

Abstract
Angiotensin II receptor blockade has been shown to inhibit atherosclerosis in several different animal models. We sought to determine if this effect was the result of blood pressure reduction per se or a result of the anti-inflammatory effects of receptor blockade. ApoE-deficient mice were fed a high fat diet and treated with either an angiotensin II receptor antagonist, candesartan (0.5 mg/kg/day, SC) or a calcium channel blocker, amlodipine (7.5 mg/kg/day, mixed with food). Atherosclerotic lesion area, aortic inflammatory gene expression as well as aortic H2O2 and superoxide production were assayed. We found that candesartan but not amlodipine treatment dramatically attenuated the development of atherosclerosis despite a similar reduction in blood pressure. Similarly, candesartan treatment inhibited aortic expression of inflammatory genes and production of reactive oxygen species, effects not seen with amlodipine. These data demonstrate that angiotensin II receptor blockade inhibits atherosclerosis by reducing vascular oxidative stress and inflammatory gene production independent of blood pressure reduction.
Keywords: Atherosclerosis, angiotensin II, angiotensin II receptor antagonists, oxidative stress, vascular biology
Introduction
Both humoral and mechanical factors appear to be involved in the pathogenesis of atherosclerosis. The renin-angiotensin system has been implicated as a major contributing factor to the progression of atherosclerosis in apoE-deficient mice.1-13 Smooth muscle cells exposed to angiotensin II also demonstrate an increase in MAP kinase activation, upregulation of NAD(P)H oxidase components, and increased expression of inflammatory markers such as MCP-1, VCAM-1, and M-CSF.6,14,15 Hypertension and the biomechanical effects can lead to endothelial dysfunction,16 increased MMP and inflammatory gene expression,17-19 and accelerated atherosclerosis.2 The magnitude of the relative contributions of humoral and mechanical factors to atherosclerosis remain unclear.
The renin angiotensin system had been implicated in the pathogenesis of atherosclerosis based on both clinical and experimental studies.1,3,4,20-24 Thus, it has been proposed that inhibition of the renin angiotensin system may have anti-atherosclerotic effects independent of blood pressure reduction. This hypothesis remains controversial as there are data available that both support and refute this concept.25-29 Therefore, we attempted to compare the relative effects of blood pressure reduction with an angiotensin II type-I receptor (AT1) blocker and a calcium channel blocker on atherosclerosis, inflammatory gene expression, and reactive oxygen species (ROS) generation in apoE-deficient mice while controlling for an equivalent degree of blood pressure reduction.
Materials and Methods
Animals, Drugs, and Diets
Male apolipoprotein E-deficient mice on a C57BL/6 background were purchased from The Jackson Laboratory (Bar Harbor, ME) and housed individually in ventilated micro-isolator units on a 12 hour light/dark schedule. The mice were given free access to water and food. The animals were housed and cared for according to the guidelines proposed by the National Institutes of Health for the care and use of experimental animals. All experiments in the present study were conducted on mice beginning at 6 and 8 weeks of age.
Candesartan was a kind gift from Astra-Zeneca. Dosages of candesartan and amlodipine used were determined by preliminary studies such that systolic blood pressure was reduced by approximately 30 mmHg. Candesartan was delivered via subcutaneous mini-osmotic pumps (Alzet, model 1002) implanted in a dorsal subcutaneous pocket after the mice were anaesthetized with 375 mg/kg 2,2,2-tribromoethanol (Avertin, Sigma Chemical Co.). The soluble and bio-available form of candesartan (CV-11974) was used for all experiments, which was dissolved in 0.9% NaCl and 50 mM Na2CO3. Amlodipine was mixed with the powdered high fat diet using a food blender (Fisher Scientific). The final dose of amlodipine administered to the mice was 7.5 mg/kg/day.
The Western-type or saturated fat enriched diet (total caloric content 0.15% cholesterol, 42% fat) used in all experiments was purchased from Teklad, Inc. (TD 88137) in either pellet or powder form. The components per kilogram as listed by the manufacturer are as follows: 195 g high protein casein, 3 g DL-methionine, 341.46 g sucrose, 150 g corn starch, 210 g anhydrous milkfat, 1.5 g cholesterol, 50 g cellulose, 35 g mineral mix (AIN-76), 4 g calcium carbonate, 10 g vitamin mix, and 0.04 g ethoxyquin.
Systolic blood pressure was measured before the start of treatment, once a month thereafter, and just prior to sacrifice using a computerized, non-invasive, tail-cuff method (BP2000, Visitech). One set of 10 measurements was acquired for all animals and the mean blood pressure was calculated. All animals were acclimated to the machine before taking measurements to ensure accuracy.
Morphological Evaluation
For the morphological endpoint we divided 50 apoE-deficient mice into five weight-matched groups. The first three groups were treated for 4 months as follows: 1) standard chow diet (Purina, Certified Rodent Diet), 2) pelleted high fat diet, and 3) pelleted high fat diet with Candesartan treatment (0.5 mg/kg/day SC). The remaining 20 mice were split into two groups and treated for 6 months as follows: 4) pelleted high fat diet, 5) pelleted high fat diet with candesartan treatment (0.5 mg/kg/day SC) for the last two months only. In a similar experiment we divided 30 apoE-deficient mice into three groups as follows: 1) standard chow diet, 2) pulverized high fat diet fed through individual feeder/mesh cylinders (Allentown Caging Equipment Co., Allentown, NJ), and 3) pulverized high fat diet blended with amlodipine (7.5 mg/kg/day) using individual feeders.
Animals were euthanized by CO2 inhalation at each designated time point. The animals were then perfused with 0.9% saline by cardiac puncture, followed by pressure fixation at 100 mm Hg with a 4% phosphate buffered formalin solution. Atherosclerotic plaque area in the descending aortas was measured as previously described.2 Briefly, the aorta was opened longitudinally from the origin of the descending thoracic aorta to the renal arteries. Digital images were acquired and the extent of atherosclerosis was expressed as the percentage of the lumen surface area covered with atherosclerotic plaque.
Gene Expression
Using the same experimental groups described above, mice were treated for two weeks, with blood pressures assessed at 0, 7 and 14 days. The mice were euthanized by CO2 and their aortas were quickly harvested, snap frozen in liquid nitrogen, homogenized in Buffer RLT (Qiagen) and the homogenates were passed through a QiaShredder spin column to ensure complete homogenization of the samples. RNA was extracted and purified using the Qiagen Rneasy Mini protocol for animal tissue. All gene expression was quantified using real-time PCR (Roche light-cycler). The primers used for real-time PCR experiments were obtained from Sigma/Genosys and are as follows: MCP1-upsream primer: 5′- ACTCTCACTGAAGCCAGCTC -3′; MCP-1 downstream primer: 5′-CAGAGAGGGAAAAATGGATC -3′; PAI-1 upstream, 5′-ACAGCCAACAAGAGCCAATC-3′, downstream primer, 5′-GACACGCCATAGGGAGAGAA--3′; 18S rRNA PCR upstream primer, 5′-GAACGTCTGCCCTATCAACT -3′; downstream primer, 5′-CCAAGATCCAACTACGAGCT -3′. Superscript II, Platinum Taq polymerase, RnaseOut, and all other PCR components were purchased from Invitrogen/Life Technologies. cDNA was purified using the QIAquick PCR Purification kit (Qiagen), with an additional ethanol washing step added. Standards for each gene were created using conventional PCR and tested for purity by gel electrophoresis. All gene expression was normalized to 18s mRNA levels and all samples were run in triplicate and averaged.
ROS Production in Aortic Ring Segments
H2O2 production was measured using an Amplex Red assay kit purchased from Molecular Probes, Inc using the manufacturer’s instructions except that Krebs-Ringers-Phosphate-Glucose (KRPG) buffer (145 mM NaCl, 5.7 mM NaH2PO4, 4.9 mM KCl, 0.5 mM CaCl2, 1.2 mM MgSO4, 5.5 mM anhydrous glucose) was used. ApoE-deficient mice were treated for six weeks with chow diet, high fat diet, high fat diet + candesartan (0.5 mg/kg/day), or high fat diet + amlodipine (7.5 mg/kg/day) as described above. The mouse aortas were carefully harvested and the adventitia removed under a dissecting microscope. The proximal halves of the aortas were then cut into 4 equal 5 mm segments and incubated two per tube in the reaction mixture for 15 hours in the dark at 37°C. Care was taken to insure minimal manipulation of the rings to maintain endothelial integrity. The supernatant was then read in a microplate reader (Cytofluor series 4000, PerSeptive Biosystems). The fluorescent values were normalized to the dry weights of the ring segments. Similarly, superoxide production by isolated aortic rings was measured using the lucigenin assay as previously described.30
Statistical Analysis
All data are presented as mean ± SEM. Statistical Significance was determined by ANOVA. The Duncan New Multiple Range Test was used to determine the significance of individual time points.
Results
Effect of angiotensin II receptor antagonism on atherosclerotic lesion development in apoE-deficient mice fed a high fat diet for 4 months
ApoE-deficient mice treated with candesartan infused via osmotic pumps at a dose of 0.5 mg/kg/day and fed a high fat diet for 4 months demonstrated a significant reduction in systolic blood pressure from 100.1 ± 2.1 to 72.9 ± 1.6 mmHg (p<0.0001, Figure 1A). Treatment with candesartan completely blocked the effect of the high fat diet on atherosclerotic lesion area in the descending thoraco-abdominal aorta. Mice that were fed a high fat diet for 4 months had an atherosclerotic lesion area of 18.6 ± 2.4%. Conversely, those fed a high fat diet for four months and simultaneously treated with candesartan had only 3.8 ± 0.5% of their descending aortas covered with lesions (p<0.001, Figure 1B and 1C). The mean lesion area in the candesartan-treated animals was not statistically different from the lesion area in the mice maintained on the standard chow diet (3.8 ± 0.5% vs. 3.5 ± 1.0%) indicating a complete inhibition of the atherogenic effect of the high fat diet.
Figure 1.
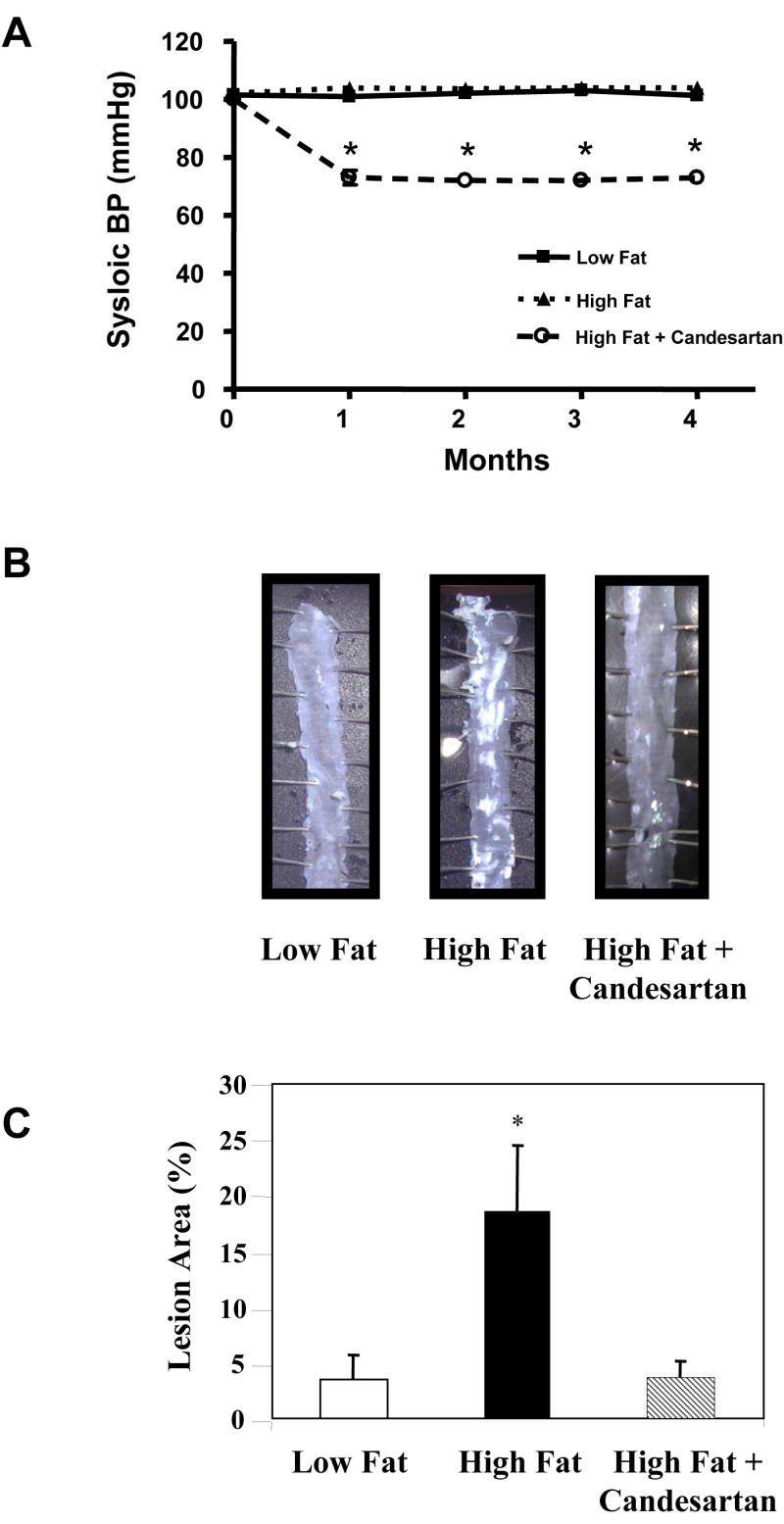
Inhibition of atherosclerosis by treatment with candesartan. Male apoE-deficient mice were fed either a standard chow diet (Low Fat) or a high fat diet for 4 months. Some animals were treated with candesartan for the entire 4 month experimental period. Panel A shows blood pressure measurements obtained using the tail cuff technique. Panel B contains representative en face preparations of the descending aortas from animals from each of the 3 experimental groups. Note the marked inhibition of atherosclerosis in the animals fed the high fat diet and treated with candesartan. Panel C summarizes the mean atherosclerotic lesion area for all 3 experimental groups. Eight -12 animals were included in each experimental group. * = P <0.01
Effect of calcium channel blockade on atherosclerosis in apoE-deficient mice
In order to determine the effects of blood pressure reduction per se vs. the specific effects of blocking the angiotensin II receptor, we also treated apoE-deficient mice that were fed a high fat diet with the calcium channel blocker amlodipine (7.5 mg/kg/day). The dose of amlodipine was selected so as to produce a reduction in blood pressure that similar to that seen in the candesartan-treated group. ApoE-deficient mice treated with amlodipine and fed a high fat diet for 4 months demonstrated a significant reduction in systolic blood pressure from 99.5 ± 1.3 to 70.4 ± 3.7 mmHg (p<0.001, Figure 2A). Note that this reduction was similar in magnitude to the one seen in the candesartan-treated mice.
Figure 2.

Lack of Inhibition of atherosclerosis by treatment with amlodipine. Male apoE-deficient mice were fed a high fat diet for 4 months. Some animals were treated with amlodipine for the entire 4 month experimental period. Panel A shows blood pressure measurements obtained using the tail cuff technique. Note that the blood pressure reduction was similar to that seen with candesartan treatment. Panel B contains representative en face preparations of the descending aortas from animals from both of the experimental groups. Note the lack of effect of amlodipine on atherosclerotic lesion area. Mean data are resented in panel C. Eight -12 animals were included in each experimental group. * = p <0.01
Despite the commensurate hypotensive effects of amlodipine, there was no significant effect on atherosclerotic lesion area (24.2 ± 2.7% vs. 20.6 ± 2.4%, p = 0.354). This finding suggests that while mechanical factors may play a role in atherosclerosis, the effect of candesartan treatment on atherosclerotic lesion area may be more related to the direct effects of angiotensin II receptor antagonism as opposed to blood pressure reduction alone.
Effects of angiotensin II receptor antagonism on pre-existing lesions in apoE-deficient mice
To determine the effects of candesartan treatment on previously-formed lesions in apoE-deficient mice, we performed additional studies in animals with established atherosclerosis. Two groups of animals were used. Both were fed a high fat diet for 6 months, but one was treated with candesartan (0.5 mg/kg/day) during the last two months. The systolic blood pressures of these mice were reduced to the same extent as we observed in the animals treated with candesartan for 4 months (figures 1 and 3).
Figure 3.

Angiotensin II receptor blockade halts atherosclerotic disease progression in animals with established atherosclerosis. Male apoE-deficient mice were fed a high fat diet for 6 months. Some animals were treated with candesartan for the months 5 and 6. Panel A shows blood pressure measurements obtained using the tail cuff technique. Panel B contains representative en face preparation from both treatment groups. Note that treatment with candesartan resulted in a halt in the progression of atherosclerosis. Mean data are presented in panel C. Eight - 10 animals were included in each experimental group. * = p <0.01
We found that mice fed a high fat diet for 6 months had an average atherosclerotic lesion area of 52.4 ± 4.5%. In contrast, the mice that were fed a high fat diet for 6 months with candesartan treatment beginning after month four had an atherosclerotic lesion area of only 22.3 ± 4.0%, (p < 0.0002). This was not significantly different from the mice that were fed a high fat diet for four months only, (18.6 ± 2.43%, p = 0.448). Although treatment with candesartan did not induce atherosclerotic lesion regression, the progression of atherosclerosis appears to have been essentially halted once treatment began.
Comparison of the effects of candesartan and amlodipine on inflammatory and fibrogenic gene expression
Although both the candesartan and amlodipine were effective in lowering blood pressure to the same degree, the two drugs had markedly differing effects on inflammatory gene expression in aortic tissue. In order to examine the relative effects of these two anti-hypertensive agents on inflammatory and fibrogenic gene expression, we treated apoE-deficient mice for two weeks under the following conditions: normal chow diet, high fat diet, high fat diet + candesartan, and high fat diet + amlodipine. We found that candesartan potently down-regulated plasminogen activator inhibitor-1 (PAI-1), a gene known for its robust response to angiotensin II stimulation in vascular smooth muscle cells. Figure 4 illustrates that when apoE-deficient mice were fed the high fat diet and treated with candesartan, aortic expression of PAI-1 is reduced by approximately 60% compared to animals on high fat diet alone. Conversely, amlodipine had no significant effect on basal PAI-1 expression.
Figure 4.

Candesartan but not amlodipine inhibited inflammatory gene expression in apoE-deficient mice fed a high fat diet. Male apoE-deficient mice were fed either a standard chow diet (Low Fat) or a high fat diet. Some animals were treated with amlodipine or candesartan. MCP-1 and PAI-1 mRNA expression were measured using quantitative real time PCR. Shown are mean relative expression levels from 10 animals per treatment group. * = p<0.01 vs. normal chow (low fat) control group. # = p <0.05 vs. normal chow (low fat) control group.
Another gene of interest involved in vascular disease that we focused on is monocyte chemoattractant protein-1 (MCP-1). Known to be important in the pathogenesis of atherosclerosis,31,32 MCP-1 mRNA expression was found to be elevated approximately 3 fold in mice maintained on a high fat diet compared to those on normal chow (see Figure 4). Aortic tissue from animals on a high fat diet and treated with candesartan for two weeks demonstrated an approximate 40% reduction in MCP-1 expression. Amlodipine, however, had no significant effect on MCP-1 mRNA expression.
Effect of candesartan and amlodipine on aortic ROS production
It has been well established that experimental atherosclerosis is due in part to the production of reactive oxygen species. In order to explore possible differential effects of candesartan and amlodipine on vascular production of reactive oxygen species, we measured H2O2 production using the amplex red assay. ApoE-deficient mice were divided into four groups containing 6-8 mice as follows: 1) low fat chow diet, 2) high fat diet, 3) high fat diet + candesartan (0.5 mg/kg/day), and 4) high fat diet + amlodipine (7.5 mg/kg/day). The mice were treated with the aforementioned regimen for six weeks before their aortas were harvested and processed. These studies showed that the high fat diet significantly increased H2O2 production. Treatment with Candesartan partially normalized this increase whereas amlodipine had no significant effect on the diet-mediated increase in H2O2 production (Figure 5). Similarly, superoxide production was increased in the animals fed a high fat diet and the administration of candesartan significantly reduced superoxide production in animals fed either the low fat or high fat diet (figure 5).
Figure 5.


Inhibition of production of ROS by candesartan. Panel A: Candesartan but not amlodipine inhibits diet induced increases in aortic H2O2 production. Treatment groups were the same as used in figure 4. H2O2 was measured using the amplex red assay. * = p<0.01 vs. normal chow (low fat) control group. Panel B: Candesartan also inhibited the diet-induced increase in superoxide production. * = p<0.01 vs. normal chow (low fat) control group.
Discussion
The data presented here demonstrate that the anti-atherosclerotic effects of AT1 receptor antagonism are not a result of the concomitant reduction in blood pressure. We have shown that atherosclerosis of the descending thoracoabdominal aorta can be completely inhibited in apoE-deficient mice fed a high fat diet by infusing candesartan, a potent angiotensin II receptor type-1 blocker. Conversely, when apoE-deficient mice were treated with the calcium channel blocker amlodipine, we found that there was no significant reduction in atherosclerotic lesion formation. This occurred despite a commensurate reduction in systolic blood pressure. Importantly, we also found that treatment with the angiotensin II receptor blocker dramatically decreased aortic expression of prototypical pro-inflammatory and fibrogenic genes and vascular production of ROS. This effect was not seen in the animals treated with the calcium channel blocker. Taken together, these findings demonstrate that the anti-atherosclerotic, anti-inflammatory effects of the angiotensin receptor blocker, candesartan, are largely due to effects independent of blood pressure reduction.
We and others2,4 have shown that infusing apoE-deficient mice on a high cholesterol diet with angiotensin II had an extremely potent pro-atherogenic effect. Conversely, raising blood pressure to the same degree with norepinephrine had only a minimal effect on atherosclerotic lesion formation.2 In the current study, using two different pharmacological approaches to lowering blood pressure, we have shown in an analogous fashion, that AT1 receptor antagonism results in an anti-atherosclerotic effect independent of blood pressure reduction. Clearly, atherosclerosis is a multifactorial disease that cannot be solely linked to one receptor. In our particular setting, however, it appears that endogenous angiotensin II may be more important than mechanical factors in stimulating the progression of atherosclerosis in apoE-deficient mice.
To examine the possible effects of AT1 receptor blockade on established atherosclerotic lesions, we studied the effects of candesartan on the progression of atherosclerotic disease extent in animals that had been maintained on a high fat diet for 4 months. These mice were then treated with candesartan for two months while continuing on a high fat diet regimen. Although the AT1 antagonist did not cause regression of pre-existing lesions, atherosclerotic development was completely halted when compared to the four month high fat diet controls. This suggests that although no notable regression occurred during this time, the angiotensin II receptor blocker was able to blunt further lesion formation during a period of accelerated lesion development.
The potential pro-atherosclerotic effects of angiotensin II have been well described.1-4,14,33-35 Angiotensin II is capable of upregulating a myriad of growth factors, cytokines, chemokines, and other pro-inflammatory gene products. Central to this process is the production of reactive oxygen species.35,36 Thus, there are significant mechanistic data supporting a functional role for endogenous angiotensin II in the pathogenesis of atherosclerosis.
The contribution of blood pressure reduction to the anti-atherosclerotic effects of interfering with the renin-angiotensin system (RAS) remains controversial. Several groups have demonstrated that inhibition of the RAS can reduce atherosclerosis in a range of animal models including apoE-deficient mice,7,37-39 Watanabe rabbits,21,24,40 and monkeys8,41 despite having variable effects on blood pressure. Several of these studies utilized lower doses of ACE inhibitors that did not cause a reduction in blood pressure in an attempt to define the blood pressure-independent effects of ACE inhibition.24,37,39,41 While one of these studies showed equivalent anti-atherosclerotic effects of the low dose ACE inhibitor,39 others showed that the magnitude of the reduction in atherosclerosis seen with low dose ACE inhibitors was less than that seen when doses sufficient to lower blood pressure were used.24,37,41 These inconsistencies leave in doubt the precise contribution of blood pressure reduction to the anti-atherosclerotic effects of interfering with the RAS. However, the data presented here lend support to the concept that the blood pressure-independent effects of angiotensin II may have a dominant role in preventing atherosclerosis.
Although the effect of amlodpine and other calcium channel blockers on atherosclerosis has not been studied as vigorously as drugs that affect the RAS in animal models, the data in general do not suggest the drug has a dramatic effect on the development of atherosclerosis.42-44 An exception to this is an apparent strain-dependent effect in rabbits.45,46 Amlodipine specifically has been shown to reduce calcification of atherosclerotic plaques, but did not reduce arterial cholesterol content in APOE*3 Leiden mice fed a high fat diet.47 Amlodipine has, however been shown to have some antioxidant48,49 and anti-proliferative properties49 and reduced serum LDL oxidation in rabbits on a high cholesterol diet.50 While there may be less convincing evidence for a significant anti-atherosclerotic effect of this class of drugs, we elected to use amlodipine in these studies as there is considerable debate over the relative efficacy of these 2 classes of drugs in inhibiting atherosclerosis.
We found that the angiotensin receptor antagonist and not the calcium channel antagonist inhibited expression of prototypical pro-atherosclerotic genes. It should be noted that we assayed expression of these genes at a very early time point in an effort to determine the consequences of drug treatment prior to the development of overt atherosclerotic lesions. This was done to try to distinguish changes that promoted atherosclerosis as opposed to changes in expression that were a consequences of the disease.
We selected PAI-1 and MCP-1 because of their known involvement in atherosclerosis. PAI-1 has been implicated in stroke, myocardial infarction, and restenosis. Data from genetic models show that PAI-1-deficiency protects against atherosclerosis in the mouse carotid artery, suggesting a possible role in accelerating lesion formation. PAI-1 mRNA is elevated in human atheromatous plaques51 and its expression has been shown to be increased in macrophages, smooth muscle, and endothelial cells in response to angiotensin II.52,53 Elevated PAI-1 levels in rat carotid arteries following balloon-injury has been shown to correlate with increased smooth muscle cell proliferation as well. Similarly, it has been shown that MCP-1 is a potent chemoattractant for inflammatory cells and that arterial MCP-1 expression is upregulated in the setting of atherosclerosis and hypertension.18 Genetic models of animals deficient in the receptor for MCP-1 demonstrate decreased atherosclerosis.31,32 Thus, the differential effects of candesartan and amlodipine on inflammatory and fibrogenic gene expression are consistent with the morphological findings demonstrating a more favorable effect with the AT1 receptor antagonist.
In addition to the differential gene expression seen between mice treated with the two drugs in question, we sought to determine if differences in oxidative stress existed in aortas of mice treated with either drug. We found that mice treated with amlodipine did not normalize the diet-induced increase in ROS production whereas treatment with candesartan returned blunted the production of ROS. This correlates with our finding showing differential lesion formation and inflammatory gene expression between the two drugs. Furthermore, the fact that high fat diet alone increased production of ROS compared to low fat controls is consistent with the concept that ROS may play a critical role in the progression of atherosclerosis.
Altered oxidative stress likely has additional effects on the arterial wall that are not limited to proinflammatory and fibrogenic genes. A decrease in the production of ROS could also directly alter smooth muscle cell proliferation and migration.54,55 In addition, other redox-sensitive genes, known to be involved in atherogenesis such as LOX-156 may be down-regulated in the setting of angiotensin receptor blockade.
In summary, we have shown that anti-inflammatory and anti-atherosclerotic effects of the AT1 receptor antagonist candesartan occur independently of its blood pressure lowering effects. Animals treated with amlodipine demonstrated an equal reduction in systolic blood pressure but there was no significant reduction in atherosclerotic lesion extent, inflammatory gene expression, or production of ROS. Candesartan did not induce regression of established atherosclerotic lesions, but was able to halt further progression. The results presented here further underline the potent role of angiotensin II in atherosclerosis and demonstrate the lesser mechanistic importance of blood pressure reduction per se as opposed to the anti-inflammatory effects of AT1 receptor antagonism.
Acknowledgments
This work was supported by NIH P01 HL58000, RO1 HL70531, R01 DK059499, VA Merit Funding and a grant from Astra Zeneca, Inc.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Weiss D, Sorescu D, Taylor WR. Angiotensin II and atherosclerosis. Am J Cardiol. 2001;87:25C–32C. doi: 10.1016/s0002-9149(01)01539-9. [DOI] [PubMed] [Google Scholar]
- 2.Weiss D, Kools JJ, Taylor WR. Angiotensin II-induced hypertension accelerates the development of atherosclerosis in apoE-deficient mice. Circulation. 2001;103:448–454. doi: 10.1161/01.cir.103.3.448. [DOI] [PubMed] [Google Scholar]
- 3.Daugherty A, Cassis L. Chronic angiotensin II infusion promotes atherogenesis in low density lipoprotein receptor -/- mice. Ann N Y Acad Sci. 1999;892:108–118. doi: 10.1111/j.1749-6632.1999.tb07789.x. [DOI] [PubMed] [Google Scholar]
- 4.Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest. 2000;105:1605–1612. doi: 10.1172/JCI7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ihara M, Urata H, Kinoshita A, Suzumiya J, Sasaguri M, Kikuchi M, Ideishi M, Arakawa K. Increased chymase-dependent angiotensin II formation in human atherosclerotic aorta. Hypertension. 1999;33:1399–1405. doi: 10.1161/01.hyp.33.6.1399. [DOI] [PubMed] [Google Scholar]
- 6.Ushio-Fukai M, Alexander RW, Akers M, Griendling KK. p38 Mitogen-activated protein kinase is a critical component of the redox-sensitive signaling pathways activated by angiotensin II. Role in vascular smooth muscle cell hypertrophy. J Biol Chem. 1998;273:15022–15029. doi: 10.1074/jbc.273.24.15022. [DOI] [PubMed] [Google Scholar]
- 7.Candido R, Jandeleit-Dahm KA, Cao Z, Nesteroff SP, Burns WC, Twigg SM, Dilley RJ, Cooper ME, Allen TJ. Prevention of accelerated atherosclerosis by angiotensin-converting enzyme inhibition in diabetic apolipoprotein E-deficient mice. Circulation. 2002;106:246–253. doi: 10.1161/01.cir.0000021122.63813.32. [DOI] [PubMed] [Google Scholar]
- 8.Miyazaki M, Sakonjo H, Takai S. Anti-atherosclerotic effects of an angiotensin converting enzyme inhibitor and an angiotensin II antagonist in Cynomolgus monkeys fed a high-cholesterol diet. Br J Pharmacol. 1999;128:523–529. doi: 10.1038/sj.bjp.0702833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee MA, Bohm M, Paul M, Ganten D. Tissue renin-angiotensin systems. Their role in cardiovascular disease. Circulation. 1993;87:IV7–13. [PubMed] [Google Scholar]
- 10.Dzau VJ. Theodore Cooper Lecture: Tissue angiotensin and pathobiology of vascular disease: a unifying hypothesis. Hypertension. 2001;37:1047–1052. doi: 10.1161/01.hyp.37.4.1047. [DOI] [PubMed] [Google Scholar]
- 11.Gibbons GH. The pathophysiology of hypertension: the importance of angiotensin II in cardiovascular remodeling. Am J Hypertens. 1998;11:177S–181S. doi: 10.1016/s0895-7061(98)00198-8. [DOI] [PubMed] [Google Scholar]
- 12.Prasad A, Tupas-Habib T, Schenke WH, Mincemoyer R, Panza JA, Waclawin MA, Ellahham S, Quyyumi AA. Acute and chronic angiotensin-1 receptor antagonism reverses endothelial dysfunction in atherosclerosis. Circulation. 2000;101:2349–2354. doi: 10.1161/01.cir.101.20.2349. [DOI] [PubMed] [Google Scholar]
- 13.da Cunha V, Tham DM, Martin-McNulty B, Deng G, Ho JJ, Wilson DW, Rutledge JC, Vergona R, Sullivan ME, Wang YX. Enalapril attenuates angiotensin II-induced atherosclerosis and vascular inflammation. Atherosclerosis. 2005;178:9–17. doi: 10.1016/j.atherosclerosis.2004.08.023. [DOI] [PubMed] [Google Scholar]
- 14.Griendling KK, Ushio-Fukai M, Lassegue B, Alexander RW. Angiotensin II signaling in vascular smooth muscle. New concepts. Hypertension. 1997;29:366–373. doi: 10.1161/01.hyp.29.1.366. [DOI] [PubMed] [Google Scholar]
- 15.Griendling KK, Ushio-Fukai M. Reactive oxygen species as mediators of angiotensin II signaling. Regul Pept. 2000;91:21–27. doi: 10.1016/s0167-0115(00)00136-1. [DOI] [PubMed] [Google Scholar]
- 16.Taylor WR. Mechanical deformation of the arterial wall in hypertension: a mechanism for vascular pathology. Am J Med Sci. 1998;316:156–161. doi: 10.1097/00000441-199809000-00002. [DOI] [PubMed] [Google Scholar]
- 17.Li-Saw-Hee FL, Edmunds E, Blann AD, Beevers DG, Lip GY. Matrix metalloproteinase-9 and tissue inhibitor metalloproteinase-1 levels in essential hypertension. Relationship to left ventricular mass and anti-hypertensive therapy. Int J Cardiol. 2000;75:43–47. doi: 10.1016/s0167-5273(00)00274-6. [DOI] [PubMed] [Google Scholar]
- 18.Capers Qt, Alexander RW, Lou P, De Leon H, Wilcox JN, Ishizaka N, Howard AB, Taylor WR. Monocyte chemoattractant protein-1 expression in aortic tissues of hypertensive rats. Hypertension. 1997;30:1397–1402. doi: 10.1161/01.hyp.30.6.1397. [DOI] [PubMed] [Google Scholar]
- 19.Capers Qt, Laursen JB, Fukui T, Rajagopalan S, Mori I, Lou P, Freeman BA, Berrington WR, Griendling KK, Harrison DG, Runge MS, Alexander RW, Taylor WR. Vascular thrombin receptor regulation in hypertensive rats. Circ Res. 1997;80:838–844. doi: 10.1161/01.res.80.6.838. [DOI] [PubMed] [Google Scholar]
- 20.Brunner HR, Laragh JH, Baer L, Newton MA, Goodwin FT, Krakoff LR, Bard RH, Buhler FR. Essential hypertension: renin and aldosterone, heart attack and stroke. N Engl J Med. 1972;286:441–449. doi: 10.1056/NEJM197203022860901. [DOI] [PubMed] [Google Scholar]
- 21.Schuh JR, Blehm DJ, Frierdich GE, McMahon EG, Blaine EH. Differential effects of renin-angiotensin system blockade on atherogenesis in cholesterol-fed rabbits. J Clin Invest. 1993;91:1453–1458. doi: 10.1172/JCI116350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andersson RG, Jacobsson L, Persson K. Angiotensin converting enzyme inhibitors and atherosclerosis. J Physiol Pharmacol. 1994;45:13–25. [PubMed] [Google Scholar]
- 23.Kowala MC, Grove RI, Aberg G. Inhibitors of angiotensin converting enzyme decrease early atherosclerosis in hyperlipidemic hamsters. Fosinopril reduces plasma cholesterol and captopril inhibits macrophage-foam cell accumulation independently of blood pressure and plasma lipids. Atherosclerosis. 1994;108:61–72. doi: 10.1016/0021-9150(94)90037-x. [DOI] [PubMed] [Google Scholar]
- 24.Chobanian AV, Hope S, Brecher P. Dissociation between the antiatherosclerotic effect of trandolapril and suppression of serum and aortic angiotensin-converting enzyme activity in the Watanabe heritable hyperlipidemic rabbit. Hypertension. 1995;25:1306–1310. doi: 10.1161/01.hyp.25.6.1306. [DOI] [PubMed] [Google Scholar]
- 25.Julius S, Kjeldsen SE, Weber M, Brunner HR, Ekman S, Hansson L, Hua T, Laragh J, McInnes GT, Mitchell L, Plat F, Schork A, Smith B, Zanchetti A. Outcomes in hypertensive patients at high cardiovascular risk treated with regimens based on valsartan or amlodipine: the VALUE randomised trial. Lancet. 2004;363:2022–2031. doi: 10.1016/S0140-6736(04)16451-9. [DOI] [PubMed] [Google Scholar]
- 26.Yusuf S, Sleight P, Pogue J, Bosch J, Davies R, Dagenais G. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342:145–153. doi: 10.1056/NEJM200001203420301. [DOI] [PubMed] [Google Scholar]
- 27.Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT) Jama. 2002;288:2981–2997. doi: 10.1001/jama.288.23.2981. [DOI] [PubMed] [Google Scholar]
- 28.Braunwald E, Domanski MJ, Fowler SE, Geller NL, Gersh BJ, Hsia J, Pfeffer MA, Rice MM, Rosenberg YD, Rouleau JL. Angiotensin-converting-enzyme inhibition in stable coronary artery disease. N Engl J Med. 2004;351:2058–2068. doi: 10.1056/NEJMoa042739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wing LM, Reid CM, Ryan P, Beilin LJ, Brown MA, Jennings GL, Johnston CI, McNeil JJ, Macdonald GJ, Marley JE, Morgan TO, West MJ. A comparison of outcomes with angiotensin-converting--enzyme inhibitors and diuretics for hypertension in the elderly. N Engl J Med. 2003;348:583–592. doi: 10.1056/NEJMoa021716. [DOI] [PubMed] [Google Scholar]
- 30.Warnholtz A, Nickenig G, Schulz E, Macharzina R, Brasen JH, Skatchkov M, Heitzer T, Stasch JP, Griendling KK, Harrison DG, Bohm M, Meinertz T, Munzel T. Increased NADH-oxidase-mediated superoxide production in the early stages of atherosclerosis: evidence for involvement of the renin-angiotensin system. Circulation. 1999;99:2027–2033. doi: 10.1161/01.cir.99.15.2027. [DOI] [PubMed] [Google Scholar]
- 31.Dawson TC, Kuziel WA, Osahar TA, Maeda N. Absence of CC chemokine receptor-2 reduces atherosclerosis in apolipoprotein E-deficient mice. Atherosclerosis. 1999;143:205–211. doi: 10.1016/s0021-9150(98)00318-9. [DOI] [PubMed] [Google Scholar]
- 32.Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in CCR2-/- mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394:894–897. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- 33.Griendling KK, Alexander RW. The angiotensin (AT1) receptor. Semin Nephrol. 1993;13:558–566. [PubMed] [Google Scholar]
- 34.Griendling KK, Lassegue B, Alexander RW. Angiotensin receptors and their therapeutic implications. Annu Rev Pharmacol Toxicol. 1996;36:281–306. doi: 10.1146/annurev.pa.36.040196.001433. [DOI] [PubMed] [Google Scholar]
- 35.Griendling KK, Alexander RW. Oxidative stress and cardiovascular disease. Circulation. 1997;96:3264–3265. [PubMed] [Google Scholar]
- 36.Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res. 1994;74:1141–1148. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- 37.Hayek T, Attias J, Coleman R, Brodsky S, Smith J, Breslow JL, Keidar S. The angiotensin-converting enzyme inhibitor, fosinopril, and the angiotensin II receptor antagonist, losartan, inhibit LDL oxidation and attenuate atherosclerosis independent of lowering blood pressure in apolipoprotein E deficient mice. Cardiovasc Res. 1999;44:579–587. doi: 10.1016/s0008-6363(99)00239-4. [DOI] [PubMed] [Google Scholar]
- 38.Hayek T, Attias J, Smith J, Breslow JL, Keidar S. Antiatherosclerotic and antioxidative effects of captopril in apolipoprotein E-deficient mice. J Cardiovasc Pharmacol. 1998;31:540–544. doi: 10.1097/00005344-199804000-00011. [DOI] [PubMed] [Google Scholar]
- 39.Keidar S, Attias J, Coleman R, Wirth K, Scholkens B, Hayek T. Attenuation of atherosclerosis in apolipoprotein E-deficient mice by ramipril is dissociated from its antihypertensive effect and from potentiation of bradykinin. J Cardiovasc Pharmacol. 2000;35:64–72. doi: 10.1097/00005344-200001000-00008. [DOI] [PubMed] [Google Scholar]
- 40.Chobanian AV, Haudenschild CC, Nickerson C, Hope S. Trandolapril inhibits atherosclerosis in the Watanabe heritable hyperlipidemic rabbit. Hypertension. 1992;20:473–477. doi: 10.1161/01.hyp.20.4.473. [DOI] [PubMed] [Google Scholar]
- 41.Miyazaki M, Takai S. Antiatherosclerotic effect of alacepril, an angiotensin-converting enzyme inhibitor, in monkeys fed a high-cholesterol diet. Hypertens Res. 1999;22:49–54. doi: 10.1291/hypres.22.49. [DOI] [PubMed] [Google Scholar]
- 42.Overturf M, Sybers H, Schaper J, Taegtmeyer H. Hypertension and atherosclerosis in cholesterol-fed rabbits. II. One-kidney, one clip Goldblatt hypertension treated with nifedipine. Atherosclerosis. 1987;66:63–76. doi: 10.1016/0021-9150(87)90180-8. [DOI] [PubMed] [Google Scholar]
- 43.Kritchevsky D, Tepper SA, Klurfeld DM. Flordipine, a calcium channel blocker, which does not influence lipidemia or atherosclerosis in cholesterol-fed rabbits. Atherosclerosis. 1988;69:89–92. doi: 10.1016/0021-9150(88)90292-4. [DOI] [PubMed] [Google Scholar]
- 44.Waters D, Lesperance J, Francetich M, Causey D, Theroux P, Chiang YK, Hudon G, Lemarbre L, Reitman M, Joyal M, et al. A controlled clinical trial to assess the effect of a calcium channel blocker on the progression of coronary atherosclerosis. Circulation. 1990;82:1940–1953. doi: 10.1161/01.cir.82.6.1940. [DOI] [PubMed] [Google Scholar]
- 45.Watanabe N, Ishikawa Y, Okamoto R, Watanabe Y, Fukuzaki H. Nifedipine suppressed atherosclerosis in cholesterol-fed rabbits but not in Watanabe heritable hyperlipidemic rabbits. Artery. 1987;14:283–294. [PubMed] [Google Scholar]
- 46.Thiery J, Niedmann PD, Seidel D. The beneficial influence of nifedipine on the regression of the cholesterol-induced atherosclerosis in rabbits. Res Exp Med (Berl) 1987;187:359–367. doi: 10.1007/BF01855662. [DOI] [PubMed] [Google Scholar]
- 47.van de Poll SW, Delsing DJ, Jukema JW, Princen HM, Havekes LM, Puppels GJ, van der Laarse A. Raman spectroscopic investigation of atorvastatin, amlodipine, and both on atherosclerotic plaque development in APOE*3 Leiden transgenic mice. Atherosclerosis. 2002;164:65–71. doi: 10.1016/s0021-9150(02)00055-2. [DOI] [PubMed] [Google Scholar]
- 48.Franzoni F, Santoro G, Regoli F, Plantinga Y, Femia FR, Carpi A, Galetta F. An in vitro study of the peroxyl and hydroxyl radical scavenging capacity of the calcium antagonist amlodipine. Biomed Pharmacother. 2004;58:423–426. doi: 10.1016/j.biopha.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 49.Mason RP, Marche P, Hintze TH. Novel vascular biology of third-generation L-type calcium channel antagonists: ancillary actions of amlodipine. Arterioscler Thromb Vasc Biol. 2003;23:2155–2163. doi: 10.1161/01.ATV.0000097770.66965.2A. [DOI] [PubMed] [Google Scholar]
- 50.Turgan N, Habif S, Kabaroglu CG, Mutaf I, Ozmen D, Bayindir O, Uysal A. Effects of the calcium channel blocker amlodipine on serum and aortic cholesterol, lipid peroxidation, antioxidant status and aortic histology in cholesterol-fed rabbits. J Biomed Sci. 2003;10:65–72. doi: 10.1007/BF02255999. [DOI] [PubMed] [Google Scholar]
- 51.Tipping PG, Davenport P, Gallicchio M, Filonzi EL, Apostolopoulos J, Wojta J. Atheromatous plaque macrophages produce plasminogen activator inhibitor type-1 and stimulate its production by endothelial cells and vascular smooth muscle cells. Am J Pathol. 1993;143:875–885. [PMC free article] [PubMed] [Google Scholar]
- 52.Vaughan DE, Lazos SA, Tong K. Angiotensin II regulates the expression of plasminogen activator inhibitor-1 in cultured endothelial cells. A potential link between the renin-angiotensin system and thrombosis. J Clin Invest. 1995;95:995–1001. doi: 10.1172/JCI117809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vaughan DE. Angiotensin, fibrinolysis, and vascular homeostasis. Am J Cardiol. 2001;87:18C–24C. doi: 10.1016/s0002-9149(01)01509-0. [DOI] [PubMed] [Google Scholar]
- 54.Zhang Y, Griendling KK, Dikalova A, Owens GK, Taylor WR. Vascular hypertrophy in angiotensin II-induced hypertension is mediated by vascular smooth muscle cell-derived H2O2. Hypertension. 2005;46:732–737. doi: 10.1161/01.HYP.0000182660.74266.6d. [DOI] [PubMed] [Google Scholar]
- 55.Zafari AM, Ushio-Fukai M, Akers M, Yin Q, Shah A, Harrison DG, Taylor WR, Griendling KK. Role of NADH/NADPH oxidase-derived H2O2 in angiotensin II-induced vascular hypertrophy. Hypertension. 1998;32:488–495. doi: 10.1161/01.hyp.32.3.488. [DOI] [PubMed] [Google Scholar]
- 56.Chen J, Liu Y, Liu H, Hermonat PL, Mehta JL. Molecular dissection of angiotensin II-activated human LOX-1 promoter. Arterioscler Thromb Vasc Biol. 2006;26:1163–1168. doi: 10.1161/01.ATV.0000209998.73303.b5. [DOI] [PubMed] [Google Scholar]