAdexCAw1 at the SwaI site and cotransfected with viral genome fragments into HEK 293 cells (CRL 1573; American Type Culture Collection [ATCC], Rockville, MD; Miyake et al., 1996). The recombinant adenoviruses were amplified and CsCl-purified (Kanegae et al., 1994). They were then transfected into mouse dorsal root ganglion (DRG) cells over a period of 60 min, 3 h after plating of the cells.
Figure 1.

Schematic diagrams of membrane protein–GFP fusion proteins used in this study. GAP-43 and SNAP-25 do not contain transmembrane domain, but are associated with the plasma membrane by lipid moiety at their NH2-terminal and central regions, respectively. Synaptophysin has four transmembrane domains and its COOH-terminal region is located on the cytoplasmic side of the synaptic vesicles. TrkA and TGN-38 contain one transmembrane domain and their COOH-terminal regions are located on the cytoplasmic side of the plasma membrane and the TGN. GFPs were fused with the COOH-terminal region of these membrane proteins, and thus located on the cytoplasmic sides.
Cell Culture
DRG cells were isolated from adult mice according to the method that was originally developed for rabbit DRG neurons (Goldenberg and De Boni, 1983). In brief, DRGs were dissected from the lumbar region of a 4-wk-old C57 black mouse, dissociated by sequential protease treatments (0.25% collagenase for 1 h and 0.25% trypsin for 15 min at 37°C) followed by trituration with a Pasture pipette, and then plated onto glass coverslips coated with laminin. The culture medium consisted of DME medium containing 5% new calf serum, 5% horse serum, and 1 ng/ml NGF (Takara, Shiga, Japan). For synaptic stimulation, 3 nM of latrotoxin (Alomone Labs, Jerusalem, Israel) was added to normal Ringer solution containing 4 mM EGTA (Ceccarelli and Hurlbut, 1980).
Microscopy and Data Analysis
The cells were examined 40 h after infection. The medium was changed to DME medium, not containing phenol red, but supplemented with 10 mM Hepes (pH 7.3). The coverslips were mounted onto slide glasses using Scotch tape as a spacer and sealed with Valap (vaseline/lanolin/paraffin 1:1:1). Scanning fluorescence images were acquired using the MRC1000-confocal laser scanning unit (Bio-Rad Labs., Hercules, CA) coupled to a Zeiss Axioplan microscope equipped with a Zeiss ×40, 1.3 NA oil-immersed objective, or the MRC1024 confocal laser scanning unit coupled to a Zeiss Axiovert100 microscope equipped with a Zeiss ×40, C-APO (Carl Zeiss, Inc., Thornwood, NY). The axons were excited using the 488-nm wavelength of a krypton–argon laser. Typically, the axons were first bleached by continuous scanning for ∼1 min with 100% laser power at zoom 6.0 and then observed under 30 or 10% laser power at zoom 4.0. Data were collected without averaging at a single focal plane with the confocal aperture fully open at a resolution of 768 × 512 pixels, and the time required between frames was 3.45 s unless otherwise specified. The cells were examined at room temperature.
The speed of the vesicles was determined by measuring the distance moved between two successive frames (3.45 s). When we follow the vesicle movements, the vesicles change their speed. But we chose two successive frames from one-time series data for the statistical analysis of the speed of vesicles to avoid double counting of the same vesicles.
To estimate the anterograde membrane flux, we measured the increase in the area of the axon occupied by the transporting vesicles between two successive frames. Considering that two-dimensional images obtained by microscopy are the projection of images of tubular and spherical vesicles to the horizontal plane, we could estimate the increase in the total surface area of vesicles from the two-dimensional images. For this purpose, the vesicles were considered to be cylindrical in shape. If each vesicle is a cylinder with diameter di and height hi (i = 1, 2,...), then the area occupied by the vesicles on the monitor (S) is approximated as S = Σ (di × hi), while the total surface area T = Σ (lateral surface of cylinder + upper and lower surfaces) = Σ (πdi × hi + πdi 2/4) = πS + Σ (πdi 2/4) > πS. If we assume that all of the vesicles are spherical, T = 4S. Thus the actual value for the net surface area (T) should be larger than πS, because the vesicles comprised a mixture of cylinder-like and spherical vesicles. We calculated the net membrane flow as the increase rate of πS.
Double Label Experiments
For the double labeling of DRG cells with transporting vesicles and mitochondria, the cells were treated with Mitotracker for 10 min, and then washed with the culture medium (Nakata and Hirokawa, 1995). For FM1-43 labeling, cells were treated with 15 μM FM1-43 (Molecular Probes, Inc., Eugene, OR) in high potassium Ringer solution (31.5 mM NaCl, 90 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 30 mM glucose, 25 mM Hepes, pH 7.3) for 10 min, and then washed 3× over a 15-min period with normal Ringer solution (119 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 30 mM glucose, 25 mM Hepes, pH 7.3; Betz et al., 1992; Liu and Tsien, 1995). In some experiments, DRG cells were incubated with culture medium containing 15 μM FM1-43 overnight and then washed with the culture medium not containing FM1-43. For the double labeling with endocytotic tracers, the cells were treated with 1 mg/ml Texas red–dextran (mol wt 3,000; Molecular Probes, Inc.) overnight and washed 2× over 10 min with the culture medium. For double labeling with the SV2 antibody, the cells were fixed with 2% paraformaldehyde in PBS for 30 min, rinsed with PBS, and then permeabilized with 0.02% saponin in PBS for 10 min. After incubation with 2% bovine serum albumin in PBS, the cells were incubated with a 1:200 dilution of anti-SV2 monoclonal antibody followed by incubation with a rhodamine-conjugated secondary antibody. For all the double labeling experiments, the cells were excited using a Kr/Ar laser (488 + 568 nm) and detected using FITC and Texas red filters. Except for the fixed samples, the movements of the vesicles that were labeled by either or both markers were observed simultaneously in real time.
Correlative Electron Microscopy
For correlative electron microscopy, DRG neurons were cultured on cellocate (Eppendorf-Netheler-Hinz GmbH, Hamburg, Germany). They were infected with adenovectors and observed by laser scan microscopy. A living axon was photobleached and the extent of vesicle transport was ascertained. The axon was again photobleached and the medium was exchanged for a solution of 2% paraformaldehyde and 0.1% glutaraldehyde in 0.1 M phosphate buffer, pH 7.3. After 10 min, the fixative was washed away by PBS, and the axon was observed by laser scan microscopy. Cells were again fixed with a solution of 2% paraformaldehyde and 2.5% glutaraldehyde in 0.1 M phosphate buffer, pH 7.3, for 1 h and processed for conventional electron microscopy. The photobleached axon was identified using the marks on the cellocate, and the same axon was observed using a JEOL 2000EX electron microscope (JEOL, Inc., Tokyo, Japan) at 80 kV.
Immunogold Electron Microscopy
DRG neurons 40 h after the infection with adenovectors were fixed with a solution of 3% paraformaldehyde in 0.1 M phosphate buffer, pH 7.3, for 30 min at room temperature. The cells were cryoprotected with 4% sucrose and 0.1 M phosphate buffer for 10 min; 10% sucrose and 0.1 M phosphate buffer for 30 min; 20% sucrose and 0.1 M phosphate buffer for 30 min; and 30% sucrose and 0.1 M phosphate buffer for 2 h at room temperature. Then the cells were freeze thawed with liquid freon and incubated with 20% sucrose and 0.1 M phosphate buffer for 10 min; 10% sucrose and 0.1 M phosphate buffer for 10 min; and 4% sucrose and 0.1 M phosphate buffer for 10 min. After washing with PBS, the cells were blocked with 2% BSA–PBS for 30 min, and incubated with 1:20 anti-flag M2 monoclonal antibody (Eastman Kodak Co., Rochester, NY) in the blocking buffer for 3 h at 37°C. The cells were washed 4× with PBS, blocked with 2% BSA–TBS, pH 8.2, for 30 min, and incubated with 1:10 5-nm colloidal gold–conjugated anti–mouse IgG (Amersham Corp., Arlington Heights, IL) in 2% BSA-TBS for overnight at 4°C and 2 h at 37°C. After extensive washing with TBS, the cells were fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer, pH 7.4. The cells were postfixed with 0.5% osmium tertaoxide and 0.8% potassium ferrocyanide in 0.1 M cacodylate buffer for 10 min on ice. Then they were stained with 2.5% uranyl-acetate for 2 h at room temperature, dehydrated, and then embedded in Epon 812. Thin sections were stained with uranyl acetate and lead citrate and examined with a JEOL 2000EX electron microscope at 80 kV.
Results
Targeting of GFP Fusion Proteins in Mouse DRG Neurons
First, we tested whether the membrane protein–GFP fusion proteins are correctly targeted in mouse DRG neurons. 40 h after infection with recombinant adenovirus, we examined DRG neurons by confocal laser scan microscopy. Adenovirus vectors transduced DRG neurons in tissue culture without significant effects on neuronal viability. GAP-43 is a growth cone–associated protein that is localized on the plasma membrane of growth cones (Meiri et al., 1986; Skene, 1989). The GAP-43–GFP fusion protein was targeted to the growth cone (Fig. 2 a), and a confocal slice observed under higher magnification revealed that the protein was localized on the plasma membrane at the growing neurites (Fig. 2 d). Synaptosome-associated protein 25 (SNAP-25) is a soluble NSF attachment protein receptor at the target membrane (t-SNARE) that is localized on the plasma membrane and involved in the docking and fusion of synaptic vesicles to the plasma membrane (Sollner et al., 1993). The SNAP-25–GFP fusion protein was localized in the distal axon (Fig. 2 b), quite similar to the reported localization of endogenous SNAP-25 in nerve cells (Garcia et al., 1995). TrkA, a high affinity receptor for NGF, is expressed on the plasma membrane and internalized after binding to NGF (Klein et al., 1991; Meakin et al., 1992; Ehlers et al., 1995). The trkA-GFP fusion protein was localized on the plasma membrane and accumulated in the growing tips of axons (Fig. 2 c).
Figure 2.
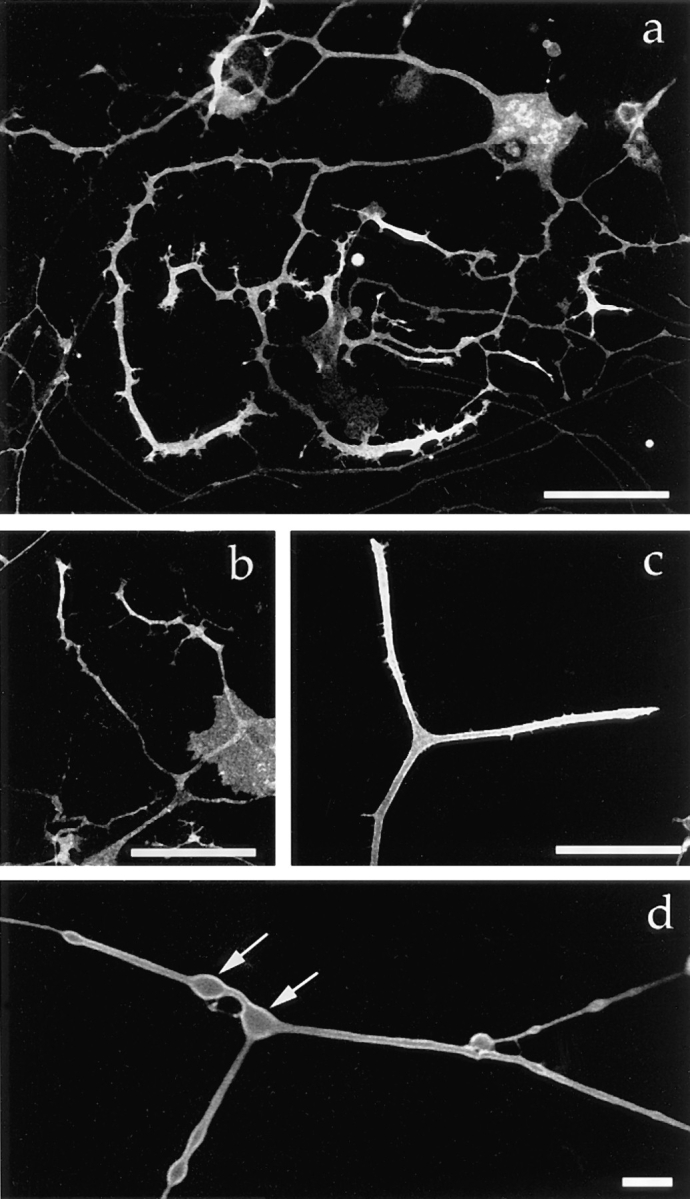
Targeting of the plasma membrane protein–GFP fusion proteins in mouse DRG neurons. 40 h after infection with each recombinant virus, living cells were examined by confocal laser scan microscopy. (a) GAP-43–GFP fusion protein. (b) SNAP-25–GFP fusion protein. (c) TrkA–GFP fusion protein. The proteins were concentrated in the growthcones. (d) Higher magnification of a confocal slice showing that GAP-43–GFP is localized on the plasma membrane. Arrows indicate varicosities. Bars: (a–c) 50 μm; (d) 5 μm.
In contrast to the above proteins, synaptophysin is a synaptic vesicle integral membrane protein (Sudhof et al., 1987). The synaptophysin–GFP fusion protein was abundant in varicosities (Fig. 3 a) and was colocalized with endogenous synaptic vesicle protein SV2 (Fig. 3, c and d; Bajjalieh et al. 1992; Feany et al., 1992). Observation of a confocal slice at high magnification confirmed that synaptophysin– GFP was localized inside neurites, especially in varicosities (Fig. 3 b). To test whether synaptophysin–GFP is exocytosed on stimulation, we added latrotoxin in the presence of EGTA to the medium, causing massive synaptic vesicle secretion and inhibiting endocytotic recycling (Ceccarelli and Hurlbut, 1980), as a result of which almost all the synaptic vesicles were fused with the plasma membrane (Valtorta et al., 1988; Torri-Tarelli et al., 1990). Synaptophysin–GFP redistributed onto the plasma membrane upon stimulation, resulting in an increase in the surface area of varicosities (Fig. 3, e and f), demonstrating that synaptophysin–GFP was exocytosed on the stimulation that is specific to synaptic vesicle secretion (Ushkaryov et al., 1992).
Figure 3.
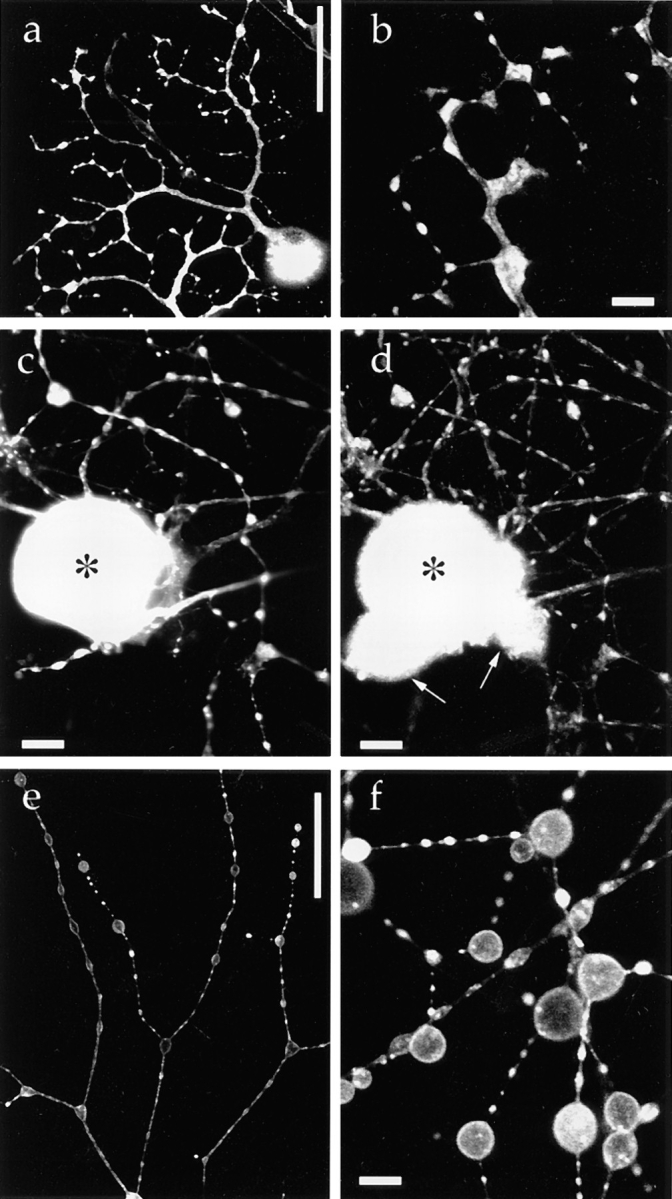
Targeting of a synaptic vesicle protein–GFP fusion protein in mouse DRG neurons. 40 h after infection with adenovirus vector carrying synaptophysin–GFP chimeric DNA, living cells were examined using confocal laser scan microscopy unless otherwise indicated. (a) Low magnification. Synaptophysin–GFP is accumulated in varicosities. (b) Confocal slice observed at higher magnification revealed that synaptophysin–GFP is localized in the cytoplasm in varicosities. (c and d) Double labeling of fixed DRG cells with synaptophysin–GFP (c) and endogenous synaptic vesicle protein SV2 (d). In the clump of cell bodies, only the cell indicated by (*) expressed synaptophysin–GFP, whereas the other cell bodies (arrows) were not labeled by GFP. Axons and cell bodies that expressed synaptophysin–GFP exhibited the colocalization of synaptophysin–GFP and SV2. (e and f) Synaptophysin–GFP was exocytosed onto the plasma membrane after stimulation with 3 nM latrotoxin and EGTA. Synaptophysin– GFP redistributed in the plasma membrane of varicosities. Because synaptic vesicles were fused with the plasma membrane, the total surface area of the varicosities was markedly increased. Bars: (a and e) 50 μm; (c and d) 10 μm; (b and f) 5 μm.
TGN-38 is an integral membrane protein that is mainly present on the TGN (Reaves et al., 1992; Banting and Ponnambalam, 1997). It is known that the protein recycles between the TGN and the cell surface, and a small fraction of it is localized on the cell surface (Reaves et al., 1993). TGN-38–GFP has been shown to be localized on the TGN (Girotti and Banting, 1996). TGN-38–GFP was localized as clusters of patches within the cell body (Fig. 4), which is quite similar to the reported distribution of TGN in neurons (Krijnse-Locker et al., 1995). We found that a small fraction of TGN-38–GFP is localized on the plasma membrane of DRG neurons. These results demonstrate that GAP-43–GFP, SNAP-25–GFP, trkA–GFP, synaptophysin– GFP, and TGN-38–GFP are correctly targeted in mouse DRG neurons, and that these fusion proteins can be used to further study the transport of membrane proteins in neurons.
Figure 4.

Targeting of the TGN residual protein–GFP fusion protein in mouse DRG neurons. 40 h after infection with the adenovirus vector carrying TGN-38–GFP chimeric DNA, the living cells were examined by serial Z-sectioning and the maximum Z projection was reconstituted. Most of the TGN-38–GFP molecules were localized within the cell body as perinuclear clusters. In addition, we observed fine dots in the cytoplasm (note that they are not the transporting vesicles). We found that a small amount of the TGN-38–GFP was expressed on the plasma membrane of the cell body and the axons. Bar, 50 μm.
Transporting Vesicles Were Visualized by Laser Scan Microscopy after Photobleaching
To visualize transporting vesicles, we examined the axons using laser scan microscopy with photomultiplier detection. Vesicles will move at a speed of ∼1 μm/s in the fast axonal transport, and it takes ∼1 s to scan a full-frame image of 768 × 512 pixels at normal scan speed by the laser scan unit. Thus, integration of images is not appropriate because vesicles move several micrometers during collecting the images. However, a single scan by normal scan speed is fast enough to get the image of moving vesicles free from the obvious effect of the vesicle movement. For example, a vesicle that is 1 μm in the longitudinal direction of the monitor (this direction will cause the maximum errors due to slow scan speed) can be scanned within 20 ms at normal scan speed with a full-frame image at zoom factor 4.0 with ×40 objectives (in this typical setting in this study, 512 pixels on the monitor approximately correspond to 75 μm). Then, if the vesicle was moving at a speed of 1 μm/s, the error of the measurement due to the vesicle motion during the scan is 1 μm/s × 0.02 s = 0.02 μm, which is below the resolution of the microscopy. Thus, efforts were made to get less noisy and more bright image by a single scan.
We fully opened the confocal aperture to obtain the maximum amount of light from the sample (Terasaki and Dailey, 1995). When we amplified the gain of the photomultiplier, signals from the GFP-fusion proteins on the plasma membrane etc. interfered with the detection of the transporting vesicles in the cytoplasm. To overcome this problem, we photobleached a small area of an axon at 100% laser power and then performed a time series observation of the area at lower laser power. The vesicles that initially existed in the irradiated area became invisible as a result of the photobleaching, but fluorescent vesicles continuously moved into the area from the adjacent unirradiated area of the axon. This enabled us to detect moving vesicles that were only slightly fluorescent. The photobleaching method has merit above other possible solutions to the problem such as accumulating the marker proteins in the TGN or the Golgi apparatus, because it does not require the perturbation of the cellular membrane trafficking and it can be applied to any kind of the membrane proteins, thus it is possible to observe natural transporting phenomena in cells. After a few minutes, the observed area became densely packed with the vesicles that continuously moved into the area (Fig. 5, o–r). Then, we again photobleached the area and observed the vesicles newly invading the area. Repeated photobleaching did not affect the transport of the vesicles. Simultaneous DIC observation showed normal vesicle movement (Fig. 5, e–l). Axonal growth was confirmed in the photobleached axon (Fig. 5, a–d). Electron microscopy of the photobleached area did not reveal any trace of structural damage (see Fig. 12). Because only a small part of the cell was irradiated, it did not affect cell viability, and vigorous transport of vesicles was observed in the same area of the axon over a period of 2–3 h or more during which time several hundred images were taken and photobleaching was carried out more than 10 times.
Figure 5.
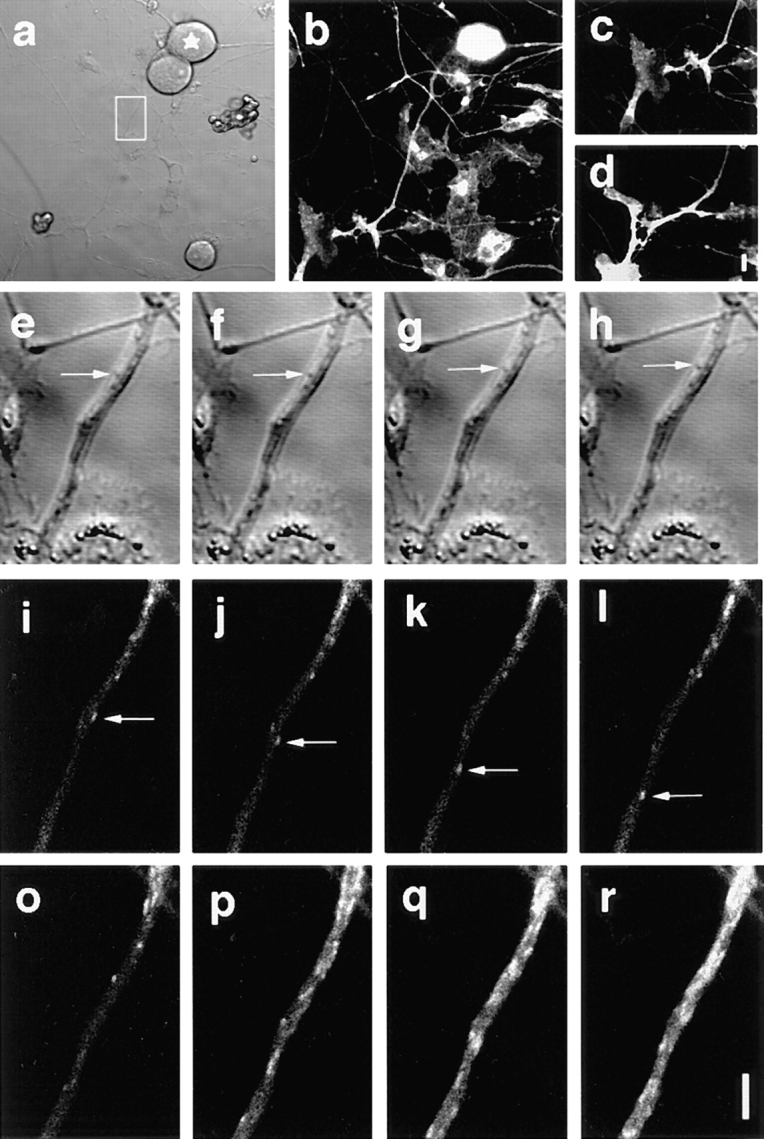
Visualization of the GAP-43–GFP-transporting vesicles after photobleaching of axons. Mouse DRG neurons were cultured for 40 h after infection with the recombinant adenovirus that carry GAP-43–GFP fusion construct. DIC image (a) and simultaneous GFP image (b) of a DRG neuron before photobleaching. The axon in the boxed area in a was repeatedly photobleached and observed (e–r) for ∼ 1 h. The star in a indicates the cell body of the neuron. c presents the lower left side of b. The growth-cone of the axon was brightly stained with GAP-43–GFP. d shows the same area after 1 h observartion. Note that the axon grows more than 10 μm in spite of the repeated photobleaching and the image collection (the data in e–r were collected while the axon was growing from c to d). e–h and i–l show the simultaneous DIC and GFP images of the boxed area of a immediately after photobleaching. Interval between the frames was 2.35 s. Vesicle movements were observed in both DIC images (e–h, arrows) and GFP images (i–l, arrows). Note that the vesicle movement that was observed by GFP images was not seen in DIC images, although the images were collected simultaneously. o–r show that the significant amount of the vesicles are transported in the axon. Intervals between the frames were 60 s. o is immediately after photobleaching. Several vesicles moved into the photobleached area from the proximal side. After 3 min (r), the photobleached area was filled with the transporting vesicles, and it became difficult to trace each individual vesicle. When the observing area was jammed with vesicles as seen in r, the area was photobleached completely by 100% laser, and again observed. These series of experiments (o–r) were repeated for more than 10× within the 1 h. Bars: (a–d) 10 μm; (e–r) 5 μm.
Figure 12.
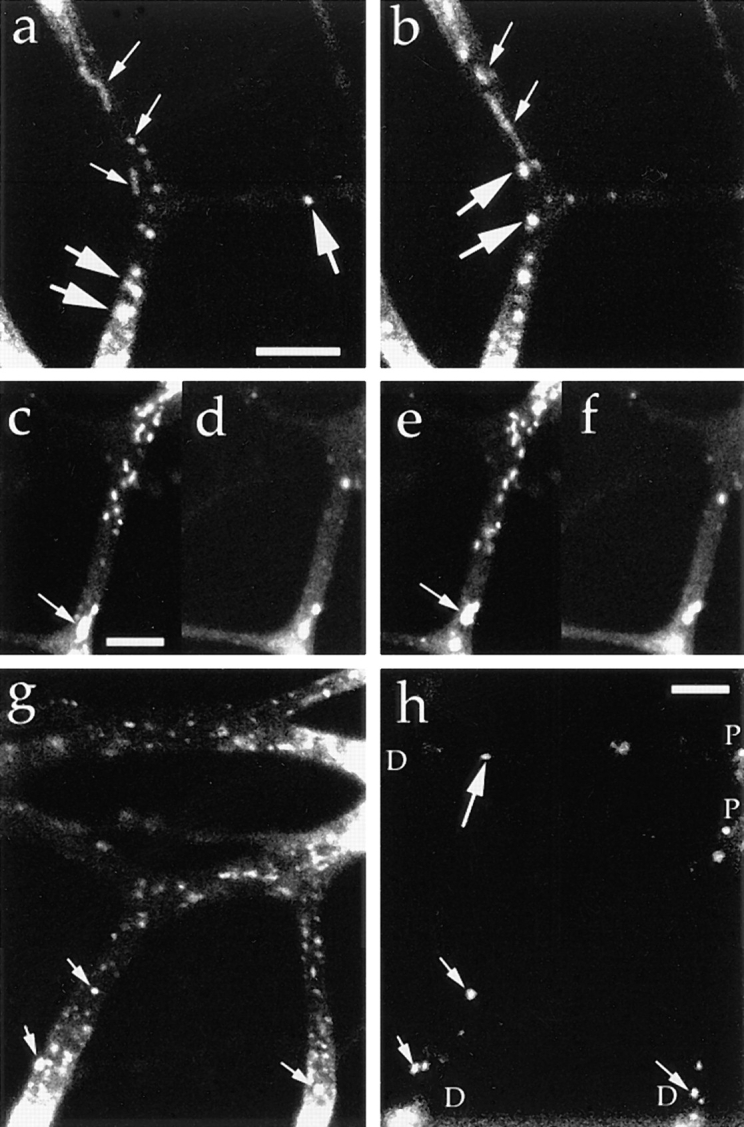
Synaptophysin is transported by the tubulovesicular organelles from the cell body and by the endosomes from the distal axons. Neurons were transfected with adenovirus vector carrying synaptophysin–GFP chimeric DNA. (a and b) Tubular and vesicular organelles with relatively low fluorescence intensity (small arrows) moved anterogradely from the proximal side of the bleached area, whereas large globular vesicles with high fluorescence intensity (large arrows) moved retrogradely from the distal sides. The upper side is proximal to the cell body, and the interval between the frames (a and b) is 20.7 s. (c–f) Double labeling with synaptophysin–GFP (c and e) and FM1-43 (d and f). Tubulovesicular organelles from the proximal side (c and e) were not labeled with FM1-43 (d and f). Signals from retrogradely moving large vesicles were observed using both green (c and e) and red (d and f) filters. Arrowheads indicate the vesicles whose green signal was brighter than the red signal (thus double labeled by both GFP and FM1-43). Interval between the frames in c and d and e and f was 1.65 s. The upper side is proximal to the cell body. (g and h) Double labeling with synaptophysin–GFP and Texas red–dextran (h). P indicates the proximal side of the axons, and D indicates the distal side of the axons. Anterogradely moving tubulovesicular organelles were not labeled with Texas red– dextran, whereas retrogradely moving bright globular vesicles were double labeled with both GFP and Texas red–dextran (small arrows). Note the endosome that was not labeled with GFP (large arrows). Bars, 5 μm.
Generation of the Vesicles Transporting Newly Synthesized Proteins in the Cell Body
Using the above approach, we investigated the generation of transporting vesicles in the cell body (Fig. 6). We found that tubules arose, protruded, and pinched off from the bright staining of GAP-43–GFP within the cell body, the distribution of which was similar to that of TGN-38 (Fig. 4). The vesicles moved at the speed of fast axonal transport as soon as they were generated in the cell body, and moved directly into the axon.
Figure 6.

Generation of a transporting vesicle in the cell body. (a) The cell was transfected with adenovirus vector carrying GAP-43–GFP chimeric DNA. A bright label within the cell body was observed. (b) Time series imaging of a part of the cell body in a. A bright punctuate staining emerged from static intense GFP-staining in the cell body (2nd frame). From there, a tubular vesicle protruded at the speed of fast axonal transport (2nd–4th frames), separated from the static intense GFP-staining (5th frame), and proceeded into the axon (6th and 7th frames). Diamonds indicate the distal edge of the tubule. Intervals between frames are 3.45 s. Bar, 5μm.
Tubulovesicular Organelles that Vary in Size and Shapes Transport the Plasma Membrane Proteins in Axons
We found that the transporting vesicles varied in shape, from tubules to spheres in the axons of GAP-43– or SNAP-25–transfected neurons (see Figs. 5–11). We checked that the observed tubular structures were not mitochondria by double labeling of DRG axons with GFP and Mitotracker. We found that the tubulovesicular organelles were distinct from mitochondria (Fig. 7). These vesicles moved mainly in the anterograde direction at an average speed of 0.76 ± 0.26 μm/s. The speed was independent of vesicle size and was distributed ∼0.9 μm/s with a single peak (see Fig. 9 b). The size distribution of these vesicles varied between neurons. The shape of the vesicles was flexible: for example, tubular vesicles were more elongated in the longitudinal direction when they started to move, and bent when they changed their direction of movement. We often found that tubular vesicles broke into smaller vesicles while they were moving down in axon (Fig. 8 d). Judging from the movement and the elongated shape of the tubular vesicles before they broke down, it was unlikely that the tubular vesicles had consisted of several small independent vesicles. Furthermore, in some neurons, the longitudinal size of vesicles decreased as the vesicles proceeded in the same axons (Fig. 8, a and b). For example, it was 1.71 ± 0.94 μm in length and 0.55 ± 0.22 μm in width in the proximal axon (∼40 μm from the cell body), and 1.16 ± 0.68 μm in length in the distal part (∼110 μm from the cell body) of the same axon (Fig. 9 a). As shown in Fig. 6 a and b, the fluorescent intensity of each vesicle in the distal axon is not as high as that in the proximal axons. This eliminates the possibility that long tubules are simply compressed into small vesicles, because total fluorescence intensity of the vesicles will be unchanged in such case. These results suggest that tubular vesicles divided into smaller vesicles while moving down axons.
Figure 11.

Interbranch transport of vesicles within an axon. Neurons were transfected with adenovirus vector carrying SNAP-25–GFP chimeric DNA. The cell body is indicated by (*) and axons growing to right of the cell body are also stained. In the boxed area, the axon grows from the left side and diverges to the upper and lower right branches. The boxed area was observed at higher magnification. Under higher magnification, the tubular vesicle that is indicated with a star moved retrogradely from the upper branch and then anterogradely into the lower branch (a–e). Intervals between frames are 3.45 s. Bar, 5μm.
Figure 7.

The tubular structure identified here was not a mitochondrion. Neurons were transfected with adenovirus vectors carrying GAP-43–GFP chimeric DNA after 3 h in culture and were observed using a confocal laser scan microscope 40 h later. 15 min before observation Mitotracker was added to the medium for vital staining of mitochondria. The medium was changed just before observation. The axon was observed in both FITC mode and Texas red mode simultaneously, and the movements of both GAP-43 transporting vesicles (a and c) and mitochondria (b and d) were recorded. a–d are the simultaneous double labeling, and interval between a and b and c and d is 17.25 s. The upper side is proximal to the cell body. The central area of the axon was photobleached before observation. Note that transporting vesicles cannot be identified in the peripheral area of the axon because of the high level of background staining. The GAP-43–containing vesicles identified in the central area were transported from the peripheral area during observation. Most of the mitochondria were stationary and only a few moved into the central region (arrows). Bar, 5 μm.
Figure 9.

Quantitative analysis of vesicle size and speed. (a) Histogram of longitudinal size of vesicles in proximal (n = 69) and distal (n = 39) areas of the same axon. (b) Distribution of speed of GAP-43–transporting vesicles. For this purpose, two adjacent frames from >20 different cells of observations were chosen (interval between the frames is 3.45 s). Then, the anterograde displacement of identified vesicles between the two frames was measured (n = 86). Note the single peak distribution ∼0.9 μm/s in the histogram. The speed was independent of vesicle size or shape (data not shown).
Figure 8.
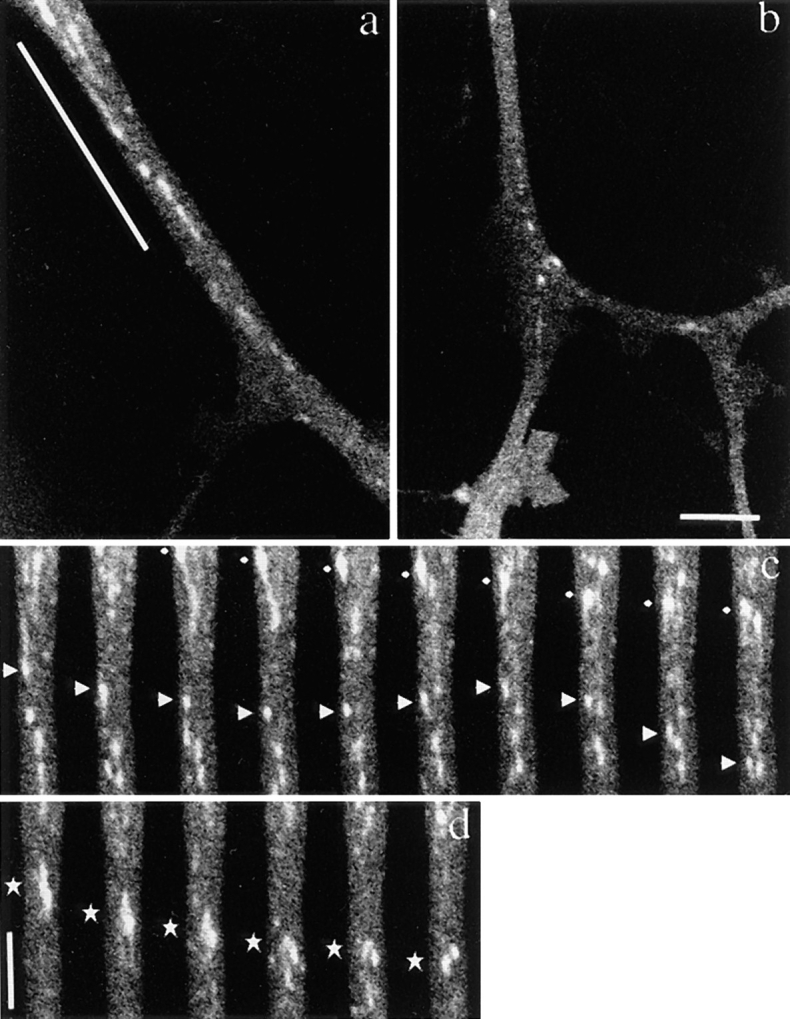
Axonal transport of vesicles containing GAP-43–GFP chimeric protein in mouse DRG neurons. Neurons were transfected with adenovirus vectors carrying GAP-43–GFP chimeric DNA after 3 h in culture and were observed using a laser scan microscope 40 h later. Note that numerous vesicles of various sizes were transported from the proximal side of the axon. (a and b) Proximal and distal area of the same axon. a shows an axon ∼40 μm from the cell body, b shows the same axon ∼110 μm from the cell body. The upper side of both micrographs is proximal to the cell body. Note the difference in the sizes of GAP-43–transporting vesicles even though the degree of magnification of both figures is the same. (c and d) Time course images of the underlined area of the axon in a. The intervals between frames are 3.45 s. The upper side of both micrographs is proximal to the cell body. In c, tubular and spherical vesicles of various sizes and shapes are moving. The vesicle indicated by diamonds showed representative continuous anterograde movement. The vesicle indicated by arrowheads switched back to retrograde movement, and again switched direction anterogradely. In d, the vesicle indicated by stars broke down into smaller spheres while moving anterogradely in the axon. Bars: (b and d) 5 μm.
Tubulovesicular Organelles Are Distinct from Endosomes
We detected the bidirectional movement of transporting vesicles in GAP-43– or SNAP-25–transfected neurons, and we considered that the retrogradely moving vesicles were not endosomes endocytosed from the plasma membrane, but were anterogradely moving tubulovesicular organelles that changed their direction of movement. Several lines of evidence support this. First, we often observed the switch of anterogradely moving vesicles to retrogradely moving ones (Fig. 8 c). Second, the retrogradely moving vesicles were quite similar to the anterogradely moving vesicles in shape, size, and fluorescence intensity.
To further confirm that the retrograde tubulovesiclar organelles are not endosomes, we performed double labeling experiments with a membrane protein–GFP fusion protein and Texas red–dextran. If our interpretation is correct, the tubulovesicular organelles that transport GAP-43 should not be labeled with Texas red–dextran. In contrast, the cell surface receptor proteins that are endocytosed upon binding to their ligands should be transported by two types of vesicle: tubulovesicular organelles and endosomes. TrkA is a high affinity receptor for NGF (Klein et al., 1991) and is endocytosed on binding to NGF (Ehlers et al., 1995). We found that trkA was transported by both tubulovesicular organelles and endosomes by double labeling with trkA–GFP and Texas red–dextran (Fig. 10, a and b). Tubulovesicular organelles were not labeled with Texas red–dextran while large globular vesicles were double labeled by GFP and Texas red–dextran. This was in clear contrast with the GAP-43–transporting vesicles that consisted of tubulovesicular organelles only and were not stained by Texas red–dextran (Fig. 10, c and d).
Figure 10.
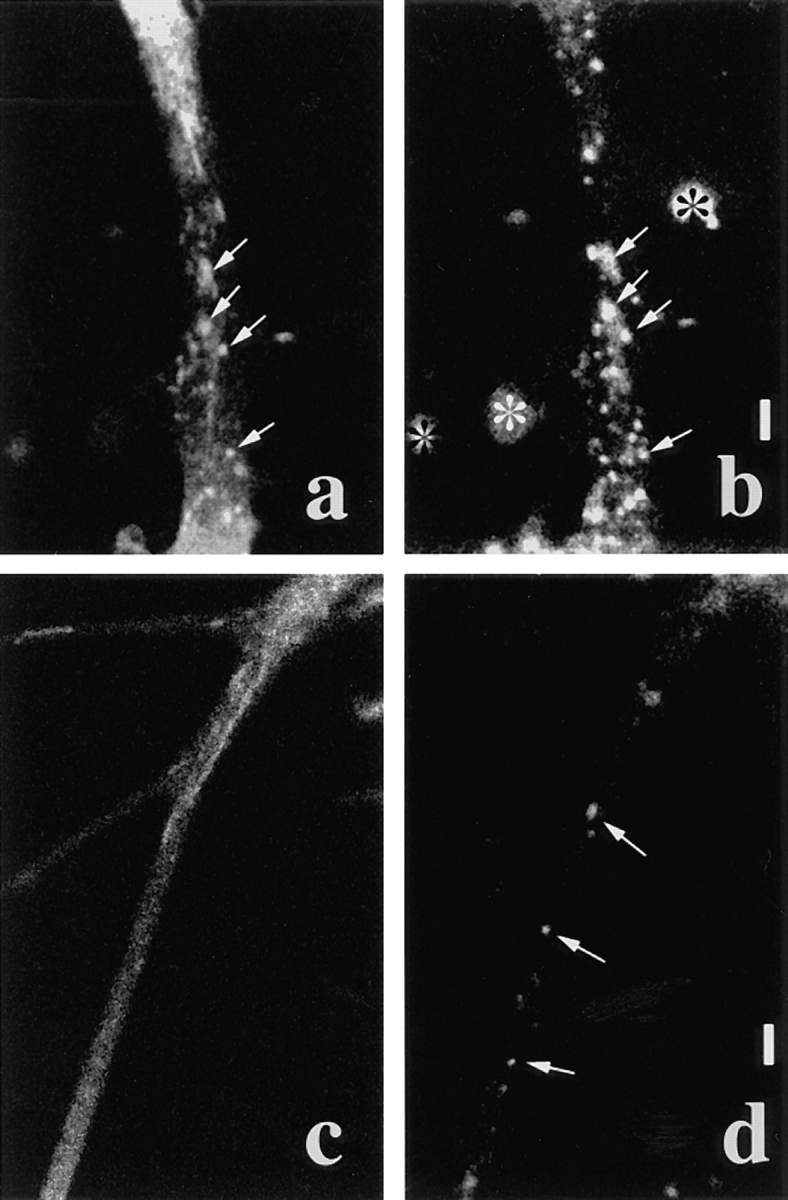
Tubulovesicular organelles were distinct from endosomes. TrkA was transported by both tubulovesicular organelles and endosomes, but GAP-43 was transported only by tubulovesicular organelles. (a and b) Double labeling with trkA–GFP (a) and Texas red–dextran (b). The upper side is proximal to the cell body. Tubulovesicular organelles from the proximal axon were labeled only by GFP. Retrogradely moving large globular vesicles were labeled by both GFP and Texas red–dextran (arrows). Note that some vesicles were labeled by Texas red–dextran but not GFP. Bright stainings outside of the cell (*) in b were aggregates of Texas red–dextran attached to the coverslip. (c and d) Double labeling with GAP-43–GFP (c) and Texas red–dextran (d). Upper-right side is proximal to the cell body. The axon bifurcates, and the upper left side and lower left side are distal to the cell body. Note that endosomes were labeled by Texas red– dextran (arrows) but not by GFP, whereas tubulovesicular organelles were labeled only by GFP. Bars, 5 μm.
Tubulovesicular Organelles Move Directly from Branch to Branch in Axon
We detected the transport of vesicles from one distal branch of an axon to the other distal branch at a branching point (Fig. 11). Previously, transport between a cell body or dendrites and axons has been proposed, but not between the branches of the same axon. These vesicles moved retrogradely from one branch and then moved anterogradely to the other branch. This phenomenon is interpreted as follows: these vesicles might be transported from the cell body into a branch of the axon, but if they are not used there, they might switch back, and at the branching point, turn into the other branch of the axon instead of back to the cell body. They might fuse with the plasma membrane in the distal area of the new branch. The result, together with the bidirectional movement of the vesicles, indicates that the tubulovesicular organelles circulate among the branches of axons.
Net Membrane Transport by the GAP-43–containing Tubulovesicular Organelles Will be Sufficient for Axonal Outgrowth
It was unknown whether the supply of plasma membrane precursors by internal vesicular transport is sufficient for axonal growth. Because our present method detects the vesicles that moved into the photobleached area, it is suitable to measure the amount of the net transport as well as to observe individual vesicle movement. Culture of DRG cells under this condition can be used to clarify this, because these cells vigorously extend their axons by as much as 1 μm/min. Thus, the increase in surface area is at most ∼10 μm2/min if the diameter of the axon is 3 μm. We calculated the net anterograde membrane flow of the GAP-43–containing vesicles in vigorously transporting axons, by measuring the time course of the increase in the area occupied by the GAP-43–transporting vesicles after the complete photobleaching of the area. We found that the net membrane flow was 59 ± 37 μm2/min. This value is sufficient for supplying the membranes required for the axonal growth. Fig. 5 (o–r) visually supports the present results. The interval between each frame is 1 min and the diameter of the axon is 2.5 μm. Increase of the tubulovesicular organelles in the photobleached area is so vigorous that the photobleached area >20 μm in length was considerably filled with the tubulovesicular organelles within 3 min.
Synaptic Vesicle Protein Is Transported by Tubulovesicular Organelles from the Cell Body and by Endosomes from Distal Axons
We found two types of organelles that moved in synaptophysin–GFP-transfected cells (Fig. 12, a and b): first, tubulovesicular organelles similar to those transporting plasma membrane proteins in size, shape (1.48 ± 1.01 μm in longitudinal length, 0.46 ± 0.13 μm in width, see Table I), speed (0.69 ± 0.33 μm/s), and behavior in axons; and second, organelles that were much wider and in most cases globular in shape (1.33 ± 0.48 μm in length, and 0.89 ± 0.26 μm in width), more intensely stained with GFP (fluorescence intensity of tubulovesicular organelles was 105 ± 22 [arbitrary units], whereas that of large globular vesicles was 240 ± 21.1), but fewer in number (in most cases, the number of tubulovesicular organelles was 8–14 times higher than that of the large globular vesicles) than the tubulovesicular organelles. They changed their direction of movement more frequently than did the tubulovesicular organelles (25% of large vesicles stop or change their direction of movement within three successive frames [6.9 s], whereas only 3% of tubulovesicular organelles stop or change their direction), and accordingly, their movement was bidirectional, but the net transport was retrograde (Fig. 13).
Table I.
Sizes and Speeds of the Transporting Vesicles of Membrane Proteins in Axons
| Membrane proteins | Tubulovesicular organelles | Large globular vesicles* | ||||||
|---|---|---|---|---|---|---|---|---|
| vesicle size‡ | speed§ | |||||||
| length | width | |||||||
| μm | μm/s | |||||||
| GAP-43 | 1.51 ± 0.89 | 0.54 ± 0.22 (n = 110) | 0.76 ± 0.26 (n = 86) | (−) | ||||
| SNAP-25 | 1.09 ± 0.51 | 0.47 ± 0.15 (n = 83) | 0.98 ± 0.74 (n = 40) | (−) | ||||
| TrkA | 1.79 ± 2.78 | 0.53 ± 0.12 (n = 98) | 0.88 ± 0.58 (n = 98) | (+) | ||||
| synaptophysin | 1.48 ± 1.01 | 0.46 ± 0.13 (n = 101) | 0.69 ± 0.33 (n = 101) | (+) | ||||
| TGN-38 | 1.15 ± 0.74 | 0.65 ± 0.19 (n = 53) | 0.76 ± 0.50 (n = 53) | (+) | ||||
The existence of large globular vesicles that colocalized with endocytotic markers was indicated as (+).
The large globular vesicles were not included.
Retrogradely moving or stationary vesicles were not included.
Figure 13.



Movements of several representative transporting vesicles of GAP-43 (A), and synaptophysin (B, tubulovesicular organelles, and C, large globular vesicles that colocalize with FM1-43 or Texas red–dextran). Anterograde movement (from the cell body to the periphery) was set as plus direction. The net translocation (μm) of each vesicle from the starting point was plotted against the time (s). Intervals between the frames were 3.3 s. Comparison with the tubulovesicular organelles that transport either GAP-43 (A) or synaptophysin (B) revealed that the large globular vesicles often change the direction of the movement (C). Nevertheless, the large globular vesicles have a tendency to move retrogradely.
To further confirm that these two types of vesicle are different, we labeled the endosomal compartment of the synaptophysin–GFP-transfected cells by FM1-43 and Texas red–dextran. FM1-43 is an amphiphatic dye used to monitor endosomal traffic and especially synaptic vesicle recycling at nerve terminals (Betz et al., 1992; Smith and Betz, 1996), thus the dye is suitable for use to test whether synaptophysin-containing vesicles are derived from the plasma membrane by membrane recycling. When we loaded 15 μM FM1-43 into the culture medium (Liu and Tsien, 1995), we found that a number of FM1-43–labeled vesicles were transported in DRG axons. To take advantage of the fact that FM1-43 can be observed using both green and red filters (Betz et al., 1992), we double labeled the transporting vesicles with synaptophysin–GFP and FM1-43. For this purpose, we set the electronic gains of the photomultipliers so that the green signal of FM1-43 is of a much lower intensity than the red signal. Then, if the vesicles can be viewed only through a green filter, the vesicles are labeled only by GFP, and if the green signal is as bright as or brighter than the red signal, the vesicles are labeled by both GFP and FM1-43. We found that the tubulovesicluar organelles were not labeled by FM1-43, and several large globular vesicles were labeled by both FM1-43 and GFP (Fig. 12, c–f). Because we could not detect the GFP signal if it was of a lower intensity than the green signal of FM1-43, we used another endosomal marker to achieve more complete double labeling. Texas red–dextran is a fluid phase marker, and is detected using a red filter only. We confirmed that tubulovesicular organelles were not labeled by Texas red–dextran, while large globular vesicles were labeled by it (Fig. 12, g and h). We noted endosomal vesicles labeled by Texas red–dextran but not by synaptophysin– GFP (Fig. 12 h, large arrow). These results demonstrate that most of the newly synthesized synaptophysin was transported by tubulovesicular organelles that were distinct from the endosomal compartments, whereas recycled synaptophysin was transported by endosomal compartments.
Although both vesicles moved bidirectionally, the net transport by the tubulovesicular organelles was anterograde, and the net transport by the endosomal compartments was retrograde. In varicosities, some of the tubulovesicular organelles passed through them, while others ceased moving just like in the other area of axons, and, at present, we did not find evidence on the selective release of tubulovesicular organelles in varicosities, presumably because the varicosities are already filled with synaptic vesicles, and the turnover rate of the synaptic vesicles proteins with newly synthesized ones may be relatively slow.
Tubulovesicular Organelles Mediate Transport from the TGN to the Cell Surface
Membrane proteins are sorted at the TGN (Kelly, 1993a ; Bauerfeind and Huttner, 1993; Huttner et al., 1995; Banting and Ponnambalam, 1997). Thus, the TGN is considered to be where the transporting vesicles are generated. Thus, tubulovesicular organelles should transport the proteins that shuttle between the TGN and the cell surface. To test this hypothesis, we investigated the transport of TGN-38–GFP in DRG neurons. We found that TGN-38 was transported by the tubulovesicular organelles (Fig. 14, a and b), demonstrating that tubulovesicular organelles play a role in the transport of proteins from the TGN to the cell surface. TGN-38–GFP was transported back from the cell surface by endocytotic vesicles (Fig. 14, c and d), but it was segregated from the endocytotic compartment in the cell body (Fig. 14, e and f).
Figure 14.
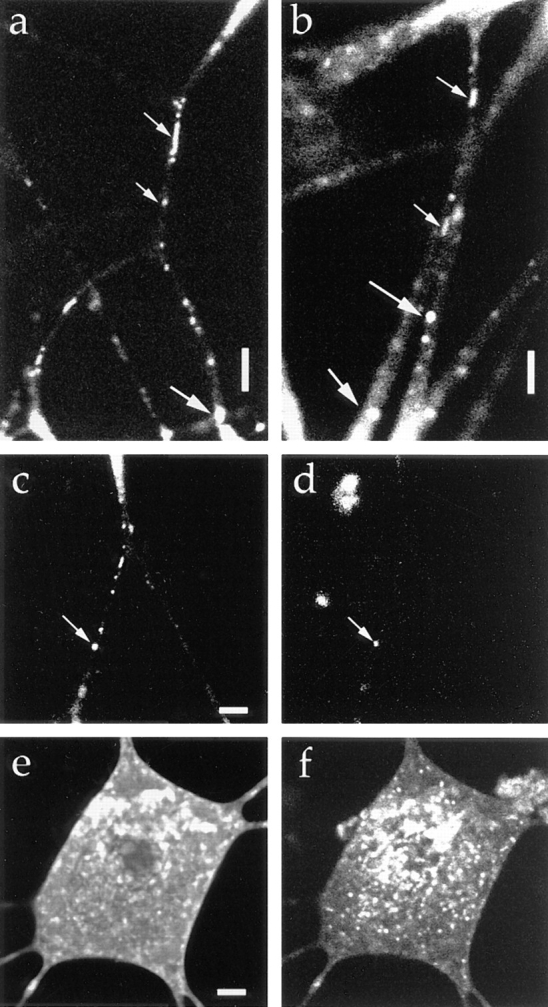
Vesicles transporting the TGN residual protein. The cells were transfected with adenovirus vector carrying TGN-38–GFP chimeric DNA. (a and b) The upper side is proximal to the cell body. Tubulovesicular organelles moved anterogradely (small arrows), and large globular vesicles moved retrogradely (large arrows). (c and d) Double labeling with TGN-38–GFP (c) and Texas red–dextran (d). Note that the retrogradely moving vesicle (arrow) was labeled by both GFP and Texas red–dextran. (e and f) Double labeling with TGN-GFP (e) and Texas red–dextran (f). Confocal slice not photobleached. TGN-38–GFP and Texas red– dextran were segregated in the cell body. Bars, 5 μm.
Correlative Electron Microscopy of the Tubulovesicular Organelles
To correlate the tubulovesicular organelles observed by laser scan microscopy with those observed by electron microscopy, we examined by electron microscopy the area of an axon in which we had observed extensive vesicle transport. Fig. 15 a shows the numerous transporting vesicles that moved into the photobleached area of a living axon. Fig. 15 shows the same axon after the axon was photobleached again and the medium was changed to the fixative, showing that the transporting vesicles moved into the photobleached area before fixation. Fig. 15 c shows an electron micrograph of the area corresponding precisely to that in Fig. 15 b. This micrograph shows that photobleaching did not damage the fine structure of the axon. Observation at higher magnification revealed numerous tubular or vesicular organelles in the area that correspond to the transporting vesicles observed by laser scan microscopy in number and shapes (Fig. 15, d and e, arrows). However, the widths of the vesicles observed by electron microscopy were smaller than the vesicles observed by laser scan microscopy. Presumably, this is due to the fact that a brightly fluorescent object has a halo that makes the object appear bigger than its actual size. For example, microtubules are 24 nm in diameter, but they appear much wider under fluorescence microscopy. In fact, no other membrane structures that may correspond to the light microscopic image were observed in the electron micrographs. Thus the actual diameter of most of the tubulovesicular organelles will be <0.1 μm. In contrast, the length of tubular vesicles may well reflect on the real length of the vesicles, because the errors arising due to the fluorescent halos are assumed to be the same in both the longitudinal and lateral directions. In fact, long tubules were previously identified in electron microscopic studies of axons (Lindsley and Ellisman, 1985). Whether these tubules were derived from one continuous system from the cell body to the terminal or a system of discontinuous components that move within the axon is a matter of debate (Peters et al., 1991). In this study, we found tubules as long as 25 μm moving down axons.
Figure 15.
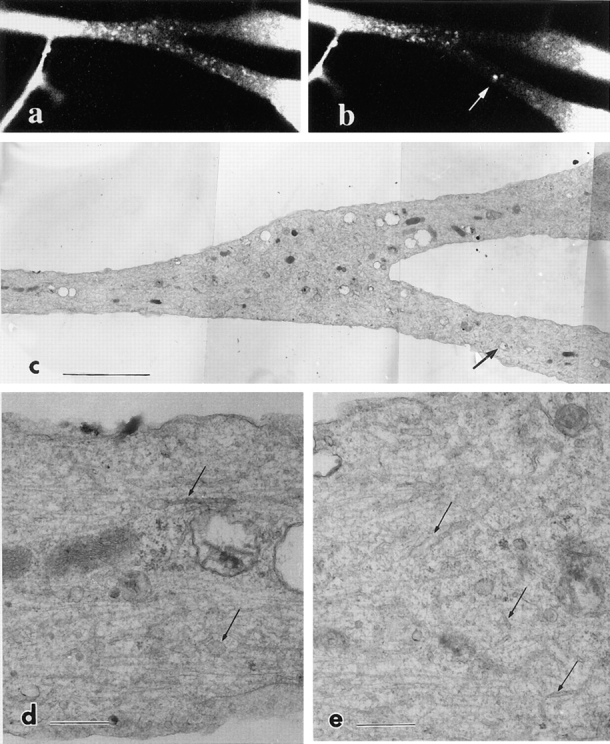
Correlative electron microscopy of trkA–GFP-transporting vesicles. The axon was photobleached and moving vesicles were observed in the living cell (a). Then the axon was again photobleached and fixed with 2% paraformaldehyde and 0.1% glutaraldehyde. Numerous anterogradely moving vesicles as well as a retrogradely moving large vesicle (arrow) moved into the area before fixation (b). (c) The same area of the axon was prepared for electron microscopy. Arrow in c indicates the corresponding endosome in b. (d and e) Higher magnification of c. Numerous tubules and vesicles were observed (arrows). Bars: (c) 10 μm; (d and e) 1 μm.
Immunogold Electron Microscopy of the Tubulovesicular Organelles
Because there was a discrepancy between the width of the tubulovesicular organelles observed in laser scan microscopy and electron microscopy, we next examined whether the GFP-fusion proteins are localized on the tubulovesicular organelles in electron microscopy. We designed GAP-43–GFP construct to contain flag epitope tag in its COOH terminus. Using antiflag monoclonal antibody, we performed immunogold electron microscopy of DRG neurons 40 h after infection. 5-nm colloidal gold stained the plasma membrane of the infected neurons (Fig. 16 a), consistent with the light microscopic observation (Fig. 2 d). In addition, tubulovesicular organelles were stained with the gold (Fig. 16, a and b), while areas enriched in cytoskeletons or mitochondria were not (Fig. 16 a). A few gold labels were observed in noninfected neurons (Fig. 16 c). The results indicate that the tubulovesicular organelles that were observed in the laser scan microscopy were identical to the tubulovesicular organelles observed in the electron microscopy (Figs. 15 and 16).
Figure 16.
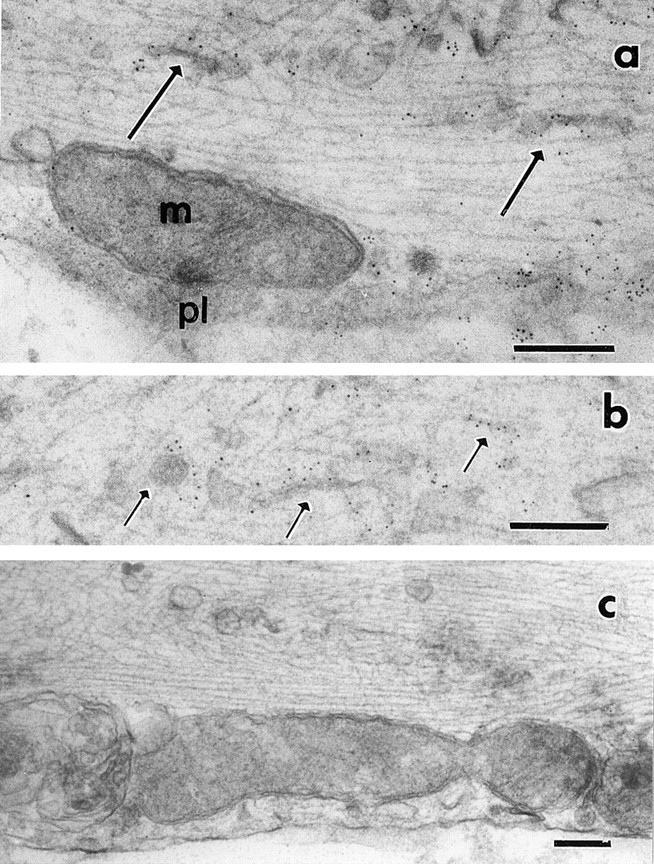
Immunogold electron microscopy of GAP-43–GFP-transporting vesicles. The axons were incubated with antiflag antibody followed by 5-nm colloidal gold conjugated second antibody. pl and m in a indicate plasma membrane and mitochondria, respectively. Arrows indicate some of the tubulovesicular organelles. Plasma membrane and tubular or vesicular organelles were labeled with gold particles. (c) Noninfected DRG neuron as a control. Only a few gold particles were seen. Bars, 0.2 μm.
Discussion
Identification of Tubulovesicular Organelles as Vesicles Transporting Newly Synthesized Proteins from the TGN to the Plasma Membrane
We have visualized the vesicles that transport GAP-43–GFP, SNAP-25–GFP, trkA–GFP, synaptophysin–GFP, and TGN-38–GFP in axons and found that they all share common features as follows: (a) The shape of the vesicles is heterogeneous (Table I; note that standard deviations of the length of the vesicles are much bigger than those of the width of the vesicles) and flexible, varying from spheres to tubules. Thus, vesicles may become globular or fragmented when the cells are homogenized and fractionated. (b) The vesicles move bidirectionally and switch their direction of movement, but the net transport was anterograde. (c) The speed of movement was as much as ∼1 μm/s. (d) Their movement could not be observed well by simultaneous DIC images. (e) They were not stained by endosomal tracers such as Texas red–dextran or FM1-43, or by the mitochondrial marker Mitotracker.
It is surprising that the transporting vesicles were so similar and indistinguishable (Table I), when we consider the different features of the proteins transported (Fig. 1). GAP-43 and SNAP-25 are synthesized in free ribosomes and then attached to the membrane by a lipid moiety (Liu et al., 1994; Veit et al., 1996), whereas TrkA is a integral membrane protein. Synaptophysin is a synaptic vesicle integral membrane protein. TGN-38 is a protein that cycles between the TGN and the cell surface but mainly resides in the TGN and has been used as a marker for the TGN (Banting and Ponnambalam, 1997). The only common feature of these proteins is that they are transported from the TGN to the plasma membrane (synaptic vesicle proteins are considered to be delivered to the plasma membrane and then endocytosed to form mature synaptic vesicles, see Discussion below). Thus, we would like to propose that these vesicles are organelles whose function is the transport of newly synthesized membrane proteins to the plasma membrane.
Our observation that tubules and spheres various in size and shape transport the newly synthesized proteins in axons well fits the results of electron microscopic studies in early days (Lindsley and Ellisman, 1985; Peters et al., 1991; Porter et al., 1979). When axonal transport was blocked, tubular organelles, spherical organelles, and smooth ER-like network structures accumulated on the proximal side of axons (Smith 1980; Tsukita and Ishikawa, 1980; Hirokawa et al., 1990, 1991). Because various-sized vesicles were identified in electron microscopic studies, we speculated that the vesicles transporting a newly synthesized protein may be a subset of them, and may be homogeneous in size and shapes. To our surprise, the vesicles that were labeled only by one GFP-fusion protein varied considerably in size and shape. Correlative electron microscopic study revealed that numerous tubules and vesicles existed in the axon, corresponding to the moving vesicles seen using laser scan microscopy. Immunogold electron microscopy confirmed that the GFP fusion proteins were really localized on these tubules and vesicles. These vesicles are the organelles that had been morphologically identified as the axoplasmic reticulum and the discrete element (vesicles and vesiculotubular bodies) in previous electron microscopic studies (Lindsley and Ellisman, 1985). Laser scan microscopy did detect moving tubules as long as 25 μm, but did not detect the complicated network membrane structures detected in previous electron microscopic studies, presumably because the present photobleaching method only detects moving vesicles. Complicated axoplasmic reticulum will be relatively stationary in axons, and there is a possibility that the tubulovesicular organelles may fuse with or bud from it. Previous electron microscopic studies revealed structural details of the tubulovesicular organelles (Lindsley and Ellisman, 1985), but the molecular compositions, or functions of these organelles were not elucidated. With the subsequent development of light microscopic techniques, many aspects of fast axonal transport were revealed (Allen et al., 1982), but the resolution achieved using these techniques was not sufficiently high to visualize the tubulovesicular organelles in intact axons, and thus, these organelles have been somehow ignored in recent studies. The functions of and some molecular components comprising the tubulovesicular organelles are known, and it is possible to detect these organelles in living cells. Thus, we hope that further study on these organelles will be carried out in future.
Fast axonal transport has been measured in situ by isotope-labeling stuides (50–400 mm/d) (Grafestein and Forman, 1980). Considering the facts that (a) fast axonal transport is temperature-dependent and (b) fast axonal transport of the growing axons is two to three times slower than that in mature axons (Grafestein and Forman, 1980), our present data that tubulovesicular organelles are transported as fast as ∼1 μm/s (∼86.4 mm/d) at room temperature in growing DRG axons is consistent with the speed of the fast axonal transport in the radioactive labeling studies. In these studies, which had established the rate of the axonal transport, isotope-labeled transmitters or amino acids were injected into the cell bodies, and the axons (nerves) were cut into pieces several hours later. Then the amount of isotopes in axon fragments were plotted against the distance from the cell bodies. We were interested in whether the isotope-labeling study had detected a fraction of the vesicles that ceased moving or changed the direction of the movement as we found in the present study. For this purpose, we reevaluated the original isotope labeling data (for example see Goldberg et al., 1978). In these studies, a single peak definitely moved to the distal end of nerves at a speed of fast axonal transport. However, within several hours, the initial sharp high peak became broad low peak as the peak moved to the distal ends. Especially, a large fraction of isotopes was left behind the peak and formed a long slope from the peak to the cell body. It was not known why the peak decayed so fast and much isotope was left behind the peak, but there was a discussion that intermittent movement or retrograde movement of the vesicles may result in these phenomena (Goldberg et al., 1978). The peak fraction in the isotope study corresponds to the vesicles that moved continuously anterogradely. The delayed fraction corresponds to the vesicles that ceased moving or changed the direction of the movement. Thus the previous isotope-labeling experiments clearly support our present data.
At present, we do not know whether these tubulovesicular organelles can be classified into subgroups based on the molecules they transport. One possibility is that all membrane proteins (except for those on the peptide-containing secretory vesicles) are transported by the same transporting vesicles as bulk flow. The other possibility is that these newly synthesized proteins are sorted into different vesicles that are morphologically or behaviorally indistinguishable and transported. Identification of a motor protein (Kif1A) that appears specific to the transport of synaptic vesicle proteins may suggest the latter possibility (Okada et al., 1995), but direct evidence will be obtained using differently colored fluorescent proteins (Heim et al., 1994) and the method described in this paper.
Implication for Synaptic Vesicle Formation
Neurons develop specialized membrane organelles in their terminals for synaptic transmission. These organelles, synaptic vesicles, are well characterized with respect to their protein components and the dynamics involved in neurotransmitter release (Sudhof and Jahn, 1991; Trimble et al., 1991). However, it is largely unknown how synaptic vesicles are formed and in which form they are transported in the axons. In this study, we demonstrated that synaptic vesicle proteins are transported in the axon by tubulovesicular organelles, and not by small clear vesicles as are synaptic vesicles. This indicates that mature synaptic vesicles must be produced at the synaptic area. Our observation is supported by the results of a previous cell fractionation study using PC12 cells that demonstrated that small clear vesicles (which may correspond to synaptic vesicles in neurons) are formed by endocytosis after newly synthesized proteins are first transported to the plasma membrane (Regnier-Vigouroux et al., 1991; Huttner et al., 1995).
In fact, recent studies on the origin of synaptic vesicles suggest that synaptic vesicles are specialized endosomes and are maturated by recycling between plasma membrane and endosomes (Regnier-Vigouroux et al., 1991; Kelly, 1993a ; Huttner et al., 1995). The question then arises regarding whether the synaptic vesicle proteins (especially synaptophysin that is targeted to endosomes when the gene encoding it is transfected to nonneuronal cells [Cameron et al., 1991; Lindstedt and Kelly, 1991]) are targeted to axonal endosomes via the cell body and then transported in axons, or are directly transported from the TGN to the nerve terminal plasma membrane and then recycled (Kelly, 1993b ). Our results demonstrate that most synaptophysin molecules, in a similar manner to plasma membrane proteins, are transported by tubulovesicular organelles in DRG neurons, supporting the latter possibility.
Synaptophysin was transported retrogradely by endosomal compartments. These vesicles were wider and of a more globular shape than the tubulovesicular organelles, and switched their direction of movement more frequently. These vesicles were double labeled with Texas red–dextran and FM1-43, both of which are endocytotic markers. These vesicles were not labeled with GAP-43–GFP. This is in accordance with the in vivo observation that when a peripheral nerve was ligated, GAP-43 accumulated only in the proximal side of the ligated portion, but synaptophysin accumulated in both sides (Dahlstrom and Li, 1994), indicating that synaptophysin in the distal side of axons in vivo was located on the endosomal compartments. We do not know whether synaptophysin in large globular endosomes is again recruited for the biogenesis of synaptic vesicles, or degraded in the cell body.
Implication for Axonal Growth
It was unknown whether the supply of plasma membrane precursors by internal vesicular transport is sufficient for axonal growth. We calculated the net membrane flow by the GAP-43 containing tubulovesicular organelles, and estimated that the anterograde vesicular transport is sufficient for supplying the membranes required for the axonal growth. It is worth critically analyzing the factors that will cause the errors of our present estimation. Overestimation may occur due to the difference between the fluorescence image and the real size of the vesicles. According to the comparison between the confocal images and electron microscopic images of individual tubulovesicular organelles, this factor would be about five to six times, so the net membrane transport by GAP-43 containing vesicles is still sufficient for the axonal growth. Underestimation will occur due to the facts that (a) we do not count for the vesicles out of focus, (b) we do not count for the anterograde vesicles that do not contain GAP-43 proteins if such vesicles exist, and (c) the method of calculation of the total surface area is underestimation (see Materials and Methods). Thus, in spite of these factors, the internal membrane transport will be sufficient for the axonal growth in DRG neurons.
We detected the direct transport of vesicles from one distal branch of an axon to the other at a branching point. We also found that tubulovesicular organelles move bidirectionally in axons, and often switch the direction of movement. This shows that newly synthesized proteins are not transported unidirectionally, but are circulated within axons. These movements appear to be an inefficient way of transporting proteins, but in fact, they are an efficient and simple way to make use of the newly synthesized proteins in branching axons. If the transport of the plasma membrane precursors were rate limiting and unidirectional, when one branch of the axon grows and the others do not, selective transport of the precursors to the growing branch should occur in the axon. If this were the case, we should hypothesize that a signal to preferentially provide the growing branch with precursors would be delivered from the growing tips to the cell body or proximal axons, and according to the signal, the plasma membrane precursors should be sorted at every branching point. This would be a complex and difficult task for neurons. If transport of the plasma membrane is not rate limiting but the transport is unidirectional, it is not necessary to hypothesize the existence of a signal regulating axonal transport. However, in this case, because the transport is assumed to be nonselective at branching points, all the plasma membrane precursors that are transported into nongrowing branches are wasted. Thus, newly synthesized proteins are not efficiently targeted. In our circulation model based on this study (Fig. 17), unused tubulovesicular organelles in nongrowing branches will switch direction and be transported to the growing branch of the axon where the plasma membrane precursors are used efficiently. The growing tips of neurites do not have to transmit a signal to inform the cell body or proximal axon that they are growing, all they have to do is use the precursors that arrive at the growing tips. The cell body does not have to know the position of the neurites that are growing, it need only synthesize a sufficient amount of the precursors for the total neurite outgrowth. Thus, axonal growth can be controlled locally at the growing tip of the axon (Campenot, 1977) by the rate at which the growing tips incorporate the precursors into the plasma membrane.
Figure 17.

Circulation model of axonal transport of plasma membrane and synaptic vesicle proteins. (1) Vesicles protrude from the TGN in the cell body at the speed of fast axonal transport, separate from the network, and proceed into axons. (2) In axons, vesicles are tubular or spherical, and move at an average speed of ∼0.8 μm/s. Net membrane flow was as high as 50 μm2/min. (3) The vesicles break down into smaller vesicles while moving down the axon. (4) Some vesicles switch direction and move retrogradely in axons. (5) Vesicles move from one branch to another branch of an axon at a branching point. (6) Endosomes also move bidirectionally. Tubulovesicular organelles circulate within axons to deliver newly synthesized plasma membrane and synaptic vesicle proteins to the area where they are required.
Acknowledgments
We thank Dr. R. Tsien for the GFP clones, Drs. P. Kahle and E. Shooter and Dr. T. Hunter for the trkA cDNAs; T. Funakoshi for advice on cell culture; S. Nonaka for advice on computer analysis; K. Kaida (Student of the University of Tokyo, School of Medicine) for the statistical analysis of TrkA-vesicles.
This work was supported by a COE Research grant from the Ministry of Education, Science, Sports, and Culture, Japan to N. Hirokawa.
Abbreviations used in this paper
- DRG
dorsal root ganglion
- GFP
green fluorescent protein
- VEC-DIC
video-enhanced contrast differential interference contrast
- SNAP-25
synaptosome-associated protein 25
- t-SNARE
soluble NSF attachment protein receptor at the target membrane
References
- Allen RD, Metuzals J, Tasaki I, Brady ST, Gilbert SP. Fast axonal transport in squid giant axon. Science. 1982;218:1127–1129. doi: 10.1126/science.6183744. [DOI] [PubMed] [Google Scholar]
- Bajjalieh SM, Peterson K, Shinghal R, Scheller RH. SV2, a brain synaptic vesicle protein homologous to bacterial transporters. Science. 1992;257:1271–1273. doi: 10.1126/science.1519064. [DOI] [PubMed] [Google Scholar]
- Banting G, Ponnambalam S. TGN38 and its orthologues: roles in post-TGN vesicle formation and maintenance of TGN morphology. Biochim Biophys Acta. 1997;1355:209–217. doi: 10.1016/s0167-4889(96)00146-2. [DOI] [PubMed] [Google Scholar]
- Bauerfeind R, Huttner WB. Biogenesis of constitutive secretory vesicles, secretory granules and synaptic vesicles. Curr Opin Cell Biol. 1993;5:628–635. doi: 10.1016/0955-0674(93)90132-a. [DOI] [PubMed] [Google Scholar]
- Bennett MK, Wandinger-Ness A, Simons K. Release of putative exocytotic transport vesicles from perforated MDCK cells. EMBO (Eur Mol Biol Organ) J. 1988;7:4075–4085. doi: 10.1002/j.1460-2075.1988.tb03301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz WJ, Mao F, Bewick GS. Activity-dependent fluorescent staining and destaining of living vertebrate motor nerve terminals. J Neurosci. 1992;12:363–375. doi: 10.1523/JNEUROSCI.12-02-00363.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady ST. A novel brain ATPase with properties expected for the fast axonal transport motor. Nature. 1985;37:73–75. doi: 10.1038/317073a0. [DOI] [PubMed] [Google Scholar]
- Cameron PL, Sudhof TC, Jahn R, De Camilli P. Colocalization of synaptophysin with transferrin receptors: implications for synaptic vesicle biogenesis. J Cell Biol. 1991;115:151–164. doi: 10.1083/jcb.115.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campenot RB. Localization of neurite development by nerve growth factor. Proc Natl Acad Sci USA. 1977;74:4516–4519. doi: 10.1073/pnas.74.10.4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccarelli B, Hurlbut WP. Ca2+-dependent recycling of synaptic vesicles at the frog neuromuscular junction. J Cell Biol. 1980;87:297–303. doi: 10.1083/jcb.87.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlstorm AB, Li JY. Fast and slow axonal transport-different methodological approaches give complementary information: contributions of the stop-flow/crush approach. Neurochem Res. 1994;19:1413–1419. doi: 10.1007/BF00972470. [DOI] [PubMed] [Google Scholar]
- de Curtis I, Simons K. Isolation of exocytic carrier vesicles from BHK cells. Cell. 1989;58:719–727. doi: 10.1016/0092-8674(89)90106-2. [DOI] [PubMed] [Google Scholar]
- Ehlers MD, Kaplan DR, Price DL, Koliatsos VE. NGF-stimulated retrograde transport of trkA in the mammalian nervous system. J Cell Biol. 1995;130:149–156. doi: 10.1083/jcb.130.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feany MB, Lee S, Edwards RH, Buckley KM. The synaptic vesicle protein SV2 is a novel type of transmembrane transporter. Cell. 1992;70:861–867. doi: 10.1016/0092-8674(92)90319-8. [DOI] [PubMed] [Google Scholar]
- Garcia EP, McPherson PS, Chilcote TJ, Takei K, De Camilli P. rbSec1A and B colocalize with syntaxin 1 and SNAP-25 throughout the axon, but are not in a stable complex with syntaxin. J Cell Biol. 1995;129:105–20. doi: 10.1083/jcb.129.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girotti M, Banting G. TGN-38-green fluorescent protein hybrid proteins expressed in stably transfected eukaryotic cells provide a tool for the real-time, in vivo study of membrane traffic pathways and suggest a possible role for rat TGN38. J Cell Sci. 1996;109:2915–2926. doi: 10.1242/jcs.109.12.2915. [DOI] [PubMed] [Google Scholar]
- Goldberg DJ, Schwartz JH, Sherbany AA. Kinetic properties of normal and perturbed axonal transport of serotonin in a single identified axon. J Physiol. 1978;281:559–579. doi: 10.1113/jphysiol.1978.sp012439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldenberg SS, De Boni U. Pure population of viable neurons from rabbit dorsal root ganglia, using gradients of Percoll. J Neurobiol. 1983;14:195–206. doi: 10.1002/neu.480140304. [DOI] [PubMed] [Google Scholar]
- Grafstein B, Forman DS. Intracellular transport in neurons. Physiol Rev. 1980;60:1167–1283. doi: 10.1152/physrev.1980.60.4.1167. [DOI] [PubMed] [Google Scholar]
- Grote E, Hao JC, Bennett MK, Kelly RB. A targeting signal in VAMP regulating transport to synaptic vesicles. Cell. 1995;81:581–589. doi: 10.1016/0092-8674(95)90079-9. [DOI] [PubMed] [Google Scholar]
- Heim R, Prasher DC, Tsien RY. Wavelength mutations and posttranslational autoxidation of green fluorescent protein. Proc Natl Acad Sci USA. 1994;91:12501–12504. doi: 10.1073/pnas.91.26.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N. Organelle transport along microtubules-the role of KIFs. Trends Cell Biol. 1996;6:135–141. doi: 10.1016/0962-8924(96)10003-9. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Sato-Yoshitake R, Yoshida T, Kawashima T. Brain dynein (MAP1C) localizes on both anterogradely and retrogradely transported membranous organelles in vivo. J Cell Biol. 1990;111:1027–1037. doi: 10.1083/jcb.111.3.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Sato-Yoshitake R, Kobayashi N, Pfister KK, Bloom GS, Brady ST. Kinesin associates with anterogradely transported membranous organelles in vivo. J Cell Biol. 1991;114:295–302. doi: 10.1083/jcb.114.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbeck PJ. Products of endocytosis and autophagy are retrieved from axons by regulated retrograde organelle transport. J Cell Biol. 1993;121:305–315. doi: 10.1083/jcb.121.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbeck PJ, Bray D. Rapidly transported organelles containing membrane and cytoskeletal components: their relation to axonal growth. J Cell Biol. 1987;105:2827–2835. doi: 10.1083/jcb.105.6.2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttner, W.B., M. Ohashi, R.H. Kehlenbach, F.A. Barr, R. Bauerfeind, O. Braunling, D. Corbeil, M. Hannah, H.A. Pasolli, A. Schmidt et al. 1995. Biogenesis of neurosecretory vesicles. Cold Spring Harbor Symposia on Quantitative Biology. LX:315–327. [DOI] [PubMed]
- Jung LJ, Scheller RH. Peptide processing and targeting in the neuronal secretory pathway. Science. 1991;251:1330–1335. doi: 10.1126/science.2003219. [DOI] [PubMed] [Google Scholar]
- Kanegae Y, Makimura M, Saito I. A simple and efficient method for purification of infectious recombinant adenovirus. Jpn J Med Sci Biol. 1994;47:157–166. doi: 10.7883/yoken1952.47.157. [DOI] [PubMed] [Google Scholar]
- Karns LR, Ng S-C, Freeman JA, Fishman MC. Cloning of complementary DNA for GAP-43, a neuronal growth-related protein. Science. 1987;236:597–600. doi: 10.1126/science.2437653. [DOI] [PubMed] [Google Scholar]
- Kelly, R.B. 1993a. Storage and release of neurotransmitters. Cell72/Neuron. 10(Suppl.):43–53. [DOI] [PubMed]
- Kelly RB. A question of endosomes. Nature. 1993b;364:487–488. doi: 10.1038/364487a0. [DOI] [PubMed] [Google Scholar]
- Klein R, Jing SQ, Nanduri V, O'Rourke E, Barbacid M. The trk proto-oncogene encodes a receptor for nerve growth factor. Cell. 1991;65:189–197. doi: 10.1016/0092-8674(91)90419-y. [DOI] [PubMed] [Google Scholar]
- Krijnse-Locker J, Parton RG, Fuller SD, Griffiths G, Dotti CG. The organization of the endoplasmic reticulum and the intermediate compartment in cultured rat hippocampal neurons. Mol Biol Cell. 1995;6:1315–1332. doi: 10.1091/mbc.6.10.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey JD, Ellisman MH. The neuronal endomembrane system: I. Direct links between rough endoplasmic reticulum and the cis element of the Golgi apparatus. J Neurosci. 1985;5:3124–3134. doi: 10.1523/JNEUROSCI.05-12-03111.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linstedt AD, Kelly RB. Synaptophysin is sorted from endocytotic markers in neuroendocrine PC12 cells but not transfected fibroblasts. Neuron. 1991;7:309–317. doi: 10.1016/0896-6273(91)90269-6. [DOI] [PubMed] [Google Scholar]
- Liu G, Tsien RW. Properties of synaptic transmission at single hippocampal synaptic boutons. Nature. 1995;375:404–408. doi: 10.1038/375404a0. [DOI] [PubMed] [Google Scholar]
- Liu Y, Fisher DA, Storm DR. Intracellular sorting of neuromodulin (GAP-43) mutants modified in the membrane targeting domain. J Neurosci. 1994;14:5807–5817. doi: 10.1523/JNEUROSCI.14-10-05807.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzio JP, Brake B, Banting G, Howell KE, Braghetta P, Stanley KK. Identification, sequencing and expression of an integral membrane protein of the trans-Golgi network (TGN38) Biochem J. 1990;270:97–102. doi: 10.1042/bj2700097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meakin SO, Suter U, Drinkwater CC, Welcher AA, Shooter EM. The rat trk protooncogene product exhibits properties characertistic of the slow nerve growth factor receptor. Proc Natl Acad Sci USA. 1992;89:2374–2378. doi: 10.1073/pnas.89.6.2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiri KF, Pfenninger KH, Willard MB. Growth-associated protein, GAP-43, a polypeptide that is induced when neurons extend axons, is a component of growth cones and corresponds to pp46, a major polypeptide of a subcellular fraction enriched in growth cones. Proc Natl Acad Sci USA. 1986;83:3537–3541. doi: 10.1073/pnas.83.10.3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake S, Makimura M, Kanegae Y, Harada S, Sato Y, Takamori K, Tokuda C, Saito I. Efficient generation of recombinant adenoviruses using adenovirus DNA-terminal protein complex and a cosmid bearing the full-length virus genome. Proc Natl Acad Sci USA. 1996;93:1320–1324. doi: 10.1073/pnas.93.3.1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakata T, Hirokawa N. Point mutation of adenosine triphosphate-binding motif generated rigor kinesin that selectively blocks anterograde lysosome membrane transport. J Cell Biol. 1995;131:1039–1053. doi: 10.1083/jcb.131.4.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Yamazaki H, Sekine-Aizawa Y, Hirokawa N. The neuron-specific kinesin superfamily protein KIF1A is a unique monomeric motor for anterograde axonal transport of synaptic vesicle precursors. Cell. 1995;81:769–780. doi: 10.1016/0092-8674(95)90538-3. [DOI] [PubMed] [Google Scholar]
- Oyler GA, Higgins GA, Hart RA, Battenberg E, Billingsley M, Bloom FE, Wilson MC. The identification of a novel synaptosomal-associated proein, SNAP-25, differentially expressed by neuronal subpopulations. J Cell Biol. 1989;109:3039–3052. doi: 10.1083/jcb.109.6.3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters, A., S. Palay, and H.D. Webster. 1991. The axon. In The Fine Structure of the Nervous System. 3rd edition. Oxford University Press, New York. 101–137.
- Porter, K.R., H.R. Byers, and M.H. Ellisman. 1979. The cytoskeleton. In The Neurosciences. Fourth Study Program. F.O. Schmitt and F.G. Worden, editors. MIT Press, Cambridge, MA. 703–722.
- Reaves B, Wilde A, Banting G. Identification, molecular characterization and immunolocalization of an isoform of the trans-Golgi-network (TGN)-specific integral membrane protein TGN38. Biochem J. 1992;283:313–316. doi: 10.1042/bj2830313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaves B, Horn M, Banting G. TGN38/41 recycles between the cell surface and the TGN: brefeldin A affects its rate of return to the TGN. Mol Biol Cell. 1993;4:93–105. doi: 10.1091/mbc.4.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regnier-Vigouroux A, Tooze S, Huttner WB. Newly synthesized synaptophysin is transported to synaptic-like microvesicles via constitutive secretory vesicles and the plasma membrane. EMBO (Eur Mol Biol Organ) J. 1991;10:3589–3601. doi: 10.1002/j.1460-2075.1991.tb04925.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler S, Nargang FE, Steinberg G, Schliwa M. Kinesin is essential for cell morphogenesis and polarized secretion in Neurospora crassa. EMBO (Eur Mol Biol Organ) J. 1997;16:3025–3034. doi: 10.1093/emboj/16.11.3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene JH. Axonal growth-associated proteins. Annu Rev Neurosci. 1989;12:127–156. doi: 10.1146/annurev.ne.12.030189.001015. [DOI] [PubMed] [Google Scholar]
- Smith CB, Betz WJ. Simultaneous independent measurement of endocytosis and exocytosis. Nature. 1996;380:531–534. doi: 10.1038/380531a0. [DOI] [PubMed] [Google Scholar]
- Smith RS. The short term accumulation of axonally transported organelles in the region of localized lesions of single myelinated axons. J Neurocytol. 1980;9:39–65. doi: 10.1007/BF01205226. [DOI] [PubMed] [Google Scholar]
- Sollner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell. 1993;75:409–418. doi: 10.1016/0092-8674(93)90376-2. [DOI] [PubMed] [Google Scholar]
- Sudhof TC, Jahn R. Proteins of synaptic vesicles involved in exocytosis and membrane recycling. Neuron. 1991;6:665–677. doi: 10.1016/0896-6273(91)90165-v. [DOI] [PubMed] [Google Scholar]
- Sudhof TC, Lottspeich F, Greengard P, Mehl E, Jahn R. The cDNA and derived amino acid sequences for rat and human synaptophysin. Nucleic Acids Res. 1987;15:9607. doi: 10.1093/nar/15.22.9607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terasaki, M., and M.E. Dailey. 1995. Confocal microscopy of living cells. In Handbook of Biological Confocal Microscopy. J.B. Pawley, editor. Plenum Press, New York. 327–346.
- Torri-Tarelli F, Villa A, Valtorta F, De Camilli P, Greengard P, Ceccarelli B. Redistribution of synaptophysin and synapsin I during α-latrotoxin-induced release of neurotransmitter at the neuromuscular junction. J Cell Biol. 1990;110:449–459. doi: 10.1083/jcb.110.2.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimble WS, Linial M, Scheller RH. Cellular and molecular biology of the presynaptic nerve terminal. Annu Rev Neurosci. 1991;14:93–122. doi: 10.1146/annurev.ne.14.030191.000521. [DOI] [PubMed] [Google Scholar]
- Tsukita S, Ishikawa H. The movement of membranous organelles in axons. Electron microscopic identification of anterogradely and retrogradely transported organelles. J Cell Biol. 1980;84:513–530. doi: 10.1083/jcb.84.3.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushkaryov YA, Petrenko AG, Geppert M, Sudhof TC. Neurexins: synaptic cell surfce proteins related to the a-latrotoxin receptor and laminin. Science. 1992;257:50–56. doi: 10.1126/science.1621094. [DOI] [PubMed] [Google Scholar]
- Vale RD, Reese TS, Sheetz MP. Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell. 1985;42:39–50. doi: 10.1016/s0092-8674(85)80099-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valtorta F, Jahn R, Fesce R, Greengard P, Ceccarelli B. Synaptophysin (p38) at the frog neuromuscular junction: its incorporation into the axolemma and recycling after intense quantal secretion. J Cell Biol. 1988;107:2717–2727. doi: 10.1083/jcb.107.6.2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veit M, Sollner TH, Rothman JE. Multiple palmitoylation of synaptotagmin and the t-SNARE SNAP-25. FEBS Lett. 1996;385:119–123. doi: 10.1016/0014-5793(96)00362-6. [DOI] [PubMed] [Google Scholar]
- Walworth NC, Novick PJ. Purification and characterization of constitutive secretory vesicles from yeast. J Cell Biol. 1987;105:163–174. doi: 10.1083/jcb.105.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]