

Abstract
Oxidative stress is widely recognized as a key mediator of degenerative processes in Parkinson’s disease (PD). Recently, we demonstrated that the dopaminergic toxin MPP+ initiates oxidative stress to cause caspase-3-dependent apoptotic cell death in mesencephalic dopaminergic neuronal (N27) cells. In this study, we determined the source of reactive oxygen species (ROS) produced during MPP+-induced apoptotic cell death. In addition to mitochondria, plasma membrane NADPH oxidase is considered a major producer of ROS inside the cell. Here, we show that N27 cells express key NADPH oxidase subunits gp91phox and p67phox. We used structurally diverse NADPH oxidase inhibitors, aminoethyl-benzenesulfonylfluoride (AEBSF, 100-1000 μM), apocynin (100-1000 μM), and diphenylene iodonium (DPI, 3-30 μM), to inhibit intrinsic NADPH oxidase activity in N27 cells. Flow cytometric analysis using the ROS-sensitive dye hydroethidine revealed that AEBSF blocked 300 μM MPP+-induced ROS production for over 45 min in N27 cells, in a dose-dependent manner. Further treatment with DPI, apocynin, and SOD also blocked MPP+-induced ROS production. In Sytox cell death assays, co-treatment with AEBSF, apocynin, or DPI for 24 hr significantly suppressed MPP+-induced cytotoxic cell death. Similarly, co-treatment with these inhibitors also significantly attenuated MPP+-induced increases in caspase-3 enzymatic activity. Furthermore, quantitative DNA fragmentation ELISA assays revealed that AEBSF, DPI, and apocynin rescue N27 cells from MPP+-induced apoptotic cell death. Together, these results indicate for the first time that intracellular ROS generated by NAPDH oxidase are present within the mesencephalic neuronal cells, and are a key determinant of MPP+-mediated dopaminergic degeneration in in vitro models of dopaminergic degeneration. This study supports a critical role of NADPH oxidase in the oxidative damage in PD; targeting this enzyme may lead to novel therapies for PD.
Keywords: Oxidative damage, Dopamine, NADPH oxidase inhibitor, Neurotoxicity, Neuroprotection, Parkinson’s disease
1. Introduction
Parkinson’s disease (PD) is a common neurodegenerative disorder characterized by progressive motor dysfunction and variable cognitive impairment (Schapira, 1997; Sherer et al., 2001; Schulz and Falkenburger, 2004; Gandhi and Wood, 2005; Przedborski, 2005). Its key neuropathological features are the loss of substantia nigra pars compacta dopaminergic neurons and loss of striatal dopamine content, which together lead to bradykinesia, tremors, and postural instability in PD (Schapira, 1997; Sherer et al., 2001; Schulz and Falkenburger, 2004; Gandhi and Wood, 2005; Przedborski, 2005). Recent studies have demonstrated oxidative stress as the major initiator of apoptotic cell death in several neurodegenerative disorders, including PD (Zigmond et al., 2002; Dawson and Dawson, 2003; Di Monte, 2003; Jenner, 2003; Kanthasamy et al., 2003; Thiruchelvam et al., 2003; Greenamyre and Hastings, 2004; Maguire-Zeiss et al., 2005; Przedborski and Ischiropoulos, 2005; McCormack et al., 2006). The potent dopaminergic toxin MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydro- -pyridine) causes an irreversible PD-like syndrome in humans, non-human primates, and in animals, and reproduces most of the neurochemical and pathological hallmarks, including the substantial degeneration of dopaminergic neurons; consequently, MPTP has been used extensively in experimental PD models (Dauer and Przedborski, 2003; Hirsch et al., 2003b; Przedborski et al., 2004; Bove et al., 2005; Smeyne and Jackson-Lewis, 2005; Watanabe et al., 2005).
Several studies have implicated oxidative stress in the pathogenesis of PD. Reactive oxygen species (ROS) generated from mitochondrial and/or extra-mitochondrial sources appear to be the main contributor of oxidative stress-mediated neurodegeneration in PD models (Koutsilieri et al., 2002; Tikka et al., 2002; Beal, 2003; Ischiropoulos and Beckman, 2003; Jenner, 2003; Kanthasamy et al., 2003; Greenamyre and Hastings, 2004; Love, 2004; Przedborski and Ischiropoulos, 2005). One of the well-recognized pathways responsible for generation of oxidative radicals is mitochondrial toxicity induced by accumulation of MPP+ in the inner mitochondrial membrane, and the subsequent disruption of complex I in the electron transport chain (Cassarino et al., 1999; Fiskum et al., 2003; Kalivendi et al., 2003). Studies have also shown auto-oxidation of the neurotransmitter dopamine (Obata, 2002; Sidhu et al., 2004) or the interaction of MPP+ with iron stores within the pigmented substantia nigra cells as possible sources of oxidative stress (Andersen, 2004; Mandel et al., 2004; Youdim et al., 2004; Mancuso et al., 2007).
Recently, inflammation has also been suggested to contribute to the pathogenesis of PD (Beal, 2003; Hald and Lotharius, 2005; Sawada et al., 2006; Wersinger and Sidhu, 2006). ROS are among the inflammatory mediators capable of promoting neurodegeneration, which are derived from activation of microglial NADPH oxidase (Serrano et al., 2003; Infanger et al., 2006; Sawada et al., 2006; Ushio-Fukai, 2006). NADPH oxidase is a multisubunit enzyme that catalyzes the reduction of molecular oxygen to form superoxide radicals, and is composed of gp91phox, p22phox, p47phox, p67phox, and p40phox subunits. Under basal conditions, the p47phox, p67phox, and p40phox subunits are present in the cystosol as a complex, and are separated from the transmembrane gp91phox and p22phox subunits (Serrano et al., 2003; Infanger et al., 2006; Sawada et al., 2006; Ushio-Fukai, 2006). Upon activation, the p47phox subunit gets phosphorylated, and translocates to the membrane as a complex to assemble with gp91phox and p22phox to form an active NADPH oxidase capable of reducing oxygen to a superoxide radical(O2-) to generate microglial and/or extra-mitochondrial-derived ROS (Serrano et al., 2003; Infanger et al., 2006; Sawada et al., 2006; Ushio-Fukai, 2006).
NADPH oxidase is ubiquitously expressed in a wide variety of cells and organ systems, including brain regions such as hippocampus, cortex, striatum, thalamus, and amygdala (Serrano et al., 2003; Geiszt, 2006; Infanger et al., 2006; Takeya and Sumimoto, 2006). Immunohistochemistry studies have identified NADPH oxidase subunits in different brain regions and in different cell types including neurons, astrocytes, and microglial cells (Sun et al., 2007). Several non-neuronal, neuronal, and glial cell lines including PC12 (Ibi et al., 2006), SH-SY5Y (Nikolova et al., 2005), GT1-7 (Schneider et al., 2003), IC11 (Schneider et al., 2003), Neuro2A (Reis et al., 2006), and BV-2 (Reis et al., 2006) have also been shown to express various NADPH oxidase subunit proteins.
We recently established that N27 cells are a superior cell culture model for studying dopaminergic neurodegeneration, compared to PC12 and SH-SY5Y cells, because N27 cells are derived from the mesencephalon, a brain region directly affected by Parkinson’s disease, and they represent a homogenous population of tyrosine hydroxylase-positive cells with functional characteristics resembling dopaminergic neurons (Anantharam et al., 2002; Kaul et al., 2003; Yang et al., 2004; Kaul et al., 2005b; Kanthasamy et al., 2006; Sun et al., 2006). We showed that MPP+ treatment in N27 cells induces acute generation of ROS in a time- and dose-dependent manner (Kaul et al., 2003; Kaul et al., 2005a), and that ROS generation precedes changes in mitochondrial membrane potential or cytochrome C release. Using pharmacological inhibitors, herein we determined whether NADPH oxidase is an upstream source of reactive oxygen species that might be involved in propagating MPP+-induced apoptotic cell death of N27 cells.
2. Materials and methods
2.1. Chemicals
MPP+ (1-methyl-4-phenylpyridinium), superoxide dismutase, DPI, AEBSF, apocynin,and β-Actin antibody (mouse monoclonal) were purchased from Sigma-Aldrich (St. Louis, MO); Ac-DEVD-AFC (Acetyl-Leu-Glu-His-Asp-7-amino-4-fluorocoumarin) was obtained from MP Biomedicals (Livermore, CA). NADPH oxidase antibodies against gp91phox and p67phox were purchased from Santacruz labs (Santa Cruz, CA). ECL chemiluminescence kit was purchased from Amersham Pharmacia Biotech (Piscataway, NJ). RPMI-1640, fetal bovine serum, L-glutamine, penicillin and streptomycin, dihydroethidine (DhEt) and Sytox were purchased from Invitrogen/Molecular Probes (Eugene, OR). Cell Death Detection ELISA Plus Assay Kit was purchased from Roche Molecular Biochemicals (Indianapolis, IN). Bradford protein assay kit was purchased from Bio-Rad Laboratories (Hercules, CA).
2.2. Cell Cultures
The immortalized rat mesencephalic dopaminergic neuronal cells (N27) were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 50 units penicillin, and 50 μg/ml Cell cultures were maintained in a humidified atmosphere of 5% CO2 at 37°C, as previously described (Kaul et al., 2003).
2.3. Treatment Paradigm
N27 cells were exposed to 300 μM MPP+ in the presence or absence of AEBSF (100-1000 μM), apocynin (100-1000 μM), and DPI (3-30 μM) for up to 24 hrs. For ROS and cytotoxicity measurements, untreated and treated N27 cells were directly used for measurements. For caspase-3 and DNA fragmentation assays, cells were harvested and lysed, and lysates were used for measurements. For Western blots, membrane and cytosolic fractions were obtained from cell lysates as described previously (Anantharam et al., 2002). Protein concentration was determined by the Bradford protein assay.
2.4. Measurement of ROS Generation
Flow cytometry analysis was performed using a Becton Dickenson FACScan™ flow cytometer (Becton Dickenson, San Francisco, CA) with a ROS-sensitive dye, hydroethidine, as described previously (Kaul et al., 2003; Kaul et al., 2005a). Briefly, N27 cells were re-suspended with Earle’s balanced salt solution (EBSS) with 2 mM calcium, then incubated with 10 μM hydroethidine for 15 min at 37°C in the dark, and then exposed to 300 μM MPP+, in the presence or absence of either AEBSF (100-1000 μM), apocynin (300 μM), DPI (10 μM), or SOD (100 units). Next, ROS generation was measured over 45 min. All the flow cytometric data were analyzed by Cellquest™ data analysis software (Becton Dickenson, San Francisco, CA) to determine significant increases in fluorescence intensity, indicating increases in ROS generation.
2.5. Cytotoxicity Assay with Sytox Green
Cytotoxicity measurements were performed using Sytox green assay, as described previously (Latchoumycandane et al., 2005). Membrane-impermeable DNA dye Sytox green readily enters dying cells, resulting in increased fluorescence. The intensity of fluorescence is directly proportional to the amount of dead cells. After growing N27 cells in 24-well plates for 24 hr, cells were immediately exposed to 300 μM MPP+ in the presence of NADPH oxidase inhibitors (100-1000 μM AEBSF, 100-1000 μM apocynin and 3-30 μM DPI) in a 1 μM Sytox-containing growth media. After 24 hr, cytotoxic cell death was quantified by measuring DNA-bound Sytox green using a Gemini fluorescence microplate reader (Ex 485 nm and Em 538 nm) (Molecular Devices Corporation). Fluorescent images of Sytox-positive cells were taken after 24 hr exposure with a NIKON TE2000 microscope, and pictures were captured with a SPOT digital camera.
2.6. Caspase Enzymatic Activity Assay
Assessment of caspase activation was conducted as described previously (Kaul et al., 2003) using Ac-DEVD-AFC as the substrate for the enzymatic activity assay. The caspase-3-cleaved product was measured using a fluorescence plate reader (Ex 400 nm and Em 505 nm). Bradford protein assay was used for determination of protein concentration.
2.7. Western Blot Analysis
Western blot analysis was performed as described previously (Anantharam et al., 2002). Cells were collected and washed once with ice-cold PBS before lysis with buffer (protease inhibitors and 0.5% Triton X-100 in PBS). The membrane and cytosolic fractions containing equal amounts of protein were resolved on 10% SDS-PAGE and transferred onto nitrocellulose membrane. A standard Western blot procedure was followed for immunoblot with either polyclonal antibodies directed against gp91phox or p67phox NADPH oxidase subunits followed by treatment with HRP-conjugated secondary anti-rabbit antibody, and then ECL detection. The nitrocellulose membrane blots were also reprobed with monoclonal β-actin antibody to confirm equal protein loading.
2.8. DNA Fragmentation Assay
Cell Death Detection ELISA Plus Assay Kit (Roche Molecular Biochemicals, Indianapolis, IN) was used for analysis of DNA fragmentation by quantification of histone-associated low molecular weight DNA in the cytoplasm of cells (Anantharam et al., 2002). Briefly, cell pellets were lysed and subjected to centrifugation, and then the supernatants were incubated with the HRP-conjugated antibody-recognizing histones. Bound HRP-conjugates were assessed colorimetrically with ABTS as substrate at 405 nM using a plate reader (Spectramax, Molecular Devices). The optical density at 490 nm was used as a blank. The data were normalized to protein concentration.
2.9. Data Analysis
All data analysis was performed with Prism 4.0 software (GraphPad software, San Diego). One-way ANOVA was used for multiple comparisons. A significant difference was accepted if p<0.05.
3. Results
3.1. NADPH oxidase inhibitor significantly blocks MPP+-induced increases in ROS generation in a dose-dependent manner
Previously, we showed that ROS mediate dopaminergic toxicity, including MPP+-induced apoptotic cell death, in mesencephalic clonal neuronal N27 cells (Anantharam et al., 2002; Kaul et al., 2003; Kitazawa et al., 2003). We also demonstrated that MPP+ treatment induces a time- and dose-dependent increase in ROS production in N27 cells, and that SOD-mimetic MnTBAP almost completely reversed MPP+-induced increases in ROS generation (Anantharam et al., 2002; Kaul et al., 2003; Kitazawa et al., 2003). In this study, we examined whether NADPH oxidase inhibitors AEBSF, apocynin, and DPI attenuate MPP+-induced ROS generation. SOD was used to demonstrate specificity of MPP+-induced ROS, which can be scavenged by SOD treatment. We and others have shown addition of exogenous SOD or the cell permeable SOD mimetic MnTBAP attenuated both generation of ROS and apoptosis in neuronal cells (Drukarch et al., 1998; Choi et al., 1999; Luetjens et al., 2000; Kitazawa et al., 2001; Anantharam et al., 2002; Kaul et al., 2003); hence, we used SOD not as an NADPH oxidase inhibitor but as a ROS inhibitor. Using flow cytometric analysis with the ROS-sensitive fluorescence probe hydroethidine, we show that the NADPH oxidase inhibitor AEBSF significantly blocked MPP+-induced increases in ROS production. Figure 1A is a representative flow cytometric histogram showing an inhibitory effect of 500 μM AEBSF on 300 μm MPP+-induced ROS generation exposed for 45 min. Figure 1B shows a dose-dependent inhibition of MPP+-induced ROS generation by AEBSF. Exposure to 300 μM MPP+ for 45 min induced a 77% increase in ROS production compared to untreated control cells, whereas in the presence of 0.3 mM AEBSF, MPP+ induced an increase in ROS generation of only 58%, while MPP+ induced ROS generation was almost completely blocked in the presence of 0.5 and 1 mM AEBSF. Similarly, cotreatment with apocynin (300 μM), DPI (10 μM), and SOD (100 units) also significantly reduced MPP+-induced increases in ROS generation (Fig. 1C). Together, these data indicate that MPP+ treatment generates superoxide species in dopaminergic cells.
Fig. 1.


Effect of AEBSF on MPP+-induced ROS generation. (A) Representative flow cytometric histogram of dihydroethidine (DhEt) fluorescence in N27 treated cells with 300 μM MPP+. 10 μM DhEt was added to the cells and incubated for 15 min at 37°C in the dark, then DhEt loaded N27 cells were exposed to MPP+ in the presence or absence of 500 μM AEBSF and fluorescence intensity was measured at 45 min by flow cytometry. The shift of the curve to the right in MPP+-treated cells indicates an increase in ROS generation and shift to the left by AEBSF indicates its inhibitory effect on MPP+-induced ROS generation. The X-axis shows the log scale of fluorescence intensity and the Y-axis represents the cell count. (B) AEBSF blocks MPP+-induced increases in ROS generation in a dose-dependent manner. ROS generation was measured using flow cytometry and hydroethidine, a ROS-sensitive dye. Increases in fluorescence intensity indicate increases in ROS. Hydroethidine (10 μM) was added to the N27 cells and incubated for 15 min at 37°C in the dark. After incubation with hydroethidine, cells were pretreated with AEBSF (100-1000 μM), for an additional 15 min, and then exposed to 300 μM MPP+ for 45 min, as described in the methods section. Data represent the mean ± SEM (n=4). (C) NADPH oxidase inhibitors, apocynin and DPI, and free radical scavenger SOD suppress MPP+-induced increases in ROS generation. DhEt (10 μM) was added to the N27 cells and incubated for 15 min at 37°C in the dark. After incubation with hydroethidine, cells were pretreated with DPI (10 μM) or apocynin (300 μM) or SOD (100 units), for an additional 15 min, and then exposed to 300 μM MPP+ for 45 min, as described in the methods section. Data represent the mean ± SEM (n=3). *p<0.05 and **p<0.01 indicate significant differences compared to untreated control cells. ## p<0.01 indicates differences compared to MPP+-treated cells.
3.2. Identification of gp91phox and p67phox expression in mesencephalic neuronal N27 cells
Since the NADPH oxidase inhibitor AEBSF significantly blocked MPP+-induced superoxide radical formation, we determined whether N27 cells express NADPH oxidase using Western blot analyses. As shown in Fig. 2A, the Western blot analysis using anti-gp91phox antibody revealed a 91-kDa band corresponding to the gp91phox NADPH oxidase subunit. Further, the gp91phox protein was predominantly present in the plasma membrane fraction. Similarly, anti-p67phox antibody revealed a 67-kDa band corresponding to the p67phox NADPH oxidase subunit (Fig. 2B) and was mainly localized to the cytosol. A weak expression was noted in the membrane fraction, which may be attributed to basal activation of NADPH oxidase in the cells. These data strongly suggest that the major NADPH oxidase subunits are expressed in N27 dopaminergic neuronal cells, and that these cells could be used to evaluate the role of neuronal NADPH oxidase in MPP+-induced oxidative damage.
Fig. 2.

Western blot: (A) gp91phox expression. (B) p67phox expression. Membrane and cytosolic fractions were isolated from N27 cells as described in the methods section, and were separated by 10% SDS-polyacrylamide gel electrophoresis, transferred to nitrocellulose membrane, and gp91phox and p67phox were detected using a rabbit polyclonal antibody. A 91 kDa band corresponding to gp91phox was predominantly expressed in the membrane fraction and was absent in the cytosolic fraction, whereas a 67 kDa band corresponding to p67phox was mainly expressed in the cytosolic fraction. To confirm equal protein loading in each lane, the membranes were reprobed with β-actin antibody. The immunoblots were visualized using Amersham’s ECL detection agents.
3.3. NADPH oxidase inhibitors dose-dependently block MPP+-induced increases in cytotoxic cell death
In this experiment, we examined the effect of many NADPH oxidase inhibitors on MPP+-induced cell death by Sytox green fluorescence, which stains only dead/dying cells both qualitatively and quantitatively. N27 cells were exposed to 300 μM MPP+ for 24 hr in the presence or absence of structurally diverse NADPH oxidase inhibitors: AEBSF, apocynin, and DPI. Figure 3 is representative of untreated, MPP+-treated, and inhibitor-cotreated N27 cells at the end of a 24 hr treatment in phase-contrast (right panels) and Sytox FITC fluorescence imaging (left panels). An increase in the number of Sytox-positive green cells indicates the increase in cell death because the Sytox green dye permeates compromised cell membranes to stain nuclear chromatin. The number of Sytox-positive cells was many fold higher in MPP+-treated cells compared to untreated controls, or 500 μM AEBSF, 10 μM DPI, or 300 μM apocynin. Quantitative analysis of Sytox fluorescence using a fluorescence plate reader revealed a dose-dependent inhibition of MPP+-induced cytotoxic cell death by NADPH oxidase inhibitors (Fig. 4). Exposure to 300 μM MPP+ for 24 hr resulted in a 2.5-fold increase in the number of Sytox-positive cells compared to untreated control cells. As shown in Fig. 4A, 0.3 and 1 mM AEBSF blocked MPP+-induced increases in Sytox-positive cells by >50% and 95%, respectively, compared to MPP+-treated N27 cells. Similarly, apocynin at 0.3 and 1 mM almost completely prevented MPP+-induced increases in Sytox-positive cells (Fig. 4B). Furthermore, MPP+-induced increase in Sytox-positive cells was also significantly reduced in 10 μM and 30 μM DPI co-treated cells (Fig. 4C). Exposure of N27 cells to apocynin (0.1-1 mM), AEBSF (0.1-1mM), and DPI (3-30 μM) alone for 24 hr did not result in statistically significant increases in the number of Sytox-positive cells compared to untreated control cells (data not shown). Together, these data strongly suggest that structurally diverse NADPH oxidase-specific inhibitors significantly prevented MPP+-induced cytotoxic cell death. Based on results from this experiment, we used 500 μM AEBSF, 300 μM apocynin, and 10 μM DPI in subsequent experiments to further characterize their effect on MPP+-induced apoptotic cell death mechanisms.
Fig. 3.
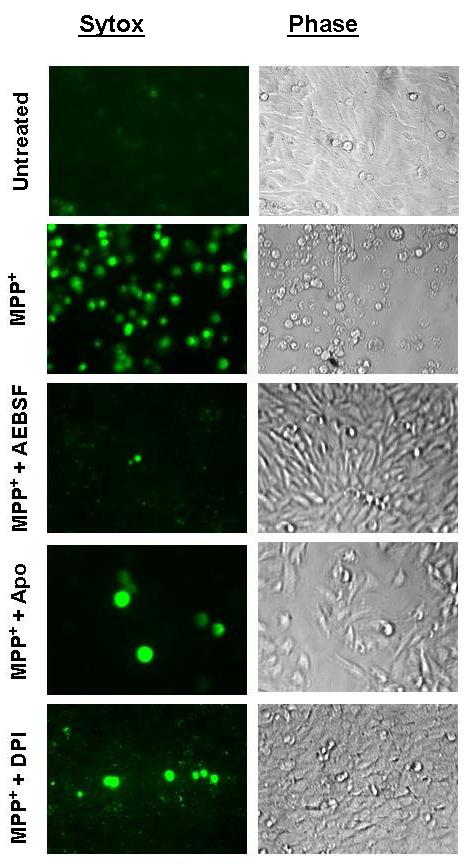
NADPH oxidase inhibitors block MPP+-induced increases in cytotoxic cell death. Sytox green assay was used to determine cytotoxicity, since Sytox is a DNA binding membrane impermeable dye that can only penetrate dying cells; increased fluorescence indicates cell death. N27 cells were treated with 300 μM MPP+ in the presence or absence of either 500 μM AEBSF, 300 μM apocynin, or 10 μM DPI in Sytox-containing medium, as described in the methods section. The two panels indicate representative fluorescence Sytox-positive images (left) and phase contrast images (right) for indicated treatments, and to demonstrate the extent of cytotoxic cell death.
Fig. 4.

NADPH oxidase inhibitors dose-dependently attenuate MPP+-induced increases in cytotoxic cell death. Quantitative Sytox green assay was used to determine the extent of cytotoxicity. N27 cells were treated with 300 μM MPP+ in the presence or absence of either 100-1000 μM AEBSF, 100-1000 μM apocynin, or 3-30 μM DPI in Sytox-containing medium, as described in the methods section. After 24 hr, cytotoxic cell death was quantified by measuring DNA-bound Sytox green in florescent microplate reader as described in the methods section. Data represent the mean ± SEM (n=6). **p<0.01 indicates significant differences compared to control cells and ##p<0.01 indicates significant differences compared to MPP+-treated cells.
3.4. NADPH oxidase inhibitors attenuate MPP+-induced increases in caspase-3 enzyme activity
Previously, we showed that ROS mediate MPP+-induced activation of multiple caspases, including caspase-9 and caspase-3, in neuronal N27 cells (Kaul et al., 2003; Kaul et al., 2005a). Here, we examined the effect of structurally diverse NADPH oxidase inhibitors on MPP+-induced increases in caspase-3 enzyme activity. N27 cells were exposed to 300 μM MPP+ for 24 hr in the presence or absence of AEBSF, apocynin, and DPI. Quantitative analysis of caspase-3 enzyme activation using fluorescence substrates revealed that MPP+-induced caspase-3 activation was significantly reduced in the presence of NADPH oxidase inhibitors (Fig. 5). Exposure to 300 μM MPP+ for 24 hr resulted in a 4-fold increase in caspase-3 activation compared to untreated control cells, whereas co-treatment with 500 μM AEBSF, 300 μM apocynin, and 10 μM DPI suppressed MPP+-induced increases in caspase-3 activation by 114%, 30%, and 38% of the control, respectively, compared to 300% in MPP+-treated N27 cells (Fig. 5). These results suggest that NADPH oxidase mediates MPP+-induced caspase-3 activation in N27 neuronal cells.
Fig. 5.

NADPH oxidase inhibitors attenuate MPP+-induced increases in caspase-3 enzyme activity. N27 cells were treated with 300 μM MPP+ in the presence or absence of either 500 μM AEBSF, 300 μM apocynin, or 10 μM DPI. Cells were lysed after 24 hr treatment and assayed for caspase-3 activity, as described in the methods section. Data represent the mean ± SEM (n=4). *p<0.01 and **p<0.01 indicate significant differences compared to untreated control cells. ## p<0.01 indicates significant differences compared to MPP+-treated cells.
3.5. NADPH oxidase inhibitors suppress N27 cells from MPP+-induced apoptotic cell death
Recently, we showed that oxidative stress-induced caspase-3-dependent proteolytic activation of PKCδ mediates MPP+-induced apoptotic cell death in neuronal N27 cells (Kaul et al., 2003; Kaul et al., 2005a). We used a quantitative DNA fragmentation ELISA assay to determine the effect of NADPH oxidase inhibitors on MPP+-induced apoptotic cell death. N27 cells were exposed to 300 μM MPP+ for 24 hr in the presence or absence of AEBSF, apocynin, and DPI. DNA fragmentation analysis revealed that NADPH oxidase inhibitors significantly suppressed MPP+-induced apoptotic cell death (Fig. 6). Exposure to 300 μM MPP+ for 24 hr resulted in a 2.5-fold increase in DNA fragmentation compared to untreated control cells. Co-treatment with 500 μM AEBSF, and 10 μM DPI suppressed MPP+-induced increases in DNA fragmentation by 63% and 22% of the control, respectively, compared to 143% in MPP+-treated N27 cells. 300 μM apocynin treatment completely prevented MPP+ induced DNA fragmentation. These results demonstrate that NADPH oxidase mediates MPP+-induced apoptotic cell death in dopaminergic neuronal cells.
Fig. 6.

NADPH oxidase inhibitors block N27 cells from MPP+-induced apoptotic cell death. DNA fragmentation, an indicator of cell death, was measured using a cell death detection ELISA kit. Briefly, N27 cells were treated with 300 μM MPP+ in the presence or absence of either 500 μM AEBSF, 300 μM apocynin or 10 μM DPI. Cells were lysed after 24 hr treatment and assayed for DNA fragmentation, as described in the methods section. Data represent the mean ± SEM (n=4). *p<0.01 and **p<0.01 indicate significant differences compared to untreated control cells. ## p<0.01 indicates significant differences compared to MPP+-treated cells.
4. Discussion
The present study demonstrates that pharmacological inhibitors of NADPH oxidase protect dopaminergic neuronal cells from MPP+-induced apoptotic cell death. Notably, NADPH oxidase inhibitors apocynin, DPI, and AEBSF effectively block MPP+-induced ROS production, caspase-3 activation, DNA fragmentation, and cytotoxic cell death in mesencephalic dopaminergic neuronal cells. N27 dopaminergic neuronal cells are devoid of glial cells and serve as a useful model to examine whether NADPH oxidase of neuronal origin contributes to MPP+-induced oxidative damage. To our knowledge, this is the first demonstration of the role of neuronal NADPH oxidase in MPP+-induced apoptotic cell death in dopaminergic cells.
Oxidative stress, caspases, and apoptotic cell death have all been implicated in Parkinson’s disease (Zigmond et al., 2002; Dawson and Dawson, 2003; Jenner, 2003; Greenamyre and Hastings, 2004; Maguire-Zeiss et al., 2005; Przedborski and Ischiropoulos, 2005). Both mitochondrial and extra-mitochondrial ROS have been shown to contribute to the degenerative process in cell cultures and animal models of PD. Several studies have shown plasma membrane NADPH oxidase is the primary enzyme responsible for generating extra-mitochondrial or cytosolic ROS (Serrano et al., 2003; Geiszt, 2006; Infanger et al., 2006; Sawada et al., 2006; Ushio-Fukai, 2006). NADPH oxidase and its protein subunits are mainly present in astrocytes and microglial cells (Serrano et al., 2003; Infanger et al., 2006; Takeya and Sumimoto, 2006). Several studies have provided evidence for the involvement of microglial NADPH oxidase in the inflammatory responses associated with the neurodegenerative process in MPTP-PD models (Beal, 2003; Serrano et al., 2003; Hald and Lotharius, 2005; Infanger et al., 2006; Sawada et al., 2006; Ushio-Fukai, 2006; Wersinger and Sidhu, 2006). Activated microglial cells exert quite different functions, including production of inflammatory cytokines, chemokines, and reactive superoxide ions. In the microglia, NADPH oxidase seems to be the primary enzyme responsible for synthesis of reactive superoxide ions, which in turn mediates functionally-relevant crosstalk between different inflammatory events, as well as within nigra-striatal systems (Serrano et al., 2003; Infanger et al., 2006; Sawada et al., 2006; Ushio-Fukai, 2006).
We recently demonstrated ROS is associated with MPP+-induced increases in mitochondrial dysfunction, cytochrome C release, caspase-3 activation, and apoptotic cell death in N27 cells (Kaul et al., 2003; Kaul et al., 2005a; Kaul et al., 2005b). MPP+-induced ROS production in these cells preceded changes in cytochrome C release, suggesting an extra-mitochondrial source for ROS (Kaul et al., 2003; Kaul et al., 2005a; Kaul et al., 2005b). In the present study, we demonstrate expression of the plasma membrane NADPH oxidase gp91phox and p67phox subunits in N27 cells by Western blot analysis. Thus, NADPH oxidase may be a principal source of MPP+-induced extra-mitochondrial ROS in N27 cells. Recent studies show other dopamine-producing cell lines, PC12 (Ibi et al., 2006) and SH-SY5Y (Nikolova et al., 2005), also express NADPH oxidase subunit proteins. In the present study, pretreatment with the NADPH oxidase-specific inhibitor AESBF blocked MPP+-induced ROS production in a dose-dependent manner, suggesting that NADPH oxidase is the enzyme responsible for ROS production in N27 cells. We show that AEBSF, apocynin, and DPI all blocked MPP+-induced cytotoxic cell death in a dose-dependent manner. AEBSF, DPI, and apocynin treatment also significantly attenuated MPP+-induced increases in caspase-3 activation in N27 cells. Co-treatment with apocynin, DPI, and AEBSF all rescued N27 cells from MPP+-induced DNA fragmentation, suggesting ROS derived from NADPH oxidase may mediate most of MPP+-induced apoptotic cell death in N27 cells. Upon comparison, AEBSF was most potent in blocking MPP+-induced ROS generation, whereas all three inhibitors, AEBSF, apocynin, and DPI, were equally potent in blocking MPP+-induced cytotoxicity. Apocynin and DPI were equally potent in blocking MPP+-induced caspase-3 activation and DNA fragmentation. Our results are in agreement with recent studies demonstrating apocynin and DPI promote the survival of primary striatal neurons (Ma and Zhou, 2006) and protect against glutamate-induced apoptosis in SHSY5Y cells (Nikolova et al., 2005) and amyloid-beta precursor peptide-induced cytotoxicity in primary neurons (Qin et al., 2006). These studies demonstrate that the neuroprotective properties of DPI and apocynin are not due to inhibition of microglial NADPH oxidase, but rather are due to inhibition of endogenously expressed NADPH oxidase. In animal studies, DPI and apocynin protect against global cerebral ischemia (Wang et al., 2006), and rotenone- (Gao et al., 2003a), paraquat- (Purisai et al., 2006), 6-OHDA- (Yasuhara et al., 2004), MPTP- (Gao et al., 2003b) and IFN-gamma/LPS- (Hirsch et al., 2003a) induced dopaminergic degeneration. In these studies, the neuroprotective properties of apocynin and DPI were associated with inhibition of microglial NADPH oxidase activity, but the role of neuronal NADPH oxidase was not investigated. The neuroprotective effects observed in this study with AEBSF, another NADPH oxidase inhibitor, are also in agreement with studies demonstrating AEBSF prevents NADPH oxidase-induced ROS generation, and cytotoxic and apoptotic cell death in non-neuronal tissues (He et al., 2005; Polytarchou and Papadimitriou, 2005).
We previously demonstrated the critical role of caspase-3-mediated proteolytic activation of PKCδ in in vitro and in vivo models of dopaminergic degeneration (Anantharam et al., 2002; Kanthasamy et al., 2003; Kaul et al., 2003; Kitazawa et al., 2003; Anantharam et al., 2004; Yang et al., 2004; Kaul et al., 2005a; Kaul et al., 2005b; Latchoumycandane et al., 2005; Kanthasamy et al., 2006). PKCδ was recently shown to play a central role in the regulation of NADPH oxidase activation in non-neuronal systems (Fan et al., 2005; Zhao et al., 2005; Iaccio et al., 2006; Waki et al., 2006) in addition to Rac1 and Rac2. In these studies, PKCδ regulated NADPH oxidase activity by up-regulation of NOX1 subunit, a homologue of the catalytic subunit gp91phox (NOX1), at the mRNA level via epidermal growth factor. PKCδ also regulates the phosphorylation and translocation of the p67phox subunit to the plasma membrane to activate the gp91phox catalytic subunit (Fan et al., 2005; Zhao et al., 2005; Iaccio et al., 2006; Waki et al., 2006). Taken together, PKCδ and NADPH oxidase possibly interact to accelerate oxidative damage in the nigral dopaminergic system. Future studies should address the relative contribution of neuronal and glial forms of NADPH oxidase in the oxidative damage of dopaminergic neurons using mixed glial and neuronal cultures.
In conclusion, we show that diverse NADPH oxidase inhibitors AEBSF, DPI, and apocynin significantly attenuate MPP+-induced ROS production, caspase-3 activation, and DNA fragmentation. These results suggest that extra-mitochondrial ROS produced by neuronal NADPH oxidase, in part, contribute to oxidative stress and apoptotic cell death in the dopaminergic cell line. Elucidating the role of neuronal NADPH oxidase in oxidative stress-induced dopaminergic cell models may help to define the mechanisms of oxidative damage in PD.
Acknowledgments
This work was supported by National Institute of Health (NIH) grants NS38644, ES10586 and NS45133. W. Eugene and Linda Lloyd Endowed Professorship to AGK is also acknowledged. The authors acknowledge Ms. Keri Henderson for her assistance in the preparation of this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anantharam V, Kitazawa M, Wagner J, Kaul S, Kanthasamy AG. Caspase-3-dependent proteolytic cleavage of protein kinase Cdelta is essential for oxidative stress-mediated dopaminergic cell death after exposure to methylcyclopentadienyl manganese tricarbonyl. J Neurosci. 2002;22:1738–51. doi: 10.1523/JNEUROSCI.22-05-01738.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantharam V, Kitazawa M, Latchoumycandane C, Kanthasamy A, Kanthasamy AG. Blockade of PKCdelta proteolytic activation by loss of function mutants rescues mesencephalic dopaminergic neurons from methylcyclopentadienyl manganese tricarbonyl (MMT)-induced apoptotic cell death. Ann N Y Acad Sci. 2004;1035:271–89. doi: 10.1196/annals.1332.017. [DOI] [PubMed] [Google Scholar]
- Andersen JK. Iron dysregulation and Parkinson’s disease. J Alzheimers Dis. 2004;6:S47–52. doi: 10.3233/jad-2004-6s602. [DOI] [PubMed] [Google Scholar]
- Beal MF. Mitochondria, oxidative damage, and inflammation in Parkinson’s disease. Ann N Y Acad Sci. 2003;991:120–31. doi: 10.1111/j.1749-6632.2003.tb07470.x. [DOI] [PubMed] [Google Scholar]
- Bove J, Prou D, Perier C, Przedborski S. Toxin-induced models of Parkinson’s disease. NeuroRx. 2005;2:484–94. doi: 10.1602/neurorx.2.3.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassarino DS, Parks JK, Parker WD, Jr., Bennett JP., Jr. The parkinsonian neurotoxin MPP+ opens the mitochondrial permeability transition pore and releases cytochrome c in isolated mitochondria via an oxidative mechanism. Biochim Biophys Acta. 1999;1453:49–62. doi: 10.1016/s0925-4439(98)00083-0. [DOI] [PubMed] [Google Scholar]
- Choi WS, Yoon SY, Oh TH, Choi EJ, O’Malley KL, Oh YJ. Two distinct mechanisms are involved in 6-hydroxydopamine- and MPP+-induced dopaminergic neuronal cell death: role of caspases, ROS, and JNK. J Neurosci Res. 1999;57:86–94. doi: 10.1002/(SICI)1097-4547(19990701)57:1<86::AID-JNR9>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson’s disease. Science. 2003;302:819–22. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- Di Monte DA. The environment and Parkinson’s disease: is the nigrostriatal system preferentially targeted by neurotoxins? Lancet Neurol. 2003;2:531–8. doi: 10.1016/s1474-4422(03)00501-5. [DOI] [PubMed] [Google Scholar]
- Drukarch B, Schepens E, Stoof JC, Langeveld CH, Van Muiswinkel FL. Astrocyte-enhanced neuronal survival is mediated by scavenging of extracellular reactive oxygen species. Free Radic Biol Med. 1998;25:217–20. doi: 10.1016/s0891-5849(98)00050-1. [DOI] [PubMed] [Google Scholar]
- Fan CY, Katsuyama M, Yabe-Nishimura C. PKCdelta mediates up-regulation of NOX1, a catalytic subunit of NADPH oxidase, via transactivation of the EGF receptor: possible involvement of PKCdelta in vascular hypertrophy. Biochem J. 2005;390:761–7. doi: 10.1042/BJ20050287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiskum G, Starkov A, Polster BM, Chinopoulos C. Mitochondrial mechanisms of neural cell death and neuroprotective interventions in Parkinson’s disease. Ann N Y Acad Sci. 2003;991:111–9. doi: 10.1111/j.1749-6632.2003.tb07469.x. [DOI] [PubMed] [Google Scholar]
- Gandhi S, Wood NW. Molecular pathogenesis of Parkinson’s disease. Hum Mol Genet. 2005;14:2749–55. [PubMed] [Google Scholar]
- Gao HM, Liu B, Hong JS. Critical role for microglial NADPH oxidase in rotenone-induced degeneration of dopaminergic neurons. J Neurosci. 2003a;23:6181–7. doi: 10.1523/JNEUROSCI.23-15-06181.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao HM, Liu B, Zhang W, Hong JS. Critical role of microglial NADPH oxidase-derived free radicals in the in vitro MPTP model of Parkinson’s disease. Faseb J. 2003b;17:1954–6. doi: 10.1096/fj.03-0109fje. [DOI] [PubMed] [Google Scholar]
- Geiszt M. NADPH oxidases: new kids on the block. Cardiovasc Res. 2006;71:289–99. doi: 10.1016/j.cardiores.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Greenamyre JT, Hastings TG. Biomedicine. Parkinson’s--divergent causes, convergent mechanisms. Science. 2004;304:1120–2. doi: 10.1126/science.1098966. [DOI] [PubMed] [Google Scholar]
- Hald A, Lotharius J. Oxidative stress and inflammation in Parkinson’s disease: is there a causal link? Exp Neurol. 2005;193:279–90. doi: 10.1016/j.expneurol.2005.01.013. [DOI] [PubMed] [Google Scholar]
- He L, Dinger B, Sanders K, Hoidal J, Obeso A, Stensaas L, Fidone S, Gonzalez C. Effect of p47phox gene deletion on ROS production and oxygen sensing in mouse carotid body chemoreceptor cells. Am J Physiol Lung Cell Mol Physiol. 2005;289:L916–24. doi: 10.1152/ajplung.00015.2005. [DOI] [PubMed] [Google Scholar]
- Hirsch EC, Breidert T, Rousselet E, Hunot S, Hartmann A, Michel PP. The role of glial reaction and inflammation in Parkinson’s disease. Ann N Y Acad Sci. 2003a;991:214–28. doi: 10.1111/j.1749-6632.2003.tb07478.x. [DOI] [PubMed] [Google Scholar]
- Hirsch EC, Hoglinger G, Rousselet E, Breidert T, Parain K, Feger J, Ruberg M, Prigent A, Cohen-Salmon C, Launay JM. Animal models of Parkinson’s disease in rodents induced by toxins: an update. J Neural Transm Suppl. 2003b:89–100. doi: 10.1007/978-3-7091-0643-3_6. [DOI] [PubMed] [Google Scholar]
- Iaccio A, Collinet C, Gesualdi NM, Ammendola R. Protein kinase C-alpha and -delta are required for NADPH oxidase activation in WKYMVm-stimulated IMR90 human fibroblasts. Arch Biochem Biophys. 2006 doi: 10.1016/j.abb.2006.11.009. [DOI] [PubMed] [Google Scholar]
- Ibi M, Katsuyama M, Fan C, Iwata K, Nishinaka T, Yokoyama T, Yabe-Nishimura C. NOX1/NADPH oxidase negatively regulates nerve growth factor-induced neurite outgrowth. Free Radic Biol Med. 2006;40:1785–95. doi: 10.1016/j.freeradbiomed.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Infanger DW, Sharma RV, Davisson RL. NADPH oxidases of the brain: distribution, regulation, and function. Antioxid Redox Signal. 2006;8:1583–96. doi: 10.1089/ars.2006.8.1583. [DOI] [PubMed] [Google Scholar]
- Ischiropoulos H, Beckman JS. Oxidative stress and nitration in neurodegeneration: cause, effect, or association? J Clin Invest. 2003;111:163–9. doi: 10.1172/JCI17638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner P. Oxidative stress in Parkinson’s disease. Ann Neurol. 2003;53(Suppl 3):S26–36. doi: 10.1002/ana.10483. discussion S36-8. [DOI] [PubMed] [Google Scholar]
- Kalivendi SV, Kotamraju S, Cunningham S, Shang T, Hillard CJ, Kalyanaraman B. 1-Methyl-4-phenylpyridinium (MPP+)-induced apoptosis and mitochondrial oxidant generation: role of transferrin-receptor-dependent iron and hydrogen peroxide. Biochem J. 2003;371:151–64. doi: 10.1042/BJ20021525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanthasamy AG, Kitazawa M, Kanthasamy A, Anantharam V. Role of proteolytic activation of protein kinase Cdelta in oxidative stress-induced apoptosis. Antioxid Redox Signal. 2003;5:609–20. doi: 10.1089/152308603770310275. [DOI] [PubMed] [Google Scholar]
- Kanthasamy AG, Anantharam V, Zhang D, Latchoumycandane C, Jin H, Kaul S, Kanthasamy A. A novel peptide inhibitor targeted to caspase-3 cleavage site of a proapoptotic kinase protein kinase C delta (PKCdelta) protects against dopaminergic neuronal degeneration in Parkinson’s disease models. Free Radic Biol Med. 2006;41:1578–89. doi: 10.1016/j.freeradbiomed.2006.08.016. [DOI] [PubMed] [Google Scholar]
- Kaul S, Anantharam V, Kanthasamy A, Kanthasamy AG. Wild-type alpha-synuclein interacts with pro-apoptotic proteins PKCdelta and BAD to protect dopaminergic neuronal cells against MPP+-induced apoptotic cell death. Brain Res Mol Brain Res. 2005a;139:137–52. doi: 10.1016/j.molbrainres.2005.05.022. [DOI] [PubMed] [Google Scholar]
- Kaul S, Kanthasamy A, Kitazawa M, Anantharam V, Kanthasamy AG. Caspase-3 dependent proteolytic activation of protein kinase C delta mediates and regulates 1-methyl-4-phenylpyridinium (MPP+)-induced apoptotic cell death in dopaminergic cells: relevance to oxidative stress in dopaminergic degeneration. Eur J Neurosci. 2003;18:1387–401. doi: 10.1046/j.1460-9568.2003.02864.x. [DOI] [PubMed] [Google Scholar]
- Kaul S, Anantharam V, Yang Y, Choi CJ, Kanthasamy A, Kanthasamy AG. Tyrosine phosphorylation regulates the proteolytic activation of protein kinase Cdelta in dopaminergic neuronal cells. J Biol Chem. 2005b;280:28721–30. doi: 10.1074/jbc.M501092200. [DOI] [PubMed] [Google Scholar]
- Kitazawa M, Anantharam V, Kanthasamy AG. Dieldrin-induced oxidative stress and neurochemical changes contribute to apoptopic cell death in dopaminergic cells. Free Radic Biol Med. 2001;31:1473–85. doi: 10.1016/s0891-5849(01)00726-2. [DOI] [PubMed] [Google Scholar]
- Kitazawa M, Anantharam V, Kanthasamy AG. Dieldrin induces apoptosis by promoting caspase-3-dependent proteolytic cleavage of protein kinase Cdelta in dopaminergic cells: relevance to oxidative stress and dopaminergic degeneration. Neuroscience. 2003;119:945–64. doi: 10.1016/s0306-4522(03)00226-4. [DOI] [PubMed] [Google Scholar]
- Koutsilieri E, Scheller C, Grunblatt E, Nara K, Li J, Riederer P. Free radicals in Parkinson’s disease. J Neurol. 2002;249(Suppl 2):II1–5. doi: 10.1007/s00415-002-1201-7. [DOI] [PubMed] [Google Scholar]
- Latchoumycandane C, Anantharam V, Kitazawa M, Yang Y, Kanthasamy A, Kanthasamy AG. Protein kinase Cdelta is a key downstream mediator of manganese-induced apoptosis in dopaminergic neuronal cells. J Pharmacol Exp Ther. 2005;313:46–55. doi: 10.1124/jpet.104.078469. [DOI] [PubMed] [Google Scholar]
- Love R. Mitochondria back in the spotlight in Parkinson’s disease. Lancet Neurol. 2004;3:326. doi: 10.1016/s1474-4422(04)00782-3. [DOI] [PubMed] [Google Scholar]
- Luetjens CM, Bui NT, Sengpiel B, Munstermann G, Poppe M, Krohn AJ, Bauerbach E, Krieglstein J, Prehn JH. Delayed mitochondrial dysfunction in excitotoxic neuron death: cytochrome c release and a secondary increase in superoxide production. J Neurosci. 2000;20:5715–23. doi: 10.1523/JNEUROSCI.20-15-05715.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Zhou J. Dopamine Promotes the Survival of Embryonic Striatal Cells: Involvement of Superoxide and Endogenous NADPH Oxidase. Neurochem Res. 2006 doi: 10.1007/s11064-006-9038-6. [DOI] [PubMed] [Google Scholar]
- Maguire-Zeiss KA, Short DW, Federoff HJ. Synuclein, dopamine and oxidative stress: co-conspirators in Parkinson’s disease? Brain Res Mol Brain Res. 2005;134:18–23. doi: 10.1016/j.molbrainres.2004.09.014. [DOI] [PubMed] [Google Scholar]
- Mancuso C, Scapagini G, Curro D, Giuffrida Stella AM, De Marco C, Butterfield DA, Calabrese V. Mitochondrial dysfunction, free radical generation and cellular stress response in neurodegenerative disorders. Front Biosci. 2007;12:1107–23. doi: 10.2741/2130. [DOI] [PubMed] [Google Scholar]
- Mandel S, Maor G, Youdim MB. Iron and alpha-synuclein in the substantia nigra of MPTP-treated mice: effect of neuroprotective drugs R-apomorphine and green tea polyphenol (-)-epigallocatechin-3-gallate. J Mol Neurosci. 2004;24:401–16. doi: 10.1385/JMN:24:3:401. [DOI] [PubMed] [Google Scholar]
- McCormack AL, Atienza JG, Langston JW, Di Monte DA. Decreased susceptibility to oxidative stress underlies the resistance of specific dopaminergic cell populations to paraquat-induced degeneration. Neuroscience. 2006;141:929–37. doi: 10.1016/j.neuroscience.2006.03.069. [DOI] [PubMed] [Google Scholar]
- Nikolova S, Lee YS, Kim JA. Rac1-NADPH oxidase-regulated generation of reactive oxygen species mediates glutamate-induced apoptosis in SH-SY5Y human neuroblastoma cells. Free Radic Res. 2005;39:1295–304. doi: 10.1080/10715760500176866. [DOI] [PubMed] [Google Scholar]
- Obata T. Role of hydroxyl radical formation in neurotoxicity as revealed by in vivo free radical trapping. Toxicol Lett. 2002;132:83–93. doi: 10.1016/s0378-4274(02)00076-0. [DOI] [PubMed] [Google Scholar]
- Polytarchou C, Papadimitriou E. Antioxidants inhibit human endothelial cell functions through down-regulation of endothelial nitric oxide synthase activity. Eur J Pharmacol. 2005;510:31–8. doi: 10.1016/j.ejphar.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Przedborski S. Pathogenesis of nigral cell death in Parkinson’s disease. Parkinsonism Relat Disord. 2005;11(Suppl 1):S3–7. doi: 10.1016/j.parkreldis.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Ischiropoulos H. Reactive oxygen and nitrogen species: weapons of neuronal destruction in models of Parkinson’s disease. Antioxid Redox Signal. 2005;7:685–93. doi: 10.1089/ars.2005.7.685. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Tieu K, Perier C, Vila M. MPTP as a mitochondrial neurotoxic model of Parkinson’s disease. J Bioenerg Biomembr. 2004;36:375–9. doi: 10.1023/B:JOBB.0000041771.66775.d5. [DOI] [PubMed] [Google Scholar]
- Purisai MG, McCormack AL, Cumine S, Li J, Isla MZ, Di Monte DA. Microglial activation as a priming event leading to paraquat-induced dopaminergic cell degeneration. Neurobiol Dis. 2006 doi: 10.1016/j.nbd.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin B, Cartier L, Dubois-Dauphin M, Li B, Serrander L, Krause KH. A key role for the microglial NADPH oxidase in APP-dependent killing of neurons. Neurobiol Aging. 2006;27:1577–87. doi: 10.1016/j.neurobiolaging.2005.09.036. [DOI] [PubMed] [Google Scholar]
- Reis K, Halldin J, Fernaeus S, Pettersson C, Land T. NADPH oxidase inhibitor diphenyliodonium abolishes lipopolysaccharide-induced down-regulation of transferrin receptor expression in N2a and BV-2 cells. J Neurosci Res. 2006;84:1047–52. doi: 10.1002/jnr.21005. [DOI] [PubMed] [Google Scholar]
- Sawada M, Imamura K, Nagatsu T. Role of cytokines in inflammatory process in Parkinson’s disease. J Neural Transm Suppl. 2006:373–81. doi: 10.1007/978-3-211-45295-0_57. [DOI] [PubMed] [Google Scholar]
- Schapira AH. Pathogenesis of Parkinson’s disease. Baillieres Clin Neurol. 1997;6:15–36. [PubMed] [Google Scholar]
- Schneider B, Mutel V, Pietri M, Ermonval M, Mouillet-Richard S, Kellermann O. NADPH oxidase and extracellular regulated kinases 1/2 are targets of prion protein signaling in neuronal and nonneuronal cells. Proc Natl Acad Sci U S A. 2003;100:13326–31. doi: 10.1073/pnas.2235648100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz JB, Falkenburger BH. Neuronal pathology in Parkinson’s disease. Cell Tissue Res. 2004;318:135–47. doi: 10.1007/s00441-004-0954-y. [DOI] [PubMed] [Google Scholar]
- Serrano F, Kolluri NS, Wientjes FB, Card JP, Klann E. NADPH oxidase immunoreactivity in the mouse brain. Brain Res. 2003;988:193–8. doi: 10.1016/s0006-8993(03)03364-x. [DOI] [PubMed] [Google Scholar]
- Sherer TB, Betarbet R, Greenamyre JT. Pathogenesis of Parkinson’s disease. Curr Opin Investig Drugs. 2001;2:657–62. [PubMed] [Google Scholar]
- Sidhu A, Wersinger C, Moussa CE, Vernier P. The role of alpha-synuclein in both neuroprotection and neurodegeneration. Ann N Y Acad Sci. 2004;1035:250–70. doi: 10.1196/annals.1332.016. [DOI] [PubMed] [Google Scholar]
- Smeyne RJ, Jackson-Lewis V. The MPTP model of Parkinson’s disease. Brain Res Mol Brain Res. 2005;134:57–66. doi: 10.1016/j.molbrainres.2004.09.017. [DOI] [PubMed] [Google Scholar]
- Sun F, Anantharam V, Zhang D, Latchoumycandane C, Kanthasamy A, Kanthasamy AG. Proteasome inhibitor MG-132 induces dopaminergic degeneration in cell culture and animal models. Neurotoxicology. 2006;27:807–15. doi: 10.1016/j.neuro.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Sun GY, Horrocks LA, Farooqui AA. The roles of NADPH oxidase and phospholipases A(2) in oxidative and inflammatory responses in neurodegenerative diseases. J Neurochem. 2007 doi: 10.1111/j.1471-4159.2007.04670.x. [DOI] [PubMed] [Google Scholar]
- Takeya R, Sumimoto H. Regulation of novel superoxide-producing NAD(P)H oxidases. Antioxid Redox Signal. 2006;8:1523–32. doi: 10.1089/ars.2006.8.1523. [DOI] [PubMed] [Google Scholar]
- Thiruchelvam M, McCormack A, Richfield EK, Baggs RB, Tank AW, Di Monte DA, Cory-Slechta DA. Age-related irreversible progressive nigrostriatal dopaminergic neurotoxicity in the paraquat and maneb model of the Parkinson’s disease phenotype. Eur J Neurosci. 2003;18:589–600. doi: 10.1046/j.1460-9568.2003.02781.x. [DOI] [PubMed] [Google Scholar]
- Tikka TM, Vartiainen NE, Goldsteins G, Oja SS, Andersen PM, Marklund SL, Koistinaho J. Minocycline prevents neurotoxicity induced by cerebrospinal fluid from patients with motor neurone disease. Brain. 2002;125:722–31. doi: 10.1093/brain/awf068. [DOI] [PubMed] [Google Scholar]
- Ushio-Fukai M. Localizing NADPH oxidase-derived ROS. Sci STKE. 2006;349:re8. doi: 10.1126/stke.3492006re8. [DOI] [PubMed] [Google Scholar]
- Waki K, Inanami O, Yamamori T, Nagahata H, Kuwabara M. Involvement of protein kinase Cdelta in the activation of NADPH oxidase and the phagocytosis of neutrophils. Free Radic Res. 2006;40:359–67. doi: 10.1080/10715760500539121. [DOI] [PubMed] [Google Scholar]
- Wang Q, Tompkins KD, Simonyi A, Korthuis RJ, Sun AY, Sun GY. Apocynin protects against global cerebral ischemia-reperfusion-induced oxidative stress and injury in the gerbil hippocampus. Brain Res. 2006;1090:182–9. doi: 10.1016/j.brainres.2006.03.060. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Himeda T, Araki T. Mechanisms of MPTP toxicity and their implications for therapy of Parkinson’s disease. Med Sci Monit. 2005;11:RA17–23. [PubMed] [Google Scholar]
- Wersinger C, Sidhu A. An inflammatory pathomechanism for Parkinson’s disease? Curr Med Chem. 2006;13:591–602. doi: 10.2174/092986706776055760. [DOI] [PubMed] [Google Scholar]
- Yang Y, Kaul S, Zhang D, Anantharam V, Kanthasamy AG. Suppression of caspase-3-dependent proteolytic activation of protein kinase C delta by small interfering RNA prevents MPP+-induced dopaminergic degeneration. Mol Cell Neurosci. 2004;25:406–21. doi: 10.1016/j.mcn.2003.11.011. [DOI] [PubMed] [Google Scholar]
- Yasuhara T, Shingo T, Kobayashi K, Takeuchi A, Yano A, Muraoka K, Matsui T, Miyoshi Y, Hamada H, Date I. Neuroprotective effects of vascular endothelial growth factor (VEGF) upon dopaminergic neurons in a rat model of Parkinson’s disease. Eur J Neurosci. 2004;19:1494–504. doi: 10.1111/j.1460-9568.2004.03254.x. [DOI] [PubMed] [Google Scholar]
- Youdim MB, Stephenson G, Ben Shachar D. Ironing iron out in Parkinson’s disease and other neurodegenerative diseases with iron chelators: a lesson from 6-hydroxydopamine and iron chelators, desferal and VK-28. Ann N Y Acad Sci. 2004;1012:306–25. doi: 10.1196/annals.1306.025. [DOI] [PubMed] [Google Scholar]
- Zhao X, Xu B, Bhattacharjee A, Oldfield CM, Wientjes FB, Feldman GM, Cathcart MK. Protein kinase Cdelta regulates p67phox phosphorylation in human monocytes. J Leukoc Biol. 2005;77:414–20. doi: 10.1189/jlb.0504284. [DOI] [PubMed] [Google Scholar]
- Zigmond MJ, Hastings TG, Perez RG. Increased dopamine turnover after partial loss of dopaminergic neurons: compensation or toxicity? Parkinsonism Relat Disord. 2002;8:389–93. doi: 10.1016/s1353-8020(02)00019-6. [DOI] [PubMed] [Google Scholar]