

Abstract
Xenobiotic metabolism, a ubiquitous natural response to foreign compounds, elicits initiating signals for many pathophysiological events. Currently, most widely used techniques for identifying xenobiotic metabolites and metabolic pathways are empirical and largely based on in vitro incubation assays and in vivo radiotracing experiments. Recent work in our lab has shown that LC-MS-based metabolomic techniques are useful tools for xenobiotic metabolism research since multivariate data analysis in metabolomics can significantly rationalize the processes of xenobiotic metabolite identification and metabolic pathway analysis. In this review, the technological elements of LC-MS-based metabolomics for constructing high-quality datasets and conducting comprehensive data analysis are examined. Four novel approaches of using LC-MS-based metabolomic techniques in xenobiotic metabolism research are proposed and illustrated by case studies and proof-of-concept experiments, and the perspective on their application is further discussed.
Keywords: Metabolomics, Xenobiotic metabolism, Drug metabolism, LC-MS, Multivariate data analysis
INTRODUCTION
In response to xenobiotic exposure, all living creatures, from bacteria to humans, perform a certain level of metabolism to facilitate the elimination and/or utilization of exogenous chemicals. Observations of positive and negative impacts of xenobiotic metabolism have led to the numerous discoveries in contemporary biomedical sciences. In fact, identification of xenobiotic metabolites and metabolic pathways has become an indispensable part of drug metabolism and chemical toxicology research since the results from these studies establish the scientific basis for understanding molecular mechanisms of chemical-induced beneficial or toxicological effects. In modern drug discovery and development, the role of drug metabolism is not limited to satisfying the regulatory requirement of identifying the major metabolites of new chemical entity, ascertaining the major enzymes responsible for the biotransformation, and predicting potential drug-drug interactions and pharmacogenetic idiosyncrasies. In fact, identification of biologically active metabolites led to the development of safer and more potent drugs, such as acetaminophen from acetanilide (Brodie and Axelrod, 1948) and fexofenadine from terfenadine (Yun et al., 1993). As for toxicological science, studying the biotransformation of xenobiotics has significantly enhanced our knowledge on chemical-induced toxicity and carcinogenesis.
Currently, identification of metabolites and metabolic pathways in xenobiotic metabolism research is largely empirical and based on the data obtained from in vitro incubation systems and in vivo animal and human trials, even though some in silico methodologies have been developed in the recent years (Ekins et al., 2005). In vitro assays, including hepatocyte, microsome and recombinant human P450s incubations, as well as organ (mainly liver) perfusion, provide efficient and convenient venues to investigate xenobiotic metabolism, since the dominant concentrations of xenobiotic and its metabolites in an in vitro system makes the identification of new metabolites much easier than with in vivo systems (Venkatakrishnan et al., 2003). Furthermore, screening assays using highly purified xenobiotic metabolizing enzymes (XME) and their corresponding inhibitors (chemical inhibitors or antibodies) can generate information regarding potential metabolic routes and the contributing XMEs in vivo. However, compared to an in vivo system, the limitations of an in vitro approach are also apparent, due to the inability to conduct comprehensive phase I and II investigations, the artificially high xenobiotic concentration, the lack of gene and protein regulation (such as hormonal regulation) as well as the absence of sufficient adsorption, distribution, and elimination mechanisms. Therefore, to validate the conclusion from in vitro studies and to delineate the complete ADME processes, in vivo investigation of xenobiotic metabolism is inevitable for both drug metabolism and chemical toxicology research. Because bioactive metabolites or the metabolites formed by reactive intermediates are frequently not the most abundant metabolite species, one major challenge in an in vivo study is how to identify xenobiotic metabolites unequivocally and thoroughly by the analysis of urine and serum, which contain thousands of chemical species. A common solution is to use a radiolabeled xenobiotic to trace and to identify xenobiotic metabolites in complex biological matrices. In general, radiotracing is a very efficient method for conducting in vivo metabolism because of its sensitivity and ready quantitation. However, the time and cost for synthesizing appropriate quantity and purity of radiolabeled compound, as well as the concerns for environmental contamination and human or animal health, hamper its wide application.
Recently, accompanying the enhanced resolution of mass spectrometry (MS), several algorithm-based methods, such as mass defect filtering techniques, have been developed in the drug metabolism field (Mortishire-Smith et al., 2005; Zhu et al., 2006). In essence, these metabolite searching methods use the numerical values of mass increase or decrease caused by known metabolic reactions or an artificial mass defect window as the screening filter to identify the metabolites formed by single or even multiple reactions. The major issue with this approach is that the filter cannot cover all biotransformation reactions in vivo, especially many uncommon reactions that may cause dramatic mass changes, such as hydrolysis, dealkylation, peroxidation as well as structural rearrangement (Guengerich, 2001). In addition, the results obtained from mass filtering are frequently plagued with false-positive entries. Therefore, a methodology that can circumvent the drawbacks and limitations of current in vivo metabolite identification methods will definitely improve the data quality and enhance the efficiency of analytical processes in xeno-biotic metabolism research.
TECHNICAL PLATFORMS OF METABOLOMICS
Metabolomics, a methodology for measuring small-molecule metabolite profiles and fluxes in biological matrices, following genetic modification or exogenous challenges, has become an important component of systems biology, complementing genomics, transcriptomics and proteomics (Nicholson and Wilson, 2003; Fernie et al., 2004; Griffin, 2006). Because of the comprehensive nature of metabolite measurement and the capacity to detect subtle changes in a large dataset, metabolomics has found broad application. Examples include the identification of biomarkers, and the unravelling of pathophysiological mechanisms in many scientific fields, such as plant biology (Schauer and Fernie, 2006), microbiology (van der Werf et al., 2005), toxicology (Nicholson et al., 2002) and disease diagnosis (Brindle et al., 2002). While metabolomics is often used to refer to studies of the endogenous metabolites, in our view, this term should also be extended to the environmental chemicals and drugs to which we are exposed. The human metabolome, for example, is many times bigger than the 2,180 or so calculated endogenous chemicals that are purely the products of the genome, transcriptome and proteome in isolation (Wishart et al., 2007) since food, medicines, bacterial metabolites (gut flora), and environmental chemicals (diet, air, pollution) furnish a far greater proportion of this metabolite pool. For example, the food mutagen PhIP and its metabolites (see below), which are not purely of endogenous origin, are surely present in the tissues of any person who consumes cooked, as opposed to boiled, food. Therefore, it will never be possible in the immediate future to distinguish pure endobiotics from pure xenobiotics and xenobiotics that have been modified by the proteome. In order not to involve ourselves in futile labeling of constituents of the metabolome, we take the word “metabolomics” to cover all small molecule constituents of biological fluids/tissues that can be identified and quantitated, irrespective of their origin. This approach of broadening the coverage of metabolomics has a clear advantage than endogenous compound-only approach for solving some dilemma of defining whether a chemical is a component of the metabolome or not, such as acetaminophen glucuronide, which has approximately half of the mass from exogenous parent drug and the other half (glucuronic acid moiety) from endogenous glucose metabolism, and life-essential sodium ions present in body fluids, which is purely a xenobiotic.
Most widely-used analytical instruments in metabolomic research are nuclear magnetic resonance (NMR) spectrometers and MS. Other techniques, such as electrochemistry or infrared spectroscopy, have also been adopted, but their application is limited by the lack of detailed structural information that they provide (Kristal et al., 2007). The pros and cons of using NMR or MS in metabolomic research have been discussed extensively in several recent reviews (Schlotterbeck et al., 2006; Pan and Raftery, 2007). Overall, NMR has advantages such as the non-destructive nature of sample preparation and the comprehensive coverage of chemical species, while MS possesses much better sensitivity and resolution as well as high-throughput capacity.
Attempts to improve the limiting sensitivity of 1H NMR for biological samples into the nanomolar range have included the application of extremely high static magnetic field strengths, up to 21.2 Tesla (920 MHz 1H spectrometer; Hashi et al., 2002) and the usage of a probe that is cryogenically cooled to 4.5 K in order to increase signal to noise ratios (Kikuchi et al., 2004). However, the metabolomic implementation of such cutting-edge instrumentation is still awaited. Another and exciting application of NMR in metabolomic research has been the use of so-called hetero-nuclear NMR methodologies, which are particularly pertinent to plant metabolomics. Here, plants grown exclusively on 13C- and 15N-labeled nutrient sources have been investigated using two-dimensional heteronuclear single quantum correlation spectroscopy (2D-HSQC NMR). 1H-13C and 1H-15N 2D-HSQC have furnished valuable insights into the effects of xenobiotic stresses on the systems biology of both wild-type and mutant Arabidopsis thaliana plants (Kikuchi et al., 2004). Such technologies, however, have so far found limited application in mammalian drug metabolism studies.
Although many seminal metabolomic studies were conducted using NMR, an increasing number of studies based on MS technology have been published recently, dependent upon the abovementioned advantages and the wide availability of MS instruments (Want et al., 2007). Introduction of prepared biological samples into a mass spectrometer can be through direct injection, gas chromatography (GC), liquid chromatography (LC) as well as capillary electrophoresis (Dettmer et al., 2007). Among them, LC-MS is the most widely used instrument since LC-based sample introduction results in lesser ion suppression and higher resolution than direct infusion, and it generally avoids the chemical derivatization that is generally required for GC-MS. Therefore, this review will focus on the LC-MS-based metabolomic techniques and their application to xenobiotic metabolism.
Traditionally, HPLC has been main staple of LC instruments. Recent development on ultra-performance liquid chromatography (UPLC) using 1–2 micron-size particles brings a much higher resolution for analyte separation and a lower limit of detection for ions (Wilson et al., 2005). MS instruments with high-resolution mass measurement, such as time-of-flight (TOF) or Fourier transform (FT) mass spectrometers are preferred, since accurate masses of xenobiotic metabolites are not only the prerequisite for peak identification and collection across samples, but also readily furnish empirical formulae, and thus candidate chemical structures. Furthermore, the sensitivity of metabolite detection can be improved by optimizing the gradient or composition of mobile phase to improve ion spray and ionization. It should also be noted that LC-MS measurement in the absence of authentic standards is non-quantitative for the measurement of either major or minor metabolites. However, it can provide an estimation of relative abundances of identified metabolites if metabolites are ionized with comparable efficiency and the compounds elute at positions in the chromatogram that are subject to similar ion suppression effects. Overall, high-resolution and reproducible LC-MS measurement sets up the basis for subsequent data processing and multivariate data analysis (MDA).
Appropriate data processing is required to prepare chromatographic and spectral data for MDA (Katajamaa and Oresic, 2007). General procedures include data condensation and reduction by centroiding and deisotoping mass spectra; chromatographic alignment to reduce the variation in retention time; filtering to remove noise or background signals; and peak recognition and collection by setting threshold windows for mass (m/z) and retention time (RT). To decrease the influence of systematic and sample biases (such as the degree of urine dilution), MS data should also be normalized by either the parameters of the complete dataset (such as total ion count, median ion count, etc.) or the intensities of single or multiple internal standards (such as creatinine in the case of urine; Sysi-Aho et al., 2007). Consequentially, a multivariate dataset containing information about sample identities, ion identities (RT and m/z values) and relative ion intensities can be generated (Fig. 1). Processed datasets can be directly used for MDA, or be further statistically transformed and scaled according to the properties of data and the purpose of MDA analysis. To identify latent components or principal components (PC) in a complex dataset, data are projected onto a new coordinate system based on pattern recognition algorithm or MDA method (Schlotterbeck et al., 2006). Thereafter, a model containing one or multiple PCs can be established to represent a large portion of examined dataset. In contrast to other statistical techniques, such as t-test and ANOVA, an established MDA model and its PCs can be presented in the scores plot, in which sample-PC and sample-sample relationships can be visualized. In LC-MS-based metabolomics, the spatial distance between two samples in the scores plot reflects their differences in chemical composition. When a clear sample clustering is observed in the scores plot, the contribution of individual ions to PCs and to the group separation can be further examined in the loadings plot, in which the relationships between ions and PCs are depicted. With appropriate MDA modeling, ions contributing to the sample separation can be detected in the loadings plot and be further characterized.
Figure 1.
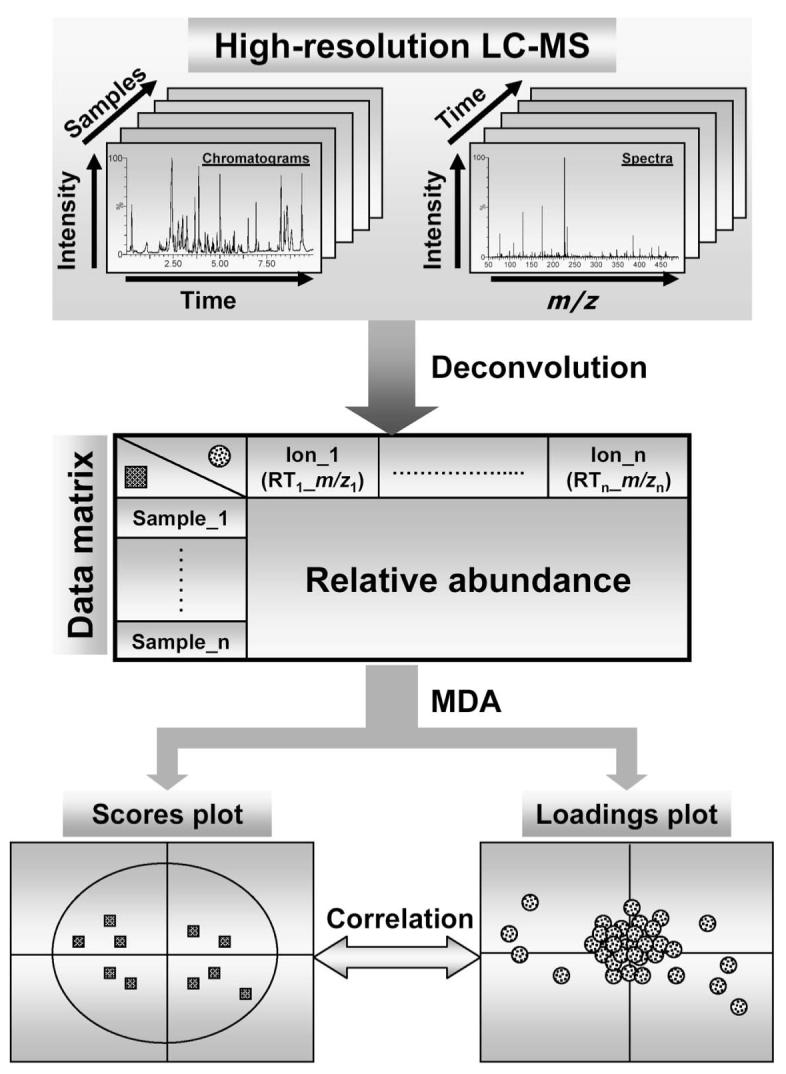
Procedures of LC-MS-based metabolomics. Chromatographic and spectral data are acquired by high-resolution LC-MS. Subsequent data processing, such as centroiding, deisotoping, filtering, peak recognition, yields a data matrix containing information on sample identity, ion identity (RT and m/z) and ion abundance. With appropriate data transformation and scaling, a multivariate model can be established through unsupervised or supervised MDA. The scores plot illustrates the principal or latent components of the model and sample classification, while the loadings plot presents the contribution of each ion to each principal component of the MDA model.
Two major categories of MDA methods, unsupervised and supervised MDA, have been widely used in metabolomic data analysis. In unsupervised MDA, sample classification is unknown or intentionally blinded to the analytical software, while in supervised MDA this information is provided to the software for the purpose of model construction. The most popular unsupervised method is principal components analysis (PCA). Because of its indiscriminate nature, the markers identified in a robust PCA model can usually be validated. Supervised MDA encompasses many methods, including partial least squares (PLS); orthogonal partial least squares (OPLS); soft-independent modeling of class analogy (SIMCA) and partial least squares-discriminant analysis (PLS-DA) (Trygg et al., 2007). The selection of supervised MDA method is determined by the data properties and the aim of MDA analysis.
METABOLOMIC APPROACHES FOR IDENTIFYING NOVEL METABOLITES AND METABOLISM PATHWAYS
Detecting xenobiotic metabolites through the combination of LC-MS and MDA was first proposed by Plumb et al. (Plumb et al., 2003), but has not been fully explored. Recently, the results from our studies on in vivo metabolism of several xenobiotics indicated that this metabolomic strategy is very efficient in identifying metabolites and elucidating in vivo metabolic pathways and overall metabolic maps (Chen et al., 2006; Giri et al., 2006; Chen et al., 2007; Giri et al., 2007). Based on these experiences, four approaches on the utilization of LC-MS-based metabolomics techniques in xenobiotic metabolism research are proposed in this review, and their potential applications in resolving practical issues in drug metabolism and toxicology are illustrated by case studies and proof-of-concept experiments.
Identification of Metabolites through Metabolomic Comparison between Vehicle Treatment and Xenobiotic Treatment
A straightforward approach for identifying in vivo xenobiotic metabolites is to compare the LC-MS chromatograms of biological samples (urine or feces) collected after vehicle treatment and xenobiotic treatment (Fig. 2A). However, without using radiolabeled compound, it is difficult to identify xenobiotic metabolites through visual examination of LC-MS chromatograms that contain information from thousands of chemical species. Therefore, how to facilitate the identification of xenobiotic metabolites through graphic presentation is a major challenge in this strategy. A xenobiotic and its metabolites only appear in the samples from xenobiotic treatment, and so when using metabolomic techniques, the differences between vehicle treatment and xenobiotic treatment are mainly defined by the presence of the xenobiotic and its metabolites. With appropriate data processing, the separation of vehicle-treated and xenobiotic-treated sample groups can be achieved in the scores plot of a multivariate model, and xenobiotic metabolite ions can be conveniently identified by analyzing ions contributing to the separation of xenobiotic treatment from vehicle treatment. Employing this approach, urine samples from mice treated with vehicle and the chemotherapeutic agent aminoflavone are clearly separated in the scores plot generated by an unsupervised PCA analysis (Fig. 2B). Thirteen aminoflavone metabolites appear prominently in the loadings plot as the ions that greatly contribute to the separation of aminoflavone treatment from vehicle treatment in the first PC (Fig. 2C; Chen et al., 2006).
Figure 2.

Metabolite identification through metabolomic comparison between vehicle treatment and xenobiotic treatment. A. Experimental scheme. Samples for LC-MS measurement are collected after dosing animals with vehicle or xenobiotic. Metabolite identification is conducted after MDA (in most cases, unsupervised PCA) of LC-MS data. B. A scores plot of a PCA analysis on 24-h mouse urine samples from vehicle (△) and 50 mg/kg aminoflavone (▲) treatment. The t[1] and t[2] values represent the scores of each sample in principal component 1 and 2, respectively. LC-MS measurement was conducted using UPLC-QTOFMS (Waters), data processing using MarkerLynx™ (Waters), and MDA using SIMCA-P+ software (Umetrics). Data are adopted from an aminoflavone metabolism study (Chen et al., 2006). C. Loadings plot of chemical ions in the urine samples from vehicle and aminoflavone treatment. The p[1] and p[2] values represent the contributing weights of each ion to principal components 1 and 2 of the PCA model, respectively.
In addition, the in vivo metabolic map of the areca alkaloids arecoline and arecaidine as well as the principal arecoline metabolite arecoline 1-oxide were also elucidated using this approach (Fig. 3; Giri et al., 2006; Giri et al., 2007). Until the metabolomic investigation of arecoline ([I] in Fig. 3), only the three primary metabolites of [I] had been elucidated in the rat, namely arecoline 1-oxide [II], the hydrolysis product arecaidine [III], and the N-acetylcysteine conjugate [IV]. By carrying out vehicle versus xenobiotic metabolomic comparisons individually for [I], [II], and [III], a whole new vista of metabolites was revealed, including the secondary metabolic pathways of [II], specifically, reduction back to [I], C=C double-bond reduction to N-methylnipecotic acid 1-oxide methyl ester [V], excretion of the N-acetylcysteine conjugate of the N-oxide, [VII] and its subsequent methylthiol [XIV] and thiol [XV] metabolites. Of interest was the detection of the corresponding aldehyde of arecoline, [VI]. This metabolite was not detected after administration of either [I] or [III], and so is shown in the map as arising from the N-oxide by an unknown mechanism. Moreover, these studies also revealed that [III] formed several novel metabolites, including an N-oxide [VIII], the C=C double-bond reduced metabolite N-methylnipecotic acid [XII] and its glycine conjugate [XIII], an N-acetylcysteine conjugate [IX], plus a glycerol [X] and a glycine [XI] conjugate. Finally, it can be seen that many of these metabolites have two or more chiral centers, and the resolving power of the UPLC was such that diastereomeric metabolite pairs were clearly visible for a number of the metabolites arising [II], which is itself chiral (Giri et al., 2007). Thus, the metabolic map, as shown, has been simplified to exclude diastereomeric metabolites, which can be found elsewhere (Giri et al., 2007). Since arecoline is the principal alkaloid present in areca nut, which is chewed by some 200 million users in Asia, the unique insights revealed through metabolomics in this metabolic map may prove useful in the search for a better understanding of the clinical toxicology of these alkaloids (Giri et al., 2006; Giri et al., 2007).
Figure 3.

Metabolic map of the areca alkaloid arecoline constructed from metabolomic studies in mice with arecoline, arecaidine, and the arecoline principal metabolite arecoline 1-oxide. The metabolites of arecoline [I] depicted are arecoline 1-oxide [II], arecaidine [III], arecoline N-acetylcysteine conjugate [IV], N-methylnipecotic acid 1-oxide methyl ester [V], the aldehyde metabolite of arecoline [VI], arecoline 1-oxide N-acetylcysteine conjugate [VII], arecaidine 1-oxide [VIII], arecaidine N-acetylcysteine conjugate [IX], arecaidine glycerol conjugate [X], arecaidine glycine conjugate [XI], N-methylnipecotic acid [XII], N-methylnipecotic acid glycine conjugate [XIII], the 4-methylthiol of arecoline 1-oxide [XIV], and the 4-thiol of arecoline 1-oxide [XV]. Details of diastereomeric metabolites have been published elsewhere (Giri et al., 2007).
Identification of Metabolites through Metabolomic Comparison between Unlabeled Xenobiotic Treatment and Stable Isotope-labeled Xenobiotic Treatment
Using a metabolomics-based comparison between vehicle treatment and xenobiotic treatment can dramatically facilitate the metabolite identification process (Figs. 2 and 3), especially for the xenobiotic treatments that yield no significant change in endogenous metabolic processes in the test subjects or animals. However, for studying the metabolism of xenobiotics that can also dramatically alter or disrupt the endogenous metabolic processes, such as the treatments eliciting strong toxic responses, the ions contributing to the separation of vehicle and xenobiotic treatment in the loadings plot include both xenobiotic metabolites and endogenous ions induced by the xenobiotic treatment. Therefore, a stable isotope-based metabolomics approach is proposed to facilitate the metabolite identification in this situation. Because of its non-radioactive nature and abundant choices for chemical synthesis, stable isotopes, such as 2H, 13C, 15N, or 18O, have been widely used for xenobiotic metabolism research (VandenHeuvel, 1986). For example, xenobiotic metabolites can be identified using the isotope cluster formed after dosing test subjects with an equal-molar mixture of labeled and unlabeled xenobiotic, though the process is still largely empirical. In the proposed stable isotope-based metabolomic approach, the equal doses of labeled and unlabeled xenobiotic are administered to the same types of test animals (Fig. 4A). Since the endogenous ions affected by labeled or unlabeled xenobiotic treatment are the same or very similar, the ions contributing to the separation of labeled group from unlabeled group in the scores plot should mainly be due to the xenobiotic and its metabolites. To examine this hypothesis, a proof-of-concept experiment was conducted, in which one group of mice were treated with a hepatotoxic dose of acetaminophen (APAP) and the other with an equimolar amount of deuterated APAPa. Following a PCA analysis, samples from APAP and deuterated APAP treatment were distinctively separated in the scores plots (Fig. 4B). Correspondingly, unlabeled APAP metabolites and their labeled counterparts appear symmetrically in the loadings plot (Fig. 4C). The top three ranking ions contributing to the separation of two treatments were identified as APAP (I), APAP glucuronide (II) and cysteinyl-APAP (III).
Figure 4.

Metabolite identification through metabolomic comparison between unlabeled xenobiotic treatment and stable isotope-labeled xenobiotic treatment. A. Experimental scheme. Samples for LC-MS measurement are collected after dosing animals with unlabeled or stable isotope-labeled xenobiotic. Metabolite identification is conducted after MDA (in most cases, unsupervised PCA) of LC-MS data. B. Scores plot of a proof-of-concept PCA analysis on 24-h mouse urine samples from 400 mg/kg APAP (△) and 400 mg/kg deuterated APAP (▲) treatment. The t[1] and t[2] values represent the scores of each sample in principal components 1 and 2, respectively. LC-MS measurement was conducted using UPLC-QTOFMS (Waters), data processing using Marker-Lynx™ (Waters), and MDA using SIMCA-P+ software (Umetrics). C. Loadings plot of chemical ions in the urine samples from APAP and deuterated APAP treatment. The p[1] and p[2] values represent the contributing weights of each ion to principal components 1 and 2 of PCA model, respectively.
Identification of Metabolic Pathways through Metabolomic Comparison among Wild-type and Genetically-modified Mice
Structural elucidation of xenobiotic metabolites identified by traditional in vitro or in vivo methods, as well as the aforementioned metabolomic approaches, in most cases, can provide clear clues about the biotransformation reactions occurring for a xenobiotic and thence help to establish the metabolic pathways involved. The role of individual XME in xenobiotic metabolism can be evaluated by using recombinant human XME and/or a specific inhibitor in vitro. The major issue with such in vitro approaches is the lack of a physiological context with regard to xenobiotic concentration and enzyme reaction system. Genetically-modified mouse models, including gene knockout and transgenic mice, provide alternative tools for determining drug metabolism pathways in vivo. Several major XMEs have their corresponding knockouts (including CYP1A1, CYP1A2, CYP1B1, and CYP2E1) and humanized (including CYP1A1, CYP1A2, CYP1B1, CYP2D6, CYP2E1, and CYP3A4) mouse lines established (Gonzalez, 2003; Gonzalez, 2004; Gonzalez and Yu, 2006). Gene knockout models are suitable for studying the endogenous function of XMEs as well as their role in xenobiotic metabolism, while humanized models are ideal for studying the underlying mechanisms of interspecies differences in xenobiotic metabolism or using as surrogate models for predicting xenobiotic metabolism in humans. Furthermore, a mouse model with all P450 activities abolished by conditional knockout of cytochrome P450 reductase has been established, which can be used to determine whether P450s have any role in the metabolism of individual xenobiotic (Henderson et al., 2003). As for studying the influence of drug-drug interaction on xenobiotic metabolism, knockout and humanized mouse models of several important nuclear receptors (including AhR, PXR, CAR) have also been established (Gonzalez and Yu, 2006). Recently, chimeric mice with liver comprised of more than 80% human hepatocytes have been generated, which further expand the available tools to simulate human-specific xenobiotic metabolism in animal models (Tateno et al., 2004; Katoh and Yokoi, 2007).
Genetically-modified animal models are powerful tools for exploiting the mechanism of xenobiotic metabolism, especially genotype-dependent metabolism. However, how to conduct a comprehensive and efficient analysis on the metabolism data acquired from the treatments on multiple animal models is a major challenge. The combination of genetically-modified mouse models with LC-MS-based metabolomics provide a rational solution since the graphic presentation of genotype-dependent grouping in the scores plot of MDA model can directly reflect the influence of a targeted gene (XME or nuclear receptor) on xenobiotic metabolism. In addition, structural elucidation of metabolite ions contributing to the separation among the genotypes can further yield important information on the in vivo metabolic pathways (Fig. 5A). This approach was applied to determine the role of the mouse-human difference in CYP1A2 activity in a recent study on the metabolism of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP), a potent rodent carcinogen and a potential human carcinogen (Chen et al., 2007). After a PCA analysis on the LC-MS data from the vehicle and PhIP treatment on wild-type, Cyp1a2-null, and CYP1A2-humanized mice, the control urine samples from three mouse lines cluster together while the PhIP-treated urine samples from three mouse lines appear in the different positions in a three-PC scores plot, indicating PhIP displays a distinctive metabolic pattern in each CYP1A2-related mouse line (Fig. 5B). In the loadings plot, the metabolites existing in high abundance in each genotype were conveniently identified by their spatial positions (correlating to the spatial positions of each PhIP-treated genotype in the scores plot). Subsequent structural elucidation based on accurate mass measurement and tandem MS/MS fragmentation resulted in the identification of 17 PhIP metabolites, including 8 novel metabolites, and the clear conclusions regarding PhIP metabolism were as follows: i) mouse CYP1A2 has higher PhIP 4-hydroxylase activity while human CYP1A2 higher PhIP N2-hydroxylase activity, which is mainly responsible for the genotoxicity of PhIP; ii) in Cyp1a2-null mice, PhIP metabolism shifts from the hydroxylation reaction to the N-methylation reaction; iii) other P450s (CYP2Cs) are also involved in the PhIP metabolism since PhIP was still extensively metabolized in Cyp1a2-null mice. This study highlights the capability of LC-MS-based metabolomic techniques to elucidate the in vivo metabolic map and constituent pathways for a xenobiotic.
Figure 5.

Identification of metabolic pathways through metabolomic comparison among wild-type and genetically-modified mice. A. Experimental scheme. Samples for LC-MS measurement are collected after dosing wild-type, knockout and transgenic mice with vehicle or xenobiotic. Identification of metabolites and analysis of metabolic pathways are conducted after MDA (supervised or unsupervised) of LC-MS data. B. Scores plot of a PCA analysis on 24-h mouse urine samples from vehicle treatment (wild-type: △; Cyp1a2-null: ◇; CYP1A2-humanized:□) and 10 mg/kg PhIP treatment (wild-type:▲ Cyp1a2-null: ◆; CYP1A2-humanized: ■). The t[1], t[2], and t[3] values represent the scores of each sample in principal components 1, 2, 3, respectively. LC-MS measurement was conducted using UPLC-QTOFMS (Waters), data processing using MarkerLynx™ (Waters), and MDA using SIMCA-P+ software (Umetrics). Data are adopted from an PhIP metabolism study (Chen et al., 2007). C. Loadings plot of ions in the urine samples from wild-type, Cyp1a2-null and CYP1A2-humanized mice treated with vehicle and PhIP. The p[1], p[2] and p[3] values represent the contributing weights of each ion to principal components 1, 2, 3 of PCA model, respectively.
Metabolomic Approach for Identifying Human XME Polymorphism Responsible for Adverse Drug Reactions (ADRs)
ADR is a detrimental event occurring for a limited group of patients after taking drug within clinically recommended dose limits. The occurrence of ADRs is not uncommon for many prescription drugs, especially medicines that target life-threatening diseases such as cancer, but it also can occur for drugs that are well-tolerated in patients and widely recognized as safe (Giacomini et al., 2007). This latter type of ADR has been largely attributed to genetic polymorphisms of XMEs, although other factors such as health status (cancer, hepatic dysfunction, inflammation and infection), drug-drug interactions, or exposure to environmental contaminants (such as heavy metals and nuclear receptor ligands). All these factors can significantly alter enzymatic activities or expression levels of XMEs responsible for the biotransformation of a therapeutic agent, causing changes in metabolic activity in ADR patients. Therefore, the information obtained from studying the metabolic pathways related to ADRs may help to identify the patients susceptible to ADR and promote the transformation of empirical medication to individualized medication (Clayton et al., 2006; Nebert and Vesell, 2006). When ADRs observed in a clinical study are suspected to be related to polymorphism in drug metabolism, a common practice is to conduct an investigation into the pharmacokinetics and metabolite profiles of the suspected agent. Subsequent pharmacogenetic or pharmacogenomic investigation of the affected metabolic pathways and XMEs can pinpoint the genetic factors responsible for an ADR (Evans and Relling, 1999). Application of this strategy first resulted in the identification of a correlation between CYP2D6 polymorphism (Mahgoub et al., 1977; Gonzalez et al., 1988) and an excessive hypotensive response to debrisoquine (Idle et al., 1978). Other examples of pharmacogenetic determinants of ADRs include UDP-glucuronosyltransferase 1A1 (UGT1A1) and irinotecan (Ando et al., 2000) and thiopurine methyltransferase (TPMT) and 6-mercaptopurine (Wang and Weinshilboum, 2006).
Currently, the identification of ADR-related metabolites and metabolic pathways remains empirical. Because of its multivariate nature based on the chemical complexity of human biological samples and the genetic polymorphism of human subjects, ADR research can be an ideal field for applying LC-MS-based metabolomic techniques (Fig. 6A). In fact, the feasibility of this metabolomic approach was validated in a proof-of-concept experiment, in which the urine samples from a panel of human subjects of known CYP2D6 genotype, acquired from a previous study in our lab (Zhen et al., 2006), were analyzed by LC-MS and supervised MDAa. In debrisoquine-treated subjects, urine samples from poor metabolizers (PMs) were clearly separated from urine samples of extensive metabolizers (EMs) in the scores plot of an OPLS model (Fig. 6B). Not unexpectedly, in the loadings plot, the ion mainly responsible for the separation of PMs from EMs was debrisoquine, which is high in PMs, while ions responsible for the separation of EMs from PMs are 4-hydroxy-debrisoquine (4-OH-Deb) and two ring-open products of debrisoquine, putatively as 2-(guanidinoethyl)benzoic acid (I) and 2-(guanidinomethyl)phenyl acetic acid (II) (Allen et al., 1975; Allen et al., 1976; Idle et al., 1979; Eiermann et al., 1998) (Fig. 6C). Thus, the pharmacogenetic phenotypes of CYP2D6 EMs and PMs can now be visualized through metabolomic phenotyping, rather than by calculating metabolic ratios of debrisoquine/4-OH-Deb, as first defined 30 years ago (Mahgoub et al., 1977).
Figure 6.

Metabolomic approach for identifying human XME polymorphism responsible for ADR. A. Experi-mental scheme. Samples for LC-MS measurement are collected from the patients with normal response to the drug and the patients with ADR to the drug. Identification of metabolites and analysis of metabolism pathway are conducted after MDA (in most cases, supervised) of LC-MS data. B. Scores plot of a proof-of-concept OPLS analysis on 8-h urine samples from extensive metabolizers (△) and poor metabolizers (▲) treated with 10 mg debrisoquine. The t[1]P and t[2]O values represent the scores of each sample in principal component 1 (predictive component) and 2 (orthogonal component), respectively. LC-MS measurement was conducted using UPLCQTOFMS (Waters), data processing using MarkerLynx™ (Waters), and MDA using SIMCA-P+ software (Umetrics). Data are adopted from a debrisoquine metabolism study (Zhen et al., 2006). C. Loadings plot of ions in the human urine samples after debrisoquine treatment. The w*c[1]P and w*c[2]O values represent the contributing weights of each ion to principal components 1 (predictive component) and 2 (orthogonal component) of the OPLS model, respectively.
CONCLUSION AND FUTURE PERSPECTIVE
In this review, we describe and discuss four applicable approaches of using LC-MS-base metabolomic techniques to resolve practical issues in drug metabolism. Unambiguous results from our case studies and proof-of-concept experiments indicate that LC-MS-based metabolomic techniques possess the great promise of becoming standard tools for identifying xenobiotic metabolites and elucidating xenobiotic metabolic maps. By comparison to traditional in vitro and in vivo methods in drug metabolism, LC-MS-based metabolomics techniques show clear advantages in the capacity of handling large datasets and in the graphical representation of metabolism-related sample classification, as well as in the indiscriminative nature of metabolite identification. To achieve the objective of understanding the mechanism of xenobiotic metabolism-related events using this particular technological platform, careful experimental design, high-resolution data acquisition, robust data processing and stringent data analysis are required. Overall, with the increasing availability of high-resolution LC-MS instruments and enhanced functions of MDA software, we expect that LC-MS-based metabolomics technologies will be widely adopted and utilized in the xenobiotic metabolism research field.
ACKNOWLEDGMENT
We thank Kristopher Krausz and Yueying Zhen for providing debrisoquine data for multivariate data analysis. Research described in this manuscript was supported by the NCI Intramural Research Program of the NIH. JRI is grateful to US Smokeless Tobacco Company for a grant for collaborative research.
ABBREVIATIONS
- XME
xenobiotic metabolizing enzyme
- P450
cytochrome P450
- MDA
multivariate data analysis
- PCA
principal components analysis
- MS
mass spectrometry
- LC-MS
liquid chromatography-mass spectrometry
- UPLC
ultra-performance liquid chromatography
- TOFMS
time-of-flight mass spectrometry
- ADR
adverse drug reaction
Footnotes
Details of this study will be reported in a separate manuscript.
REFERENCES
- Allen JG, Brown AN, Marten TR. Metabolism of debrisoquine sulphate in rat, dog and man. Xenobiotica. 1976;6:405–409. doi: 10.3109/00498257609151653. [DOI] [PubMed] [Google Scholar]
- Allen JG, East PB, Francis RJ, Haigh JL. Metabolism of debrisoquine sulfate. Identification of some urinary metabolites in rat and man. Drug Metab. Dispos. 1975;3:332–337. [PubMed] [Google Scholar]
- Ando Y, Saka H, Ando M, Sawa T, Muro K, Ueoka H, Yokoyama A, Saitoh S, Shimokata K, Hasegawa Y. Polymorphisms of UDP-glucuronosyltransferase gene and irinotecan toxicity: a pharmacogenetic analysis. Cancer Res. 2000;60:6921–6926. [PubMed] [Google Scholar]
- Brindle JT, Antti H, Holmes E, Tranter G, Nicholson JK, Bethell HW, Clarke S, Schofield PM, McKilligin E, Mosedale DE, Grainger DJ. Rapid and noninvasive diagnosis of the presence and severity of coronary heart disease using 1H-NMR-based metabonomics. Nat. Med. 2002;8:1439–1444. doi: 10.1038/nm1202-802. [DOI] [PubMed] [Google Scholar]
- Brodie BB, Axelrod J. The Fate of Acetanilide in Man. J. Pharmacol. Exp. Ther. 1948;94:29. [PubMed] [Google Scholar]
- Chen C, Ma X, Malfatti MA, Krausz KW, Kimura S, Felton JS, Idle JR, Gonzalez FJ. A comprehensive investigation of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) metabolism in the mouse using a multivariate data analysis approach. Chem. Res. Toxicol. 2007;20:531–542. doi: 10.1021/tx600320w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Meng L, Ma X, Krausz KW, Pommier Y, Idle JR, Gonzalez FJ. Urinary metabolite profiling reveals CYP1A2-mediated metabolism of NSC686288 (aminoflavone) J. Pharmacol. Exp. Ther. 2006;318:1330–1342. doi: 10.1124/jpet.106.105213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton TA, Lindon JC, Cloarec O, Antti H, Charuel C, Hanton G, Provost JP, Le Net JL, Baker D, Walley RJ, Everett JR, Nicholson JK. Pharmaco-metabonomic phenotyping and personalized drug treatment. Nature. 2006;440:1073–1077. doi: 10.1038/nature04648. [DOI] [PubMed] [Google Scholar]
- Dettmer K, Aronov PA, Hammock BD. Mass spectrometry-based metabolomics. Mass Spectrom Rev. 2007;26:51–78. doi: 10.1002/mas.20108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiermann B, Edlund PO, Tjernberg A, Dalen P, Dahl ML, Bertilsson L. 1- and 3-hydroxylations, in addition to 4-hydroxylation, of debrisoquine are catalyzed by cytochrome P450 2D6 in humans. Drug Metab. Dispos. 1998;26:1096–1101. [PubMed] [Google Scholar]
- Ekins S, Andreyev S, Ryabov A, Kirillov E, Rakhmatulin EA, Bugrim A, Nikolskaya T. Computational prediction of human drug metabolism. Expert Opin. Drug Metab. Toxicol. 2005;1:303–324. doi: 10.1517/17425255.1.2.303. [DOI] [PubMed] [Google Scholar]
- Evans WE, Relling MV. Pharmacogenomics: translating functional genomics into rational therapeutics. Science. 1999;286:487–491. doi: 10.1126/science.286.5439.487. [DOI] [PubMed] [Google Scholar]
- Fernie AR, Trethewey RN, Krotzky AJ, Willmitzer L. Metabolite profiling: from diagnostics to systems biology. Nat. Rev. Mol. Cell. Biol. 2004;5:763–769. doi: 10.1038/nrm1451. [DOI] [PubMed] [Google Scholar]
- Giacomini KM, Krauss RM, Roden DM, Eichelbaum M, Hayden MR, Nakamura Y. When good drugs go bad. Nature. 2007;446:975–977. doi: 10.1038/446975a. [DOI] [PubMed] [Google Scholar]
- Giri S, Idle JR, Chen C, Zabriskie TM, Krausz KW, Gonzalez FJ. A metabolomic approach to the metabolism of the areca nut alkaloids arecoline and arecaidine in the mouse. Chem. Res. Toxicol. 2006;19:818–827. doi: 10.1021/tx0600402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri S, Krausz KW, Idle JR, Gonzalez FJ. The metabolomics of (+/−)-arecoline 1-oxide in the mouse and its formation by human flavin-containing monooxygenases. Biochem. Pharmacol. 2007;73:561–573. doi: 10.1016/j.bcp.2006.10.017. [DOI] [PubMed] [Google Scholar]
- Gonzalez FJ. Role of gene knockout and transgenic mice in the study of xenobiotic metabolism. Drug Metab. Rev. 2003;35:319–335. doi: 10.1081/dmr-120026496. [DOI] [PubMed] [Google Scholar]
- Gonzalez FJ. Cytochrome P450 humanised mice. Hum. Genomics. 2004;1:300–306. doi: 10.1186/1479-7364-1-4-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez FJ, Skoda RC, Kimura S, Umeno M, Zanger UM, Nebert DW, Gelboin HV, Hardwick JP, Meyer UA. Characterization of the common genetic defect in humans deficient in debrisoquine metabolism. Nature. 1988;331:442–446. doi: 10.1038/331442a0. [DOI] [PubMed] [Google Scholar]
- Gonzalez FJ, Yu AM. Cytochrome P450 and xenobiotic receptor humanized mice. Annu. Rev. Pharmacol. Toxicol. 2006;46:41–64. doi: 10.1146/annurev.pharmtox.45.120403.100007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin JL. The Cinderella story of metabolic profiling: does metabolomics get to go to the functional genomics ball? Philos. Trans. R Soc. Lond. B Biol. Sci. 2006;361:147–161. doi: 10.1098/rstb.2005.1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guengerich FP. Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem. Res. Toxicol. 2001;14:611–650. doi: 10.1021/tx0002583. [DOI] [PubMed] [Google Scholar]
- Hashi K, Shimizu T, Goto A, Kiyoshi T, Matsumoto S, Wada H, Fujito T, Hasegawa K, Yoshikawa M, Miki T, Ito S, Hamada M, Hayashi S. Achievement of a 920-MHz high resolution NMR. J. Magn. Reson. 2002;156:318–321. doi: 10.1006/jmre.2002.2559. [DOI] [PubMed] [Google Scholar]
- Henderson CJ, Otto DM, Carrie D, Magnuson MA, McLaren AW, Rosewell I, Wolf CR. Inactivation of the hepatic cytochrome P450 system by conditional deletion of hepatic cytochrome P450 reductase. J. Biol. Chem. 2003;278:13480–13486. doi: 10.1074/jbc.M212087200. [DOI] [PubMed] [Google Scholar]
- Idle JR, Mahgoub A, Angelo MM, Dring LG, Lancaster R, Smith RL. The metabolism of [14C]-debrisoquine in man. Br. J. Clin. Pharmacol. 1979;7:257–266. doi: 10.1111/j.1365-2125.1979.tb00930.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idle JR, Mahgoub A, Lancaster R, Smith RL. Hypotensive response to debrisoquine and hydroxylation phenotype. Life Sci. 1978;22:979–983. doi: 10.1016/0024-3205(78)90363-6. [DOI] [PubMed] [Google Scholar]
- Katajamaa M, Oresic M. Data processing for mass spectrometry-based metabolomics. J. Chromatogr. A. 2007;1158:318–328. doi: 10.1016/j.chroma.2007.04.021. [DOI] [PubMed] [Google Scholar]
- Katoh M, Yokoi T. Application of chimeric mice with humanized liver for predictive ADME. Drug Metab. Rev. 2007;39:145–157. doi: 10.1080/03602530601021340. [DOI] [PubMed] [Google Scholar]
- Kikuchi J, Shinozaki K, Hirayama T. Stable isotope labeling of Arabidopsis thaliana for an NMR-based metabolomics approach. Plant Cell Physiol. 2004;45:1099–1104. doi: 10.1093/pcp/pch117. [DOI] [PubMed] [Google Scholar]
- Kristal BS, Shurubor YI, Kaddurah-Daouk R, Matson WR. High-performance liquid chromatography separations coupled with coulometric electrode array detectors: a unique approach to metabolomics. Methods Mol. Biol. 2007;358:159–174. doi: 10.1007/978-1-59745-244-1_10. [DOI] [PubMed] [Google Scholar]
- Mahgoub A, Idle JR, Dring LG, Lancaster R, Smith RL. Polymorphic hydroxylation of Debrisoquine in man. Lancet. 1977;2:584–586. doi: 10.1016/s0140-6736(77)91430-1. [DOI] [PubMed] [Google Scholar]
- Mortishire-Smith RJ, O'Connor D, Castro-Perez JM, Kirby J. Accelerated throughput metabolic route screening in early drug discovery using high-resolution liquid chromatography/quadrupole time-of-flight mass spectrometry and automated data analysis. Rapid Commun. Mass Spectrom. 2005;19:2659–2670. doi: 10.1002/rcm.2111. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Vesell ES. Can personalized drug therapy be achieved? A closer look at pharmaco-metabonomics. Trends Pharmacol. Sci. 2006;27:580–586. doi: 10.1016/j.tips.2006.09.008. [DOI] [PubMed] [Google Scholar]
- Nicholson JK, Connelly J, Lindon JC, Holmes E. Metabonomics: a platform for studying drug toxicity and gene function. Nat. Rev. Drug Discov. 2002;1:153–161. doi: 10.1038/nrd728. [DOI] [PubMed] [Google Scholar]
- Nicholson JK, Wilson ID. Opinion: understanding ‘global’ systems biology: metabonomics and the continuum of metabolism. Nat. Rev. Drug Discov. 2003;2:668–676. doi: 10.1038/nrd1157. [DOI] [PubMed] [Google Scholar]
- Pan Z, Raftery D. Comparing and combining NMR spectroscopy and mass spectrometry in metabolomics. Anal. Bioanal. Chem. 2007;387:525–527. doi: 10.1007/s00216-006-0687-8. [DOI] [PubMed] [Google Scholar]
- Plumb RS, Stumpf CL, Granger JH, Castro-Perez J, Haselden JN, Dear GJ. Use of liquid chromatography/time-of-flight mass spectrometry and multivariate statistical analysis shows promise for the detection of drug metabolites in biological fluids. Rapid Commun. Mass Spectrom. 2003;17:2632–2638. doi: 10.1002/rcm.1250. [DOI] [PubMed] [Google Scholar]
- Schauer N, Fernie AR. Plant metabolomics: towards biological function and mechanism. Trends Plant Sci. 2006;11:508–516. doi: 10.1016/j.tplants.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Schlotterbeck G, Ross A, Dieterle F, Senn H. Metabolic profiling technologies for biomarker discovery in biomedicine and drug development. Pharmacogenomics. 2006;7:1055–1075. doi: 10.2217/14622416.7.7.1055. [DOI] [PubMed] [Google Scholar]
- Sysi-Aho M, Katajamaa M, Yetukuri L, Oresic M. Normalization method for metabolomics data using optimal selection of multiple internal standards. BMC Bioinformatics. 2007;8:93. doi: 10.1186/1471-2105-8-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateno C, Yoshizane Y, Saito N, Kataoka M, Utoh R, Yamasaki C, Tachibana A, Soeno Y, Asahina K, Hino H, Asahara T, Yokoi T, Furukawa T, Yoshizato K. Near completely humanized liver in mice shows human-type metabolic responses to drugs. Am. J. Pathol. 2004;165:901–912. doi: 10.1016/S0002-9440(10)63352-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trygg J, Holmes E, Lundstedt T. Chemometrics in metabonomics. J. Proteome Res. 2007;6:469–479. doi: 10.1021/pr060594q. [DOI] [PubMed] [Google Scholar]
- van der Werf MJ, Jellema RH, Hankemeier T. Microbial metabolomics: replacing trial-and-error by the unbiased selection and ranking of targets. J. Ind. Microbiol. Biotechnol. 2005;32:234–252. doi: 10.1007/s10295-005-0231-4. [DOI] [PubMed] [Google Scholar]
- VandenHeuvel WJ. Drug metabolite identification: stable isotope methods. J. Clin. Pharmacol. 1986;26:427–434. doi: 10.1002/j.1552-4604.1986.tb03553.x. [DOI] [PubMed] [Google Scholar]
- Venkatakrishnan K, von Moltke LL, Obach RS, Greenblatt DJ. Drug metabolism and drug interactions: application and clinical value of in vitro models. Curr. Drug Metab. 2003;4:423–459. doi: 10.2174/1389200033489361. [DOI] [PubMed] [Google Scholar]
- Wang L, Weinshilboum R. Thiopurine S-methyltransferase pharmacogenetics: insights, challenges and future directions. Oncogene. 2006;25:1629–1638. doi: 10.1038/sj.onc.1209372. [DOI] [PubMed] [Google Scholar]
- Want EJ, Nordstrom A, Morita H, Siuzdak G. From exogenous to endogenous: the inevitable imprint of mass spectrometry in metabolomics. J. Proteome Res. 2007;6:459–468. doi: 10.1021/pr060505+. [DOI] [PubMed] [Google Scholar]
- Wilson ID, Nicholson JK, Castro-Perez J, Granger JH, Johnson KA, Smith BW, Plumb RS. High resolution “ultra performance” liquid chromatography coupled to oa-TOF mass spectrometry as a tool for differential metabolic pathway profiling in functional genomic studies. J. Proteome Res. 2005;4:591–598. doi: 10.1021/pr049769r. [DOI] [PubMed] [Google Scholar]
- Wishart DS, Tzur D, Knox C, Eisner R, Guo AC, Young N, Cheng D, Jewell K, Arndt D, Sawhney S, Fung C, Nikolai L, Lewis M, Coutouly MA, Forsythe I, Tang P, Shrivastava S, Jeroncic K, Stothard P, Amegbey G, Block D, Hau DD, Wagner J, Miniaci J, Clements M, Gebremedhin M, Guo N, Zhang Y, Duggan GE, Macinnis GD, Weljie AM, Dowlatabadi R, Bamforth F, Clive D, Greiner R, Li L, Marrie T, Sykes BD, Vogel HJ, Querengesser L. HMDB: the Human Metabolome Database. Nucleic Acids Res. 2007;35:D521–526. doi: 10.1093/nar/gkl923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun CH, Okerholm RA, Guengerich FP. Oxidation of the antihistaminic drug terfena-dine in human liver microsomes. Role of cytochrome P-450 3A(4) in N-dealkylation and C-hydroxylation. Drug Metab. Dispos. 1993;21:403–409. [PubMed] [Google Scholar]
- Zhen Y, Slanar O, Krausz KW, Chen C, Slavik J, McPhail KL, Zabriskie TM, Perlik F, Gonzalez FJ, Idle JR. 3,4-Dehydrodebrisoquine, a novel debrisoquine metabolite formed from 4-hydroxydebrisoquine that affects the CYP2D6 metabolic ratio. Drug Metab. Dispos. 2006;34:1563–1574. doi: 10.1124/dmd.105.008920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M, Ma L, Zhang D, Ray K, Zhao W, Humphreys WG, Skiles G, Sanders M, Zhang H. Detection and characterization of metabolites in biological matrices using mass defect filtering of liquid chromatography/high resolution mass spectrometry data. Drug Metab. Dispos. 2006;34:1722–1733. doi: 10.1124/dmd.106.009241. [DOI] [PubMed] [Google Scholar]