

Abstract
Dysregulated sphingolipid metabolism causes neuronal cell death and is associated with insulin resistance and diseases. Thus, we hypothesized that diabetes-induced changes in retinal sphingolipid metabolism may contribute to neuronal pathologies in diabetic retinopathy. ESIMS/MS was used to measure ceramide content and ceramide metabolites in whole retinas after 2, 4, and 8 weeks of streptozotocin-induced diabetes. After 4 and 8 weeks of diabetes, a ∼30% decrease in total ceramide content was observed, concomitant with a significant ∼30% increase in glucosylceramide levels in fed diabetic rats compared with their age-matched controls. Acute insulin therapy as well as a short-term lowering of glucose via fasting did not affect the increase in glucosylceramide composition. To assess the putative biological consequences of the increase in glucosylceramide composition, R28 retinal neurons were treated with glucosylceramide synthase inhibitors. Inhibiting glycosphingolipid metabolism increased insulin sensitivity in retinal neurons. Glycosphingolipid inhibitors augmented insulin-stimulated p70 S6kinase activity in the presence of inhibitory concentrations of high glucose or glucosamine. Inhibition of glycosphingolipid synthesis also suppressed glucosamine- and interleukin-1β-induced death. Consistent with these inhibitor studies, pharmacological accumulation of glycosphingolipids increased activation of the endoplasmic reticulum stress response, a putative modulator of insulin resistance and neuronal apoptosis. It is speculated that an increase in glucosylceramide, and possibly higher-order glycosphingolipids, could contribute to the pathogenesis of diabetic retinopathy by contributing to local insulin resistance, resulting in neuronal cell death. Thus, dysfunctional glycosphingolipid metabolism may contribute to metabolic stress in diabetes, and therapeutic strategies to restore normal sphingolipid metabolism may be a viable approach for treatment of diabetic retinopathy.
Vision loss from diabetic retinopathy results from the cellular dysfunction of multiple cell types of the retina. This multifaceted disease affects the vascular, glia (micro and macro), and neurons of the retina (1). The effect of neuronal apoptosis, which occurs early and is chronic in diabetes, is just now being fully appreciated. We and others (2-6) have reported that the neurons of the retina undergo apoptosis in both human and experimental diabetes models. However, the direct and indirect causes of neuronal dysfunction remain poorly defined. We recently demonstrated that the insulin receptors, as well as downstream prosurvival cascades including phosphatidylinositol 3-kinase/Akt and p70 S6 kinases, are impaired in the diabetic retina (7), which may underlie the neuronal apoptosis.
In addition to loss of neurotrophic input, metabolic stresses may also be a causative factor in diabetic retinopathy. Sphingolipid metabolites have been demonstrated to regulate cellular stress and fate via a balance between proapoptotic/growth-arresting lipids and prosurvival/proliferative lipids and their resulting effect on signaling pathways (8). Ceramides are generally considered proapoptotic sphingolipids that accumulate in response to stress and proapoptotic stimuli, such as interleukin (IL)-1β and tumor necrosis factor (TNF)-α. Ceramides contribute to apoptosis/growth arrest at the biochemical level by inhibiting phosphatidylinositol 3-kinase/Akt (9,10) and extracellular signal-related kinase (11) signaling cascades and at the biophysical level by regulating mitochondrial permeability (12) and Golgi fragmentation (13).
Glycosphingolipids are metabolites of ceramide that have been implicated in cellular immunity, inflammation, and multidrug resistance to cancer (14). Simple glycosphingolipids, such as glucosyl and galactosylceramide (cerebrosides or monohexosylceramides), serve as building blocks for more complex glycosphingolipids, including sulfatides, globosides, and gangliosides. Recent reports (15-21) suggest that these glycosphingolipids can mediate apoptosis, insulin resistance, and cellular stress. In addition, altered sphingolipid and glycosphingolipid metabolism causes several retinal diseases. Lysosomal storagediseases, which often are a consequence of dysregulated sphingolipid metabolism, are associated with retinal impairment. As examples, patients with Farber’s disease (acid ceramidase), Tay-Sachs/Sandhoff (hexosaminidase A or B), Gaucher’s (glucosylceramidase), Krabbe’s (galactoslyceramidase), and Niemann Pick (sphingomyelinase) disease lose vision due to retinal neuronal cell death. Furthermore, overexpression of a neutral ceramidase gene in Drosophila abrogates retinal degeneration (22). Thus, understanding the roles that (glyco)sphingolipid enzymes and their metabolites have in the retina may offer new targets for retinal diseases. Herein, we hypothesize that diabetes alters retinal sphingolipid metabolism and may contribute to the pathogenesis of diabetic retinopathy. The data indicate that increased glycosphingolipid composition may contribute to the metabolic stress that leads to retinal inflammation and neurodegeneration in diabetes.
RESEARCH DESIGN AND METHODS
Bovine insulin was purchased from Sigma (St. Louis, MO). Laminin and cell-permeable cAMP were purchased from BD Biosciences (Franklin Lakes, NJ) and MP Biomedicals (Irvine, CA), respectively. Anti-phospho-p70 S6K (Thr389) and total p70 S6K were obtained from Cell Signaling Technology (Beverly, MA). Anti-GRP78 was purchased from Assay Designs (Ann Arbor, MI). Glucosylceramide synthase rabbit antisera was a generous gift from Drs. R.E. Pagano and D.L. Marks, Mayo Clinic and Foundation (Rochester, MN) (23). Anti-C/EBP homologous protein (CHOP), anti-rabbit, and anti-mouse IgG-horseradish peroxidase were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). N-butyldeoxygalactonojirimycin (NB-DGJ) was purchased from Toronto Research Chemicals (North York, ON, Canada) and conduritol B epoxide (CBE) and dL-threo-1-phenyl-2-palmitoylamino-3-morpholino-1-propanol (PPMP) from Biomol (Plymouth Meeting, PA). IL-1β was purchased from Promega (Madison, WI).
Cell culture and mouse model
R28 cells, an E1A immortalized model of retinal neurons, were a generous gift from Dr. Gail M. Seigel, State University of New York, Buffalo, New York (24). These cells were grown in Dulbecco’s modified Eagle’s medium containing 5 mmol/l glucose supplemented with 10% newborn calf serum (Hyclone, Logan, UT) and were differentiated on laminin-coated plates or coverslips with the addition of 25 μmol/l cell-permeable cAMP as previously described (25). Cells were treated as described in the text.
Male Sprague-Dawley rats (Charles River Laboratories, Wilmington, MA) were fasted overnight and given a single intraperitoneal injection of streptozotocin (STZ) (65 mg/kg; Sigma) freshly dissolved in 10 mmol/l sodium citrate buffer (pH 4.5). Diabetes was confirmed 6 days later by blood glucose >250 mg/dl (Lifescan, Milpitas, CA). Age-matched control and diabetic rats were monitored regularly by weight and blood glucose tests. Rats were housed in accordance with the institutional animal care and use committee guidelines, and the study protocol adhered to the Association for Research in Vision and Ophthalmology statement for the use of animals in ophthalmic and vision research. Rats were maintained by the Juvenile Diabetes Research Foundation Animal Core Facility at Penn State University and group housed in solid plastic-bottom cages with bedding as well as ad libitum food (Teklad Global 18% protein rodent diet) and water under a normal 12-h light-dark schedule. In some cases, groups of animals were fasted for 16 h before they were killed. Another group of animals received subcutaneous insulin injections with 5 units Humulin Regular/5 units Humulin Ultralente twice daily for 3 days before they were killed as previously described (26). Retinas were dissected at the indicated times and snap frozen in liquid N2. The average weight and blood glucose of the animals on the days they were killed are indicated in Table 1.
TABLE 1.
Average weight and blood glucose
| Study | Group | n | Weight (g) | Blood glucose (mg/dl) |
|---|---|---|---|---|
| Fig. 1A and C | Control (2 weeks) | 6 | 288.7 ± 4.6 | 109.1 ± 3.2 |
| Diabetic (2 weeks) | 6 | 236.7 ± 5.6 | 440.8 ± 16.4 | |
| Control (4 weeks) | 6 | 398.0 ± 7.8 | 107.7 ± 14.9 | |
| Diabetic (4 weeks) | 6 | 293.1 ± 13.6 | 371.2 ± 6.5 | |
| Control (8 weeks) | 7 | 517.0 ± 18.5 | 89.0 ± 1.6 | |
| Diabetic (8 weeks) | 6 | 342.8 ± 16.1 | 426.0 ± 19.3 | |
| Fig. 1B | Control (4 weeks) | 8 | 412.7 ± 11.3 | 101.9 ± 2.7 |
| Diabetic (4 weeks) | 7 | 317.5 ± 14.7 | 362.6 ± 31.1 | |
| Diabetic + insulin (4 weeks) | 6 | 307.4 ± 13.8 | 73.8 ± 8.4 | |
| Fig. 2 | Control fasted (4 weeks) | 8 | 346.0 ± 5.1 | 79.0 ± 2.0 |
| Control fed (4 weeks) | 7 | 391.1 ± 14.7 | 108.6 ± 4.3 | |
| Diabetic fasted (4 weeks) | 8 | 260.4 ± 6.0 | 227.6 ± 22.6 | |
| Diabetic fed (4 weeks) | 7 | 303.6 ± 9.6 | 409.3 ± 17.6 |
Data are means ± SE.
Ceramide and sphingolipid metabolites quantification
To measure sphingolipid metabolites, whole retinas were homogenized in 10 mmol/l Tris, pH 7.2. An aliquot was utilized for protein concentration determination. The lysates were then subjected to lipid extraction. Sphingolipid metabolites were detected by mass spectrometry and normalized to total protein by methods described by the authors by liquid chromatography and electrospray ionization-tandem mass spectrometry (27,28). Internal standards were obtained from Avanti Polar Lipids (Alabaster, AL).
Western blot analysis
Western blot analyses were performed essentially as previously described (9). Briefly, treated R28 cells were washed in ice-cold Dulbecco’s PBS solution, and lysis buffer (50 mmol/l HEPES, 137 mmol/l NaCl, 5 mmol/l NaF, 1 mmol/l EDTA, 1 mmol/l EGTA, 1 mmol/l NaVO4, 1% NP-40, protease inhibitor cocktail; Roche, Indianapolis, IN) was added. Cell lysates were cleared by centrifugation, and the Bio-Rad DC protein assay was utilized to determine protein concentration. Typically, 30 μg of protein lysate per sample was separated on 4-12% NuPAGE gels (Invitrogen, Carlsbad, CA) and transferred to hybond nitrocellulose membranes (GE Healthcare). The membranes were blocked in 5% nonfat milk in Tris-buffered saline with 0.1% Tween-20 (TBST) for 1 h and then incubated with the primary antibodies (1:1,000 dilution in 5% nonfat milk TBST or 5% BSA in TBST) overnight at 4°C. After incubation, the membranes were washed three times with TBST for 10 min each. The blots were then incubated with secondary horseradish peroxidase-conjugated goat anti-rabbit IgG (Santa Cruz) antibody at a 1:5,000 dilution in 5% nonfat milk in TBST for 2 h at room temperature. The membranes were then washed three times with TBST. The bands were visualized by enhanced chemiluminescence and quantified using ImageQuant (Molecular Dynamics, Sunnyvale, CA) or GeneTools SynGene (K&R Technology, Frederick, MD) software.
Immunohistochemistry and confocal microscopy
Retina sections (10 μm) on glass slides were fixed in 2% paraformaldehyde for 10 min at room temperature and subsequently rinsed in PBS (2 × 10 min). Slides were blocked in 10% donkey serum (Jackson ImmunoResearch, West Grove, PA) for 1 h before incubation with anti-glucosylceramide synthase antibody (1:100) overnight at 4°C. Slides were subsequently washed with PBS containing 0.1% Triton X-100 (3 × 20 min). The slides were then incubated with the secondary antibody (CY3-conjugated donkey anti-rabbit, 1:1,000; Jackson ImmunoResearch). Slides were then washed in PBS with Tween (3 × 20 min) and mounted with aqueous medium (Aqua/Polymount; Polysciences). All images were obtained with a confocal microscope (TCS SP2 AOBS; Leica, Deerfield, IL) at 512 × 512-pixel resolution. Images were maximum projections of z-stacks.
Ganglioside determination
A colormetric assay was utilized to determine the relative amount of lipid-bound sialic acid on gangliosides from R28 cells by a resorcinol-HCl-Cu2+ assay after periodate treatment utilizing methods described by others (29).
Cell viability/death assays
R28 cells were cultured on glass coverslips and treated as described in the text. After treatment, cells were washed in PBS and fixed in 2% paraformaldehyde for 10 min before staining/mounting with Vectashield mounting medium with DAPI (4′,6-diamidino-2-phenylindole) (Vector Laboratories, Burlingame, CA). Five randomly sampled fields from each coverslip were observed to determine the percent of pyknotic nuclei as previously described (25). In other cases, total cell counts were used to determine viability.
Statistical methods
One-way ANOVA with Bonferroni multiple comparison posttest and t test analysis were performed using GraphPad Prism 4.0 software, with statistical significance considered if P < 0.05. Data are reported as the means ± SE from at least three replicate experiments.
RESULTS
Diabetes decreases total ceramide content in retinas of STZ-induced diabetic rats
Proinflammatory cytokines, such as IL-1β and TNF-α, are increased in diabetic rat retinas (30-32), and these cytokines have been demonstrated to increase ceramide content through the activation of the de novo or salvage pathways. Furthermore, ceramide content is increased in the skeletal muscle of insulin-resistant rats (33,34) and humans with diabetes (35,36). We therefore hypothesized that ceramide content would also be increased in these retinas. Ceramide content was measured after 2, 4, and 8 weeks of STZ-induced diabetes by ESI-MS/MS (27). The total of all ceramide molecular species is shown in Fig. 1A. After 2 weeks of diabetes, ceramide levels remained constant; however, surprisingly, after 4 and 8 weeks of diabetes there was a ∼30% decrease in total ceramide content. We have previously shown that acute insulin therapy in diabetic rats can restore or partially restore impaired signaling in this model (7,26); thus, it was determined whether 72 h of insulin therapy would restore ceramide levels to control levels. We again observed that after 4 weeks of diabetes, ceramide mass was decreased ∼30%, but insulin did not restore ceramides to basal levels (Fig. 1B). These data demonstrate that retinal ceramide content is diminished by diabetes and suggest that the altered lipid metabolism may impart retinal resistance to insulin action.
FIG. 1.
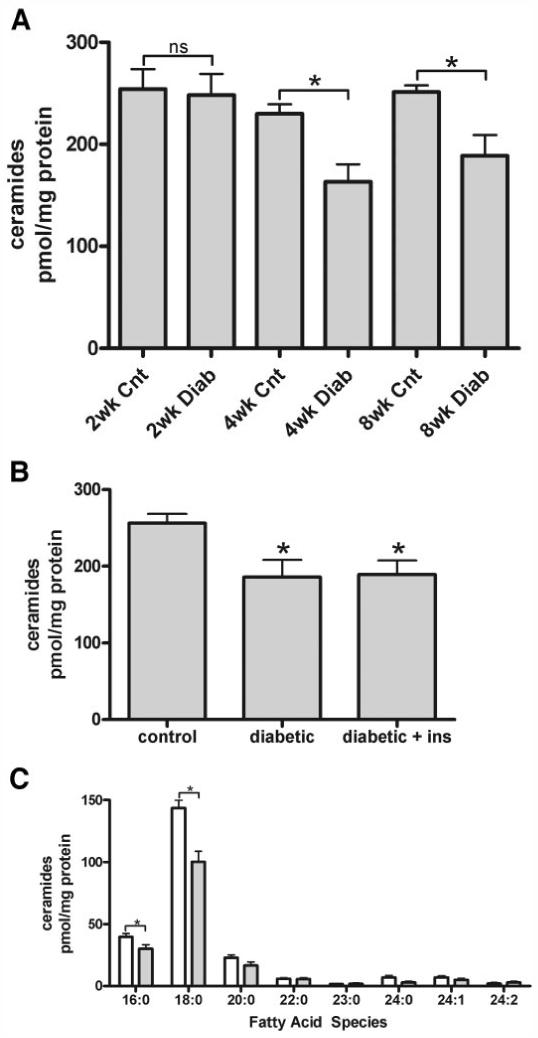
Ceramide mass is decreased in the diabetic retina. A: Ceramides were measured by ESI-MS/MS after lipid extraction from control (Cnt) and diabetic (Diab) animals at the indicated time points; 2 weeks P > 0.05, 4 weeks P < 0.01, and 8 weeks P < 0.01. B: Additional retinas after 4 weeks of diabetes were also assessed for ceramide content after short-term systemic insulin treatment. C: Ceramide fatty acid species after 4 weeks of diabetes was assessed by ESI-MS/MS. Means ± SE, n = 6-8 animals per group. *P ≤ 0.01. See Table 1 for individual n values.  , 4-week control;
, 4-week control;  , 4-week diabetic.
, 4-week diabetic.
In light of the 30% reduction in total ceramide content, we next analyzed the different molecular species of ceramides (Fig. 1C). In the normal retina, 79% of the ceramides contained long-chain saturated C16:0 or C18:0 fatty acids, whereas 21% contained very-long-chain fatty acyl species (C20:0 or longer). This ratio was unaltered by 4 weeks of diabetes, even though the total amount of C16:0 and C18:0 ceramide species diminished by 30%, paralleling the decrease in total ceramide content in diabetes. Moreover, total retinal lipid fatty acid composition, as assessed by gas-liquid chromatography, did not change after 4 weeks of diabetes (data not shown). These data further suggest a unique fatty acid composition of ceramide, as total retinal fatty acid composition also contains unsaturated oleic, arachidonic, and docosahexaenoic acids, which were not observed in the ceramide species from the retina.
Diabetes increases retinal glucosylceramide content in diabetic rats
Decreased ceramide mass suggests an alteration in sphingolipid metabolism, so additional sphingolipids were also measured. Quantification of sphingomyelin, sphinganine, and sphingosine by ESI-MS/MS (28) after 4 weeks of diabetes revealed no significant changes compared with age-matched control retinas (Table 2). Phosphorylated sphingoid metabolites (ceramide-1-phosphate, sphingosine-1-phosphate, and sphinganine-1-phosphate) were present in small amounts, and no overt alterations were observed (data not shown). Increased monohexosylceramide has been implicated in renal complications of diabetes (37), so monohexosylceramide content was also assessed. We observed a significant increase of ∼30% in monohexosylceramide levels in fed diabetic rats compared with their age-matched controls (Fig. 2A). These monohexosylceramides were further identified by ESI-MS/MS to be glucosylceramide with no detectable levels of galactosylceramides. This is not surprising as the retina is devoid of the oligodendrocytes and myelin, which are rich in galactosylceramide and sulfated galactosylceramide (sulfatides). After fasting rats for 16 h, which diminished the blood glucose concentration of the diabetic animals by 55.6% (227.63 ± 22.55 vs. 409.29 ± 17.63 mg/dl), the levels of glucosylceramide in the retinas of fasted diabetic animals were at the same levels as their fed diabetic counterparts, still 30% above basal. Thus, a short-term decrease in glucose concentration did not diminish the higher levels of glucosylceramide, suggesting that the increase in glucosylceramide may reflect changes in glucosylceramide synthase and not mass action. We also assessed the fatty acid composition of the glucosylceramide (Fig. 2B). Similar to ceramide, the majority of glucosylceramide contains saturated palmitic and stearic fatty acids. These C16:0 and C18:0 molecular species of glucosylceramide are significantly increased in diabetes (P < 0.01) and comprise between 63 and 70% of the total glucosylceramide species. We conclude that the elevated metabolism of ceramide in diabetic rat retinas generates glyco(glucosyl)sphingolipids but not other sphingoid derivatives.
TABLE 2.
Sphingolipid content in the rat retina
| Sphingolipid | Control (pmol/mg) | Diabetes (pmol/mg) | P value |
|---|---|---|---|
| Sphinganine | 132.2 ± 16.59 | 113.6 ± 11.26 | 0.37 |
| Sphingosine | 235.9 ± 14.89 | 227.4 ± 13.54 | 0.68 |
| Sphingomyelin | 4854.1 ± 184 | 4835.9 ± 207 | 0.94 |
Data are means ± SE.
FIG. 2.
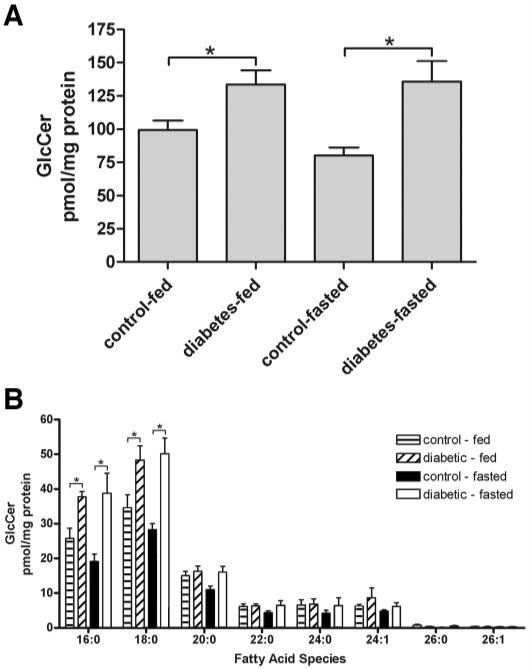
Glucosylceramide is increased in diabetic retinas. A: Glucosylceramides were also assessed after 4 weeks from control and diabetic animals under fed and fasted conditions. B: The fatty acid composition of the glucosylceramides from A. Means ± SE, n = 7-8 animals per group. *P < 0.05.
Glucosylceramide synthase is expressed in the retina
The first committed step to the formation of many glycosphingolipids is catalyzed by glucosylceramide synthase, which forms glucosylceramide through the addition of glucose from uridine diphosphate (UDP)-glucose to ceramide. By immunohistochemistry, we demonstrated that the retina expressed glucosylceramide synthase, with strongest immunoreactivity observed within the neuronal plexiform layers and the outer segments of the photoreceptors, which was unaltered by diabetes (Fig. 3) (23). In addition we have validated these results using another antibody to GCS obtained from Exalpha Biologicals (data not shown). This result demonstrates that the intact rat retina expresses glucosylceramide synthase, a finding consistent with our ESI-MS/MS data (Fig. 2B), and has the capability of metabolizing ceramide to glucosylceramide metabolites.
FIG. 3.
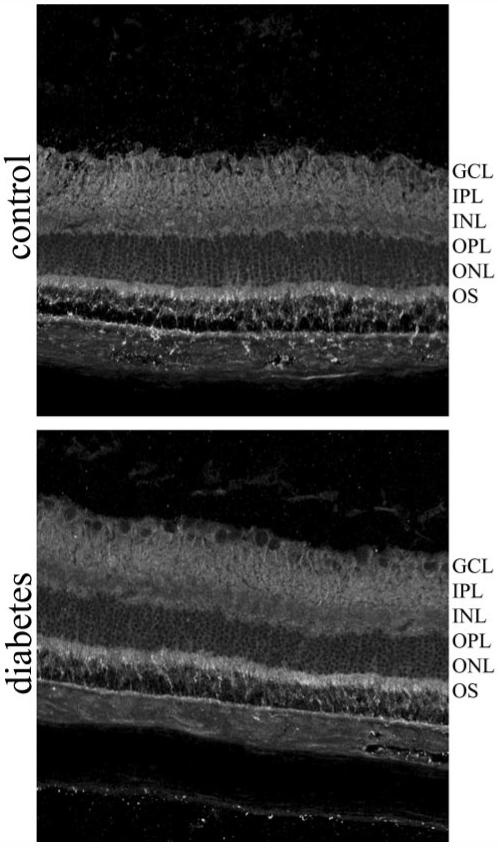
Glucosylceramide synthase is unaltered in the diabetic retina. Glucosylceramide synthase localization was assessed between control and diabetic rat retinas by immunohistochemistry. GCL, retinal ganglion cell layer; INL, inner nuclear layer; IPL, inner plexiform layer; ONL, outer nuclear layer; OPL, outer plexiform layer; OS, photoreceptor outer segments.
Since glucosylceramides are the first committed step to the formation of most gangliosides, it was next determined whether hyperglycemic conditions would increase gangliosides formation in vitro. R28 cells, a model of retinal neurons, were treated with an elevated glucose concentration for 72 h. Gangliosides were purified, and a colorimetric assay was utilized to determine the relative amount of sialic acid-containing sphingolipids. We noted a significant 69% increase in gangliosides treated with elevated levels of glucose (control 1.00 ± 0.07 vs. high glucose [50 mmol/l] 1.69 ± 0.23, n = 3; P < 0.05). Thus, hyperglycemic conditions may be sufficient to increase glycosphingolipid metabolism.
Inhibition of glycosphingolipids metabolism increases insulin sensitivity in retinal neurons
To investigate the potential consequence of this increase in glucosylceramide, an in vitro model of retinal neurons was utilized to determine the putative effects of altering glycosphingolipid metabolism on insulin signaling. R28 cells were pretreated for 24 h with NB-DGJ, a selective imino sugar inhibitor of glucosylceramide synthase (38,39), or PPMP, a morpholino ceramide analog inhibitor of glucosylceramide synthase. Then, R28 cells were treated with high-glucose conditions or glucosamine for an additional 24 h still in the presence of these inhibitors, with the last 2 h under serum-free conditions. R28 cells were subsequently treated with insulin (10 nmol/l for 15 min) and the phosphorylation of p70 S6K, an insulin-responsive prosurvival enzyme whose activity is decreased in the diabetic retina (7), was analyzed by Western blotting (Fig. 4). In the absence of insulin, these glucosylceramide synthase inhibitors caused a modest increase in basal phosphorylation of p70 S6K. When R28 cells were stimulated with insulin, there was a marked increase in Thr389 phosphorylation of this enzyme. Pretreatment with NB-DGJ or PPMP induced a significant increase in insulin-stimulated phosphorylation of p70 S6K. Also of importance, high glucose and glucosamine suppressed insulin-stimulated p70 S6K. In the presence of NB-DGJ or PPMP, insulin further stimulated p70 S6K phosphorylation in the presence of inhibiting concentrations of high glucose or glucosamine. Thus, inhibition of glycosphingolipid metabolism increases insulin sensitivity in retinal neurons.
FIG. 4.
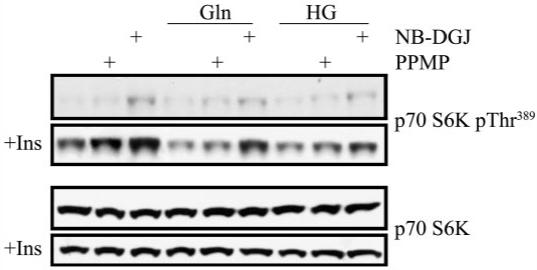
Inhibition of glycosphingolipid metabolism increases insulin sensitivity in retinal neurons. R28 cells were pretreated with glucosylceramide synthase inhibitors NB-DGJ (100 μmol/l) or PPMP (1 μmol/l) for 24 h before treatments with high glucose (HG, 30 mmol/l) or glucosamine (Gln, 5 mmol/l) for an additional 24 h. Cells were then serum starved for 2 h under the same conditions and treated with insulin (10 nmol/l) for 15 min. Western blots were then performed, and the phosphorylation state of p70 S6K, a kinase whose activity is diminished in the diabetic retina, was assessed. A representative blot of three separate experiments. The differences in basal activity of these drugs may reflect different pharmacokinetic and pharmacodynamic characteristics of these drugs.
Glycosphingolipids augment the endoplasmic reticulum stress response in retinal neurons
Diabetes-induced stress and insulin resistance have been associated with induction of the endoplasmic reticulum (ER) stress response (40-43). Stress-induced abnormal protein processing and folding is associated with neurodegeneration (44,45). We investigated the ability of accumulating glycosphingolipids to regulate ER stress in cultured R28 cells. R28 cells were pretreated with CBE, an inhibitor of glucosylceramidase, to limit the catabolism of glucosylceramides and subsequently treated with or without glucosamine. Glucosamine alone induced the expression of glucose-related protein 78 (GRP78) and CHOP, chaperone proteins that are markers of ER stress (Fig. 5). In the presence of CBE, this induction of GRP78 and CHOP was significantly increased. These data suggest that glycoconjugated ceramide metabolites may contribute to ER stress responses in retinal neurons.
FIG. 5.
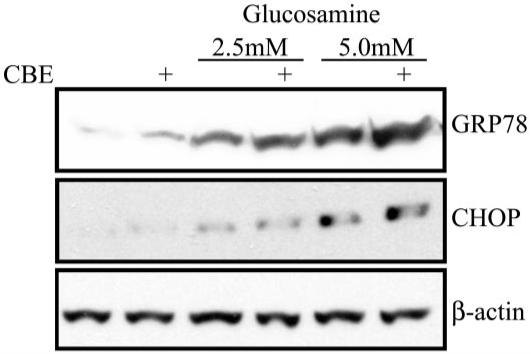
Inhibition of glycosphingolipid catabolism augments glucosamine-induced ER stress responses. A: R28 cells were treated with CBE, an inhibitor of glucosylceramidase, for 2 weeks before plating for experimentation. Untreated and CBE-treated cells were then treated with glucosamine at the indicated dose for 24 h. Cellular lysate was subjected to Western blotting and analyzed for the expression of GRP78, CHOP, or β-actin. A representative blot of three separate experiments.
Glycosphingolipids regulate glucosamine- and inflammatory cytokine-induced cell death in retinal neurons
Treatment of retinal pericytes with glucosamine increases glycosphingolipid formation, which suppresses pericyte proliferation (46). We therefore investigated if glucosamine-induced R28 retinal neuronal cell death (47) can be inhibited by suppressing glycosphingolipid metabolism (Fig. 6A). Treatment of R28 cells with glucosamine for 24 h under serum-free conditions induced 25% pyknosis of the remaining adhered cells, which was significantly higher than basal cell death (P < 0.01). This apoptosis was suppressed to ∼14% for cells treated with either NB-DGJ (P < 0.01) or PPMP (P < 0.01).
FIG. 6.
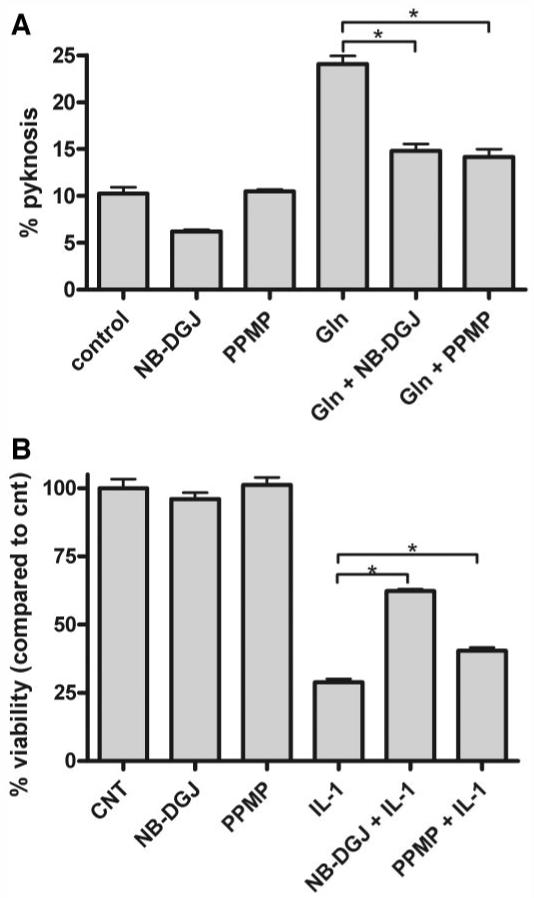
Inhibition of glycosphingolipid metabolism increases R28 retinal neuronal cell viability. A: R28 cells were pretreated with the glucosylceramide synthase inhibitors NB-DGJ (100 μmol/l) or PPMP (1 μmol/l) for 48 h before stimulation with 5 mmol/l glucosamine (Gln) for an additional 24 h. Cells were fixed and stained with DAPI, and pyknotic nuclei were counted and are expressed relative to the total number of cells in each field of view. B: Similarly, R28 cells were treated with NB-DGJ or PPMP and subsequently treated with IL-1β (10 ng/ml) for 24 h. Total cell counts were assessed. Mean ± SE. Three coverslips per treatment. *P < 0.05.
Similarly, as inflammatory cytokines have been demonstrated to mediate their apoptotic effects through altered glycosphingolipid metabolism (19,48), we determined whether inhibition of glycosphingolipid metabolism could suppress IL-1β-induced death in R28 cells (Fig. 6B). We observed that IL-1β significantly reduced cellular viability to 30% of untreated cells (P < 0.01). Importantly, both NB-DGJ and PPMP increased R28 cell viability in the presence of IL-1β (P < 0.01 and P < 0.05, respectively). Therefore, we conclude that inhibition of glycosphingolipid synthesis inhibits the apoptotic effects of hexosamine pathway metabolites and/or inflammatory cytokines, which may be involved in the pathogenesis of diabetes complications.
DISCUSSION
In this study, we demonstrate for the first time that increased glycosphingolipid synthesis through glucosylceramides may contribute to cell death in diabetic retinopathy. In the diabetic rat retina, cellular ceramide levels are decreased (Fig. 1), with a corresponding increase in glucosylceramides (Fig. 2). Pharmacological strategies were utilized to determine the biological consequences of altered glycosphingolipid metabolism in cultured retinal neurons. In vitro, inhibition of glucosylceramide catabolism augments glucosamine-induced stress response (Fig. 5) and inhibition of glycosylceramide synthesis increased insulin sensitivity in retinal neurons (Fig. 4). Decreased glycosphingolipid synthesis also reduced glucosamine- and IL-1β-induced retinal neuronal cell death (Fig. 6). Thus, elevated glucosylceramides composition can putatively mediate stress responses and/or insulin resistance, contributing to the pathogenesis of diabetic retinopathy.
Our data demonstrate that increased glycosphingolipids may be a major mediator of diabetic retinopathy and are consistent with several studies that implicate glycoconjugated ceramide metabolites in the complications of diabetes. Increases in glycosphingolipids also have been implicated in renal hypertrophy/diabetic nephropathy, possibly mediated by advanced glycation end products (37,49) and in the diabetic liver (50,51). In fact, autoantibodies to sulfatides, GT3, GD3, and GM2-1 have been identified in patients with type 1 diabetes (52). Consistent with our data (Figs. 4-6), glycosphingolipids may facilitate proapoptotic, inflammatory cytokine signaling cascades and exacerbate diabetes complications. Our fatty acid data also indicate a primarily saturated composition for the accumulating glucosylceramide, which is consistent with localization within highly structured membrane domains associated with insulin signaling cascades. It may be that augmented glycosphingolipid metabolism contributes to neuronal dysfunction through altered lipid microdomains. In fact, we have recently found that cholesterol depletion, which disrupts lipid microdomains, reduces insulin receptor autophosphorylation by insulin in R28 retinal neurons (T.E.F., M.K., unpublished data). In addition, GM3 within microdomains (18) has been implicated in insulin resistance in response to TNF-α (15), and, conversely, mice deficient in GM3 synthase exhibit greater insulin sensitivity (21). Recent reports suggest that glycosphingolipids can also mediate apoptosis and cellular stress. GD3 has been implicated in TNF-α and CD95 (Fas)-induced apoptosis (16,17,19,48), and GT1b has been demonstrated to inhibit prosurvival Akt activation (53). Lastly, in neuronal tissues, glycosphingolipids have been implicated in increasing sensitivity to neurotoxic agents such as the excitatory amino acid neurotransmitter, glutamate (20), a potential contributor to diabetic retinopathy (54,55). Taken together, glycosphingolipids may be responsible, in part, for insulin resistance in diabetes. Alternatively, diabetic retinopathy could, in part, result from the inability to process augmented glycosphingolipid metabolism, instead of being the result of a specific glycosphingolipid.
We demonstrated that inhibition of glucosylceramide catabolism further sensitized cultured retinal neurons to glucosamine-induced stress. Supporting our studies demonstrating glycosphingolipid-enhanced stress responses and cell death in retinal neurons are studies from the Futerman laboratory, in which glutamate-induced calcium release and neuronal cell death can be reversed by imiglucerase, a recombinant glucosylceramidase (20). In addition, a glucosylceramide synthase inhibitor NB-DGJ reduced hippocampal neuronal death caused by thapsigargin treatment, an inducer of intracellular calcium release and ER stress (56). Furthermore, GM1 accumulation induces the ER stress response in vitro and in vivo in neurons (57), consistent with data not shown, demonstrating that exogenous GM1 induces the ER stress protein GRP78 in retinal neurons. The ability of glycosphingolipids to mediate stress responses, possibly through the regulation of calcium homeostasis, provides a mechanism by which increased glycosphingolipid composition may contribute to neuronal cell death in diabetic retinopathy. Taken together, glycosphingolipids may be a causative factor for diminished insulin signaling and neuronal cell death in diabetic retinas.
While we have emphasized the possible mechanisms by which glycosphingolipids may affect the neurons, we are cognizant of the fact that the effects of this increase may be manifested through other cell types in the retina. One characteristic of diabetic retinopathy is pericyte dropout. El Bawab and colleagues (46) laboratory has recently demonstrated that glucosamine-induced pericyte growth inhibition is mediated through increased ganglioside production. Furthermore, glycosphingolipids have been demonstrated to activate microglial cell lines to produce the inflammatory mediators inducible nitric oxide synthase, TNF-α, and IL-1β through protein kinase C and NADPH oxidase (58) and protein kinase A (59). Lastly, glycosphingolipids can stimulate vascular endothelial growth factor production (60), which in turn could possibly modulate vascular permeability and/or angiogenesis. Interestingly, vascular endothelial growth factor can be produced in the retina under ER stress conditions (61). Thus, activated glial, vascular, and neuronal tissue may synergistically be the consequence of, or exacerbated by, altered glycosphingolipid metabolism.
The increase in retinal glucosylceramide content in diabetes may reflect augmented UDP-glucose production through the pentose pathway, suggesting that hyperglycemia in diabetes drives glucosylceramide production. The fact that glucosamine induced ER stress and cellular death may also suggest a role for increased glucose metabolism through the hexosamine pathway to exacerbate glycosphingolipid-induced diabetes complications. The increase in glucosylceramide mass does not appear to reflect a change in glucosylceramide synthase mass or localization, as demonstrated by immunohistochemistry. Even though we have not ruled out a change in enzymatic activity of glucosylceramide synthase, our data support studies from the Shayman Laboratory (37), where an increase in renal glucosylceramide mass at the expense of ceramide was associated with an increase in UDP-glucose and not glucosylceramide synthase activity (37,62). Alternatively, the inability of short-term fasting or insulin treatment to diminish glucosylceramide mass may reflect an inability of glucosylceramidase to reduce these levels within this time period.
Our studies are, to the best of our knowledge, the first to demonstrate that augmented glycosphingolipid metabolism may contribute to the neuronal pathology of diabetic retinopathy. This increased glycosphingolipid content may be a consequence of multiple factors. Several putative causes of diabetes complications include hyperglycemia-induced increases in the pentose pathway, hexosamine pathway, and/or advanced-glycated end products, all capable of amplifying glycosphingolipid synthesis and/or contributing to stress-induced pathology. Taken together, these studies identify glycosphingolipids as potential therapeutic targets with clinical significance in diabetes complications including retinopathy.
ACKNOWLEDGMENTS
This work was supported by the Juvenile Diabetes Research Foundation (JDRF) Diabetic Retinopathy Center at Penn State University (to M.K. and T.W.G.) and EY015800 (to M.K. and T.E.F.) from the National Eye Institute. T.W.G. is the Jack and Nancy Turner Professor of Ophthalmology. Additional support was provided by GM PA-02-132 (to A.H.M.) and AG23168 (to X.H.). R.E.A.’s research is supported by grants from the National Eye Institute (EY04149, EY00871, and EY12190), the National Center for Research Resources (RR17703), Research to Prevent Blindness, Inc., and the Foundation Fighting Blindness.
We acknowledge Neelam Desai and Wendy Dunton of the JDRF Animal Core and Rhona Ellis of the JDRF Imaging Core facility for their assistance. We also thank Drs. Richard E. Pagano and David L. Marks (Mayo Clinic and Foundation, Rochester, MN) for providing the glucosylceramide synthase antibody and Allen Kunselman for statistical consultation.
Glossary
- CBE
conduritol B epoxide
- CHOP
C/EBP homologous protein
- ER
enoplasmic reticulum
- IL
interleukin
- NB-DGJ
N-butyldeoxygalactonojirimycin
- PPMP
dL-threo-1-phenyl-2-palmitoylamino-3-morpholino-1-propanol
- STZ
streptozotocin
- TBST
Tris-buffered saline with 0.1% Tween-20
- TNF
tumor necrosis factor
- UDP
uridine diphosphate
REFERENCES
- 1.Gardner TW, Antonetti DA, Barber AJ, LaNoue KF, Levison SW. Diabetic retinopathy: more than meets the eye. Surv Ophthalmol. 2002;47(Suppl 2):S253–S262. doi: 10.1016/s0039-6257(02)00387-9. [DOI] [PubMed] [Google Scholar]
- 2.Wolter JR. Diabetic retinopathy. Am J Ophthalmol. 1961;51:1123–1141. doi: 10.1016/0002-9394(61)91802-5. [DOI] [PubMed] [Google Scholar]
- 3.Bloodworth JM., Jr Diabetic retinopathy. Diabetes. 1962;11:1–22. [PubMed] [Google Scholar]
- 4.Hammes HP, Federoff HJ, Brownlee M. Nerve growth factor prevents both neuroretinal programmed cell death and capillary pathology in experimental diabetes. Mol Med. 1995;1:527–534. [PMC free article] [PubMed] [Google Scholar]
- 5.Barber AJ, Lieth E, Khin SA, Antonetti DA, Buchanan AG, Gardner TW. Neural apoptosis in the retina during experimental and human diabetes: early onset and effect of insulin. J Clin Invest. 1998;102:783–791. doi: 10.1172/JCI2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barber AJ, Antonetti DA, Kern TS, Reiter CE, Soans RS, Krady JK, Levison SW, Gardner TW, Bronson SK. The Ins2Akita mouse as a model of early retinal complications in diabetes. Invest Ophthalmol Vis Sci. 2005;46:2210–2218. doi: 10.1167/iovs.04-1340. [DOI] [PubMed] [Google Scholar]
- 7.Reiter CE, Wu X, Sandirasegarane L, Nakamura M, Gilbert KA, Singh RS, Fort PE, Antonetti DA, Gardner TW. Diabetes reduces basal retinal insulin receptor signaling: reversal with systemic and local insulin. Diabetes. 2005;55:1148–1156. doi: 10.2337/diabetes.55.04.06.db05-0744. [DOI] [PubMed] [Google Scholar]
- 8.Kolesnick R. The therapeutic potential of modulating the ceramide/sphingomyelin pathway. J Clin Invest. 2002;110:3–8. doi: 10.1172/JCI16127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bourbon NA, Sandirasegarane L, Kester M. Ceramide-induced inhibition of Akt is mediated through protein kinase Czeta: implications for growth arrest. J Biol Chem. 2002;277:3286–3292. doi: 10.1074/jbc.M110541200. [DOI] [PubMed] [Google Scholar]
- 10.Stover T, Kester M. Liposomal delivery enhances short-chain ceramide-induced apoptosis of breast cancer cells. J Pharmacol Exp Ther. 2003;307:468–475. doi: 10.1124/jpet.103.054056. [DOI] [PubMed] [Google Scholar]
- 11.Bourbon NA, Yun J, Berkey D, Wang Y, Kester M. Inhibitory actions of ceramide upon PKC-epsilon/ERK interactions. Am J Physiol Cell Physiol. 2001;280:C1403–C1411. doi: 10.1152/ajpcell.2001.280.6.C1403. [DOI] [PubMed] [Google Scholar]
- 12.Birbes H, Bawab SE, Obeid LM, Hannun YA. Mitochondria and ceramide: intertwined roles in regulation of apoptosis. Adv Enzyme Regul. 2002;42:113–129. doi: 10.1016/s0065-2571(01)00026-7. [DOI] [PubMed] [Google Scholar]
- 13.Hu W, Xu R, Zhang G, Jin J, Szulc ZM, Bielawski J, Hannun YA, Obeid LM, Mao C. Golgi fragmentation is associated with ceramide-induced cellular effects. Mol Biol Cell. 2005;16:1555–1567. doi: 10.1091/mbc.E04-07-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bleicher RJ, Cabot MC. Glucosylceramide synthase and apoptosis. Biochim Biophys Acta. 2002;1585:172–178. doi: 10.1016/s1388-1981(02)00338-4. [DOI] [PubMed] [Google Scholar]
- 15.Tagami S, Inokuchi Ji J, Kabayama K, Yoshimura H, Kitamura F, Uemura S, Ogawa C, Ishii A, Saito M, Ohtsuka Y, Sakaue S, Igarashi Y. Ganglioside GM3 participates in the pathological conditions of insulin resistance. J Biol Chem. 2002;277:3085–3092. doi: 10.1074/jbc.M103705200. [DOI] [PubMed] [Google Scholar]
- 16.Kristal BS, Brown AM. Apoptogenic ganglioside GD3 directly induces the mitochondrial permeability transition. J Biol Chem. 1999;274:23169–23175. doi: 10.1074/jbc.274.33.23169. [DOI] [PubMed] [Google Scholar]
- 17.De Maria R, Lenti L, Malisan F, d’Agostino F, Tomassini B, Zeuner A, Rippo MR, Testi R. Requirement for GD3 ganglioside in CD95- and ceramide-induced apoptosis. Science. 1997;277:1652–1655. doi: 10.1126/science.277.5332.1652. [DOI] [PubMed] [Google Scholar]
- 18.Kabayama K, Sato T, Kitamura F, Uemura S, Kang BW, Igarashi Y, Inokuchi J. TNFalpha-induced insulin resistance in adipocytes as a membrane microdomain disorder: involvement of ganglioside GM3. Glycobiology. 2005;15:21–29. doi: 10.1093/glycob/cwh135. [DOI] [PubMed] [Google Scholar]
- 19.Garcia-Ruiz C, Colell A, Morales A, Calvo M, Enrich C, Fernandez-Checa JC. Trafficking of ganglioside GD3 to mitochondria by tumor necrosis factor-alpha. J Biol Chem. 2002;277:36443–36448. doi: 10.1074/jbc.M206021200. [DOI] [PubMed] [Google Scholar]
- 20.Pelled D, Shogomori H, Futerman AH. The increased sensitivity of neurons with elevated glucocerebroside to neurotoxic agents can be reversed by imiglucerase. J Inherit Metab Dis. 2000;23:175–184. doi: 10.1023/a:1005622001239. [DOI] [PubMed] [Google Scholar]
- 21.Yamashita T, Hashiramoto A, Haluzik M, Mizukami H, Beck S, Norton A, Kono M, Tsuji S, Daniotti JL, Werth N, Sandhoff R, Sandhoff K, Proia RL. Enhanced insulin sensitivity in mice lacking ganglioside GM3. Proc Natl Acad Sci U S A. 2003;100:3445–3449. doi: 10.1073/pnas.0635898100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Acharya U, Patel S, Koundakjian E, Nagashima K, Han X, Acharya JK. Modulating sphingolipid biosynthetic pathway rescues photoreceptor degeneration. Science. 2003;299:1740–1743. doi: 10.1126/science.1080549. [DOI] [PubMed] [Google Scholar]
- 23.Marks DL, Wu K, Paul P, Kamisaka Y, Watanabe R, Pagano RE. Oligomerization and topology of the Golgi membrane protein glucosylceramide synthase. J Biol Chem. 1999;274:451–456. doi: 10.1074/jbc.274.1.451. [DOI] [PubMed] [Google Scholar]
- 24.Seigel GM. Establishment of an E1A-immortalized retinal cell culture. In Vitro Cell Dev Biol Anim. 1996;32:66–68. doi: 10.1007/BF02723034. [DOI] [PubMed] [Google Scholar]
- 25.Barber AJ, Nakamura M, Wolpert EB, Reiter CE, Seigel GM, Antonetti DA, Gardner TW. Insulin rescues retinal neurons from apoptosis by a phosphatidylinositol 3-kinase/Akt-mediated mechanism that reduces the activation of caspase-3. J Biol Chem. 2001;276:32814–32821. doi: 10.1074/jbc.M104738200. [DOI] [PubMed] [Google Scholar]
- 26.Barber AJ, Antonetti DA, Gardner TW. Altered expression of retinal occludin and glial fibrillary acidic protein in experimental diabetes: the Penn State Retina Research Group. Invest Ophthalmol Vis Sci. 2000;41:3561–3568. [PubMed] [Google Scholar]
- 27.Han X. Characterization and direct quantitation of ceramide molecular species from lipid extracts of biological samples by electrospray ionization tandem mass spectrometry. Anal Biochem. 2002;302:199–212. doi: 10.1006/abio.2001.5536. [DOI] [PubMed] [Google Scholar]
- 28.Merrill AH, Jr, Sullards MC, Allegood JC, Kelly S, Wang E. Sphingolipidomics: high-throughput, structure-specific, and quantitative analysis of sphingolipids by liquid chromatography tandem mass spectrometry. Methods. 2005;36:207–224. doi: 10.1016/j.ymeth.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 29.Jourdian GW, Dean L, Roseman S. The sialic acids. XI. A periodate-resorcinol method for the quantitative estimation of free sialic acids and their glycosides. J Biol Chem. 1971;246:430–435. [PubMed] [Google Scholar]
- 30.Carmo A, Cunha-Vaz JG, Carvalho AP, Lopes MC. L-arginine transport in retinas from streptozotocin diabetic rats: correlation with the level of IL-1 beta and NO synthase activity. Vision Res. 1999;39:3817–3823. doi: 10.1016/s0042-6989(99)00117-0. [DOI] [PubMed] [Google Scholar]
- 31.Gerhardinger C, Costa MB, Coulombe MC, Toth I, Hoehn T, Grosu P. Expression of acute-phase response proteins in retinal Muller cells in diabetes. Invest Ophthalmol Vis Sci. 2005;46:349–357. doi: 10.1167/iovs.04-0860. [DOI] [PubMed] [Google Scholar]
- 32.Krady JK, Basu A, Allen CM, Xu Y, LaNoue KF, Gardner TW, Levison SW. Minocycline reduces proinflammatory cytokine expression, microglial activation, and caspase-3 activation in a rodent model of diabetic retinopathy. Diabetes. 2005;54:1559–1565. doi: 10.2337/diabetes.54.5.1559. [DOI] [PubMed] [Google Scholar]
- 33.Turinsky J, O’Sullivan DM, Bayly BP. 1,2-Diacylglycerol and ceramide levels in insulin-resistant tissues of the rat in vivo. J Biol Chem. 1990;265:16880–16885. [PubMed] [Google Scholar]
- 34.Gorska M, Dobrzyn A, Zendzian-Piotrowska M, Gorski J. Effect of streptozotocin-diabetes on the functioning of the sphingomyelin-signalling pathway in skeletal muscles of the rat. Horm Metab Res. 2004;36:14–21. doi: 10.1055/s-2004-814197. [DOI] [PubMed] [Google Scholar]
- 35.Straczkowski M, Kowalska I, Nikolajuk A, Dzienis-Straczkowska S, Kinalska I, Baranowski M, Zendzian-Piotrowska M, Brzezinska Z, Gorski J. Relationship between insulin sensitivity and sphingomyelin signaling pathway in human skeletal muscle. Diabetes. 2004;53:1215–1221. doi: 10.2337/diabetes.53.5.1215. [DOI] [PubMed] [Google Scholar]
- 36.Adams JM, 2nd, Pratipanawatr T, Berria R, Wang E, DeFronzo RA, Sullards MC, Mandarino LJ. Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes. 2004;53:25–31. doi: 10.2337/diabetes.53.1.25. [DOI] [PubMed] [Google Scholar]
- 37.Zador IZ, Deshmukh GD, Kunkel R, Johnson K, Radin NS, Shayman JA. A role for glycosphingolipid accumulation in the renal hypertrophy of streptozotocin-induced diabetes mellitus. J Clin Invest. 1993;91:797–803. doi: 10.1172/JCI116299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Platt FM, Neises GR, Karlsson GB, Dwek RA, Butters TD. N-butyldeoxyga-lactonojirimycin inhibits glycolipid biosynthesis but does not affect N-linked oligosaccharide processing. J Biol Chem. 1994;269:27108–27114. [PubMed] [Google Scholar]
- 39.Andersson U, Butters TD, Dwek RA, Platt FM. N-butyldeoxygalactonojirimycin: a more selective inhibitor of glycosphingolipid biosynthesis than N-butyldeoxynojirimycin, in vitro and in vivo. Biochem Pharmacol. 2000;59:821–829. doi: 10.1016/s0006-2952(99)00384-6. [DOI] [PubMed] [Google Scholar]
- 40.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 41.Ozawa K, Miyazaki M, Matsuhisa M, Takano K, Nakatani Y, Hatazaki M, Tamatani T, Yamagata K, Miyagawa J, Kitao Y, Hori O, Yamasaki Y, Ogawa S. The endoplasmic reticulum chaperone improves insulin resistance in type 2 diabetes. Diabetes. 2005;54:657–663. doi: 10.2337/diabetes.54.3.657. [DOI] [PubMed] [Google Scholar]
- 42.Nakatani Y, Kaneto H, Kawamori D, Yoshiuchi K, Hatazaki M, Matsuoka TA, Ozawa K, Ogawa S, Hori M, Yamasaki Y, Matsuhisa M. Involvement of endoplasmic reticulum stress in insulin resistance and diabetes. J Biol Chem. 2005;280:847–851. doi: 10.1074/jbc.M411860200. [DOI] [PubMed] [Google Scholar]
- 43.Cardozo AK, Ortis F, Storling J, Feng YM, Rasschaert J, Tonnesen M, Van Eylen F, Mandrup-Poulsen T, Herchuelz A, Eizirik DL. Cytokines down-regulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic β-cells. Diabetes. 2005;54:452–456. doi: 10.2337/diabetes.54.2.452. [DOI] [PubMed] [Google Scholar]
- 44.Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y, Takahashi R. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell. 2001;105:891–902. doi: 10.1016/s0092-8674(01)00407-x. [DOI] [PubMed] [Google Scholar]
- 45.Paschen W, Frandsen A. Endoplasmic reticulum dysfunction: a common denominator for cell injury in acute and degenerative diseases of the brain? J Neurochem. 2001;79:719–725. doi: 10.1046/j.1471-4159.2001.00623.x. [DOI] [PubMed] [Google Scholar]
- 46.Masson E, Wiernsperger N, Lagarde M, El Bawab S. Involvement of gangliosides in glucosamine-induced proliferation decrease of retinal pericytes. Glycobiology. 2005;15:585–591. doi: 10.1093/glycob/cwi039. [DOI] [PubMed] [Google Scholar]
- 47.Nakamura M, Barber AJ, Antonetti DA, LaNoue KF, Robinson KA, Buse MG, Gardner TW. Excessive hexosamines block the neuroprotective effect of insulin and induce apoptosis in retinal neurons. J Biol Chem. 2001;276:43748–43755. doi: 10.1074/jbc.M108594200. [DOI] [PubMed] [Google Scholar]
- 48.Garcia-Ruiz C, Colell A, Mari M, Morales A, Calvo M, Enrich C, Fernandez-Checa JC. Defective TNF-alpha-mediated hepatocellular apoptosis and liver damage in acidic sphingomyelinase knockout mice. J Clin Invest. 2003;111:197–208. doi: 10.1172/JCI16010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Masson E, Troncy L, Ruggiero D, Wiernsperger N, Lagarde M, Bawab SE. a-Series gangliosides mediate the effects of advanced glycation end products on pericyte and mesangial cell proliferation: a common mediator for retinal and renal microangiopathy? Diabetes. 2005;54:220–227. doi: 10.2337/diabetes.54.1.220. [DOI] [PubMed] [Google Scholar]
- 50.Abregu AV, Genta SB, Sanchez Riera AN, Sanchez SS. Immunohistochemical detection of hepatic GM1 and GM2 gangliosides in streptozotocin-induced diabetic rats. Hepatol Res. 2002;24:256. doi: 10.1016/s1386-6346(02)00094-3. [DOI] [PubMed] [Google Scholar]
- 51.Sanchez SS, Abregu AV, Aybar MJ, Sanchez Riera AN. Changes in liver gangliosides in streptozotocin-induced diabetic rats. Cell Biol Int. 2000;24:897–904. doi: 10.1006/cbir.1999.0452. [DOI] [PubMed] [Google Scholar]
- 52.Misasi R, Dionisi S, Farilla L, Carabba B, Lenti L, Di Mario U, Dotta F. Gangliosides and autoimmune diabetes. Diabetes Metab Rev. 1997;13:163–179. doi: 10.1002/(sici)1099-0895(199709)13:3<163::aid-dmr189>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 53.Wang XQ, Sun P, Paller AS. Inhibition of integrin-linked kinase/protein kinase B/Akt signaling: mechanism for ganglioside-induced apoptosis. J Biol Chem. 2001;276:44504–44511. doi: 10.1074/jbc.M106563200. [DOI] [PubMed] [Google Scholar]
- 54.Lieth E, Barber AJ, Xu B, Dice C, Ratz MJ, Tanase D, Strother JM. Glial reactivity and impaired glutamate metabolism in short-term experimental diabetic retinopathy: Penn State Retina Research Group. Diabetes. 1998;47:815–820. doi: 10.2337/diabetes.47.5.815. [DOI] [PubMed] [Google Scholar]
- 55.Ambati J, Chalam KV, Chawla DK, D’Angio CT, Guillet EG, Rose SJ, Vanderlinde RE, Ambati BK. Elevated gamma-aminobutyric acid, glutamate, and vascular endothelial growth factor levels in the vitreous of patients with proliferative diabetic retinopathy. Arch Ophthalmol. 1997;115:1161–1166. doi: 10.1001/archopht.1997.01100160331011. [DOI] [PubMed] [Google Scholar]
- 56.Pelled D, Lloyd-Evans E, Riebeling C, Jeyakumar M, Platt FM, Futerman AH. Inhibition of calcium uptake via the sarco/endoplasmic reticulum Ca2+-ATPase in a mouse model of Sandhoff disease and prevention by treatment with N-butyldeoxynojirimycin. J Biol Chem. 2003;278:29496–29501. doi: 10.1074/jbc.M302964200. [DOI] [PubMed] [Google Scholar]
- 57.Tessitore A, del P Martin M, Sano R, Ma Y, Mann L, Ingrassia A, Laywell ED, Steindler DA, Hendershot LM, d’Azzo A. GM1-ganglioside-mediated activation of the unfolded protein response causes neuronal death in a neurodegenerative gangliosidosis. Mol Cell. 2004;15:753–766. doi: 10.1016/j.molcel.2004.08.029. [DOI] [PubMed] [Google Scholar]
- 58.Min KJ, Pyo HK, Yang MS, Ji KA, Jou I, Joe EH. Gangliosides activate microglia via protein kinase C and NADPH oxidase. Glia. 2004;48:197–206. doi: 10.1002/glia.20069. [DOI] [PubMed] [Google Scholar]
- 59.Min KJ, Yang MS, Jou I, Joe EH. Protein kinase A mediates microglial activation induced by plasminogen and gangliosides. Exp Mol Med. 2004;36:461–467. doi: 10.1038/emm.2004.58. [DOI] [PubMed] [Google Scholar]
- 60.Koochekpour S, Merzak A, Pilkington GJ. Vascular endothelial growth factor production is stimulated by gangliosides and TGF-beta isoforms in human glioma cells in vitro. Cancer Lett. 1996;102:209–215. doi: 10.1016/0304-3835(96)04161-4. [DOI] [PubMed] [Google Scholar]
- 61.Abcouwer SF, Marjon PL, Loper RK, Vander Jagt DL. Response of VEGF expression to amino acid deprivation and inducers of endoplasmic reticulum stress. Invest Ophthalmol Vis Sci. 2002;43:2791–2798. [PubMed] [Google Scholar]
- 62.el-Khatib M, Radin NS, Shayman JA. Glycosphingolipid synthesis and proliferation in a renal cell line grown in high glucose. Am J Physiol. 1996;270:F476–F484. doi: 10.1152/ajprenal.1996.270.3.F476. [DOI] [PubMed] [Google Scholar]