

Abstract
While many cell types express receptors for the Fc domain of IgG (FcγR), only primate polymorphonuclear neutrophils (PMN) express an FcγR linked to the membrane via a glycan phosphoinositol (GPI) anchor. Previous studies have demonstrated that this GPI-linked FcγR (FcγRIIIB) cooperates with the transmembrane FcγR (FcγRIIA) to mediate many of the functional effects of immune complex binding. To determine the role of the GPI anchor in Fcγ receptor synergy, we have developed a model system in Jurkat T cells, which lack endogenously expressed Fcγ receptors. Jurkat T cells were stably transfected with cDNA encoding FcγRIIA and/or FcγRIIIB. Cocrosslinking the two receptors produced a synergistic rise in intracytoplasmic calcium ([Ca2+]i) to levels not reached by stimulation of either FcγRIIA or FcγRIIIB alone. Synergy was achieved by prolonged entry of extracellular Ca2+. Cocrosslinking FcγRIIA with CD59 or CD48, two other GPI-linked proteins on Jurkat T cells also led to a synergistic [Ca2+]i rise, as did crosslinking CD59 with FcγRIIA on PMN, suggesting that interactions between the extracellular domains of the two Fcγ receptors are not required for synergy. Replacement of the GPI anchor of FcγRIIIB with a transmembrane anchor abolished synergy. In addition, tyrosine to phenylalanine substitutions in the immunoreceptor tyrosine-based activation motif (ITAM) of the FcγRIIA cytoplasmic tail abolished synergy. While the ITAM of FcγRIIA was required for the increase in [Ca2+]i, tyrosine phosphorylation of crosslinked FcγRIIA was diminished when cocrosslinked with FcγRIIIB. These data demonstrate that FcγRIIA association with GPI-linked proteins facilitates FcγR signal transduction and suggest that this may be a physiologically significant role for the unusual GPI-anchored FcγR of human PMN.
The binding of immune complexes by polymorphonuclear neutrophils (PMN)1 receptors for the Fc domain of IgG (Fcγ receptors) induces essential host defense and inflammatory responses such as adhesion, phagocytosis of antibody-coated microorganisms, degranulation, and the respiratory burst (33, 38). PMN activation by immune complexes is important in the pathology of serum sickness, the Arthus reaction, acute glomerulonephritis, rheumatoid arthritis, and other idiopathic inflammatory disorders as well as in host defense against infection. The Fcγ receptors are a family of hematopoietic cell receptors that share structurally related ligand-binding domains for the Fc portion of immunoglobulins, but which differ in their transmembrane and intracellular domains (for review see 16, 33). These varying cytoplasmic tails presumably give rise to distinct intracellular signals to provide diversity of function.
Primate PMN are unique, because in addition to the transmembrane FcγR, FcγRIIA, they express the only known eukaryotic nontransmembrane FcγR, the glycan phosphoinositol (GPI)-linked FcγRIIIB. Ligand binding by transmembrane FcγRIIA initiates a tyrosine kinase cascade dependent upon the cytoplasmic tail of this receptor, which contains one copy of an immunoreceptor tyrosine-based activation motif (ITAM) (11, 27), a substrate for phosphorylation by members of the src tyrosine kinase family. The phosphorylated ITAM of FcγRIIA can bind to and activate syk tyrosine kinase, which subsequently activates a number of effector pathways (16). In contrast, little is known about the signaling mechanisms of FcγRIIIB, the most abundant PMN Fcγ receptor. Some studies have suggested an inability of FcγRIIIB to transduce signals independently. These studies, taken together with this receptor's lack of a cytoplasmic domain, have led to the concept that FcγRIIIB is primarily an Fc-binding molecule that aids in immune complex presentation to FcγRIIA (1, 13). However, evidence now suggests that FcγRIIIB is able to mediate intracellular signaling events, such as the activation of the src family member hck and induction of intracellular calcium fluxes (14, 19, 39, 49). Moreover, FcγRIIIB cooperates with FcγRIIA in PMN activation. When ligated together, as would occur when PMN bind immune complexes, FcγRIIA and FcγRIIIB synergize to activate the respiratory burst and to increase intracytoplasmic calcium (44, 47).
Despite the importance of the cooperation between FcγRIIA and FcγRIIIB for PMN function, its mechanism is not understood. As primary, terminally differentiated, nondividing cells, PMN are exceedingly resistant to genetic and cell biological manipulations which have aided characterization of receptor function in other systems. We developed a model system to dissect the functional roles and domains of FcγRIIA and FcγRIIIB in Jurkat T cells, which lack endogenous Fcγ receptors but are fully competent for tyrosine kinase signaling. In transfected Jurkat T cells, the PMN Fcγ receptors synergized to induce a rise in intracytoplasmic Ca2+ concentration ([Ca2+]i) that was greater and more prolonged than from ligation of either receptor individually. This was identical to the effect of coligation of these receptors in PMN (44). The synergistic calcium rise required the influx of extracellular calcium and depended upon the GPI anchor of FcγRIIIB, since a mutant in which the GPI anchor was replaced by the transmembrane domain of CD7 was unable to synergize with FcγRIIA. Moreover, crosslinking other GPI-linked proteins on Jurkat T cells with FcγRIIA also led to a synergistic increase in [Ca2+]i. The increase in [Ca2+]i also required the tyrosines of the FcγRIIA ITAM. Surprisingly, we found that phosphorylation of the ITAM was diminished under conditions that led to the synergistic calcium flux and that the kinetics of PLC-γ1 phosphorylation was not altered by the replacement of the GPI anchor of FcγRIIIB with the transmembrane domain of CD7. Thus, synergy between FcγR requires the GPI anchor of FcγRIIIB, but not for an increase in FcγRIIA-dependent tyrosine kinase signaling. We hypothesize instead that the role for the GPI anchor of FcγRIIIB is to sequester FcγRIIA into specialized membrane domains where signal transduction by the ITAM is altered. This could provide a further level of modulation of activation signals from immune complex binding and may explain many of the functions of the unusual GPI-linked FcγR of primate PMN. Moreover, this could be a general mechanism by which GPI anchored proteins affect signal transduction from transmembrane receptors.
Materials and Methods
Cells and Antibodies
The human Jurkat T cells (American Type Culture Collection, Rockville, MD) were maintained in RPMI 1640 medium (Gibco Laboratories, Grand Island, NY) containing 10% heat-inactivated FCS (Hyclone, Logan, UT), 2 mM l-glutamine, 0.1 mM NEAA, 50 mM 2-mercaptoethanol, and 100 μg/ml penicillin and streptomycin under a 5% CO2 atmosphere. The bulk population was cloned before transfection to minimize heterogeneity of the population. Human PMN were freshly purified from the peripheral blood of healthy donors as described (5). The following mAbs were used in this study: IV.3 (anti-CD32, anti-FcγRII; 26), 3G8 (anti-CD16, anti-FcγRIII; 9), IH4 (anti-CD55, anti-DAF; 8), MEM-43 (anti-CD59, anti-Protectin), 10G10 (anti-CD59; kindly provided by Dr. Marilyn Telen, Duke University, Durham, NC), MEM-102 (anti-CD48; Harlan Bioproducts, Indianapolis, IN), II1A5 (anti-FcγRII; kindly provided by Dr. Jurgen Frey, Universität Bielefeld), and mouse IgG2b isotype control (Sigma Chemical Co., St. Louis, MO). To crosslink primary antibodies, goat F(ab′)2 fragments specific for mouse F(ab′) or goat F(ab′)2 fragments specific for mouse IgG1 or mouse IgG2b (Sigma Chemical Co) were used. Antibody fragments of IV.3, 3G8, or 10G10 were made by standard methods or purchased (Medarex, Annandale, NJ). For FACS® analysis, bound mAbs were detected using FITC-conjugated goat F(ab′)2 fragments specific for mouse F(ab′) (Sigma Chemical Co.). Anti-phospholipase C γ-1 (PLC-γ1) was purchased from Upstate Biotechnology (Lake Placid, NY) or Transduction Laboratories (Lexington, KY). Anti-phosphotyrosine (Upstate Biotechnology) was detected with HRP-conjugated goat antibodies specific for mouse IgG2b (Caltag Laboratories, So. San Francisco, CA).
FcγRIIA and FcγRIIIB Expression Constructs and Transfection into Jurkat T Cells
The oligos 5′-CCTGAATTCCTCCGGATATCTTTGGTGAC-3′ and 5′-AGAGGATCCGCTGCCACTGCTCTTATTAC-3′ were used to amplify the human FcγRIIIB (CD16) cDNA by RT-PCR of human PMN mRNA (24). The resulting product was digested with EcoRI and HindIII and ligated into similarly digested vectors, pBluescript II SK+/−, pRcCMV, and pCEP4 (Invitrogen, San Diego CA). The intactness of the cDNA was verified by DNA sequencing (ABI PRISM Dye Terminator Cycle Sequencing Ready Reaction Kit; Perkin Elmer, Foster City, CA). The FcγRIIIB/CD7 construct was made by ligating a HindIII/MluI fragment of the CD16/CD7/syk construct (kindly provided by Dr. Brian Seed, Harvard Medical School, Boston, MA; (20) and a MluI/NotI adaptor (annealed oligonuclotides 5′-CGCGTTAATAGATCGATGC-3′ and 5′-GGCCGCATCGATCTATTAA-3′ [stop codons underlined]) into HindIII/NotI-digested pRcCMV. This construct encodes the FcγRIIIB extracellular domain joined with a CD7 transmembrane domain. The cDNA was verified by DNA sequencing. The cDNAs encoding FcγRIIA and FcγRIIA with both ITAM tyrosines in the cytoplasmic tail mutated to phenylalanine were prepared as described (7, 27) and cloned into pRcCMV and pCEP4.
The resulting plasmids were introduced into clones of Jurkat T cells by electroporation. Cells (107) in 400 μl HEBS (25 mM Hepes, pH 7.05, 140 mM NaCl, 750 mM Na2HPO4) and plasmid (30 μg in 100 μl HEBS) were added to a 0.4-mm-gap width cuvette and electroporated at 1,000 μF, 330 v (Electroporator II; Invitrogen). After electroporation, cells were cultured for 36 to 48 h in normal propagation media. Cells were transferred to selective media (propagation media plus 1.4 mg/ml geneticin/G418 [Gibco Laboratories] and/or 600 μg/ml hygromycin B [Boehringer Mannheim, Indianapolis, IN]) and cultured for 2 to 3 wk. High protein-expressing cell populations were selected by fluorescence-activated cell sorting using mAb IV.3 or mAb 3G8. Briefly, cells (106) were resuspended in 50 μl PBS/5% FCS with 1 μg antibody and incubated on ice for 45 min. Cells were washed and then incubated an additional 30 min with F(ab′)2 fragments of goat anti–mouse IgG-FITC (Sigma Chemical Co.). Cells were analyzed on a flow cytometer (Coulter Electronics, Hialeah, FL) or sorted using a fluorescence-activated cell sorter (Becton Dickenson, Palo Alto, CA). All cDNAs were introduced into at least two different Jurkat clones and all experiments yielded equivalent results in all clones.
[Ca2+]i Measurements
Jurkat transfectants were loaded with 3 μM Fura 2-AM (Molecular Probes, Eugene, OR) in RPMI 1640/10% FCS for 40 min in the dark at 37°C. PMN were loaded with 5 μM Fura-2 AM in Hanks Balanced Salt Solution (HBSS; Gibco Laboratories), 1 mM MgCl2, 1 mM CaCl2, and 1% vol/vol human serum albumin (HBSS++) for 25 min in the dark at 37°C. Cells (6 × 106) were washed once, resuspended in RPMI 1640/10% FCS or HBSS++ containing the appropriate mAbs, and incubated 30 min on ice. Cells were washed three times and resuspended in 2 ml calcium buffer (25 mM Hepes, pH 7.4, 125 mM NaCl, 5 mM KCl, 1 mM Na2HPO4, 1 mg/ml d-glucose, 1 mg/ml BSA, 1 mM CaCl2, 0.5 mM MgCl2). Changes in fluorescence, using excitation wavelengths of 340 and 380 nm and the emission wavelength of 510 nm, were measured with a spectrofluorimeter (F-2000; Hitachi Instruments, Danbury, CT) equipped with a thermostatic cuvette holder maintained at 37°C. Cells were warmed to 37°C for 5 min and added to the cuvette; then 10 μl mouse F(ab′) specific goat F(ab′)2 fragments were added. Intracellular calcium concentrations were calculated as described (36).
Receptor Crosslinking, Immunoprecipitation, and Western Blots
Cells (1–2 × 107) were incubated in RPMI 1640/10% FCS containing the mAb IV.3 (15 μg/ml) or the mAbs IV.3 and 3G8 (15 μg/ml each) for 30 min on ice. Cells were washed three times, resuspended in 0.5 ml RPMI 1690 with 10% FCS, and then warmed to 37°C for 10 min. Crosslinking mouse F(ab′) specific goat F(ab′)2 fragments (20 μl) were added for various times. Cells were lysed with an equal volume of 2× lysis buffer (100 mM Tris-HCl, pH 7.4, 2% NP-40, 0.5% deoxycholate, 300 mM NaCl, 2 mM EDTA, 2 mM NaF, 250 μM Na3VO4, 1 mM Na2MoO4, 1 mM Na2H2P2O7, 10 ng/ml calyculin, 25 μg/ml aprotinin, 25 μg/ml leupeptin, 15 μg/ml pepstatin A, 1 mM phenylmethylsulfonyl fluoride) at 4°C. Samples were centrifuged 5 min at 14,000 g. Resulting supernatants were rotated overnight with 75 μl of a 1:1 slurry of Gamma Bind plus Sepharose (Pharmacia Biotech, Piscataway, NJ). For PLC γ-1 immunoprecipitations, 10 μl of polyclonal antibodies were added to each sample. Beads were washed extensively and resuspended in reducing cocktail (50% vol/vol glycerol, 250 mM Tris-HCl, pH 6.8, 5% wt/vol SDS, 570 mM 2-mercaptoethanol, bromphenol blue). Samples were boiled for 5 min and then subjected to SDS-PAGE and electrotransfer onto Immobilon-P (Milipore, Bedford, MA) membranes. Blots were probed with anti-phosphotyrosine, anti-FcγRII (II1A5), or anti-PLC γ-1. Bound antibodies were detected with HRP-conjugated mouse specific goat antibodies. Antibody reactive protein was visualized using enhanced chemiluminescence (ECL; Amersham Intl., Arlington Heights, IL). Tyrosine phosphorylation of FcγRIIA or PLC-γ1 under different conditions was compared by normalizing the amount of phosphorylation, determined by densitometry of the anti-phosphotyrosine blots, to the amount of protein precipitated, as determined by reprobing the same blots with antibodies to the relevant protein. Multiple experiments were combined for analysis by comparing all experimental conditions to the ratio obtained for wild-type receptors in the same experiment.
Results
Cocrosslinking FcγRIIA and FcγRIIIB Results in a Synergistic [Ca2+]i Rise
Jurkat T cells, which do not express endogenous Fcγ receptors, were stably transfected with the cDNAs encoding FcγRIIA and FcγRIIIB (J2/3; Fig. 1, top). In addition, stable transfectants were made which express FcγRIIA along with a chimeric receptor consisting of the extracellular portion of FcγRIIIB coupled to the transmembrane domain of CD7 (J2/3-CD7; Fig. 1, middle). A third transfectant was made that expresses FcγRIIIB and an FcγRIIA receptor in which the tyrosines (Y282 and Y298) of the ITAM have been mutated to phenylalanines (27; J2Y→ F/3, Fig. 1, bottom). FACS® analysis indicated that each mutant receptor is expressed at a level at least comparable to that of the corresponding wild-type receptor (Fig. 1).
Figure 1.
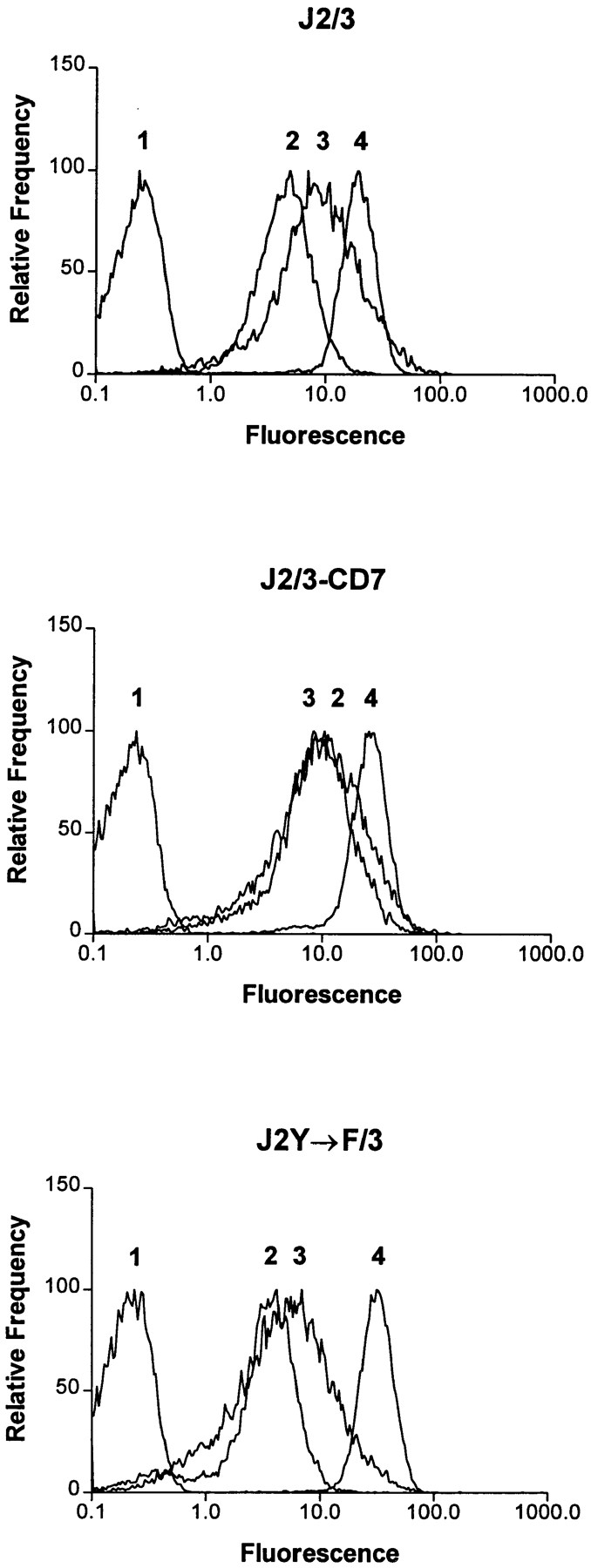
Fluorescent flow cytometric analysis of FcγR expression. Jurkat T cells (106) expressing various Fcγ receptors were resuspended in 50 μl PBS/5% FCS with 1 μg of the mAb IV.3 (2), specific for FcγRIIA, mAb 3G8 (3), specific for FcγRIIIB, or the mAb MEM-43 (4), specific for CD59. Cells were also stained with a negative control antibody (1). Cells were washed and then stained with F(ab′)2 fragments of FITC-conjugated goat anti–mouse antibodies and then analyzed by FACS®. Cells expressing wild-type FcγRIIA and FcγRIIIB (J2/3; top), wild-type FcγRIIA and the chimeric FcγRIIIB/CD7 (J2/3-CD7; middle), or wild-type FcγRIIIB and the mutant FcγRIIA where the tyrosines within the ITAM (Y282 and Y298) are changed to phenylalanine (J2Y→ F/3; bottom) are shown.
Previous studies in PMN have shown that FcγRIIA and FcγRIIIB in PMN cooperate to generate a calcium flux that is greater than the sum of the calcium fluxes generated by crosslinking either receptor individually (44). In addition, it has been shown that Jurkat cells that were stably transfected with FcγRIIA are able to flux calcium after receptor ligation (15), suggesting the signaling machinery used by Fcγ receptors is functional in these cells. Therefore we compared [Ca2+]i in J2/3 cells after crosslinking FcγRIIA and FcγRIIIB individually or after crosslinking both receptors together, using a F(ab′)2 crosslinking antibody. Crosslinking FcγRIIA resulted in a significant, short lived rise in [Ca2+]i (Fig. 2, top). In contrast, crosslinking FcγRIIIB alone resulted in a slow rise in [Ca2+]i with a magnitude lower than for FcγRIIA (Fig. 2, top). When both FcγR were crosslinked together, there was an increase in the maximum [Ca2+]i rise and a prolongation of the increase (Fig. 2, top). Synergy did not require the Fc fragment of either anti-FcγRII or -FcγRIII mAb, since similar results were obtained by using the F(ab) fragment of the mAb IV.3 and the F(ab′)2 fragment of the mAb 3G8 (data not shown). Neither the addition of antibodies specific for Fcγ receptors alone nor the crosslinking goat F(ab′)2 fragments alone induced a rise in [Ca2+]i (Fig. 2, top and data not shown). In PMN, crosslinking FcγRIIIB is able to mediate a rise in intracellular calcium by itself. This difference between the Jurkat transfectants and PMN is most likely due to the level of FcγRIIIB expression. In PMN, FcγRIIIB is extremely abundant on the cell surface (12, 13). Phosphatidylinositol-specific phospholipase C (PLC) treatment of PMN, an enzyme that cleaves GPI-linked proteins and that removes 80% of the FcγRIIIB from the cell surface, abolishes the rise in [Ca2+]i after FcγRIIIB crosslinking (35, and data not shown). Nonetheless, the expression level of FcγRIIIB in the transfected Jurkat cells was sufficient to produce a synergistic rise in [Ca2+]i.
Figure 2.
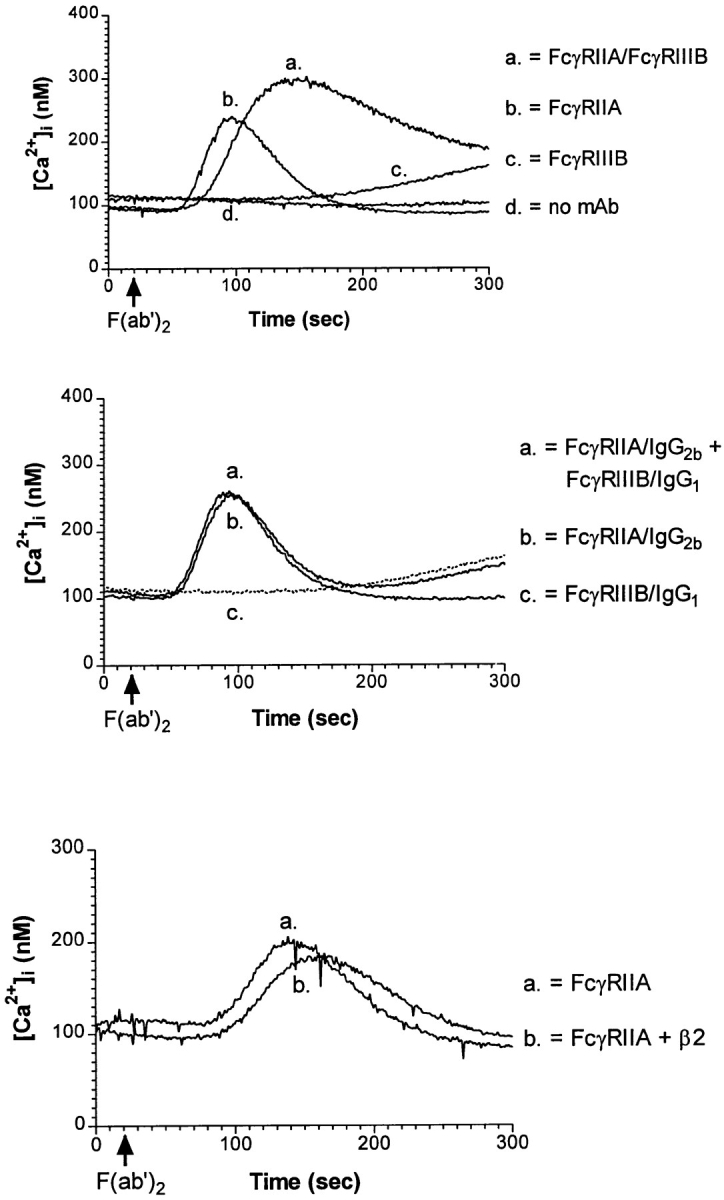
Changes in the [Ca2+]i after crosslinking FcγR. Fura 2-AM pre-loaded J2/3 cells were incubated 30 min with the mAb IV.3 (anti-FcγRII, IgG2b), the mAb 3G8 (anti-FcγRIIIB, IgG1), or both these mAbs (top and middle). J2/3 cells also were incubated with mAb IV.3 and the mAb IB4, specific for β2 integrins (bottom). F(ab′)2 fragments of goat anti–mouse antibodies (top and bottom), F(ab′)2 fragments of goat anti–mouse IgG1 (middle), or F(ab′)2 fragments of goat anti–mouse IgG2b (middle) were added to crosslink Fcγ receptors at 20 s. Each curve is representative of at least three independent experiments. When FcγRIIA was crosslinked with mAb IV.3/anti-IgG1 or FcγRIIIB was crosslinked with mAb 3G8/anti-IgG2b, no rise in [Ca2+]i resulted, demonstrating specificity of the secondary antibodies (data not shown). No rise in [Ca2+]i resulted from the addition of secondary antibodies alone (data not shown).
To determine if the synergistic calcium response required bridging of FcγRIIA and FcγRIIIB together or whether the augmentation in [Ca2+]i could be achieved by simultaneously crosslinking each Fcγ receptor individually, isotype-specific secondary crosslinking antibodies were used (Fig. 2, middle). FcγRIIA was crosslinked with IV.3, an IgG2b mAb, and goat F(ab′)2 fragments specific for mouse IgG2b and FcγRIIIB was crosslinked with 3G8, an IgG1 mAb, and goat F(ab′)2 fragments specific for mouse IgG1. When both Fcγ receptors were individually and simultaneously crosslinked, no synergistic rise in [Ca2+]i was found (Fig. 2, middle), paralleling results found in PMN (44). In fact, the resulting rise in [Ca2+]i appeared to be additive of the rises obtained by crosslinking both Fcγ receptors individually (Fig. 2, middle).
To show specificity of the synergy, cells were incubated with anti-FcγRII mAb IV.3 and the mAb IB4, specific for β2 (CD18) integrins (Fig. 2, bottom). The β2 integrin LFA-1 is expressed at a level similar to the transfected FcγRIIIB (data not shown). Moreover, LFA-1 synergizes with the ITAM-containing T cell antigen receptor to prolong an increase in [Ca2+]i (45). However, there was no synergy between LFA-1 and FcγRIIA for [Ca2+]i rise. This result indicates that signaling through FcγRIIA is augmented when cocrosslinked to FcγRIIIB, as would occur under physiological conditions where both Fcγ receptors are ligated by immune complexes.
The GPI Anchor Is Necessary and Sufficient for the Contribution of FcγRIIIB to Synergy
Primate PMN are the only cells that express a GPI-anchored Fcγ receptor (32). To determine whether the GPI anchor was necessary for FcγRIIIB contribution to the synergistic increase in [Ca2+]i, stable transfectants were made expressing FcγRIIA and a chimeric FcγRIIIB with the GPI anchor replaced by the transmembrane domain of CD7 (J2/3-CD7; Fig. 1, middle). When FcγRIIA and FcγRIIIB/ CD7 were crosslinked together in these cells, the [Ca2+]i rise was similar to the rise generated when FcγRIIA was crosslinked alone without any synergy from FcγRIIIB (Fig. 3, middle). The inability of the chimeric FcγRIIIB/ CD7 molecule to contribute to the synergistic [Ca2+]i rise was not due to inadequate expression of this protein, since the FcγRIIIB/CD7 molecule was expressed at a greater level than the wild-type FcγRIIIB (Fig. 1, top and middle). This experiment demonstrates that the GPI anchor is necessary for the synergistic [Ca2+]i rise.
Figure 3.

[Ca2+]i in cells expressing the chimeric FcγRIIIB/CD7. J2/3 cells (top), J2/3-CD7 cells (middle), or PMN (bottom) were preloaded with Fura 2-AM. J2/3 and J2/3-CD7 cells were then incubated for 30 min with the mAb IV.3 (anti-FcγRII), mAb 3G8 (anti-FcγRIII), mAb MEM-43 (anti-CD59), or combinations of these mAbs. PMN were incubated with mAb IV.3 F(ab), mAb 10G10 F(ab′)2 (anti-CD59), or combinations of these mAbs. Experiments were performed as described in Fig. 2. Each curve is representative of at least three independent experiments. For PMN, the change in [Ca2+]i at 140 s after the addition of crosslinking antibody was calculated and results are shown as the mean ± SEM for three independent experiments (bottom).
To determine whether any aspect of the extracellular Ig domains of FcγRIIIB rise were required for the synergistic [Ca2+]i rise, other GPI-linked proteins expressed by Jurkat cells were cocrosslinked with FcγRIIA. CD48 (not shown) and CD59 (protectin) (Fig. 1) are both expressed by parental Jurkat cells and by each of the transfectants at levels equal to or greater than FcγRIIIB. When these GPI-linked proteins, CD59 (Fig. 3, top) and CD48 (not shown), were cocrosslinked with FcγRIIA, a synergistic rise in [Ca2+]i also occurred in Jurkat cells transfected with FcγRIIA alone (data not shown), in J2/3 cells (Fig. 3, top), and in J2/3-CD7 cells (Fig. 3, middle). In all of these cells, ligation of CD59 alone produced a [Ca2+]i rise similar to that elicited by crosslinking FcγRIIIB alone (Fig. 3, top, and data not shown).
These experiments demonstrate that the GPI anchor of FcγRIIIB is required for FcγR cooperation but that other extracellular domains will substitute for FcγRIIIB when cocrosslinked with FcγRIIA. This is strong evidence against the hypothesis that interaction between the extracellular domains of the receptors is required for synergy, as has been proposed for FcγRIIA and FcγRIIIB interaction with the β2 integrin CR3 (for review see 30). Moreover, since these cells do not express CR3, this experiment shows that FcγR synergy can occur without this PMN integrin.
Synergy in PMN between FcγRIIA and FcγRIIIB was found for the rise in [Ca2+]i (data not shown and 44), the respiratory burst (data not shown and 44, 47, 49), and degranulation (data not shown). To determine if the synergistic rise in [Ca2+]i could also be obtained in PMN with other GPI-anchored proteins, FcγRIIA and CD59 were cocrosslinked and a prolongation in the rise [Ca2+]i was found (Fig. 3, bottom). The synergistic rise in [Ca2+]i with FcγRIIA and CD59 was not as pronounced as with FcγRIIIB and FcγRIIA. No significant synergy between FcγRIIA and CD59 was found in assays of degranulation or respiratory burst. This was true for CD48, CD55, and CD66b, other GPI-linked proteins on PMN, as well (data not shown). This is most likely due to a lower level of expression of these GPI-anchored proteins on PMN as compared to FcγRIIIB (CD59 has ∼13% of the expression of FcγRIIIB, CD48 has 1%, CD55 has 6%, and CD66b has 9%; data not shown). This is consistent with the lack of a synergistic rise in [Ca2+]i obtained in PMN treated with phosphatidylinositol-specific PLC, which reduces the amount of FcγRIIIB on the cell surface by 80% (35 and data not shown).
The ITAM of FcγRIIA Is Required for Calcium Flux
Activation of tyrosine phosphorylation and propagation of a tyrosine kinase cascade by receptor associated ITAMs is thought to be essential for Fcγ receptor signaling (16, 43). To determine whether this cascade had a role in Fcγ receptor synergy, Jurkat cells were transfected with FcγRIIIB and a mutant FcγRIIA in which tyrosines Y282 and Y298 contained within the ITAM were mutated to phenylalanines (J2Y→ F/3; Fig. 1, bottom). It has been shown in model systems that these tyrosines are required for [Ca2+]i flux when FcγRIIA is ligated alone (27, 28). No synergistic [Ca2+]i flux occurred in J2Y→ F/3 cells when FcγRIIA was ligated either alone or together with FcγRIIIB, although these cells were fully competent to increase [Ca2+]i in response to antigen receptor ligation (Fig. 4). Therefore, these tyrosines in the cytoplasmic tail of FcγRIIA are required for the synergistic [Ca2+]i rise. Thus both the GPI anchor of FcγRIIIB and the ITAM motif of FcγRIIA are required for synergy in calcium signaling.
Figure 4.

[Ca2+]i flux in cells expressing FcγRIIA containing the ITAM mutation. Fura 2-AM preloaded J2Y→ F/3 cells were incubated with the mAbs IV.3 (anti-FcγRII) and 3G8 (anti-FcγRIII), then analyzed by fluorimetry as described in Fig 2. The mAb C305, specific for the TCR/CD3 complex, was added at 300 sec to demonstrate that these cells are competent to flux [Ca2+]i.
The Synergistic Signal Does Not Result in Increased Tyrosine Phosphorylation of FcγRIIA
Because of the requirement for the ITAM in synergy and the association of GPI-linked proteins with src family kinases (4, 43), we hypothesized that an early step in this synergistic interaction might be an increased tyrosine phosphorylation of the ITAM of FcγRIIA. When FcγRIIA was immunoprecipitated from J2/3 cells after crosslinking FcγRIIA alone, its tyrosine phosphorylation peaked at 1 min and was diminished by 5 min (Fig. 5 A, top). Surprisingly, crosslinking FcγRIIA and FcγRIIIB together did not enhance tyrosine phosphorylation of FcγRIIA as expected but actually diminished detection of the tyrosine phosphorylation of FcγRIIA (Fig. 5 A, top). Averages from three experiments after normalization for the amount of receptor immunoprecipitated showed that FcγRIIA was phosphorylated ∼10-fold less under synergistic conditions as compared to ligation of FcγRIIA alone. We also analyzed the tyrosine phosphorylation of FcγRIIA in J2/3-CD7 cells. Ligation of FcγRIIA without FcγRIIIB induced tyrosine phosphorylation of itself to a similar extent and with similar kinetics as in cells expressing both wild-type Fcγ receptors (Fig. 5 B, bottom). In striking contrast to the results obtained in J2/3 cells by crosslinking both wild-type Fc receptors, cocrosslinking FcγRIIA and FcγRIIIB/CD7 did not significantly diminish the extent or alter the kinetics of FcγRIIA phosphorylation (Fig. 5 A, bottom). To determine if the marked diminution of FcγRIIA tyrosine phosphorylation also occurred when it was crosslinked with other GPI-anchored proteins, FcγRIIA was crosslinked with CD48 or CD59 (Fig. 5 B). Cocrosslinking any GPI-anchored protein with FcγRIIA markedly diminished its tyrosine phosphorylation. In addition, we analyzed the extent of tyrosine phosphorylation of FcγRIIA in PMN after ligating FcγRIIA, individually or together with FcγRIIIB, by using the F(ab) fragment of mAb IV.3 and the F(ab′)2 of mAb 3G8. Crosslinking both Fcγ receptors resulted in ∼2–3-fold diminished tyrosine phosphorylation of FcγRIIA when compared to ligating FcγRIIA alone (data not shown).
Figure 5.

Tyrosine phosphorylation of FcγRIIA after crosslinking FcγR. (A) J2/3 (top) or J2/3-CD7 (bottom) cells were incubated with mAb IV.3 (anti-FcγRII) or with mAbs IV.3 and 3G8 (anti-FcγRIII) for 30 min on ice and then warmed 10 min to 37°C. (B) J2/3 cells were incubated with various combinations of mAbs specific for FcγRII, FcγRIII, CD48, or CD59. In both panels, crosslinking F(ab′)2 fragments of goat anti–mouse antibodies were added for various amounts of time. At each time point, an aliquot was removed, lysed, and FcγRIIA immunoprecipitated. Proteins were separated by SDS-PAGE, and blots were probed with anti-phosphotyrosine. Cocrosslinking of GPI- but not transmembrane-anchored FcγRIIIB diminishes tyrosine phosphorylation of FcγRIIA. Blots shown are representative of at least five experiments.
The Synergistic Calcium Rise Does Not Result from the Prolonged Tyrosine Phosphorylation of PLC-γ1
PLC-γ1 is one of several PLC isoforms that converts phosphatidylinositol 4,5-bisphosphate to diacylglycerol and inositol 1,4,5-triphosphate leading to the release of intracellular stores of calcium. In several cell types, crosslinking FcγRIIA induces the tyrosine phosphorylation of PLC-γ1, which leads to its activation (25, 42). To determine whether prolonged activation of PLC-γ1 could account for the synergistic increase in [Ca2+]i, its tyrosine phosphorylation was examined. In agreement with previous studies, crosslinking FcγRIIA in the transfected Jurkat cells resulted in tyrosine phosphorylation of PLC-γ1 that was visible by 1 min (data not shown, and 42). Crosslinking FcγRIIIB and FcγRIIA in J2/3 cells resulted in tyrosine phosphorylation of PLC-γ1, which was not different from cocrosslinking FcγRIIA and the chimeric FcγRIIIB/CD7 in J2/3-CD7 cells (Fig. 6). Thus, Fcγ receptor synergy is independent of the tyrosine phosphorylation of PLC-γ1.
Figure 6.

The tyrosine phosphorylation of PLC-γ1 after crosslinking various FcγR. J2/3 (squares) or J2/3-CD7 (triangles) cells were incubated with mAbs IV.3 (anti-FcγRII) and 3G8 (anti- FcγRIII), warmed to 37°C, and crosslinking initiated by addition of F(ab′)2 fragments of goat anti–mouse antibodies. At each time point, an aliquot was removed, PLC-γ1 was immunoprecipitated, and proteins were separated by SDS-PAGE. Blots were probed with anti-phosphotyrosine and subsequently with anti–PLC-γ1 antibodies to determine the relative phosphorylation of the immunoprecipitated enzyme, as described in Materials and Methods. Three independent experiments from both cell types were analyzed by densitometry, and the mean and SEM of the three experiments are shown.
The Synergistic Rise in [Ca2+]i Requires the Influx of Extracellular Calcium
To determine the source of Ca2+ for the synergistic [Ca2+]i rise in the J2/3 cells, changes in Fura-2 fluorescence were measured in the presence of extracellular EGTA to prevent calcium influx from the medium. The synergistic [Ca2+]i rise was inhibited almost immediately after addition of EGTA, indicating that calcium influx through plasma membrane channels is largely responsible for the prolonged [Ca2+]i rise (Fig. 7 A, left) as found in PMN (44). Similarly, the synergistic [Ca2+]i rise induced by cocrosslinking FcγRIIA and CD59 was abolished by the addition of EGTA (Fig. 7 A, middle). As a control, the changes in intracellular calcium were measured after the T-cell receptor complex (TCR/CD3) was crosslinked with the mAb C305 (Fig. 7 A, right). Previous studies have shown that the rise in intracellular calcium after TCR crosslinking results from an initial rise derived from intracellular stores followed by a secondary sustained calcium influx through plasma membrane channels that can be abolished by the addition of EGTA (41). The addition of EGTA to Jurkat cells treated only with crosslinking secondary antibody does cause a small decrease in the amount of intracellular calcium, but this small depletion does not account for the large loss in the synergistic calcium influx from extracellular stores, as previously shown in PMN (37; Fig. 7, A and C, left). The changes in intracellular calcium also were measured when EGTA was added immediately before Fcγ receptor crosslinking (Fig. 7 B, left). Crosslinking led to an initial rise in [Ca2+]i, but the synergistic [Ca2+]i rise was substantially diminished after cocrosslinking FcγRIIA with FcγRIIIB or CD59 (Fig. 7 B, middle and right). The magnitude of the [Ca2+]i rise also was diminished in the presence of EGTA, again demonstrating that a significant contribution to the [Ca2+]i rise is due to the influx of extracellular calcium (Fig. 7 B). The slow rise in [Ca2+]i after crosslinking either FcγRIIIB or CD59 alone was abolished in the presence of EGTA (Fig. 7 C, right, and data not shown). EGTA treated cells do not produce a flux in [Ca2+]i after the addition of crosslinking secondary antibodies alone (Fig. 7 C, left).
Figure 7.

The synergistic rise in [Ca2+]i requires the influx of extracellular calcium. Changes in Fura 2-AM fluorescence after receptor crosslinking in J2/3 cells was measured as in Fig. 2 in the absence or presence of 2 mM EGTA to prevent calcium influx from the medium. (A) 2 mM EGTA was added 280 s after crosslinking. (B) 2 mM EGTA was added immediately before receptor crosslinking. Also shown is no added EGTA. (C) 2 mM EGTA was added at 0 or 300 s.
Discussion
Since the discovery that GPI-linked proteins can transduce proliferative signals, attention has focused on the mechanism by which these proteins, anchored into the outer leaflet of the plasma membrane by their fatty acyl chains, can signal to the cell cytoplasm. Two distinct but not mutually exclusive paradigms have developed. One model suggests that GPI-linked proteins can sequester into specialized membrane domains, especially after clustering (for review see 29, 34). These domains, which are defined by their insolubility in Triton X-100, contain characteristic lipid components, such as glycosphingolipids and cholesterol, but may be depleted in certain phospholipids. GPI-linked proteins are enriched ∼200-fold in these domains, and there is evidence for concentration of Src kinases, G protein–coupled receptors, and heterotrimeric G proteins in these membrane domains as well. This has led some investigators to hypothesize that these domains function in signal transduction, and indeed crosslinking of GPI-linked proteins leads to rapid induction of tyrosine phosphorylation (43). On the other hand, some src family kinases sequestered in these domains have low specific activity, suggesting that these glycolipid domains function not in signaling but as a reservoir of signaling molecules that can be recruited to other parts of the membrane (34).
The second model for signal transduction by GPI-linked proteins involves their physical association with transmembrane proteins. For example, FcγRIIIB has been shown to associate with the integrin Mac-1, as has the GPI-linked urokinase receptor (uPAR), which also can associate with another integrin, αvβ3 (21, 46). These physical associations have functional consequences, for example, induction of IgG-mediated phagocytosis in transfected 3T3 cells (21), or cellular adhesion to vitronectin (46). Thus, it is possible that GPI-linked proteins transduce information to the cytoplasm through physical interaction with transmembrane proteins.
The interaction of FcγRIIA and FcγRIIIB on human PMN presents an opportunity to test these hypotheses concerning signal transduction by GPI-linked proteins. When immune complexes bind to PMN, FcγRIIA and FcγRIIIB are brought into proximity. While synergy between the receptors in signal transduction in response to immune complexes has been shown, interpretation is complicated by the interaction of both receptors with other membrane proteins such as Mac-1 (40, 48), and by the inability to use molecular genetic techniques to probe receptor function in these primary cells. For these reasons, we have developed a model system to understand Fcγ receptor synergy on PMN. In Jurkat cells without Mac-1, FcγRIIA and FcγRIIIB can synergize to increase [Ca2+]i, demonstrating that extracellular domain association with Mac-1 is not required for at least this aspect of synergy. Indeed, since coligation of two other GPI-linked proteins, otherwise structurally unrelated to FcγRIIIB, also can synergize with FcγRIIA to increase [Ca2+]i, it is unlikely that extracellular domain interactions other than with multivalent ligands are required to induce synergy between the transmembrane and GPI-linked Fcγ receptors. The synergistic increase in [Ca2+]i may be important in numerous PMN functions, including degranulation (3, 23), actin polymerization (2), and phagocytosis (17, 18).
Our data support the hypothesis that association of FcγRIIA with glycolipid domains enriched in GPI-linked proteins fundamentally alters subsequent signaling. Cocrosslinking FcγRIIA with any of the GPI-linked proteins induced the synergistic increase in [Ca2+]i and, surprisingly, decreased the extent of FcγRIIA tyrosine phosphorylation. When FcγRIIIB was expressed with a transmembrane domain, its synergy with FcγRIIA was abolished, as was its effect on FcγRIIA tyrosine phosphorylation. These data support the hypothesis that the membrane environment of FcγRIIA is altered by crosslinking it with GPI- anchored proteins. This altered environment modulates the FcγRIIA-generated signal in fundamental ways. We initially expected that the synergistic [Ca2+]i rise would be associated with increased phosphorylation of the ITAM of FcγRIIA, because src family kinases, which phosphorylate ITAMs, have been found to be concentrated in these domains. However, our finding of decreased tyrosine phosphorylation is consistent with the report that CD45, the major transmembrane tyrosine phosphatase present on lymphocytes, is excluded from glycolipid-enriched membrane domains, resulting in lower specific activity of the lymphocyte src kinases in these domains (34). We propose that FcγRIIA has diminished tyrosine phosphorylation after cocrosslinking with FcγRIIIB, because ligation with GPI-linked proteins causes FcγRIIA to be brought into membrane domains with less-active src kinases. It is also possible that an additional signaling pathway is used to mediate synergistic calcium signaling, since the prolonged rise in intracellular calcium is not due to the prolonged tyrosine phosphorylation of PLC-γ1. Calcium mobilization after crosslinking FcεRI activates a sphingosine kinase that produces sphingosine-1-phosphate as a second messenger for intracellular calcium mobilization (6). Alternatively, localization of the Fcγ receptors within specialized membrane domains may activate the synergistic influx of extracellular calcium. Indeed, a plasma membrane calcium pump has been identified in caveolae (10).
Our data further extend the observations made with several receptors, including Fcγ receptors, that there may be interaction on the cell surface between receptors recognizing the same ligand. For example, T cells express two distinct receptors that interact with MHC class I molecules, one that mediates the positive signal, the T cell receptor, and a second receptor, NKB1, that mediates an inhibitory signal (22, 31). It has been observed in phagocytic cells that the Fcγ receptor, FcγRIIB, inhibits phagocytosis mediated by FcγRIIA. Decreased tyrosine phosphorylation induced by FcγRIIB after interaction with IgG ligand may be responsible for this inhibition of FcγRIIA-mediated phagocytosis (Hunter, S., and A.D. Schreiber, unpublished results).
In summary, transfection of human PMN Fcγ receptors into the Jurkat cell line has allowed for the further dissection of the mechanism by which these receptors cooperate in immune complex–induced PMN activation. We have defined two essential structural components of the synergistic signal, the GPI-anchor of FcγRIIIB and the ITAM of FcγRIIA. Moreover, we have shown that synergy can occur in the absence of the phagocyte integrin Mac-1, previously postulated to be an essential component for synergy. In PMN, 10,000 to 20,000 FcγRIIA molecules are expressed on the cell surface together with 10 to 20 times more FcγRIIIB (12, 13). Thus it is highly likely that whenever FcγRIIA is ligated by an immune complex, it is in association with several GPI-linked FcγRIIIB and that the modulated signal which occurs because of association with GPI domains is the major mechanism of immune complex-mediated PMN activation.
Acknowledgments
We thank Dr. Ming-jie Zhou (Molecular Probes, Inc.) for the PCR clone of CD16, Dr. Brian Seed for the CD16/CD7/ζ cDNA, Dr. Andrew Chan for the C305 mAb, Dr. Jurgen Frey for the II1A5 mAb, and Drs. Doug Lublin and Scott Blystone (Washington University, St. Louis, MO) for helpful discussions.
This work was supported by grants from the National Institutes of Health and the Arthritis Foundation to E.J. Brown. J.M. Green is supported as a Lucille P. Markey Pathway postdoctoral fellow.
Abbreviations used in this paper
- [Ca2+]i
intracytoplasmic Ca2+ concentration
- GPI
glycan phosphoinositol
- ITAM
immunoreceptor tyrosine-based activation motif
- PLC
phospholipase C
- PMN
polymorphonuclear neutrophils
Footnotes
Address all correspondence to Dr. Eric J. Brown, Division of Infectious Diseases, Washington University School of Medicine, 660 S. Euclid Ave., Box 8051, St. Louis, MO 63110. Tel.: (314)362-2125. Fax: (314) 362-9230. E-mail: ebrown@id.wustl.edu
References
- 1.Anderson CL, Shen L, Eicher DM, Wewers MD, Gill JK. Phagocytosis mediated by three distinct Fc γ receptor classes on human leukocytes. J Exp Med. 1990;171:1333–1345. doi: 10.1084/jem.171.4.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bengtsson T, Jaconi ME, Gustafson M, Magnusson K-E, Theler J-M, Lew DP, Stendahl O. Actin dynamics in human neutrophils during adhesion and phagocytosis is controlled by changes in intracellular free calcium. Eur J Cell Biol. 1993;62:49–58. [PubMed] [Google Scholar]
- 3.Berger M, Birx DL, Wetzler EM, O'Shea JJ, Brown EJ, Cross AS. Calcium requirements for increased complement receptor expression during neutrophil activation. J Immunol. 1985;135:1342–1348. [PubMed] [Google Scholar]
- 4.Brown D. The tyrosine kinase connection: how GPI anchored proteins activate T cells. Curr Opin Immunol. 1993;5:349–354. doi: 10.1016/0952-7915(93)90052-t. [DOI] [PubMed] [Google Scholar]
- 5.Brown EJ. In vitro assays of phagocytic function of human peripheral blood leukocytes: receptor modulation and signal transduction. Methods Cell Biol. 1994;45:147–164. doi: 10.1016/s0091-679x(08)61850-6. [DOI] [PubMed] [Google Scholar]
- 6.Choi HU, Kim J, Kinet J. Calcium mobilization via sphingosine kinase in signaling by the FcεRI antigen receptor. Nature. 1996;380:634–636. doi: 10.1038/380634a0. [DOI] [PubMed] [Google Scholar]
- 7.Clark MR, Stuart SG, Kimberly RP, Ory PA, Goldstein IM. A single amino acid distinguishes the high-responder from the low-responder form of Fc receptor II on human monocytes. Eur J Immunol. 1991;21:1911–1916. doi: 10.1002/eji.1830210820. [DOI] [PubMed] [Google Scholar]
- 8.Coyne KE, Hall SE, Thompson S, Arce MA, Kinoshita T, Fujita T, Anstee DJ, Rosse W, Lublin DM. Mapping of epitopes, glycosylation sites, and complement regulatory domains in human decay accelerating factor. J Immunol. 1992;149:2906–2913. [PubMed] [Google Scholar]
- 9.Fleit HB, Wright SD, Unkeless JC. Human neutrophil Fc γ receptor distribution and structure. Proc Natl Acad Sci USA. 1982;79:3275–3279. doi: 10.1073/pnas.79.10.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fujimoto T. Calcium pump of the plasma membrane is localized in caveolae. J Cell Biol. 1993;120:1147–1157. doi: 10.1083/jcb.120.5.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang M-M, Indik Z, Brass LF, Hoxie JA, Schreiber AD, Brugge JS. Activation of FcγRII induces tyrosine phosphorylation of multiple proteins including FcγRII. J Biol Chem. 1992;267:5467–5473. [PubMed] [Google Scholar]
- 12.Huizinga, T.W.J., C.E. Van der Schoot, C. Jost, R. Klaassen, M. Kleijer, A.E.G.K. van dem Borne, D. Roos, and P.A.T. Tetteroo. 1988. The PI-linked receptor FcγRIII is released on stimulation of neutrophils. Nature. 333:667–669. [DOI] [PubMed]
- 13.Huizinga TW, van Kemenade F, Koenderman L, Dolman KM, von dem Borne AE, Tetteroo P A, Roos D. The 40-kDa Fc γ receptor (FcγRII) on human neutrophils is essential for the IgG-induced respiratory burst and IgG-induced phagocytosis. J Immunol. 1989;142:2365–2369. [PubMed] [Google Scholar]
- 14.Hundt M, Schmidt RE. The glycosylphosphatidylinositol-linked Fcγ receptor III represents the dominant receptor structure for immune complex activation of neutrophils. Eur J Immunol. 1992;22:811–816. doi: 10.1002/eji.1830220327. [DOI] [PubMed] [Google Scholar]
- 15.Hunter S, Kamoun M, Schreiber AD. Transfection of an Fcγ receptor cDNA induces T cells to become phagocytic. Proc Natl Acad Sci USA. 1994;91:10232–10236. doi: 10.1073/pnas.91.21.10232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Indik ZK, Park JG, Hunter S, Schreiber AD. The molecular dissection of Fcγ receptor mediated phagocytosis. Blood. 1995;86:4389–4399. [PubMed] [Google Scholar]
- 17.Jaconi MEE, Lew DP, Carpentier J-L, Magnusson KE, Sjogren M, Stendahl O. Cytosolic free calcium elevation mediates the phagosome-lysosome fusion during phagocytosis in human neutrophils. J Cell Biol. 1990;110:1555–1564. doi: 10.1083/jcb.110.5.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jaconi ME, Theler JM, Schlegel W, Lew PD. Cytosolic free Ca2+signals in single adherent human neutrophils: generation and functional role. Eur J Pediatr. 1993;152:S26–S32. doi: 10.1007/BF02072084. [DOI] [PubMed] [Google Scholar]
- 19.Kimberly RP, Ahlstrom JW, Click ME, Edberg JC. The glycosyl phosphatidylinositol-linked FcγRIII on PMN mediates transmembrane signaling events distinct from FcγRII. J Exp Med. 1990;171:1239–1255. doi: 10.1084/jem.171.4.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kolanus W, Romeo C, Seed B. T cell activation by clustered tyrosine kinases. Cell. 1993;74:171–183. doi: 10.1016/0092-8674(93)90304-9. [DOI] [PubMed] [Google Scholar]
- 21.Krauss JC, Poo H, Xue W, Mayo-Bond L, Todd RF, Petty HR. Reconstitution of antibody-dependent phagocytosis in fibroblasts expressing FcγRIIIB and the complement receptor type 3. J Immunol. 1994;153:1769–1777. [PubMed] [Google Scholar]
- 22.Lanier LL, Phillips JH. Inhibitory MHC class I receptors on NK cells and T cells. Immunol Today. 1996;17:86–91. doi: 10.1016/0167-5699(96)80585-8. [DOI] [PubMed] [Google Scholar]
- 23.Lew PD, Monod A, Waldvogel FA, Dewald B, Baggiolini M, Pozzan T. Quantitative analysis of the cytosolic free calcium dependency of exocytosis from three subcellular compartments in intact human neutrophils. J Cell Biol. 1986;102:2197–2204. doi: 10.1083/jcb.102.6.2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lindberg FP, Gresham HD, Schwarz E, Brown EJ. Molecular cloning of integrin-associated protein: an immunoglobulin family member with multiple membrane spanning domains implicated in αvβ3-dependent ligand binding. J Cell Biol. 1993;123:485–496. doi: 10.1083/jcb.123.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liscovitch M, Cantley LC. Lipid second messengers. Cell. 1994;77:329–334. doi: 10.1016/0092-8674(94)90148-1. [DOI] [PubMed] [Google Scholar]
- 26.Looney RJ, Ryan DH, Takahashi K, Fleit HB, Cohen HJ, Abraham GN, Anderson CL. Identification of a second class of IgG Fc receptors on human neutrophils. A 40-kilodalton molecule also found on eosinophils. J Exp Med. 1986;163:826–836. doi: 10.1084/jem.163.4.826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mitchell MA, Huang M-M, Chien P, Indik ZK, Pan XQ, Schreiber AD. Substitutions and deletions in the cytoplasmic domain of the phagocytic receptor FcγRIIA: effect on receptor tyrosine phosphorylation and phagocytosis. Blood. 1994;84:1753–1759. [PubMed] [Google Scholar]
- 28.Odin JA, Edberg JC, Painter CJ, Kimberly RP, Unkeless JC. Regulation of phagocytosis and [Ca2+]iflux by distinct regions of an Fc receptor. Science. 1991;254:1785–1788. doi: 10.1126/science.1837175. [DOI] [PubMed] [Google Scholar]
- 29.Parton RG, Simons K. Digging into caveolae. Science. 1995;269:1398–1399. doi: 10.1126/science.7660120. [DOI] [PubMed] [Google Scholar]
- 30.Petty HR, Todd R., III Integrins as promiscuous signal transduction devices. Immunol Today. 1996;17:209–212. doi: 10.1016/0167-5699(96)30013-3. [DOI] [PubMed] [Google Scholar]
- 31.Phillips JH, Gumperz JE, Parham P, Lanier LL. Superantigen-dependent, cell-mediated cytotoxicty inhibited by MHC class I receptors on T lymphocytes. Science. 1995;268:403–405. doi: 10.1126/science.7716542. [DOI] [PubMed] [Google Scholar]
- 32.Ravetch JV, Perussia B. Alternative membrane forms of FcγRIII (CD16) on human natural killer cells and neutrophils: cell type-specific expression of two genes that differ in single nucleotide substitutions. J Exp Med. 1989;170:481–497. doi: 10.1084/jem.170.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ravetch JV, Kinet JP. Fc receptors. Annu Rev Immunol. 1991;9:457–492. doi: 10.1146/annurev.iy.09.040191.002325. [DOI] [PubMed] [Google Scholar]
- 34.Rodgers W, Rose JK. Exclusion of CD45 inhibits activity of p56 associated with glycolipid-enriched membrane domains. J Cell Biol. 1996;135:1515–1523. doi: 10.1083/jcb.135.6.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rosales C, Brown EJ. Two mechanisms for IgG Fc-receptor-mediated phagocytosis by human neutrophils. J Immunol. 1991;146:3937–3944. [PubMed] [Google Scholar]
- 36.Rosales C, Brown EJ. Signal transduction by neutrophil immunoglobulin G Fc receptors. Dissociation of [Ca2+] rise from IP3 . J Biol Chem. 1992;267:5265–5271. [PubMed] [Google Scholar]
- 37.Rosales C, Brown EJ. Calcium channel blockers nifedipine and diltiazem inhibit Ca2+release from intracellular stores in neutrophils. J Biol Chem. 1992;267:1443–1448. [PubMed] [Google Scholar]
- 38.Rosales, C., and E.J. Brown. 1993. Neutrophil receptors and modulation of the immune response. In The Neutrophil. J.S. Abramson and J.G. Wheeler, editors. IRL Press, Oxford. 23–62.
- 39.Salmon JE, Brogle NL, Edberg JC, Kimberly RP. Fcγ receptor III induces actin polymerization in human neutrophils and primes phagocytosis mediated by Fcγ receptor II. J Immunol. 1991;146:997–1004. [PubMed] [Google Scholar]
- 40.Sehgal G, Zhang K, Todd RF, Boxer LA, Petty HR. Lectin-like inhibition of immune-complex receptor-mediated stimulation of neutrophils: effects on cytosolic calcium release and superoxide production. J Immunol. 1993;150:4571–4580. [PubMed] [Google Scholar]
- 41.Sei Y, Takemura M, Gusovsky F, Skolnick P, Basile A. Distinct mechanisms for Ca2+ entry induced by OKT3 and Ca2+depletion in Jurkat T cells. Exp Cell Res. 1995;216:222–231. doi: 10.1006/excr.1995.1028. [DOI] [PubMed] [Google Scholar]
- 42.Shen Z, Lin CT, Unkeless JC. Correlations among tyrosine phosphorylation of Shc, p72syk, PLC-γ1, and [Ca2+]iflux in FcγRIIA signaling. J Immunol. 1994;152:3017–3023. [PubMed] [Google Scholar]
- 43.Stefanova I, Horejsi V, Ansotegui IJ, Knapp W, Stockinger H. GPI-anchored cell-surface molecules complexed to protein tyrosine kinases. Science. 1991;254:1016–1019. doi: 10.1126/science.1719635. [DOI] [PubMed] [Google Scholar]
- 44.Vossebeld PJM, Kessler J, Von dem Borne AEGK, Roos D, Verhoeven AJ. Heterotypic FcγR clusters evoke a synergistic Ca2+response in human neutrophils. J Biol Chem. 1995;270:10671–10679. doi: 10.1074/jbc.270.18.10671. [DOI] [PubMed] [Google Scholar]
- 45.Wacholtz MC, Patel SS, Lipsky PE. Leukocyte function- associated antigen 1 is an activation molecule for human T cells. J Exp Med. 1989;170:431–448. doi: 10.1084/jem.170.2.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wei Y, Lukashev M, Simon DI, Bodary SC, Rosenberg S, Doyle MV, Chapman HA. Regulation of integrin function by the urokinase receptor. Science. 1996;273:1551–1554. doi: 10.1126/science.273.5281.1551. [DOI] [PubMed] [Google Scholar]
- 47.Zhou M-J, Brown EJ. CR3 (Mac-1, aMb2, CD11b/CD18) and FcγRIII cooperate in generation of a neutrophil respiratory burst: requirement for FcγRII and tyrosine phosphorylation. J Cell Biol. 1994;125:1407–1416. doi: 10.1083/jcb.125.6.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou M, Todd RF, III, Van de Winkel JGJ, Petty HR. Cocapping of the leukoadhesin molecules complement receptor type 3 and lymphocyte function-associated antigen-1 with Fcγ receptor III on human neutrophils: possible role of lectin-like interactions. J Immunol. 1993;150:3030–3041. [PubMed] [Google Scholar]
- 49.Zhou M-J, Lublin DM, Link DC, Brown EJ. Distinct tyrosine kinase activation and Triton X-100 insolubility upon FcγRII or FcγRIIIB ligation in human polymorphonuclear leukocytes: implications for immune complex activation of the respiratory burst. J Biol Chem. 1995;270:13553–13560. doi: 10.1074/jbc.270.22.13553. [DOI] [PubMed] [Google Scholar]