

Abstract
Mutations of the otoferlin gene have been shown to underlie deafness disorders in humans and mice. Analysis of genetically engineered mice lacking otoferlin have demonstrated an essential role for this protein in vesicle exocytosis at the inner hair cell afferent synapse. Here, we report on the molecular and phenotypic characterization of a new ENU-induced missense mutation of the mouse otoferlin gene designated Otofdeaf5Jcs. The mutation is a single T to A base substitution in exon 10 of Otof that causes a non-conservative amino acid change of isoleucine to asparagine in the C2B domain of the protein. Although strong immunoreactivity with an otoferlin-specific antibody was detected in cochlear hair cells of wild type mice, no expression was detected in mutant mice, indicating that the missense mutation has a severe effect on the stability of the protein and potentially its localization. Auditory brainstem response (ABR) analysis demonstrated that mice homozygous for the missense mutation are profoundly deaf, consistent with an essential role for otoferlin in inner hair cell neurotransmission. Vestibular-evoked potentials (VsEPs) of mutant mice, however, were equivalent to those of wild type mice, indicating that otoferlin is unnecessary for vestibular function even though it is highly expressed in both vestibular and cochlear hair cells.
Keywords: Otoferlin, Mutation, Deafness, Mouse, ABR, VsEP
1. Introduction
Studies of human and mouse deafness mutations have proven to be of great value in identifying genes and proteins critical to the hearing process. A mutation in the human otoferlin gene (OTOF) was shown to underlie the nonsyndromic prelingual deafness disorder DFNB9 (Yasunaga et al., 1999), and a genetically engineered null mutation of the orthologous mouse gene (Otof) also was shown to cause deafness in homozygous mutant mice (Roux et al., 2006). Otoferlin is a member of the mammalian ferlin family of proteins that share homology with the C. elegans FER-1 protein. Otoferlin and the two other mammalian ferlin family members, dysferlin and myoferlin, are membrane-anchored cytosolic proteins that contain six predicted C2 (protein kinase C conserved region 2) domains and appear to be involved in membrane fusion processes (Yasunaga et al., 2000). C2 domains have been implicated in Ca2+ and phospholipid binding and are found in a variety of membrane-associated proteins. The C2-domain containing protein synaptotagmin I localizes to synaptic vesicles and is thought to be essential for the fast, Ca2+-dependent component of neurotransmitter release (Rizo et al., 1998).
On the basis of its similarity to synaptotagmin and its expression in hair cells of the inner ear, otoferlin was hypothesized to be involved in synaptic vesicle trafficking in the auditory system (Yasunaga et al., 1999). Subsequent analyses of otoferlin deficient mice provided evidence that otoferlin is indeed essential for exocytosis and neurotransmitter release at the inner hair cell ribbon synapse (Roux et al., 2006). Although otoferlin is clearly crucial to hearing and likely involved in inner hair cell synaptic vesicle exocytosis, questions still remain about its specific function in this process (Brandt et al., 2007; Parsons, 2006; Roberts, 2006). The calcium binding kinetics of otoferlin have not yet been determined, and definitive evidence for its role as a calcium sensor requires further experimentation. One approach for assessing the functional roles of the putative Ca2+-binding C2 domains and other parts of the otoferlin protein is to analyze the phenotypes of animal models with mutations in these domains.
We report here on the molecular and phenotypic characterization of an ENU-induced mouse mutation, designated deaf5Jcs (henceforth abbreviated deaf5), that was discovered in a mutagenesis screen of proximal mouse Chromosome 5 (Wilson et al., 2005). We show that the deaf5 mutation is a single T to A base substitution in exon 10 of Otof that causes a nonconservative amino acid change of isoleucine to asparagine in the C2B domain of the protein. Auditory brainstem response (ABR) analysis demonstrates that mice homozygous for this missense mutation are profoundly deaf. Otoferlin is strongly expressed in vestibular as well as cochlear hair cells (Roux et al., 2006; Schug et al., 2006), yet mutant mice do not exhibit circling or head bobbing behaviors typical of vestibular dysfunction in mice. To directly assess receptor function, we measured vestibular-evoked potentials (VsEPs) (Jones et al., 2005), but found no evidence of vestibular deficits in deaf5/deaf5 mutant mice compared with controls.
2. Materials and methods
2.1. PCR, DNA sequencing, and genotyping
To screen for the deaf5 mutation, total RNA was isolated from inner ear tissue using TriZol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions, and cDNA was synthesized using the iScript cDNA syntesis kit (BioRad, Hercules, CA). Overlapping primer sets (Table 1) spanning the entire mouse Otof cDNA sequence (NM_031875) were used to amplify RT-PCR products for DNA sequence analysis. To confirm that the cDNA single base substitution of T to A was the mutation, genomic DNA for PCR was prepared from mouse tail samples using the Hot Shot method (Truett et al., 2000), and PCR primers flanking exon 10 of the Otof gene (Table 1) were used to amplify products for DNA sequence analysis. PCR products were purified with the QIAquick PCR Purification Kit or gel purified using the QIAquick gel cleanup kit (Qiagen Inc., Valencia, CA) according to the manufacturer’s protocol and then sequenced using an Applied Biosystems (Foster City, CA) 3700 DNA sequencer with an optimized Big Dye Terminator Cycle Sequencing method.
Table 1. PCR primers used for DNA sequence analysis of the Otof gene.
Primer names begin with “wex” if they correspond to “within exon” locations or “ex” if they flank an exon, followed by the exon number. “F” indicates a forward primer and “R” a reverse primer, relative to the direction of transcription.
| primer name | sequence 5′ - 3′ | expected product sizes (bp) |
|---|---|---|
| primers for sequencing Otof cDNA: | ||
| wex1F
wex7R |
TTGGTTGCCTTGGTCTCTGT
CGAGGCTAGAGACACGGAGT |
806 |
| wex6F
wex13R |
CCATGAAACTCGGCAAAACT
ATGAACGCCTTCTTCACGTT |
802 |
| wex12F
wex17R |
CAACATCAAGACACCCCACA
CTTCTTCATCCCCAGGCTCT |
839 |
| wex16F
wex22R |
ATGATTGACCGGAAAAATGG
GGAAGAGGGTCTTGACTTTGG |
811 |
| wex20/21F
wex26R |
AGTTGGAGAGCATGGGACAG
AAGGCAGCCAGTAGGTCTCC |
874 |
| wex26F
wex33R |
CCAGATCTACCGAGGCAGTG
CCACAGGTCTCATGTTGTCG |
825 |
| wex32F
wex38R |
AGGAGCCAGAGGAGGAAGAG
GCTTGCTGTAGAAGCGGTTT |
822 |
| wex38F
wex43R |
ATGTTGACAGTGGCCGTGTA
GGGATCTTGTACTCCGTCTCA |
875 |
| wex43F
wex46R |
AGCAGGAGGACAAACAGGAC
CAGGCCCCAAGGATCTTCT |
688 |
|
| ||
| genomic DNA primers for deaf5 mutation confirmation and genotyping: | ||
| ex10F
ex10R |
TACTGCCCACATGAGCTTTG
CAGAGGAATCCAGCTGAAGG |
349 wild type; 186 and 163 mutant |
To genotype mice, genomic DNA from mouse tail samples was amplified with PCR primers flanking exon 10 (Table 1). The PCR products were then digested with the restriction endonuclease BglII. The PCR product from wild type DNA, which contains a pallindromic BglII recognition sequence, is cleaved into 186 and 163 bp fragments, whereas the 349 bp product from mutant DNA is not cleaved because the BglII restriction site is destroyed by the deaf5 mutation (Fig. 1B).
Fig. 1. The deaf5 missense mutation of Otof.
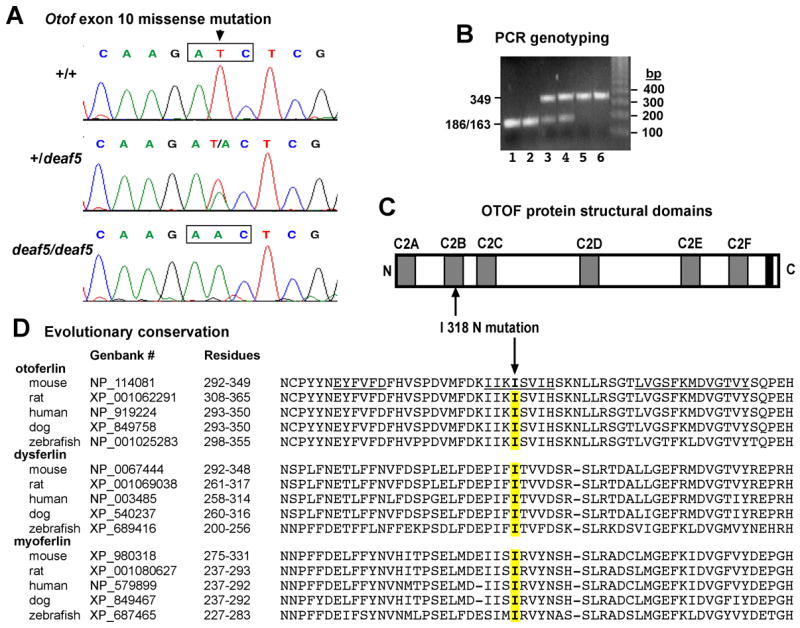
A. Sequence chromatograms of genomic DNA from +/+ wildtype mice, +/deaf5 heterozygotes, and deaf5/deaf5 mutants illustrating a T to A transversion in exon 10 of Otof that changes an ATC codon for isoleucine (I) to an AAC codon for asparagine (N). B. PCR-based assay to distinguish +/+ (lanes 1 and 2), +/deaf5 (lanes 3 and 4), and deaf5/deaf5 (lanes 5 and 6) genotypes of mice. The deaf5 mutation is a T to A transversion that destroys a BglII restriction site (AGATCT) in exon 10 of Otof, so that PCR amplification of genomic DNA with flanking primers followed by BglII digestion of the PCR products results in a single 349 bp fragment in mutant DNA and two small fragments of 186 and 163 bp (constituting one unresolved band in the gel shown) in wildtype DNA. C. Schematic representation of the mouse otoferlin protein with its C2 domains shown as gray rectangles and its single transmembrane domain shown as a black rectangle. The I to N amino acid substitution at residue 318 (I318N, indicated by upward pointing arrow), occurs within the C2B domain. D. Evolutionary conservation of the mutated isoleucine residue. ClustalW alignment comparing the partial C2B domain of mouse otoferlin with the homologous C2 domain regions of otoferlin, dysferlin, and myoferlin from mouse, rat, human, dog, and zebrafish. The mutated isoleucine (I) is indicated by the downward pointing arrow, and the corresponding amino acids of the homologous sequences are highlighted in yellow. The underlined residues in the mouse otoferlin sequence are predicted beta strands (BETApro Protein Beta Sheet Prediction, http://www.ics.uci.edu/~baldig/betasheet.html).
2.2. Auditory-evoked brainstem response (ABR)
Hearing assessment was performed as previously described (Zheng et al., 1999). Mice were anesthetized by intraperitoneal injection with tribromoethanol given at a dose of 2.5 mg per 10 g of body weight. Body temperature was maintained by placing mice on a heating pad in a sound-attenuating chamber. Needle electrodes were placed just under the skin, with the active electrode placed between the ears just above the vertex of the skull, the ground electrode between the eyes, and the reference electrode underneath the left ear. High frequency transducers were placed just inside the ear canal and computer-generated sound stimuli were presented at defined intervals. Thresholds were determined for a broad-band click and for 8, 16 and 32 kHz pure-tone stimuli by increasing the sound pressure level (SPL) in 10 dB increments followed by 5 dB increases and decreases to determine the lowest level at which a distinct ABR wave pattern could be recognized. Stimulus presentation and data acquisition were performed using the Smart EP evoked potential system (Intelligent Hearing Systems, Miami, FL).
2.3. Vestibular-evoked potentials (VsEPs)
Previously published methods for VsEP recordings in mice (Jones et al., 1999; Jones et al., 2004; Jones et al., 2006; Jones et al., 2005) were modified for the present study to utilize a noninvasive coupling system (developed by S. Jones, personal communication) for securing the mouse head to the mechanical shaker. Mice were anesthetized with 5 μl per gram body weight of a ketamine (18 mg/ml) and xylazine (2 mg/ml) solution injected intraperitoneally. Core body temperature was maintained at 37.0 ± 0.2°C using a homeothermic heating blanket system (FHC, Inc., Bowdoin, ME). The head was coupled to a custom platform with a head clip consisting of a lightweight plastic spring hair clip with tines modified to encircle the head anterior to the pinnae. Stimuli were delivered to the head using a voltage-controlled mechanical shaker. Linear acceleration pulses, of 2 ms duration, were presented to the cranium in the naso-occipital axis in an alternating manner between normal and inverted polarities. Stimuli were presented at a rate of 17 pulses/sec. Stimulus amplitude ranged from +6 dB to −18 dB re: 1.0 g/ms (where 1 g = 9.8m/s2) adjusted in 3 or 6 dB steps.
Stainless steel wire was placed subcutaneously at the nuchal crest to serve as the noninverting electrode. Needle electrodes were placed posterior to the right pinna and at the ventral neck for inverting and ground electrodes, respectively. Traditional signal averaging was used to resolve responses in electrophysiological recordings. Ongoing electroencephalographic activity was amplified (200,000X), filtered (300 to 3000Hz, −6 dB amplitude points) and digitized (1024 points, 8 μs/point). Multiple primary responses (256) were averaged for each VsEP response waveform. All responses were replicated. A bracketing threshold procedure was used wherein recordings began at the maximum stimulus intensity (i.e., +6 dB re: 1.0 g/ms) with and without acoustic masking, then intensity was decreased in 3 dB steps down to −18 dB. A final recording was conducted at +6 dB to confirm the quality of the preparation. A broad-band forward masker (50 to 50,000 Hz, 97 dB SPL) was presented during VsEP measurements to verify absence of cochlear responses. VsEP threshold, measured in dB re: 1.0 g/ms, was determined from the intensity series. Threshold was defined as the stimulus level midway between the level just producing a discernible response and the stimulus level that did not.
2.4. Immunofluorescence and phalloidin staining of hair cells
Immunofluorescence was carried out as described (Safieddine et al., 1997) on cochleae from three deaf5/deaf5 mutants and two +/deaf5 littermate controls at postnatal day 6 (P6). Inner ear tissues were fixed overnight at 4°C in 4% paraformaldehyde in PBS followed by sinking in 25% sucrose. Cochlear cryosections were cut 14 μm and mounted on Superfrost plus slides (Fisher Scientific, Pittsburgh, PA). Sections were blocked for 30 minutes at room temperature in 5% normal goat serum in PBS. Tissue sections were incubated overnight at 4° C with primary antibody diluted 1:250 in blocking solution. Slides were washed three times in PBS followed by a 30 minute incubation with goat anti-rabbit alexa fluor 488 (Invitrogen) diluted 1:500. Samples were washed with PBS and coverslipped with Fluoromount G (Electron Microscopy Sciences, Hatfield, PA). Fluorescent images were visualized using a Leitz (Wetzlar, Germany) DMRXE microscope with the appropriate fluorescent cubes and captured using a Leica DC300FX digital camera with Leica Firecam (v1.3) software.
Organ of Corti surface preparations and phalloidin staining of hair cell actin filaments were adapted from previously described methods (Beyer et al., 2000). Heads from four P6 deaf5/deaf5 mutants and three +/deaf5 littermate controls were bissected and fixed overnight in 4% paraformaldehyde. Inner ears were dissected away from the head and bone and stria vascularis were removed from the cochlea to expose the organ of Corti. Inner ear tissue was permeabilized in 0.3% Triton-X-100 for 5 minutes followed by a 30 minute incubation in rhodamine phalloidin (Invitrogen) diluted 1:100 in PBS. Samples were rinsed several times in PBS. The cochlear turns were separated from the modiolus and mounted on glass slides with Vectashield mounting media (Vector Laboratories, Burlingame, CA).
2.5 Mice
The deaf5 mutation was the only mutation with an associated hearing deficit that was identified in a previously described region-specific ENU mutagenesis screen (Wilson, et al., 2005). An F2 deaf5/deaf5 mouse from the original screen (on a mixed C57BL/6J, C3H strain background) was bred to a BALB mouse, and the resulting F1 mice were intercrossed. A homozygous mutant male from this cross was imported to The Jackson Laboratory from John Schimenti at Cornell University. Oocytes from female C3HeB/FeJ mice were fertilized in vitro with sperm from the imported deaf5/deaf5 male, and the resulting progeny were intercrossed to establish a mouse colony that is maintained by sibling mating, while keeping the deaf5 mutant allele segregating. This strain (an admixture of C57BL/6J, C3H, and BALB strains) is now called STOCK Otofdeaf5Jcs (Jackson Laboratory Stock Number 6128). Heterozygous and homozygous animals are identified by DNA genotyping as described above.
All experimental mice were housed in the Mouse Mutant Resource at the Jackson Laboratory, and all procedures involving their use were approved by the institutional Animal Care and Use Committee.
3. Results
The recessive, ENU-induced deaf5 mutation was discovered in a random mutagenesis screen of mouse Chr 5 (Wilson et al., 2005). Mutant mice were identified by their failure to exhibit a Preyer reflex (flick of the pinnae) in response to a loud clickbox sound burst. Non-complementation with a known Chr 5 deletion localized the deaf5 mutation between Shh (position 28.8 Mb, NCBI build 36) and D5Mit229 (position 31.9 Mb) (Wilson et al., 2005). This 3.1 Mb interval contains the otoferlin gene (Otof, position 30.7 Mb), and because mutations of this gene and its homolog are known to cause deafness in mice (Roux et al., 2006) and humans (Yasunaga et al., 1999), it was examined as a candidate for deaf5.
RT-PCR analysis of RNA extracted from inner ears showed that Otof transcripts are present in deaf5/deaf5 mutant mice. Otof mRNA from mutant mice was then screened for mutations by sequencing overlapping RT-PCR products that covered the entire cDNA (mouse reference sequence NM_031875). A single base substitution (T to A) at nucleotide 953 of the protein coding portion of the cDNA was the only alteration discovered in the mutant cDNA. The T to A mutation was confirmed by sequencing genomic DNA from four mutants, three controls and two heterozygotes for the exon 10 region of Otof (Fig. 1A). The T to A mutation destroys a BglII restriction site (AGATCT), which allows for an easy genotyping assay of genomic DNA (Fig. 1B). The deaf5 T to A mutation changes codon ATC to AAC, causing a nonconservative amino acid change from isoleucine (Ile, I) to asparagine (Asn, N) at residue 318 of the mouse OTOF protein (I318N missense mutation; NP_114081 reference sequence), which corresponds to residue 319 of human OTOF (NP_919224). The isoleucine residue at position 318 of the mouse OTOF protein is conserved in rat, human, dog, and zebrafish, and it is also conserved in the paralogous dysferlin and myoferlin proteins of these species (Fig. 1D).
To examine the effect of the deaf5 missense mutation on auditory function, we assessed hearing by ABR in 8 mutant mice (deaf5/deaf5), 8 homozygous controls (+/+), and 11 littermate heterozygotes (+/deaf5) at 24–31 days of age. All +/+ and +/deaf5 mice exhibited normal ABR thresholds to broad-band click and 8 kHz, 16 kHz, and 32 kHz pure-tone stimuli according to previously established criteria (Zheng et al., 1999). In contrast, ABRs could not be detected at all in deaf5/deaf5 mutant mice, even for stimuli intensities up to 100 dB SPL (Fig. 2A), indicating profound deafness and consistent with results reported for Otof knockout mice (Roux et al., 2006). Behavioral tests (movement, righting response and swim tests) indicated normal vestibular function in deaf5 mutant mice, consistent with results obtained from similar tests of Otof knockout mice (Roux et al., 2006). To more directly test for vestibular deficits in deaf5 mutant mice, we used a quantitative electrophysiological measure of gravity receptor function, the vestibular-evoked potential (VsEP), which has previously been used successfully to assess vestibular function in other mutant mice (Jones et al., 2005). VsEP thresholds of deaf5/deaf5 mutant mice were not different from those of +/deaf5 littermate controls (Fig. 2B).
Fig. 2. Auditory but not vestibular dysfunction in deaf5/deaf5 mutant mice.
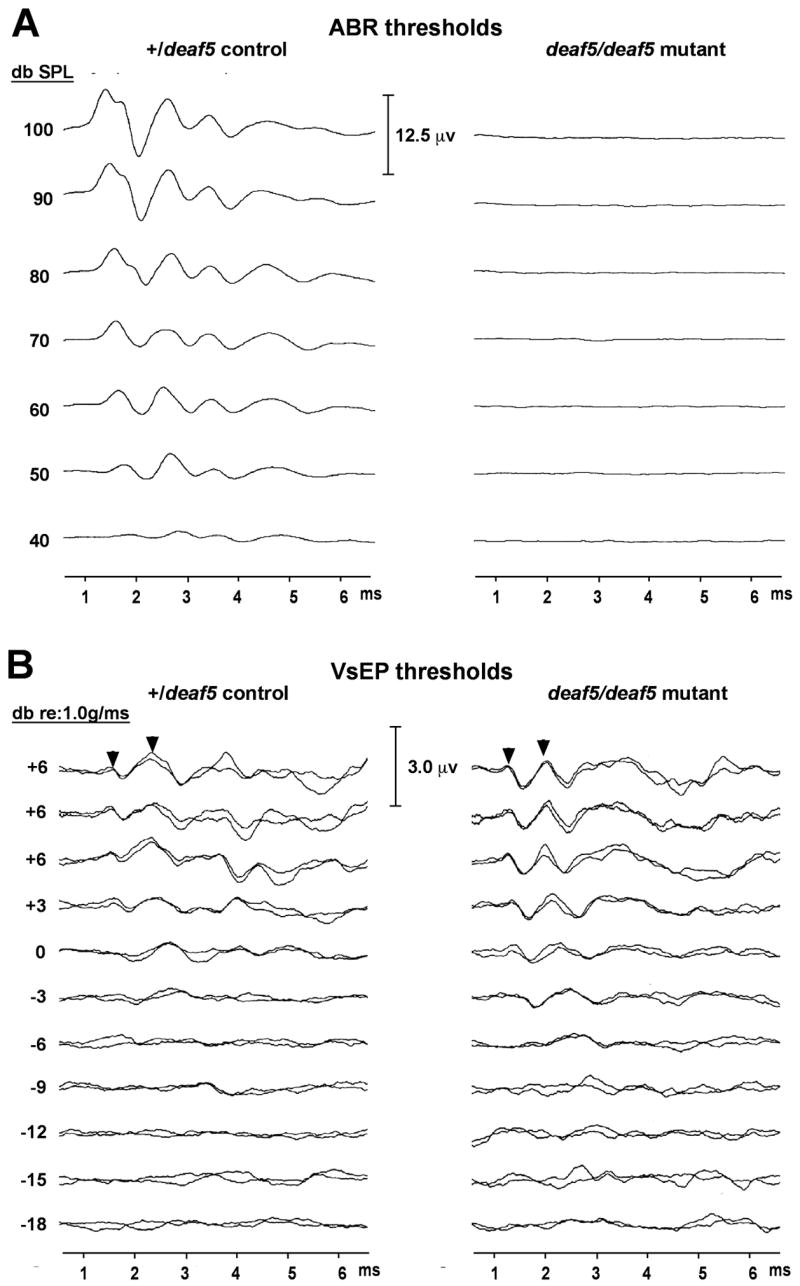
A. Auditory brainstem response (ABR) recordings show normal wave patterns and thresholds in +/deaf5 heterozygous control mice, but no evoked responses at all in deaf5/deaf5 homozygous mutant mice tested at 4 weeks of age. ABRs for broad-band click stimuli are shown here, but similar results were obtained for 8 kHz, 16 kHz, and 32 kHz pure tone stimuli. B. Vestibular evoked potential (VsEP) recordings of +/deaf5 control mice and deaf5/deaf5 mutant mice show nearly identical response amplitudes and threshold levels. Diagnostic peaks are indicated by arrowheads.
The deaf5 missense mutation causes profound deafness in mutant mice that could be the result of either partial (domain specific) or complete loss (null) of function. To distinguish between these possibities, we examined cochleae of three mutant and two heterozygous control mice at P6 for the presence of OTOF using a previously described OTOF-specific polyclonal antibody (Roux et al., 2006). OTOF-specific immunofluorescence was pronounced in cochlear hair cells of control mice but absent in mutant mice (Fig. 3A,B). To verify that hair cells are present in mutant inner ears, organ of Corti surface preparations from P6 mice were stained with phalloidin, which showed that hair cell numbers and hair bundle morphology are the same in +/deaf5 and deaf5/deaf5 mice (Fig. 3C,D). The lack of OTOF in hair cells of mutant mice indicates that the deaf5 missense mutation has a very severe effect on protein conformation or stability, which results in its degradation.
Fig. 3. Absence of OTOF in cochlear hair cells of deaf5/deaf5 mutant mice.
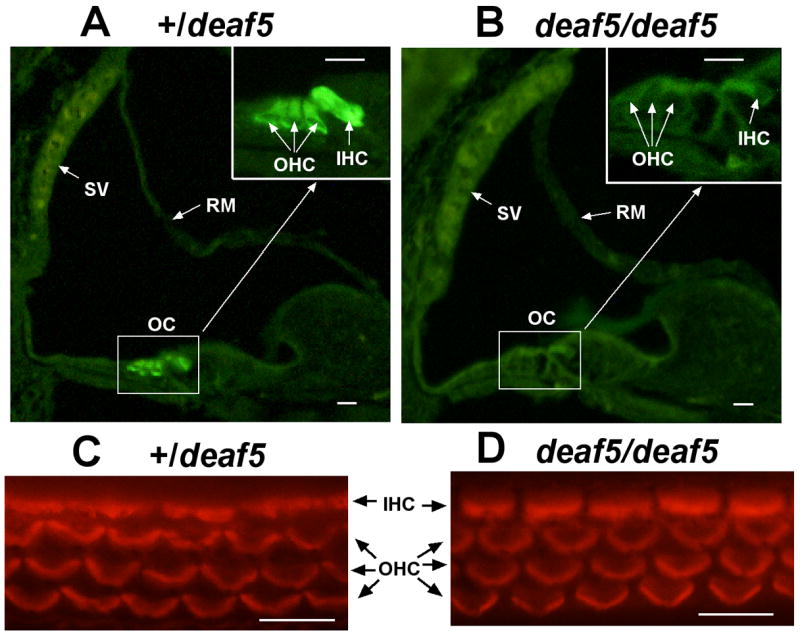
Cross sections through one turn of the cochlea from a P6 +/deaf5 control mouse (A) and a P6 deaf5/deaf5 mutant (B). The boxed region enclosing the organ of Corti (OC) is enlarged in the upper righthand corner in both A and B, and the stria vascularis (SV) and Reissner’s membrane (RM) are indicated by arrows. OTOF-specific immunofluorescence was present in inner hair cells (IHC) and outer hair cells (OHC) of the +/deaf5 control but was not detected in hair cells of the deaf5deaf5 mutant. Phalloidin staining of organ of Corti surface preparations from P6 +/deaf5 controls (C) and littermate deaf5/deaf5 mutants (D) verify that inner and outer hair cells are intact in mutant mice. All scale bars, 10 μm.
4. Discussion
We provide the following evidence that a missense mutation of Otof underlies the deafness phenotype of the ENU-induced deaf5 mutation: (1) the Otof gene lies within the 3.1 Mb candidate interval for the deaf5 mutation, (2) mutations of the otoferlin gene have been shown to underlie both human and mouse deafness disorders, (3) the deaf5 mutation (a T to A substitution) discovered in exon 10 of Otof is predicted to cause a change in a highly conserved amino acid residue (I318N), and (4) immunohistofluorescence analysis of cochlear cross sections demonstrates that deaf5 mutant mice lack otoferlin protein. Isoleucine is a nonpolar and hydrophobic amino acid, whereas asparagine is polar and uncharged. The change in side chain polarity induced by the I318N mutation, which occurs in a predicted beta strand of the C2B domain (Fig. 1D), may disrupt the normal conformation of the protein and lead to its instability.
Missense mutations in C2 domains of otoferlin have been reported to underlie several cases of human deafness, including the P490Q and I515T mutations in the C2C domain (Mirghomizadeh et al., 2002), the L1011P mutation in the C2D domain (Tekin et al., 2005), and the P1825A mutation in the C2F domain (Migliosi et al., 2002). Thus, ablations of individual C2 domains by themselves can disrupt the overall function of the protein, indicating that these domains are not simply redundant components that additively increase the Ca2+ binding property of the protein. Other deafness-causing missense mutations of the human otoferlin gene have been reported in the C-terminus (Varga et al., 2003) and in the coiled coil domain (Varga et al., 2006), demonstrating the importance of those regions as well. Human patients with congenital recessive deafness caused by mutations in the C2B domain of otoferlin have not yet been described, but the results presented here for deaf5 mutant mice suggest that clinical screening for mutations in this domain is warranted.
Phenotypes caused by mutations of otoferlin have been classified as auditory neuropathies because patients lack auditory brainstem responses but exhibit normal otoacoustic emissions (Rodriguez-Ballesteros et al., 2003; Varga et al., 2003). This phenotype also has been demonstrated in genetically engineered Otof-deficient mice (Roux et al., 2006) and in mice with an ENU-induced D1772G missense mutation in the C2F domain of otoferlin (Schwander et al., 2007). DPOAE testing has not yet been performed on deaf5 mutant mice, but this analysis will be necessary for a full phenotypic charactierization of this mutant. Our immunofluorescence results showing a lack of OTOF protein in cochlear hair cells of deaf5 mutant mice (Fig. 3) demonstrate that the effect of the I318N mutation on the overall conformation or stability of the protein is severe enough to cause its degradation or mislocalization. Unfortunately, the lack of protein in mutant hair cells precludes an in vivo assessment of the specific functional properties of the C2B domain, such as the effects of the mutation on Ca2+ binding or SNARE protein interactions or other vesicular membrane fusion events. It will be interesting to see if other deafness-causing missense mutations of otoferlin also result in a loss of protein from hair cells, or if this is a unique feature of the I318N mutation.
OTOF protein is highly expressed in both cochlear and vestibular hair cells of rat (Schug et al., 2006) and mouse (Roux et al., 2006) inner ears, yet mice with otoferlin mutations, although deaf, appear to have normal vestibular function on the basis of behavioral assessments. Results from behavioral tests, however, cannot exclude the possiblility that normal balance is achieved by compensation from visual or proprioceptive inputs. Thus, behavior by itself may not be a reliable indicator of vestibular deficits (de Caprona et al., 2004; Jones et al., 2004). To more directly test for vestibular dysfunction, we compared VsEP thresholds of mutant and control mice (Fig. 2B). These results show a normal electrophysiological response to linear accelerations in deaf5/deaf5 mutant mice and exclude the necessity of invoking compensatory sensory inputs to account for their normal balance behavior. If otoferlin is essential for vesicle exocytosis at afferent synapses of cochlear inner hair cells (Roux et al., 2006), then why is it not needed for vestibular hair cell neurotransmission? A possible explanation is that there may be other otoferlin-like proteins in synapses of vestibular, but not cochlear, hair cells that compensate for the loss of otoferlin. Another possible explanation for normal vestibular function in otoferlin deficient mice is that the large K+ conductance of type I vestibular hair cells may facilitate ephatic transmission, in which hair cell currents directly influence the calyx membrane potential without the involvement of synaptic vesicles (Goldberg, 1996). Alternatively, otoferlin’s crucial role in exocytosis may be restricted to the unique, rapid response properties of the cochlear inner hair cell and may not be necessary for neurotransmission in vestibular hair cells, which function at subacoustic frequencies and thus do not require an ultrafast rate of exocytosis.
Acknowledgments
We especially thank John Schimenti (Cornell University) for providing us with the deaf5Jcs mutant mice and Saaid Safieddine (Institut Pasteur) for providing us with the otoferlin antibody. We also thank Rob Burgess and Greg Cox (The Jackson Laboratory) for their careful review of the manuscript and Sandra Gray for mouse colony management. This work was supported by National Institutes of Health (NIH) Grants DC004301 and RR01183. The Jackson Laboratory institutional shared services are supported by NIH Grant CA34196.
LIST of ABBREVIATIONS
- OTOF
otoferlin
- C2 domain
protein kinase C conserved region 2
- ABR
auditory brain stem response
- VsEP
vestibular-evoked potential
- DPOAE
distortion product otoacoustic emission
- PCR
polymerase chain reaction
- RT-PCR
reverse transcriptase polymerase chain reaction
- kHz
kilohertz
- dB
decibel
- SPL
sound pressure level
- P6
postnatal day 6
- PBS
phosphate buffered saline
- ENU
N-ethyl-N-nitrosourea
- Chr
chromosome
- I or Ile
isoleucine
- N or Asn
asparagine
- SNARE
“soluble NSF (N-ethylmaleimide sensitive factor) attachment receptor” protein superfamily
- OC
organ of Corti
- SV
stria vascularis
- RM
Reissner’s membrane
- IHC
inner hair cell
- OHC
outer hair cell
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Beyer LA, Odeh H, Probst FJ, Lambert EH, Dolan DF, Camper SA, Kohrman DC, Raphael Y. Hair cells in the inner ear of the pirouette and shaker 2 mutant mice. J Neurocytol. 2000;29:227–40. doi: 10.1023/a:1026515619443. [DOI] [PubMed] [Google Scholar]
- Brandt N, Kuhn S, Munkner S, Braig C, Winter H, Blin N, Vonthein R, Knipper M, Engel J. Thyroid hormone deficiency affects postnatal spiking activity and expression of Ca2+ and K+ channels in rodent inner hair cells. J Neurosci. 2007;27:3174–86. doi: 10.1523/JNEUROSCI.3965-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Caprona MD, Beisel KW, Nichols DH, Fritzsch B. Partial behavioral compensation is revealed in balance tasked mutant mice lacking otoconia. Brain Res Bull. 2004;64:289–301. doi: 10.1016/j.brainresbull.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Goldberg JM. Theoretical analysis of intercellular communication between the vestibular type I hair cell and its calyx ending. J Neurophysiol. 1996;76:1942–57. doi: 10.1152/jn.1996.76.3.1942. [DOI] [PubMed] [Google Scholar]
- Jones SM, Erway LC, Bergstrom RA, Schimenti JC, Jones TA. Vestibular responses to linear acceleration are absent in otoconia- deficient C57BL/6JEi-het mice. Hear Res. 1999;135:56–60. doi: 10.1016/s0378-5955(99)00090-8. [DOI] [PubMed] [Google Scholar]
- Jones SM, Erway LC, Johnson KR, Yu H, Jones TA. Gravity receptor function in mice with graded otoconial deficiencies. Hear Res. 2004;191:34–40. doi: 10.1016/j.heares.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Jones SM, Jones TA, Johnson KR, Yu H, Erway LC, Zheng QY. A comparison of vestibular and auditory phenotypes in inbred mouse strains. Brain Res. 2006;1091:40–46. doi: 10.1016/j.brainres.2006.01.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SM, Johnson KR, Yu H, Erway LC, Alagramam KN, Pollak N, Jones TA. A quantitative survey of gravity receptor function in mutant mouse strains. J Assoc Res Otolaryngol. 2005;6:297–310. doi: 10.1007/s10162-005-0009-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliosi V, Modamio-Hoybjor S, Moreno-Pelayo MA, Rodriguez-Ballesteros M, Villamar M, Telleria D, Menendez I, Moreno F, Del Castillo I. Q829X, a novel mutation in the gene encoding otoferlin (OTOF), is frequently found in Spanish patients with prelingual non-syndromic hearing loss. J Med Genet. 2002;39:502–6. doi: 10.1136/jmg.39.7.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirghomizadeh F, Pfister M, Apaydin F, Petit C, Kupka S, Pusch CM, Zenner HP, Blin N. Substitutions in the conserved C2C domain of otoferlin cause DFNB9, a form of nonsyndromic autosomal recessive deafness. Neurobiol Dis. 2002;10:157–64. doi: 10.1006/nbdi.2002.0488. [DOI] [PubMed] [Google Scholar]
- Parsons TD. Neurobiology: auditory fidelity. Nature. 2006;444:1013–4. doi: 10.1038/4441013a. [DOI] [PubMed] [Google Scholar]
- Rizo J, Sudhof TC. C2-domains, structure and function of a universal Ca2+-binding domain. J Biol Chem. 1998;273:15879–82. doi: 10.1074/jbc.273.26.15879. [DOI] [PubMed] [Google Scholar]
- Roberts WM. Snaring otoferlin’s role in deafness. Cell. 2006;127:258–60. doi: 10.1016/j.cell.2006.10.010. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Ballesteros M, del Castillo FJ, Martin Y, Moreno-Pelayo MA, Morera C, Prieto F, Marco J, Morant A, Gallo-Teran J, Morales-Angulo C, Navas C, Trinidad G, Tapia MC, Moreno F, del Castillo I. Auditory neuropathy in patients carrying mutations in the otoferlin gene (OTOF) Hum Mutat. 2003;22:451–6. doi: 10.1002/humu.10274. [DOI] [PubMed] [Google Scholar]
- Roux I, Safieddine S, Nouvian R, Grati M, Simmler MC, Bahloul A, Perfettini I, Le Gall M, Rostaing P, Hamard G, Triller A, Avan P, Moser T, Petit C. Otoferlin, defective in a human deafness form, is essential for exocytosis at the auditory ribbon synapse. Cell. 2006;127:277–89. doi: 10.1016/j.cell.2006.08.040. [DOI] [PubMed] [Google Scholar]
- Safieddine S, Wenthold RJ. The glutamate receptor subunit delta1 is highly expressed in hair cells of the auditory and vestibular systems. J Neurosci. 1997;17:7523–31. doi: 10.1523/JNEUROSCI.17-19-07523.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schug N, Braig C, Zimmermann U, Engel J, Winter H, Ruth P, Blin N, Pfister M, Kalbacher H, Knipper M. Differential expression of otoferlin in brain, vestibular system, immature and mature cochlea of the rat. Eur J Neurosci. 2006;24:3372–80. doi: 10.1111/j.1460-9568.2006.05225.x. [DOI] [PubMed] [Google Scholar]
- Schwander M, Sczaniecka A, Grillet N, Bailey JS, Avenarius M, Najmabadi H, Steffy BM, Federe GC, Lagler EA, Banan R, Hice R, Grabowski-Boase L, Keithley EM, Ryan AF, Housley GD, Wiltshire T, Smith RJ, Tarantino LM, Muller U. A forward genetics screen in mice identifies recessive deafness traits and reveals that pejvakin is essential for outer hair cell function. J Neurosci. 2007;27:2163–75. doi: 10.1523/JNEUROSCI.4975-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekin M, Akcayoz D, Incesulu A. A novel missense mutation in a C2 domain of OTOF results in autosomal recessive auditory neuropathy. Am J Med Genet A. 2005;138:6–10. doi: 10.1002/ajmg.a.30907. [DOI] [PubMed] [Google Scholar]
- Truett GE, Heeger P, Mynatt RL, Truett AA, Walker JA, Warman ML. Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT) Biotechniques. 2000;29:52–54. doi: 10.2144/00291bm09. [DOI] [PubMed] [Google Scholar]
- Varga R, Kelley PM, Keats BJ, Starr A, Leal SM, Cohn E, Kimberling WJ. Non-syndromic recessive auditory neuropathy is the result of mutations in the otoferlin (OTOF) gene. J Med Genet. 2003;40:45–50. doi: 10.1136/jmg.40.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga R, Avenarius MR, Kelley PM, Keats BJ, Berlin CI, Hood LJ, Morlet TG, Brashears SM, Starr A, Cohn ES, Smith RJ, Kimberling WJ. OTOF mutations revealed by genetic analysis of hearing loss families including a potential temperature sensitive auditory neuropathy allele. J Med Genet. 2006;43:576–81. doi: 10.1136/jmg.2005.038612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson L, Ching YH, Farias M, Hartford SA, Howell G, Shao H, Bucan M, Schimenti JC. Random mutagenesis of proximal mouse chromosome 5 uncovers predominantly embryonic lethal mutations. Genome Res. 2005;15:1095–105. doi: 10.1101/gr.3826505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasunaga S, Grati M, Chardenoux S, Smith TN, Friedman TB, Lalwani AK, Wilcox ER, Petit C. OTOF encodes multiple long and short isoforms: genetic evidence that the long ones underlie recessive deafness DFNB9. Am J Hum Genet. 2000;67:591–600. doi: 10.1086/303049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasunaga S, Grati M, Cohen-Salmon M, El-Amraoui A, Mustapha M, Salem N, El-Zir E, Loiselet J, Petit C. A mutation in OTOF, encoding otoferlin, a FER-1-like protein, causes DFNB9, a nonsyndromic form of deafness. Nat Genet. 1999;21:363–9. doi: 10.1038/7693. [see comments] [DOI] [PubMed] [Google Scholar]
- Zheng QY, Johnson KR, Erway LC. Assessment of hearing in 80 inbred strains of mice by ABR threshold analyses. Hear Res. 1999;130:94–107. doi: 10.1016/s0378-5955(99)00003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]