

Abstract
Metabolomics is the systematic identification and quantitation of all metabolites in a given organism or biological sample. The enhanced resolution provided by nuclear magnetic resonance (NMR) spectroscopy and mass spectrometry (MS), along with powerful chemometric software, allows the simultaneous determination and comparison of thousands of chemical entities, which has lead to an expansion of small molecule biochemistry studies in bacteria, plants, and mammals. Continued development of these analytical platforms will accelerate the widespread use of metabolomics and allow further integration of small molecules into systems biology. Here, recent studies using metabolomics in xenobiotic metabolism and genetically modified mice are highlighted.
Before the advent of molecular medicine, unraveling the mechanisms of disease was essentially a biochemical endeavor. The pioneering contributions of Sir Archibald Garrod in defining the chemical nature of alkaptonuria in 1902 and Ivar Asbjørn Følling in uncovering the underlying biochemical basis of inherited impaired cognitive function and development in 1934 relied solely upon the systematic study of urine constituents and of metabolism, using methods that would be viewed as alien today. Nevertheless, these studies highlighted important concepts about chemical individuality and led to simple diagnostic tests, dietary interventions, and treatments for these diseases. In this current age of molecular medicine, it is now within the reach of most laboratories to investigate the structure and functional expression of genomes on a global scale. However, the study of proteins at a global level, proteomics, has not yet achieved such widespread use. In contrast, metabolomics is emerging as a field with tremendous promise in extending “omics” from the gene to the small molecule.
Metabolomics, the global metabolic profiling of cells, tissues, or organisms in relation to genetic variation or external stimuli, is now sufficiently mature to join the ranks of the other omics to close the systems biology loop. Small molecule biochemistry, the bedrock of medical research for over a century, is now reemerging from the shadows where it has been eclipsed by molecular genetics, reverse genetics, genomics, proteomics, and other contemporary fields that bring with them technologies of immense power and insight. Via the rapid development of metabolomics technology platforms and by incorporating metabolomics into research efforts, it is now possible to answer key questions that could not be fully addressed by the other omics alone. From bacteria to humans, examples of this principle are accruing at a rapid pace that has been made possible by remarkable recent developments in analytical chemistry, such as high-field nuclear magnetic resonance (NMR) and mass spectrometry (MS) platforms for small molecule separation, detection, and characterization, together with the availability of relatively user-friendly multivariate data analysis software packages that are able to deconvolute the huge data matrices generated in a metabolomic experiment. Such analysis of the metabolome is being applied to an ever-increasing number of biological and medical research topics, from bacteria to human.
One important area in which metabolomics is making a quintessential contribution is prokaryotic genome annotation. In the postgenomic age, it has become apparent that at least 30%–50% of genes in a newly sequenced bacterial genome were incorrectly annotated or are of unknown function (Siew et al., 2004). Indeed, the function of one in five genes in the best understood bacterial genome, E. coli, is unknown (Liang et al., 2002). Small molecule signatures could allow the function of these genes to be determined.
It has also recently become apparent that the microbiota of the human distal gut may play a key role in health and disease. The collective genome of these microorganisms contains at least 100 times as many genes as our own genome (Gill et al., 2006), and data are beginning to emerge regarding their role in human health (Ley et al., 2006). A recent metabolomics study harnessed both NMR and MS technologies to compare normal gut flora colonization in conventional mice to human infant gut flora colonization in germ-free mice (Martin et al., 2007). These authors suggested a role of the microbiome in the absorption and storage of lipids, as well as in the extraction of energy from the diet. They also proposed that manipulation of the microbiome to produce “a balanced and well-adapted microflora” may prevent certain human gastrointestinal and hepatic diseases (Martin et al., 2007). In the future, if we are to understand more fully the relationship between dietary nutrients, dietary xenobiotics, gut flora, and human health and disease, it will be a priority to have correct genomic annotations of the main species of bacteria and archaea that inhabit the human gut.
The biochemistry of plants is of particular relevance to the health and well-being of humans. The metabolomes of plants are unexpectedly complex, and it has been estimated that some 200,000 molecules will eventually be detected (Fiehn, 2002). Although the complete genome of the well-studied flowering plant Arabidopsis thaliana has been sequenced, over 30% of its genes are of unknown function, and only 9% have been studied experimentally (Seki et al., 2002), which calls for a metabolomic approach for gene annotation. Understanding metabolic networks in plants has already delivered many important outcomes, such as the detection of unintended effects of genetically modified (GM) food crops using an unbiased global screening approach instead of traditional targeted metabolite detection, including exposure to known toxins (Dixon et al., 2006).
In mammals, the potential of metabolomics is enormous, particularly with respect to studying the interface between our chemical universe and human biology. Humans are probably exposed to some 1–3 million discrete chemicals in our lifetimes, and most of these xenobiotics are metabolized in the body to render them less toxic. However, in a significant number of cases, the detoxication apparatus renders a xenobiotic more chemically reactive, potentially more toxic, and, in some cases, carcinogenic. Understanding the role of human genetic differences in xenobiotic metabolism and its relationship to interindividual susceptibility to chemical toxicities and carcinogenesis is an area of intense interest (Wogan et al., 2004). A convenient, cost-effective means to determine metabolic potential would be of great value in understanding the molecular epidemiology of cancer and is an area in which metabolomics can be brought to bear. In addition, drug-induced toxicities have a major impact on the clinical use of drugs, and the pharmaceutical industry has sought to develop methods to predict the potential adverse effects of compounds early in drug development. For this reason, metabolomics is being developed as a platform to study and predict drug toxicities during preclinical drug development. After administration of a drug, urinary metabolites can serve as a fingerprint for organ-specific toxicities, thus potentially eliminating the need for laborious pathological analysis of tissues and enhancing throughput (Lindon et al., 2007).
Another area of drug development in which metabolomics can have a major role is pharmacogenetics, the study of person-to-person variability in drug metabolism and toxicity. Genetic differences in drug metabolism have historically been studied in clinical pharmacology by direct genotyping for known variant alleles of genes that encode drug-metabolizing enzymes and transporters. For a more global analysis of interindividual variation in metabolism and susceptibility to toxicity, metabolomics can be employed. A proof-of-principle study in an animal model revealed the potential for metabolomics profiling in predicting responses to drugs (Clayton et al., 2006). However, it remains to be determined whether this approach can be translated to humans due to large interindividual variations in the urinary metabolome, resulting from genetic and dietary diversity.
Recently, the use of transgenic mouse lines has yielded important results to better understand pathways that lead to drug and chemical toxicity, drug-drug interactions, mechanisms of carcinogenesis, and the role of metabolism in cancer chemotherapy as well as to develop precise biomarkers that can be translated to humans. In all of these areas, the unique combination of transgenic mouse generation and metabolomic analysis is furnishing answers to important questions relevant to human health and disease. For example, metabolomic technology, in particular the power of multivariate analysis of liquid chromatography-mass spectrometry (LC-MS) data, has been applied in combination with cytochrome P450 (a gene important in xenobiotic detoxication, among other functions) knockout mice to reveal a fuller panoply of the metabolites of the ubiquitous dietary carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP), revealing 17 urinary metabolites, 8 of which were novel (Chen et al., 2007). PhIP, a suspected human dietary carcinogen, is of potential importance as a food mutagen found in overcooked and pan-fried meats. Use of CYP1A2-humanized mice has demonstrated that the human CYP1A2 is far more active than the corresponding mouse enzyme in producing a metabolite that ultimately leads to the DNA-binding derivative of the carcinogen. Similarly, by carrying out a metabolomic study on mice given the experimental anticancer drug aminoflavone, 13 mouse urinary metabolites were observed, 12 of which were novel (Chen et al., 2006). The use of CYP1A2-humanized mice revealed an important species difference in the metabolism of aminoflavone (Chen et al., 2006). It is conceivable that similar studies, aimed at assessing human-specific genetic differences in metabolism, could be performed in drug-treated humans.
Metabolomics can be used to find molecular fingerprints for gene deficiency and activation of nuclear receptors. For example, molecular finger-prints have been uncovered in urine for both the presence of an active nuclear receptor, peroxisome proliferator-activated receptor α (PPARα), and the activation of PPARα by its ligands (Zhen et al., 2007) (Figure 1A). While metabolic alterations would be expected upon ligand activation, quite surprisingly, a number of changes in levels of metabolites were noted in the absence of PPARα. Using these data, metabolites can be found that distinguish between groups, and these metabolites can then serve as bio-markers for PPARα expression and activation. The differences between genotypes can also be visualized by novel software that displays the global differences in the urinary metabolome between Pparα+/+ and Pparα−/− mice (Figure 1B). One of the principal findings of the PPARα metabolomics study was that novel steroid metabolites of unknown function were highly elevated upon ligand activation of PPARα (Zhen et al., 2007).
Figure 1. PPARα Metabolomics.
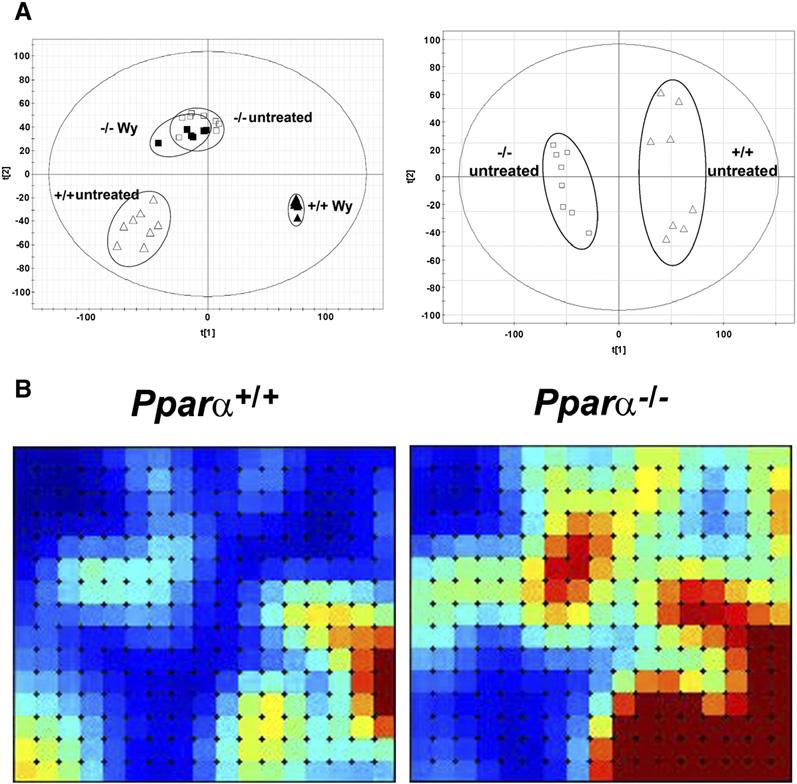
(A) Metabolomics analysis of ligand-treated and untreated wild-type (+/+) and Pparα null (−/−) mice by ultra-performance liquid chromatography/time-of-flight mass spectrometry. Left panel: principal components analysis (PCA) scores plot of principle component 1 versus principle component 2 for four groups of mice, wild-type and Pparα null mice both untreated and treated with Wy-14,643, a PPARα agonist ligand. Right panel: PCA scores plot of component 1 versus component 2 for two groups of mice, untreated wild-type and Pparα null mice. Note that the treated and untreated null mice cluster together.
(B) Analyses and visualization of patterns in MS data matrices. Self-organizing maps showing a holistic view of the mouse urinary metabolome for groups of male C57BL/6 mice with Pparα+/+ and Pparα−/− genotypes. Maps were constructed using Gene Expression Dynamics Inspector (GEDI) software (Guo et al., 2006) and published data (Zhen et al., 2007). Each tile comprises a group of metabolites that covary across the whole data set, on average 12. Dark blue is the lowest intensity of metabolites; dark red is the highest intensity. The two urinary biomarkers for the Pparα+/+ genotype that we have reported (Zhen et al., 2007), 2,8-dihydroxyquinoline and xanthurenic acid, are both located in the bottom right tiles and are thus in the region that is considerably elevated by the presence of the Pparα gene. Figure 1B kindly provided by A. Patterson (Laboratory of Metabolism, Center for Cancer Research, NCI, NIH).
Metabolomics also holds great promise for the discovery of chemical biomarkers for diseases that are amenable to measurement by urine analysis. Indeed, 1H-NMR analysis of human serum has been used to search for chemical signatures of coronary artery disease (Brindle et al., 2002). This task is quite daunting due to the genetic differences, dietary diversity, and other problems inherent in the human population that lead to marked interindividual differences in the human serum metabolome. While these difficulties can be overcome by studying large populations of cases and controls with powerful statistical multivariate data analysis software that is able to mine large data sets, a better delineated approach may be the use of animal disease models to identify candidate biomarkers that can then be validated in humans. Biomarkers that can be used to diagnose prediabetes, cancer, and coronary artery disease would be of great value in clinical medicine.
As has been discussed, metabolomics is playing a unique role in the systems biological study of health, disease, the environment, and agriculture. Small molecule biochemistry, relegated to a lesser role over the past two decades by genomics, transcriptomics, and proteomics, is now undergoing a renaissance of sorts. With the advent of high-end chemical analytical instrumentation, combined with chemometric analysis of the data-rich outputs, small molecule biochemistry is now fully integrated with the other omics to more fully complete systems biology. While we fully expect that metabolomics will find a place in all aspects of biological and medical research, its contributions could be accelerated by the development of quantum leap technologies. At present, due to their diverse physical properties, the global profiling of small molecules requires multiple analytical platforms, including NMR and a range of MS technologies, in order to gain maximum coverage of the metabolome. This situation, requiring many instruments, is cumbersome. We look forward to a day when metabolomic analysis that can capture the whole metabolome is truly accessible and widely available in the way that PCR, SNP mapping, and microarray analysis are now. This will require a commitment of the best minds in academic research, as well as a willingness of industrial manufacturers to fulfill the collective vision of a world with simple, cheap, and reproducible metabolomic profiling, a true electronic dog's nose.
REFERENCES
- Brindle JT, Antti H, Holmes E, Tranter G, Nicholson JK, Bethell HW, Clarke S, Schofield PM, McKilligin E, Mosedale DE, Grainger DJ. Nat. Med. 2002;8:1439–1444. doi: 10.1038/nm1202-802. [DOI] [PubMed] [Google Scholar]
- Chen C, Meng L, Ma X, Krausz KW, Pommier Y, Idle JR, Gonzalez FJ. J. Pharmacol. Exp. Ther. 2006;318:1330–1342. doi: 10.1124/jpet.106.105213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Ma X, Malfatti MA, Krausz KW, Kimura S, Felton JS, Idle JR, Gonzalez FJ. Chem. Res. Toxicol. 2007;20:531–542. doi: 10.1021/tx600320w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton TA, Lindon JC, Cloarec O, Antti H, Charuel C, Hanton G, Provost JP, Le Net JL, Baker D, Walley RJ, et al. Nature. 2006;440:1073–1077. doi: 10.1038/nature04648. [DOI] [PubMed] [Google Scholar]
- Dixon RA, Gang DR, Charlton AJ, Fiehn O, Kuiper HA, Reynolds TL, Tjeerdema RS, Jeffery EH, German JB, Ridley WP, Seiber JN. J. Agric. Food Chem. 2006;54:8984–8994. doi: 10.1021/jf061218t. [DOI] [PubMed] [Google Scholar]
- Fiehn O. Plant Mol. Biol. 2002;48:155–171. [PubMed] [Google Scholar]
- Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, Gordon JI, Relman DA, Fraser-Liggett CM, Nelson KE. Science. 2006;312:1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Eichler GS, Feng Y, Ingber DE, Huang S. J. Biomed. Biotechnol. 2006;2006:69141. doi: 10.1155/JBB/2006/69141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- Liang P, Labedan B, Riley M. Physiol. Genomics. 2002;9:15–26. doi: 10.1152/physiolgenomics.00086.2001. [DOI] [PubMed] [Google Scholar]
- Lindon JC, Holmes E, Nicholson JK. FEBS J. 2007;274:1140–1151. doi: 10.1111/j.1742-4658.2007.05673.x. [DOI] [PubMed] [Google Scholar]
- Martin FP, Dumas ME, Wang Y, LegidoQuigley C, Yap IK, Tang H, Zirah S, Murphy GM, Cloarec O, Lindon JC, et al. Mol. Syst. Biol. 2007;3:112. doi: 10.1038/msb4100153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki M, Narusaka M, Kamiya A, Ishida J, Satou M, Sakurai T, Nakajima M, Enju A, Akiyama K, Oono Y, et al. Science. 2002;296:141–145. doi: 10.1126/science.1071006. [DOI] [PubMed] [Google Scholar]
- Siew N, Azaria Y, Fischer D. Nucleic Acids Res. 2004;32:D281–D283. doi: 10.1093/nar/gkh116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wogan GN, Hecht SS, Felton JS, Conney AH, Loeb LA. Semin. Cancer Biol. 2004;14:473–486. doi: 10.1016/j.semcancer.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Zhen Y, Krausz KW, Chen C, Idle JR, Gonzalez FJ. Mol. Endocrinol. 2007;21:2136–2151. doi: 10.1210/me.2007-0150. [DOI] [PMC free article] [PubMed] [Google Scholar]