

Abstract
Objective
To report the clinical outcomes and molecular genetics of nongermline retinal hemangioblastoma managed by surgical resection.
Methods
Retrospective case series of 3 patients aged 16 to 46 years treated at a tertiary care referral center (Casey Eye Institute, Portland, Oregon). Tumors 7 to 9 mm in diameter were removed from 3 consecutive eyes (in 3 patients) via internal en bloc surgical resection using a bimanual technique. Samples of DNA from 2 of 3 tumors were tested for von Hippel-Lindau gene (VHL) mutations as a clue to the molecular basis for spontaneously occurring hemangioblastoma. Main outcome measures were morbidity, visual acuity, resolution of macular exudates, and presence of VHL markers.
Results
Visual acuity improved or remained stable in all patients. All 3 developed cataracts, extracted in 2 instances. Histopathological findings were typical of retinal hemangioblastoma in all cases. The cells from one patient’s DNA sample showed loss of heterozygosity for the VHL gene, while no genetic abnormalities were detected in the other patient’s DNA sample.
Conclusions
Our patients’ favorable outcomes suggest that surgical resection is an option for patients with large retinal hemangioblastomas. In addition, ours may be the first report of retinal hemangioblastoma unassociated with a VHL mutation.
Retinal hemangioblastoma is a clinically and histopathologically distinct tumor that may occur sporadically or as a manifestation of von Hippel-Lindau (VHL) disease.1 von Hippel-Lindau disease is transmitted in an autosomal dominant fashion due to germline mutations in the VHL gene, a tumor suppressor gene on the short arm of chromosome 3 (3p25.5).2 It consists of 3 exons, which encode a messenger RNA (mRNA) transcript expressed in many tissues and translated into a 213-amino acid protein.
Originally identified as a tumor suppressor, the VHL protein is now known to repress expression of mRNAs that are normally induced under hypoxic conditions.3,4 The hypoxia-inducible factor is a key transcription factor responsible for up-regulation of expression of hypoxia-inducible genes. When mutated, the VHL gene produces a protein that is unable to regulate hypoxia-inducible factor, permitting accumulation of hypoxia-inducible factor and subsequent activation of vascular endothelial growth factor and other hypoxiainducible genes.5,6 This can result in growth of retinal hemangioblastomas and other tumors associated with VHL disease.
Clinically, retinal hemangioblastomas are characterized by their vascular appearance, often with dilated feeder vessels. They may increase in size with endophytic, exophytic, or sessile growth patterns. Vision loss may occur due to leakage of fluid and proteins from the tumor and traction retinal detachment secondary to associated vitreous and retinal changes. Various treatment options have been used, including observation, thermal laser, cryotherapy, brachytherapy, photodynamic therapy, transpupillary thermotherapy, external beam radiation therapy, and systemic and intravitreal vascular endothelial growth factor inhibitors.7-18 Treatment depends on the size and location of the tumor. Smaller tumors are generally treated using laser if posterior in location or using cryotherapy if in the retinal periphery. These methods are less effective on larger tumors (>4 mm in diameter), which may be more effectively treated using radiation therapy. However, tumors greater than 5 mm in diameter or with exudative or tractional retinal detachment have a poor prognosis even using radiation therapy.
We describe the treatment of 3 patients with large solitary retinal hemangioblastomas using internal en bloc surgical resection. All patients lack a germline mutation in the VHL gene, as determined by sequence analysis of leukocyte cell DNA. To analyze the molecular basis for sporadically occurring retinal hemangioblastoma, 2 of the resected tumors were evaluated for loss of heterozygosity (LOH) and for VHL gene mutations.
METHODS
SURGICAL TECHNIQUE
Patients or their guardians had a thorough explanation of the procedure, risks, benefits, and complications before surgery. All questions were answered, and consent was obtained.
Surgery was performed under general anesthesia by one of us (D.J.W.) in all patients. A retrobulbar injection of 4 mL of a 50:50 mixture of 2% lidocaine hydrochloride (Xylocaine; Abraxis Pharmaceutical Products, Schaumburg, Illinois) with epinephrine and 0.75% bupivacaine hydrochloride (Marcaine; Astra-Zeneca, London, United Kingdom) was used to aid intraoperative hemostasis and postoperative pain control. All patients were treated using a standard 3-port pars plana vitrectomy. In addition to conventional handheld fiber optic illumination, a separate fiber optic illumination source was used (Alcon Inc, Fort Worth, Texas) to permit bimanual surgical procedures within the eye. After complete pars plana vitrectomy and membrane dissection when indicated, diathermy was applied on either side of the large feeder vessels. An angulated subretinal forcep was passed through the first diathermy mark into the subretinal space and back into the vitreous cavity through the second mark. A second forcep was used to transfer an 8-0 to 10-0 polypropylene suture (Prolene; Ethicon Inc, Somerville, New Jersey) into the grasp of the subretinal forcep. The suture was then drawn through the subretinal space and was used to ligate the feeding vessels. Intraocular diathermy was used to cauterize the retina, and diathermy and scissor dissection were used to section the retina 1 mm to 2 mm peripheral to the tumor. The feeder vessels were sectioned using scissors. After the tumor was dissected free from the adjacent retina, the superotemporal sclerotomy was enlarged to allow removal of the tumor en bloc. Air-fluid exchange, laser photocoagulation, and gas tamponade were then accomplished in the standard fashion.
HISTOPATHOLOGICAL EXAMINATION
Tissue was fixed in 10% formalin and was then embedded in paraffin or epoxy resin (Epon-Araldite; Electron Microscopy Sciences, Fort Washington, Pennsylvania). Five-micrometer (paraffin) sections and 1-μm (epoxy resin) sections were cut for staining and analysis.
MOLECULAR GENETIC STUDIES
Peripheral blood was obtained from each patient, and genomic DNA was isolated. The exonic regions of the VHL gene were sequenced for detection of abnormalities using a standard protocol (Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania). Tissue was obtained from fresh tumor tissue and from hematoxylin-eosin-stained slides, from which tumor cells were microdissected. DNA was extracted from these tissue samples using a tissue DNA isolation kit (DNeasy; Qiagen Inc, Valencia, California). Polymerase chain reaction to amplify VHL exon sequences, gel electrophoresis, and dideoxynucleotide DNA sequencing were performed using standard methods. The following primers were used to amplify the protein coding portions of all 3 exons of the VHL gene: exon 1 forward 5′-TTAACAACAGCCTACGGTGCTG-3′ and reverse 5′-CTTCAGACCGTGCTATCGTC-3′; exon 2 forward 5′-AGCCAGGACGGTCTTGATCTC-3′ and reverse 5′-GGCAAAAATTGAGAACTGGGC-3′; and exon 3 forward 5′-ACAGGTAGTTGTTGGCAAAGC-3′ and reverse 5′-CCTAAACATCACAATGCCTAG-3′.
LOSS OF HETEROZYGOSITY
Microdissection was performed as previously described.19,20 Briefly, unstained 5-μm sections that had been embedded in paraffin were deparaffinized in 10% xylene, rinsed with ethanol of decreasing concentrations (100% to 80%), hematoxylin-eosin stained, and rinsed in 10% glycerol in EDTA buffer. Manual microdissection into areas with predominantly tumor cells (“foamy” vacuolated stromal cells) and into areas with predominantly normal cells was performed under light microscopy. Microdissected cells were subjected to DNA isolation and subsequent polymerase chain reaction to amplify the VHL gene as previously described.20,21 Satellite markers were used in quantitative Southern blot analysis to examine the samples for LOH, as previously described.21 The satellite markers used were D3S1038 and D3S1110 (Research Genetics, Huntsville, Alabama). Testing for LOH was performed as previously described.21 Briefly, satellite markers D3S1038 and D3S1110, situated near the VHL gene, were amplified by polymerase chain reaction using the incorporation of radioactive nucleotides. Amplified products were electrophoresed on a 6% acrylamide gel and were transferred and dried onto a 3-mm Whatmann paper (Whatmann International Ltd, Maidstone, England). Autoradiography was performed to establish if alleles were informative (heterozygous), and LOH was defined when 1 allele was completely or partially absent, as determined by comparing intensities of the autoradiographic signal for each allele.
RESULTS
CLINICAL FINDINGS
Preoperative and postoperative findings are given in the Table. Visual acuity improved or remained stable in all patients. The intraretinal lipid present in the maculas of patients 1 and 2 gradually resolved after removal of the tumors, while some lipid remained in the macula of patient 3 at this point in follow-up (Figure 1; efigure 1E-L is available at http://www.archophthalmol.com). The marked dilation of the retinal feeder vessels resolved quickly 1 to 2 weeks following resection of the hemangioblastoma.
Table.
Demographic, Preoperative, and Postoperative Data From 3 Patients Undergoing En Bloc Resection of Retinal Hemangioblastomas
| Patient No. |
|||
|---|---|---|---|
| Variable | 1 | 2 | 3 |
| Age, y | 16 | 44 | 46 |
| Preoperative VA | 20/60 | 20/20 | 20/40 |
| Preoperative findings | Exudative retinal detachment, macular exudates | Normal other than tumor | Tractional and exudative retinal detachment, macular exudates |
| Tumor diameter, mm | 9 | 7 | 9 |
| Postoperative VA | 20/30 With correction 20/25 Pinhole | 20/30 Without correction 20/20 Best-corrected vision | 20/40 Without correction 20/25 Pinhole |
| Postoperative findings | Mostly resolved exudate | None | Mostly resolved exudate |
| Complications | Cataract | Cataract, retinal tear | Cataract, retinal tear |
| Subsequent surgery | None | Cataract extraction, intraocular lens | None |
Abbreviation: VA, visual acuity.
Figure 1.
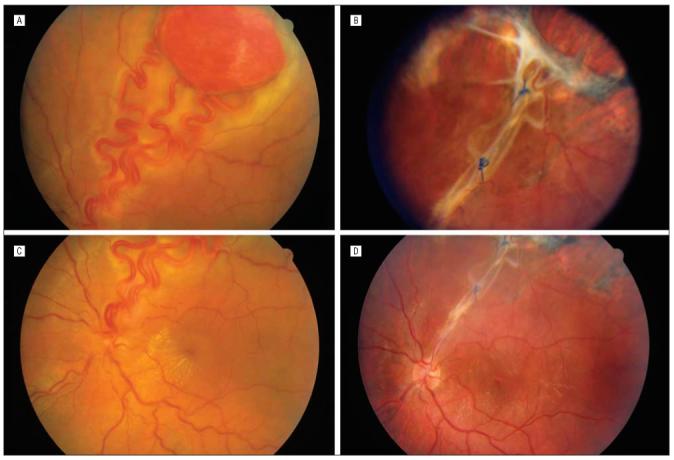
Patient 1. A, Large superotemporal retinal hemangioblastoma before surgery. B, At 4 months after surgery, polypropylene sutures ligate retinal vessels in the retinal defect, with some fibrosis at the resection site. C, Before surgery. D, After surgery, with resolution of macular exudates.
eFigure.
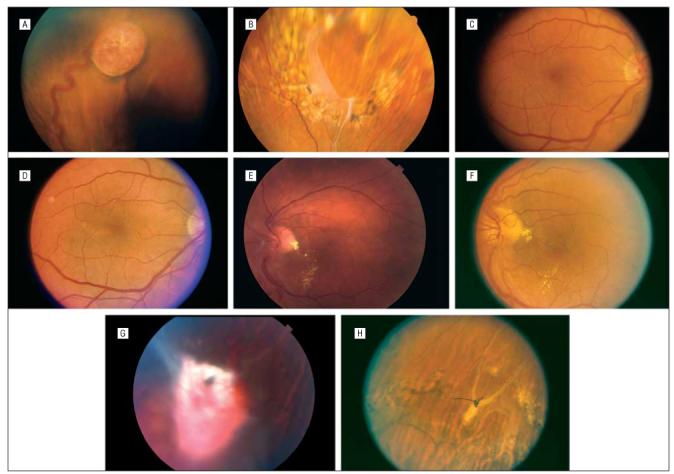
A-H, Views of patients 2 and 3. Patient 2, A-D, Before and after surgery. E, View of the resection site, with surrounding photocoagulation. Fibrosis hides the 10-0 polypropylene sutures. Patient 3, F-H. Before surgery and at 4 months after surgery, with partial resolution of macular exudates (F). Prominent fibrosis overlies the retinal hemangioblastoma (G). After surgery, the retina is attached via laser photocoagulation around the resection site (H).
All patients developed some degree of cataract, and this was severe enough to be visually substantial in patients 2 and 3. Patient 2 has undergone uneventful cataract extraction. In 2 patients, retinal breaks were made through surgical manipulation adjacent to the site of tumor resection. These were treated during the course of the primary resection.
HISTOPATHOLOGICAL FINDINGS
All 3 patients had findings typical of retinal hemangioblastoma. There were small capillary-sized vessels with morphologically normal endothelial lining. In the interstitial space, there were vacuolated “stromal” cells, typical of retinal hemangioblastoma (Figure 2).20-22
Figure 2.
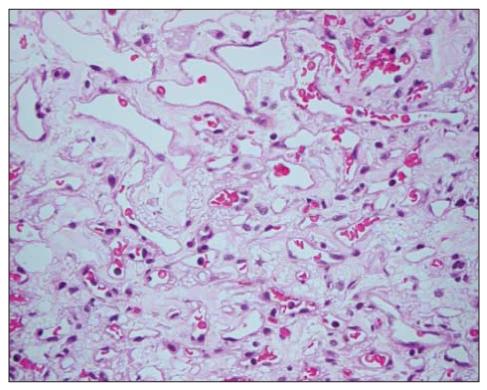
Histopathological specimen from the tumor of patient 1 stained with toluidine blue shows the typical foamy stromal cells of retinal capillary hemangioblastoma.
In patient 3, who had tractional and exudative retinal detachment, there was collagenous tissue adherent to the tumor on the vitreous side of the lesion. This correlated with the fibrous tissue noted on preoperative evaluation.
MOLECULAR GENETIC STUDIES
Molecular analysis of the VHL gene isolated from fresh tumor tissue from patients 1 and 2 demonstrated no sequence alterations within the protein coding portion of VHL exons. Patient 1 was shown not to have LOH. Results of testing for LOH suggest that patient 2 has only 1 copy of the VHL gene within the tumor cells, as the tumor tissue microdissected from this patient’s pathological specimen proved to have LOH (Figure 3) for satellite marker D3S1038. Marker D3S1110 was noninformative. Sequencing data obtained from DNA extracted from stromal cell DNA after microdissection were unreliable and yielded no meaningful results for patient 1 or for patient 2.
Figure 3.
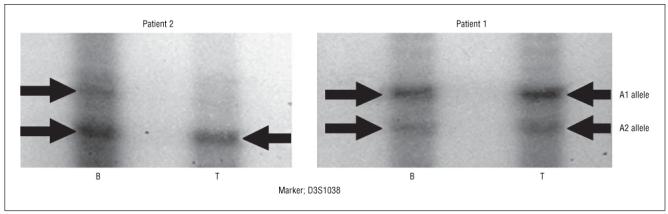
Loss of heterozygosity (LOH) in the DNA from tumor tissue (T) compared with DNA from blood (B) is apparent in patient 2, whereas there is no LOH in patient 1.
COMMENT
Retinal hemangioblastomas greater than 5 mm in diameter or associated with tractional or exudative retinal detachment have a poor prognosis using nonsurgical modes of treatment. Surgical resection of retinal hemangioblastomas has been described previously. Peyman et al23 reported resection of retinal hemangioblastomas using an external approach in 2 patients. Transretinal feeder vessel ligation in conjunction with intraocular thermal laser has also been reported in 1 case of retinal hemangioblastoma.24 With the improvement in separate illumination sources for vitreoretinal surgery, procedures requiring bimanual manipulation are possible and were used in this series. We chose to ligate the large feeder vessels because of the risk of intraocular or postoperative bleeding using diathermy alone. It is possible that this step is not necessary. As this is a small uncontrolled series, it is impossible to compare surgical resection with other potential treatments in this group of patients, but the favorable outcomes suggest that surgical resection should be considered for patients with large retinal hemangioblastomas. The complications encountered (cataract, epiretinal membrane, and intraoperative retinal breaks) are not unsubstantial but are acceptable given the expected natural history.25
von Hippel-Lindau disease is a dominantly inherited disease in which loss of a tumor suppressor leads to a high rate of formation of several tumors, the most common of which is retinal hemangioblastoma.25 However, the prevalence of truly sporadic retinal hemangioblastoma is unclear. As VHL disease is known to be the result of a germline defect in a tumor suppressor gene on chromosome 3p25-26, it is appealing to suggest that the sporadic cases of retinal hemangioblastoma, like sporadic retinoblastoma, conform to the Knudsen hypothesis. In this study, an objective was to investigate whether VHL gene defects were responsible for the sporadic retinal hemangioblastomas excised from the eyes of our patients.
We tested this by 3 methods. In method 1, we isolated DNA and sequenced the VHL exons from segments of excised whole tumor. We were unable to detect a mutation from any of the 3 exons from patient 1 or from patient 2. These samples were not purified for stromal cells (the cells reported to contain the VHL mutation)21; therefore, because of cell type heterogeneity the DNA from normal cells in the tissue analyzed may have obscured the presence of a mutation. To bypass this possibility, foamy stromal cells, the cells implicated as causative for the tumors and shown to harbor VHL gene mutations, were microdissected in method 2 for DNA sequencing from paraffin-embedded tumor tissue. Meaningful results could not be obtained. DNA isolated from fixed tissue is known to be susceptible to degradation, and depending on fixation and staining procedures, DNA can be fragmented to less than 200 base pairs, which may affect purification and subsequent analysis. Therefore, method 3 of investigation was undertaken, namely, analysis for LOH in the microdissected cells. This standard method of analysis showed that stromal cells from patient 1 did not have LOH, while the equivalent sample from patient 2 did have LOH (Figure 3).
These results are intriguing and are worthy of further discussion, considering that LOH has been found in all tumors analyzed in retinal hemangioblastomas from VHL disease.26 However, analyses of sporadic hemangioblastomas from the cerebellum have not always shown abnormalities of the VHL gene.26-28 Similar to these cerebellar hemangioblastoma cases, patient 1 did not have a detectable mutation in the exons of the VHL gene from a fresh tissue sample and did not have LOH in a microdissected sample. Therefore, in a tumor that was clinically and pathologically identical to that caused by VHL mutations, our analysis was unable to define a deleterious change within exonic sequences. This tumor may still be a result of malfunction of the VHL gene in the case of a disruptive mutation in an intron or in the promoter region, as these regions were not analyzed. Theoretically, LOH testing may also misinform because it relies on detection of the deletion of a satellite marker proximal to the VHL gene and on the assumption that absence of this marker indicates a microdeletion that includes the VHL gene.
Patient 2 had no detectable mutation in the VHL gene from a sample of whole tumor but had positive LOH from the isolated tumor stromal cells. These results may indicate a masking of the mutation in stromal cells by the normal cells in the sample that then was detected as an LOH in the purified homogeneous cell sample. This still does not explain why we did not detect a mutation with in the other VHL allele. The possibility also exists that a mutation is present in an intervening sequence, which causes some abnormality with processing of the precursor mRNA into the mature mRNA, thereby perturbing protein production. This would not have been detected in our sequencing analysis, as only the exons were sequenced. More than 500 different VHL mutations have been reported, including familial cases, sporadic cases, and cell lines, and all these mutations are contained with in the protein coding portion of the mature mRNA (VHL universal mutation database; http://www.vhl.org/research/beroud.htm). Although it is possible that mutations outside of the mature mRNA may disrupt the function of VHL protein, it is also plausible that an alternative protein or a VHL-independent pathway may be involved in the formation of retinal hemangioblastomas.
In conclusion, we demonstrated successful internal en bloc surgical resection of large retinal hemangioblastomas with acceptable intraoperative and postoperative morbidity. This technique has the potential for maintaining excellent visual acuity in this group of patients. It also permits removal of the tumor in a fashion that allows comprehensive study, including molecular genetics and molecular pathology. To our knowledge, this is the first finding of a retinal hemangioblastoma unassociated with a VHL gene mutation, indicating that a novel mechanism may be involved in the evolution of this particular retinal hemangioblastoma.
Acknowledgments
Funding/Support: This study was supported in part by an unrestricted grant to Casey Eye Institute from Research to Prevent Blindness and the Clayton Foundation for Research. Addition support was received from the Clayton Foundation for Research (Dr Stout) and from the Heed Foundation and the Ronald G. Michels Foundation (Dr Schlesinger).
Footnotes
Financial Disclosure: None reported.
Additional Information: The eFigure is available at http://www.archophthalmol.com.
REFERENCES
- 1.Singh A, Shields JA, Shields CL. Solitary retinal capillary hemangioma: hereditary (von Hippel-Lindau disease) or non-hereditary? Arch Ophthalmol. 2001;119(2):232–234. [a published correction appears in Arch Ophthalmol. 2001;119(8):1226] [PubMed] [Google Scholar]
- 2.Latif F, Tory K, Gnarra J, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260(5112):1317–1320. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- 3.Stratmann R, Krieg M, Haas R, Plate KH. Putative control of angiogenesis in hemangioblastomas by the von Hippel-Lindau tumor suppressor gene. J Neuropathol Exp Neurol. 1997;56(11):1242–1252. doi: 10.1097/00005072-199711000-00009. [DOI] [PubMed] [Google Scholar]
- 4.Iliopoulos O, Levy AP, Jiang C, Kaelin WG, Goldberg MA. Negative regulation of hypoxia-inducible genes by the von Hippel-Lindau protein. Proc Natl Acad Sci U S A. 1996;93(20):10595–10599. doi: 10.1073/pnas.93.20.10595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Min JH, Yang H, Ivan M, Gertler F, Kaelin WG, Pavletich NP. Structure of an HIF-1α-pVHL complex: hydroxyproline recognition in signaling [published online ahead of print May 9, 2002] Science. 2002;296(5574):1886–1889. doi: 10.1126/science.1073440. [DOI] [PubMed] [Google Scholar]
- 6.Hon WC, Wilson MI, Harlos K, et al. Structural basis for the recognition of hydroxyproline in HIF-1α by pVHL [published online ahead of print June 5, 2002] Nature. 2002;417(6892):975–978. doi: 10.1038/nature00767. [DOI] [PubMed] [Google Scholar]
- 7.Annesley WJ, Jr, Leonard BC, Shields JA, Tasman WS. Fifteen year review of treated cases of retinal angiomatosis. Trans Sect Ophthalmol Am Acad Ophthalmol Otolaryngol. 1977;83(3 pt 1):OP446–OP453. [PubMed] [Google Scholar]
- 8.Lane CM, Turner G, Gregor DJ, Bird AC. Laser treatment of retinal angiomatosis. Eye. 1989;3(pt 1):33–38. doi: 10.1038/eye.1989.5. [DOI] [PubMed] [Google Scholar]
- 9.Welch RB. von Hippel-Lindau disease: the recognition and treatment of early angiomatosis retinae and the use of cryosurgery as an adjunct to therapy. Trans Am Ophthalmol Soc. 1970;68:367–424. [PMC free article] [PubMed] [Google Scholar]
- 10.Kreusel KM, Bornfeld N, Lommatzsch A, et al. Ruthenium-106 brachytherapy for peripheral retinal capillary hemangioma. Ophthalmology. 1998;105(8):1386–1392. doi: 10.1016/S0161-6420(98)98017-4. [DOI] [PubMed] [Google Scholar]
- 11.Raja D, Benz MS, Murray TG, Escalona-Benz EM, Markoe A. Salvage external beam radiotherapy of retinal capillary hemangiomas secondary to von Hippel-Lindau disease: visual and anatomic outcomes. Ophthalmology. 2004;111(1):150–153. doi: 10.1016/j.ophtha.2003.04.003. [DOI] [PubMed] [Google Scholar]
- 12.Mochizuki Y, Noda Y, Enaida H, et al. Retinal capillary hemangioma managed by transpupillary thermotherapy. Retina. 2004;24(6):981–984. doi: 10.1097/00006982-200412000-00028. [DOI] [PubMed] [Google Scholar]
- 13.Bakri SJ, Sears JE, Singh AD. Transient closure of a retinal capillary hemangioma with verteporfin photodynamic therapy. Retina. 2005;25(8):1103–1104. doi: 10.1097/00006982-200512000-00024. [DOI] [PubMed] [Google Scholar]
- 14.Szabó A, Gehl Z, Seres A. Photodynamic (verteporfin) therapy for retinal capillary haemangioma, with monitoring of feeder and draining blood vessel diameters. Acta Ophthalmol Scand. 2005;83(4):512–513. doi: 10.1111/j.1600-0420.2005.00476.x. [DOI] [PubMed] [Google Scholar]
- 15.Aaberg TM, Jr, Aaberg TM, Sr, Martin DF, Gilman JP, Myles R. Three cases of large retinal capillary hemangiomas treated with verteporfin and photodynamic therapy. Arch Ophthalmol. 2005;123(3):328–332. doi: 10.1001/archopht.123.3.328. [DOI] [PubMed] [Google Scholar]
- 16.Girmens JF, Erginay A, Massin P, Scigalla P, Gaudric A, Richard S. Treatment of von Hippel-Lindau retinal hemangioblastoma by the vascular endothelial growth factor receptor inhibitor SU5416 is more effective for associated macular edema than for hemangioblastomas. Am J Ophthalmol. 2003;136(1):194–196. doi: 10.1016/s0002-9394(03)00101-6. [DOI] [PubMed] [Google Scholar]
- 17.Aiello LP, George DJ, Cahill MT, et al. Rapid and durable recovery of visual function in a patient with von Hippel-Lindau syndrome after systemic therapy with vascular endothelial growth factor receptor inhibitor SU5416. Ophthalmology. 2002;109(9):1745–1751. doi: 10.1016/s0161-6420(02)01159-4. [DOI] [PubMed] [Google Scholar]
- 18.Maher ER, Yates JR, Harries R, et al. Clinical features and natural history of von Hippel-Lindau disease. Q J Med. 1990;77(283):1151–1163. doi: 10.1093/qjmed/77.2.1151. [DOI] [PubMed] [Google Scholar]
- 19.Vortmeyer AO, Gnarra JR, Emmert-Buck MR, et al. von Hippel-Lindau gene deletion detected in the stromal cell component of a cerebellar hemangioblastoma associated with von Hippel-Lindau disease. Hum Pathol. 1997;28(5):540–543. doi: 10.1016/s0046-8177(97)90075-7. [DOI] [PubMed] [Google Scholar]
- 20.Zhuang Z, Bertheau P, Emmert-Buck MR, et al. A microdissection technique for archival DNA analysis of specific cell populations in lesions < 1 mm in size. Am J Pathol. 1995;146(3):620–625. [PMC free article] [PubMed] [Google Scholar]
- 21.Chan CC, Vortmeyer AO, Chew EY, et al. VHL gene deletion and enhanced VEGF gene expression detected in the stromal cells of retinal angioma. Arch Ophthalmol. 1999;117(5):625–630. doi: 10.1001/archopht.117.5.625. [DOI] [PubMed] [Google Scholar]
- 22.Chan CC, Lee YS, Zhuang Z, Hackett J, Chew EY. von Hippel-Lindau gene deletion and expression of hypoxia-inducible factor and ubiquitin in optic nerve hemangioma. Trans Am Ophthalmol Soc. 2004;102:75–81. [PMC free article] [PubMed] [Google Scholar]
- 23.Peyman GA, Rednam KR, Mottow-Lippa L, Flood T. Treatment of large von Hippel tumors by eye wall resection. Ophthalmology. 1983;90(7):840–847. doi: 10.1016/s0161-6420(83)34481-x. [DOI] [PubMed] [Google Scholar]
- 24.Farah ME, Uno F, Hofling-Lima AL, et al. Transretinal feeder vessel ligature in von Hippel-Lindau disease. Eur J Ophthalmol. 2001;11(4):386–388. doi: 10.1177/112067210101100414. [DOI] [PubMed] [Google Scholar]
- 25.Fisher PG, Tontiplaphol A, Pearlman EM, et al. Childhood cerebellar hemangioblastoma does not predict germline or somatic mutations in the von Hippel-Lindau tumor suppressor gene. Ann Neurol. 2002;51(2):257–260. doi: 10.1002/ana.10107. [DOI] [PubMed] [Google Scholar]
- 26.Lee JY, Dong SM, Park WS, et al. Loss of heterozygosity and somatic mutations of the VHL tumor suppressor gene in sporadic cerebellar hemangioblastomas. Cancer Res. 1998;58(3):504–508. [PubMed] [Google Scholar]
- 27.Oberstrass J, Reifenberger G, Reifenberger J, Wechsler W, Collins VP. Mutation of the von Hippel-Lindau tumour suppressor gene in capillary hemangioblastomas of the central nervous system. J Pathol. 1996;179(2):151–156. doi: 10.1002/(sici)1096-9896(199606)179:2<151::aid-path556>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 28.Kanno H, Kondo K, Ito S, Yamamoto I. Somatic mutations of the von Hippel-Lindau tumor suppressor gene in sporadic central nervous system hemangioblastomas. Cancer Res. 1994;54(18):4845–4847. [PubMed] [Google Scholar]