

Abstract
The UP element, a component of bacterial promoters located upstream of the −35 hexamer, increases transcription by interacting with the RNA polymerase α-subunit. By using a modification of the SELEX procedure for identification of protein-binding sites, we selected in vitro and subsequently screened in vivo for sequences that greatly increased promoter activity when situated upstream of the Escherichia coli rrnB P1 core promoter. A set of 31 of these upstream sequences increased transcription from 136- to 326-fold in vivo, considerably more than the natural rrnB P1 UP element, and was used to derive a consensus sequence: −59 nnAAA(A/T)(A/T)T(A/T)TTTTnnAAAAnnn −38. The most active selected sequence contained the derived consensus, displayed all of the properties of an UP element, and the interaction of this sequence with the α C-terminal domain was similar to that of previously characterized UP elements. The identification of the UP element consensus should facilitate a detailed understanding of the α–DNA interaction. Based on the evolutionary conservation of the residues in α responsible for interaction with UP elements, we suggest that the UP element consensus sequence should be applicable throughout eubacteria, should generally facilitate promoter prediction, and may be of use for biotechnological applications.
Escherichia coli promoters recognized by the major form of RNA polymerase (RNAP Eσ70, subunit composition α2ββ′σ) contain up to three recognition elements. Two elements, hexamers centered approximately 10 and 35 bp upstream of the transcription start site (1, 2), interact with σ70 (3). The third element, the UP element, located upstream of the −35 hexamer, binds the C-terminal domain of the RNAP α-subunit (αCTD) (4, 5). The most extensively characterized UP element is an adenine (A) and thymine (T)-rich sequence located between −40 and −60 in the rrnB P1 promoter that stimulates promoter activity at least 30-fold by increasing the initial equilibrium constant (KB) and possibly a later step(s) in the transcription initiation pathway (kf) (4, 6). UP elements have also been described in other promoters and can function with holoenzymes containing different σ factors (4, 7–11).
The 8-kDa αCTD interacts with activator proteins as well as with DNA; the 28-kDa α N-terminal domain contains determinants for dimerization, assembly with the β- and β′-subunits, and also interacts with transcription factors (4, 5, 12–15). The two domains are connected by a flexible linker, which permits the αCTD to bind DNA and interact with activators at different sites upstream of the core promoter (5, 12, 16–18). The αCTD residues involved in DNA binding are highly conserved among eubacterial α-subunits (19, 20); therefore, the DNA sequences recognized by α are also very likely to be conserved.
Consensus sequences derived previously from E. coli promoters contain highly conserved −10 and −35 hexamers, but no highly conserved upstream sequences (1, 2, 21), suggesting that UP elements are not crucial for transcription of all promoters. However, UP elements may not be uncommon: upstream A+T-rich sequences in several E. coli and Bacillus subtilis promoters were found to interact with RNAP and/or to increase transcription in vitro in the absence of added factors (22–24). Many promoters contain A+T-rich sequences upstream of the −35 hexamer, and RNAP frequently protects DNA upstream of the −35 hexamer (25–28), although in most cases the functional significance of the A+T-rich sequences has not been established.
The number of upstream sequences proven to interact with α is insufficient to derive an accurate UP element consensus sequence. Therefore, we developed an in vitro selection followed by an in vivo screen to identify upstream sequences from a random DNA population that greatly increased promoter activity. We identified 31 upstream sequences conferring larger increases on rrnB P1 core promoter activity than any previously identified UP elements, characterized a representative for its effects on transcription and interactions with α in vitro, and derived a consensus UP element sequence.
MATERIALS AND METHODS
rrnB P1 Promoter Fragments with Random Upstream Sequences.
Promoter fragments used in the first round of selection were synthesized in vitro from two partially complementary oligonucleotides (Fig. 1A). Oligonucleotides [2.4 ng each; Genosys (The Woodlands, TX)/NSC Technologies (Mt. Prospect, IL)] were incubated in 40 mM Tris⋅HCl (pH 7.5), 20 mM MgCl2, and 50 mM NaCl for 5 min at 95°C, then annealed by slow cooling to 30°C. The 3′ ends were extended with 0.5 mM dNTPs and T7 DNA polymerase (Sequenase, Amersham) at 37°C for 1 hr, and the resulting fragments were extracted with phenol, then chloroform, and dNTPs were removed by using a Microcon 30K filter (Amicon). Eighteen randomly chosen fragments were sequenced after cloning into phage λ; the frequency of base pairs at each position in the randomized region (−59 to −38) was approximately equal. Promoter fragments (1 μg) were digested with HindIII and labeled with 5 μCi [α-32P]-dATP and T7 DNA polymerase for 15 min at 37°C in NEBuffer 2 (New England Biolabs).
Figure 1.

In vitro selection. (A) Synthesis of rrnB P1 promoter fragments with a randomized upstream region. Oligonucleotides were annealed and extended with T7 DNA polymerase to form a library of double-stranded DNA fragments with different UP element regions (−59 to −38). The top strand oligonucleotide (80 nt) contained (from 5′ to 3′) an EcoRI site (RI), rrnB P1 sequence from −66 to −60, random sequence from −59 to −38, and rrnB P1 sequence from −37 to +1. The bottom strand oligonucleotide (81 nt) contained (from 5′ to 3′) a HindIII site (H3) and rrnB P1 sequence from +50 to −17. Each contained a short additional sequence 5′ to the restriction site to ensure enzyme digestion. The randomized region is indicated by a hatched box, and −10 and −35 hexamers of rrnB P1 by open boxes. (B) Theoretical time course of RNAP binding to promoters containing (UP+) or lacking (UP−) an UP element. Broken line represents a time at which RNAP-promoter binding reactions were stopped to enrich for UP element-containing fragments.
In Vitro Selection.
Labeled promoter fragments were incubated with 4 nM RNAP (a gift from R. Landick, Univ. of Wisconsin) in 30 mM KCl, 10 mM Tris⋅Cl (pH 7.9), 10 mM MgCl2, 1 mM dithiothreitol, 0.1 mg/ml bovine serum albumin, 500 μM ATP, and 50 μM CTP for 4 min at 22°C, followed by addition of heparin (10 μg/ml). [ATP and CTP were required to stabilize the RNAP–rrnB P1 open complex (29, 30)]. RNAP–promoter complexes were separated from unbound DNA on 5% nondenaturing polyacrylamide gels, eluted by diffusion into Tris⋅HCl (10 mM)/EDTA (1 mM), pH 8.0, and the purified DNA was amplified by PCR by using Pfu DNA polymerase (Stratagene). Primers [Integrated DNA Technologies (Coralville, IA)/NSC Technologies] contained all nonrandomized promoter positions to reduce the frequency of PCR-generated mutations that might increase core promoter binding by RNAP. The downstream primer (101 nt) contained a HindIII site and rrnB P1 sequence from +50 to −37, and the upstream primer (21 nt) contained an EcoRI site and rrnB P1 sequence from −66 to −60. PCRs were monitored on gels and were stopped before heteroduplexes (resulting from annealing of noncomplementary products) could become a significant component in the population (31). The second and subsequent rounds of selection used PCR-amplified promoter fragments from the previous round, and the RNAP binding reactions were carried out under progressively more stringent conditions (lower RNAP concentration and shorter reaction times). Twenty-four rounds of in vitro selection were performed, and the most active promoters in vivo (see below) were sequenced after rounds 5, 9, 14, 19, 22, and 24.
In Vivo Activity Determination.
PCR-amplified DNAs were digested with EcoRI and HindIII and ligated to purified arms of phage λ to construct promoter–lacZ fusions (32, 33). Phage DNAs were packaged in vitro, and phage were plated on E. coli NK5031 on pH 6 MacConkey agar plates containing 3% lactose (33). Promoter DNAs from phage producing dark red plaques [i.e., higher β-galactosidase (β-gal) activity than from the rrnB P1 promoter (−66 to +50)] were sequenced using T7 DNA polymerase (Sequenase, Amersham) after PCR amplification directly from plaques. Strains monolysogenic for λ prophages carrying the promoter–lacZ fusions were distinguished from multilysogens by a PCR-based assay similar to that described previously (34), and β-gal activities were measured from cells grown in Luria-Bertani medium (33).
Lac and Hybrid-lac Promoters.
Strains containing lac or hybrid-lac promoters were derivatives of NK5031 containing promoter–lacZ fusions constructed in λ phage system II (6). The rrnB P1-lac hybrid promoter was described previously (6). The lac promoter (−40 to +52) with substituted upstream sequence from −59 to −41 (“SUB”; see Table 1 legend and ref. 6), and the 4192-lac hybrid promoter (containing the 4192 upstream sequence; Fig. 2A), were constructed by PCR using a lac promoter-containing plasmid as a template (11). The 4192-lac hybrid promoter contained (from 5′ to 3′) an EcoRI site, rrnB P1 sequence from −66 to −60, upstream sequence 4192 from −59 to −38, and lac promoter sequence from −37 to +52.
Table 1.
Effects of upstream sequences on lac core promoter activity
| Strain | Promoter | Miller units† | Relative activity |
|---|---|---|---|
| RLG 4288 | lac (−40 to +52)* | 50 | 1 |
| RLG 4208 | 4192-lac hybrid | 5,390 | 108 |
| RLG 4282 | rrnB P1-lac hybrid | 1,940 | 39 |
The sequence from position −59 to −38 in this construct is 5′-gac tgc agt ggt acc tag gag g-3′.
Average activity (two experiments with less than 2% variability).
Figure 2.

Upstream sequences and relative transcription activities of 31 in vitro-selected promoters used in defining an UP element consensus sequence. (A) Promoters contained wild-type rrnB P1 sequences (solid line; open boxes indicate the −10 and −35 hexamers) and different upstream regions (dotted line). Sequences of the nontemplate strand in the upstream region (−59 to −38) from 31 selected promoters, wild-type rrnB P1, and the rrnB P1 core promoter are shown. Wild-type rrnB P1 contained its natural UP element sequence, and the core rrnB P1 promoter contained an upstream sequence with no UP element function [the SUB sequence with A residues at positions −39 and −40 (6)]. Upstream sequence names are the strain numbers of λ lysogens carrying the promoter–lacZ fusions. Asterisks indicate promoters with single base-pair mutations (probably introduced during PCR amplification) downstream of the transcription start site (between +2 and +17). Sequence variation in this region of rrnB P1 does not affect promoter activity (35). Promoter activities are expressed relative to the activity of the core rrnB P1 promoter (activity = 1; strain RLG3097) and were determined from β-gal measurements in λ lysogens containing promoter–lacZ fusions. Relative activities differed by less than 10% in at least two different experiments. (B) Nucleotide frequencies (percentage of 31 sequences) at each position, −59 to −38, in the set of selected sequences shown in A. (C) Frequency diagram of the data in B. Each nucleotide is represented as a letter proportional in size to its frequency at that position in the selected population. (D) Consensus UP element sequence. One nucleotide is indicated when it is present in more than 55% of the population and two when together they represent more than 95% of the population.
In Vitro Transcription.
Promoter fragments were cloned into pRLG770 (36) and contained rrnB P1 sequences from −66 to −60 and from −37 to +50, and one of three different sequences from −59 to −38: the rrnB P1 UP element (pRLG4238); the SUB sequence, which has no UP element function (pRLG4210; see Fig. 2A and ref. 6), or the 4192 upstream sequence (pWR68). Supercoiled DNA concentrations were determined both spectrophotometrically and by quantitation of the amount of (vector-encoded) RNA-1 transcription under conditions of RNAP excess (40 nM). Transcription was carried out as described (4) except that reactions contained 170 mM NaCl. Reconstituted RNAPs (19, 37) were used at concentrations that resulted in equivalent transcription from the lacUV5 promoter (2.7 nM for RNAP containing wild-type α, 9 nM for αR265A, and 17.4 nM for αΔ235). Gels were analyzed by PhosphorImaging (Molecular Dynamics).
Footprinting.
Promoter fragments were generated by PCR from plasmids pRLG4238 (rrnB P1) or pWR68 (4192-rrnB P1) by using vector-specific primers, digested with HindIII (at position +50), and end-labeled with [α-32P]-dATP (DuPont) (38). Labeled fragments were purified on 5% acrylamide/7 M urea gels to eliminate nicked DNAs, eluted by diffusion, purified by using an Elutip (Schleicher & Schuell), incubated at 95°C for 4 min in 20 mM NaCl, 20 mM Tris⋅HCl, 1 mM EDTA (pH 7.4), and reannealed at 65°C for 30 min, followed by slow cooling to 30°C. Footprints were performed essentially as described (4, 19, 38). DNase I footprint reactions were done at 37°C with 59 nM wild-type or 82 nM αR265A RNAP. Hydroxyl radical footprint reactions were done at 22°C with 16 nM wild-type RNAP or 5 μM purified α. Gels were analyzed by PhosphorImaging.
RESULTS
Selection of Optimal Upstream Sequences.
An in vitro selection was used to identify sequences from a random DNA population fused upstream of the rrnB P1 −35 hexamer that would increase the rate of formation of complexes with RNAP (Fig. 1). The procedure was modeled after selections for binding sites for other proteins [e.g., SELEX (39), see also refs. 31, 40, 41]. In addition, the incubation time of the DNA fragments with RNAP was limited to enrich for promoters that bound RNAP rapidly, because we showed previously that the rrnB P1 UP element increased the rate of RNAP binding (6) (Fig. 1B).
We used RNAP holoenzyme, rather than purified α, so that the selected sequences would be positioned correctly with respect to the rest of the promoter. Correct alignment of upstream sequences with respect to the core promoter is necessary for function (24, 42–44). Although purified α binds to UP element DNA (4, 19), the stoichiometry and binding orientation of α in α–DNA complexes is not certain and could differ, in theory, from α bound in the context of holoenzyme.
The randomized section (−59 to −38) in the initial fragment population was limited to 22 bp to ensure inclusion of nearly all possible variants; a longer randomized sequence would have greatly increased the required amount of DNA and, for technical reasons, would have necessitated exclusion of a large majority of sequence variants. We did not randomize the wild-type residue at position −37 because the cytosine adjacent to the −35 hexamer is strongly favored in rrnB P1 (45).
The initial round of selection contained approximately 7 × 1012 promoter DNA fragments, representing about 40% of the 422 possible upstream sequence combinations. Complexes were separated from unbound DNA on gels, amplified by PCR, and subjected to 23 additional rounds of selection. The selection was considered complete after 24 rounds, because the sequence composition of fragments with high promoter activity did not vary substantially after round 22 (see below).
Selected Upstream Sequences Strongly Increase Promoter Activity in Vivo.
Promoters obtained by in vitro selection were fused to lacZ on a phage λ vector and screened by plaque color on indicator plates. Fifty to sixty percent of the fusions exhibited plaque phenotypes stronger than rrnB P1, and lysogens were constructed from 31 of these phages (24 from round 24 and seven from round 19). β-gal activities were compared with those from rrnB P1 promoter–lacZ fusions containing or lacking the natural rrnB P1 UP element. All 31 promoters were more active in vivo than the natural rrnB P1 promoter (Fig. 2A). The upstream sequence with the greatest effect increased transcription 326-fold, about 5-fold more than the rrnB P1 UP element. [The rrnB P1 UP element increased transcription 69-fold, about 2-fold more than that reported previously (6), most likely as a result of minor differences in the endpoints of the constructs used for comparison (see also Fig. 2 legend).]
Upstream Consensus Sequence.
The sequences of the 31 upstream regions (Fig. 2A) share common features. All are A+T-rich (64–91%), and most contain two A-tracts and an intervening T-tract. As illustrated in the frequency distribution (Fig. 2 B and C), almost all have A residues at positions −41 to −43, and greater than 50% have A at position −44. In addition, nearly all contain A residues at −56 and −57, and 81% contain an A at −55. Nearly all contain a T-tract from −47 to −50 and A or T residues between −51 and −54. Positions −38 to −40 contain only 8 of the 64 possible triplets, suggesting that there are also constraints for UP element function at these positions. There is little preference for specific residues at −45, −46, −58, and −59, in agreement with site-directed mutagenesis studies on the rrnB P1 UP element (S.T.E., W.R., S. Chen, T.G., W. Niu, R. H. Ebright, and R.L.G., unpublished work). The consensus determined from these 31 upstream sequences contains two conserved regions, a proximal region (−44 to −41, AAAA) and a distal region [−57 to −47, AAA(A/T)(A/T)T(A/T)TTTT] separated by two nonconserved positions (−45 and −46; Fig. 2D).
A Selected Upstream Sequence Increases lac Promoter Activity.
We fused a representative selected upstream sequence (4192, containing a perfect match to the consensus; Fig. 2A) to another core promoter, lac, to determine whether, like the rrnB P1 UP element (4, 6), it functioned as a separable promoter element. Upstream sequence 4192 increased transcription from the lac core promoter 108-fold in vivo, greater than the 39-fold effect of the rrnB P1 UP element on the lac core promoter (Table 1), consistent with the relative effects of the two upstream sequences on the rrnB P1 core promoter (Fig. 2). The two upstream sequences increased transcription of rrnB P1 slightly more than lac (Fig. 2), reflecting differences in the kinetic characteristics and/or sequences of positions −39 and −38 in the two core promoters (Table 1). It has been proposed that upstream sequences can confer different effects on promoters with different kinetic characteristics (46, 47).
Upstream Sequence 4192 Increases Transcription by Purified RNAP in Vitro and Requires the αCTD for Function.
We compared transcription in vitro of the rrnB P1 core promoter, the rrnB P1 promoter with its natural UP element, and the rrnB P1 promoter with upstream sequence 4192 (Fig. 3). Like the rrnB P1 UP element, upstream sequence 4192 stimulated transcription in the absence of factors other than RNAP (lanes 1, 4, and 7), and it had a greater effect than the wild-type rrnB P1 UP element, consistent with the relative effects of the two sequences in vivo.
Figure 3.

In vitro transcription of plasmid templates containing rrnB P1 promoters with either the wild-type (WT) UP element (lanes 1–3), no UP element (the SUB sequence, see Fig. 2A, lanes 4–6), or the 4192 selected upstream sequence (lanes 7–9). Plasmids were transcribed with WT RNAP (lanes 1, 4, and 7), αΔ235 RNAP (lanes 2, 5, and 8), or αR265A RNAP (lanes 3, 6, and 9). rrnB P1 transcripts (terminated at rrnB T1 ≈220 nt downstream of the transcription start site) and vector-encoded RNA I transcripts (≈110 nt) are indicated with arrows.
We also tested whether this stimulation depended on the DNA binding domain of the RNAP α-subunit by transcribing the same templates with RNAP lacking the αCTD (Fig. 3, lanes 2, 5, and 8) or with RNAP containing an alanine substituted at position 265 of α (R265A; lanes 3, 6, and 9). As with the wild-type rrnB P1 UP element (lanes 1–3; refs. 4 and 19), upstream sequence 4192 increased transcription only with RNAP containing the wild-type αCTD (lanes 7–9). Upstream sequence 4192 therefore has the characteristics of an UP element.
UP Element Sequences Protected by RNAP and by Purified α.
We used DNase I footprinting to determine whether UP element 4192 was protected by RNAP, and whether the interaction depended on amino acid residue R265 in α, as was observed for the rrnB P1 UP element (4, 19). (Because DNase I cleaves A+T-rich sequences inefficiently, only a subset of UP element positions could be monitored for RNAP binding.) As expected, both the core promoter region and positions in UP element 4192 were protected by wild-type RNAP (Fig. 4, lane 3, small arrows; positions −46, −47, −59, and −60). Position −44 (large arrow) was hypersensitive to DNase I. αR265A RNAP protected only the core promoter and did not display the characteristic enhancement at position −44. We conclude that the αCTD is required for binding of RNAP to UP element 4192.
Figure 4.
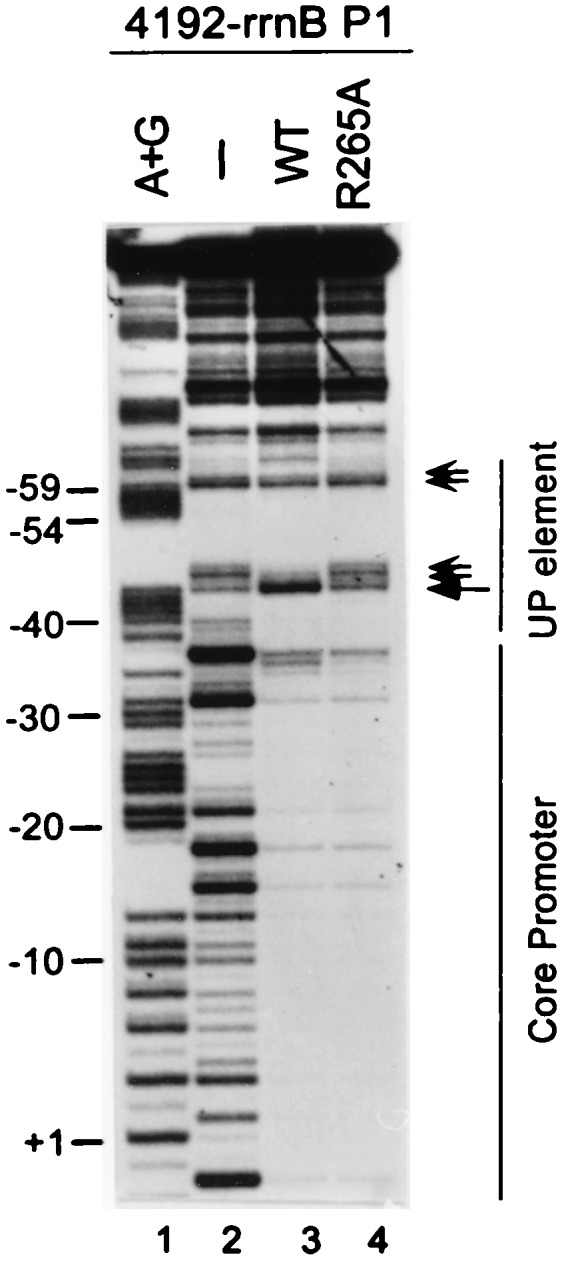
DNase I footprints of RNAP bound to the 4192-rrnB P1 promoter. The DNA fragment was labeled at promoter position +50 in the nontemplate strand. Lanes: 1, A+G sequence markers; 2, no RNAP; 3, wild-type (WT) RNAP; 4, αR265A RNAP. Small arrows indicate upstream region positions protected by WT RNAP, but not by αR265A RNAP. Large arrow indicates a position of enhanced DNase I cleavage in the WT RNAP footprint.
The positions protected by RNAP and by purified α in the 4192 and rrnB P1 UP elements were investigated in finer detail by using hydroxyl radical footprints (Fig. 5). Both RNAP and α protected regions centered at about −40 and −51 in each UP element (Fig. 5, lanes 3, 4, 8, and 9). We conclude that the 4192 UP element interacts with α similarly to previously characterized UP elements.
Figure 5.

Hydroxyl radical footprints of RNAP or purified α bound to rrnB P1 promoters containing either the 4192 UP element (A) or the rrnB P1 UP element (B). DNA fragments were labeled in the nontemplate strand at position +50. Lanes 1 and 6, A+G sequence markers; lanes 2, 5, 7, and 10, no protein; lanes 3 and 7, purified α; lanes 4 and 9, WT RNAP. The protected regions in the UP element and core promoter are indicated by vertical bars at the right of each panel.
The most obvious difference in the footprints of the two UP elements was that the proximal (−40) region was protected less fully in the 4192 UP element than in the rrnB P1 UP element, especially with purified α (Fig. 5 and PhosphorImager scans, not shown). Purified α bound to the distal region of each UP element with approximately equal affinity in titration experiments (data not shown). Thus, the strength of the 4192 UP element cannot be attributed solely to an increased affinity for α (see Discussion). We conclude that α recognizes both UP elements similarly, but not identically, and it is not clear whether the minor differences in the footprints are related to the relative effects of the two UP elements on transcription.
DISCUSSION
An UP Element Consensus Sequence.
We identified upstream sequences that increased promoter activity as much as 326-fold, much more than any previously discovered UP element. Characterization of a representative sequence indicated that it had the features of an UP element, functioning through interactions with the RNAP αCTD. The upstream sequences allowed us to derive an UP element consensus that should facilitate identification of bacterial promoters in diverse eubacteria, aid our understanding of promoter recognition by RNAP, and may be of interest for biotechnological applications.
The consensus UP element sequence contains 15 highly conserved positions in two DNA regions, each of which can function independently and may comprise a binding site for an αCTD monomer (S.T.E., W.R., S. Chen, T.G., W. Niu, R. H. Ebright, and R.L.G., unpublished work). In support of this model, promoters deleted for the distal (−51) region retain substantial UP element activity (6), and the two regions function with wild-type efficiency when separated by an 11-bp insertion (42). Initial studies indicate that positions −41 to −43 and −51 to −53 are most crucial for function, and that the proximal (−40) region increases transcription more than the distal region (S.T.E., W.R., S. Chen, T.G., W. Niu, R. H. Ebright, and R.L.G., unpublished work). Flexible and independent positioning of αCTD monomers is also supported by recent studies indicating that α can interact simultaneously with DNA on either side of the catabolite activator protein (CAP) in RNAP–CAP–DNA complexes (17). Thus, in some cases, UP elements may be positioned at least one DNA turn further upstream than those examined here (refs. 8 and 42; S. E. Aiyar, R.L.G., and W.R., unpublished work).
Because each αCTD monomer would be predicted to use the same protein surface for DNA binding, the “two site” model suggests that similar α recognition sequences should be found in each UP element region. A-tracts present in each subsite may constitute part of the α recognition sequence. Because A-tract DNA displays noncanonical B-form features (48), characteristics of DNA structure could play a role in recognition by α. Nevertheless, it is not clear whether α recognizes a backbone conformation determined by the sequence, functional groups in the major or minor grooves, or a combination of these features. Furthermore, differences between the proximal and distal binding sequences could result from constraints placed on the proximal site by the presence of the −35 hexamer and its interaction with the RNAP σ-subunit.
Our goal was to identify the upstream DNA sequences best able to stimulate transcription. The sequence with the largest effect, UP element 4192, increased promoter activity about 5-fold more than the rrnB P1 UP element, but did not bind purifed α with higher affinity. It is possible that UP element 4192 might have a larger effect on later steps in initiation than the rrnB P1 UP element and/or it might position an αCTD monomer(s) more effectively than the rrnB P1 UP element. Because interactions between RNAP and either upstream A-tracts (presumably UP elements; see below) or transcription factors can reduce the activities of certain promoters by inhibiting promoter escape (46, 49, 50), it is possible that sequences with higher affinity for α might exist but have been eliminated in our in vivo screen for high promoter activity. Some promoters in the final population selected in vitro did not result in high β-gal activity, and in theory some of these promoters might contain tighter binding sites for α. Further studies will be required to determine whether there are DNA sequences with higher affinity for α and whether they lead to less than maximal transcription of rrnB P1.
Relationship Between UP Elements and Sequences That Result in DNA Bending.
The UP element consensus sequence defined here contains A-tracts (Fig. 2). Because A-tracts phased with the helical repeat lead to DNA bending and can increase transcription (43, 46, 51), it has been proposed that upstream DNA curvature per se can increase promoter activity (52). In other work (S. E. Aiyar, R.L.G., and W.R., unpublished results) we found that phased A-tracts fused to the rrnB P1 or lac core promoters stimulated transcription 15- to 20-fold by binding to the αCTD. However, this stimulation was less than 20% of that observed with the most active UP element described here. In addition, some UP elements (e.g., rrnB P1) do not display obvious curvature (53). Thus, we suggest that the macroscopic curvature conferred by phased A-tract sequences is not the major feature responsible for α binding. Further genetic and structural analyses will be required to define the critical contacts between individual nucleotides and amino acids in the αCTD–DNA complex and the role (if any) of other localized DNA structural features in DNA recognition by α.
Occurrence of UP Elements in Bacterial Promoters.
UP element sequences characterized thus far suggest a general correlation between the extent of similarity to the consensus and the ability to stimulate transcription. The rrnB P1 and rrnD P1 UP elements both greatly increase transcription and contain relatively good matches to the consensus (Fig. 6 and ref. 11). UP elements in certain other promoters exhibit poorer matches to the consensus and increase transcription only 2- to 10-fold [λ PL2 (9); phage Mu Pe (10); rrnB P2, RNA II, and merT (ref. 11); see also ref. 55].
Figure 6.

Comparison of upstream sequences with large effects on promoter activity to the UP element consensus sequence (see Discussion). Sequences were aligned by their −35 hexamers where applicable [the spoVG promoter is recognized by a holoenzyme with an alternative σ-subunit (54)]. Sequence numbering refers to the rrnB P1 promoter.
The residues in αCTD responsible for DNA binding (L262, R265, N268, C269, G296, K298, and S299) are highly conserved (19, 20), suggesting that the UP element sequences in most eubacteria will be the same as in E. coli. A compilation of B. subtilis promoter sequences (26) previously indicated a preference for A- and T-tracts at precisely the positions established here for the E. coli consensus UP element. It was suggested that these sequences probably represent binding sites for the αCTD (26), and in fact the B. subtilis flagellin promoter was shown to contain an upstream sequence that increases transcription 20-fold in vivo by interacting with α (8). Two other B. subtilis promoters, spoVG and the bacteriophage SP82 promoter AluI56, contain A+T-rich upstream sequences that are almost perfect matches to the consensus (Fig. 6) and dramatically increase promoter activity in vivo and in vitro (22, 56). Although the role of the αCTD has not been confirmed in most cases, it appears that UP element-like sequences occur frequently in promoters from Gram-positive bacteria (26, 57).
We searched known E. coli promoters and putative promoters from the entire E. coli genome sequence for matches to the UP element sequences identified in our selection (S.T.E., A. Huerta, J. Collado-Vides, and R.L.G., unpublished data). Approximately 3% of promoters for mRNAs and 19% of promoters for stable RNAs had sequences upstream of the −35 hexamer with at least 11 of 15 matches to the consensus between −57 and −41 (positions −59, −58, −46, −45, −40, −39, and −38 were excluded from the search, see Results). Because sequences with lesser similarity to the consensus also have some UP element function (11), we suggest that UP elements are a common component of E. coli promoters.
Concluding Remarks.
We favor a view of bacterial promoter structure in which three RNAP recognition elements (the −10 and −35 hexamers and the UP element) function as semiindependent modules (see also ref. 23). In this general view, promoter activity correlates positively with the number of promoter elements present and positioned correctly, with the extent of similarity of each to the consensus, and with the relative importance of individual matching positions within each module. In addition, there may be negative contributions from nucleotides least favored at specific positions. In this context, the effectiveness of a particular UP element will be determined not only by its similarity to the UP element consensus, but also by the strength and kinetic characteristics of the core promoter. In extreme cases, an increased match to consensus may decrease transcription by reducing promoter clearance.
Acknowledgments
We thank L. Gold for suggestions early in the course of this work, J. Collado-Vides and A. Huerta for computer analyses, NSC Technologies for providing oligonucleotides, and R. Ebright for comments on the manuscript. This work was supported by National Institutes of Health (NIH) Grant GM37048 (to R.L.G.). S.T.E. was supported by an NIH Genetics Predoctoral Training Grant and by a Hatch grant from the U.S. Department of Agriculture.
ABBREVIATIONS
- RNAP
RNA polymerase
- αCTD
C-terminal domain of the RNAP α-subunit
- β-gal
β-galactosidase
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Hawley D K, McClure W R. Nucleic Acids Res. 1983;11:2237–2255. doi: 10.1093/nar/11.8.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harley C B, Reynolds R P. Nucleic Acids Res. 1987;15:2343–2361. doi: 10.1093/nar/15.5.2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dombroski A J, Walter W A, Record M T J, Siegele D A, Gross C A. Cell. 1992;70:501–512. doi: 10.1016/0092-8674(92)90174-b. [DOI] [PubMed] [Google Scholar]
- 4.Ross W, Gosink K K, Salomon J, Igarashi K, Zou C, Ishihama A, Severinov K, Gourse R L. Science. 1993;262:1407–1413. doi: 10.1126/science.8248780. [DOI] [PubMed] [Google Scholar]
- 5.Blatter E E, Ross W, Tang H, Gourse R L, Ebright R H. Cell. 1994;78:889–896. doi: 10.1016/s0092-8674(94)90682-3. [DOI] [PubMed] [Google Scholar]
- 6.Rao L, Ross W, Appleman J A, Gaal T, Leirmo S, Schlax P J, Gourse R L. J Mol Biol. 1994;235:1421–1435. doi: 10.1006/jmbi.1994.1098. [DOI] [PubMed] [Google Scholar]
- 7.Newlands J T, Gaal T, Mecsas J, Gourse R L. J Bacteriol. 1993;175:661–668. doi: 10.1128/jb.175.3.661-668.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fredrick K, Caramori T, Chen Y-C, Galizzi A, Helmann J D. Proc Natl Acad Sci USA. 1995;92:2582–2586. doi: 10.1073/pnas.92.7.2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giladi H, Murakami K, Ishihama A, Oppenheim A B. J Mol Biol. 1996;260:484–491. doi: 10.1006/jmbi.1996.0416. [DOI] [PubMed] [Google Scholar]
- 10.van Ulsen P, Hillebrand M, Kainz M, Collard R, Zulianello L, van de Putte P, Gourse R L, Goosen N. J Bacteriol. 1997;179:530–537. doi: 10.1128/jb.179.2.530-537.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ross, W., Aiyar, S. E., Salomon, J. & Gourse, R. L. (1998) J. Bacteriol. in press. [DOI] [PMC free article] [PubMed]
- 12.Busby S, Ebright R H. Cell. 1994;79:743–746. doi: 10.1016/0092-8674(94)90063-9. [DOI] [PubMed] [Google Scholar]
- 13.Ishihama A. J Bacteriol. 1993;175:2483–2489. doi: 10.1128/jb.175.9.2483-2489.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Negishi T, Fujita N, Ishihama A. J Mol Biol. 1995;248:723–728. doi: 10.1006/jmbi.1995.0254. [DOI] [PubMed] [Google Scholar]
- 15.Niu W, Kim Y, Dong Q, Ebright Y W, Ebright R H. Cell. 1996;87:1123–1134. doi: 10.1016/s0092-8674(00)81806-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jeon Y H, Yamazaki T, Otomo T, Ishihama A, Kyogoku Y. J Mol Biol. 1997;267:953–962. doi: 10.1006/jmbi.1997.0902. [DOI] [PubMed] [Google Scholar]
- 17.Murakami K, Owens J T, Belyaeva T A, Meares C F, Busby S J, Ishihama A. Proc Natl Acad Sci USA. 1997;94:11274–11278. doi: 10.1073/pnas.94.21.11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Belyaeva T A, Bown J A, Fujita N, Ishihama A, Busby S J. Nucleic Acids Res. 1996;24:2242–2251. doi: 10.1093/nar/24.12.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gaal T, Ross W, Blatter E E, Tang H, Jia X, Krishnan V V, Assa-Munt N, Ebright R H, Gourse R L. Genes Dev. 1996;10:16–26. doi: 10.1101/gad.10.1.16. [DOI] [PubMed] [Google Scholar]
- 20.Murakami K, Fujita N, Ishihama A. EMBO J. 1996;15:4358–4367. [PMC free article] [PubMed] [Google Scholar]
- 21.Lisser S, Margalit H. Nucleic Acids Res. 1993;21:1507–1516. doi: 10.1093/nar/21.7.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Banner C D, Moran C P J, Losick R. J Mol Biol. 1983;168:351–365. doi: 10.1016/s0022-2836(83)80023-0. [DOI] [PubMed] [Google Scholar]
- 23.Bujard H, Brenner M, Deuschle U, Kammerer W, Knaus R. In: RNA Polymerase and the Regulation of Transcription. Reznikoff W S, Burgess R R, Dahlberg J E, Gross C A, Record Jr M T, Wickens M P, editors. New York: Elsevier; 1987. pp. 95–103. [Google Scholar]
- 24.Frisby D, Zuber P. J Bacteriol. 1991;173:7557–7564. doi: 10.1128/jb.173.23.7557-7564.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Galas D J, Eggert M, Waterman M S. J Mol Biol. 1985;186:117–128. doi: 10.1016/0022-2836(85)90262-1. [DOI] [PubMed] [Google Scholar]
- 26.Helmann J D. Nucleic Acids Res. 1995;23:2351–2360. doi: 10.1093/nar/23.13.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ozoline O N, Deev A A, Arkhipova M V. Nucleic Acids Res. 1997;25:4703–4709. doi: 10.1093/nar/25.23.4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ozoline O N, Tsyganov M A. Nucleic Acids Res. 1995;23:4533–4541. doi: 10.1093/nar/23.22.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gourse R L. Nucleic Acids Res. 1988;16:9789–9809. doi: 10.1093/nar/16.20.9789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Borukhov S, Sagitov V, Josaitis C A, Gourse R L, Goldfarb A. J Biol Chem. 1993;268:23477–23482. [PubMed] [Google Scholar]
- 31.Pollock R, Treisman R. Nucleic Acids Res. 1990;18:6197–6204. doi: 10.1093/nar/18.21.6197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miura A, Krueger J H, Itoh S, deBoer H A, Nomura M. Cell. 1981;25:773–782. doi: 10.1016/0092-8674(81)90185-9. [DOI] [PubMed] [Google Scholar]
- 33.Gaal T, Barkei J, Dickson R R, deBoer H A, deHaseth P L, Alavi H, Gourse R L. J Bacteriol. 1989;171:4852–4861. doi: 10.1128/jb.171.9.4852-4861.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Powell B S, Rivas M P, Court D L, Nakamura Y, Rivas M P, Turnbough C L., Jr Nucleic Acids Res. 1994;22:5765–5766. doi: 10.1093/nar/22.25.5765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bartlett M S, Gourse R L. J Bacteriol. 1994;176:5560–5564. doi: 10.1128/jb.176.17.5560-5564.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ross W, Thompson J F, Newlands J T, Gourse R L. EMBO J. 1990;9:3733–3742. doi: 10.1002/j.1460-2075.1990.tb07586.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang H, Kim Y, Severinov K, Goldfarb A, Ebright R H. Methods Enzymol. 1996;273:130–134. doi: 10.1016/s0076-6879(96)73012-4. [DOI] [PubMed] [Google Scholar]
- 38.Newlands J T, Ross W, Gosink K, Gourse R L. J Mol Biol. 1991;220:569–583. doi: 10.1016/0022-2836(91)90101-b. [DOI] [PubMed] [Google Scholar]
- 39.Tuerk C, Gold L. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 40.Blackwell T K, Weintraub H. Science. 1990;250:1104–1110. doi: 10.1126/science.2174572. [DOI] [PubMed] [Google Scholar]
- 41.Wright W E, Binder M, Funk W. Mol Cell Biol. 1991;11:4104–4110. doi: 10.1128/mcb.11.8.4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Newlands J T, Josaitis C A, Ross W, Gourse R L. Nucleic Acids Res. 1992;29:719–726. doi: 10.1093/nar/20.4.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bracco L, Kotlarz D, Kolb A, Diekmann S, Buc H. EMBO J. 1989;8:4289–4296. doi: 10.1002/j.1460-2075.1989.tb08615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McAllister C F, Achberger E C. J Biol Chem. 1989;264:10451–10456. [PubMed] [Google Scholar]
- 45.Josaitis C, Gaal T, Ross W, Gourse R L. Biochim Biophys Acta. 1990;1050:307–311. doi: 10.1016/0167-4781(90)90186-6. [DOI] [PubMed] [Google Scholar]
- 46.Ellinger T, Behnke D, Knaus R, Bujard H, Gralla J D. J Mol Biol. 1994;239:466–475. doi: 10.1006/jmbi.1994.1389. [DOI] [PubMed] [Google Scholar]
- 47.Tang Y, Murakami K, Ishihama A, deHaseth P L. J Bacteriol. 1996;178:6945–6951. doi: 10.1128/jb.178.23.6945-6951.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Young M A, Srinivasan J, Goljer I, Kumar S, Beveridge D L, Bolton P H. Methods Enzymol. 1995;261:121–144. doi: 10.1016/s0076-6879(95)61007-3. [DOI] [PubMed] [Google Scholar]
- 49.Choy H E, Park S W, Aki T, Parrack P, Fujita N, Ishihama A, Adhya S. EMBO J. 1995;14:4523–4529. doi: 10.1002/j.1460-2075.1995.tb00131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Monsalve M, Calles B, Mencia M, Salas M, Rojo F. Mol Cell. 1997;1:99–107. doi: 10.1016/s1097-2765(00)80011-8. [DOI] [PubMed] [Google Scholar]
- 51.Gartenberg M R, Crothers D M. J Mol Biol. 1990;219:217–230. doi: 10.1016/0022-2836(91)90563-l. [DOI] [PubMed] [Google Scholar]
- 52.Perez-Martin J, Rojo F, deLorenzo V. Microbiol Rev. 1994;58:268–290. doi: 10.1128/mr.58.2.268-290.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gaal T, Rao L, Estrem S T, Yang J, Wartell R M, Gourse R L. Nucleic Acids Res. 1994;22:2344–2350. doi: 10.1093/nar/22.12.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moran C P, Jr, Lang N, Banner C D, Haldenwang W G, Losick R. Cell. 1981;25:783–791. doi: 10.1016/0092-8674(81)90186-0. [DOI] [PubMed] [Google Scholar]
- 55.Czarniecki D, Noel R J, Jr, Reznikoff W S. J Bacteriol. 1997;179:423–429. doi: 10.1128/jb.179.2.423-429.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McAllister C F, Achberger E C. J Biol Chem. 1988;263:11743–11749. [PubMed] [Google Scholar]
- 57.Graves M C, Rabinowitz J C. J Biol Chem. 1986;261:11409–11415. [PubMed] [Google Scholar]