

Abstract
We have learned a great deal over the last several years about the molecular mechanisms that govern cell growth, cell division and cell death. Normal cells pass through cell cycle (growth) and divide in response to mitogenic signals that are transduced through their cognate cell surface receptors to the nucleus. Despite the fact that cellular growth and division are mechanistically distinct steps, they are usually coordinately regulated, which is critical for normal cellular proliferation. The precise mechanistic basis for this coordinated regulation is unclear. TFII-I is a unique, signal induced multifunctional transcription factor that is activated upon a variety of signaling pathways and appears to participate in distinct phases of cell growth. For instance, TFII-I is required for growth factor-induced transcriptional activation of the c-fos gene, which is essential for cell cycle entry. Two alternatively spliced isoforms of TFII-I exhibit opposing but necessary functions for mitogen-induced transcriptional activation of c-fos. Besides transcriptional activation of the c-fos proto-oncogene and eventual entry into cell cycle, TFII-I also appears to have a role in later phases of the cell cycle and cell division. Here we discuss how a multitude of signaling inputs target TFII-I isoforms, which may exert their functions in distinct phases of the cell cycle and play a key role in the coordinated regulation of cellular proliferation.
Keywords: Transcription, Signaling, TFII-I, PLC-γ, Cell Cycle, Cell Division
Introduction
Genetic and epigenetic re-programming plays a critical role in the cellular response to environmental cues (1, 2). For example, a resting, non-cycling fibroblast enters cell cycle and begins to activate pro-proliferate genes in response to growth-promoting signals. Similarly, an uncommitted white blood cell makes lineage choices by upregulating lineage specific genes and proceeding along a defined path of hemopoiesis in response to particular immune modulatory signals. It is clear that, in both instances, there is an alteration in transcriptional programming upon introduction of the signal. Although all somatic cells have identical genome, it is the alteration in gene expression profile in response to extra-cellular cues that results in distinct biological outcomes.
The faithful passage of genetic information from one cell to its progeny is essential for growth and development (3,4). Thus, cell division requires a precisely timed process, the cell cycle, during which error-free replication of DNA, followed by division of the nucleus and partitioning of the parent cytoplasm gives rise to two daughter cells. Before committing to reproduce, cells sense growth factors/mitogen in their immediate environment (3,4). Engagement of these mitogens to their cognate cell surface receptors results in transduction of signals to the nucleus through a series of biochemical steps. Spatial and/or temporal activation of target downstream genes ensue to drive cell cycle progression past the restriction point and initiate DNA replication. Thus, it is likely that a set of signal-induced transcription factors would be available within the cell to coordinately regulate transcription and cell cycle events.
The prerequisite to switching on/activation of a particular gene or a set of genes is that the targeted transcriptional unit in the chromosome becomes “remodeled” and accessible for transcription (5). Once the natural constraints imposed by chromatin are obviated, the next step in gene expression is recruitment of gene/tissue/stage specific transcription factors to their cognate sites on the promoter (5, 6). These transcription factors, in turn, facilitate recruitment of the transcriptional machinery at the core promoter, the rate and/or stability of which dictate the rate of RNA syhtnesis. Therefore, a proper communication between site-specific transcription factors recruited at upstream promoters and/or enhancers and the transcriptional machinery, consisting of RNA polymerase and general transcription factors, is crucial in transcriptional regulation of a given gene (6). Because of these bewildering complexities associated with transcriptional regulation of gene expression, tractable model systems are necessary to dissect the molecular mechanisms that control signal-induced transcription. Here we present studies on transcription factor TFII-I, which led to unexpected observations regarding signal-induced transcription in various cell culture systems and further suggests a direct link between transcription and cell cycle.
Transcriptional Regulation of c-fos by TFII-I
Growth factor induced transcriptional activation of the immediate early (IE) c-fos proto-oncogene is essential for quiescent cells to re-enter cell cycle and eventually undergo proliferation (7-12). The c-fos gene expression is virtually undetectable upon serum starvation but it is elevated within minutes upon growth factor stimulation. The rapid kinetics and the dynamic histone modifications of IE genes means that these genes do not exhibit the highly stable chromatin modification state, normally associated with most active genes (13-15). Lack of this constraint together with rapid signal-induced transcriptional response also necessitates an unusual specificity at the level of promoter occupancy. The promoter region contains binding sites for several transcription factors, some of which are constitutively active in the nucleus while others are signal induced (7-15). Despite the fact that c-fos has served as a paradigm for understanding signal-induced transcription for two decades, it is still unclear as to how various signaling pathways converge on this gene simultaneously. For example, the transcriptional activation of c-fos is dependent on both the Ras and Rho signaling pathways (7-16). While Ras and Rho belong to the GTP-binding protein (G-protein) super family, the Ras pathway involves Raf/Mek1/ERK activation and Rho signaling predominantly involves cytoskeletal reorganization (17-20). It is mechanistically unclear how these two distinct but necessary pathways lead to activation of the c-fos gene.
TFII-I is a ubiquitously expressed transcription factor that is postulated to facilitate communication between upstream regulatory factors and the basal machinery (21-25). TFII-I belongs to a family of vertebrate specific transcription factors, each characterized by the presence of reiterated 90 amino acids domains (designated as R1-R6 in Figure 1) and multiple alternatively spliced isoforms (26, 27 reviewed in 25). The most conserved region within these repeats is called the I-repeat (23). There are four characterized alternatively spliced isoforms of TFII-I: Δ (957 amino acids), α (977 amino acids), β (978 amino acids) and γ (998 amino acids) (26, 27). Of the four, the γ-isoform is most likely expressed predominantly in neuronal cells and the α-isoform is lacking in murine cells (26). Each isoform contains all the repeats, the basic region (BR, also the DNA binding domain), a putative leucine zipper (LZ) and a functional nuclear localization signal (amino acids 297-304 with respect to the Δ-isoform) (27, 28) (Figure 1A).
Figure 1. Schematic structure of TFII-I isoforms.
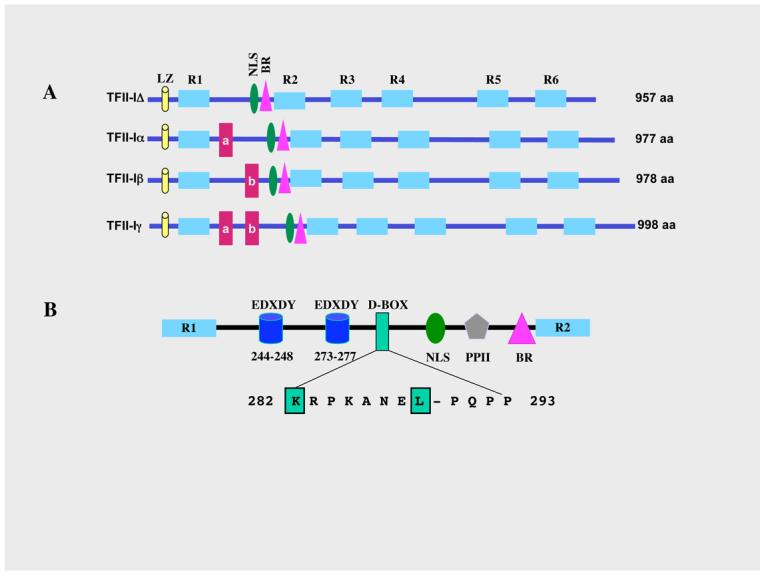
(A) Schematics of the four TFII-I isoforms and their respective amino acid lengths. LZ: leucine zipper; NLS: nuclear localization signal; BR: basic region/DNA binding domain; R1-R6: repeat domains; a, b: exons a (21 aa) and b (22 aa) encoded regions. (B) The region between R1 and R2 is expanded to show key regulatory features. Shown are the two Src auto phosphorylation sites: EDXDY at positons 244-248 and 273-277; D-box: MAPK interaction site between aa 282-293; and PPII: polyproline II domain, SH3 binding motif. The sequence of the D-box is also shown with most conserved residues boxed and highlighted in red.
Although TFII-I was functionally identified and characterized based on its ability to bind the core promoter sequence element (Inr) and activate transcription (21-24), later on it was demonstrated that it also functions as a signal-induced multifunctional transcription factor that is tyrosine phosphorylated in response to B cell receptor (BCR) and growth factor signaling (29-34). Importantly, induced tyrosine phosphorylation of TFII-I is necessary for its transcriptional function (32). A variety of growth-promoting and mitogenic stimuli (e.g., EGF, PDGF, serum and TPA) can enhance tyrosine phosphorylation of TFII-I and subsequent activation of the c-fos promoter, which harbors multiple TFII-I binding sites (29-32). Transcriptional activity of TFII-I requires an intact Ras pathway, since a dominant negative Ras can block TFII-I dependent transcriptional activation of c-fos (31, 35). It has also been shown that the MAPK pathway is required for TFII-I-dependent activation of the c-fos promoter and TFII-I physically interacts with Erk/MAPK through its conserved D-box (31, 35) (Figure 1B). Additionally, there are several consensus Src-phosphorylation sites, two putative YXXP sites and a proline-rich PPII helix that can potentially bind to SH2 and SH3 domains respectively (25) (Figure 1B). One of the tyrosine-phosphorylation sites (Y248) has been demonstrated to be required for transcriptional activity of TFII-I at several promoters (32). The integrity of Y248 is also required for interaction with MAPK, suggesting that tyrosine phosphorylation of TFII-I is critical for its downstream function (35). Intriguingly, TFII-I is downstream of both the Ras and Rho pathways, raising the possibility that besides SRF and Elk-1, TFII-I could provide the necessary regulatory input for c-fos activation (36).
Opposing action of TFII-I isoforms regulate c-fos transcription
Although each isoform individually binds to DNA and activates transcription both from the Inr-dependent (Vβ) and Inr-independent (c-fos) promoters in vitro and in vivo, co-expression of different combinations of TFII-I isoforms leads to enhanced basal activity of the Vβ promoter and attenuated signal responsive activity of the c-fos promoter (27). In addition, while upon ectopic over-expression all isoforms localize preferentially to the nucleus and exhibit similar transcriptional activity, the distribution of the endogenous isoforms in various cells suggests distinct/non-redundant functional roles in vivo (27).
Subcellular staining indicated that the TFII-IΔ isoform exhibits a predominantly cytoplasmic localization in serum-starved cells (32). Growth factor signaling leads to rapid tyrosine phosphorylation and nuclear translocation of TFII-IΔ in murine fibroblasts, which is necessary for the transcriptional activation of c-fos (28, 36). However, in addition to the Δ-isoform, murine fibroblasts also express the β-isoform. Surprisingly, while the endogenous TFII-IΔ isoform is localized predominantly in the cytoplasm in basal state, TFII-Iβ is localized in the nucleus (37). Interestingly, mitogenic stimulation leads to tyrosine phosphorylation and nuclear import of TFII-IΔ and concomitant nuclear export of TFII-Iβ. Consistent with the nuclear localization of TFII-Iβ in unstimulated cells, it is recruited to the c-fos promoter in the absence of signaling and the recruitment is undetectable in the presence of signaling (37).
To address how TFII-IΔ transduces mitogenic signals to the nucleus, it was shown that the tyrosine phosphorylated TFII-IΔ physically interacts with Erk1/2 in the cytoplasm and translocates to the nucleus together with Erk1/2 (37). Consistent with this observation, the nuclear localization of TFII-IΔ was required for nuclear localization of Erk because a ΔNLS mutant of TFII-IΔ retained Erk in the cytoplasm, even in the presence of mitogenic signaling. Using both gain of function and loss of function analyses, it was clearly demonstrated that signal-induced nuclear localization of Erk requires a functional TFII-I (37).
As mentioned earlier, c-fos is an IE gene the expression of which is basally off but turned on very rapidly after signaling. The rapid kinetics, together with the dynamic histone modifications of IE genes implies that these genes do not exhibit the highly stable chromatin modification state, normally associated with most active genes(13-15). Because the constitutive nuclear localization of TFII-Iβ allows it to be recruited to the c-fos promoter in the absence of signaling, this phenomenon may serve two purposes (Figure 1). First, it allows c-fos expression to be silenced in the absence of signaling, which is a necessary requirement for quiescence. In this regard, it has been shown that TFII-I might serve to negatively regulate c-fos gene expression, although the precise mechanism or the usage of specific isoforms was not addressed (38). Second, it perhaps ensures specificity such that the same promoter site can be utilized for signal-induced transcriptional activation mediated by TFII-IΔ. Given that the two isoforms bind to the same site on the c-fos promoter in vitro with identical DNA binding specificity (35), they are likely to be recruited to the same site in vivo. Additionally, there exists an independent and dynamic regulation of histone phosphorylation and acetylation on immediate-early (IE) gene nucleosomes (13-15). While the histone phosphorylation is inducible and aided by activation of the MAPK cascade, histone acetylation is largely preexisting and mediated by a combined effect of HATs and HDACs (13-15). However, the precise mechanism by which these dynamics are achieved upon mitogenic signaling remains unknown. The alteration in promoter occupancy by the two isoforms of TFII-I, together with their respective interactions with MAPK and HDACs (37-40), perhaps provides a reasonable mechanism to explain the unusual dynamics of c-fos regulation. Moreover, TFII-I is also shown to be associated with histone demethylase co-repressor complex, LSD1/BHC110 (38). Our own observation indicates a preferential interaction of TFII-Iβ with LSD1 (M.I. Tussie-Luna and A.L.R, unpublished) further providing a functional distinction between the isoforms in regulating c-fos expression (Figure 2).
Figure 2. Growth factor mediated gene regulation by TFII-I isoforms.

In the absence of signaling, TFII-Iβ remains in the nucleus, bound to the c-fos promoter. Because it appears to repress c-fos transcription and preferentially interacts with HDAC and LSD1, we surmise that promoter bound TFII-Iβ is associated with these co-repressors. In contrast, the TFII-IΔ isoform remains in the cytoplasm in the absence of signaling where it interacts with p190 RhoGAP. Upon mitogenic signaling, TFII-IΔ under goes tyrosine phosphorylation, interacts with activated MAPK/Erk and translocates to the nucleus. Nuclear TFII-I has been shown to bind to the same site on the c-fos promoter. Given that it interacts with G-kinase and MAPK, it is likely that promoter bound TFII-IΔ remains associated with these kinases.
Role of TFII-I in Rho/GAP pathway
Although transcriptional activation of c-fos requires an intact Rho and Ras pathways, it remains to be precisely elucidated how these distinct pathways coordinately control activation of the gene. The p190RhoGAP proteins are potent Rho inhibitors, which are implicated in the regulation of Rho-dependent cytoskeletal reorganization and subsequent transcriptional activation in response to mitogens (19, 20). In a search for proteins that might interact with p190RhoGAP, TFII-I was identified as an interacting partner of the p190A RhoGAP protein (36). The interaction was mediated via the FF-domain in p190RhoGAP and the N-terminal 90 amino acids in TFII-I (36). Although FF domains are usually present in splicing and transcription factors, utilization of FF domains for signal-induced transcriptional regulation was not known (36). Stimulation through the PDGF receptor resulted in tyrosine phosphorylation of TFII-I and its subsequent release from p190RhoGAP to activate c-fos. Interestingly, PDGF signaling also leads to tyrosine phosphorylation of p190 at its FF domain, which inhibits interaction with TFII-I (36). Consistent with these observations TFII-I was found to be constitutively nuclear in p190 null cells, resulting in an enhanced c-fos transcription in response to growth factors. Most importantly, ectopic expression of p190 rescued both cytoplasmic localization of TFII-I and subsequent downregulation of c-fos (36). These results suggest that regulated association of TFII-I with p190RhoGAP in the cytoplasm is an important step in mitogen-induced transcription regulation of c-fos, although the precise involvement and contribution of individual isoforms of TFII-I in the p190 RhoGAP pathway is not yet determined.
ER-stress induced regulation of TFII-I
Recent data suggests that TFII-I is also an important regulator in the endoplasmic reticulum (ER) stress response pathway (41, 42). TFII-I is recruited to the ER-stress response element (ERSE) of the Grp78 promoter, which belongs to the family of stress-induced chaperones (41, 42). Interestingly, it has been shown that Src-dependent tyrosine phosphorylation of TFII-I is required for its transcriptional activation of the Grp78. ER stress induces tyrosine phosphorylation of TFII-I at Y248, nuclear translocation and subsequent recruitment to the promoter (42). The transcriptional activation of ER chaperones, for example Grp78, upon ER stress is a general pathway that triggers unfolded protein response (UPR), which can induce cell death as well as pro-survival mechanisms (42). Identification of TFII-I as an important regulator of ER stress-mediated activation of Grp78 response is thus significant for the UPR pathway. These results further suggest that critical tyrosine residues in TFII-I can be targeted by different kinases upon distinct signaling pathways and thus, provide a rationale for its multifunctional activity.
Role of TFII-I in G-kinase pathway
The nitric oxide (NO)/cGMP/cGMP-dependent protein kinase (G-kinase) signal transduction pathway is important for regulation of cell growth, differentiation, and apoptosis (43). There are two major types of G-kinase: a soluble type I and a membrane-bound type II. While type I G-kinase is widely expressed and present in smooth muscle and endothelial and neuronal cells, type II G-kinase is more limited in expression (43). The G-kinase signaling pathway regulates gene expression in various cell types, both at the transcriptional as well at the post-transcriptional level. The resulting transcriptional effects are either positive or negative (43). For example, transcriptional regulation of c-fos by nitric oxide and cGMP can occur by nuclear translocation of G-kinase I (43, 44). Surprisingly, TFII-I was found to specifically interact with G-kinase Iβ in vitro and in vivo (44). cGMP analogs enhanced association between TFII-I and G-kinase Iβ in the nucleus (44). Importantly, from a regulatory standpoint, G-kinase was shown to phosphorylate TFII-I on two serine residues (S371 and S743) in vitro and in vivo, which potentiated TFII-I dependent transcriptional activation of c-fos. Consequently, mutation of these two serine residues resulted in a loss of TFII-I dependent transcriptional activation of c-fos (44). Whether the physical and functional interactions between TFII-I and G-kinase Iβ leads to additional interactions of TFII-I with other transcriptional regulators or whether G-kinase Iβ is recruited to the c-fos promoter together with TFII-I remains to be determined.
Regulation of TFII-I by Btk
TFII-I was shown to physically and functionally interact with the non-receptor Bruton's tyrosine kinase (Btk) that is activated upon B cell receptor (BCR) engagement (33, 34). Btk is the target of multiple mutations in humans, each of which result in X-linked agammaglobulinemia (XLA) (45, 46). A spontaneous mutation in mice (R28C) produces X-linked immunodeficiency (xid) (47). Btk belongs to the Tec family of non-receptor tyrosine kinases that includes TecI, TecII, Itk, Bmx/Etk and DSrc28C, (found in Drosophila) (33, 34). A distinctive feature of these kinases is the presence of a pleckstrin homology (PH) domain in the N-terminal region followed by a unique Tec homology (TH) domain (33, 48).
Despite the biological importance of Btk in B cell differentiation and its important role in calcium signaling and phospholipid metabolism, the precise downstream function of this kinase remains to be determined at the biochemical level. TFII-I was shown to interact with Btk in vivo, via the TH and PH domains (33). TFII-I is tyrosine phosphorylated by Btk in vitro, and transiently tyrosine phosphorylated after IgM stimulation of B cells in vivo, suggesting that TFII-I may be a physiological target of Btk (33). It was shown further that ectopic expression of wild type Btk enhances TFII-I mediated transcriptional activation and its tyrosine phosphorylation in transient transfection assays (34, 49). Mutation of Btk in either the PH domain (R28C, xid mutation) or the kinase domain (K430E) compromises its ability to enhance both the tyrosine phosphorylation and the transcriptional activity of TFII-I (34). TFII-I associates constitutively in vivo with wild type but not xid Btk. However, membrane IgM cross-linking in B cells leads to dissociation of TFII-I from Btk. Furthermore, while TFII-I is found both in the nucleus and cytoplasm of wild type and xid primary resting B cells, nuclear TFII-I is greater in xid B cells (34). Most strikingly, receptor crosslinking of wild type (but not xid) B cells results in increased nuclear import of TFII-I (34). However, consistent with its ubiquitous expression pattern, TFII-I is also tyrosine phosphorylated by Src family kinases upon growth factor stimulation (32). Together, these observations led to the model that TFII-I is latently found in the cytoplasm but upon activation (tyrosine phosphorylation) by signaling through surface receptors it translocates to the nucleus for transcriptional activation (49) (Figure 3).
Figure 3. TFII-I isoforms may connect lipid metabolism to gene expression.
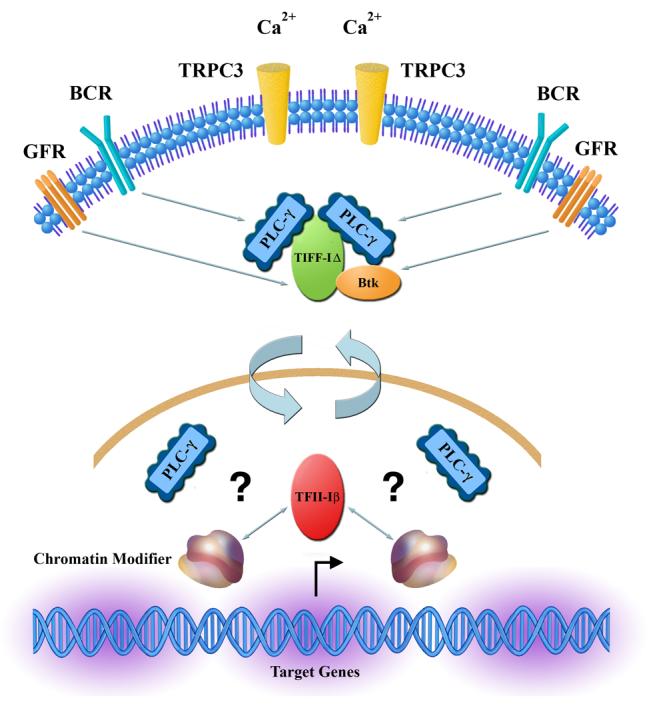
Because both isoforms of TFII-I have been described to interact with PLC-γ, we propose that TFII-IΔ interacts with (soluble) PLC-γ in the cytoplasm to regulate TRPC-3 mediated Ca2+ entry, while TFII-Iβ interacts with PLC-γ in the nucleus. In addition, TFII-I also interacts with Btk in the cytoplasm. Because TFII-Iβ interacts with chromatin modifiers such as HDACs and LSD1 and PLC-γ has been shown to play a role in transcription, it is possible that TFII-Iβ might connect phopsholipid metabolism to signal-induced gene regulation. Given the signal-induced reverse translocation of TFII-I isoforms, such a process might also lead to alteration in subcellular PLC-γ.
Cytoplasmic function of TFII-I
Engagement of cell surface receptor tyrosine kinases by their cognate ligands is associated with Ca2+ influx into the cell, resulting in generation of IP3 (inositol 1,4,5-trisphosphate) and DAG (diacylglycerol) from PIP2 (phosphatidylinositol 4,5-bisphosphate) via the enzymatic action of phospholipase C (PLC) (50). For instance, activation of the growth factor receptors or immune (B and T cell receptors) receptors results in activation of PLC-γ and transient Ca2+ influx (50, 51). Recent studies however, uncovered a novel lipase-independent role of PLC-γ that is necessary for intracellular Ca2+ entry via transient receptor potential channel 3 (TRPC3) (50). In an unexpected recent development, TFII-I was identified as an interacting partner of PLC-γ (53). It was demonstrated that tyrosine phosphorylated TFII-I interacts with the Src-homology (SH)-2 domain of PLC-γ (53). Agonist-induced calcium entry (ACE) into the cell by TRPC3 requires its binding to phospholipase C-gamma (PLC-γ) (52). The cytoplasmic interaction of TFII-I to PLC-γ via a split pleckstrin homology (PH) domain in a mutually exclusive fashion with TRPC3 results in reduced TRPC3 mediated ACE (53). PH domains are found in a variety of signaling intermediates and generally believed that they mediate membrane proximal lipid binding (48). A recent gestalt algorithm search, however, revealed that a PH-like domain (also called a split PH domain) is more widely spread and present in a variety of both nuclear and cytoplasmic proteins, including PLC-γ and TRPC3 (52). Indeed, association of PLC-γ with TRPC3 is mediated through its split PH domain where the two split PH domain from two proteins are brought into close proximity to result in a structural complementation and enhanced surface expression of TRPC3, both of which are necessary for ACE (53).
Interestingly, it was shown that TFII-I has a signal-inducible split PH domain, which mimics the split PH domain of TRPC3. The interaction of TFII-I with PLC-γ is via its split PH domain, thereby providing a rationale of how TFII-I can effectively compete with TRPC3 for PLC-γ binding (53). The “basally-closed” split PH domain in TFII-I undergoes a conformational “opening” upon signal-induced tyrosine phosphorylation, resulting in its interaction with PLC-γ via both the phosphotyrosine and split PH domains (53, 54). Because TRPC3 surface expression and function requires its binding to PLC-γ, competitive association of TFII-I with PLC-γ lowers the surface expression of TRPC3 and reduces its Ca2+ channeling activity (Figure 3). Interestingly, this phenomenon does not require either the nuclear localization or transcriptional function of TFII-I because a nuclear localization (and consequently a transcription) deficient mutant of TFII-I was fully competent in PLC-γ binding and inhibition of Ca2+ influx into the cell (53).
Double life of TFII-I
A number of factors, originally shown to exclusively have either cytoplasmic function or nuclear function, have now been demonstrated to have both functions. These include voltage-gated calcium channels (VGCC) or the derived fragment called calcium channel-associated transcriptional regulator (CCAT), DREAM and TFII-I (55-58). Interestingly, in all three cases, the transcriptional activity appears to be regulated directly or indirectly by signaling via calcium channels (56). While, the first two factors do not seem to be directly regulated by phosphorylation, TFII-I activity associated with both its cytoplasmic as well as nuclear residency requires phosphorylation. It is interesting to ponder how tyrosine phosphorylated TFII-I is simultaneously associated with PLC-γ in the cytoplasm and is also required for transcription in the nucleus (54). There are several potential explanations for this conundrum. First, it is likely that there are distinct pools of cytoplasmic TFII-I. Each pool could be associated with different cytoplasmic tethers and/or signaling intermediates and thus responsive to distinct signals via different cell surface receptors (54). For instance, the pool of TFII-I associated with PLC-γ (and perhaps Btk) might be responsive to BCR signaling. In contrast, the pool of TFII-I associated with p190RhoGAP and/or Erk/MAPK might be downstream of the growth factor receptors (GFRs) (54, Figure 3). Regardless of the pool it is associated with, TFII-I would undergo signal-induced tyrosine phosphorylation, dissociate from the complex and translocate to the nucleus for gene activation (54). However, it is equally likely that there is a dynamic equilibrium between these pools and upon signaling there might be a conformational alteration in TFII-I, allowing it to move from one pool to another. Given that TFII-I exhibits multiple potential phosphorylation sites, a distinct second possibility is that there is only one pool of cytoplasmic TFII-I but that different signals (and corresponding kinases) target these sites in TFII-I under different conditions. The plasticity of this complex might depend on the specificity of the signal and the particular phospho-tyrosine moiety. Accordingly, TFII-I might interact with different partners and thereby modulate distinct downstream pathways (54). Thirdly, it is possible that different subcellular localizations (and thus functions) of TFII-I are kinetically distinct. Because the PLC-γ mediated Ca2+ influx occurs within 2-5 min, receptor-induced tyrosine phosphorylation of TFII-I and its subsequent membrane proximal interactions with PLC-γ (and Btk and/or other cytoplasmic tethers) should occur within this time frame. However, since the nuclear translocation of TFII-I occurs at later time points (12-20 min) (36, 37), release of tyrosine phosphorylated TFII-I from the cytoplasmic tether and subsequent dimerization and nuclear translocation is kinetically distinct from the membrane proximal step (54). Last but not in the least, it is conceivable that while a subset of the events occurs near the plasma membrane, others occur in the proximity of the nuclear membrane (54). In this model, the two isoforms of TFII-I, by virtue of being in different subcellular compartments, might have distinct lipid-mediated interactions and consequently different functions in signaling and transcription (Figure 3). Because phosphoinositols are thought to control chromatin dynamics and the fact that PLC-γ possibly plays an important role in chromatin remodeling and gene regulation (59), an interesting thought is that TFII-I isoforms might connect phospholipid metabolism to signal induced gene regulation (Figure 3).
Role of TFII-I in cell cycle
Mammalian cells search for the availability of growth factors and whether the environment is favorable for proliferation. If these criteria are met, the critical cell cycle regulators cyclins and their associated kinases (CDKs) integrate extracellular signals to drive cells through G1 phase of the cell cycle and initiate replication. Subsequently, activation of cyclin-dependent kinases is also important for G1-S phase transition (3, 4). The expression of D type cyclins depends largely on growth regulatory signals, thus enabling these cyclins to serve as key molecular links between growth factor stimulation and cell cycle machinery (60).
It has been shown recently that TFII-I is a key regulator of cyclin D1 (61). TFII-I is recruited to the cyclin D1 core-promoter in vivo and transcriptionally activates this gene, which results in accelerated entry into and exit from S phase. In contrast, regulated destruction of TFII-I appears to be necessary for cell cycle arrest induced by ionizing radiation (IR)-mediated DNA damage and concomitant p53 activation. Genetoxic stress results in ubiquitination and targeted proteasomal destruction of TFII-I that is dependent on the presence of intact p53 and ATM (Ataxia Telangiectasia Mutated) pathways (61). Consistent with the fact that TFII-I delivers the growth factor dependent transcriptional signal to the cyclin D1 promoter, mutation of tyrosine residues in TFII-I, which are required for its mitogen mediated transcriptional activity, abolishes cyclin D1 gene expression (61). Thus, TFII-I may provide a molecular link between mitogen-dependent signaling and nuclear gene expression to control cell division and proliferation (62).
Although ectopic expression of TFII-I results in elevated cyclin D1 levels and bypass radiation induced cell cycle block, it is undetermined whether these cells progress through cycle despite DNA damage or whether they undergo DNA repair (61, 62). Moreover, it also remains to be shown whether TFII-I undergoes preferential degradation during particular phases of the cell cycle and what molecular mechanism(s) might govern this timed destruction (62). Because the TFII-I protein can undergo various post-translational modifications (e.g., phosphorylation, sumoylation and ubiquitination), it is likely that some of these modifications selectively occur at particular phase of the cell cycle (62). It is tempting to conjecture that the ability to undergo different kinds of post-translational modifications might endow TFII-I with significant plasticity, allowing it to differentially respond to a variety of signals in a cell cycle and differentiation stage specific fashion (Figure 4).
Figure 4. Function of TFII-I in distinct phases of cell cycle via differential modification.

TFII-I appears to play distinct roles in distinct phases of cell cycle and cell division. We conjecture that this is achieved via differential post-translational modification of TFII-I, which might result in different interactions with different factors. The modifications might include tyrosine and serine/threonine phosphorylation as well as sumoylation and ubiquitylation. The function of individual isoforms of TFII-I in this process is currently unknown.
Recent unpublished data also suggest that TFII-I might have a function in centrosome integrity and cell division (Hakre, S and A.L.R., unpublished). It appears that silencing TFII-I (both isoforms) results in a significant early G1/S block as well as a late G2/M block. Interestingly, preliminary data with TFII-Iβ specific knockdown suggest primarily a late G2/M block (Hakre, S and A.L.R., unpublished). Furthermore, analysis of multiple clones of a cell line stably expressing the ΔNLSTFII-I revealed that they exhibit cytokinetic defects–as evidenced by enhanced number of binucleated cells and fragmented centrosomes (Hakre, S and A.L.R., unpublished). We speculate that perhaps control of cellular proliferation by TFII-I isoforms not only involves regulation of mitogenic signaling to transcriptionally activate c-fos and subsequent cell cycle entry but also regulation of latter phases of the cell cycle and cell division (Figure 4).
Future Perspectives
The mechanism of TFII-IΔ nuclear import is beginning to be worked out. But the mechanism of TFII-Iβ nuclear export is completely unknown. To begin to understand signal-induced gene regulation by TFII-I, both mechanisms must be worked out. Further, while we have gained some insight into the function of two of the isoforms of TFII-I, virtually nothing is known about the other two. Do these have distinct functions? The relative abundance of these isoforms in a given cell and/or their cell type specific expression perhaps determines the functional effects of TFII-I. Moreover, different studies in different cell culture systems preclude us to conclude whether the signal-induced translocation of and transcriptional regulation by TFII-I isoforms are universal. Nevertheless, it is anticipated that TFII-I coordinately regulates cell growth and division by utilizing its different isoforms and their differential phosphorylation patterns in distinct phases of cell cycle. Identification and characterization of these sites will be necessary to validate this notion.
The TFII-I family consists of three related genes that are closely located on human chromosome 7 (7q11.23), a portion of which is deleted in a haplo-insufficient manner in Williams-Beuren syndrome, WBS (34, 63). WBS is a neuro-developmental disorder with multisystem manifestations, including supravalvular aortic stenosis (SVAS), hypercalcemia in infancy. These patients also exhibit mild to moderate mental retardation, cognitive defects and characteristic craniofacial features (26, 63). The frequency of this genetic haploinsuffiency is estimated to be 1 in 20,000 live births. Although most cases are sporadic, WBS is inherited as an autosomal dominant trait in a select few families. To date, 17 open reading frames have been identified within this region (26, 63). Despite the fact that WBS is a multisystem dysfunction most likely caused as a result of haplo-insufficiency in several genes, it is anticipated that there will be a strong genotype-phenotype correlation. Indeed, targeted deletions in TFII-I family genes in murine models suggested that craniofacial defects observed in WBS are likely caused by defects in TFII-I family genes (64). However, it remains to be seen which functions of TFII-I are affected in this disease and whether the isoforms play any distinct roles. Moreover, given TFII-I is a downstream target of Btk, mutations in which are associated with XLA, it would be interesting to test whether subcellular localization, tyrosine phosphorylation and/or function of TFII-I isoforms are altered in the B cells of these patients. Finally, the structure and function of other family members must be determined to have a better understanding of the physiological role of this group of transcription factors.
Acknowledgement
The work done in my laboratory has been made possible by support from the National Institutes of Health: AI45150 and HD046034. I gratefully acknowledge my colleagues and collaborators, whose important contributions have made these studies significant. I thank Dr. Peter Brodeur for critically reading this review.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brivanlou AH, Darnell JE. Signal transduction and the control of gene expression. Science. 2002;295:813–818. doi: 10.1126/science.1066355. [DOI] [PubMed] [Google Scholar]
- 2.Sears RC, Nevins JR. Signaling networks that link cell proliferation and cell fate. J Biol Chem. 2002;277:11617–20. doi: 10.1074/jbc.R100063200. [DOI] [PubMed] [Google Scholar]
- 3.Coffman JA. Cell cycle development. Dev Cell. 2004;6:321–327. doi: 10.1016/s1534-5807(04)00067-x. [DOI] [PubMed] [Google Scholar]
- 4.Pardee AB. G1 events and regulation of cell proliferation. Science. 1989;246:603–608. doi: 10.1126/science.2683075. [DOI] [PubMed] [Google Scholar]
- 5.Struhl K. Fundamentally different logic of gene regulation in eukaryotes and prokaryotes. Cell. 1999;98:1–4. doi: 10.1016/S0092-8674(00)80599-1. [DOI] [PubMed] [Google Scholar]
- 6.Roeder RG. Transcriptional regulation and the role of diverse coactivators in animal cells. FEBS Lett. 2005;579:909–915. doi: 10.1016/j.febslet.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 7.Hill CS, Treisman R. Transcriptional regulation by extracellular signals: mechanisms and specificity. Cell. 1995;80:199–211. doi: 10.1016/0092-8674(95)90403-4. [DOI] [PubMed] [Google Scholar]
- 8.Hill CS, Wynne J, Treisman R. The Rho family GTPases RhoA, Rac1, and CDC42 regulate transcriptional activation by SRF. Cell. 1995;81:1159–1170. doi: 10.1016/s0092-8674(05)80020-0. [DOI] [PubMed] [Google Scholar]
- 9.Murai K, Treisman R. R, Interaction of serum response factor (SRF) with the Elk-1 B box inhibits RhoA-actin signaling to SRF and potentiates transcriptional activation by Elk-1. Mol Cell Biol. 2002;22:7083–7092. doi: 10.1128/MCB.22.20.7083-7092.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schratt G, Weinhold B, Lundberg AS, Schuck S, Berger J, Schwarz H, Weinberg RA, Ruther U, Nordheim A. Serum response factor is required for immediate-early gene activation yet is dispensable for proliferation of embryonic stem cells. Mol. Cell Biol. 2001;21:2933–43. doi: 10.1128/MCB.21.8.2933-2943.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vickers ER, Kasza A, Kurnaz IA, Seifert A, Zeef LA, O'donnell A, Hayes A, Sharrocks AD. Ternary complex factor-serum response factor complex-regulated gene activity is required for cellular proliferation and inhibition of apoptotic cell death. Mol. Cell Biol. 2004;23:10340–10351. doi: 10.1128/MCB.24.23.10340-10351.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y, Falasca M, Schlessinger J, Malstrom S, Tsichlis P, Settleman J, Hu W, Lim B, Prywes R. Activation of the c-fos serum response element by phosphatidyl inositol 3-kinase and rho pathways in HeLa cells. Cell Growth Differ. 1998;9:513–522. [PubMed] [Google Scholar]
- 13.Thomson S, Clayton AL, Mahadevan LC. Independent dynamic regulation of histone phosphorylation and acetylation during immediate-early gene induction. Mol Cell. 2001;8:1231–1241. doi: 10.1016/s1097-2765(01)00404-x. [DOI] [PubMed] [Google Scholar]
- 14.Hazzalin CA, Mahadevan LC. Dynamic acetylation of all lysine 4-methylated histone H3 in the mouse nucleus: analysis at c-fos and c-jun. PLoS Biol. 2005;3:393–397. doi: 10.1371/journal.pbio.0030393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dyson MH, Thomson S, Inagaki M, Goto H, Arthur SJ, Nightingale K, Iborra FJ, Mahadevan LC. MAP kinase-mediated phosphorylation of distinct pools of histone H3 at S10 or S28 via mitogen- and stress-activated kinase 1/2. J. Cell Sci. 2005;118:2247–2259. doi: 10.1242/jcs.02373. [DOI] [PubMed] [Google Scholar]
- 16.Tominaga T, Sahai E, Chardin P, McCormick F, Courtneidge SA, Alberts AS. Diaphanous-related formins bridge Rho GTPase and Src tyrosine kinase signaling. Mol. Cell. 2000;5:13–25. doi: 10.1016/s1097-2765(00)80399-8. [DOI] [PubMed] [Google Scholar]
- 17.Chen Z, Gibson TB, Robinson F, Silvestro L, Pearson G, Xu B, Wright A, Vanderbilt C, Cobb MH. MAP kinases. Chem Rev. 2001;101:2449–2476. doi: 10.1021/cr000241p. [DOI] [PubMed] [Google Scholar]
- 18.Morrison DK, Davis RJ. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu Rev Cell Dev Biol. 2003;19:91–118. doi: 10.1146/annurev.cellbio.19.111401.091942. [DOI] [PubMed] [Google Scholar]
- 19.Hall A. Small GTP-binding proteins and the regulation of actin cytoskeleton. Annu. Rev. Cell Biol. 1995;19:31–54. doi: 10.1146/annurev.cb.10.110194.000335. [DOI] [PubMed] [Google Scholar]
- 20.Symons M, Settleman J. Rho family GTPases: more than simple switches. Trends Cell Biol. 2000;10:415–419. doi: 10.1016/s0962-8924(00)01832-8. [DOI] [PubMed] [Google Scholar]
- 21.Roy AL, Meisterernst M, Pognonec P, Roeder RG. Cooperative interaction of an initiator-binding transcription initiation factor and the helix-loop-helix activator USF. Nature. 1991;354:245–248. doi: 10.1038/354245a0. [DOI] [PubMed] [Google Scholar]
- 22.Roy AL, Malik S, Meisterernst M, Roeder RG. An alternative pathway for transcription initiation involving TFII-I. Nature. 1993;365:355–359. doi: 10.1038/365355a0. [DOI] [PubMed] [Google Scholar]
- 23.Roy AL, Du H, Gregor PD, Novina CD, Martinez E, Roeder RG. Cloning of an Inr- and E-box binding protein, TFII-I, that interacts physically and functionally with USF1. EMBO J. 1997;16:101–115. doi: 10.1093/emboj/16.23.7091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheriyath V, Novina CD, Roy AL. TFII-I regulates Vβ promoter activity through an initator element. Mol. Cell. Biol. 1998;18:4444–4454. doi: 10.1128/mcb.18.8.4444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roy AL. Biology and biochemistry of the signal induced transcription factor TFII-I. Gene. 2001;262:1–13. doi: 10.1016/s0378-1119(01)00625-4. [DOI] [PubMed] [Google Scholar]
- 26.Perez-Jurado LA, Wang Y-K, Peoples R, Coloma A, Cruces J, Francke U. A duplicated gene in the breakpoint regions of the 7q11.23 Williams-Beuren syndrome deletion encodes the initiator binding protein TFII-I and BAP-135, a phosphorylation target of Btk. Hum. Mol. Genet. 1998;7:325–334. doi: 10.1093/hmg/7.3.325. [DOI] [PubMed] [Google Scholar]
- 27.Cheriyath V, Roy AL. Alternatively spliced isoforms of TFII-I: Complex formation, nuclear translocation and differential gene regulation. J. Biol. Chem. 2000;275:26300–26308. doi: 10.1074/jbc.M002980200. [DOI] [PubMed] [Google Scholar]
- 28.Cheriyath V, Roy AL. Structure-function analysis of TFII-I: Roles of the N-terminal end, basic region, and I-repeats. J. Biol. Chem. 2001;276:8377–8383. doi: 10.1074/jbc.M008411200. [DOI] [PubMed] [Google Scholar]
- 29.Grueneberg DA, Henry RW, Brauer A, Novina CD, Cheriyath V, Roy AL, Gilman M. A multifunctional DNA-binding protein that promotes the formation of serum response factor/homeodomain complexes: identity to TFII-I. Genes Dev. 1997;11:2482–2493. doi: 10.1101/gad.11.19.2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Novina CD, Cheriyath V, Roy AL. Regulation of TFII-I activity by phosphorylation. J. Biol. Chem. 1998;273:33443–33448. doi: 10.1074/jbc.273.50.33443. [DOI] [PubMed] [Google Scholar]
- 31.Kim DW, Cheriyath V, Roy AL, Cochran BH. TFII-I enhances activation of the c-fos promoter through interactions with upstream elements. Mol Cell Biol. 1998;18:3310–3320. doi: 10.1128/mcb.18.6.3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheriyath V, Desgranges ZP, Roy AL. c-Src dependent transcriptional activation of TFII-I. J. Biol. Chem. 2002;277:22798–22805. doi: 10.1074/jbc.M202956200. [DOI] [PubMed] [Google Scholar]
- 33.Yang W, Desiderio S. BAP-135, a target for Bruton's tyrosine kinase in response to B cell receptor engagement. Proc. Natl. Acad. Sci. USA. 1997;94:604–609. doi: 10.1073/pnas.94.2.604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Novina CD, Kumar S, Bajpai U, Cheriyath V, Zhang K, Pillai S, Wortis HH, Roy AL. Regulation of nuclear translocation and transcriptional activity of TFII-I by Bruton's tyrosine kinase. Mol. Cell. Biol. 1999;19:5014–5024. doi: 10.1128/mcb.19.7.5014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim DW, Cochran BH. Extracellular signal-regulated kinase binds to TFII-I and regulates its activation of the c-fos promoter. Mol Cell Biol. 2000;20:1140–1148. doi: 10.1128/mcb.20.4.1140-1148.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang W, Sordella R, Chen GC, Hakre S, Roy AL, Settleman J. An FF domain-dependent protein interaction mediates a signaling pathway for growth factor-induced gene expression. Mol. Cell. 2005;17:23–35. doi: 10.1016/j.molcel.2004.11.024. [DOI] [PubMed] [Google Scholar]
- 37.Hakre S, Tussie-Luna I, Ashworth T, Novina CD, Settleman J, Sharp PA, Roy AL. Opposing Functions of TFII-I Spliced Isoforms in Growth Factor Induced Gene Expression. Mol Cell. 2006;24:301–308. doi: 10.1016/j.molcel.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 38.Hakimi MA, Dong Y, Lane WS, Speicher DW, Shiekhattar R. A candidate X-linked mental retardation gene is a component of a new family of histone deacetylase-containing complexes. J Biol Chem. 2003;278:7234–7239. doi: 10.1074/jbc.M208992200. [DOI] [PubMed] [Google Scholar]
- 39.Tussie-Luna MI, Bayarsaihan D, Seto E, Ruddle FH, Roy AL. Physical and functional interactions of histone deacetylase 3 with TFII-I family proteins and PIASxb. Proc. Natl. Acad. Sci. USA. 2002;99:12807–12812. doi: 10.1073/pnas.192464499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wen Y, Cress D, Roy AL, Seto E. Histone deacetylase 3 binds to and regulates the multifunctional transcription factor TFII-I. J. Biol. Chem. 2003;278:1841–1847. doi: 10.1074/jbc.M206528200. [DOI] [PubMed] [Google Scholar]
- 41.Parker R, Phan T, Baumeister P, Roy B, Cheriyath V, Roy AL, Lee AS. Identification of TFII-I as the endoplasmic reticulum stress response element binding factor ERSF: its autoregulation by stress and interaction with ATF6. Mol. Cell. Biol. 2001;21:3220–3233. doi: 10.1128/MCB.21.9.3220-3233.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hong M, Lin M, Huang JM, Baumeister P, Hakre S, Roy AL, Lee AS. Transcriptional regulation of the Grp78 promoter by endoplasmic reticulum stress: Role of TFII-I and its tyrosine phosphorylation. J Biol Chem. 2005;280:16821–16828. doi: 10.1074/jbc.M413753200. [DOI] [PubMed] [Google Scholar]
- 43.Eigenthaler M, Lohmann SM, Walter U, Pilz RB. Signal transduction by cGMP-dependent protein kinases and their emerging roles in the regulation of cell adhesion and gene expression. Rev Physiol Biochem Pharmacol. 1999;135:173–209. doi: 10.1007/BFb0033673. [DOI] [PubMed] [Google Scholar]
- 44.Casteel DE, Zhuang S, Gudi T, Tang J, Vuica M, Desiderio S, Pilz RB. cGMP-dependent protein kinase I beta physically and functionally interacts with the transcriptional regulator TFII-I. J Biol Chem. 2002;277:32003–32014. doi: 10.1074/jbc.M112332200. [DOI] [PubMed] [Google Scholar]
- 45.Thomas JD, Sideras P, Smith CIE, Vorechovsky I, Chapman V, Paul WE. Colocalization of X-linked agammaglobulinemia and X-linked immunodeficiency genes. Science. 1993;261:355–358. doi: 10.1126/science.8332900. [DOI] [PubMed] [Google Scholar]
- 46.Tsukada S, Saffran DC, Rawlings DJ, Parolini O, Allen RC, Klisak I, Sparkes RS, Kubagawa H, Mohandas T, Quan S, Belmont JW, Cooper MD, Conley ME, Witte ON. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell. 1993;72:279–290. doi: 10.1016/0092-8674(93)90667-f. [DOI] [PubMed] [Google Scholar]
- 47.Rawlings DJ, Saffran DC, Tsukada S, Largaespada DA, Grimaldi JC, Cohen L, Mohr RN, Bazan JF, Howard M, Copeland NG, Jenkins NA, Witte ON. Mutation of the unique region of Bruton's tyrosine kinase in immunodeficient XID mice. Science. 1993;261:358–361. doi: 10.1126/science.8332901. [DOI] [PubMed] [Google Scholar]
- 48.Lemmon MA. Plecstrin Homology Domains: Two Halves Make a Hole? Cell. 2005;120:574–576. doi: 10.1016/j.cell.2005.02.023. [DOI] [PubMed] [Google Scholar]
- 49.Sacristan C, Tussie-Luna MI, Logan SM, Roy AL. Mechanism of Bruton's tyrosine kinase-mediated recruitment and regulation of TFII-I. J. Biol. Chem. 2004;279:7147–7158. doi: 10.1074/jbc.M303724200. [DOI] [PubMed] [Google Scholar]
- 50.Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signaling. Nat. Rev. Mol. Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- 51.Clapham DE. Calcium signaling. Cell. 1995;80:259–268. doi: 10.1016/0092-8674(95)90408-5. [DOI] [PubMed] [Google Scholar]
- 52.van Rossum DB, Patterson RL, Sharma S, Barrow RK, Kornberg M, Gill DL, Snyder SH. Phospholipace Cγ1 controls surface expression of TRPC3 through an intermolecular PH domain. Nature. 2005;434:99–104. doi: 10.1038/nature03340. [DOI] [PubMed] [Google Scholar]
- 53.Caraveo G, van Rossum DB, Patterson RL, Snyder SH, Desiderio S. Action of TFII-I outside the nucleus as an inhibitor of agonist-induced calcium entry. Science. 2006;314:122–125. doi: 10.1126/science.1127815. [DOI] [PubMed] [Google Scholar]
- 54.Roy AL. Transcription factor TFII-I conducts a cytoplasmic orchestra. ACS Chem. Biol. 2006;1:619–622. doi: 10.1021/cb6004323. [DOI] [PubMed] [Google Scholar]
- 55.Gomez-Ospina N, Tsuruta F, Barreto-Chang O, Hu L, Dolmetsch R. The C terminus of the L-type voltage-gated calcium channel Ca(V)1.2 encodes a transcription factor. Cell. 2006;127:591–606. doi: 10.1016/j.cell.2006.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Naranjo JR, Mellstrom B. Split personality of transcription factors inside and outside the nuclear border. Science STKE. 2007;371:pe5. doi: 10.1126/stke.3712007pe5. [DOI] [PubMed] [Google Scholar]
- 57.Carrion AM, Link WA, Ledo F, Mellstrom B, Naranjo JR. DREAM is a Ca2+-regulated transcriptional repressor. Nature. 1999;398:80–84. doi: 10.1038/18044. [DOI] [PubMed] [Google Scholar]
- 58.Park CY, Dolmetsch R. The double life of a transcription factor takes it outside the nucleus. Science. 2006;314:64–65. doi: 10.1126/science.1133757. [DOI] [PubMed] [Google Scholar]
- 59.Rando OJ, Chi TH, Crabtree GR. Second messenger control of chromatin remodeling. Nat. Structrl. Biol. 2003;10:81–83. doi: 10.1038/nsb0203-81. [DOI] [PubMed] [Google Scholar]
- 60.Fu M, Wang C, Li Z, Sakamaki T, Pestell RG. Cyclin D1: normal and abnormal functions. Endocrinology. 2004;145:5439–5447. doi: 10.1210/en.2004-0959. [DOI] [PubMed] [Google Scholar]
- 61.Desgranges ZP, Ahn J, Lazebnik MB, Ashworth T, Lee C, Pestell RC, Rosenberg N, Prives C, Roy AL. Inhibition of TFII-I-dependent cell cycle regulation by p53. Mol Cell Biol. 2005;24:10940–10952. doi: 10.1128/MCB.25.24.10940-10952.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Desgranges ZP, Roy AL. TFII-I: Connecting Mitogenic Signals to Cell Cycle Regulation. Cell Cycle. 2006;5:356–359. doi: 10.4161/cc.5.4.2442. [DOI] [PubMed] [Google Scholar]
- 63.Hirota H, Matsuoka R, Chen XN, Salandanan LS, Lincoln A, Rose FE, Sunahara M, Osawa M, Bellugi U, Korenberg JR. Williams syndrome deficits in visual spatial processing linked to GTF2IRD1 and GTF2I on chromosome 7q11.23. Genet Med. 2003;5:311–321. doi: 10.1097/01.GIM.0000076975.10224.67. [DOI] [PubMed] [Google Scholar]
- 64.Tassabehji M, Hammond P, Karmiloff-Smith A, Thompson P, Thorgeirsson SS, Durkin ME, Popescu NC, Hutton T, Metcalfe K, Rucka A, Stewart H, Read AP, Maconochie M, Donnai D. GTF2IRD1 in craniofacial development of humans and mice. Science. 2005;310:1184–1187. doi: 10.1126/science.1116142. [DOI] [PubMed] [Google Scholar]