

Abstract
We have investigated the role of the ADP- ribosylation induced by brefeldin A (BFA) in the mechanisms controlling the architecture of the Golgi complex. BFA causes the rapid disassembly of this organelle into a network of tubules, prevents the association of coatomer and other proteins to Golgi membranes, and stimulates the ADP-ribosylation of two cytosolic proteins of 38 and 50 kD (GAPDH and BARS-50; De Matteis, M.A., M. DiGirolamo, A. Colanzi, M. Pallas, G. Di Tullio, L.J. McDonald, J. Moss, G. Santini, S. Bannykh, D. Corda, and A. Luini. 1994. Proc. Natl. Acad. Sci. USA. 91:1114–1118; Di Girolamo, M., M.G. Silletta, M.A. De Matteis, A. Braca, A. Colanzi, D. Pawlak, M.M. Rasenick, A. Luini, and D. Corda. 1995. Proc. Natl. Acad. Sci. USA. 92:7065–7069). To study the role of ADP-ribosylation, this reaction was inhibited by depletion of NAD+ (the ADP-ribose donor) or by using selective pharmacological blockers in permeabilized cells. In NAD+-depleted cells and in the presence of dialized cytosol, BFA detached coat proteins from Golgi membranes with normal potency but failed to alter the organelle's structure. Readdition of NAD+ triggered Golgi disassembly by BFA. This effect of NAD+ was mimicked by the use of pre–ADP- ribosylated cytosol. The further addition of extracts enriched in native BARS-50 abolished the ability of ADP-ribosylated cytosol to support the effect of BFA. Pharmacological blockers of the BFA-dependent ADP-ribosylation (Weigert, R., A. Colanzi, A. Mironov, R. Buccione, C. Cericola, M.G. Sciulli, G. Santini, S. Flati, A. Fusella, J. Donaldson, M. DiGirolamo, D. Corda, M.A. De Matteis, and A. Luini. 1997. J. Biol. Chem. 272:14200–14207) prevented Golgi disassembly by BFA in permeabilized cells. These inhibitors became inactive in the presence of pre–ADP-ribosylated cytosol, and their activity was rescued by supplementing the cytosol with a native BARS-50–enriched fraction. These results indicate that ADP-ribosylation plays a role in the Golgi disassembling activity of BFA, and suggest that the ADP-ribosylated substrates are components of the machinery controlling the structure of the Golgi apparatus.
The Golgi apparatus is a complex structure that can be schematically viewed as composed of two basic elements: flat disc-shaped cisternae and tubular- reticular networks. Groups of three to eight cisternae piled in stacks are in continuity with cisternae of adjacent stacks through tubular-reticular elements. The overall tridimensional appearance of the Golgi complex is therefore ribbon-like, with alternating compact (stacked cisternae) and noncompact (tubular-reticular) zones; the cis and trans poles of the complex are made mostly of tubular networks (Tanaka et al., 1986; Rambourg and Clermont, 1990; Clermont et al., 1994). A notable feature of these structures is that despite their complexity they are highly dynamic: stacks can rapidly change shape and tubules can be seen to emanate from, or retract to, the cisternae under a variety of conditions (Lippincott-Schwartz et al., 1989; Cole et al., 1996). Given the central role of the Golgi complex in the secretory process, there is much interest in understanding the molecular mechanisms responsible for generating and maintaining the organelle's structure as well as the relationships existing between such structure and the organelle's functions. However, although recent significant progress mainly based on studies of Golgi reassembly after fragmentation induced by the toxin ilimaquinone or during mitosis (Lucocq and Warren, 1987; Lucocq et al., 1987, 1989; Moskalewski and Thyberg, 1990; Souter et al., 1993; Acharya et al., 1995a ,b; Rabouille et al., 1995a ,b; Warren et al., 1995; Kondo et al., 1997), these mechanisms are still largely obscure.
A potentially important tool to approach the problem of the Golgi structure is brefeldin A (BFA)1, a fungal macrocyclic lactone originally discovered as an antiviral agent and well known for its ability to inhibit constitutive protein secretion (Takatsuki and Tamura, 1985; Misumi et al., 1986; Fujiwara et al., 1988; Magner and Papagiannes, 1988; Doms et al., 1989). BFA causes an impressively rapid and extensive disruption of the Golgi morphology, consisting of the transformation of Golgi stacks into a tubular-reticular network (Lippincott-Schwartz et al., 1989, 1990; Orci et al., 1991) as early as 30 s after its application (Pavelka and Ellinger, 1993). This is followed by the formation of long microtubule-dependent tubules connecting the Golgi region with the cell periphery, and by the redistribution of most of the Golgi resident proteins into the ER (Lippincott-Schwartz et al., 1989, 1990; Alcalde et al., 1992; Pavelka and Ellinger, 1993). It seems likely that understanding how BFA disorganizes the Golgi complex would provide important clues as to how the organelle's structure is normally maintained. It has been hypothesized that the structural effects of BFA are due to the release of coat proteins (COP) from Golgi membranes (Donaldson et al., 1991; Klausner et al., 1992). Indeed, a well-characterized molecular action of BFA is to release a set of polypeptides from the Golgi complex including the coatomer (a major protein complex involved in COPI-coated vesicle formation) and the small GTP binding protein ARF (ADP-ribosylation factor) (Donaldson et al., 1992; Helms and Rothman, 1992). Other proteins released by BFA from Golgi membranes are γ-adaptin, p200, the low-density lipoprotein C (LDLC)-encoded protein, spectrin, and cyclin B (Narula et al., 1992; Robinson and Kreis, 1992; Beck et al., 1994; Podos et al., 1994; Jackman et al., 1995; Erickson et al., 1996). Although the coatomer is indeed likely to play a role in Golgi structure (Guo et al., 1994; Misteli and Warren, 1994, 1995a ,b), the evidence that its detachment from the organelle is the sole cause of BFA-induced Golgi disruption is far from direct or clear. It is not easy to understand, for instance, how coatomer dissociation could mediate the very rapid BFA-induced tubular transformation of Golgi stacks, since the coatomer is localized almost exclusively on cisternal rims, vesicles, buds, and tubule tips, whereas it is virtually absent from the core of Golgi cisternae (Oprins et al., 1993). It seems more likely that multiple factors are involved in the dynamic control of the Golgi shape.
We have recently reported that BFA potently stimulates an endogenous ADP-ribosylation reaction that selectively modifies two cytosolic proteins of 38 kD (GAPDH), and 50 kD (De Matteis et al., 1994). The 50-kD ADP-ribosylation substrate (BARS-50; Di Girolamo et al., 1995) binds GTP and is regulated by βγ subunits of trimeric G proteins; it has been proposed, therefore, to be a novel G protein involved in membrane transport (Di Girolamo et al., 1995). BFA activates ADP-ribosylation both in intact and Triton-solubilized Golgi membranes through a site with a ligand selectivity identical to that involved in the BFA- dependent inhibition of ARF binding (Di Girolamo et al., 1995). Moreover, a series of chemical inhibitors of ADP-ribosylation antagonizes the BFA-induced redistribution of the Golgi complex in intact cells (Weigert et al., 1997). Thus, there is correlative evidence suggesting that ADP- ribosylation plays a role in the cellular actions of BFA. In this study, we use a direct approach to investigate whether ADP-ribosylation is involved in the BFA-induced disassembly of the Golgi complex by using manipulations aimed at controlling the ADP-ribosylated state of BARS-50 and GAPDH in the intracellular space to then examine if the structural effects of BFA are modified. We find that treatments designed to inhibit ADP-ribosylation by depleting permeabilized cells of NAD+ (the ADP-ribose donor in ADP-ribosylation reactions) strongly inhibit the ability of BFA to rapidly transform Golgi stacks into a tubular-reticular network, and that NAD+ restores the ability of BFA to disassemble Golgi stacks. Moreover, the use of ADP-ribosylated cytosol in permeabilized cells mimicked this effect of NAD+ and prevented the effects of ADP-ribosylation inhibitors on Golgi morphology. These results implicate NAD+, ADP-ribosylation, and the proteins involved in this reaction in the mechanisms controlling the structure of the Golgi complex.
Materials and Methods
Cell Culture
Rat basophilic leukemia (RBL)-2H3 cells were grown in DME supplemented with 16% FCS and 1 mM l-glutamine. CHO cells were cultured in DME supplemented with 10% FCS.
Antibodies and Other Reagents
NAD+, NADP+, NADH, BFA, and GAPDH from skeletal rabbit muscles were obtained from Sigma Chemical Co. (St. Louis, MO). Tissue culture materials were from GIBCO BRL (Grand Island, NY) and Seromed (Berlin, Germany). GTP and ATP were from Boehringer Mannheim (Mannheim, Germany). Rabbit anti–α-mannosidase II (Man II) antibody was provided by K. Moremen (University of Georgia, Athens, GA), and a rabbit anti–β-COP antibody by J. Donaldson and J. Lippincott-Schwartz (National Institutes of Health, Bethesda, MD). All other chemicals were obtained from commercial sources at the highest available purity. BFA was stored at −20°C in stock solutions in DMSO. Dicumarol was prepared before use as an aqueous solution.
Cell Permeabilization
RBL (grown in glass chamber slides) were placed on ice and immediately washed with the permeabilization buffer (PB: 25 mM Hepes-Koh, pH 6.95, 125 mM KOAc, 2.5 mM Mg[OAc]2, 10 mM glucose, 1 mM DTT, 1 mM EGTA, and 0.5 μM taxol). Cells were then incubated with 3 U/ml of streptolycin O (SLO) (Biomerieux, Marcy l'Etoile, France), previously activated for 5 min at room temperature in PB for 8 min on ice. Unbound SLO was removed and cell monolayer was washed with cold PB, and then treated with permeabilization buffer supplemented with 1 mg/ml rat brain cytosol, 1 mM ATP, 250 μM UTP, 2 mM creatine phosphate, 7.3 U/ml creatine phosphokinase at 37°C for between 20-30 min (in the presence of the indicated treatments). To check the extent of permeabilization, cells were stained with Trypan blue (and propidium iodide) and the leakage of the cytosolic enzyme lactic dehydrogenase was measured. With the adopted schedule of SLO treatment, 95% of cells were stained with Trypan blue or propidium iodide and >80% of the lactic dehydrogenase activity was recovered in the supernatant of the permeabilized cell monolayer. Rat brain cytosol was prepared according to Malhotra et al. (1989).
BFA-dependent ADP-Ribosylation
ADP-Ribosylation in Permeabilized Cells.
RBL cells were plated in 24-well plates and used after 24 h at 90% confluency (300,000 cells/well per 250 μl). They were permeabilized as described above and then exposed for 20 or 60 min to PB containing 500 μM tymidine, 30 μM 32P-NAD+ (3 μCi/sample) and, where specified, BFA. At the end of the incubations the supernatant and the cell proteins were precipitated with 10% TCA, dissolved in sample buffer, and separated on SDS-PAGE. The radioactivity bound to BARS-50 and GAPDH was evaluated by fluorography.
ADP-Ribosylation of Cytosol.
Cytosol and membranes were prepared from rat brain as described (De Matteis et al., 1994). Cytosol (10 mg/ml) and salt-washed membranes (2 mg/ml) were incubated in the presence or absence of 200 μM NAD+ or 100 μM BFA or both for 60 min at 37°C. Under these experimental conditions the ADP-ribosylation of BARS-50 (evaluated in parallel experiments run in the presence of 32P-NAD+) was maximal (>90%), whereas that of GAPDH was only partial (3–4%). No other proteins were detectably ADP-ribosylated by BFA (see Fig. 3). At the end of the incubation the samples were centrifuged at 100,000 g for 60 min and then the supernatants (cytosol) were dialyzed for 16 h at 4°C and used in immunofluorescence experiments in permeabilized cells as described below.
Figure 3.

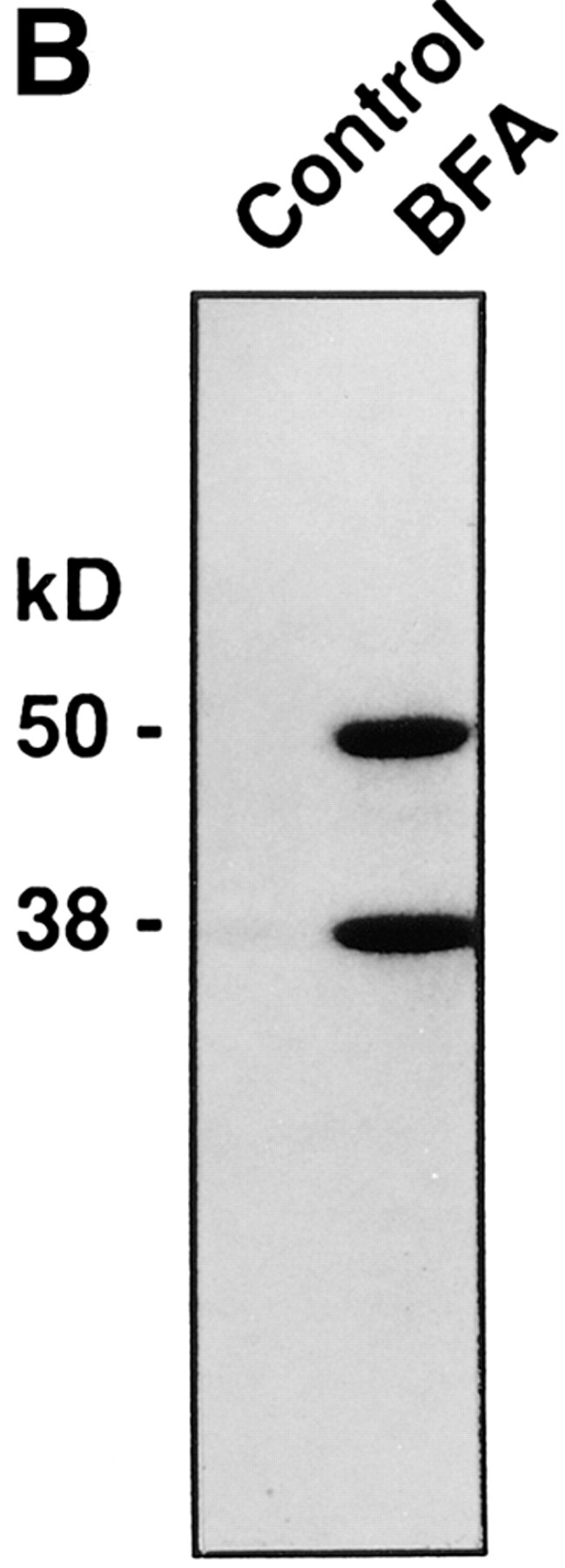
BFA induces the ADP-ribosylation of BARS-50 and GAPDH in permeabilized cells. (A) RBL cells were permeabilized with 3 U/ml SLO and exposed to 10 μg/ml BFA in the presence of 32P-NAD+ for 20 or 60 min at 37°C. At the end of the incubation, proteins were separated on SDS-PAGE, and the radioactivity bound to BARS-50 and GAPDH in the presence of BFA was evaluated by fluorography. Similar results were obtained in four experiments. (B) Cytosol was ADP-ribosylated exactly as described in Materials and Methods. Proteins were separated on SDS-PAGE and the radioactive bands revealed by fluorography. Only BARS-50 and GAPDH were detectably ADP-ribosylated by BFA. GAPDH was also weakly modified in the absence of the toxin, due to a nonenzymatic ADP-ribosylation different from that induced by BFA (De Matteis et al., 1994).
Immunofluorescence and Lectin Staining
Intact or permeabilized RBL cells were fixed in 4% paraformaldehyde in PBS at room temperature for 10 min, quenched in 10 mM NH4Cl for 10 min, washed in PBS, and permeabilized with 0.05% saponin, 0.2% BSA in PBS for 30 min at room temperature. The cells were stained with FITC-conjugated helix pomatia lectin (100 μg/ml in PBS containing 0.2% BSA) for 45 min or incubated with primary antibody for 1 h at room temperature, washed thoroughly with PBS, and incubated with specific FITC-, TRITC-, or Cy3-conjugated secondary antibody for 30 min at room temperature. After thorough washing, slides were mounted in Mowiol 4-88 (Calbiochem-Novabiochem, La Jolla, CA) and examined using a microscope equipped with a Plan-Neofluar 40× objective (Axiophot; Carl Zeiss, Thornwood, NY). RBL or CHO cells grown in glass chamber slides (Nunc, Roskilde, Denmark; intact or permeabilized as described above), were fixed in 4% paraformaldehyde in PBS (pH 7.4) at room temperature for 10 min, quenced in 10 mM NH4Cl for 10 min and then washed in PBS and permeabilized with 0.05% saponin, 0.2% BSA in PBS for 30 min at room temperature. Cells were stained with FITC-conjugated helix pomatia lectin (100 μg/ml in PBS containing 0.2% BSA) for 45 min or incubated with primary antibody for 1 h at room temperature, washed thoroughly with PBS and incubated with specific FITC-, TRITC-, or Cy3-conjuagted secondary antibody for 30 min at room temperative as described earlier (Buccione et al., 1996).
Electron Microscopy
Cells were fixed with 2% glutaraldehyde in PBS (pH 7.4), postfixed with reduced osmium (1% of OsO4 and 1.5% of potassium ferrocianide in 0.1 M cacodilate buffer, pH 7.4), and embedded in Epon 812 as described earlier (Buccione et al., 1996).
Preparation of BARS-50–enriched Cytosolic Fractions
Rat brain cytosol (Malhotra et al., 1989) was precipitated with 35% saturated (NH4)2SO4. The precipitate was dissolved in 25 mM Hepes, pH 8.0, containing 5% glycerol, 0.5 M (NH4)2SO4 and 1 mM DTT (buffer A) and applied to a phenyl sepharose HP column (Pharmacia Biotech, Piscataway, NJ) equilibrated with buffer A. Proteins were eluted with a linear gradient of buffer A minus (NH4)2SO4. The fractions containing BARS-50 were identified by the BFA-dependent ADP-ribosylation assay (De Matteis et al., 1994). These fractions (containing a 45-fold enriched BARS-50 and no GAPDH) were concentrated and dialyzed against buffer B (25 mM Hepes, pH 7.2, 50 mM K, and 1 mM Mg acetate) overnight. The final protein concentration was 2–3 mg/ml.
Results
NAD+ Is Required for the BFA-induced Tubular Reticular Transformation of the Golgi Complex and the Redistribution of Golgi Enzymes into the ER
To examine the effects of NAD+ on the Golgi structure, cells were depleted of the nucleotide by a permeabilization protocol designed to selectively porate the plasma membrane. The resulting membrane damage and cell rounding caused a less than perfect resolution of the Golgi complex at the immunofluorescence level; however, the redistribution of Golgi markers into the ER by BFA remained easily detectable. Moreover, the fine structure of the organelle was very well preserved (see Figs. 1 and 2). It is known that the permeabilization with SLO results in the rapid loss of most low molecular weight soluble molecules, including NAD+, from the cell interior (Bhakdi et al., 1993). In addition, the brain cytosol normally used in experiments with permeabilized cells was extensively dialyzed. Fig. 1 shows that whereas in intact cells BFA caused the expected rapid and complete diffuse redistribution of Golgi markers (with ∼50% effective concentration [EC50] of 0.3 μg/ml), in NAD+-depleted cells (permeabilized and exposed to dialyzed cytosol), the toxin dramatically lost activity (Fig. 1 d) even when used at concentrations up to 100-fold higher than those active in intact cells (not shown). The loss of BFA activity was not due to ATP depletion or microtubule depolymerization (both conditions are known to impede Golgi redistribution), because these experiments were routinely conducted in the presence of an ATP-regenerating system and the microtubule stabilizer taxol.
Figure 1.
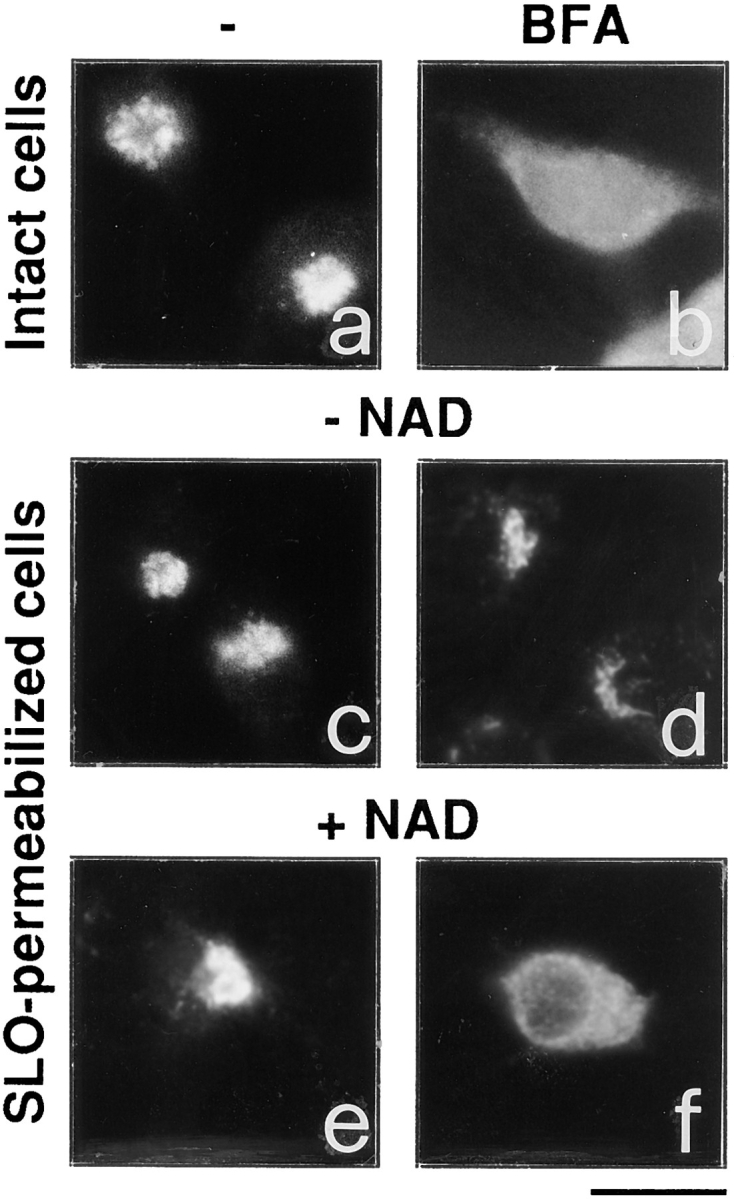
NAD+ is required for the BFA-induced redistribution of Golgi markers in permeabilized cells. Intact RBL cells (a and b) were treated with 3 μg/ml BFA for 15 min (b), or were permeabilized with 3 U/ml SLO (c–f), and incubated for 20 min at 37°C as described in Materials and Methods in the absence (c) or the presence of 150 μM NAD+ (e), or of 10 μg/ml BFA alone (d), or of BFA plus 150 μM NAD+ (f). Cells were then stained with an anti-Man II antibody. Similar results were obtained using the helix pomatia lectin, a marker of the cis Golgi compartment (not shown). Experiments were repeated four times in duplicate with similar results. Bar, 5 μm
Figure 2.
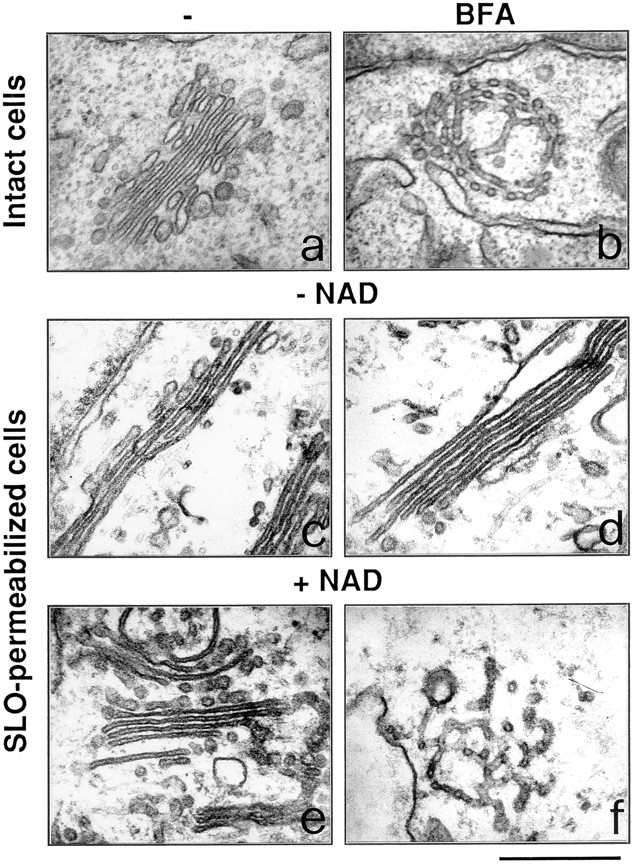
Ultrastructure of the Golgi complex in permeabilized cells: NAD+ is required for the effect of BFA. Intact RBL cells (a and b) were treated with 3 μg/ml BFA for 15 min (b), or were permeabilized with 3 U/ml SLO (c–f), and incubated for 20 min at 37°C in the absence (c) or in the presence of BFA alone (d), or of 150 μM NAD+ (e), or of BFA in combination with 150 μM NAD+ (f). The cells were then processed for electron microscopy. Experiments were repeated at least three times in duplicate with similar results. Bar, 0.5 μm.
The effect of adding NAD+ to the permeabilization medium was then examined. In the presence of the nucleotide (15–450 μM) and dialyzed cytosol (1 mg/ml), BFA strikingly regained its ability to induce the redistribution of the Golgi complex (Fig. 1 f), albeit with a lower potency than in intact cells (EC50: ∼5 μg/ml). Both NAD+ and cytosol were necessary for BFA to express its activity. NAD+ had no visible effect in the absence of BFA (Fig. 1 e). Very high concentrations of BFA (>50 μg/ml) or long incubations with the toxin were able to induce slow Golgi disassembly even in the absence of NAD+ in the incubation medium (not shown). Possibly, at high concentrations of BFA, the cellular NAD+ presumably remaining after permeabilization might be sufficient to sustain Golgi disassembly. To investigate whether this effect of NAD+ might be due to the participation of the nucleotide in redox reactions, NADH (which is inactive as a substrate of ADP- ribosylation) was added together with NAD+ at concentrations up to twice those of the oxidized nucleotide. NADH had no effect on Golgi morphology both in the presence and the absence of BFA (not shown).
The effects of BFA and NAD+ were also investigated at the ultrastructural level. In thin sections of CHO and RBL cells the Golgi complex appears as one or more stacks of cisternae and a number of associated tubular and vesicular profiles (Fig. 2 a). In intact cells, BFA rapidly produced its typical effect consisting of the conversion of the Golgi stacks into a tubular network (Fig. 2 b) and, later, of the disappearance of the Golgi complex. Upon SLO permeabilization, the cells visibly lost their cytoplasmic content but subcellular organelles maintained a nearly normal appearance. The stacked cisternae were fairly well preserved, albeit rather thin, with rare loci of detachment from each other, and with a small population of short tubular and vesicular profiles in their vicinity (Fig. 2 c). In permeabilized cells incubated in the absence of NAD+, in line with the results seen at the immunofluorescence level, BFA had no effect on Golgi morphology even when used at concentrations up to 100-fold higher than those active in intact cells (Fig. 2 d). However, the readdition of NAD+ in the permeabilization buffer strikingly restored the ability of BFA to induce its typical phenotype albeit with a potency lower than that found in intact cells (the effects were visible at 5 μg/ml) (Fig. 2 f). NADP+ and NADH did not modify the effects of NAD+ (not shown). Most of the above experiments in RBL cells were repeated in CHO cells; the findings were similar in the two cell lines (not shown), suggesting a widespread role of NAD+ in the regulation of the Golgi structure.
BFA-dependent ADP-Ribosylation in Permeabilized Cells
Since ADP-ribosylation had been previously studied only in cell homogenates, it was important to verify that the reaction also occurs in permeabilized cells. Cells were porated by SLO under the same conditions used for morphological experiments, exposed to radioactive NAD+ and BFA at concentrations twice the EC50 in permeabilized cells, and the labeling of GAPDH and BARS-50 was evaluated. Fig. 3 A shows that ADP-ribosylation (at ∼10% of the maximal level) was clearly detectable after 20 min. The lack of a stronger signal might be because of slow exchange of BARS-50 (native Mol Wt: ∼200 kD; Di Girolamo et al., 1995) through the SLO-induced pores. This would result in long permanence of BARS-50 within the cell (in close proximity of the ADP-ribosylating enzyme) and, therefore, in high intracellular concentrations of ADP-ribosylated protein, whereas the extracellular cytosol might be ADP-ribosylated to a much lower extent.
Involvement of the ADP-Ribosylation Substrates in the Effects of BFA on Golgi Morphology
If the effect of BFA on the Golgi structure requires ADP-ribosylation, pre–ADP-ribosylated cytosol should mimic the effect of NAD+. ADP-ribosylated cytosol was prepared by incubating brain cytosol with ADP-ribosylating enzyme-containing membranes in the presence of BFA and NAD+. Control cytosols were prepared with only BFA or NAD+ or their vehicles. As previously described, BARS-50 and GAPDH were selectively ADP-ribosylated when both BFA and NAD+ were present during the incubation but not under control conditions (De Matteis et al., 1994; Di Girolamo et al., 1995) (Fig. 3 B). It might be noticed that GAPDH was weakly modified also in the absence of the toxin, but this is known to be due to a nonenzymatic ADP-ribosylation reaction different from the specific modification induced by BFA (De Matteis et al., 1994). Notably, BARS-50 was ADP-ribosylated exhaustively whereas GAPDH was modified to a minor extent (2–4%). No other protein was detectably ADP-ribosylated by BFA (Fig. 3 B). ADP-ribosylated or control cytosols were then separated from membranes by centrifugation and dialyzed extensively. They were indistinguishable from each other in terms of protein composition (as determined by SDS-PAGE), and behaved identically in a functional test measuring their ability to support the vesiculation of the Golgi complex by the nonhydrolyzable analogue of GTP, GTPγS in permeabilized cells (not shown). Both control and ADP-ribosylated cytosols were then tested in experiments of BFA-dependent Golgi disassembly in the absence of NAD+. Whereas control cytosols were inactive (Fig. 4 b), pre– ADP-ribosylated cytosol supported the BFA-induced Golgi tubular transformation (Fig. 4 e). The addition of NAD+ did not noticeably change the activity of pre–ADP-ribosylated cytosol (Fig. 4 f) whereas, in agreement with previous experiments (Fig. 1 f), the nucleotide restored the ability of control cytosols to support the activity of BFA (Fig. 4 c). It was next tested whether the addition of extracts enriched in native (nonADP-ribosylated) BARS-50 and/or GAPDH would reverse the effects of the ADP- ribosylated cytosol: ADP-ribosylated cytosol was mixed with either purified GAPDH or enriched native BARS-50 (Silletta, M.G., manuscript in preparation) and assayed for its ability to support the activity of BFA (Fig. 4, g–i). GAPDH had no effect (not shown); instead, native BARS-50 (in the absence of NAD+), reversed the effects of ADP-ribosylated cytosol (Fig. 4 g). Pre–ADP-ribosylated BARS-50 or native BARS-50 with NAD+ (Fig. 4, h and i) were, as expected, without effect. Altogether, the data indicate that BARS-50, in the native form, acts to prevent the Golgi-disassembling action of BFA, and that ADP-ribosylation inactivates the protein. The role of GAPDH, if any, remains unclear.
Figure 4.

Pre–ADP-ribosylated cytosol replaces NAD+ in sustaining the Golgi-disassembling activity of BFA. Effects of BARS-50–containing extracts. RBL cells were permeabilized with 3 U/ml SLO and incubated with control (a–c) or ADP-ribosylated (d–i) cytosol (1 mg/ml) for 20 min at 37°C in the absence (a and d) or in the presence (b, c, and e–i) of 10 μg/ml BFA. Native BARS-50 (an extract prepared as described in Materials and Methods and diluted 10-fold in ADP-ribosylated cytosol) was added in i (with NAD+) and in g without NAD+. ADP-ribosylated BARS-50 (an extract identical to that containing native BARS-50 but prepared from ADP-ribosylated cytosol; see Materials and Methods) was added to h. Cells were fixed and labeled with anti-Man II antibody. Similar results were obtained in four different experiments. Bar, 5 μm.
Role of Coatomer
It is known that BFA induces the cytosolic redistribution of coatomer from Golgi membranes and that this effect precedes the disassembly of the Golgi complex. Based on this temporal association, it has been proposed that coatomer redistribution is a major cause of the effects of BFA on Golgi morphology (Donaldson et al., 1991). Therefore, we wanted to test whether the effect of BFA on coatomer dissociation requires NAD+. Fig. 5 shows, however, that BFA in permeabilized cells induces the cytosolic redistribution of the coatomer (as revealed by antibodies against β-COP), with a potency similar to that reported in intact cells (Fig. 5 b), both in the presence and in the absence of NAD+ (Fig. 5, d and f). Also, the use of pre–ADP-ribosylated cytosol did not influence the effect of BFA on coatomer (Fig. 5, g and h). The effect of BFA was clearly detectable although permeabilization itself induced partial dissociation of β-COP from the Golgi apparatus in some cells (Donaldson et al., 1991). As noted above, by contrast, BFA requires NAD+ or pre–ADP-ribosylated cytosol to cause Golgi disassembly (Figs. 1 and 4). Thus, in the absence of NAD+, BFA can dissociate coatomer from the Golgi complex without affecting the structure of the organelle; only the presence of the nucleotide or of ADP-ribosylated substrates allows the toxin to express its effects on Golgi morphology.
Figure 5.
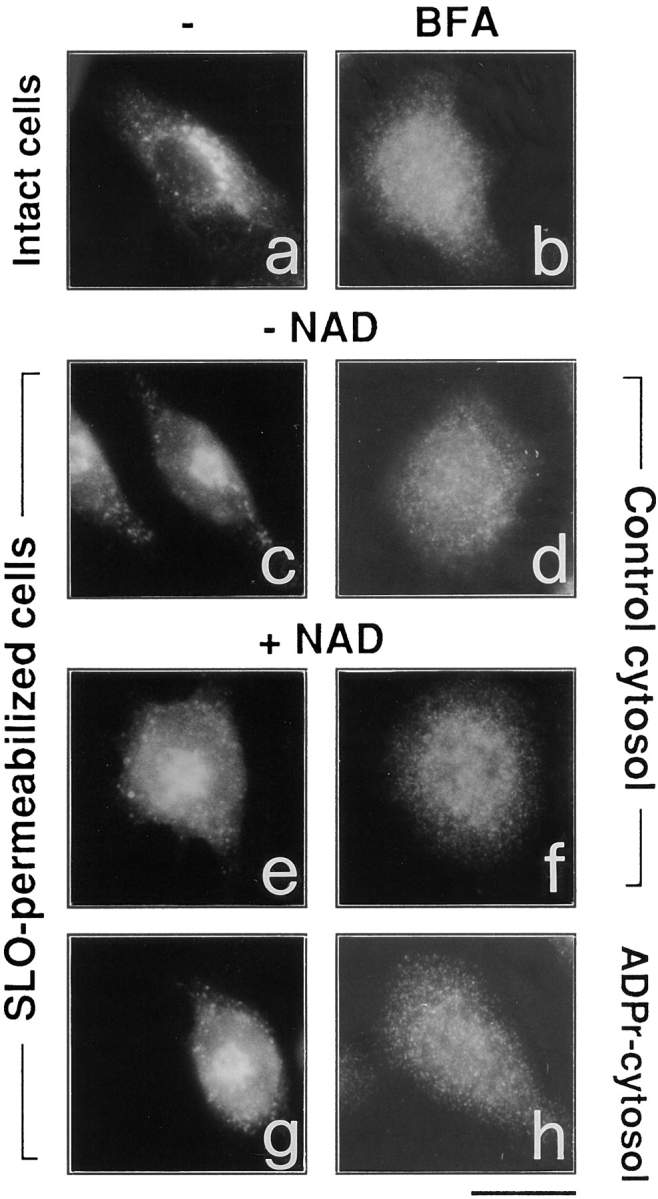
NAD+ is not required for BFA-induced coatomer dissociation from the Golgi complex in permeabilized cells. Intact RBL cells (a and b) were treated with 3 μg/ml BFA (b), or were permeabilized with 3 U/ml SLO and then exposed to control buffer (c), or to a buffer containing 150 μM NAD+ (e), or 10 μg/ml BFA alone (d), or BFA in combination with 150 μM NAD+ (f). The cells were fixed, permeabilized with saponin, and stained with anti–β-COP antibody. SLO permeabilization induces a partial detachment of β-COP from Golgi complex (c) compared to intact cells (a), but BFA is completely effective in inducing the total cytosolic redistribution of β-COP independently of the presence of NAD+ in the permeabilization buffer (d and f). Pre–ADP-ribosylated cytosol (g and h) behaves indistinguishably from control cytosol. Similar results were obtained in permeabilized CHO cells (not shown). The experiments were repeated four times in duplicate with similar results. Bar, 5 μm.
Inhibitors of BFA-dependent ADP-Ribosylation Prevent the BFA-induced Golgi Disassembly. Role of the ADP-Ribosylation Substrates
As recently reported, several compounds belonging to two different chemical groups, one containing a coumarin, and the other a quinone ring, act as inhibitors of the BFA- dependent ADP-ribosylation in vitro (Weigert et al., 1997). Dicumarol and ilimaquinone, a marine sponge metabolite that causes the gradual and reversible breakdown of the Golgi complex (Takizawa et al., 1993), are relatively potent and nontoxic representatives of the two classes of compounds. Remarkably, ADP-ribosylation inhibitors are able to antagonize the BFA-induced redistribution of Golgi enzymes into the ER in intact cells (Weigert et al., 1997), with potencies similar to those observed in assays of BFA-dependent ADP-ribosylation in vitro. This suggests that they inhibit the BFA-induced Golgi disassembly by inhibiting ADP-ribosylation. We wanted to directly test this possibility by assessing whether ADP-ribosylation inhibitors would lose their effect in permeabilized cells exposed to pre–ADP-ribosylated cytosol. The effect of these agents on the fine structure of the Golgi complex, both in intact and permeabilized cells, was first characterized. In intact cells, as expected, a prominent early effect of BFA was the disorganization and tubular-vesicular transformation of the Golgi complex (Fig. 2 b); dicumarol (Fig. 6 a) or ilimaquinone (not shown) strongly inhibited these alterations and, in fact, afforded a remarkable preservation of the stack structure. The effects of the inhibitors, in line with previous data (Weigert et al., 1997), were dose-dependent and dependent on the dose of BFA, since higher concentrations of the toxin overcame the inhibition (Fig. 6 b). In permeabilized cells, dicumarol had very similar effects to those seen in vivo in that it inhibited the BFA and NAD+-induced Golgi disassembly in the presence of control cytosol (Fig. 7 b). It was then tested whether dicumarol would maintain its ability to antagonize BFA in the presence of pre–ADP-ribosylated cytosol. Remarkably, under these conditions, the effect of dicumarol was largely prevented (Fig. 7 d). Furthermore, when the pre–ADP-ribosylated cytosol was complemented with enriched native (nonADP-ribosylated) BARS-50 and NAD+ (Fig. 7 f), dicumarol regained its property to prevent the BFA-induced redistribution of the Golgi apparatus. GAPDH had no effect (not shown). The addition of pre–ADP-ribosylated BARS-50 under the same conditions was unable to restore dicumarol activity (Fig. 7 h). These experiments indicate that dicumarol either acts by preventing the ADP-ribosylation of BARS-50 or that it requires unmodified BARS-50 to exert its effects.
Figure 6.
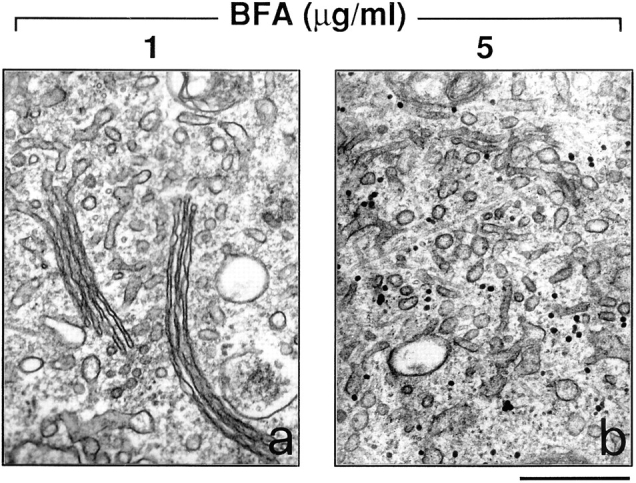
Dicumarol prevents the tubular-reticular transformation of the Golgi apparatus induced by BFA. RBL cells were treated with the indicated BFA concentrations for 15 min after a 30-min pretreatment with 200 μM dicumarol. They were then processed for electron microscopy. Dicumarol (and ilimaquinone, not shown) prevents the tubular-reticular transformation and disappearance of the Golgi stacks induced by moderate (a), but not by high concentrations of BFA (b). Similar results were obtained in three independent experiments run in duplicate. Bar, 0.5 μm.
Figure 7.
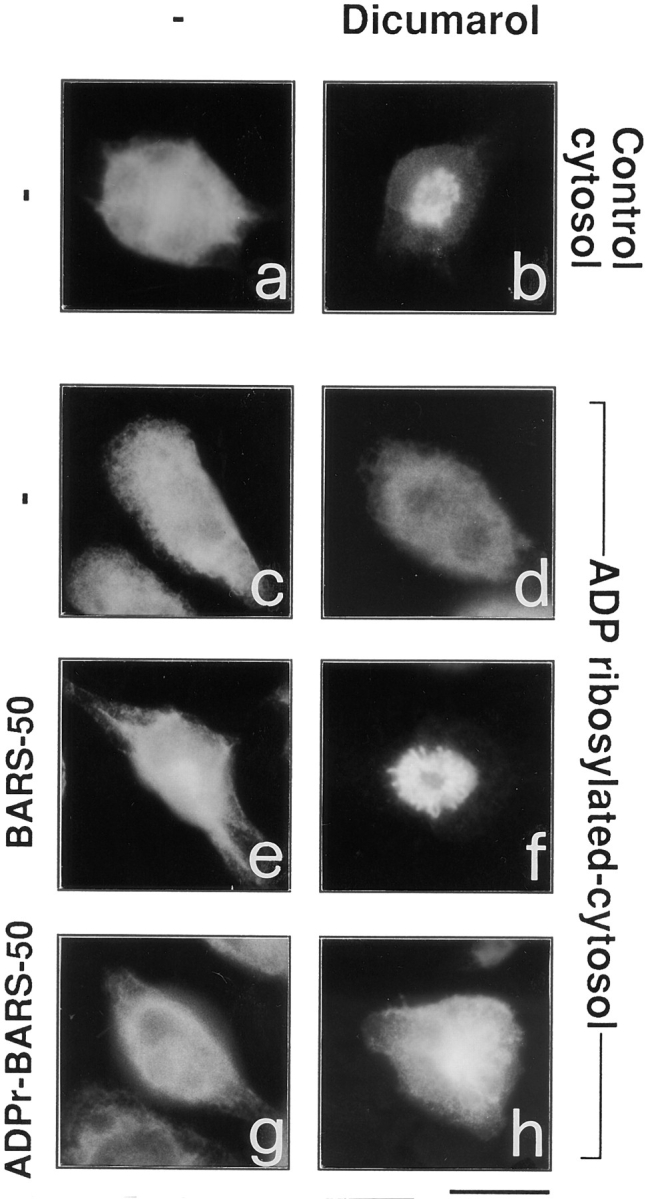
Pre–ADP-ribosylated cytosol prevents and native BARS-50 rescues the anti-BFA effects of dicumarol on the Golgi complex in permeabilized cells. RBL cells were permeabilized with 3 U/ml SLO, and incubated for 20 min at 37°C in a medium containing BFA (10 μg/ml) and NAD+ (150 μM) without (a, c, e, and g) or with 200 μM dicumarol (b, d, f, and h) in the presence of control (a and b) or pre–ADP-ribosylated (c–h) cytosol (1 mg/ml). A native BARS-50–enriched extract (see Fig. 4 legend) was added to (e) and (f), whereas ADP-ribosylated BARS-50 was added to (g) and (h). The cells were fixed and labeled with anti-Man II antibody. Similar results were obtained in three independent experiments. Bar, 5 μm.
Discussion
NAD+ Is Required for Golgi Disassembly by BFA
In permeabilized cells exposed to BFA at concentrations active in intact cells and in the presence of ATP and dialyzed cytosol, the addition of physiological concentrations of NAD+ is required for disassembly of the Golgi complex. The effect of NAD+ was not mediated by changes in ATP levels or the state of microtubules (both are factors already known to affect Golgi dynamics; for review see Klausner et al., 1992), since it was observed under conditions where both microtubules and ATP levels are kept stable. It remains unclear whether the requirement for NAD+ is absolute under all conditions, because very high concentrations of BFA (50 or more μg/ml) can cause a partial disorganization of the Golgi apparatus even without addition of the pyridine nucleotide in the permeabilization medium. This can be explained in two ways. One is that very high levels of BFA can somehow bypass the NAD+ requirement. Alternately, and, in our view, more likely, NAD+-depleted cells most probably still contain low amounts of the nucleotide. Our procedure for NAD+ depletion (cell permeabilization by SLO and dialysis of the cytosol) is unlikely to be completely effective, because of tight binding of NAD+ to cytosolic proteins and because a large fraction of the pyridine nucleotide is trapped inside organelles, mainly mitochondria, that are not porated by SLO (Bhakdi et al., 1993). Thus, conceivably, high BFA concentrations can activate ADP-ribosylation to an extent sufficient to sustain partial Golgi disorganization even in the presence of low levels of NAD+. More work is needed to establish whether NAD+ is a necessary, or a potent facilitatory component, of the machinery involved in Golgi disassembly. For brevity, however, in the rest of the discussion, the role of NAD+ will be referred to as required for Golgi disassembly. The requirement for NAD+ has been used in the past as one of the criteria to define the role of ADP-ribosylation in the mechanism of action of bacterial toxins such as cholera and pertussis toxins (Moss and Vaughan, 1988; Okazaki and Moss, 1994). Clearly, it is not sufficient alone; it must be combined with converging lines of evidence to establish the role of ADP-ribosylation in the action of BFA.
ADP-Ribosylated Cytosol Replaces NAD+ in Supporting BFA-induced Golgi Disassembly
The main such piece of evidence is that ADP-ribosylated cytosol can substitute for NAD+ in enabling BFA to alter the Golgi structure. Several observations indicate that the ADP-ribosylation of the cytosolic substrates, rather than some unknown concomitant BFA-induced modification, is responsible for the activity of ADP-ribosylated cytosol. First, only the coincubation of cytosol with both BFA and NAD+, but not with BFA or NAD+ alone, is effective in causing the cytosol to support Golgi disassembly. While BFA and NAD+ may have different effects, the only known consequence of combining the two agents is the ADP-ribosylation of BARS-50 and GAPDH. Second, the effects of BFA and NAD+ in permeabilized cells are prevented by ADP-ribosylation inhibitors. Third, these inhibitors become unable to antagonize BFA in the presence of ADP-ribosylated cytosol. A question raised by these results is whether the effects of ADP-ribosylation depend on the loss or gain of function of the target proteins. Complementing ADP-ribosylated cytosol with extracts enriched in native BARS-50 reversed the effects of the pre–ADP-ribosylated cytosol, despite the presence of the cytosolic ADP-ribosylated BARS-50. It appears, therefore, that this protein normally acts to prevent the action of BFA and that it loses its activity in the ADP-ribosylated state. Interestingly, GAPDH was inactive in its native and ADP-ribosylated forms. Thus, although a role for GAPDH cannot yet be excluded, these results also suggest that BARS-50 is likely to be the ADP-ribosylation substrate involved in modulating the action of BFA on the Golgi structure.
ADP-Ribosylation Inhibitors Prevent the Effects of BFA on Golgi Structure and Act Via the Cytosolic ADP-Ribosylation Substrates
We have previously described a series of synthetic molecules endowed with the properties to prevent the BFA- dependent ADP-ribosylation in vitro, and reported that they inhibit the effects of BFA on the Golgi in vivo (Weigert et al., 1997). Certain indirect lines of evidence including the facts that (a) the profile of activity of these drugs as inhibitors of ADP-ribosylation in vitro is similar to their profile as antagonists of BFA in vivo, and (b) they inhibit Golgi redistribution by antagonizing BFA in an apparently selective fashion, rather than through toxic or nonspecific effects, suggesting that the block of ADP-ribosylation and the inhibition of BFA in vivo are causally linked (Weigert et al., 1997). In this report, we have built on these findings to provide more direct evidence that these inhibitors indeed act through the ADP-ribosylation substrates. First, in permeabilized as well as intact cells, the inhibitors antagonized the effect of BFA and NAD+. Second and more important, when the roles of ADP-ribosylation in their action were directly tested by replacing NAD+ with pre–ADP-ribosylated cytosol as a means to support the effect of BFA, the inhibitors dramatically lost activity. Moreover, they recovered activity when native BARS- 50-containing extracts were added to the ADP-ribosylated cytosol in the presence of NAD+. Altogether, these findings indicate that the ADP-ribosylation inhibitors act as BFA antagonists by preventing the ADP-ribosylation reaction. Whether they do so by binding the enzyme or the substrates remains to be defined.
ADP-Ribosylation of the Substrate Proteins Occurs in Permeabilized Cells
Another evidence for a role of ADP-ribosylation in Golgi disassembly is that this reaction occurs under the same conditions used for morphological experiments in permeabilized cells. Thus, the ADP-ribosylated substrates are generated concomitantly with the developement of the Golgi disorganization by BFA. A difficulty concerning these experiments is the limited extent of ADP-ribosylation: BFA, at a concentration twice the EC50 for inducing Golgi disruption, caused only a fraction of of BARS-50 to become ADP-ribosylated (Fig. 3). This, in view of the fact that ADP-ribosylation seems to act via loss of function of the protein, as discussed above, would appear inconsistent with a major role for ADP-ribosylation in the action of BFA. This discrepancy may be resolved by the fact that the exchange of BARS-50 (with a native mol wt of ∼200 kD; Di Girolamo et al., 1995) through the SLO-induced pores is most probably slow. Therefore, once it has entered the cell, the protein is likely to be rapidly ADP-ribosylated and then reside for a relatively long time in intracellular compartments, in the proximity of the ADP-ribosylation enzyme, before leaking out back into the extracellular cytosol. This would result in a much higher ratio of ADP-ribosylated over nonADP-ribosylated protein molecules in the cytoplasmic space than that detected int he external or the total cytosol. The intracellular levels of ADP-ribosylation, therefore, might be sufficient to support Golgi disassembly.
Altogether, the above findings on the role of NAD+ and ADP-ribosylated cytosol strongly support a role for NAD+ and ADP-ribosylation in the BFA-sensitive machinery controlling the Golgi architecture. ADP-ribosylation, however, is not sufficient to explain the morphological effects of the toxin, since the ADP-ribosylated cytosol was unable to induce the BFA phenotype alone. Additional mechanisms, most likely including the BFA-induced dissociation of the coatomer from Golgi membranes, must be required for Golgi disruption.
Role of the Coatomer and of NAD+-dependent Mechanisms in Golgi Disassembly
In the absence of NAD+, BFA, while inactive on the morphology of the Golgi complex, preserved its ability to cause the detachment of coatomer-based coats from Golgi stacks (Figs. 1–3). The dissociation revealed that in these experiments between coatomer detachment and Golgi disassembly may seem at odds with recent literature suggesting a requirement for the coatomer to maintain the Golgi structure. It has been reported that (a) isolated Golgi stacks incubated in vitro with coatomer-depleted mitotic cytosol lose their structure and change into tubular networks (Misteli and Warren, 1994); and, (b) that CHO cells with a defective ε-COP (a coatomer component) exhibit a tubular-vesicular dissociation of the Golgi complex (Guo et al., 1994). We believe that the seeming discrepancy between these results and ours can be explained by differences between the experimental systems used in the three laboratories: for instance, in permeabilized cells (our conditions), the overall cellular structure and the cytoskeleton were preserved whereas Misteli and Warren (1994) used isolated Golgi stacks in vitro; in cells with mutated ε-COP (Guo et al., 1994), the disorganization of the Golgi structure develops over long periods of time and may involve mechanisms that are quite different from those rapidly triggered by BFA, with which we are concerned. It seems reasonable to conclude that both the coatomer and a NAD+-dependent factor(s) participate in the regulation of the Golgi structure, and that their relative importance might depend on experimental conditions. Moreover, our data do not exclude; in fact, they suggest that coatomer detachment by BFA might be necessary for NAD+ to express its disassembling action on the Golgi complex.
What might be the process affected by the NAD+ and ADP-ribosylation–dependent mechanism? The disassembly and redistribution of the Golgi apparatus induced by BFA is a complex event, the key steps of which have not been clearly identified. One possibility is that the primary effect of BFA is the induction or stabilization of tubules emanating from the Golgi stacks. Alternately, the disorganization of the organelle might result from the disruption of a protein scaffold involved in maintaining the stack, and tubulation might be secondary to the loss of structure. The nature of this putative scaffold is unknown. Morphological studies reveal that proteinaceous bridges resembling triad junctions between transverse tubules and the sarcoplasmic reticulum, as well as less structured matrix, seem to connect adjacent cisternae (Cluett and Brown, 1992). Large polymers of Golgi enzymes have been proposed to form in the flat portions of the cisternae, to bind to the intercisternal matrix, and play a role in maintaining the cisternal structure (Nilsson and Warren, 1994; Nilsson et al., 1994; Slusarewicz et al., 1994); putative components of the intercisternal matrix proteins have been identified by various means (Kooy et al., 1992; Fritzler et al., 1993; Rios et al., 1994; Slusarewicz et al., 1994; Nakamura et al., 1995). Moreover, cytoskeletal proteins (comitin, spectrin, and ankyrin) have been found to be associated to the Golgi complex (Weiner et al., 1993; Beck et al., 1994, 1997; Devarajan et al., 1996; Viel and Branton, 1996). It is possible that one or some of these putative Golgi scaffold proteins may be regulated by NAD+ and ADP-ribosylation. This hypothesis can now be tested by examining the effects of NAD+ and of the two ADP-ribosylation protein substrates (BARS-50 and GAPDH; De Matteis et al., 1994; Di Girolamo et al., 1995) on the state of the above Golgi protein complexes. The properties of BARS-50 are compatible with a regulatory role in the dynamics of the Golgi structure. BARS-50 binds GTP and is regulated by the βγ subunit of trimeric GTPases (Di Girolamo et al., 1995); it has been suggested, therefore, to be a novel G protein involved in controlling the secretory pathway. GAPDH also has interesting features in that it is a multifunctional protein that, in addition to functioning in glycolysis, displays a number of other unrelated activities: for instance, it associates with the anion exchanger (band 3) on the plasma membrane as well as with microtubules and microfilaments, and it promotes the formation of triad junctions between transverse tubules and terminal cisternae of the sarcoplasmic reticulum (Caswell and Corbett, 1985). The apparent lack of effect of pure GAPDH in our assays might be because of the inability of the commercial protein to mimic the activity of the endogenous one, or, more interestingly, the possibility that GAPDH might be involved in the effects of BFA on the endocytic pathway, which we do not follow with our assays. Intriguingly, it has been reported that CHO cells expressing a mutated form of GAPDH exhibit tubular extensions emanating from late endocytic compartments that are reminiscent of the BFA-induced tubules in the endocytic compartments (Peters et al., 1995).
The Golgi apparatus, despite its complexity, is a very dynamic organelle, as observed most dramatically by the rapid and reversible effects of BFA. This study proposes that NAD+ and ADP-ribosylation are novel factors in the machinery controlling the structure of the Golgi complex and, in particular, of its tubular-reticular transformation in response to BFA. It also opens new questions concerning the significance of the NAD+-dependent regulation in the physiology of this organelle, and the precise role(s) of the ADP-ribosylation protein substrates.
Acknowledgments
We thank G. Lenaz and M. Cavazzoni (University of Bologna, Bologna, Italy) for helpful comments and discussion, K.W. Moremen (University of Georgia) for the anti-Man II polyclonal antibody, J. Donaldson and J. Lippincott-Schwartz (National Institutes of Health) for the polyclonal antibody to β-COP, and R. Bertazzi (Consorzio Mario Negri Sud) for preparation of the figures.
Abbreviations used in this paper
- BARS-50
BFA-dependent ADP-ribosylation substrate of 50 kD
- BFA
brefeldin A
- COP
coat proteins
- EC
effective concentration
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- Man
mannosidase
- PB
permeabilization buffer
- RBL
rat basophilic leukemia cells
- SLO
streptolycin O
Footnotes
This research was supported in part by a grant from the Italian National Research Council (Convenzione Consiglio Nazionale delle Ricerche-Consorzio Mario Negri Sud and Progetto Finalizzato contract #96.00755.PF39) and the Italian Association for Cancer Research. R. Weigert is the recipient of a fellowship from the Centro di Formazione e Studi per il Mezzogiorno.
Address all correspondence to Alexander Mironov and Alberto Luini, Department of Cell Biology and Oncology, Consorzio Mario Negri Sud, Via Nazionale, 66030 S. Maria Imbaro (Chieti), Italy. Tel.: 39.872.570.334. Fax: 39.872.578.240. E-mail: mironov@cmns.mnegri.it and luini@cmns.mnegri.it
References
- Acharya U, McCaffery JM, Jacobs R, Malhotra V. Reconstitution of vesiculated Golgi membranes into stacks of cisternae: requirement of NSF in stack formation. J Cell Biol. 1995a;129:577–589. doi: 10.1083/jcb.129.3.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acharya U, Jacobs R, Peters J-M, Watson N, Farquhar MG, Malhotra V. The formation of Golgi stacks from vesiculated Golgi membranes requires two distinct fusion events. Cell. 1995b;82:895–904. doi: 10.1016/0092-8674(95)90269-4. [DOI] [PubMed] [Google Scholar]
- Alcalde J, Bonay P, Roa A, Vilaro S, Sandoval IV. Assembly and disassembly of the Golgi complex: two processes arranged in a cis-transdirection. J Cell Biol. 1992;116:69–83. doi: 10.1083/jcb.116.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck KA, Buchanan JA, Malhotra V, Nelson WJ. Golgi spectrin: identification of an erythroid β-spectrin homologue associated with the Golgi complex. J Cell Biol. 1994;127:707–723. doi: 10.1083/jcb.127.3.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck KA, Buchanan JA, Nelson WJ. Golgi membrane skeleton: identification and oligomerization of a 195-kD ankyrin isoform associated with the Golgi complex. J Cell Sci. 1997;110:1239–1249. doi: 10.1242/jcs.110.10.1239. [DOI] [PubMed] [Google Scholar]
- Bhakdi S, Weller U, Walev I, Martin E, Jonas D, Palmer M. A guide to the use of pore-forming toxins for controlled permeabilization of cell membranes. Med Microbiol Immunol. 1993;182:167–175. doi: 10.1007/BF00219946. [DOI] [PubMed] [Google Scholar]
- Buccione R, Bannykh S, Santone I, Baldassarre M, Facchiano F, Bozzi Y, Di Tullio G, Mironov A, Luini A, De Matteis MA. Regulation of constitutive exocytic transport by membrane receptors. A biochemical and morphometric study. J Biol Chem. 1996;271:3523–3533. doi: 10.1074/jbc.271.7.3523. [DOI] [PubMed] [Google Scholar]
- Caswell AH, Corbett AM. Interaction of glyceraldehyde-3-phosphate dehydrogenase with isolated microsomal subfractions of skeletal muscle. J Biol Chem. 1985;260:6892–6898. [PubMed] [Google Scholar]
- Clermont Y, Rambourg A, Hermo L. Connections between the various elements of the cis- and midcompartments of the Golgi apparatus of early rat spermatids. Anat Rec. 1994;240:469–480. doi: 10.1002/ar.1092400405. [DOI] [PubMed] [Google Scholar]
- Cluett EB, Brown WJ. Adhesion of Golgi cisternae by proteinaceous interactions: intercisternal bridges as putative adhesive structures. J Cell Sci. 1992;103:773–784. doi: 10.1242/jcs.103.3.773. [DOI] [PubMed] [Google Scholar]
- Cole NB, Smith CL, Sciaky M, Terasaki M, Edidin M, Lippincott-Schwartz J. Diffusion mobility of Golgi proteins in membranes of living cells. Science. 1996;273:797–801. doi: 10.1126/science.273.5276.797. [DOI] [PubMed] [Google Scholar]
- De Matteis MA, Di Girolamo M, Colanzi A, Pallas M, Di Tullio G, McDonald LJ, Moss J, Santini G, Bannykh S, Corda D, Luini A. Stimulation of endogenous ADP-ribosylation by brefeldin A. Proc Natl Acad Sci USA. 1994;91:1114–1118. doi: 10.1073/pnas.91.3.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devarajan P, Stabach PR, Mann AS, Arbito T, Kashagarian M, Morrow JS , Identification of a small cytoplasmic ankyrin (AnkG119) in the kidney and muscle that binds βIε* spectrin and associates with the Golgi apparatus. J Cell Biol. 1996;133:819–830. doi: 10.1083/jcb.133.4.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Girolamo M, Silletta MG, De Matteis MA, Braca A, Colanzi A, Pawlak D, Rasenick MM, Luini A, Corda D. Evidence that the 50-kD substrate of brefeldin A-dependent ADP-ribosylation binds GTP and is modulated by the G-protein βg subunit complex. Proc Natl Acad Sci USA. 1995;92:7065–7069. doi: 10.1073/pnas.92.15.7065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doms RW, Russ G, Yewdell JW. Brefeldin A redistributes resident and itinerant Golgi proteins to the endoplasmic reticulum. J Cell Biol. 1989;109:61–72. doi: 10.1083/jcb.109.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson JG, Lippincott-Schwartz J, Klausner RD. Guanine nucleotides modulate the effects of brefeldin A in semipermeable cells: regulation of the association of a 110-kD peripheral membrane protein with the Golgi apparatus. J Cell Biol. 1991;112:579–588. doi: 10.1083/jcb.112.4.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson JG, Finazzi D, Klausner RD. Brefeldin A inhibits Golgi membrane-catalyzed exchange of guanine nucleotide onto ARF protein. Nature. 1992;360:350–352. doi: 10.1038/360350a0. [DOI] [PubMed] [Google Scholar]
- Erickson JW, Zhang C, Kahan RA, Evans T, Cerione RA. Mammalian Cdc42 is a brefeldin A-sensitive component of the Golgi apparatus. J Biol Chem. 1996;271:26850–26854. doi: 10.1074/jbc.271.43.26850. [DOI] [PubMed] [Google Scholar]
- Fritzler MJ, Hamel JC, Ochs RL, Chan EKL. Molecular characterization of two human autoantigens: unique cDNAs encoding 95- and 160-kD proteins of a putative family in the Golgi complex. J Exp Med. 1993;178:49–62. doi: 10.1084/jem.178.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara T, Oda K, Yokota S, Takatsuki A, Ikehara Y. Brefeldin A causes disassembly of the Golgi complex and accumulation of secretory proteins in the endoplasmic reticulum. J Biol Chem. 1988;263:18545–18552. [PubMed] [Google Scholar]
- Guo Q, Vasile E, Krieger M. Disruptions in Golgi structure and membrane traffic in a conditional lethal mammalian cell mutant are corrected by e-COP. J Cell Biol. 1994;125:1213–1224. doi: 10.1083/jcb.125.6.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helms JB, Rothman JE. Inhibition by brefeldin A of a Golgi membrane enzyme that catalyzes exchange of guanine nucleotide bound to ARF. Nature. 1992;360:352–354. doi: 10.1038/360352a0. [DOI] [PubMed] [Google Scholar]
- Jackman M, Firth M, Pines J. Human cyclins B1 and B2 are localized to strikingly different structures: B1 to microtubules, B2 primarily to the Golgi apparatus. EMBO (Eur Mol Biol Organ) J. 1995;8:1646–1654. doi: 10.1002/j.1460-2075.1995.tb07153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausner RD, Donaldson JG, Lippincott-Schwartz J. Brefeldin A: insights into the control of membrane traffic and organelle structure. J Cell Biol. 1992;116:1071–1080. doi: 10.1083/jcb.116.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo H, Rabouille C, Newman R, Levine TP, Peppins D, Freemont P, Warren G. p47 is a cofactor for p97-mediated membrane fusion. Nature. 1997;388:75–78. doi: 10.1038/40411. [DOI] [PubMed] [Google Scholar]
- Kooy J, Toh B-H, Pettitt JM, Erlich R, Gleeson PA. Human autoantibodies as reagents to conserved Golgi components. Characterization of a peripheral, 230-kD compartment-specific Golgi protein. J Biol Chem. 1992;267:20255–20263. [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Yuan LC, Bonifacino JS, Klausner RD. Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: evidence for membrane cycling from Golgi to ER. Cell. 1989;56:801–813. doi: 10.1016/0092-8674(89)90685-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Donaldson JG, Schweizer A, Berger EG, Hauri HP, Yuan LC, Klausner RD. Microtubule-dependent retrograde transport of proteins into the ER in the presence of brefeldin A suggests an ER recycling pathway. Cell. 1990;60:821–836. doi: 10.1016/0092-8674(90)90096-w. [DOI] [PubMed] [Google Scholar]
- Lucocq JM, Warren G. Fragmentation and partitioning of the Golgi apparatus during mitosis in HeLa cells. EMBO (Eur Mol Biol Organ) J. 1987;6:3239–3246. doi: 10.1002/j.1460-2075.1987.tb02641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucocq JM, Pryde JG, Berger EG, Warren G. A mitotic form of the Golgi apparatus in HeLa cells. J Cell Biol. 1987;104:865–874. doi: 10.1083/jcb.104.4.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucocq JM, Berger EG, Warren G. Mitotic Golgi fragments in HeLa cells and their role in the reassembly pathway. J Cell Biol. 1989;109:463–474. doi: 10.1083/jcb.109.2.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magner JA, Papagiannes E. Blockade by brefeldin A of intracellular transport of secretory proteins in mouse pituitary cells: effects on the biosynthesis of thyrotropin and free a-subunits. Endocrinology. 1988;122:912–920. doi: 10.1210/endo-122-3-912. [DOI] [PubMed] [Google Scholar]
- Malhotra V, Serafini T, Orci L, Shepherd JC, Rothman JE. Purification of a novel class of coated vesicles mediating biosynthetic protein transport through the Golgi stack. Cell. 1989;58:329–336. doi: 10.1016/0092-8674(89)90847-7. [DOI] [PubMed] [Google Scholar]
- Misteli T, Warren G. COP-coated vesicles are involved in the mitotic fragmentation of Golgi stacks in a cell-free system. J Cell Biol. 1994;125:269–282. doi: 10.1083/jcb.125.2.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli T, Warren G. Mitotic disassembly of the Golgi apparatus in vivo. J Cell Sci. 1995a;108:2715–2727. doi: 10.1242/jcs.108.7.2715. [DOI] [PubMed] [Google Scholar]
- Misteli T, Warren G. A role for tubular networks and COPI-independent pathway in the mitotic fragmentation of Golgi stacks in a cell-free system. J Cell Biol. 1995b;130:1027–1039. doi: 10.1083/jcb.130.5.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misumi Y, Miki K, Takatsuki A, Tamura G, Ikehara Y. Novel blockade by brefeldin A of intracellular transport of secretory proteins in cultured rat hepatocytes. J Biol Chem. 1986;261:11398–11403. [PubMed] [Google Scholar]
- Moskalewski S, Thyberg J. Disorganization and reorganization of the Golgi complex and the lysosomal system in association with mitosis. J Submicrosc Cytol Pathol. 1990;22:159–171. [PubMed] [Google Scholar]
- Moss J, Vaughan M. ADP-ribosylation of guanyl nucleotide-binding proteins by bacterial toxins. Adv Enzymol Relat Areas Mol Biol. 1988;61:303–379. doi: 10.1002/9780470123072.ch6. [DOI] [PubMed] [Google Scholar]
- Nakamura N, Rabouille C, Watson R, Nilsson T, Hui N, Slusarewicz P, Kreis TE, Warren G. Characterization of a cis-Golgi matrix protein, GM130. J Cell Biol. 1995;131:1715–1726. doi: 10.1083/jcb.131.6.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narula N, McMorrow I, Plopper G, Doherty J, Matlin KS, Burke B, Stow JL. Identification of a 200-kD, brefeldin-sensitive protein on Golgi membranes. J Cell Biol. 1992;117:27–38. doi: 10.1083/jcb.117.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson T, Warren G. Retention and retrieval in the endoplasmic reticulum and the Golgi apparatus. Curr Opin Cell Biol. 1994;6:517–521. doi: 10.1016/0955-0674(94)90070-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson T, Hoe MH, Slusarewicz P, Rabouille C, Watson R, Hunte F, Watzele G, Berger EG, Warren G. Kin recognition between medial Golgi enzymes in HeLa cells. EMBO (Eur Mol Biol Organ) J. 1994;13:562–574. doi: 10.1002/j.1460-2075.1994.tb06294.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki IJ, Moss J. Common structure of the catalytic sites of mammalian and bacterial toxin ADP-ribosyltransferases. Mol Cell Biochem. 1994;138:177–181. doi: 10.1007/BF00928460. [DOI] [PubMed] [Google Scholar]
- Oprins A, Duden R, Kreis TE, Geuze HJ, Slot JW. β-COP localizes mainly to the cis-Golgi side in exocrine pancreas. J Cell Biol. 1993;121:49–59. doi: 10.1083/jcb.121.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orci L, Tagaya M, Amherdt M, Perrelet A, Donaldson JG, Lippincott-Schwartz J, Klausner RD, Rothman JE. Brefeldin A, a drug that blocks secretion, prevents the assembly of non–clathrin-coated buds on Golgi cisternae. Cell. 1991;64:1183–1195. doi: 10.1016/0092-8674(91)90273-2. [DOI] [PubMed] [Google Scholar]
- Pavelka M, Ellinger A. Early and late transformations occurring at organelles of the Golgi area under the influence of brefeldin A: an ultrastructural and lectin cytochemical study. J Histochem Cytochem. 1993;41:1031–1042. doi: 10.1177/41.7.8515046. [DOI] [PubMed] [Google Scholar]
- Peters PJ, Hsu VW, Ooi CE, Finazzi D, Teal SB, Oorschot V, Donaldson JG, Klausner RD. Overexpression of wild-type and mutant ARF1 and ARF6: distinct perturbations of nonoverlapping membrane compartments. J Cell Biol. 1995;128:1003–1017. doi: 10.1083/jcb.128.6.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podos SD, Reddy P, Ashkenas J, Krieger M. LDLC encodes a brefeldin A-sensitive, peripheral Golgi protein required for normal Golgi function. J Cell Biol. 1994;127:679–691. doi: 10.1083/jcb.127.3.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabouille C, Misteli T, Watson R, Warren G. Reassembly of Golgi stacks from mitotic Golgi fragments in a cell-free system. J Cell Biol. 1995a;129:605–618. doi: 10.1083/jcb.129.3.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabouille C, Levine TP, Peters J-M, Warren G. An NSF-like ATPase, p97, and NSF mediate cisternal regrowth from mitotic Golgi fragments. Cell. 1995b;82:905–914. doi: 10.1016/0092-8674(95)90270-8. [DOI] [PubMed] [Google Scholar]
- Rambourg A, Clermont Y. Three-dimensional electron microscopy: structure of the Golgi apparatus. Eur J Cell Biol. 1990;51:189–200. [PubMed] [Google Scholar]
- Rios RM, Tassin A-M, Celati C, Antony C, Boissier M-C, Homberg J-C, Bornens M. A peripheral protein associated with the cis-Golgi network redistributes in the intermediate compartment upon brefeldin A treatment. J Cell Biol. 1994;125:997–1013. doi: 10.1083/jcb.125.5.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MS, Kreis TE. Recruitment of coat proteins onto Golgi membranes in intact and permeabilized cells: effects of brefeldin A and G protein activators. Cell. 1992;69:129–138. doi: 10.1016/0092-8674(92)90124-u. [DOI] [PubMed] [Google Scholar]
- Slusarewicz P, Nilsson T, Hui N, Watson R, Warren G. Isolation of a matrix that binds medial Golgi enzymes. J Cell Biol. 1994;124:405–413. doi: 10.1083/jcb.124.4.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souter E, Pypaert M, Warren G. The Golgi stack reassembles during telophase before arrival of proteins transported from the endoplasmic reticulum. J Cell Biol. 1993;122:533–540. doi: 10.1083/jcb.122.3.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takatsuki A, Tamura G. Brefeldin A, a specific inhibitor of intracellular translocation of vesicular stomatitis virus G protein: intracellular accumulation of high-mannose type G protein and inhibition of its cell surface expression. Agric Biol Chem. 1985;49:899–902. [Google Scholar]
- Takizawa PA, Yucel JK, Veit B, Faulkner DJ, Deerinck T, Soto G, Ellisman M, Malhotra V. Complete vesiculation of Golgi membranes and inhibition of protein transport by a novel sea sponge metabolite, Ilimaquinone. Cell. 1993;73:1079–1090. doi: 10.1016/0092-8674(93)90638-7. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Mitsushima A, Fukudome H, Kashima Y. Three- dimensional architecture of the Golgi complex observed by high resolution scanning electron microscopy. J Submicrosc Cytol. 1986;18:1–9. [PubMed] [Google Scholar]
- Viel A, Branton D. Spectrin: on the path from structure to function. Curr Opin Cell Biol. 1996;8:49–55. doi: 10.1016/s0955-0674(96)80048-2. [DOI] [PubMed] [Google Scholar]
- Warren G, Levine T, Misteli T. Mitotic disassembly of the mammalian Golgi apparatus. Trends Cell Biol. 1995;5:413–416. doi: 10.1016/s0962-8924(00)89094-7. [DOI] [PubMed] [Google Scholar]
- Weigert R, Colanzi A, Mironov A, Buccione R, Cericola C, Sciulli MG, Santini G, Flati S, Fusella A, Donaldson J, et al. Characterization of chemical inhibitors of brefeldin A-activated mono-ADP-ribosylation. J Biol Chem. 1997;272(22):14200–14207. doi: 10.1074/jbc.272.22.14200. [DOI] [PubMed] [Google Scholar]
- Weiner OH, Murphy J, Griffiths G, Schleicher M, Noegel AA. The actin-binding protein comitin (p24) is a component of the Golgi apparatus. J Cell Biol. 1993;123:23–34. doi: 10.1083/jcb.123.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]