

Abstract
BACKGROUND
Persistent macrophage accumulation and alveolar enlargement are hallmark features of chronic obstructive pulmonary disease (COPD). A role for CD8+ lymphocytes in the development of COPD is suggested based on observations that this T cell subset is increased in the airways and parenchyma of smokers that develop COPD with airflow limitation. In this study, we utilize a mouse model of COPD to examine the contributions of CD8+ T cells in the persistent macrophage accumulation and airspace enlargement resulting from chronic irritant exposure.
METHODS
We analyzed pulmonary inflammation and alveolar destruction in wild-type and Cd8-deficient mice chronically exposed to acrolein, a potent respiratory tract irritant. We further examined cytokine mRNA expression levels by RNase protection assay, matrix metalloproteinase (MMP) activity by gelatin zymography, and epithelial cell apoptosis by active caspase3 immunohistochemistry in wild-type and Cd8-deficient mice exposed chronically to acrolein.
RESULTS
These studies demonstrate that CD8+ T cells, are important mediators of macrophage accumulation in the lung and the progressive airspace enlargement in response to chronic acrolein exposures. The expression of several inflammatory cytokines (IP-10, IFN-γ, IL-12, RANTES, and MCP-1), MMP2 and MMP9 gelatinase activity, and caspase3 immunoreactivity in pulmonary epithelial cells were attenuated in the Cd8-deficient mice compared to wild-type.
CONCLUSIONS
These results indicate that CD8+ T cells actively contribute to macrophage accumulation and the development of irritant-induced airspace enlargement.
BACKGROUND
Chronic obstructive pulmonary disease (COPD) is a progressive disease of the airways characterized primarily by inflammation, irreversible airflow obstruction, and alveolar enlargement. COPD primarily results from the inhalation of noxious gases, with cigarette smoking the single major cause (1). COPD afflicts more than 10 million adults and is the fourth leading cause of death in the United States (1). The pathogenesis of inflammation, airway remodeling, and destruction of the alveolar unit in COPD is complex and not completely understood. Inflammation in COPD is marked by the presence of increased numbers of macrophages, neutrophils, and lymphocytes in the lung (2). Macrophages and neutrophils have been well studied and appear to play a role in the pathogenesis of COPD through the release of proteinases that alter the extracellular matrix (2). In contrast, the current understanding of the role of lymphocytes in the pathogenesis of COPD is limited.
The presence of increased CD8+ T cells in the airways and parenchyma of the lungs of smokers who developed COPD is well established (3–5). However, data linking CD8+ T cells to COPD pathogenesis are correlative and have been obtained following the development of disease in humans. The consequences of CD8+ T cell accumulation and the effector functions of these cells that may contribute to COPD pathology have not been examined. CD8+ T cells are capable of producing several soluble mediators that have been implicated in the recruitment and activation of macrophages, including chemokines (6) and cytokines (e.g., interferon-γ [IFN-γ]) (7). One possible role for CD8+ T cells in COPD is that they represent a protective function of the innate immune system that helps maintain epithelial integrity following injury (e.g., from cigarette smoke or other irritant exposures) but when accumulation persists, the effector functions of these cells (e.g. macrophage recruitment/activation) are amplified and increase lung pathology.
Tobacco smoke contains multiple irritants including acrolein (CH2=CH-CHO) (8). Notably, second-hand tobacco smoke contains high levels of acrolein because aldehydes are enriched in side stream smoke due to the lower combustion temperatures of smoldering cigarettes. In indoor air, smoking one cigarette per m3 of room-space in 10 minutes was found to lead to acrolein vapor concentrations in excess of 3.0 parts per million (ppm) (9). Acrolein is also generated endogenously during oxidative stress and by chronic inflammation (10). Furthermore, acrolein is found in high concentrations in exhaled breath and induced sputum of patients with COPD (11). In experimental animals, acrolein can induce lesions associated with COPD. We, and others, have demonstrated that acrolein induces the hallmark features of COPD including increased macrophage accumulation, epithelial damage, airspace enlargement, and mucus hypersecretion (12–15).
In this study, we examine the role of CD8+ T cells in mediating COPD pathologies induced by acrolein. CD8+ T cell and macrophage accumulation, cytokine transcript levels, metalloproteinase activation, and caspase3 activation were assessed to gain a deeper understanding of the complex interactions between immune cells, inflammation, macrophage function and epithelial cell injury. We report that following repeated toxicant exposure, CD8+ T cells contribute to increased monocyte infiltration and the development of several pathologies associated with COPD, including airspace enlargement and epithelial cell apoptosis. These findings suggest a causative role for CD8+ T cell effector functions in the development of acrolein induced COPD pathology.
METHODS
Experimental Design
To determine the roles of the local lymphocyte populations in the lung that control of acrolein-induced pulmonary inflammation and epithelial cell pathology, wild-type (C57BL/6J) mice and mice deficient in CD8+ T cells (Cd8(−/−)) were exposed to filtered air or 2.0 ppm acrolein 6h/d, 5 d/wk for up to 12 wk.
Animals
Wild-type (C57BL/6J) mice and mice deficient in CD8+ T cells (Cd8−/−) (B6.129S2Cd8atm1Mak) were purchased from the Jackson Laboratories (Bar Harbor, ME). Cd8(−/−) mice are homozygous for the Cd8atm1Mak targeted mutation and are deficient in functional CD8+ cytotoxic T cells (16). CD4 T cell development and function are normal in these mice. Cd8(−/−) mice have been backcrossed >20 generations with C57BL/6J mice (wild-type). All procedures were conducted using mice 8–12 wk of age maintained in ventilated microisolator cages housed in an American Association for Accreditation of Laboratory Animal Care-accredited animal facility. Protocols and studies involving animals were conducted in accordance with National Institutes of Health guidelines.
Acrolein Exposures
Mice were exposed to acrolein as previously described (17, 18). Acrolein vapor is generated by passing N2 (3–15 ml/min) over a 3-ml reservoir of liquid acrolein (SigmaAldrich, St. Louis, MO). This mixture was diluted with high-efficiency particle-filtered air (400 ml/min) and introduced into a 0.32-m3 stainless steel chamber. The chamber atmosphere was sampled with a series of two glass-fritted impingers, each containing 10 ml of 96% ethanol. A fraction of each sample was mixed with 50 mM hexylresorcinol (Sigma, St. Louis, MO), 2.1 mM mercury chloride (Aldrich, Milwaukee, WI), and 29.7 M trichloroacetic acid (Fisher, Fair Lawn, NJ). Samples and known standards in equal volumes were heated (65°C for 15 min) and allowed to cool (22°C for 15 min), and the absorbance at 605 nm measured with a spectrophotometer (Beckman DU-64).
CD8 and CD4 Immunohistochemistry
Animals were killed immediately after exposure by an intraperitoneal injection of pentobarbital sodium (50 mg/kg of Nembutal; Abbott Laboratories, Chicago, IL) and the posterior abdominal aorta severed. To obtain tissue for histological analysis, a cannula was inserted in the middle of the trachea, and the lung was instilled with 20% sucrose. The lung was embedded in OCT compound (Electron Microscopy Sciences, Washington, PA) and frozen at −70°C until processing. CD8+ cells were detected using a rat antibody specific for mouse CD8α (clone 53-6.7, BD Biosciences, San Jose, CA). CD4+ positive cells were detected using a rat antibody specific for mouse CD4 (clone RM4–5, BD Biosciences). Receptor positive cells were quantitated as the number of positive cells per randomly chosen, high-power field (400x magnification) of 5 fields per section from 3 sections per mouse (n = 5–8 mice per group).
Bronchoalveolar Lavage and Cell Enumeration
After exposure, mice were anesthetized (50 mg/kg of pentobarbital sodium ip) and exsanguinated by severing the posterior abdominal aorta. The lungs were then lavaged two times with 1 ml of Hanks’ balanced salt solution (HBSS). Individual BAL returns were pooled and centrifuged at 300g × 10min. The supernatant was removed and stored at −70°C. The cell pellet was reconstituted in 1 ml HBSS containing 2% fetal bovine serum (FBS). Total cell counts were determined with a hemocytometer. Differential cell counts (>300 cells) were performed on Diff-Quik-stained (Baxter Diagnostics, McGaw Park, IL) cytospin slides (Cytospin3, Shandon Scientific).
RNase Protection Assays
Cytokine expression was assessed by RNase protection assay (RPA) analysis using total RNA prepared from whole lungs using Trizol reagent (Molecular Research Center Inc., Cincinnati, OH). RNA was hybridized with 32P-labeled antisense probes generated by the in vitro transcription of templates for lymphotactin, macrophage inflammatory protein (MIP)-2, MIP-1b, MIP-3a, MIP-1a, interleukin (IL)-10, IP-10, IFN-γ, IL-12p40, RANTES, and monocyte chemotactic protein (MCP)-1, according to the manufacturer’s instructions (RiboQuant Multiprobe RPA system, BD Biosciences). The protected probes were separated by polyacrylamide gel electrophoresis (6%) and visualized using a Storm Phosphorimager (Molecular Dynamics, Sunnyvale, CA). Individual bands were quantitated by scanning densitometry and normalized to L32 using ImageQuant software (Molecular Dynamics).
Zymogram
After exposure, mice were anesthetized (50 mg/kg of pentobarbital sodium ip) and exsanguinated by severing the posterior abdominal aorta. Lungs were removed and immersed in liquid nitrogen. Lungs were homogenized in HBSS. Gelatin zymography was performed using whole lung homogenates. Briefly, 60 μg of lung protein in 2x Tris-Glycine gel loading buffer was electrophoresed (125V; 60 minutes) in a 10% Tris-Glycine gel containing 0.1 % gelatin (Invitrogen, Carlsbad CA). Gels were washed twice in zymogram renaturing solution, preincubated (30 minutes, 37°C) in zymogram developing solution and incubated in zymogram developing solution (Invitrogen). Gels were stained in 0.5% Coomassie blue (Sigma, St. Louis, MO) in 40% methanol, 10% acetic acid (1h, 22°C), and destained in 40% methanol, 10% acetic acid (1h, 22°C) and two changes of destaining solution to visualize digested bands in the gelatin matrix. A parallel SDS-PAGE gel was loaded with 60 ug of protein and stained with Coomassie blue as a loading control. Gels were photographed using a digital camera. Individual bands were quantitated by scanning densitometry using ImageQuant software (Molecular Dynamics).
Tissue Fixation for Histology and Caspase3 Immunohistochemistry
To obtain tissue for histological analysis, a cannula was inserted in the trachea, and the lung was instilled with 10% phosphate-buffered formalin at a constant pressure (25 cm H2O). The trachea was ligated, and the inflated lung was immersed in fixative for 24 h. Mean Linear Intercept (Lm) was calculated on hematoxylin and eosin-stained sections of mouse lung as previously described (19, 20). Ten fields per section from five sections per mouse (5–8 mice per group) were evaluated, both vertically and horizontally, independently by each of two investigators. Measurements of the two investigators were not significantly different. Large airways and blood vessels were excluded from the measurements. Caspase3 immunohistochemistry was performed with a rabbit polyclonal antibody against active caspase3 (clone AF835, R&D Systems, Minneapolis, MN). Antigen-antibody complexes were detected in paraffin-embedded tissue sections (5 μm) of mouse lungs (n=5–8 per group) with the Vectastain ABC Elite IgG kit (Vector Laboratories, Burlingame, CA) using a goat anti-rabbit secondary antibody according to manufacturer’s protocol. Caspase3 positive cells were quantitated as the number of positive cells per randomly chosen, high-power field (400x magnification) of 5 fields per section from 3 sections per mouse (n = 5–8 mice per group).
Mucous Cell Metaplasia Quantitation
Mucous cell development along the airway epithelium is quantified in paraffin-embedded tissue sections (5 μm) stained with periodic acid-Schiff reagent. Parasaggital sections were analyzed by bright-field microscopy with image analysis software (ImagePro Plus, Media Cybernetics, Silver Spring, MD) to derive an airway mucus index that is reflective of both the amount of mucus per airway and the number of airways affected. The airway mucus index is calculated by summing the ratio of the periodic acid-Schiff-positive epithelial area to the total epithelial area per section and dividing by the number of airways per section (21).
BAL Mucin Assay
Muc5ac mucin in the airways was determined using an enzyme linked immunosorbent assay modified from Miller et al. (22). Briefly, 100 ul of BAL sample or isolated rat mucin standard is dried overnight at 37°C onto 96-well microtiter plates (Immulon, PGC Scientific), washed and incubated with a biotinylated primary anti-MUC5AC antibody (clone 45M1, LabVision, Fremont, CA) followed by an incubation with a secondary peroxidase-conjugated antibody.
Statistics
Parametric data was analyzed for statistical significance by one-way ANOVA followed by Student’s t tests. All pairwise multiple comparison procedures were analyzed by the Holm-Sidak method using SigmaStat software (Systat Software, San Jose, CA). Differences between means were considered significant when p < 0.05.
RESULTS
CD8+ lymphocytes accumulate in the lungs of acrolein-exposed mice
Acrolein exposure induces a time-dependent increase in the number of CD8+ cells in the lungs of wild-type mice (Figure 1). CD8+ cells primarily localized to the parenchyma of the lung, although some CD8+ cells were detected in the submucosa of the conducting airways. CD4+ cells, on the other hand, exhibited a modest and slower accumulation compared to CD8+ cells that was only significant after 12 weeks of exposures. A similar pattern of CD4+ cell accumulation was observed in Cd8(−/−) mice exposed to acrolein for up to 12 weeks.
Figure 1. CD8+ T cells preferentially accumulate in the lungs of acrolein-exposed mice.

The accumulation of CD8+ and CD4+ cells in the lungs of wild-type and Cd8(−/−) mice were assessed by immunocytochemistry on frozen sections. Tissue CD8+ cells were quantitated as the number of positive cells per field (400x original magnification). Values presented are means (± sem) of 5 fields per section from 3 sections per mouse (n = 7–8 mice per group). * Denotes value significantly greater than sham-exposed control at p<0.05.
Acrolein-induced macrophage accumulation is dependent on the presence of lymphocytes in the lung
One of the hallmark features of COPD is the persistent accumulation of macrophages in the lungs. Chronic acrolein exposure mimics this feature in mice (Figure 2). Macrophage accumulation in the bronchoalveolar lavage (BAL) compartment of wild-type mice increased rapidly (~3-fold at 2 weeks) and remained elevated for the duration of the exposure period. In contrast, increased macrophage accumulation does not occur in Cd8(−/−) mice exposed to acrolein for up to 12 weeks. Although the numbers of neutrophils were increased in the BAL of acrolein-exposed mice compared with unexposed, these cells were less than 5% of the total cells in wild-type and Cd8(−/−) mice (unexposed wild-type = 0.53 ± 0.09%, 12-week exposed wild-type = 2.41 ± 0.85%, unexposed Cd8(−/−) = 0.61 ± 0.13%, 12-week exposed Cd8(−/−) = 2.84 ± 0.96%). There were no significant differences in neutrophil accumulation in the BAL between strains of mice exposed to acrolein for up to 12 weeks.
Figure 2. Macrophages accumulate in the lungs of wild-type, but not CD8+ T cell-deficient mice, exposed to acrolein.

The accumulation of macrophage in the lungs of wild-type, and Cd8(−/−) mice were assessed by enumeration of bronchoalveolar lavage cells. The number of macrophages is a product of the total number of cells recovered and the percentage of macrophages as determined by Wright-stained cytocentrifuge preparations (counting ≥300 cells per slide). Values presented are means (± sem) of 10–15 mice per group. * Denotes value significantly greater than strain-matched sham-exposed controls at p<0.05. #Denotes value significantly different than exposure-matched wild-type at p<0.05.
Alterations in macrophage numbers were not associated with an aberrant accumulation of CD4+ cells, as there was no differences in the CD4+ cells in Cd8(−/−) mice compared to wild-type at any time points during the exposure.
Acrolein-induced airway pathologies occur independently of CD8+ cells
Epithelial cell hypertrophy and mucous cell metaplasia are also hallmark features of COPD in humans. Acrolein exposure induces results in hypertrophy of the epithelial cells lining the airways and a cumulative increase in the number of mucous cells in the conducting intrapulmonary airways of wild-type mice (Figure 3). This morphological change in the airway epithelium was not qualitatively or quantitatively different in Cd8(−/−) mice exposed to acrolein for up to 12 weeks (Figure 3A–D). Muc5ac protein was not detected by ELISA in the BAL of mice at any time suggesting that there is not significant hypersecretion of the mucous cells following irritant exposure (not shown).
Figure 3. Epithelial cell hypertrophy and mucous cell metaplasia develop in mice exposed to acrolein for 12 weeks.

(A–C) Mucous cell metaplasia was visualized by light microscopy of periodic acid-Schiff-stained lung sections. Photomicrographs (400x original magnification) are representative of 8 mice per group. Scale bar = 50μm. (D) Mucous production by airway epithelial cells in acrolein-exposed mice is not different between wild-type and Cd8(−/−) mice. Mucous index was derived from n = 5 mice per group. Values presented are means ± sem. No significant differences were observed between wild-type and Cd8(−/−) mice exposed to acrolein for 12 weeks. * denotes value significantly greater than strain-matched sham-exposed control at p<0.05.
Acrolein-induced alveolar enlargement is dependent on the presence of lymphocytes in the lung
Chronic acrolein exposure caused a progressive destruction of the alveolar walls in wild-type mice (Figure 4C) in a manner analogous to centrilobular emphysema commonly associated with cigarette smoking in humans (23). In contrast, this destructive process was attenuated in Cd8(−/−) mice exposed to acrolein for up to 12 weeks (Figure 4D). Increased alveolar size, estimated by mean linear intercept (Lm), was quantitatively greater in the wild-type mice compared to the Cd8(−/−) mice exposed to acrolein for 12 weeks (Figure 4E).
Figure 4. Airspace enlargement is attenuated in Cd8-deficient mice exposed to acrolein.
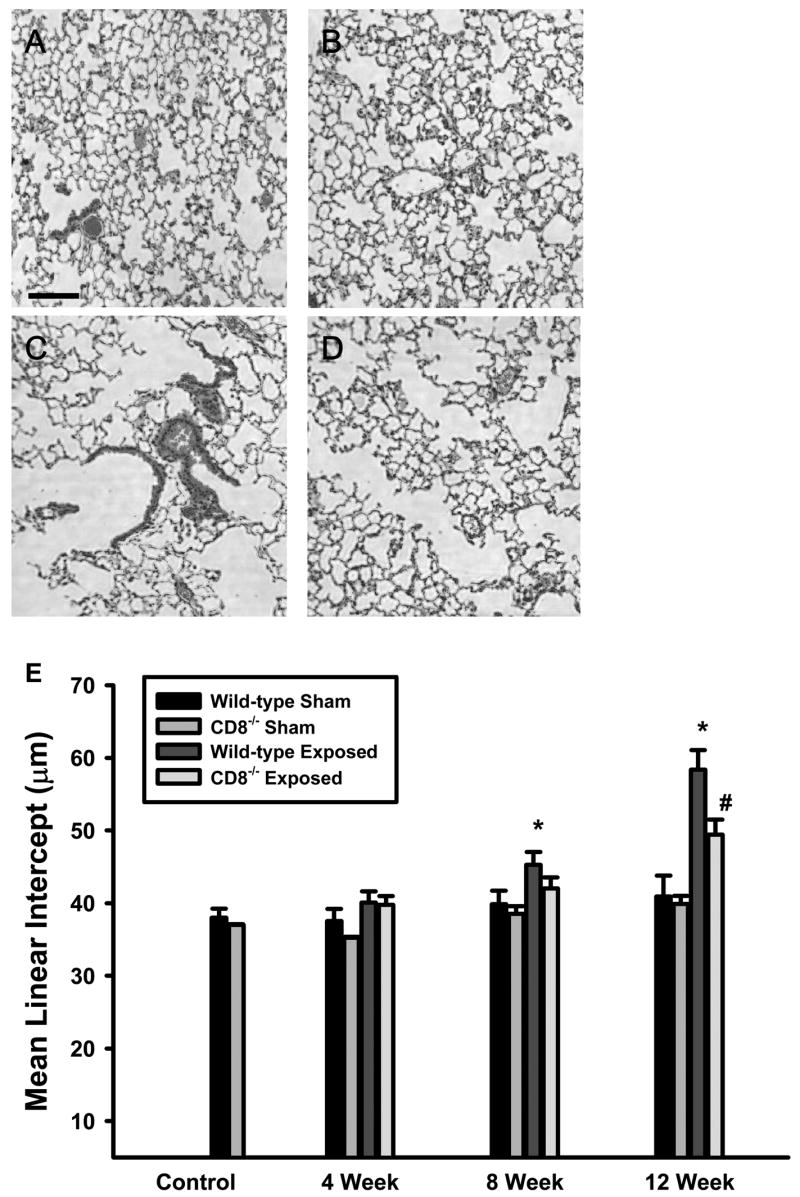
(A) Sham-exposed wild-type mice (B) Sham-exposed Cd8(−/−) mice (C) acrolein-exposed (12 weeks) wild-type mice (D) acrolein-exposed (12 weeks) Cd8(−/−) mice. Photomicrographs (400x original magnification) are representative of 8 mice per group. Scale bar = 100μm. (E) Mean linear intercept (Lm) was calculated from photomicrographs of lung parenchyma of sham- and acrolein-exposed (12 weeks) wild-type and Cd8(−/−) mice. Lm values presented are means ± sem. No significant differences were observed between sham-exposed wild-type and Cd8(−/−) mice. * denotes value significantly greater than strain-matched sham-exposed control at p<0.05. # denotes significantly different from wild-type mice exposed to acrolein at p<0.05.
Pulmonary cytokine production is attenuated in Cd8(−/−) mice
We analyzed cytokine transcripts by RPA analysis on total lung RNA isolated from wild-type and Cd8(−/−) mice exposed to acrolein for 12 weeks. Steady state levels of several cytokine messages were significantly different following acrolein exposure (FIGURE 5). IP-10, IFN-γ, and IL-12p40 were increased in wild-type but not Cd8(−/−) mice exposed to acrolein as compared to their respective controls. RANTES and MCP-1, on the other hand, were increased in both wild-type and Cd8(−/−) mice exposed to acrolein, but this increase was significantly greater in wild-type mice compared to Cd8(−/−) mice.
Figure 5. Inhibition of cytokine levels in acrolein-exposed Cd8(−/−) mice.

Cytokine transcript levels are increased in the lungs of wild-type, but not Cd8(−/−) mice, exposed to acrolein. Cytokine/chemokine transcript levels in whole lungs of mice exposed to acrolein or filtered air for 12 weeks were determined by RPA analysis. Values presented are means (± sem) of 8 mice per group. * Denotes value significantly greater than strain-matched sham-exposed control mice at p<0.05. # Denotes value significantly greater than exposure-matched Cd8(−/−) mice at p<0.05.
Acrolein-induced MMP activation in the lung is dependent on the presence of CD8+ cells
MMP2 and MMP9 were readily detectable in the lung homogenates of wild-type and Cd8(−/−) mice by gelatin zymography. The activity of MMP2 and MMP9 increased in the lung homogenates of wild-type, but not Cd8(−/−) mice, exposed to acrolein for 12 weeks as compared to unexposed controls (Figure 6A and 6B).
Figure 6. MMP-2 and MMP-9 activity were increased in the lungs of wild-type, but not Cd8(−/−) mice, exposed to acrolein.

(A) Gelatin zymography was conducted on two lung homogenates of control and acrolein-exposed (12 weeks) wild-type and Cd8(−/−) mice. Photomicrograph is representative of results obtained from 5–8 mice per group. (B) Densitometric analysis of zymograms. * Denotes value significantly greater than strain-matched control mice at p<0.05.
Active caspase3 staining in airway epithelial is dependent on the presence of CD8+ cells in the lung
Active caspase3 positive cells were rarely observed in unexposed mice. However, following exposure to acrolein, active caspase3 positive cells were observed primarily in epithelial cells of the terminal bronchioles and in alveolar epithelium. The number of active caspase3 positive cells per high powered field in the lungs of acrolein-exposed wild-type and Cd8(−/−) mice was significantly increased at 8 and 12 weeks. Furthermore, there was a significant decrease in the number of active caspase3 positive cells in Cd8(−/−) mice as compared to wild-type mice after exposure to acrolein for 12 weeks (Figure 7).
Figure 7. Increased active caspase3 is greater in the lungs of wild-type compared to Cd8(−/−) mice exposed to acrolein.
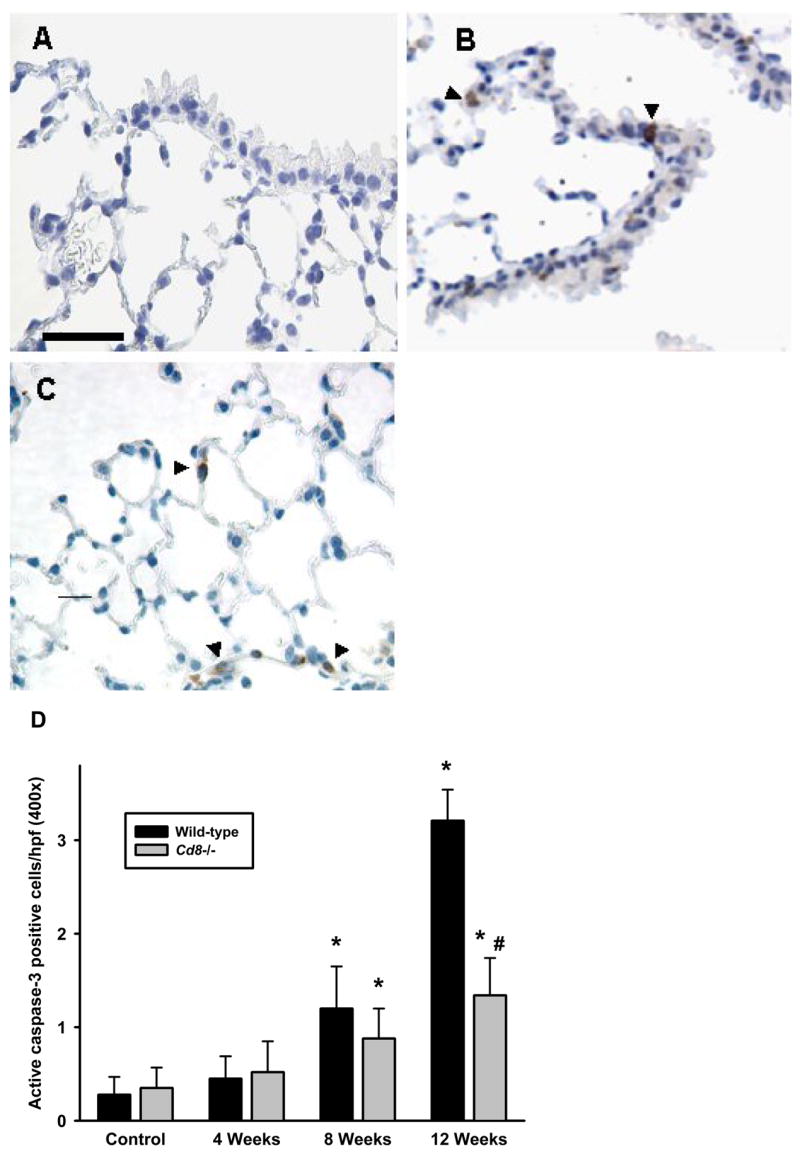
The accumulation of apoptotic cells in the lungs of wild-type and Cd8(−/−) mice were assessed by immunohistochemistry on paraffin-embedded sections using a rabbit antibody specific for active caspase3. (A) Filtered air-exposed wild-type mice (B) airway epithelium of acrolein-exposed (12 weeks) wild-type mice, and (C) alveolar epithelium of acrolein-exposed (12 weeks) wild-type mice. Photomicrographs (400x original magnification) are representative of 5–8 mice per group. Scale bar = 50μm. (D) Active caspase3 positive-stained cells were quantified from photomicrographs of lung sections of control and acrolein-exposed (12 weeks) wild-type and Cd8(−/−) mice. Values presented are means ± sem of the number of positive cells per high-powered field (400x). No significant differences were observed between filtered air-exposed wild-type and Cd8(−/−). * Denotes value significantly greater than strain-matched controls at p<0.05. # Denotes value significantly different than exposure-matched wild-type at p<0.05.
DISCUSSION
In this study, we utilize mice genetically devoid of CD8+ T cells to demonstrate that CD8+ T cells are important in the development of many pathophysiologies associated with COPD, including pulmonary monocyte accumulation, airspace enlargement, cytokine expression, protease activation, and epithelial cell apoptosis. The hypothesis that COPD is mediated, in part, by CD8+ T cell activation is based on correlative data from COPD patients. Specifically, CD8+ T cells are increased in the airways (3, 4), alveolar walls, and interstitium (5) of smokers with COPD. Furthermore, an inverse relationship exists between the numbers of CD8+ T cells in the lung and forced expiratory volume (FEV1), a critical measure of lung function (3, 5).
The present study demonstrates CD8+ T cell and macrophage accumulate in the lungs of toxicant-exposed mice, and that macrophage accumulation is dependent on the presence of CD8+ T cells. There is strong evidence to support a connection between CD8+ T cells and macrophages in the lung that contributes to the development of COPD. Saetta and coworkers demonstrated increased expression of CXCR3 chemokine receptor and its cognate ligand IP-10 (CXCL10) in the peripheral airway of smokers with COPD. These CXCR3+ cells co-expressed CD8 and IFN-γ (24). Additionally, T cells from patients with emphysema secrete more IFN-γ and IP-10 compared to control patients (7). Importantly, this study also indicates that these CXCR3 ligands, which are potent chemoattractants for cytotoxic T cells, are capable of activating alveolar macrophage to increase MMP production. Further evidence to support this paradigm comes from experiments that demonstrate IFN-γ induces alveolar macrophage production of CXCR3 ligand levels that are sufficient to induce CXCR3+ T cell migration (25). Together, these data establish a paradigm whereby the secretion of IFN-γ by CD8+ T cells following lung injury activates macrophages to express CXCR3 ligands and produce MMPs, which can lead directly to the destruction of the lung parenchyma. CXCR3 ligands also lead to the further recruitment of CD8+ T cells, as well as the increased production of IFN-γ and increased macrophage/T cell recruitment (26, 27). Most recently, Maeno et al. have demonstrated that CD8+ T cells are necessary for increased IP-10 protein and MMP-12 production in a model of cigarette smoke-induced emphysema in mice (28). Our data demonstrating that CD8+ T cells play a role in acrolein-induced IP-10 and IFN-γ expression as well as macrophage accumulation provides experimental evidence for this paradigm beyond the correlative clinical data. These studies notwithstanding, it remains to be determined whether the alveolar enlargement is attributable to the direct effects of CD8+ T cells or the indirect effect of attenuating macrophage accumulation and activation.
Several pathways could control the airspace enlargement in exposed wild-type mice that are not present in Cd8(−/−) mice. Disruption of the terminal bronchioles and parenchyma in experimentally induced COPD may be attributed to direct cytolytic effects of CD8+ T cells on bronchiolar and alveolar epithelium, the increased elastolytic burden in the lung (indirect, non-lytic, effects of CD8+ T cells), and direct cytotoxic effects of the toxicant exposure on epithelial cells. The role of CD8+ T cells in pulmonary epithelial cell turnover has not been explored in the context of emphysema. However, increased numbers of apoptotic cells in the alveolar walls correlates with the number of CD8+ cells in smokers with emphysema (29). Evidence for a direct role of the cytolytic effector function of CD8+ T cells against pulmonary epithelial cells also exists. CD8+ T cells utilize at least two independent pathways to induce lethal damage on target cells. One pathway involves degranulation of proteases, granzymes, and the pore forming protein, perforin, to induce target cell death mediated by caspase activation and apoptosis. Another major pathway involves the Fas receptor on the epithelial cell surface binding Fas ligand on cytotoxic lymphocytes, subsequently leading to apoptosis (30). These pathways are enacted in response to acute lung injury in humans (31), as well as in response to lipopolysacharride- and bleomycin-induced lung injury in mice (32, 33).
In the present study, MMP2 and MMP9 are increased in the lungs of wild-type mice, but not Cd8(−/−) mice. Decreased MMP expression is likely a result of the attenuated infiltration of macrophages into the airways as a consequence of the lack of CD8+ T cells, and could contribute to the inhibition of airspace enlargement observed in Cd8(−/−) mice. Increasing evidence suggests that matrix metalloproteinases (MMPs) may have a central role in cigarette smoking induced COPD, as several studies have shown increased concentrations and activities of MMPs in the airways of patients with COPD as compared to the controls (34–36). Macrophages derived from patients with COPD have increased levels of MMP9 mRNA, and secrete more MMP9 when stimulated with cigarette smoke conditioned medium (37). Segura-Valdez et. al. (38) showed increased MMP2 and MMP9 by immunohistochemistry in lungs of patients with COPD. Furthermore, lung MMP9 expression correlates with smoking dose and airflow obstruction (39). Animal models of COPD have also contributed to the evidence supporting a role for MMPs in the development of emphysema. In mice exposed to cigarette smoke, Hautamaki et. al. (40) demonstrated that macrophage infiltration and MMP12 was necessary for the full development of emphysema.
Chronic bronchitis, which is characterized by mucus hypersecretion and cough, represents the other hallmark feature of COPD (1). Chronic toxicant exposure results in an increase in goblet cell number and mucus production due to hyperplasia and metaplasia of the airway epithelium (41). The production of excess mucus by the increased number of goblet cells in the peripheral airways is proposed as a major contributing factor in the pathophysiology of the COPD (42). Previous data from our labs have demonstrated that repeated acrolein exposure mimics this pathology in mice and rats (15, 18). Presently, we show that the development of mucous cell metaplasia along the airways is not dependent on the presence of CD8+ T cells. These observations are consistent with previous studies that fail to show a significant correlation between the presence of these cells in the glands, epithelium or submucosa and symptoms of chronic bronchitis in smokers (43).
Although it is clear the Cd8(−/−) mice had fewer caspase3 positive cells, the number of positive cells still increased compared to control, suggesting acrolein had a direct effect on apoptosis in mice. The direct effects of acrolein on epithelial cell viability represents an additional mechanism that may contribute to the development of caspase3 activation and tissue destruction. Acrolein causes necrotic cell death of cultured pulmonary epithelial cells at high concentrations (>200 μM) apoptotic cell death at lower concentrations (<200 μM) (44, 45). Acrolein-induced apoptosis is mediated by oxidative stress and involves caspase activation (45, 46). Apoptotic cell death represents a likely mechanism of tissue destruction in the lower respiratory tract, as acrolein is demonstrated to reach concentrations up to 80 uM in the respiratory lining fluid during cigarette smoking (47). The acrolein concentrations employed in the present experiments is scaled to mimic human exposure from cigarette smoking (48).
The findings from this study do not preclude a causative role for other cell types not examined. On the contrary, the development of substantial airspace enlargement in the Cd8(−/−) mice indicates that effects of acrolein exposure on epithelial cells and the function of other leukocytes such as neutrophils, natural killer cells, and other T cell populations (e.g., CD4+ T cells) are also determinants of emphysema development in this model. Of these cell types, the neutrophil is likely the major additional determinant of airspace enlargement. The roles of neutrophils, specifically neutrophil elastase, have been implicated in humans and mouse models of emphysema (2, 49). Shapiro et al. provided empirical evidence for the contribution of neutrophils using mice genetically deficient in neutrophil elastase. This study indicated that neutrophils, although present at less than 5% of the BAL leukocytes, contribute to ~60% of the alveolar tissue destruction observed in cigarette smoke exposed mice partially via effects on macrophage metalloproteinase activation (2). We also observed that neutrophils account for only 5% of BAL leukocytes, and further observed that neutrophil numbers increased similarly regardless of phenotype. CD8 depletion resulted in an ~40% decrease in airspace enlargement in our model. This suggests that CD8+ T cells are also required for maximal macrophage effector function in toxicant-induced COPD, and that neutrophil functions represent the majority of the residual (i.e., CD8-independent) mechanisms leading to airspace enlargement.
We provide direct evidence that deficiency of CD8+ T cells attenuates the mediators, inflammation, and pathology associated with COPD. The data presented provide an initial characterization of the consequences and mechanisms of CD8+ T cell depletion, but they do not address the proximal events that lead to CD8+ T cell activation. The nature of the interactions between T cells and injured epithelial cells is also uncertain. Cosio et al. have proposed the concept that COPD is an autoimmune disease in that lung injury could lead to the alteration of self-antigens (50). Auto-reactive T cells thus could recognize the generated neo-antigens. Persistent inflammation and direct cytolysis of target cells could occur through the effector function of activated T cells (and natural killer cells) in a manner analogous to other autoimmune diseases (51). This notion is supported by experimental data that demonstrates the capacity of CD8+ T cells to recognize and lyse autoantigen-expressing alveolar type II cells in vivo (52), a response accompanied by mononuclear cell infiltration and pulmonary injury similar to interstitial pneumonia.
We speculate that an alternative mechanism underlies the role of the immune system in the development and progression of tissue injury in COPD. Epithelial cells undergoing physical or chemical stress are selectively removed to control inflammation and promote epithelial repair. Multiple mechanisms for the detection and elimination of stressed cell have been described. These mechanisms involve the de novo expression of self-antigens (e.g., MHC class I-like molecules, endogenous toll receptor ligands) that are recognized by cytotoxic T cells and natural killer cells (53, 54). The premise of this theory is that the immune system can be activated by danger or stress signals from cells exposed to pathogens, environmental stimuli, or mechanical damage, and that these signals can be constitutive, inducible, or both. Constitutive signals primarily consist of intracellular products released following cellular necrosis (as opposed to apoptosis), whereas inducible signals are molecules synthesized or modified in response to stress. In support of this theory, we have recently reported MHC class I-like molecule expression on airway epithelial cells in vitro following oxidative stress (55).
In COPD, the interactions and relationships between immune function, inflammation, proteolytic burden, and apoptosis are complex. This study takes a step toward unraveling the role of the immune system in the development of COPD pathology. The establishment of a role for CD8+ T cells in the development of inflammation and airspace enlargement provides a conceptual framework to study the role of the immune system in many pulmonary diseases that have non-pathogenic stimuli in their etiology, such as occurs with many environmental and occupational triggers.
Acknowledgments
This study was supported by the NIH/NIEHS, Center for Environmental Genetics (P30-ES06096-02), the Health Effects Institute, and National Institutes of Health grants ES006096, ES007250, ES010562, HL065612, HL077763, and AI046556.
List of Abbreviations
- COPD
chronic obstructive pulmonary disease
- MMP
matrix metalloprotease
- IP
inflammatory protein
- IFN
interferon
- IL
interleukin
- RANTES
regulated on activation normal T cell expressed and secreted
- MCP
monocyte chemotactic protein
- RPA
Rnase protection assay
Footnotes
Competing Interests. The authors declare that they have no competing interests.
Authors Contributions. MTB conceived the experiments and drafted the manuscript. SCW helped design the studies, performed the RPA analyses, and helped prepare the manuscript. NLH collected samples during the experiments and conducted the ELISAs. HD performed the gelatin zymography. EB and MV conducted the animal exposures and helped with the sample collection during experimental procedures. JWT performed the immunohistochemistry and subsequent analysis. GDL helped conceive the studies and helped draft the manuscript. All authors read and approved the final manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pauwels RA, Buist AS, Calverley PM, Jenkins CR, Hurd SS. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summary. Am J Respir Crit Care Med. 2001;163(5):1256–76. doi: 10.1164/ajrccm.163.5.2101039. [DOI] [PubMed] [Google Scholar]
- 2.Barnes PJ, Shapiro SD, Pauwels RA. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J. 2003;22(4):672–88. doi: 10.1183/09031936.03.00040703. [DOI] [PubMed] [Google Scholar]
- 3.O’Shaughnessy TC, Ansari TW, Barnes NC, Jeffery PK. Inflammation in bronchial biopsies of subjects with chronic bronchitis: inverse relationship of CD8+ T lymphocytes with FEV1. Am J Respir Crit Care Med. 1997;155(3):852–7. doi: 10.1164/ajrccm.155.3.9117016. [DOI] [PubMed] [Google Scholar]
- 4.Saetta M, Di Stefano A, Turato G, Facchini FM, Corbino L, Mapp CE, Maestrelli P, Ciaccia A, Fabbri LM. CD8+ T-lymphocytes in peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998;157(3 Pt 1):822–6. doi: 10.1164/ajrccm.157.3.9709027. [DOI] [PubMed] [Google Scholar]
- 5.Saetta M, Baraldo S, Corbino L, Turato G, Braccioni F, Rea F, Cavallesco G, Tropeano G, Mapp CE, Maestrelli P, Ciaccia A, Fabbri LM. CD8+ve cells in the lungs of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1999;160(2):711–7. doi: 10.1164/ajrccm.160.2.9812020. [DOI] [PubMed] [Google Scholar]
- 6.Small BA, Dressel SA, Lawrence CW, Drake DR, 3rd, Stoler MH, Enelow RI, Braciale TJ. CD8(+) T cell-mediated injury in vivo progresses in the absence of effector T cells. J Exp Med. 2001;194(12):1835–46. doi: 10.1084/jem.194.12.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grumelli S, Corry DB, Song LZ, Song L, Green L, Huh J, Hacken J, Espada R, Bag R, Lewis DE, Kheradmand F. An Immune Basis for Lung Parenchymal Destruction in Chronic Obstructive Pulmonary Disease and Emphysema. Plos Med. 2004;1(1):e8. doi: 10.1371/journal.pmed.0010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ayer HE, Yeager DW. Irritants in cigarette smoke plumes. Am J Public Health. 1982;72(11):1283–5. doi: 10.2105/ajph.72.11.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jermini C, Weber A, Grandjean E. [Quantitative determination of various gas-phase components of the side-stream smoke of cigarettes in the room air as a contribution to the problem of passive-smoking (author’s transl)] Int Arch Occup Environ Health. 1976;36(3):169–81. doi: 10.1007/BF00378272. [DOI] [PubMed] [Google Scholar]
- 10.Uchida K. Current status of acrolein as a lipid peroxidation product. Trends Cardiovasc Med. 1999;9(5):109–13. doi: 10.1016/s1050-1738(99)00016-x. [DOI] [PubMed] [Google Scholar]
- 11.Corradi M, Pignatti P, Manini P, Andreoli R, Goldoni M, Poppa M, Moscato G, Balbi B, Mutti A. Comparison between exhaled and sputum oxidative stress biomarkers in chronic airway inflammation. Eur Respir J. 2004;24(6):1011–7. doi: 10.1183/09031936.04.00002404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lyon JP, Jenkins LJ, Jr, Jones RA, Coon RA, Siegel J. Repeated and continuous exposure of laboratory animals to acrolein. Toxicol Appl Pharmacol. 1970;17(3):726–32. doi: 10.1016/0041-008x(70)90047-5. [DOI] [PubMed] [Google Scholar]
- 13.Feron VJ, Kruysse A, Til HP, Immel HR. Repeated exposure to acrolein vapour: subacute studies in hamsters, rats and rabbits. Toxicology. 1978;9(1–2):47–57. doi: 10.1016/0300-483x(78)90030-6. [DOI] [PubMed] [Google Scholar]
- 14.Costa DL, Kutzman RS, Lehmann JR, Drew RT. Altered lung function and structure in the rat after subchronic exposure to acrolein. Am Rev Respir Dis. 1986;133(2):286–91. doi: 10.1164/arrd.1986.133.2.286. [DOI] [PubMed] [Google Scholar]
- 15.Borchers MT, Wesselkamper S, Wert SE, Shapiro SD, Leikauf GD. Monocyte inflammation augments acrolein-induced Muc5ac expression in mouse lung. Am J Physiol. 1999;277(3 Pt 1):L489–97. doi: 10.1152/ajplung.1999.277.3.L489. [DOI] [PubMed] [Google Scholar]
- 16.Fung-Leung WP, Schilham MW, Rahemtulla A, Kundig TM, Vollenweider M, Potter J, van Ewijk W, Mak TW. CD8 is needed for development of cytotoxic T cells but not helper T cells. Cell. 1991;65(3):443–9. doi: 10.1016/0092-8674(91)90462-8. [DOI] [PubMed] [Google Scholar]
- 17.Altshuler AP, McPherson SP. Spectrophotometric analysis of aldehydes in the Los Angeles atmosphere. J Air Pollut Control Assoc. 1963;13:109–111. doi: 10.1080/00022470.1963.10468151. [DOI] [PubMed] [Google Scholar]
- 18.Borchers MT, Wert SE, Leikauf GD. Acrolein-induced MUC5ac expression in rat airways. Am J Physiol. 1998;274(4 Pt 1):L573–81. doi: 10.1152/ajplung.1998.274.4.L573. [DOI] [PubMed] [Google Scholar]
- 19.Thurlbeck WM. Measurement of pulmonary emphysema. Am Rev Respir Dis. 1967;95(5):752–64. doi: 10.1164/arrd.1967.95.5.752. [DOI] [PubMed] [Google Scholar]
- 20.Lyerla TA, Rusiniak ME, Borchers M, Jahreis G, Tan J, Ohtake P, Novak EK, Swank RT. Aberrant lung structure, composition, and function in a murine model of Hermansky-Pudlak syndrome. Am J Physiol Lung Cell Mol Physiol. 2003;285(3):L643–53. doi: 10.1152/ajplung.00024.2003. [DOI] [PubMed] [Google Scholar]
- 21.Borchers MT, Crosby J, Farmer S, Sypek J, Ansay T, Lee NA, Lee JJ. Blockade of CD49d inhibits allergic airway pathologies independent of effects on leukocyte recruitment. Am J Physiol Lung Cell Mol Physiol. 2001;280(4):L813–21. doi: 10.1152/ajplung.2001.280.4.L813. [DOI] [PubMed] [Google Scholar]
- 22.Miller LM, Foster WM, Dambach DM, Doebler D, McKinnon M, Killar L, Longphre M. A murine model of cigarette smoke-induced pulmonary inflammation using intranasally administered smoke-conditioned medium. Exp Lung Res. 2002;28(6):435–55. doi: 10.1080/01902140290096728. [DOI] [PubMed] [Google Scholar]
- 23.Hogg JC. Pathophysiology of airflow limitation in chronic obstructive pulmonary disease. Lancet. 2004;364(9435):709–21. doi: 10.1016/S0140-6736(04)16900-6. [DOI] [PubMed] [Google Scholar]
- 24.Saetta M, Mariani M, Panina-Bordignon P, Turato G, Buonsanti C, Baraldo S, Bellettato CM, Papi A, Corbetta L, Zuin R, Sinigaglia F, Fabbri LM. Increased expression of the chemokine receptor CXCR3 and its ligand CXCL10 in peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2002;165(10):1404–9. doi: 10.1164/rccm.2107139. [DOI] [PubMed] [Google Scholar]
- 25.Agostini C, Calabrese F, Poletti V, Marcer G, Facco M, Miorin M, Cabrelle A, Baesso I, Zambello R, Trentin L, Semenzato G. CXCR3/CXCL10 interactions in the development of hypersensitivity pneumonitis. Respir Res. 2005;6(1):20. doi: 10.1186/1465-9921-6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rollins BJ, Yoshimura T, Leonard EJ, Pober JS. Cytokine-activated human endothelial cells synthesize and secrete a monocyte chemoattractant, MCP-1/JE. Am J Pathol. 1990;136(6):1229–33. [PMC free article] [PubMed] [Google Scholar]
- 27.Taub DD, Lloyd AR, Conlon K, Wang JM, Ortaldo JR, Harada A, Matsushima K, Kelvin DJ, Oppenheim JJ. Recombinant human interferon-inducible protein 10 is a chemoattractant for human monocytes and T lymphocytes and promotes T cell adhesion to endothelial cells. J Exp Med. 1993;177(6):1809–14. doi: 10.1084/jem.177.6.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maeno T, Houghton AM, Quintero PA, Grumelli S, Owen CA, Shapiro SD. CD8+ T Cells are required for inflammation and destruction in cigarette smoke-induced emphysema in mice. J Immunol. 2007;178(12):8090–6. doi: 10.4049/jimmunol.178.12.8090. [DOI] [PubMed] [Google Scholar]
- 29.Majo J, Ghezzo H, Cosio MG. Lymphocyte population and apoptosis in the lungs of smokers and their relation to emphysema. Eur Respir J. 2001;17(5):946–53. doi: 10.1183/09031936.01.17509460. [DOI] [PubMed] [Google Scholar]
- 30.Henkart PA. Lymphocyte-mediated cytotoxicity: two pathways and multiple effector molecules. Immunity. 1994;1(5):343–6. doi: 10.1016/1074-7613(94)90063-9. [DOI] [PubMed] [Google Scholar]
- 31.Hashimoto S, Kobayashi A, Kooguchi K, Kitamura Y, Onodera H, Nakajima H. Upregulation of two death pathways of perforin/granzyme and FasL/Fas in septic acute respiratory distress syndrome. Am J Respir Crit Care Med. 2000;161(1):237–43. doi: 10.1164/ajrccm.161.1.9810007. [DOI] [PubMed] [Google Scholar]
- 32.Kitamura Y, Hashimoto S, Mizuta N, Kobayashi A, Kooguchi K, Fujiwara I, Nakajima H. Fas/FasL-dependent apoptosis of alveolar cells after lipopolysaccharide-induced lung injury in mice. Am J Respir Crit Care Med. 2001;163(3 Pt 1):762–9. doi: 10.1164/ajrccm.163.3.2003065. [DOI] [PubMed] [Google Scholar]
- 33.Hagimoto N, Kuwano K, Nomoto Y, Kunitake R, Hara N. Apoptosis and expression of Fas/Fas ligand mRNA in bleomycin-induced pulmonary fibrosis in mice. Am J Respir Cell Mol Biol. 1997;16(1):91–101. doi: 10.1165/ajrcmb.16.1.8998084. [DOI] [PubMed] [Google Scholar]
- 34.Finlay GA, O’Driscoll LR, Russell KJ, D’Arcy EM, Masterson JB, FitzGerald MX, O’Connor CM. Matrix metalloproteinase expression and production by alveolar macrophages in emphysema. Am J Respir Crit Care Med. 1997;156(1):240–7. doi: 10.1164/ajrccm.156.1.9612018. [DOI] [PubMed] [Google Scholar]
- 35.Vernooy JH, Lindeman JH, Jacobs JA, Hanemaaijer R, Wouters EF. Increased activity of matrix metalloproteinase-8 and matrix metalloproteinase-9 in induced sputum from patients with COPD. Chest. 2004;126(6):1802–10. doi: 10.1378/chest.126.6.1802. [DOI] [PubMed] [Google Scholar]
- 36.Molet S, Belleguic C, Lena H, Germain N, Bertrand CP, Shapiro SD, Planquois JM, Delaval P, Lagente V. Increase in macrophage elastase (MMP-12) in lungs from patients with chronic obstructive pulmonary disease. Inflamm Res. 2005;54(1):31–6. doi: 10.1007/s00011-004-1319-4. [DOI] [PubMed] [Google Scholar]
- 37.Russell RE, Culpitt SV, DeMatos C, Donnelly L, Smith M, Wiggins J, Barnes PJ. Release and activity of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2002;26(5):602–9. doi: 10.1165/ajrcmb.26.5.4685. [DOI] [PubMed] [Google Scholar]
- 38.Segura-Valdez L, Pardo A, Gaxiola M, Uhal BD, Becerril C, Selman M. Upregulation of gelatinases A and B, collagenases 1 and 2, and increased parenchymal cell death in COPD. Chest. 2000;117(3):684–94. doi: 10.1378/chest.117.3.684. [DOI] [PubMed] [Google Scholar]
- 39.Kang MJ, Oh YM, Lee JC, Kim DG, Park MJ, Lee MG, Hyun IG, Han SK, Shim YS, Jung KS. Lung matrix metalloproteinase-9 correlates with cigarette smoking and obstruction of airflow. J Korean Med Sci. 2003;18(6):821–7. doi: 10.3346/jkms.2003.18.6.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science. 1997;277(5334):2002–4. doi: 10.1126/science.277.5334.2002. [DOI] [PubMed] [Google Scholar]
- 41.Reid LM. Pathology of chronic bronchitis. Lancet. 1954;266(6806):274–8. [PubMed] [Google Scholar]
- 42.Hogg JC, Macklem PT, Thurlbeck WM. Site and nature of airway obstruction in chronic obstructive lung disease. N Engl J Med. 1968;278(25):1355–60. doi: 10.1056/NEJM196806202782501. [DOI] [PubMed] [Google Scholar]
- 43.Saetta M, Turato G, Facchini FM, Corbino L, Lucchini RE, Casoni G, Maestrelli P, Mapp CE, Ciaccia A, Fabbri LM. Inflammatory cells in the bronchial glands of smokers with chronic bronchitis. Am J Respir Crit Care Med. 1997;156(5):1633–9. doi: 10.1164/ajrccm.156.5.9701081. [DOI] [PubMed] [Google Scholar]
- 44.Hoshino Y, Mio T, Nagai S, Miki H, Ito I, Izumi T. Cytotoxic effects of cigarette smoke extract on an alveolar type II cell-derived cell line. Am J Physiol Lung Cell Mol Physiol. 2001;281(2):L509–16. doi: 10.1152/ajplung.2001.281.2.L509. [DOI] [PubMed] [Google Scholar]
- 45.Nardini M, Finkelstein EI, Reddy S, Valacchi G, Traber M, Cross CE, van der Vliet A. Acrolein-induced cytotoxicity in cultured human bronchial epithelial cells. Modulation by alpha-tocopherol and ascorbic acid. Toxicology. 2002;170(3):173–85. doi: 10.1016/s0300-483x(01)00540-6. [DOI] [PubMed] [Google Scholar]
- 46.Tanel A, Averill-Bates DA. The aldehyde acrolein induces apoptosis via activation of the mitochondrial pathway. Biochim Biophys Acta. 2005;1743(3):255–67. doi: 10.1016/j.bbamcr.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 47.Eiserich JP, van der Vliet A, Handelman GJ, Halliwell B, Cross CE. Dietary antioxidants and cigarette smoke-induced biomolecular damage: a complex interaction. Am J Clin Nutr. 1995;62(6 Suppl):1490S–1500S. doi: 10.1093/ajcn/62.6.1490S. [DOI] [PubMed] [Google Scholar]
- 48.Li L, Hamilton RF, Jr, Taylor DE, Holian A. Acrolein-induced cell death in human alveolar macrophages. Toxicol Appl Pharmacol. 1997;145(2):331–9. doi: 10.1006/taap.1997.8189. [DOI] [PubMed] [Google Scholar]
- 49.Janoff A. Elastases and emphysema. Current assessment of the protease-antiprotease hypothesis. Am Rev Respir Dis. 1985;132(2):417–33. doi: 10.1164/arrd.1985.132.2.417. [DOI] [PubMed] [Google Scholar]
- 50.Cosio MG, Majo J. Inflammation of the airways and lung parenchyma in COPD: role of T cells. Chest. 2002;121(5 Suppl):160S–165S. doi: 10.1378/chest.121.5_suppl.160s. [DOI] [PubMed] [Google Scholar]
- 51.Kagi D, Ledermann B, Burki K, Zinkernagel RM, Hengartner H. Molecular mechanisms of lymphocyte-mediated cytotoxicity and their role in immunological protection and pathogenesis in vivo. Annu Rev Immunol. 1996;14:207–32. doi: 10.1146/annurev.immunol.14.1.207. [DOI] [PubMed] [Google Scholar]
- 52.Enelow RI, Mohammed AZ, Stoler MH, Liu AN, Young JS, Lou YH, Braciale TJ. Structural and functional consequences of alveolar cell recognition by CD8(+) T lymphocytes in experimental lung disease. J Clin Invest. 1998;102(9):1653–61. doi: 10.1172/JCI4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gleimer M, Parham P. Stress management: MHC class I and class I-like molecules as reporters of cellular stress. Immunity. 2003;19(4):469–77. doi: 10.1016/s1074-7613(03)00272-3. [DOI] [PubMed] [Google Scholar]
- 54.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296(5566):301–5. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 55.Borchers MT, Harris NL, Wesselkamper SC, Vitucci M, Cosman D. NKG2D ligands are expressed on stressed human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2006;291(2):L222–31. doi: 10.1152/ajplung.00327.2005. [DOI] [PubMed] [Google Scholar]