

Abstract
Despite extensive study, there is little experimental information available as to which of the deoxyribose hydrogen atoms of duplex DNA reacts most with the hydroxyl radical. To investigate this question, we prepared a set of double-stranded DNA molecules in which deuterium had been incorporated specifically at each position in the deoxyribose of one of the four nucleotides. We then measured deuterium kinetic isotope effects on the rate of cleavage of DNA by the hydroxyl radical. These experiments demonstrate that the hydroxyl radical reacts with the various hydrogen atoms of the deoxyribose in the order 5′ H > 4′ H > 3′ H ≈ 2′ H ≈ 1′ H. This order of reactivity parallels the exposure to solvent of the deoxyribose hydrogens. Our work therefore reveals the structural basis of the reaction of the hydroxyl radical with DNA. These results also provide information on the mechanism of DNA damage caused by ionizing radiation as well as atomic-level detail for the interpretation of hydroxyl radical footprints of DNA-protein complexes and chemical probe experiments on the structure of RNA and DNA in solution.
The hydroxyl radical (⋅OH), the quintessential reactive oxygen species, is the mediator of much of the DNA damage caused by ionizing radiation (1). This damage includes strand breaks, which are initiated by abstraction of a deoxyribose hydrogen atom by the hydroxyl radical. DNA strand breaks induced by the hydroxyl radical also form the basis of a widely used method for making footprints of DNA–protein complexes (2, 3) and for studying the structure of DNA (4) and RNA (5) in solution. The key experimental advantage of the hydroxyl radical as a chemical probe is that it effects DNA cleavage with no base- or sequence-specificity (6–8). The hydroxyl radical produces highly detailed footprints that yield information about DNA structure (4, 7) and protein–DNA interactions (3, 8, 9) at single-nucleotide resolution.
Mechanistic information on the reaction of the hydroxyl radical with nucleic acids will benefit our understanding of radiation damage to DNA as well as the interpretation of chemical probe experiments. The extensive literature on the radiation chemistry of DNA (1) is a rich source of mechanistic possibilities. Not surprisingly, because of the high reactivity of the hydroxyl radical, a wide spectrum of products has been detected on treatment of the constituents of DNA (nucleic bases, nucleosides, nucleotides, or simple-sequence single-stranded DNA, for example) with ionizing radiation (1). It has been more difficult to conduct similarly detailed experiments on the biologically relevant duplex form of DNA. It is not hard to conceive, though, that the hydroxyl radical might react in a different manner with double-stranded DNA compared with simpler nucleic acid systems because the shape of the double helix would strongly influence the accessibility of the various C—H bonds in DNA.
Until now, the extent of cleavage at a particular nucleotide in a hydroxyl radical footprinting experiment only could be interpreted at the level of the entire deoxyribose because no information was available on which of the several sugar hydrogens of duplex DNA reacts most with the hydroxyl radical. Unlike DNA-cleaving drug molecules (10–12) and metal complexes (13, 14), which show a marked preference for reacting with a particular deoxyribose hydrogen atom, the hydroxyl radical, because of its high reactivity, is expected to be capable of abstracting any of the deoxyribose hydrogens. If the relative reactivity of the hydroxyl radical toward each deoxyribose hydrogen atom were known, a new level of structural detail would be gained in the interpretation of hydroxyl radical footprints.
This desire for more detailed knowledge of the mechanism of cleavage is apparent in the recent literature. For example, in their report of the crystal structure of a fragment of the Tetrahymena intervening sequence ribozyme, Doudna, Cech, and colleagues (15) compared the results of a previous hydroxyl radical cleavage study on this RNA with the solvent-accessible surface area calculated from the x-ray structure. The agreement was excellent. This comparison made use of the surface areas of the C4′ hydrogens of the ribose sugars, the generally quoted site of preferred hydroxyl radical reactivity (15). However, as demonstrated in the present paper, it will be important to reexamine assumptions regarding the reactivity of the hydroxyl radical toward particular sugar hydrogens in RNA or DNA that have been made in the absence of direct experimental determinations.
The experiments we report here had two aims: to determine which of the several deoxyribose hydrogens of duplex DNA react with the hydroxyl radical and to estimate the contributions of the individual deoxyribose hydrogens to the overall cleavage process. Our strategy involved the study of DNA molecules into which had been incorporated specifically deuterated nucleotides (12). A particular DNA molecule would, for example, have all its thymidines specifically monodeuterated at the C4′ position of the deoxyribose. Protium would be present at all other positions in thymidine and at all positions in the other three nucleotides. We compared the extent of cleavage at the deuterated nucleotides with cleavage at surrounding nondeuterated nucleotides and with a control DNA molecule containing no deuterium substitution. We anticipated that, if the hydroxyl radical abstracted a particular deoxyribose hydrogen, the corresponding deuterated nucleotide would exhibit a kinetic isotope effect (kH/kD > 1) for the cleavage reaction, and cleavage would be reduced at the deuterated nucleotide. By this means we would determine which deoxyribose hydrogen atoms were abstracted by the hydroxyl radical in the initial step of the strand breakage reaction.
MATERIALS AND METHODS
Preparation of Specifically Deuterated DNA.
[5′-dideuterio]deoxycytidine and [1′-deuterio]deoxycytidine were generous gifts from Craig A. Townsend (The Johns Hopkins University). The isotopic incorporation was >95% (11). [4′-deuterio]deoxythymidine triphosphate was the kind gift of John W. Kozarich (University of Maryland) and JoAnne Stubbe (Massachusetts Institute of Technology) (16). We synthesized [3′-deuterio]deoxyadenosine triphosphate and [2′-dideuterio]deoxyadenosine triphosphate by using published procedures (17–19). Mass spectroscopic measurements showed deuterium incorporation of >98% at the 3′ position and 89% dideuteration at the 2′ position.
For incorporation into a DNA molecule, a deuterated nucleoside triphosphate was mixed with the other three natural nucleoside triphosphates for use in a PCR. The 19-bp DNA sequence to be studied had been inserted between the BamHI and HindIII restriction sites of plasmid pUC18. The product of the PCR was cut by these two restriction endonucleases and radiolabeled at either the 5′ end or the 3′ end by standard methods.
DNA Cleavage and Electrophoretic Analysis.
The hydroxyl radical cleavage reaction was initiated by the addition of 1 μl of a solution containing 1 mM (NH4)2Fe(SO4)2⋅6H2O/2 mM Na2EDTA, 1 μl of 10 mM ascorbate, and 1 μl of 0.3% H2O2, to 10,000 dpm of radiolabeled DNA in 7 μl of 10 mM Tris⋅EDTA buffer (pH 8.0). The reaction was run at 25°C. The reaction was quenched after 2 min by the addition of 7 μl of 100 mM thiourea. The reaction mixture was either dried down in a SpeedVac (Savant) concentrator or was ethanol-precipitated. The DNA pellet was dissolved in 4 μl of formamide-containing dye mixture, was denatured at 90°C for 3 min, was quenched on ice, and was loaded on a 25% (23.5 g acrylamide, 1.25 g bisacrylamide) denaturing polyacrylamide gel. Electrophoresis was performed at 65 W for 4.5 hr. The gel was exposed to an imaging phosphor plate, which subsequently was scanned by a Model 400E PhosphorImager (Molecular Dynamics).
Measurement of Kinetic Isotope Effects.
We used the computer software package gelexplorer (20) to deconvolute and measure the integrated intensities of gel bands. We used the following procedure to calculate the apparent kinetic isotope effect on the production of the 3′-phosphate and 3′-phosphoglycolate products. The sum of the integrated intensities of the bands representing the phosphate and phosphoglycolate products for all of the nondeuterated nucleotides in a deuterated DNA sample was normalized to the corresponding sum in the all-protio (control) DNA sample. The resulting normalization factor was applied to the integrals of bands representing deuterated nucleotides, to correct for loading errors. The apparent kinetic isotope effect was calculated as the ratio of the integral of a phosphate or phosphoglycolate band in the control (protio) sample to the corresponding normalized band integral in the sample of deuterated DNA. The apparent kinetic isotope effects we report in Table 1 were obtained by averaging the isotope effects for the several deuterated nucleotides in the DNA sequence we studied.
Table 1.
Ratios of amounts of observed products of hydroxyl radical cleavage of control (all-protio) vs. deuterated DNA (H/D)
| Position deuterated | 3′-Phosphate | 3′-Phosphoglycolate* | 5′-Aldehyde* |
|---|---|---|---|
| 5′ | 1.67 ± 0.15 | 2.6 ± 0.5 | |
| 4′ | 1.09 ± 0.05 | 2.1 ± 0.3 | |
| 3′ | 1.14 ± 0.05 | ||
| 2′ | 1.11 ± 0.02 | ||
| 1′ | 1.08 ± 0.03 |
These products are produced only by abstraction of a particular deoxyribose hydrogen (see text), and so the corresponding entries represent intrinsic isotope effects. All other entries represent apparent isotope effects because several initial hydrogen abstraction events can in principle lead to production of a strand terminated by a 3′-phosphate (26).
Calculation of Solvent-Accessible Surface Area.
The solvent-accessible surface area of each deoxyribose hydrogen of the central eight bp of the self-complementary decamer duplex d(CCAACGTTGG) (21) was calculated by using the Lee and Richards algorithm (22) as implemented in the charmm module of quanta (Molecular Simulations, San Diego). For each nucleotide, the percent contribution of each deoxyribose hydrogen to the total surface area of the deoxyribose hydrogens of that nucleotide was evaluated. Reported in Table 2 is the percent accessibility for each deoxyribose hydrogen, averaged over the eight central bp of the decanucleotide duplex (21).
Table 2.
Reactivities of deoxyribose hydrogens vs. solvent accessibility
| 5′ + 5" | 4′ | 3′ | 2′ + 2" | 1′ | |
|---|---|---|---|---|---|
| % of reaction with hydroxyl radical* | 57 ± 10 | 22 ± 6 | 17 ± 5 | 13 ± 3 | 11 ± 4 |
| % of solvent-accessible surface area | 46 | 28 | 14 | 11 | 1 |
Calculated by using Eq. 6.
RESULTS AND DISCUSSION
Deuterium Kinetic Isotope Effect Experiments.
Nucleoside triphosphates containing specific deuterium substitutions were synthesized chemically and then were incorporated into DNA by PCR. The resulting oligonucleotide duplex was subjected to hydroxyl radical cleavage. We used the [Fe(EDTA)]2−/H2O2/ascorbate system (23) to generate the hydroxyl radical in solution, as is done commonly for hydroxyl radical footprinting experiments (2–4, 7–9). The products of the cleavage reaction were subjected to denaturing gel electrophoresis and were analyzed in a manner analogous to previous studies of DNA cleavage by bleomycin (16). Hydroxyl radical cleavage of DNA produces lesions in which strands have 3′-phosphate, 3′-phosphoglycolate (24), 5′-phosphate, and 5′-aldehyde termini (Fig. 1).
Figure 1.

(A) Numbering scheme for the carbon atoms of deoxyribose. (B) Structures of the products of hydroxyl radical-mediated DNA cleavage. The asterisk indicates the position of the 32P radiolabel. (Left) Structure of the 3′ end of the DNA strand at the site of cleavage (indicated by the arrow). (Right) Structure of the 5′ end of the DNA strand at the site of cleavage (indicated by the arrow).
We indeed observed reduced cleavage at deuterated nucleotides, compared with the all-protio control samples (Fig. 2). Deuteration at C1′, C2′, or C3′ leads to a small decrease in the production of the 3′-phosphate product. A larger effect is seen for deuteration at C4′. In this case, yields of both the 3′-phosphate and the 3′-phosphoglycolate products are reduced. By far the largest effect on the cleavage reaction is observed for deuteration at C5′. The amount of the 3′-phosphate product is decreased by a factor of ≈½.
Figure 2.

Comparison of hydroxyl radical cleavage patterns of control (all-protio) vs. deuterated DNA radiolabeled with 32P at the 5′ end. Broken line, control DNA. Solid line, deuterated DNA. Deuterated nucleotides are indicated by unfilled labels. The major cleavage product seen at each nucleotide is a strand terminated by a 3′-phosphate (see Fig. 1B); the minor product is the 3′-phosphoglycolate-terminated strand. Phosphoglycolate bands are marked in B by asterisks. (A) Control DNA vs. DNA containing 5′-dideuterated deoxycytidine. (B) Control DNA vs. DNA containing 4′-deuterated deoxythymidine. (C) Control DNA vs. DNA containing 3′-deuterated deoxyadenosine. (D) Control DNA vs. DNA containing 2′-dideuterated deoxyadenosine. (E) Control DNA vs. DNA containing 1′-deuterated deoxycytidine.
There are two possible explanations for our observation of different effects on the product yield for deuteration at the various deoxyribose positions. One possibility is that the intrinsic kinetic isotope effect is different for each C—H bond in the sugar. Because the hydroxyl radical is so reactive and nonselective, though, it is more likely that all deoxyribose positions would exhibit nearly the same intrinsic kinetic isotope effect. Another explanation of our results is that the effect on product yield that we observe reflects the extent of reaction of the hydroxyl radical with that deoxyribose hydrogen atom, which in turn reflects the accessibility of that hydrogen atom to the hydroxyl radical diffusing to the DNA from bulk solvent. This explanation is in good qualitative agreement with our data because the values of the apparent isotope effects we observe correspond with how exposed the site is to solvent. The 5′ hydrogens, for example, are situated on the outside of the DNA backbone, and deuteration at C5′ leads to the largest effect on cleavage.
Determination of Intrinsic Kinetic Isotope Effects.
To test this idea quantitatively, we took advantage of the chemistry that the deoxyribose undergoes after hydrogen abstraction (25, 26). The ultimate product that is observed subsequent to initial hydrogen atom abstraction from almost all positions of a DNA sugar is a DNA strand with a phosphate end that results from loss of the nucleoside that was attacked by the radical (25, 26). So the major species we observe on a denaturing gel, a DNA strand having a phosphate end at the site of cleavage (see Fig. 1 and Fig. 2), in principle could result from initial abstraction of any of several different deoxyribose hydrogens. However, it has been shown that abstraction of the C4′ hydrogen gives rise, under aerobic conditions, to a chemically unique product, a DNA strand terminated at the 3′ end with a phosphoglycolate moiety (27). 3′-phosphoglycolate is produced by bleomycin-mediated cleavage of DNA (27), for example, and bleomycin has been shown by other experiments to react exclusively with the C4′ hydrogen atom of duplex B-form DNA (12, 16, 25). No other hydrogen abstraction event yields the phosphoglycolate product. Phosphoglycolate-terminated DNA runs on a denaturing gel with a distinct mobility compared with the corresponding phosphate-ended DNA strand (6, 24), so it can be quantified individually. We also detected phosphoglycolate-terminated DNA as a product in our hydroxyl radical DNA cleavage patterns (Fig. 2). We thus were able to measure the intrinsic kinetic isotope effect for hydroxyl radical abstraction of the C4′ hydrogen atom.
A similar situation obtains for abstraction of a C5′ hydrogen atom: A unique product, a 5′-aldehyde-terminated strand, which can be separated on a gel from phosphate-terminated DNA, is produced (10). We therefore measured the intrinsic kinetic isotope effect for C5′ hydrogen abstraction by quantifying the 5′-aldehyde product for 5′-dideuterio vs. protio DNA (Fig. 3). These two intrinsic isotope effects are listed in Table 1, along with the apparent kinetic isotope effects we measured for deuteration at other positions.
Figure 3.

Comparison of hydroxyl radical cleavage patterns of control (all-protio) vs. 5′-dideuterated DNA radiolabeled with 32P at the 3′ end. Broken line, control DNA. Solid line, deuterated DNA. 5′-dideuterated deoxycytidine nucleotides are indicated by unfilled labels. The major cleavage product seen at each nucleotide is a strand terminated by a 5′-phosphate; the minor product is the 5′-aldehyde-terminated strand (see Fig. 1B). An aldehyde-terminated strand exhibits an unusually slow migration on the gel; previous work has established that there is a 2–3 nucleotide retardation in mobility compared with the corresponding Maxam–Gilbert product (10). Arrows show the correspondence between a 5′-phosphate-terminated product and the 5′-aldehyde-terminated product that is produced by reaction with that nucleotide.
The isotope effects we measure for the unique products generated by abstraction of hydrogens from C4′ (kH/kD = 2.1 ± 0.3) and C5′ (kH/kD = 2.6 ± 0.5) are similar in magnitude to each other and reasonable in value based on previously measured deuterium kinetic isotope effects for abstraction of a hydrogen atom from a C—H bond by the hydroxyl radical (28). We note that the observed kinetic isotope effect for the 5′ hydrogen, 2.6 ± 0.5, is likely the product of a primary and a secondary isotope effect because the 5′ carbon of deoxyribose is dideuterated predominantly in our experimental samples.
Calculation of the Extent of Hydrogen Abstraction from each Position in Deoxyribose.
We have used our measurements of the intrinsic and apparent isotope effects (see Table 1) to estimate the relative extent of abstraction of each deoxyribose hydrogen atom. As an example of our approach, we present the calculation for 5′ hydrogen abstraction.
Let the extent of cleavage at each nucleotide be H, which is the sum of the cleavages induced by abstraction of each deoxyribose hydrogen atom, hn (n = 1–5)
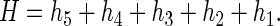 |
1 |
The fraction of the total amount of cleavage caused by abstraction of a 5′ hydrogen is then h5/H.
Similarly, the total extent of cleavage at a 5′-dideuterated nucleotide is
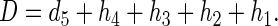 |
2 |
Then
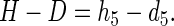 |
3 |
Let I be the (intrinsic) deuterium kinetic isotope effect for abstraction of a 5′-H atom by the hydroxyl radical. Then, for a 5′-dideuterated nucleotide,
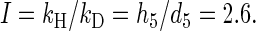 |
4 |
Substituting,
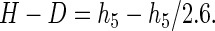 |
5 |
The fractional contribution to the total cleavage caused by 5′ hydrogen abstraction is
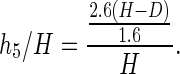 |
6 |
Because H and D, the total amount of cleavage at a particular nucleotide for the nondeuterated and deuterated samples, respectively, are experimentally measured quantities, the fractional contribution to cleavage of the 5′ deoxyribose hydrogen atom can be computed. We used similar schemes to calculate the fraction of cleavage caused by abstraction of each of the other sugar hydrogens (Table 2). These results show that, although the hydroxyl radical mediates DNA cleavage by initial abstraction of hydrogen from any of the sugar carbons, it exhibits preferential reaction with particular hydrogens. This preference is in the order 5′ H > 4′ H > 3′ H ≈ 2′ H ≈ 1′ H.
Structural Features of DNA That Affect the Reactivity of the Hydroxyl Radical.
It is immediately apparent from our calculations that the reactivity of the hydroxyl radical toward the different hydrogens of the deoxyribose varies widely. To see whether this trend in reactivity directly reflects some aspect of the structure of DNA, we calculated the solvent-accessible surface areas of the deoxyribose hydrogens from the high-resolution crystal structure of a DNA decanucleotide duplex in the B-form (21). Remarkably, the trends in reactivity toward the hydroxyl radical closely track the accessibility to solvent of the various deoxyribose hydrogens (see Table 2, second line). C5′-H and C4′-H are the primary sites of attack, and these are the most solvent-exposed hydrogen atoms in the DNA backbone. The other deoxyribose positions also show a high degree of correlation between their share of the solvent-accessible surface area and their reactivity toward the hydroxyl radical. This relationship is plotted in Fig. 4. Although a three-dimensional structure is not available for the particular DNA sequence we studied by our isotope effect experiments, the relationship between structure and reactivity shown in Fig. 4 suggests that the results of a hydroxyl radical cleavage experiment can be translated directly and quantitatively into an important structural feature of DNA: the accessibility to solvent of the DNA backbone.
Figure 4.

Solvent accessibility vs. reactivity toward the hydroxyl radical. The data in Table 2 are plotted, and the best linear fit to the data is shown. Above the graph is the equation of the best-fit line, along with the correlation coefficient.
We note here that, in previous reports in the literature, the C5′ hydrogen seldom has been considered to be the main site of hydroxyl radical reaction. C4′ is probably the most popularly suggested position (15, 29), but these speculations have been based mainly on the bleomycin mechanism (16, 25, 27) or on product analysis of DNA constituents that were treated with ionizing radiation (1, 30).
The percentages in Table 2 for percent reactivity (first line) do not add up to 100%. We have chosen not to normalize these percentages to make clear the precision of our estimates. In fact, we would not expect the reactivity values we have determined to sum to 100%, in any case. The main reason is that, for these initial kinetic isotope effect experiments, we used deoxyadenosine, deoxycytidine, and deoxythymidine, each deuterated at a different position, because these were the specifically deuterated nucleotides that we had at our disposal. There is reason to expect some degree of sequence-dependence of the apparent isotope effect, which would reflect sequence-dependent DNA structure. It would be unlikely that the apparent isotope effect measured for the 4′ position, for example, would be the same for deoxyadenosine and deoxythymidine because the structural environments of the sugar hydrogens of the A and T would likely differ. Nevertheless, the relative reactivities we report in Table 2 and their relationship to solvent accessibility represent a decided advance in understanding the chemistry of the hydroxyl radical with duplex DNA.
Acknowledgments
This research was supported by Public Health Service Grant GM40894. We thank C. A. Townsend, J. J. Hangeland, J. W. Kozarich, J. Stubbe, L. Worth, Jr., and D. Vanderwall for providing deuterated nucleotides. We also appreciate helpful discussions with C. A. Townsend, B. L. Murr, and D. Poland. We gratefully acknowledge the use of densitometry instrumentation maintained by the Institute for Biophysical Research on Macromolecular Assemblies at Johns Hopkins University, which was supported by a National Science Foundation Biological Research Centers Award and by a grant from the W. M. Keck Foundation.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.von Sonntag C. The Chemical Basis of Radiation Biology. London: Taylor & Francis; 1987. [Google Scholar]
- 2.Tullius T D. Nature (London) 1988;332:663–664. doi: 10.1038/332663a0. [DOI] [PubMed] [Google Scholar]
- 3.Lee S, Hahn S. Nature (London) 1995;376:609–612. doi: 10.1038/376609a0. [DOI] [PubMed] [Google Scholar]
- 4.Price M A, Tullius T D. Methods Enzymol. 1992;212:194–219. doi: 10.1016/0076-6879(92)12013-g. [DOI] [PubMed] [Google Scholar]
- 5.Latham J A, Cech T R. Science. 1989;245:276–282. doi: 10.1126/science.2501870. [DOI] [PubMed] [Google Scholar]
- 6.Henner W D, Grunberg S M, Haseltine W A. J Biol Chem. 1982;257:11750–11754. [PubMed] [Google Scholar]
- 7.Tullius T D, Dombroski B A. Science. 1985;230:679–681. doi: 10.1126/science.2996145. [DOI] [PubMed] [Google Scholar]
- 8.Tullius T D, Dombroski B A. Proc Natl Acad Sci USA. 1986;83:5469–5473. doi: 10.1073/pnas.83.15.5469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Draganescu A, Levin J R, Tullius T D. J Mol Biol. 1996;250:595–608. doi: 10.1006/jmbi.1995.0401. [DOI] [PubMed] [Google Scholar]
- 10.Kappen L S, Goldberg I H. Biochemistry. 1983;22:4872–4878. doi: 10.1021/bi00290a002. [DOI] [PubMed] [Google Scholar]
- 11.Hangeland J J, De Voss J J, Heath J A, Townsend C A, Ding W-D, Ashcroft J, Ellestad G A. J Am Chem Soc. 1992;114:9200–9202. [Google Scholar]
- 12.Kozarich J W, Worth L, Jr, Frank B L, Christner D F, Vanderwall D E, Stubbe J. Science. 1989;245:1396–1399. doi: 10.1126/science.2476851. [DOI] [PubMed] [Google Scholar]
- 13.Sigman D. Acc Chem Res. 1986;19:180–186. [Google Scholar]
- 14.Sitlani A, Long E C, Pyle A M, Barton J K. J Am Chem Soc. 1992;114:2303–2312. [Google Scholar]
- 15.Cate J H, Gooding A R, Podell E, Zhou K, Golden B L, Kundrot C E, Cech T R, Doudna J A. Science. 1996;273:1678–1685. doi: 10.1126/science.273.5282.1678. [DOI] [PubMed] [Google Scholar]
- 16.Worth L, Jr, Frank B L, Christner D F, Absalon M A, Stubbe J, Kozarich J W. Biochemistry. 1993;32:2601–2609. doi: 10.1021/bi00061a018. [DOI] [PubMed] [Google Scholar]
- 17.Robins M J, Samano V, Johnson M D. J Org Chem. 1990;55:410–412. [Google Scholar]
- 18.Pathak T, Bazin H, Chattopadhyaya J. Tetrahedron. 1986;19:5427–5441. [Google Scholar]
- 19.Mishra, N. C. & Broom, A. D. (1991) J. Chem. Soc. Chem. Commun 1276–1277.
- 20.Shadle S E, Allen D F, Guo H, Pogozelski W K, Bashkin J S, Tullius T D. Nucleic Acids Res. 1997;25:850–860. doi: 10.1093/nar/25.4.850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prive G G, Yanagi K, Dickerson R E. J Mol Biol. 1991;217:177–199. doi: 10.1016/0022-2836(91)90619-h. [DOI] [PubMed] [Google Scholar]
- 22.Lee B, Richards F M. J Mol Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 23.Udenfriend S, Clark C T, Axelrod J, Brodie B B. J Biol Chem. 1954;208:731–739. [PubMed] [Google Scholar]
- 24.Henner W D, Rodriguez L O, Hecht S M, Haseltine W A. J Biol Chem. 1983;258:711–713. [PubMed] [Google Scholar]
- 25.Stubbe J, Kozarich J W. Chem Rev (Washington, DC) 1987;87:1107–1136. [Google Scholar]
- 26.Pogozelski W K, Tullius T D. Chem Rev (Washington, DC) 1998;98:1089–1107. doi: 10.1021/cr960437i. [DOI] [PubMed] [Google Scholar]
- 27.Giloni L, Takeshita M, Johnson F, Iden C, Grollman A P. J Biol Chem. 1981;256:8608–8615. [PubMed] [Google Scholar]
- 28.Cunningham J, Srijaranai S. J Photochem Photobiol. 1988;43:329–335. [Google Scholar]
- 29.Westhof E, Wesolowski D, Altman S. J Mol Biol. 1996;258:600–613. doi: 10.1006/jmbi.1996.0272. [DOI] [PubMed] [Google Scholar]
- 30.von Sonntag C, Hagen U, Schon-Bopp A, Schulte-Frohlinde D. Int J Radiat Biol Relat Stud Phys Chem Med. 1981;9:109–142. [Google Scholar]