

Abstract
1-Aminocyclopropane-1-carboxylate synthase (ACC synthase, EC 4.4.1.14) catalyzes the rate-limiting step in the ethylene biosynthetic pathway in plants. To determine the amino acid residues critical for the structure and function of this enzyme, the tomato Le-ACS2 isoenzyme has been subjected to both site-directed and PCR random mutagenesis. Mutant ACC synthases with reduced enzyme activity have been selected by using a genetic screen based on the functional complementation of an Escherichia coli Ile auxotroph that has been engineered to express ACC deaminase from Pseudomonas sp. The DNA sequence of almost 1,000 clones has been determined, and 334 single missense mutations have been selected for analysis. We have identified three classes of mutants based on their activity and expression in E. coli. Class I and II mutants have the same level of protein expression as the wild type, but their enzyme activity is reduced to 0–5% and 5–50%, respectively. Class III mutants have neither activity nor detectable protein expression. The inactive mutations are clustered in regions that are highly conserved among various ACC synthases. This library of mutants will facilitate the elucidation of structure-function relationships of this regulatory enzyme.
Keywords: isoleucine auxotroph/missense mutation/growth screen/structure-function analysis
Ethylene is one of the simplest organic molecules with biological activity and is considered to be the hormone that regulates plant senescence (1). 1-Aminocyclopropane-1-carboxylic acid (ACC) synthase (S-adenosyl-l-methionine methylthioadenosine-lyase, EC 4.4.1.14) catalyzes the first committed step in the biosynthesis of ethylene by converting S-adenosylmethionine (AdoMet) to the cyclic amino acid ACC and methylthioadenosine. ACC synthase is a cytosolic enzyme that requires pyridoxal phosphate (PLP) as a cofactor (2). It has been proposed that ACC mediates an α,γ-1,3 elimination reaction that is unique among all PLP-dependent enzymes (3, 4). Biochemical characterization of this enzyme has been difficult because it has a short half-life and exists in low abundance in plant tissues (2). Its short half-life has been attributed to a suicide inactivation reaction resulting from a side β,γ-elimination reaction of AdoMet to form l-vinylglycine that inactivates the enzyme (5). The cloning and functional expression of ACC synthase in heterologous systems such as yeast and Escherichia coli have facilitated the biochemical and structural studies of this enzyme (6–11).
ACC synthase is encoded by a multigene family in every plant species examined (12). The primary sequence encoded by these genes shows sequence conservation ranging from 50% to 96% amino acid sequence identity with the highest variability at the carboxylic end of the protein (13). In addition, ACC synthase shares sequence similarity with other PLP-dependent enzymes and is most closely related to the subgroup 1 aminotransferases, which includes aspartate aminotransferase (AspAT). ACC synthase contains all 11 invariant residues in this subgroup, including four conserved residues present in all aminotransferases (13–16). Site-directed mutagenesis of some of these conserved residues in ACC synthase indicate they are essential for activity and may have similar roles in catalysis as their counterparts in AspAT (9, 10). Furthermore, complementation studies with ACC synthase mutants have indicated that like AspAT, ACC synthase functions as a dimer with shared active sites (8, 9). Deletion analysis has shown that the variable carboxyl terminus and a portion of the amino terminus of ACC synthase may be unimportant for enzyme activity but affect enzyme function and conformation (6, 17).
Because ACC synthase plays a critical role in ethylene biosynthesis, it is of great interest to understand how other domains of this protein contribute to its structure and activity. Here, we have used random and site-directed mutagenesis coupled with a genetic screen to isolate ACC synthase mutants with reduced enzymatic activity. Analysis of these mutations has allowed us to identify amino acid residues potentially important in enzyme function. We also discuss the possible correlation of residues identified as critical by mutagenesis and those identified as important based on the sequence conservation among ACC synthases and aminotransferases. Elucidation of structure-function relationships of ACC synthase has the potential to allow the design of compounds that reversibly inhibit its activity. Such compounds may be used for controlling ethylene-induced plant senescence.
MATERIALS AND METHODS
Enzymes and Chemicals.
Restriction and DNA modifying enzymes were obtained from New England Biolabs and Boehringer Mannheim. Acrylamide and SDS/PAGE gel reagents were from ICN and Bio-Rad. All other chemicals were at least reagent grade and obtained from Sigma.
Bacterial Strains and Plasmids.
The bacterial strain used for most transformations was E. coli SURE: mcrA, Δ(mcrCB-hsdSMR-mrr)171, endA1, supE-44, thi-1, gyrA-96. relA1, lac, recB, recJ, sbcC, umuC(Kanr), uvrC(Tet r) obtained from Stratagene. E. coli JHM544 (18) has been derived from E. coli K12 by UV irradiation and was obtained from the American Type Culture Collection. JAde 6 was constructed by integrating the ACC deaminase gene into the genome of JHM544 by using the method described by Hasan et al. (19). Specifically the ACC deaminase gene under the control of the recA promoter (pMON10078 from ref. 20) was subcloned as a SalI (blunt)–HindIII fragment and ligated into the EcoRV–HindIII sites of pNH64a (19). After the in vitro cre reaction, the ACC deaminase containing plasmid was digested with DraI to eliminate the ori+ plasmids and then transformed into JHM544 by electroporation. E. coli containing the integrated ACC deaminase gene were selected as Kan-resistant colonies. The integration of the ACC deaminase gene was confirmed by using PCR and Southern blot analysis of genomic DNA from the Kan-resistant colonies. The vectors used for expression of Le-ACS2 were pQE-3 and pQE-6 (Qiagen).
Oligonucleotides.
The following oligonucleotides were synthesized by using a Perkin—Elmer model 392 DNA synthesizer for making the PCR random mutants in Le-ACS2 cDNA (restriction sites used for subcloning the PCR products are underlined): region A, forward, 5′-CAGAATTCATTAAAGAGGAGAAATTAACCATGG-3′ and reverse, 5′-CTCCATAAATTTCGCAATCGCTTTTCTGAATTC-3′; region B, forward, 5′-CAACTTTCAAGATTATCATGGCTTGCCTGAATTC-3′ and reverse, 5′-GCTTTTGAAGTAATTTTGAAATTATTGGAGCTC-3′; region C, forward, 5′-GTACAACTTATTCCAATTCACTGTGAGAGCTC-3′ and reverse, 5′-GGTAACCCCATGTCTTTTGAAAGACTGTAGAC-3′; region D, forward, 5′- CTGCAACAAAGATTTAGTTCACATCGTCTAC-3′ and reverse, 5′-CAAACGAAGATCCAGGCGAGACGTTAAGCTT-3′; and region E, forward, 5′-GTTATGGAGAGTTATTATAAACGATGTTAAGCTT-3′ and reverse, 5′-GGCCGGATCCTTCCCTTTTAATTAAGTCTTA-3′.
These primers have been used for site-directed mutagenesis of Le-ACS2 (the mutated codons are underlined): P150, 5′-GGCGATGCATTTTTAGTACCTTCA(A,G,T)CATACTAC-3′; P210, 5′-CCATCAAATCGATTGGGC-3′; R286, 5′-CATGGGGTTACCAGGATTT(A,G)(C,T)AGTCGGA-3′; and G288, 5′-CATGGGGTTACCAGGATTTAGAGTCG(A,T,C)AATCATA-3′.
Mutagenesis and Screening.
The strategy for the mutagenesis and screening of Le-ACS2 is shown in Fig. 1. To facilitate the mutagenesis and sequencing, we subdivided the Le-ACS2 cDNA into five nonoverlapping segments of 300- to 400-bp length separated by six unique restriction sites. Random mutagenesis of each segment of the Le-ACS2 cDNA was performed by PCR containing a specific primer pair and using Taq DNA polymerase in the presence of an excess amount of one dNTP (21). Under these conditions, the amplified products contain substitutions correlated with the concentration of the excess nucleotide. For each segment, a pair of oligonucleotide primers were prepared as indicated above. We have selected the PCR conditions to generate on the average 2–3 nucleotide substitutions for each amplified fragment. These substitutions should occur randomly throughout the amplified region except for six nucleotides at each end constituting the restriction sites used for subcloning. To increase the probability that mutagenesis would occur randomly throughout the sequence of Le-ACS2 and all possible substitutions could occur, we carried out the PCRs by using five different protocols. These protocols differed in the identity of the nucleotide in excess and in the presence or absence of MnCl2. In particular, protocol 1 contained 3.4 mM dATP, 4.2 mM MgCl2, and 0.5 mM MnCl2; protocol 2 contained 3.4 mM dCTP, 4.2 mM MgCl2, and 0.5 mM MnCl2; protocol 3 has 3.4 mM dTTP, 4.2 mM MgCl2, and 0.5 mM MnCl2; protocol 4 has 3.4 mM dGTP and 4.7 mM MgCl2; and protocol 5 has 0.85 dGTP and 2.15 mM MgCl2. The incubation mixture (25 μl) in all five protocols contained 10 mM Tris⋅HCl (pH 8.7 at 25°C), 50 mM KCl, 5 μg/ml of BSA, 0.5 μM of each of the two primers, 600 pM of template DNA, 2 units of AmpliTaq polymerase (Perkin–Elmer), and 0.2 mM of the three nucleotides not in excess. The reaction mixtures were covered with 25 μl of mineral oil and subjected to the following PCR conditions: 94°C for 5 min, one cycle; 91°C for 1 min, 51°C for 1 min, and 72°C for 3 min for 16 cycles; followed by one cycle of incubation at 72°C for 5 min. For protocol 5 the PCR product was diluted 220-fold and used as a template for a second PCR in the presence of 3.4 mM dATP, 0.1 mM dCTP, 0.21 mM dGTP, 0.27 mM dTTP, and 3.28 mM MgCl2. The PCR-amplified fragments were digested with the appropriate restriction enzymes, gel-purified, and religated to the unmutated portion of the Le-ACS2 cDNA in the Qiagen vector. Plasmids containing mutated inserts from each of the five protocols were transformed into JAde 6 competent cells by electroporation. Transformed cells were plated on Luria–Bertani medium (LB) + 100 μg/ml amp + 25 μg/ml kan and incubated at 37°C overnight. Colonies from the LB plates were either patched on minimal media (M9) + 25 μg/ml amp + 15 μg/ml kan and LB +100 μg/ml amp + 25 μg/ml kan or replica plated on these two agar plates. To prevent cross feeding of ACC from clones expressing functional ACC synthase to nearby mutant clones, transformants were patched on 100 × 15-mm square plates (Falcon) at low density (36 transformants per plate). Mutants were selected for their inability to grow on M9 plates after 2 days of incubation at 37°C. Plasmid DNA from putative mutants was isolated and checked by restriction analysis. We isolated approximately 50 putative mutants containing fragments from each of the five PCR mutagenesis protocols for each region of Le-ACS2 except for the fifth region. Sequencing of the mutated inserts was done by using the fluorescent dye terminator kit from Applied Biosystems (ABI) and the ABI 373 or 377 automated sequencer. Analysis of the sequences obtained was achieved by using the program sequencher 3.0 (Gene Codes, Ann Arbor, MI). Mutants with single amino acid substitutions were selected and denoted by the combination of the original amino acid and its position in the Le-ACS2 amino acid sequence followed by the substituted amino acid in the one-letter notation.
Figure 1.

Strategy for screening ACC synthase mutants.
The mutants Y92L, Y92F, Y92W, K278A, and R412K were created by site-directed mutagenesis as previously described (9). Mutations at residues P150, P210, R286, and G288 were generated by PCR site-directed mutagenesis by using the oligonucleotides listed above. Routine DNA manipulations were performed as described (22).
ACC Synthase Expression and Activity Assay in E. coli.
Le-ACS2 mutants containing single missense mutations were selected and transformed into E. coli SURE to check for ACC synthase activity and expression. SURE cells containing wild-type or mutant ACC synthase cDNA were grown overnight in 3 ml of LB broth supplemented with 100 μg/ml of amp. The next day, 100 μl of the overnight culture was inoculated into a 3-ml LB + 100 μg/ml amp medium and grown for 3 hr. Aliquots from this 3-hr culture were indirectly assessed for ACC synthase activity and protein levels by measuring the amount of ACC secreted to the growth medium as described by Sato et al. (23) and expression by immunoblot analysis as previously described (9). The amount of ACC secreted after 3 hr of growth is expressed as nM ACC per 107 cells assuming that 8 × 108 cells/ml corresponds to OD600 = 1.0. E. coli lysate for immunoblot analysis was obtained by pelleting cells from a volume of the bacterial culture equivalent to OD600 = 0.75. The cell pellets then were resuspended in 60 μl of SDS/PAGE buffer and heated at 90°C for 10 min. Before loading on 10% polyacrylamide gels, the protein extracts were diluted 1:5 with SDS/PAGE buffer and 1 μl of the diluted samples were loaded for electrophoresis and immunoblot analysis. Immunoblots were probed with 1:3,000 dilution of partially purified Le-ACS2 polyclonal antibody obtained previously (13) and visualized with alkaline phosphatase conjugated to goat anti-rabbit secondary antibody (Promega).
RESULTS
Experimental Strategy and Development of the Genetic Screen.
Fig. 1 shows the mutagenesis strategy designed to identify residues critical for the activity of ACC synthase. Random mutagenesis of the Le-ACS2 isoenzyme from tomato has been carried out because the majority of the biochemical and structural studies have been done with this isoenzyme (6, 9, 17). Because our goal has been to select inactive ACC synthase mutants with single missense mutations, the PCR random mutagenesis method we have chosen has been optimized to yield a random distribution of 2–3 nucleotide substitutions in the amplified region. Because of the low number of nucleotide substitutions and the degeneracy of the genetic code, the majority of Le-ACS2 cDNAs recovered, however, will have been expected to encode functional proteins. To facilitate the selection of nonfunctional mutants, we have developed a genetic screen based on the functional complementation of E. coli Ile auxotrophic cells as shown in Figs. 1 and 2. We have used E. coli JHM-544, which has a growth requirement for Ile or its four-carbon precursor α-ketobutyrate because of a mutation in the early step of the isoleucine biosynthetic pathway that converts threonine to α-ketobutyrate (ref. 18, see also Fig. 2). An alternative pathway bypassing this mutation in JHM544 can be engineered by expressing two enzymes, ACC synthase (13) and ACC deaminase (20) in this strain (see Fig. 2). In such an E. coli strain, endogeneous AdoMet synthetase catalyzes the formation of AdoMet, which can be used as a substrate by ACC synthase to make ACC. The enzyme ACC deaminase converts ACC to α-ketobutyrate, which can be used to synthesize isoleucine, thus complementing the mutation (Fig. 2). Introduction of an active ACC synthase in this strain will complement Ile auxotrophy whereas an inactive ACC synthase will fail to do so.
Figure 2.

The biosynthetic pathway of isoleucine in the E. coli auxotroph JHM544 (shaded). The pathway was engineered to allow Ile autotrophy by integrating ACC deaminase into the genome, yielding an E. coli named JAde 6, which can metabolize ACC synthesized when transformed with ACC synthase.
The feasibility of this approach was tested by determining the growth characteristics of JHM544 expressing ACC synthase and ACC deaminase individually or in combination on minimal media or in media supplemented with Ile or ACC. As expected, JHM544 cannot grow on M9 plates without Ile supplementation (Fig. 3). JHM544 expressing ACC deaminase from either a plasmid or integrated into the genome (JAde 6) grows well on M9 plates supplemented with ACC (see Fig. 3). To assess how much ACC is required for complementing the Ile auxotrophy, we examined the growth of JHM544 expressing ACC deaminase from a plasmid, or integrated in the genome as in JAde 6 on M9 media containing different concentrations of ACC (Fig. 3). Although JHM544 cannot grow on M9 even with the addition of 3 mM ACC, JHM544 expressing ACC deaminase from a plasmid or integrated into the genome (JAde 6) grows very well on M9 media supplemented with ACC at concentrations as low as 0.05 mM. However, on M9 media containing the same concentrations of ACC, JAde 6 shows less growth compared with JHM544 expressing ACC deaminase from a plasmid. This finding is likely because of the presence of multiple copies of the plasmid containing ACC deaminase whereas JAde 6 only has one copy of the gene in the genome. Occasionally, we see some sparse growth of JHM544 on M9 + 3 mM ACC after prolonged incubation (≥3 days) at 37°C (data not shown). Because E. coli does not have an endogenous ACC deaminase, it is possible that JHM544 can use ACC to make Ile through a different pathway. Consistent with this idea, JHM544 cells expressing wild-type Le-ACS2 show some growth on M9 media without any supplementation (data not shown). Expression of both ACC synthase and ACC deaminase in JHM544 results in vigorous growth on minimal media without any supplementation (data not shown). This growth is caused by the presence of a functional ACC synthase because JHM544 expressing ACC deaminase and an inactive mutant of Le-ACS2 containing the K278A mutation is unable to grow on minimal media (data not shown). Consequently, inactive forms of ACC synthase can be identified by selecting for transformants of JHM544 expressing ACC deaminase that are unable to grow on minimal media. During this study, we used the JAde 6 strain that has an integrated ACC deaminase gene in its genome to facilitate DNA manipulations and to ensure more consistent expression. An example of the genetic screen is shown in Fig. 1.
Figure 3.

Growth of JHM544 expressing ACC deaminase either from a plasmid or integrated into the genome (JAde 6) on minimal media supplemented with different concentrations of ACC or 0.1% Ile. Plates were incubated for 2 days at 37°C.
Selection and Identification of ACC Synthase Mutants.
After screening about 8,000 transformants, we obtained almost 1,000 putative Le-ACS2 mutants (Table 1). The mutated regions from the cDNA of these clones were sequenced, and 334 mutants with single missense mutations were selected for further analysis. The location of these missense mutations are shown in Fig. 4. The mutations are found throughout the sequence of Le-ACS2 and include substitutions of five of the 11 residues conserved in ACC synthase and subgroup 1 aminotransferases. These residues are Y92, N209, G212, D237, and Y240. Our failure to isolate clones containing mutations of the other six conserved residues K278, P150, P210, R286, and G288, and R412 could be caused by a number of reasons. Although we obtained multiple independent clones containing substitutions in 188 of the 485 amino acid residues Le-ACS2, it is unlikely that we have sequenced a large enough number of mutants to be confident that all critical residues have been identified. The location and frequency of substitutions and the identity of the substituted amino acid also is limited by the PCR random mutagenesis technique we have used. For instance, the proximity of the oligoprimer to the K278 residue may have lowered the efficiency in mutating this residue. Additionally, many amino acid substitutions can arise only after two or three base changes in the same codon, which is not easily attained by using this mutagenesis procedure. Thus in addition to the mutants obtained from the random mutagenesis screen, we have used site-directed mutagenesis to generate mutants for P150, P210, R286, and G288. The mutants, K278A, Y92L, Y92W, Y92F, and R412K were available from a previous study (9).
Table 1.
Number of ACC synthase mutants isolated
| cDNA region | Clones screened | Clones sequenced | Putative mutants | Class of mutants*
|
||
|---|---|---|---|---|---|---|
| I | II | III | ||||
| A | 1,500 | 220 | 83 | 41 | 31 | 11 |
| B | 1,800 | 221 | 58 | 29 | 26 | 3 |
| C | 1,800 | 241 | 90 | 53 | 30 | 7 |
| D | 1,500 | 234 | 66 | 41 | 21 | 4 |
| E | 1,332 | 72 | 37 | 18 | 17 | 2 |
| Total | 7,932 | 988 | 334 | 182 | 125 | 27 |
Class I mutants with 0–5% wild-type activity and the same level of protein expression as wild type. Class II mutants with 5–50% wild-type activity and the same level of protein expression as wild type. Class III mutants with no activity and without detectable protein expression.
Figure 4.
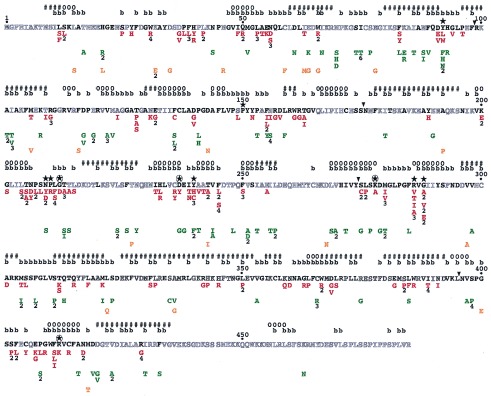
Single missense mutants of Le-ACS2 and their phenotypes. The amino acid sequence of wild-type Le-ACS2 is shown. Residues in bold are conserved in at least 75% of the aligned sequences of ACC synthase. The single amino acid substitutions are listed under the residue of Le-ACS2 that has been mutated and are shown in red for class I mutants, green for class II mutants, and orange for class III mutants. Numbers below the substituted residue indicate the number of independent clones isolated. The predicted secondary structure and solvent accessibility of the amino acid residues is shown above the amino acid sequence. The symbols used are #, helix; o, β-strand; and b, buried or with less than 10% solvent accessibility. The conserved residues between ACC synthase and subgroup 1 aminotransferases and the four invariant residues in all aminotransferases are indicated by ★ and ⊛, respectively. The ▾ shows the demarcation between the regions of Le-ACS2 subjected to PCR random mutagenesis. See Materials and Methods.
The mutant ACC synthases have been characterized by determining enzyme activity and protein expression in E. coli. Because JAde 6 contains ACC deaminase, this strain cannot be used to determine the enzymatic activity of ACC synthase. Therefore, we have isolated plasmid DNA from the clones and introduced them into the E. coli SURE. SURE has been chosen because the transformation and extraction of crude protein are easier in this strain compared with JHM544. The activity and expression of the various mutants are summarized in Fig. 4. The level of expression varies between mutants although most of them have equivalent or only slightly less protein compared with that observed in the wild-type cells. The mutants are classified into three classes based on their enzyme activity and expression in E. coli as shown in Fig. 4 and Table 1. Class I mutants have 0–5% enzyme activity and approximately the same protein expression as the wild-type Le-ACS2. Class II mutants have similar protein expression as class I mutants, but their activity varies from 6–50% of that of the wild type. Class III mutants have neither activity nor detectable protein expression. Most of the mutants isolated belong to the first two classes, whereas only 11% belong to class III (Table 1).
To provide a context for interpreting the results of the mutant screen, the secondary structure and solvent accessibility of Le-ACS2 have been predicted by using two different algorithms; PHDsec has been used for secondary structure prediction (24, 25) and PHDacc has been used to predict solvent accessibility (26). These methods have been chosen because they provide more reliable results compared with other methods. In particular PHDsec has an overall accuracy of more than 72% for most globular proteins, whereas PHDacc shows on the average a correlation of 0.53 between observed and predicted relative solvent accessibility. The location of the predicted secondary structure elements and buried residues on the Le-ACS2 amino acid sequence are shown in Fig. 4.
DISCUSSION
We have combined random mutagenesis and a genetic screen to isolate a library of single missense mutants of Le-ACS2. The mutants have been classified into three classes based on their activity and expression in E. coli (Table 1). In the following discussion, we examine the rationale of our mutational approach and the potential implications of the data on the structure and function of ACC synthase. Our proposition during this mutagenesis study is that amino acid substitutions at critical sites often will lead to nonfunctional proteins. The alignment of mutants shown in Fig. 4 identifies residues that are sensitive to substitutions. A survey of these residues, particularly for the class I and II mutants, show that they often are found in clusters that are separated by regions of the protein where none or only few mutants have been isolated. The absence of mutants in some positions may indicate that these residues are unimportant for enzyme function but also may reflect the inefficiency of the PCR mutagenesis in that region. Because the tertiary structure of ACC synthase has not yet been determined, the results of this mutagenesis must be interpreted with some caution because their effect on the overall conformation of the polypeptide is not yet known. To assist us in our analysis, we have used computer algorithms to predict the secondary structure (12, 24, 25) and solvent accessibility (26) of various residues of the protein (see Fig. 4). Based on the sequence conservation and the predicted secondary structure of ACC synthase, three different computer programs, [Fold-recognition (27), H3P3 (28), and topits (29)] suggest that the three-dimensional structure of ACC synthase can be modeled after the solved structure of AspAT (data not shown). Although the predicted structures are speculative, they provide reasonable information that can be used to explain the effects of the mutations on the structure of ACC synthase. Verification of these predictions awaits proof from more rigorous biochemical and structural studies.
The chemical nature of the amino acid substitution and the location of the amino acid residue are both important in determining whether a residue will be tolerant to mutations (30). Conservative substitutions are less likely to disrupt the conformation of a protein and thus retain protein function unless the wild-type residue makes a set of contacts that are crucial for function that cannot be replaced by another amino acid (31). This conclusion is evident in some positions in Le-ACS2 such as Y92 where conservative substitutions to Phe or Try result in mutant enzymes with some activity whereas substitutions to His or Leu inactivate the enzyme (Fig. 4). Thus, in analyzing the significance of the location of class I mutations for example, residues that are sensitive to conservative substitutions would be good candidates for playing a key role in protein function. But for class II mutations, residues that can tolerate nonconservative substitutions may indicate that the chemical nature of the amino acid is probably unimportant for function.
Most class I and class II mutants are expressed at equivalent or only slightly lower levels than wild-type protein. Because a correlation between in vivo turnover of proteins in E. coli and stability exists (32), this finding also may indicate that most of the amino acid substitutions in these mutants do not have significant effects on the stability of the protein. For class III mutants, the inactivity of the mutant enzymes is because of the lack of immunodetectable protein. The absence of the mutant enzyme can be caused by several factors. The mutations may cause misfolding or destabilize the protein, thus making the mutant enzyme more susceptible to protease degradation. It is also possible, however, that the mutant proteins are not expressed because of codon usage especially because some of them have additional nucleotide changes that result in same-sense mutations. Thus it is difficult to correctly infer the effect of the class III mutations on the structure and function of ACC synthase.
A large number of ACC synthases now have been cloned, and their alignment has been published (12). A comparison between the conserved amino acids in the alignment and the position of missense mutants in Fig. 4 shows that in general there is an agreement between the location of the mutations and the degree of conservation of a particular amino acid residue in the alignment. That the substitution map of a protein is strongly correlated with the evolution map also has been observed in a number of proteins that have been systematically mutagenized such as T4 lysozyme (33), E. coli lac repressor (34, 35), HIV-1 protease (36), β-lactamase (37), and f1 gene V protein (30, 31). This finding is particularly true for the location of class I mutations where 89% of the substitutions are in conserved residues whereas among class II mutants, 58% of the residues fall in conserved residues. Because class II mutants are still fairly active, this finding may indicate that some highly conserved residues are still tolerant of substitutions similar to what has been observed for other proteins (38).
Among the substitutions that have resulted to class I mutations are those involving the 11 invariant residues between ACC synthase and subgroup 1 aminotransferases (Fig. 4). It is interesting that Y92 and Y240 are tolerant of some substitutions. For instance the conservative substitution to Phe at both positions results to mutant enzymes that are fairly active, with 30% and 36% of wild-type activity, respectively (data not shown). The Y92 residue has been implicated in a previous study in making intersubunit contacts on the active site of ACC synthase (9). Similar to its counterpart in AspAT Y70 (39), Y92 is not essential for the activity of the enzyme though it may play a role in binding the PLP cofactor. Y240 is not absolutely conserved in all ACC synthases. An ACC synthase gene isolated from carnation shows a Phe residue at this position (40). Kinetic studies of a similar mutant in apple, Y233F, showed an active enzyme with 24-fold increase in Km for AdoMet but no change in kcat (10). White et al. (10) have suggested that Y233 plays a role in orienting the PLP cofactor in the active site and that the additional hydrogen bond from Tyr to PLP is not very important based on the kinetic properties of the Y233F mutant. Isolation of more substitutions in these invariant residues should provide information on the chemical nature of the amino acid required at each position. This work may assist in determining the roles of these critical residues in the function of ACC synthase.
The conserved tripeptide, Thr-Asn-Pro (TNP) starting at position 205 in Le-ACS2, has been implicated to be a stability determinant because its absence in the Arabidopsis ACS1 isoenzyme causes poor expression in E. coli (41). Mutations of the tripeptide in Le-ACS2 show no adverse effects on expression although all of the mutant enzymes are inactive (Fig. 4). The tripeptide is predicted to be located in a loop region that is very close to the proposed active site residue N209 (see Fig. 4). These results suggest that the individual residues of the tripeptide may be important for the proper positioning of the active site and that the deletion of the tripeptide may destabilize the enzyme.
Because the number of catalytic residues in an enzyme is expected to be small, this finding suggests that most mutations inactivate the enzyme by altering structural determinants. The mutagenesis screen used cannot delineate which residues are catalytically or structurally important. Systematic studies on the structure-function analysis of proteins have shown that two major determinants for the sensitivity of a residue to substitutions, besides conservation among homologous proteins, are solvent accessibility of the residue and whether the substituting residue is glycine or proline (30, 31). Although many amino acid substitutions can be made at surface positions with no adverse effects, substitutions at internal positions, particularly to charged or polar residues, tend to be severely destabilizing (31, 33, 42). In Le-ACS2, 54% of the residues are predicted to have less than 10% solvent accessibility and may be at buried positions (see Fig. 4). We find that 65% and 67% of the residues whose substitution leads to class I and class II mutations, respectively, are predicted to be at buried positions. Many studies have reported that substitution to either glycine or proline often is not tolerated at many positions in a protein (30, 31, 34, 35). For instance it is surprising that glycine substitutions occur in 26% of the class III mutants (Fig. 4). The ability of glycine residues to adopt a wide range of conformations compared with other amino acids can contribute to the misfolding and instability of a protein. Substitutions to proline account for 22% of class I mutations and is the most prevalent mutation of nonconserved residues (Fig. 4). The insertion of proline in a protein can disrupt the secondary structure by breaking up helical regions. Although our data are limited, Fig. 4 indicates that in several instances where a proline substitution occurs in a nonconserved residue, they occur in residues predicted to be in α-helices.
Amino- and carboxyl terminal deletion studies have been done previously to delimit the regions necessary for the activity of the Le-ACS2 isoenzyme (6, 17). Their results have shown that the deletion of 11 amino acid residues, including L12, from the initiating methionine (6) and as many as 52 amino acid residues from the carboxyl terminus of Le-ACS2 (17) still result in functional enzymes. Our data, however, show that substitutions at L12 to Ser or Phe drastically reduce enzyme activity. A possible reason for the discrepancy may be that in the previous study an alanine residue had been inserted in place of the deleted 11-residue N terminus of Le-ACS2 (6). It is possible that alanine is an acceptable substitution for L12. The amino terminus of ACC synthase isoenzymes is highly variable although L12 and S13 are highly conserved. The apparent absence of inactive mutants isolated for the last 56 amino acids of the C terminus (Fig. 4) supports the results of a previous study (17) that the carboxyl terminus is not important for enzyme activity.
There are also a number of amino acid residues that are variable among ACC synthases and yet appear to be intolerant of substitutions such as D37 and C236. Some substitutions naturally occur in other ACC synthase and yet affect Le-ACS2 activity drastically such as the Leu substitution at the highly conserved H21, which occurs naturally in Nt-ACS1 (see alignment in ref. 12). It is possible that there have been compensatory substitutions in other regions of the protein such that the environment around several of these residues may differ among isoenzymes (37). This finding also can account for why some residues like D37 and C236, though not conserved, are critical for the function of Le-ACS2. The essential role of some of these residues also may be unique to the Le-ACS2 isoenzyme.
In summary, we have combined PCR random mutagenesis and a genetic screen to isolate a collection of single missense mutants of ACC synthase. Analysis of the activity and expression of these mutant enzymes have enabled us to identify amino acid residues that may be critical for the structure and function of ACC synthase. Similar to the results of other studies, we have found some correlation between the sensitivity of a residue to substitution and its conservation among homologous proteins. The detailed characterization of these mutants have the potential to provide insight into the involvement of individual residues in catalysis and into the structure-function relationships of ACC synthase, particularly when the location of these residues in the protein has been determined from the crystal structure. Although the three-dimensional structure of ACC synthase is not yet known, preliminary crystals already have been isolated (11). The elucidation of the structure of ACC synthase will be invaluable in understanding how the activity of this important enzyme is regulated and how we may be able to control its activity to limit ethylene production. The mutants and the genetic screen also can be used to isolate second site suppressor mutations, which would provide additional information on the structure of this enzyme. Recent studies have found that the sensitivity of a site to substitution is the best predictor for the activity of additional mutants on the same site (30, 31). Thus, the data we have generated here also can be used to predict whether additional mutations in ACC synthase will be functional.
Acknowledgments
We thank Dr. Harry Klee for providing us with the ACC deaminase clone and Dr. Noaman Hasan for sending us the plasmids for cre-lox mediated integration of the ACC deaminase gene into JHM544. This work was supported by a grant from the National Science Foundation (MCB-9316475) to A.T.
ABBREVIATIONS
- ACC
1-aminocyclopropane-1-carboxylate
- AdoMet
S-adenosyl-l-methionine
- AspAT
aspartate aminotransferase
- PLP
pyridoxal 5′-phosphate
- LB
Luria–Bertani medium
References
- 1.Abeles F B, Morgan P W, Saltveit M E., Jr . Ethylene in Plant Biology. New York: Academic; 1992. [Google Scholar]
- 2.Kende H. Annu Rev Plant Physiol Plant Mol Biol. 1993;44:283–307. [Google Scholar]
- 3.Adams D O, Yang S F. Proc Natl Acad Sci USA. 1979;76:170–174. doi: 10.1073/pnas.76.1.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramalingam K, Lee K, Woodward R W, Bleecker A B, Kende H. Proc Natl Acad Sci USA. 1985;82:7820–7824. doi: 10.1073/pnas.82.23.7820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Satoh S, Yang S F. Plant Physiol. 1989;91:1036–1039. doi: 10.1104/pp.91.3.1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li N, Huxtable S, Yang S F, Kung S D. FEBS Lett. 1996;378:286–290. doi: 10.1016/0014-5793(95)01464-0. [DOI] [PubMed] [Google Scholar]
- 7.Liang X-W, Abel S, Keller J A, Shen N F, Theologis A. Proc Natl Acad Sci USA. 1992;89:11046–11050. doi: 10.1073/pnas.89.22.11046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Y, Feng L, Kirsch J F. Biochemistry. 1997;36:15477–15488. doi: 10.1021/bi971625l. [DOI] [PubMed] [Google Scholar]
- 9.Tarun A S, Theologis A. J Biol Chem. 1998;273:12509–12514. doi: 10.1074/jbc.273.20.12509. [DOI] [PubMed] [Google Scholar]
- 10.White M F, Vasquez J, Yang S F, Kirsch J F. Proc Natl Acad Sci USA. 1994;91:12428–12432. doi: 10.1073/pnas.91.26.12428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hohenester E, White M F, Kirsch J F, Jansonius J N. J Mol Biol. 1994;243:947–949. doi: 10.1006/jmbi.1994.1695. [DOI] [PubMed] [Google Scholar]
- 12.Zarembinski T I, Theologis A. Plant Mol Biol. 1994;26:1579–1597. doi: 10.1007/BF00016491. [DOI] [PubMed] [Google Scholar]
- 13.Rottmann W E, Peter G F, Oeller P W, Keller J A, Shen N F, Nagy B P, Taylor L P, Campbell A D, Theologis A. J Mol Biol. 1991;222:937–961. doi: 10.1016/0022-2836(91)90587-v. [DOI] [PubMed] [Google Scholar]
- 14.Mehta P K, Christen P. Biochem Biophys Res Commun. 1994;198:138–143. doi: 10.1006/bbrc.1994.1020. [DOI] [PubMed] [Google Scholar]
- 15.Mehta P K, Hale T I, Christen P. Eur J Biochem. 1989;186:249–253. doi: 10.1111/j.1432-1033.1989.tb15202.x. [DOI] [PubMed] [Google Scholar]
- 16.Huang P L, Parks J E, Rottmann W E, Theologis A. Proc Natl Acad Sci USA. 1991;88:7021–7025. doi: 10.1073/pnas.88.16.7021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li N, Mattoo A K. J Biol Chem. 1994;269:6908–6917. [PubMed] [Google Scholar]
- 18.Umbarger H E. J Bacteriol. 1953;65:203–209. doi: 10.1128/jb.65.2.203-209.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hasan N, Koob M, Szybalski W. Gene. 1994;150:51–56. doi: 10.1016/0378-1119(94)90856-7. [DOI] [PubMed] [Google Scholar]
- 20.Klee H J, Hayford M B, Kretzmer K A, Barry G F, Kishmore G M. Plant Cell. 1991;3:1187–1193. doi: 10.1105/tpc.3.11.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fromant M, Blanquet S, Plateau P. Anal Biochem. 1995;224:347–353. doi: 10.1006/abio.1995.1050. [DOI] [PubMed] [Google Scholar]
- 22.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 23.Sato T, Oeller P W, Theologis A. J Biol Chem. 1991;266:3752–3759. [PubMed] [Google Scholar]
- 24.Rost B, Sander C. Proteins. 1994;19:55–77. doi: 10.1002/prot.340190108. [DOI] [PubMed] [Google Scholar]
- 25.Rost B, Sander C. J Mol Biol. 1993;232:584–599. doi: 10.1006/jmbi.1993.1413. [DOI] [PubMed] [Google Scholar]
- 26.Rost B, Sander C. Proteins. 1994;20:216–226. doi: 10.1002/prot.340200303. [DOI] [PubMed] [Google Scholar]
- 27.Fischer D, Eisenberg D. Protein Sci. 1996;5:947–955. doi: 10.1002/pro.5560050516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rice D, Eisenberg D. J Mol Biol. 1997;267:1026–1038. doi: 10.1006/jmbi.1997.0924. [DOI] [PubMed] [Google Scholar]
- 29.Rost B. In: The Third International Conference on Intelligent Systems for Molecular Biology. Rawlings C, Clark D, Altman R, Hunter L, Lengauer T, Wodak S, editors. Menlo Park, CA: AAAI Press; 1995. pp. 314–321. [Google Scholar]
- 30.Zabin H B, Horvath M P, Terwilliger T C. Biochemistry. 1991;30:6230–6240. doi: 10.1021/bi00239a022. [DOI] [PubMed] [Google Scholar]
- 31.Terwilliger T C, Zabin H B, Horvath M P, Sandberg W S, Schlunk P M. J Mol Biol. 1994;236:556–571. doi: 10.1006/jmbi.1994.1165. [DOI] [PubMed] [Google Scholar]
- 32.Parsell D A, Sauer R T. J Biol Chem. 1989;264:7590–7595. [PubMed] [Google Scholar]
- 33.Rennell D, Bouvier S E, Hardy L W, Poteete A R. J Mol Biol. 1991;222:67–88. doi: 10.1016/0022-2836(91)90738-r. [DOI] [PubMed] [Google Scholar]
- 34.Markiewicz P, Kleina L G, Cruz C, Ehret S, Miller J H. J Mol Biol. 1994;240:421–433. doi: 10.1006/jmbi.1994.1458. [DOI] [PubMed] [Google Scholar]
- 35.Kleina L G, Miller J H. J Mol Biol. 1990;212:295–318. doi: 10.1016/0022-2836(90)90126-7. [DOI] [PubMed] [Google Scholar]
- 36.Loeb D D, Swanstrom R, Everitt L, Manchester M, Stamper S E, Hutchinson C A., III Nature (London) 1989;340:397–400. doi: 10.1038/340397a0. [DOI] [PubMed] [Google Scholar]
- 37.Huang W, Petrosino J, Hirsch M, Shenkin P S, Palzkill T. J Mol Biol. 1996;258:688–703. doi: 10.1006/jmbi.1996.0279. [DOI] [PubMed] [Google Scholar]
- 38.Warren M S, Marolewski A E, Benkovic S J. Biochemistry. 1996;35:8855–8862. doi: 10.1021/bi9528715. [DOI] [PubMed] [Google Scholar]
- 39.Toney M D, Kirsch J F. Biochemistry. 1991;30:7456–7461. doi: 10.1021/bi00244a013. [DOI] [PubMed] [Google Scholar]
- 40.Henskens J A M, Rouwendal G J A, Ten Have A, Woltering E J. Plant Mol Biol. 1994;26:453–458. doi: 10.1007/BF00039554. [DOI] [PubMed] [Google Scholar]
- 41.Liang X, Oono Y, Shen N F, Koehler C, Li K, Scolnick P A, Theologis A. Gene. 1995;167:17–24. doi: 10.1016/0378-1119(95)00694-x. [DOI] [PubMed] [Google Scholar]
- 42.Lim W A, Sauer R T. Nature (London) 1989;339:31–36. doi: 10.1038/339031a0. [DOI] [PubMed] [Google Scholar]