

Abstract
Carrier vesicle generation from donor membranes typically progresses through a GTP-dependent recruitment of coats to membranes. Here we explore the role of ADP ribosylation factor (ARF) 1, one of the GTP-binding proteins that recruit coats, in the production of neuroendocrine synaptic vesicles (SVs) from PC12 cell membranes. Brefeldin A (BFA) strongly and reversibly inhibited SV formation in vivo in three different PC12 cell lines expressing vesicle-associated membrane protein–T Antigen derivatives. Other membrane traffic events remained unaffected by the drug, and the BFA effects were not mimicked by drugs known to interfere with formation of other classes of vesicles. The involvement of ARF proteins in the budding of SVs was addressed in a cell-free reconstitution system (Desnos, C., L. Clift-O'Grady, and R.B. Kelly. 1995. J. Cell Biol. 130:1041–1049). A peptide spanning the effector domain of human ARF1 (2–17) and recombinant ARF1 mutated in its GTPase activity, both inhibited the formation of SVs of the correct size. During in vitro incubation in the presence of the mutant ARFs, the labeled precursor membranes acquired different densities, suggesting that the two ARF mutations block at different biosynthetic steps. Cell-free SV formation in the presence of a high molecular weight, ARF-depleted fraction from brain cytosol was significantly enhanced by the addition of recombinant myristoylated native ARF1. Thus, the generation of SVs from PC12 cell membranes requires ARF and uses its GTPase activity, probably to regulate coating phenomena.
Generation of carrier vesicles from plasma membrane or intracellular membranous compartments involves at least two components, GTP-binding proteins and coats (Rothman and Wieland, 1996; Schekman and Orci, 1996). A particularly widespread protein that regulates coat assembly on intracellular membranes is ADP ribosylation factor (ARF)1 1, a small GTP-binding protein (Donaldson and Klausner, 1994; Boman and Kahn, 1995). The budding of vesicles from Golgi cisternae can be fully reconstituted in the presence of ARF1 and coatomer (COPI) (Ostermann et al., 1993; Donaldson and Klausner, 1994; Boman and Kahn, 1995; Rothman and Wieland, 1996). ARF1 recruits coatomers to the budding vesicle and couples uncoating to fusion with target membranes (Ostermann et al., 1993; Tanigawa et al., 1993). ARF1 is also required for the recruitment of COPI to vesicles budding from the ER (Bednarek et al., 1995). A hallmark of ARF1-mediated processes is their sensitivity to the fungal metabolite brefeldin A (BFA).
The GDP–GTP exchange activity that replaces GDP bound to ARF proteins with GTP is inhibited by BFA (Donaldson et al., 1992; Helms and Rothman, 1992). The GDP form of ARF1 is unable to bind membranes and consequently, to recruit coats (Robinson and Kreis, 1992; Donaldson and Klausner, 1994). The selectivity of BFA is such that, if a membrane traffic event is sensitive to BFA, it is predicted to require ARF proteins. Inhibition of intra-Golgi and ER to Golgi traffic by BFA probably involves the COPI coatomers. BFA also interferes with coats other than COPI, especially those involved in budding from TGN. Thus it inhibits the formation of vesicles from the TGN (Simon et al., 1996) and causes the redistribution of assembly protein 1 and clathrin to the cytosol (Robinson and Kreis, 1992). Post-Golgi trafficking of the mannose-6-phosphate receptor (Wood et al., 1991) and the maturation of secretory granules (Dittie et al., 1996) are also sensitive to BFA. In addition to clathrin and COPI, BFA affects the recruitment of other “coating” molecules, such as the p47–βNAP complex (Simpson et al., 1996; Dell'Angelica et al., 1997) and p200 (Narula and Stow, 1995) to TGN membranes.
Some endocytotic pathways are also sensitive to BFA. For example, the delivery of polyimmunoglobulin A (pIgA) to plasma membrane from the specialized apical endosome in epithelial MDCK cells, or from dendritic endosomes in hippocampal neurons, is inhibited by BFA (Hunziker et al., 1991; Barroso and Sztul, 1994). BFA-sensitive recruitment of COP1-related proteins and ARF proteins to endosomes has also been reported (Whitney et al., 1995; Cavenagh et al., 1996).
The formation of synaptic vesicles at nerve terminals is a specialized endocytotic pathway that has many similarities to the formation of carrier vesicles from Golgi membranes. In this case, the donor membrane for synaptic vesicle formation is the plasma membrane or the endosome (De Camilli and Takei, 1996). Morphological evidence strongly suggests that synaptic vesicles are generated in nerve terminals through a coat-dependent mechanism (Shupliakov et al., 1997). In lysed nerve terminals, recruitment of dynamin and clathrin coats to membranous organelles is modulated by nonhydrolyzable GTP analogues (Takei et al., 1996). Cell-free reconstitution assays of neuroendocrine synaptic vesicle (SV) formation in PC12 cell extracts showed that GTPγS blocks the generation of properly sized SVs (Desnos et al., 1995), but the identity of the GTP-binding protein or proteins was not determined.
In this paper, we show that reagents that interfere with the cycling of ARF1 between cytosol and membranes block SV formation in neuroendocrine PC12 cells. SV formation was reconstituted in vitro using recombinant ARF1 and a cytosol-derived high molecular weight fraction. Since SV production in vitro is from an endocytotic pool, these results suggest that coating mechanisms associated with ER and Golgi biosynthetic pathways are also associated with at least one endocytotic pathway.
Materials and Methods
125I-labeled Na and ECL reagents were obtained from Amersham Corp. (Arlington Heights, IL). Iodogen came from Pierce Chemical Co. (Rockford, IL). ATP, GTPγS, creatine phosphate, creatine kinase, and Sephadex G25 spun columns were purchased from Boehringer Mannheim Biochemicals (Indianapolis, IN). DEAE Sephacel, Superose 6, ProtG-Sepharose 4 Fast Flow, and BL21 Escherichia coli strain were obtained from Pharmacia Biotech AB (Uppsala, Sweden). Ultrogel AcA 54 was from Biosepra (Marlborough, MA). Brefeldin A was purchased from Epicentre Technologies (Madison, WI). Bafilomycin A1 was obtained from Calbiochem-Novabiochem Corp. (LaJolla, CA). Illimaquinone and avarol were kindly provided by Dr. V. Malhotra (University of California, San Diego, CA). Cell culture media and reagents were obtained from the University of California Cell Culture Facility (San Francisco, CA). Geniticin (G418) and isopropylthio-β-d-galactoside were obtained from GIBCO BRL (Gaithersburg, MD). All the other reagent grade chemicals were purchased either from Sigma Chemical Co. (St. Louis, MO), Fisher Scientific Co. (Fairlawn, NJ), or Calbiochem-Novabiochem Corp. Female Sprague-Dawley rats were from Bantin and Kingman (Fremont, CA).
Peptides
A 16–amino acid peptide (GNIFANLFKGLFGKKE) corresponding to residues 2–17 of the human ARF1 NH2 terminus (ARF 2–17 peptide), and a scrambled peptide of identical composition (FLKANGIGLNEKKGFF) (Chen and Shields, 1996) were either a gift of Dr. D. Shields (Albert Einstein College of Medicine, New York) or were purchased from Chiron Mimotopes (San Diego, CA).
Cell Culture
PC12 cell lines stably transfected with rat vesicle-associated membrane protein–T Antigen (VAMP-TAg) and with the mutants N49A and del 61– 70 were grown in DME H-21 media supplemented with 10% horse serum, 5% FCS, 100 U/ml penicillin and 100 μg/ml streptomycin, and with 250 μg/ml G418. The cells were treated for 24 h before the experiments with 6 mM sodium butyrate to induce the expression of the different VAMP constructs as described (Grote et al., 1995).
Cell Labeling and Subcellular Fractionation
PC12 cells containing the different VAMP-TAg constructs were labeled with 125I-KT3 mAb against the TAg epitope tag following the methods of Desnos et al. (1995). Briefly, confluent dishes of cells were washed at 0°C with labeling buffer (PBS supplemented with 3% BSA, 0.3 mM CaCl2, 0.3 mM MgCl2, and 1 mg/ml glucose), labeled in the same buffer at 0°C for 15 min, and then transferred at 15°C for 40 min. After labeling, the cells were extensively washed in the same buffer, scraped, and sedimented at 800 g for 5 min. To examine the effect of reagents on the in vivo generation of SVs, cells were first labeled at 15°C and then treated in DME H-21, 10 mM Hepes, pH 7.4, for 15 min at 0°C with different drugs, and then transferred to 37°C for times indicated. Similar results were obtained by preincubating cells with the drugs for 30 min at 15°C before warming them. Labeled cells were chilled at 0°C, and then washed and pelleted as described above. Cell pellets were gently resuspended in intracellular buffer (38 mM potassium aspartate, 38 mM potassium glutamate, 38 mM potassium gluconate, 20 mM potassium MOPS, pH 7.2, 5 mM reduced glutathione, 5 mM sodium carbonate, 2.5 mM magnesium sulfate) containing protease inhibitors. Homogenizations were performed as described using a ball bearing homogenizer (cell cracker; European Molecular Biology Laboratory, Heidelberg) with 12 μm clearance. For in vivo experiments, the different conditions in the same experiment were normalized by seeding equal amounts of cells and by loading equal amount of homogenate proteins during subcellular fractionation.
A postnuclear supernatant (S1; 1,000 g for 5 min) was sedimented at 27,000 g for 35 min, and the supernatant generated (S2) was used to identify SVs by velocity sedimentation. S2 (250 μl, 3–5 mg/ml) were loaded onto 5–25% glycerol gradients prepared in intracellular buffer over a 50% sucrose cushion, and then spun at 218,000 g for 75 min in a rotor (SW55; Beckman Instruments, Inc., Palo Alto, CA). Fractions (17 and 18) were collected from the bottom and counted on a gamma counter. For either in vivo or cell-free reactions, the amount of labeled synaptic vesicles generated was determined by integration of the total cpm in the SV peak (fractions 8–13) minus the background cpm determined by integration of the same fractions in parallel reactions, kept at 4°C.
In Vitro Budding Assay
VAMP-TAg/N49A PC12 cells were labeled at 15°C as described above. The assay was performed as described by Desnos et al. (1995). Aliquots of 1 mg of homogenate were incubated for 30 min at 37° or 4°C in the presence of an ATP regenerating system (1 mM ATP, 8 mM creatine phosphate, 5 μg/ml creatine kinase), and 1 or 3 mg/ml of rat brain cytosol prepared as described (Desnos et al., 1995). ARF1 was removed from rat brain cytosol using a Superose 6 sizing column, preequilibrated in intracellular buffer, as described (Waters et al., 1991; Stamnes and Rothman, 1993). In reactions containing peptides or recombinant ARF1 proteins, mixtures were preincubated for 15 min at 0°C before warming, whereas those containing antibodies or glutathione-S-transferase (GST) fusion proteins were kept at 0°C for 3 h. The reactions were stopped by chilling to 0°C for 10 min before fractionation.
Clathrin heavy chains were quantitatively removed from cytosol using the X22 mAb (Brodsky, 1985). Briefly 60 μg of antibody were bound overnight to 40 μl of packed protein G–Sepharose in 0.5 ml of intracellular buffer. Unbound Ig was extensively washed in intracellular buffer and the X22 affinity matrix was incubated with 0.8 mg of cytosol for 2 h at 4°C with gentle rocking. The gel was spun and the cytosol recovered for in vitro reactions. The beads were washed from cytosolic proteins and the clathrin bound to them, and clathrin remaining in the cytosol was determined by immunoblotting using the TD.1 anti-clathrin heavy chain antibody (Nathke et al., 1992). Blots were performed using the enhanced chemiluminescence (Amersham Corp.) system. The amount of clathrin remaining in the cytosol was determined after exposing films for several times. Images were acquired in a digital image system (IS1000; Alpha Innotech Corp., San Leandro, CA) and quantified using the National Institutes of Health Image 1.60 program. The clathrin removed was 90–95% of the total. The cytosol protein concentration before and after the depletion remained constant.
Expression and Purification of Recombinant Proteins
Wild-type, Q71L, and T31N mutant human ARF1 cDNAs subcloned in the pET11d expression vector and yeast N-myristoyl transferase (pBB131) were kindly provided by Dr. D. Shields. Native and mutant proteins were coexpressed with N-myristoyl transferase in BL21 E. coli strain and purified as described (Randazzo et al., 1992). Myristoylated recombinant proteins were extensively dialyzed against intracellular buffer, concentrated to 2–3 mg/ml in a Centriprep 10 (Amicon Corp., Danvers, MA). Aliquots were flash frozen in liquid N2, and stored at −70°C. Purity, assessed by SDS-PAGE and Coomassie blue staining, was ∼80%. The identity of the ARF1 protein was confirmed by immunoblot using the 1D9 mAb (kindly provided by Dr. R. Kahn, Emory University, Atlanta, GA). It also bound to PC12 membranes in the presence of GTPγS (Walker et al., 1992). WBP1-GST fusion proteins were purified following manufacturer's instructions. Recombinant proteins were concentrated in a Centriprep 30 (Amicon Corp.) to 1–3 mg/ml and extensively dialyzed against intracellular buffer.
Measurements of VAMP-TAg/N49A Endocytosis and Transferrin Recycling
Endocytosis of VAMP-TAg/N49A protein was assessed as described (Grote and Kelly, 1996). Cells were plated 2 d before the assay on poly- d-lysine–coated dishes. Surface labeling was performed with 125I-KT3 (3.3 μg/ml) for 2 h at 0°C. N49A/PC12 cells were extensively washed in labeling buffer, and then incubated for 15 min at 0°C either in the absence or presence of BFA (5 μg/ml) in DME H-21, 10 mM Hepes, pH 7.4, before warming to 37°C for different times. Endocytosis was stopped at 0°C for 10 min. Antibody remaining on the cell surface was removed by acid stripping with labeling buffer supplemented with 30 mM glycine adjusted to pH 2.4. Acid-resistant antibody was collected by lysing the cells in 2M NaOH. Calculation and expression of the results were done as described (Grote and Kelly, 1996).
N49A/PC12 cells were incubated in serum-free media for 90 min at 37°C before labeling. 125I-rat transferrin (0.2 μg/ml) was bound for 15 min at 0°C and internalized at 15°C for 40 min as described above. Unbound transferrin was washed at 0°C in DME H-21 media, 0.2% BSA, 10 mM Hepes, pH 7.4. Surface-bound ligand was removed at 0°C by three washes of 6 min in mild acidic buffer (0.5 M NaCl, 50 mM MES, pH 5.0) (Martys et al., 1995), followed by three washes in PBS, 0.3 mM CaCl2, 0.3 mM MgCl2. These washes removed 70–80% of the surface-associated 125I-rat transferrin. N49A cells were then incubated at 0°C in DME H-21 media, 10 mM Hepes, pH 7.4, supplemented with 100 μg/ml of cold iron-loaded rat transferrin in the presence of either methanol (0.001%) or BFA (5 μg/ ml) for 15 min at 0°C. Cells were then warmed for different times and the reactions stopped at 0°C. 125I-rat transferrin that was released to the supernatant, and that which remained cell associated, were both determined after TCA precipitation.
Confocal Immunofluorescence Microscopy
Immunofluorescence procedures and confocal microscopy have been detailed elsewhere (Bonzelius et al., 1994). N49A/PC12 cells were plated in poly-d-lysine–coated PermanoxTM slides (Nunc Inc., Naperville, IL) 2 d before staining. Cells were loaded in vivo with KT3 antibodies (10 μg/ml) in labeling buffer and washed as mentioned above. To observe total VAMP-TAg/N49A, cells were fixed in 4% paraformaldehyde in PBS. For the endocytosis assays, cells (before fixation) were acid stripped in uptake buffer supplemented with 30 mM glycine adjusted at pH 2.4 as previously described (Grote and Kelly, 1996). Fixed cells were permeabilized in 0.02% saponin in PBS, 2% BSA, 1% fish skin gelatin, and incubated with affinity-purified fluorescent-labeled goat anti–mouse IgG (Cappel Laboratories, Malvern, PA). Observation and image acquisition were performed in a Bio Rad MRC 600 confocal laser scanning microscope (Hercules, CA).
Other Procedures
KT3 mAbs and iron-loaded rat transferrin were iodinated in iodogen-coated tubes according to Grote and Kelly (1996). Protein assays were performed using the Bio-Rad Protein Assay Dye Reagent (Hercules, CA) using BSA as standard.
Results
In Vivo SV Biogenesis is Reversibly Inhibited by BFA
To determine whether SV biogenesis is an ARF-mediated process, the effect of BFA upon synaptic vesicle biogenesis in PC12 cells was examined in vivo. The PC12 cell line was stably transfected with a luminally tagged VAMP construct bearing a point mutation in the cytoplasmic tail, N49A (N49A/PC12). This VAMP derivative shows increased targeting to SV compared with wild type (Grote et al., 1995). It shows even more specific targeting to SVs than the del61-70 mutation used in a previous study (Desnos et al., 1995). Incubating intact cells at 15°C with iodinated antibodies (125I-KT3) against the lumenal epitope labels plasma membrane and intracellular compartments without labeling synaptic vesicles (Desnos et al., 1995). Internalizing the antibody at 15°C, removing free antibody, and then incubating the cells at 37°C caused the appearance of antibody-labeled SVs, monitored by their migration on glycerol velocity gradients (Fig. 1 a, ○). Free antibody remains at the top (right) of the velocity gradients. The addition of BFA (5 μg/ml) inhibited the production of labeled vesicles upon warming to 37°C (Fig. 1 a, ⋄). After washing the drug out, the BFA-mediated block was completely reversed within 15 min (Fig. 1 a, •). The fast reversibility of the block argues against nonspecific toxicity effects.
Figure 1.

In vivo synaptic vesicle biogenesis is inhibited by BFA in a concentration dependent and reversible way. Stably transfected PC12 cells bearing the N49A VAMP-TAg construct were labeled at 15°C for 40 min in the presence of 125I-KT3 antibody, and the unbound antibody washed extensively. (a) After washing out the free antibody, cells were incubated at 0°C for 15 min either in the absence (○) or presence (⋄) of BFA (5 μg/ml). Control and BFA-treated cells were warmed for 15 min. To measure reversibility, one of the BFA-containing plates was washed thoroughly at 0°C and then reincubated at 37°C for another 15 min in the absence of BFA (•). The reactions were stopped, cells homogenized, and high speed supernatants processed for velocity sedimentation analysis as described in Methods. The data are one example of two independent experiments. (b) Cells were incubated as described in a with 0.1–10 μg/ml of BFA, and then transferred at 37°C for 15 min and processed as described. BFA inhibited vesicle production in a dose-dependent way (0% inhibition corresponds to 3550 cpm in the SV peak).
BFA was effective at concentrations as low as 0.1 μg/ml (Fig. 1 b) with a maximal inhibition (80–95%) observed at concentrations between 5–10 μg/ml (n = 25). This dose– response inhibition was similar to that described for the block of transcytosis in MDCK cells (Hunziker et al., 1991) and was slightly higher than that required to disrupt the Golgi complex in fibroblasts (Lippincott-Schwartz et al., 1991b ). In the absence of the drug, the generation of 125I-KT3–loaded vesicles from the 15°C compartment reached a plateau in less than 20 min (Fig. 2 a). Inhibition by BFA was a fast phenomenon detected as early as 7.5 min (data not shown) and maintained throughout the incubation period (Fig. 2 a). Similar inhibition was obtained in a PC12 cell line expressing del61–70 VAMP, which also shows enhanced targeting to SVs (Fig. 2 b; Grote et al., 1995). Cells transfected with wild-type VAMP showed similar inhibition (data not shown), but vesicles were labeled less efficiently and so the inhibition appeared to be less dramatic.
Figure 2.

Time course of the BFA inhibition in two VAMP-transfected PC12 cell lines. Stably transfected PC12 cells bearing the N49A (a) or del61–70 VAMP-TAg (b) constructs were labeled at 15°C for 40 min in the presence of 125I-KT3 antibody and incubated in methanol (0.001%) (○) or BFA (5 μg/ml) (⋄) as described. Cells were chased at 37°C for different times. The reactions were stopped at 0°C and the vesicle production was determined by velocity sedimentation in glycerol gradients as described. BFA blocked synaptic vesicle biogenesis in both cell lines with no detectable delay. Similar results were obtained in two independent experiments. The amount of SV production at 15 min was designated as 100%. The radioactivity corresponding to 100% was 2,000 cpm in a and 10,000 cpm in b.
The plateau in labeling (Fig. 2) could be due to accumulation of label in a stable, slowly turning over compartment. Alternatively, the plateau could mean that the vesicles recycle and a dynamic equilibrium is reached. To analyze the turnover time of the 125I-KT3–containing synaptic vesicles, N49A/PC12 cells were labeled at 15°C, and then warmed to 37°C for 15 min to allow the formation of 125I-KT3– loaded SVs. The cells were then incubated at 37°C for different times, either in the absence or presence of BFA to block the formation of newly labeled vesicles. In the absence of BFA, the amount of SVs decreased slightly after 60 min (Fig. 3, ○). In contrast, in BFA-treated cells, the amount of 125I-KT3–loaded SVs detected in velocity gradients dropped to 50% in 36 ± 2 min (n = 3) (Fig. 3, ⋄). About 60% of the labeled SVs disappeared rapidly, indicating that the majority of newly formed SVs are not inert, but are recycling through a pool that is continuously fed from a BFA-sensitive compartment.
Figure 3.

Newly formed synaptic vesicles turnover. To evaluate the life span of newly synthesized synaptic vesicles, PC12/N49A cells were labeled with 125I-KT3 antibody at 15°C as described. The free antibody was washed at 4°C and the cells were then incubated for a further 15 min at 37°C to allow SVs to form. Vesicle biogenesis was stopped by placing cells on ice followed by treatment in the absence (○) or presence (⋄) of BFA (5 μg/ml). After rewarming the cells back to 37°C for different times; the remaining vesicles were assessed by velocity sedimentation. The data are one example of three independent experiments.
In Vivo Effects of BFA on the Morphology of the KT3-labeled VAMP-containing Endosomes
To identify the morphological consequences of BFA treatment, VAMP-TAg N49A–containing endosomes were labeled by in vivo uptake of unlabeled KT3 antibody and examined by confocal immunofluorescence microscopy. Intra-cellular labeling was distinguished from plasma membrane labeling by acid stripping the cell surface. Steady-state KT3-loading at 37°C revealed fluorescent signals in vesicular structures throughout the cytoplasm, with the strongest labeling in the juxtanuclear region (Fig. 4, A and B). The internalized fluorescence signal in these compartments was acid resistant (Fig. 4 B). In contrast, after 15°C KT3 labeling, the signal resistant to acid stripping was in large vesicular structures, some of which were close to the plasma membrane, others were distributed throughout the cytosol (Fig. 4 D). No concentration of fluorescent signal was detected in the juxta- or perinuclear region. However, if cells labeled at 15°C were rewarmed to 37°C for 15 min, the label redistributed to the juxtanuclear region in a pattern similar to the steady-state labeling at 37°C (Fig. 4, E, F, I). These results show that KT3 label moves from the 15°C compartment to the perinuclear region. The addition of BFA before the warming to 37°C did not prevent the accumulation of fluorescent signal in an acid-resistant compartment around the nucleus (Fig. 4, H and J). No BFA-induced tubular endosomal structures were distinguishable. A similar juxta–perinuclear pattern was observed in cells loaded in BFA at 37°C without the 15°C preincubation (Fig. 5 b, C and D). Thus, although both 15°C and BFA inhibit SV formation, transport to the perinuclear region is sensitive only to temperature.
Figure 4.
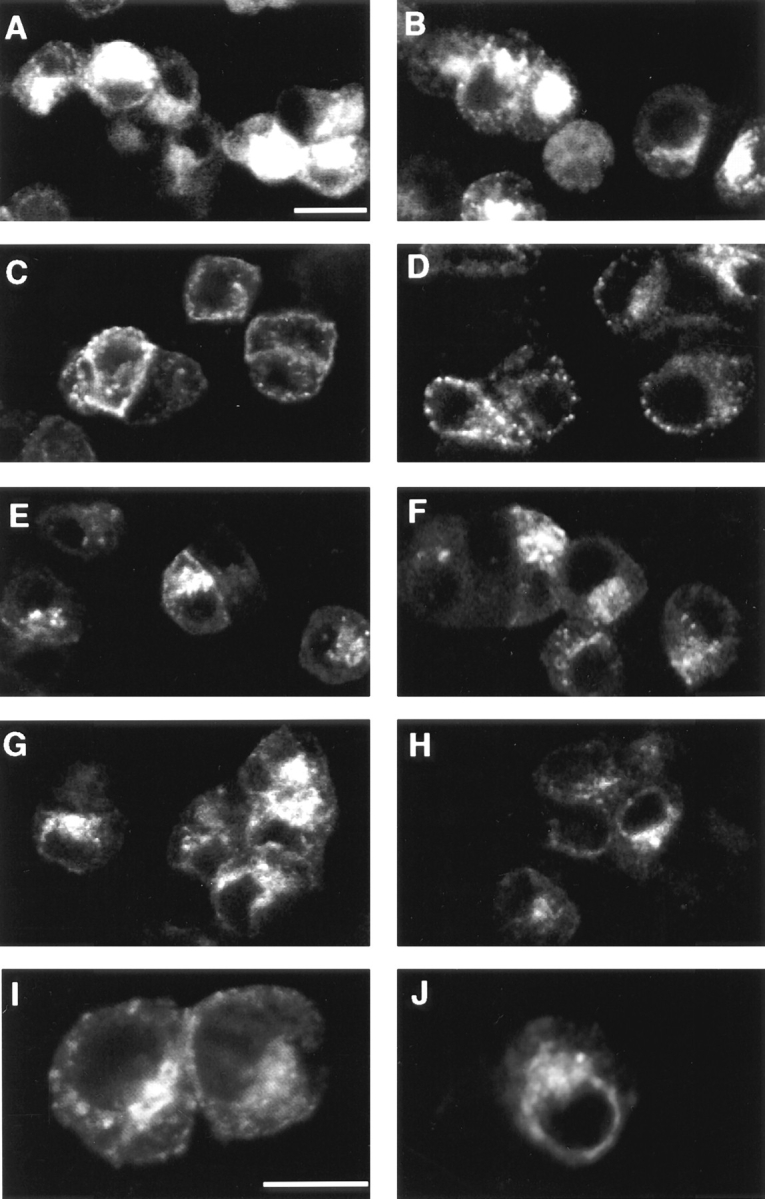
BFA does not prevent the accumulation of KT3 antibody in perinuclear endosomes. PC12 cells expressing N49A VAMP-TAg were incubated in the presence of KT3 mAb (10 μg/ml) under different conditions to label endocytotic organelles. After labeling, the cells were chilled, washed extensively, fixed and processed for immunofluorescence using a labeled secondary antibody either before (A, C, E, and G) or after (B, D, F, and H–J) stripping KT3 from the cell surface by an acid wash. A and B show the total and internal label when antibody uptake was permited for 40 min at 37°C; significantly less perinuclear staining was seen if the uptake was at 15°C for 40 min (C and D). BFA did not prevent movement to perinuclear endosomes (G, H, and J). If cells were labeled at 15°C for 40 min, and then warmed to 37°C for 15 min, the distribution of immunofluorescence was identical whether or not BFA was present (G, H, and J) or absent (E, F, and I). I and J represent higher magnifications of F and H, respectively. Bars: 10 μm.
Figure 5.


BFA does not block the 125I-KT3 internalization. (a) PC12/N49A cells were surface labeled with 125I-KT3 antibody (3.3 μg/ml) for 2 h at 0°C. The unbound label was washed away and the cells were incubated in media containing either methanol (0.001%) (○) or BFA (5 μg/ ml) (⋄) for 15 min on ice. The cells were warmed up to 37°C for different times and endocytosis was stopped by putting the cells back on ice. The internalized ligand was determined by surface-acid stripping, and expressed as fraction of the total cell-associated counts. Error bars represent standard deviation of triplicates in one of three independent experiments. (b) Internalization was assessed by immunofluorescent detection of the KT3 antibody uptake. PC12/N49A cells were incubated with unlabeled antibody for 2 h at 4°C and equilibrated in the absence (A and B) or presence (C and D) of BFA as described in a. Cells were chased at 37°C for 15 min. Reactions were stopped at 4°C and the total (A and C) and internalized VAMP-TAg (B and D) determined by acid stripping of the cell surface. Cells were fixed and processed for immunofluorescence as described. Bar, 10 μm.
The BFA-dependent SV Block Is Selective
Although formation of synaptic vesicles from the 15°C compartment was sensitive to BFA, other parts of the endocytotic pathway were not. Internalization of VAMP-TAg from the cell surface was assessed as acid-resistant bound 125I-KT3 antibody and found to be similar in the control and BFA-treated cells (Fig. 5 a). Insensitivity of internalization to BFA was also shown by immunofluorescence (Fig. 5 b). Recycling of transferrin from endosomes to the cell surface was also relatively unaffected by BFA, as shown by the kinetics of 125I-labeled rat transferrin recycling from endosomes to the plasma membrane after BFA treatment (Fig. 6). To measure recycling, 125I-labeled rat transferrin was internalized at 15°C, cells chilled to 4°C, and plasma membrane–associated ligand removed by washing. Cells were then incubated in the absence or presence of BFA for different times at 37°C. As previously reported in K562 and NRK cells (Lippincott-Schwartz et al., 1991a ; Schonhorn and Wessling-Resnick, 1994), the change in the rate of transferrin externalization was not very dramatic after BFA treatment in N49A/PC12 cells, either evaluated as cell-associated radioactivity or released radioactivity.
Figure 6.

125I-Transferrin recycling back to the cell surface is not inhibited by BFA. PC12/N49A endosomes were labeled with 125I-rat transferrin (0.2 μg/ml) at 15°C for 40 min. The free and surface-bound ligands were removed by repeated washing followed by mild acidic treatment of the cell surface. The cells were incubated in the absence (○) or presence of BFA (5 μg/ml) (⋄) at 0°C in a media containing 100 μg/ml of cold rat transferrin and chased for different times at 37°C. Transferrin in the media (•, ♦) and in the cells (○, ⋄) was determined as described. Error bars represent standard errors of triplicate points of one representative experiment from three independent ones.
The drug illimaquinone also inhibits binding of β-COP and ARF to Golgi membranes, vesicle production, and protein secretion; but it has no observable effect on the endocytic pathway (Takizawa et al., 1993). Illimaquinone also had no effect on SV formation from 15°C-labeled cells (Fig. 7, 16.3 ± 1.7% inhibition, n = 3) at a concentration that stops protein traffic through the Golgi complex. Higher illimaquinone concentrations or avarol, a structurally similar inhibitor, also did not modify SV production (data not shown).
Figure 7.

Effect of illimaquinone and bafilomycin A1 upon in vivo synaptic vesicle biogenesis. PC12/N49A cells labeled with 125I-KT3 antibody at 15°C were incubated for 15 min at 0°C either in the absence or the presence of the drugs at the concentrations indicated. Cells were then chased at 37°C for 15 min, followed by analysis of the labeled synaptic vesicles by velocity sedimentation. Illimaquinone was almost without effect (n = 2), whereas bafilomycin A1 inhibited 44 ± 6% (n = 3) at 1 μM concentration.
Bafilomycin A1, a specific inhibitor of vacuolar type ATPases, perturbs endosomal pH and membrane trafficking, in both early endosomes and from early to late endosomes (van Weert et al., 1995; Aniento et al., 1996). It inhibited SV production by 44 ± 6% (Fig. 7, n = 3). The inhibition reached a plateau at 250 nM (data not shown). A similar dose-response has been described in the inhibition of endocytosis and trafficking of transferrin receptor back to plasma membrane and in the transfer of markers from early endosomes to late endosomes (van Weert et al., 1995; Aniento et al., 1996). These results show that perturbing the pH gradients along the endocytic pathway affects SV production but does not mimic the severity of the BFA effect.
Thus, BFA affects SV formation selectively having no effect on internalization or recycling of transferrin receptor back to the cell surface. The vacuolar ATPase inhibitor, bafilomycin A1 and illimaquinone, a Golgi-specific inhibitor of β-COP accumulation, are much less effective inhibitors and have not been studied further.
Cell-free SV Biogenesis Requires ARF1 Protein
Involvement of ARF1 protein in SV biogenesis was investigated directly using a cell-free reconstitution assay. ARF1 is believed to interact with effectors through its NH2-terminal domain, since peptides corresponding to its NH2 terminus can block ARF1 function (Balch et al., 1992; Kahn et al., 1992; Randazzo et al., 1994; Boman and Kahn, 1995). The effects on SV production of an ARF1-derived, (2–17) NH2-terminus peptide were therefore examined.
In a standard cell-free assay (Desnos et al., 1995), labeled SVs are generated at 37°C, whereas no production of SV appears in a reaction at 4°C (Fig. 8, a and c). Under this condition the addition of a peptide spanning the NH2-terminus effector domain of ARF1 (2–17) at a concentration of 100 μM, inhibited the SV production by 42 ± 4%, (n = 7) (Fig. 8 a). An effect of this magnitude was not detected using a scrambled peptide of identical composition (105 ± 7%, n = 4) (Fig. 8 a). Since the (2–17) peptide has been reported to inhibit Golgi membrane traffic irreversibly by damaging membranes (Weidman and Winter, 1994), the reversibility of its effect upon SV formation was tested. Identical in vitro reactions containing low levels of brain cytosol (1 mg/ml) were run in parallel for 15 min at 37°C, in the presence of the scrambled or (2–17) peptide (100 μM). To test reversibility, additional brain cytosol was added, and the incubation continued at 37°C for another 15 min. Inhibition by (2–17) peptide was reversed by adding more cytosol (Fig. 8 b), indicating that if the peptide concentration was 100 μM or less, the inhibitory effect was not solely due to membrane damage. Since this peptide is able to inhibit ARF-independent processes (Fensome et al., 1994), we tested if the addition of wild-type ARF1 could induce a recovery of the (2–17) peptide inhibitory effect on SV formation. In standard reactions containing 100 μM of (2–17) peptide, the addition of 50 μM myristoylated wild-type ARF1 together with the peptide (Fig. 8 c) decreased the inhibition to 24 ± 10 (n = 3). These results show a competitive interaction between ARF1 and the peptide, and imply that an ARF is involved in the budding reaction.
Figure 8.

The ARF1 NH2-terminus peptide (2–17) reversibly inhibits synaptic vesicle biogenesis in a cell-free system. Standard reaction mixtures containing homogenates from PC12/N49A cells labeled with 125I-KT3 antibody at 15°C, ATP regenerating system, and rat brain cytosol (1 mg/ml or 0.35 mg/ assay) were prepared and kept on ice for 15 min, either in the absence or presence of the (2–17) NH2-terminus peptide, or the control scrambled peptide. Budding reactions were initiated by warming for 30 min at 37°C. Vesicle production was assessed by velocity sedimentation as described. (a) The (2– 17) NH2-terminus peptide (100 μM) inhibited the generation of vesicles by 42 ± 4% of control. a Shows a representative experiment out of seven experiments. The random peptide had no effect on the budding reaction at the highest concentration tested (200 μM). In b, the inhibitory effect of the (2–17) NH2-terminus peptide (100 μM) was reverted by adding additional cytosol. Reaction mixtures similar to those described in a were incubated at 0°C either with (2–17) NH2 terminus or the scrambled peptide (100 μM) for 15 min. The reactions were warmed at 37°C for 15 min, and then supplemented or not with more cytosol. The reactions were allowed to proceed for an additional 15 min before being stopped and analyzed. Dilution of peptide by the addition of cytosol did not exceed 20%. In c, the inhibition mediated by the (2–17) NH2-terminus peptide was reversed by the addition of purified myristoylated recombinant wild-type ARF1 (50 μM).
To further address ARF1 involvement, we tested the effects of purified ARF1 mutants Q71L, a GTPase null, and T31N (a GTP-binding defective mutant) on the formation of properly sized SVs, assessed by velocity gradient sedimentation. The amount of labeled SVs was unchanged by supplementing the standard reaction mixtures with 50 μM wild-type ARF1 (Fig. 9 a). In contrast, in standard reactions supplemented with 50 μM of either dominant-positive (Q71L) or dominant-negative (T31N) mutants, there was a drastic reduction in the amount of labeled vesicles detected in velocity gradients. Both recombinant mutant proteins were effective at concentrations as low as 1 μM (Fig. 9 b). This concentration is similar to the amount of ARF1 estimated to be present in the rat brain cytosol used in the standard reaction mixtures (data not shown; Kahn et al., 1988). Consistently Q71L was more potent when compared with T31N. Both mutants were also able to induce a reduction in the production of labeled SVs in the suboptimal conditions when only PC12 cell cytosol is present without added brain cytosol (Desnos et al., 1995). Even under these conditions, however, additional wild-type ARF1 had no stimulatory effect (data not shown), indicating that the endogenous ARF1 protein present in the PC12 extracts was sufficient for the vesicle budding reaction.
Figure 9.

Dominant-positive and -negative mutant ARF1 inhibit the appearance of mature synaptic vesicles in a cell-free system. Standard reaction mixtures similar to those described in Fig. 8 were performed in the absence or presence of recombinant myristoylated wild-type, Q71L (dominant positive), and T31N (dominant negative) proteins. The ARF1 proteins incubated with the reaction mixes for 15 min at 0°C and then warmed to 37°C for 30 min. (a) At 50 μM, both Q71L (▵) and T31N (---○---) inhibited the appearance of matured vesicles as assessed in velocity sedimentation compared to control (—○—). The addition of wild-type ARF1 (⋄) was without effect on the vesicle production. Vesicle production did not occur at 4°C (◫̶ ). In b dominant-negative and -positive mutant forms of ARF1 were added at concentrations ranging from 1 to 50 μM. The maximal inhibitory effect was reached by 5 μM for both of them (0% inhibition corresponds to 8,157 cpm).
To demonstrate ARF dependence directly, the cell-free assay was changed by removing the ARF1 present in rat brain cytosol. This was done by passing the cytosol through a Superose 6 sizing column and taking high molecular weight (HMW) fractions reported to be enriched in coat complexes (Stamnes and Rothman, 1993; data not shown). These HMW fractions were pooled and added to the cell-free reaction in the absence of added rat brain cytosol. The addition of HMW fractions alone stimulated the formation of vesicles by 1.7 ± 0.15 times (n = 5). However the combined addition of HMW fractions plus myristoylated wild-type ARF1 increased the vesicle production 2.6 ± 0.15 times. Further, both mutant ARFs inhibited the appearance of vesicles in the presence of HMW (Fig. 10). These results show that ARF1 is required for SV biogenesis. A likely possibility is that it recruits coating molecules present in the HMW fraction.
Figure 10.
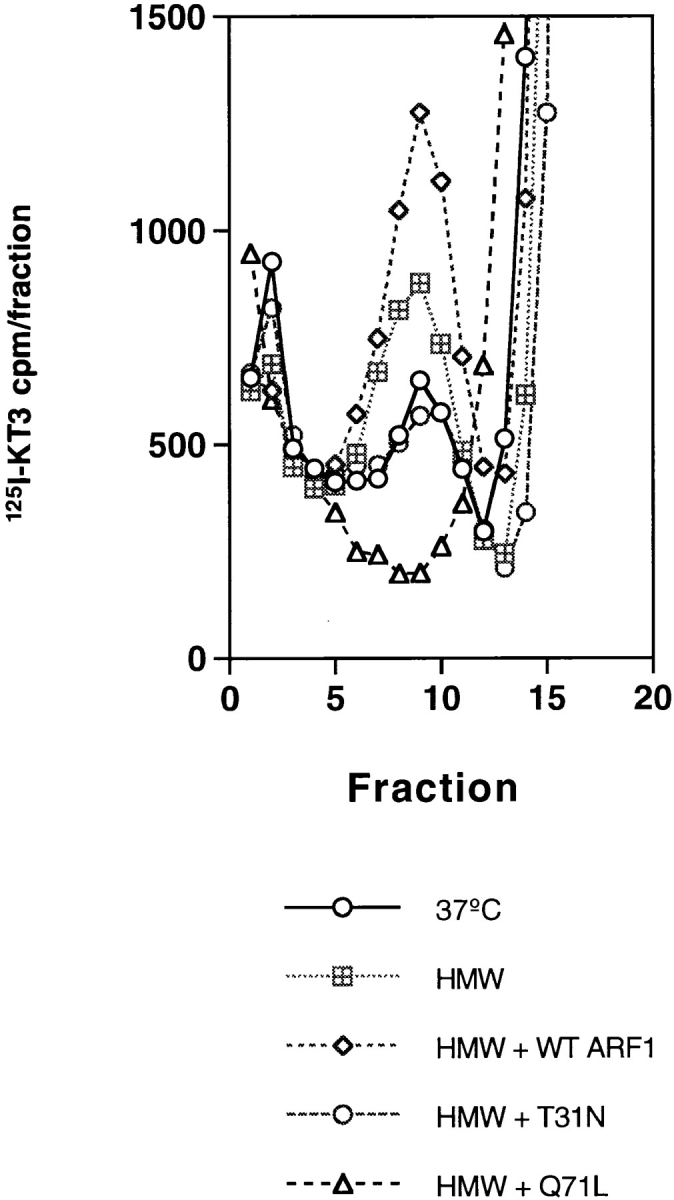
ARF1 stimulates the SV budding activity in a cell-free system in the presence of a HMW fraction from rat brain cytosol. If brain cytosol was omitted from standard reaction mixtures that contained labeled N49A/PC12 cell homogenates and ATP, little SV production was observed during a 30 min incubation at 37°C (○). The cytosol-free reaction was stimulated slightly (◫̶ ) by the addition of ARF1-depleted, HMW fractions (650 μg/assay) from Superose 6–fractionated rat brain cytosol. Under these conditions a direct stimulation of SV production by 15 μM of wild-type, myristoylated ARF1 could be observed (⋄) added before the cell-free budding reaction. Addition of 15 μM T31N ARF1 (○) inhibited the reaction to approximately the cytosol-free level whereas Q71L ARF1 (▵) inhibited the appearance of mature vesicles.
ARF molecules have been implicated in recruiting coats made up either of coatomers or of adaptor/clathrin complexes. To test if clathrin was involved in the budding process, it was quantitatively removed from rat brain cytosol using mAb to clathrin (Fig. 11 a). No significant reduction in budding efficiency was observed (Fig. 11 b). To examine the involvement of COPI coatomers in vesicle formation, we used conditions that are known to inhibit COPI function during endosomal sorting (Whitney et al., 1995) or binding of COPI to membranes (Lowe and Kreis, 1995). Addition of antibodies to β-COP, a fusion protein corresponding to the KKXX signal motif of WBPI (GST-KK) (Cosson and Letourneur, 1994), or a combination of the antibody and the inhibitory peptide had no effect on vesicle formation.
Figure 11.


Cytosolic clathrin and COPI are not needed for the cell-free budding of SV. (a) Normal rat brain cytosol was immunodepleted of clathrin heavy chains using the X22 mAb bound to protein G–Sepharose. The extent of depletion was confirmed by immunoblotting equal amounts of rat brain cytosol protein before (lane 1) or after (lane 2) the immunoabsorption with the TD.1 anti-clathrin antibody. Lane 3 corresponds to clathrin heavy chains retained on the beads. (b) In vitro reaction mixtures were prepared containing labeled N49A/PC12 cell homogenates, ATP regenerating system in the presence of normal or clathrin- depleted rat brain cytosol. Reactions containing normal rat brain cytosol (supplemented with either 11–22 μg/ml of affinity-purified anti–β-COP antipeptide antibody EAGE, 1 μM of either GST-WBP1-SS or GST-WBP1-KK, or a combination of EAGE antibody plus GST-WBP1-KK) were kept for at least 3 h at 4°C before warming to 37°C. None of the treatments substantially modified the budding of SV.
Q71L and T31N ARF1 Mutants Define Different Intermediates in SV Biogenesis
A current model for the mechanism of action of the GTPase-defective mutant Q71L on Golgi membranes predicts that both ARF and coats remain constitutively associated with the membranes coming from the donor compartment, while the GTP-binding defect in T31N precludes the association of both ARF and coats to the donor membranes (Donaldson and Klausner, 1994; Rothman and Wieland, 1996; Schekman and Orci, 1996). Thus the T31N mutation should prevent coating of the donor compartment and the Q71L mutant should cause extensive coating of a donor compartment or the accumulation of a coated vesicular intermediate. The nature of the 15°C donor compartment has been extensively studied (Lichtenstein, Y., C. Desnos, V. Faúndez, R.B. Kelly, and L. Clift-O'Grady, manuscript in preparation). From a postnuclear supernatant of labeled PC12 cells the donor compartment could be recovered at 31.4 ± 0.3% (n = 13) sucrose. About 40% of the donor peak was converted in vitro to SVs, which are recovered at 24% sucrose under the same conditions. When the in vitro reaction was performed, in the presence of Q71L or T31N ARF1, no peak of label was observed at the density of SVs (24% sucrose). Instead, the donor membranes became lighter in the presence of T31N and denser in the presence of Q71L. The donor membranes acquired a density of 28.6 ± 0.8% sucrose (n = 3) in the presence of T31N and 33.3 ± 0.7% sucrose (n = 3) in the presence of Q71L. This would be consistent with a model in which the donor compartment lost some coat during incubation with T31N. Donor membranes from cells labeled at 37°C in the presence of BFA are also light (27.5 ± 0.7, n = 2) (data not shown). The increase in density of the donor membranes in the presence of Q71L could be attributed to either the recruitment of additional coat to the donor endosomal compartment, or the formation of a coated vesicle intermediate.
Discussion
By using BFA, wild-type, and mutant ARF1 proteins, we have demonstrated that ARF1 is required for the formation of SVs in PC12 cells from the 15°C compartment. The physiological relevance of ARF1 involvement in PC12 SV formation is supported by several arguments. In vivo SV formation is reversibly sensitive to BFA at concentrations known to affect budding from Golgi membranes. The inhibition occurs in three PC12 clones expressing different forms of VAMP. The number of SVs is reduced in PC12 cells incubated in the presence of BFA. ARF1 and a cytosol-derived HMW fraction are sufficient to promote SV production from a PC12 homogenate. Micromolar concentrations of mutant recombinant ARF1s block SV formation; and cell-free incubation of homogenates in the presence of a GTP-binding defective mutant of ARF1 generates antibody-loaded compartments of density different to those formed in the presence of a GTPase-defective mutant. Since most of the ARF1 isoforms are functionally equivalent (Kahn et al., 1991; Boman and Kahn, 1995), however, we cannot be certain that only ARF1 is involved in SV formation.
Specialized Endosomal Compartments as a Source of SV?
BFA has been useful in characterizing functionally specialized endosomes in polarized cells. In MDCK cells and in primary hippocampal neuron cultures, transfer of pIgA from apical/perikaryal endosomes to the apical/axonal membrane exhibits a BFA sensitivity similar to that obtained in SV production (Hunziker et al., 1991; Barroso and Sztul, 1994; de Hoop et al., 1995). However, BFA effects on nonspecialized endocytic routes are subtle. Although BFA induces morphological alterations in endosomes, it does not block the transit of dyes to lysosomes or the recycling of transferrin back to the cell surface (Lippincott-Schwartz et al., 1991a ; Tooze and Holinshead, 1992; Wood and Wood, 1992; Schonhorn and Wessling-Resnick, 1994). The transferrin externalization rate is slowed but not blocked by BFA (Schonhorn and Wessling-Resnick, 1994; Fig. 6). In addition to being BFA sensitive, the transit of proteins through apical/perikaryal endosomes and through synaptic vesicle-generating endosomes are both unusually sensitive to low temperature blocks (15–17°C) (Barroso and Sztul, 1994; Desnos et al., 1995). This suggests that the specialized pathways in epithelia and neuronal cells may use a temperature-sensitive “coating” function, absent from nonspecialized or “housekeeping” pathways.
The endosomes of nerve terminals in primary hippocampal neurons (Mundigl et al., 1993) and of processes in differentiated PC12 cells (Bonzelius et al., 1994) are specialized compared with those in the cell body, particularly in their lack of transferrin receptor. Although it would be tempting to conjecture that these axonal endosomes might be more BFA sensitive, morphological data show that BFA causes tubulation of cell body but not axonal endosomes (Mundigl et al., 1993). One interpretation of such data is that the BFA-sensitive pathway of SV biogenesis in PC12 cells represents a more elementary form of biogenesis that is replaced in nerve terminal maturation with a more efficient and uniquely neuronal process. Alternative explanations are that the two pathways exist side by side in neurons, or the biogenesis of neuroendocrine SVs might be mechanistically unrelated to the biogenesis of neuronal SVs. Further experiments are required to distinguish between these alternatives.
PC12 SVs Are Dynamic Organelles
In immature hippocampal nerve cells, SVs go through a spontaneous rate of fusion with the plasma membrane in the absence of stimulation (Matteoli et al., 1992). Our results provide evidence that newly formed SVs in PC12 cells also cycle in the absence of stimulation. After loading the cells at 15°C, the production of vesicles at 37°C reaches a maximum in about 15 min. This equilibrium represents a balance between the kinetics of formation and disappearance. When the SV pool of PC12 cells is labeled at 37°C, and BFA added to suppress further SV formation, about half the vesicles disappear in 30 min. Although this reduction of labeled vesicles is probably due to exocytosis, it could also be due to fusion back to the donor or to other intracellular membrane compartments. BFA inhibition in vivo (Figs. 1 and 2) could be attributed to an enhancement of exocytosis rather than fusion. This is unlikely however, since BFA (data not shown) and ARF mutants block SV formation in cell-free assays, where fusion with the plasma membrane is not probable.
A Possible Mechanism of SV Biogenesis Revealed by ARF1 Mutants
It has been proposed that ARF-GTP recruits coats to the nascent vesicle that is budding from precursor Golgi membranes (Serafini et al., 1991; Rothman and Wieland, 1996). In Golgi- and ER-derived membranes, the ARF-GTPase activity is required for the uncoating of already formed vesicles (Tanigawa et al., 1993; Bednarek et al., 1995; Rothman and Wieland, 1996). Preventing GTP hydrolysis results in the formation of carrier vesicles that contain mature cargo and fusion proteins but retain their coats (Ostermann et al., 1993; Oka and Nakano, 1994; Bednarek et al., 1995; Rothman and Wieland, 1996). Since GTPγS effects are pleiotropic and can induce mistargeting of the adaptor (Seaman et al., 1993), the Q71L mutant is more informative for in vitro studies. For example, Q71L ARF can discriminate COPI- and II-dependent budding events simultaneously occurring from yeast ER (Bednarek et al., 1995).
Formation of SVs in a cell-free system resembles the production of Golgi-derived vesicles in its sensitivity to GTPγS (Desnos et al., 1995), its sensitivity to BFA and the need for ARF and a HMW protein fraction. If the inhibition of SV formation by the ARF mutants is interpreted in light of the Golgi results, then the mutant T31N ARF is predicted to prevent the binding of coat and ARF to membranes, and the Q71L ARF mutant would allow coated vesicles to form, but prevent their uncoating. Consistent with those predictions, the donor membranes became less dense when incubated in vitro with T31N ARF, and denser when incubated with Q71L. Unfortunately, the quantities of membrane currently available and their contamination with nondonor membranes preclude biochemical analyses of the two intermediate forms.
If ARFs were recruiting a coat to the SV precursor membranes, the coat would be expected to be clathrin (Heuser and Reese, 1973; Shupliakov et al., 1997). We could, however, find no evidence that the factor or factors contributed by rat brain cytosol were either clathrin or COPI. The data do not eliminate the involvement of either coat in budding, however, since sufficient coat molecules may remain attached to the donor membranes. Peripheral proteins removed from PC12 donor membranes by Tris-stripping are able to support the in vitro budding reaction (Horng, J.-T., unpublished observations). Unfortunately the quantities of proteins extractable from PC12 membranes using Tris are too small for detailed analysis. The active components in the rat brain cytosol are unlikely to include dynamin because extensive depletion using a grb2 affinity column also has no effect on SV formation in vitro (Horng, J.-T., unpublished observations). One possible coat that remains to be tested is the AP-3 coat, implicated in post-Golgi, and probably endosomal sorting events (Simpson et al., 1996; Dell'Angelica et al., 1997). It is also possible that the donor compartments contain partially coated vesicles. The addition of ARF and the high molecular weight fraction could complete the coat, allowing uncoating to proceed. Alternatively, the synaptic vesicle precursor could be a complete coated vesicle that accumulated at 15°C. For this model to be plausible, the T31N dominant negative ARF1 mutant and BFA must inhibit uncoating as well as coating, a property that has not heretofore been attributed to ARF and to BFA.
It has been noted that Golgi budding in some cell types, such as MDCK cells, is BFA-resistant and does not require the addition of exogenous ARF (Ktistakis et al., 1995, 1996). The formation of SVs more closely resembles budding from CHO Golgi membranes in its BFA and exogenous ARF sensitivity. Examination of Golgi budding in the BFA-resistant cell types generated evidence that the role of ARF is catalytic, not stoichiometric and involves the activation of phospholipase D activity by ARF (Brown et al., 1993). The role, if any, of phospholipiase D in SV formation needs to be examined further.
In summary, our data show that SV biogenesis in PC12 cells is a process that requires ARF and that the GTP/GDP status of the ARF regulates the intermediates with which it is associated. Because the ARF family is so strongly linked to coating events, our data strongly suggest the participation of coat proteins in the budding of synaptic vesicles from PC12 membranes. The next step is to identify the proteins to which ARF1 binds, and the coating molecules that are recruited.
Acknowledgments
We thank Dr. F. Brodsky (University of California, San Francisco, CA [UCSF]) for the generous gift of antibodies to clathrin (X-22) and TD.1. Dr. T. Kreis (University of Geneva, Switzerland) kindly provided antibodies to β-COP (EAGE), and Dr. F. Letourneur (Basel Institute for Immunology, Switzerland) provided fusion proteins GST-WBP1-KK and GST-WBP1-SS. We are also grateful to Drs. L. Nagy (University of Guelph, Ontario, Canada) and J. Roos (UCSF) for their helpful comments in the initial preparation of the manuscript.
Abbreviations used in this paper
- ARF
ADP ribosylation factor
- BFA
brefeldin A
- COPI and COPII
coat proteins I-II
- HMW
high molecular weight
- SV
synaptic vesicle
- TAg
T Antigen
- VAMP
vesicle-associated membrane protein
Footnotes
Please address all correspondence to Regis B. Kelly, Department of Biochemistry and Biophysics, and the Hormone Research Institute, University of California, San Francisco, CA 94143-0534. Tel.: (415) 476-4095. Fax: (415) 731-3612. e-mail: kelly@cgl.ucsf.edu
Funded by National Institutes of Health (NIH) grants NS09878, NS15927, and DA10154 to R.B. Kelly. V. Faundez is the recipient of a NIH Fogarty International Postdoctoral Fellowship.
References
- Aniento F, Gu F, Parton RG, Gruenberg J. An endosomal βCOP is involved in the pH-dependent formation of transport vesicles destined for late endosomes. J Cell Biol. 1996;133:29–41. doi: 10.1083/jcb.133.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch WE, Kahn RA, Schwaninger R. ADP-ribosylation factor is required for vesicular trafficking between the endoplasmic reticulum and the cis-Golgi compartment. J Biol Chem. 1992;267:13053–13061. [PubMed] [Google Scholar]
- Barroso M, Sztul ES. Basolateral to apical transcytosis in polarized cells is indirect and involves BFA and trimeric G protein sensitive passage through the apical endosome. J Cell Biol. 1994;124:83–100. doi: 10.1083/jcb.124.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednarek SY, Ravazzola M, Hosobuchi M, Amherdt M, Perrelet A, Schekman R, Orci L. COPI- and COPII-coated vesicles bud directly from the endoplasmic reticulum in yeast. Cell. 1995;83:1183–1195. doi: 10.1016/0092-8674(95)90144-2. [DOI] [PubMed] [Google Scholar]
- Boman AL, Kahn RA. Arf proteins: the membrane traffic police? . Trends Biochem Sci. 1995;20:147–150. doi: 10.1016/s0968-0004(00)88991-4. [DOI] [PubMed] [Google Scholar]
- Bonzelius F, Herman GA, Cardone MH, Mostov KE, Kelly RB. The polymeric immunoglobulin receptor accumulates in specialized endosomes but not synaptic vesicles within the neurites of transfected neuroendocrine PC12 cells. J Cell Biol. 1994;127:1603–1616. doi: 10.1083/jcb.127.6.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky FM. Clathrin structure characterized with monoclonal antibodies. I. Analysis of multiple antigenic sites. J Cell Biol. 1985;101:2047–2054. doi: 10.1083/jcb.101.6.2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown HA, Gutowski S, Moomaw CR, Slaughter C, Sternweis PC. ADP-ribosylation factor, a small GTP-dependent regulatory protein, stimulates phospholipase D activity. Cell. 1993;75:1137–1144. doi: 10.1016/0092-8674(93)90323-i. [DOI] [PubMed] [Google Scholar]
- Cavenagh MM, Whitney JA, Carroll K, Zhang JJ, Boman AL, Rosenwald AG, Mellman I, Kahn RA. Intracellular distribution of Arf proteins in mammalian cells. J Biol Chem. 1996;271:21767–21774. doi: 10.1074/jbc.271.36.21767. [DOI] [PubMed] [Google Scholar]
- Chen Y-G, Shields D. ADP-ribosylation factor-1 stimulates formation of nascent secretory vesicles from the trans-Golgi network of endocrine cells. J Biol Chem. 1996;271:5297–5300. doi: 10.1074/jbc.271.10.5297. [DOI] [PubMed] [Google Scholar]
- Cosson P, Letourneur F. Coatomer interaction with di-lysine endoplasmic reticulum retention motifs. Science (Wash DC) 1994;263:1629–1631. doi: 10.1126/science.8128252. [DOI] [PubMed] [Google Scholar]
- De Camilli P, Takei K. Molecular mechanisms in synaptic vesicle endocytosis and recycling. Neuron. 1996;16:481–486. doi: 10.1016/s0896-6273(00)80068-9. [DOI] [PubMed] [Google Scholar]
- de Hoop M, von Poser C, Lange C, Ikonen E, Hunziker W, Dotti CG. Intracellular routing of wild type and mutated polymeric immunoglobulin receptor in hippocampal neurons in culture. J Cell Biol. 1995;130:1447–1459. doi: 10.1083/jcb.130.6.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell'Angelica EC, Ohno H, Ooi CE, Rabinovich E, Roche KW, Bonifacino JS. AP-3: an adaptor-like protein complex with ubiquitous expression. EMBO (Eur Mol Biol Organ) J. 1997;16:917–928. doi: 10.1093/emboj/16.5.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desnos C, Clift-O'Grady L, Kelly RB. Biogenesis of synaptic vesicles in vitro. J Cell Biol. 1995;130:1041–1049. doi: 10.1083/jcb.130.5.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittie AS, Hajibagheri N, Tooze SA. The AP-1 adaptor complex binds to immature secretory granules from PC12 cells and is regulated by ADP-ribosylation factor. J Cell Biol. 1996;132:523–536. doi: 10.1083/jcb.132.4.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson JG, Klausner RD. ARF-a key regulatory switch in membrane traffic and organelle structure. Curr Opin Cell Biol. 1994;6:527–532. doi: 10.1016/0955-0674(94)90072-8. [DOI] [PubMed] [Google Scholar]
- Donaldson JG, Finazzi D, Klausner RD. Brefeldin A inhibits Golgi membrane-catalysed exchange of guanine nucleotide onto ARF protein. Nature (Lond) 1992;360:350–352. doi: 10.1038/360350a0. [DOI] [PubMed] [Google Scholar]
- Fensome A, Cunningham E, Troung O, Cockcroft S. ARF(2-17) does not specifically interact with ARF1-dependent pathways. Inhibition by peptide of phospholipases Cβ, D and exocytosis in HL60 cells. FEBS (Fed Eur Biochem Soc) Letts. 1994;349:34–38. doi: 10.1016/0014-5793(94)00634-2. [DOI] [PubMed] [Google Scholar]
- Grote E, Kelly RB. Endocytosis of VAMP is facilitated by a synaptic vesicle targeting signal. J Cell Biol. 1996;132:537–549. doi: 10.1083/jcb.132.4.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grote E, Hao JC, Bennett MK, Kelly RB. A targeting signal in VAMP regulating transport to synaptic vesicles. Cell. 1995;81:581–589. doi: 10.1016/0092-8674(95)90079-9. [DOI] [PubMed] [Google Scholar]
- Helms JB, Rothman JE. Inhibition of brefeldin A of a Golgi membrane enzyme that catalyses exchange of guanine nucleotide bound to ARF. Nature (Lond) 1992;360:352–354. doi: 10.1038/360352a0. [DOI] [PubMed] [Google Scholar]
- Heuser JE, Reese TS. Evidence for recycling of synaptic vesicle membrane during transmitter release at the frog neuromuscular junction. J Cell Biol. 1973;37:315–344. doi: 10.1083/jcb.57.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunziker W, Whitney JA, Mellman I. Selective inhibition of transcytosis of brefeldin A in MDCK cells. Cell. 1991;67:617–627. doi: 10.1016/0092-8674(91)90535-7. [DOI] [PubMed] [Google Scholar]
- Kahn RA, Goddard C, Newkirk M. Chemical and immunological characterization of the 21-kDa ADP-ribosylation factor of adenylate cyclase. J Biol Chem. 1988;263:8282–8287. [PubMed] [Google Scholar]
- Kahn RA, Kern FG, Clark J, Gelmann EP, Rulka C. Human ADP-ribosylation factors. A functionally conserved family of GTP-binding proteins. J Biol Chem. 1991;266:2606–2614. [PubMed] [Google Scholar]
- Kahn RA, Randazzo P, Serafini T, Weiss O, Rulka C, Clark J, Amherdt M, Roller P, Orci L, Rothman JE. The amino terminus of ADP-ribosylation factor (ARF) is a critical determinant of ARF activities and is a potent and specific inhibitor of protein transport. J Biol Chem. 1992;267:13039–13046. [PubMed] [Google Scholar]
- Ktistakis NT, Brown HA, Sternweis PC, Roth MG. Phospholipase D is present on Golgi-enriched membranes and its activation by ADP ribosylation factor is sensitive to brefeldin A. Proc Natl Acad Sci USA. 1995;92:4952–4956. doi: 10.1073/pnas.92.11.4952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ktistakis NT, Brown HA, Waters MG, Sternweis PC, Roth MG. Evidence that phospholipase D mediates ADP ribosylation factor- dependent formation of Golgi coated vesicles. J Cell Biol. 1996;134:295–306. doi: 10.1083/jcb.134.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Yuan L, Tipper C, Amherdt M, Orci L, Klausner RD. Brefeldin A's effects on endosomes, lysosomes and the TGN suggest a general mechanism for regulating organelle structure and membrane traffic. Cell. 1991a;67:601–616. doi: 10.1016/0092-8674(91)90534-6. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Glickman J, Donaldson JG, Robbins J, Kreis TE, Seamon KB, Sheetz MP, Klausner RD. Forskolin inhibits and reverses the effects of brefeldin A on Golgi morphology by a cAMP-independent mechanism. J Cell Biol. 1991b;112:567–577. doi: 10.1083/jcb.112.4.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe M, Kreis TE. In vitro assembly and disassembly of coatomer. J Biol Chem. 1995;270:31364–31371. doi: 10.1074/jbc.270.52.31364. [DOI] [PubMed] [Google Scholar]
- Martys JL, Shevell T, McGraw TE. Studies of transferrin recycling reconstituted in streptolysin O permeabilized chinese hamster ovary cells. J Biol Chem. 1995;270:25976–25984. doi: 10.1074/jbc.270.43.25976. [DOI] [PubMed] [Google Scholar]
- Matteoli M, Takei K, Perin MS, Sudhof TC, De Camilli P. Exo-endocytotic recycling of synaptic vesicles in developing processes of cultured hippocampal neurons. J Cell Biol. 1992;117:849–861. doi: 10.1083/jcb.117.4.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundigl O, Matteoli M, Daniell L, Thomas-Reetz A, Metcalf A, Jahn R, De Camilli P. Synaptic vesicle proteins and early endosomes in cultured hippocampal neurons: differential effects of brefeldin A in axon and dendrites. J Cell Biol. 1993;122:1207–1221. doi: 10.1083/jcb.122.6.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narula N, Stow JL. Distinct coated vesicles labeled for p200 bud from trans-Golgi network membranes. Proc Natl Acad Sci USA. 1995;92:2874–2878. doi: 10.1073/pnas.92.7.2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathke IS, Heuser J, Kupas A, Stock J, Turck CW, Brodsky FM. Folding and trimerization of clathrin subunits at the triskelion hub. Cell. 1992;68:899–910. doi: 10.1016/0092-8674(92)90033-9. [DOI] [PubMed] [Google Scholar]
- Oka T, Nakano A. Inhibition of GTP hydrolysis by Sar1p causes accumulation of vesicles that are a functional intermediate of the ER-to-Golgi transport in yeast. J Cell Biol. 1994;124:425–434. doi: 10.1083/jcb.124.4.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostermann J, Orci L, Tani K, Amherdt M, Ravazzola M, Elazar Z, Rothman JE. Stepwise assembly of functionally active transport vesicles. Cell. 1993;75:1015–1025. doi: 10.1016/0092-8674(93)90545-2. [DOI] [PubMed] [Google Scholar]
- Randazzo PA, Weiss O, Kahn RA. Preparation of recombinant ADP-ribosylation factor. Methods Enzymol. 1992;219:362–369. doi: 10.1016/0076-6879(92)19036-6. [DOI] [PubMed] [Google Scholar]
- Randazzo PA, Terui T, Sturch S, Kahn RA. The amino terminus of ADP-ribosylation factor (ARF) I is essential for interaction with Gs and ARF GTPase-activating protein. J Biol Chem. 1994;269:29490–29494. [PubMed] [Google Scholar]
- Robinson MS, Kreis TE. Recruitment of coat proteins onto Golgi membranes in intact and permeabilized cells: effects of brefeldin A and G protein activators. Cell. 1992;69:129–138. doi: 10.1016/0092-8674(92)90124-u. [DOI] [PubMed] [Google Scholar]
- Rothman JE, Wieland FT. Protein sorting by transport vesicles. Science (Wash DC) 1996;272:227–234. doi: 10.1126/science.272.5259.227. [DOI] [PubMed] [Google Scholar]
- Salama NR, Yeung T, Schekman RW. The Sec13p complex and reconstitution of vesicle budding from the ER with purified cytosolic proteins. EMBO (Eur Mol Biol Organ) J. 1993;12:4073–4082. doi: 10.1002/j.1460-2075.1993.tb06091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schekman R, Orci L. Coat proteins and vesicle budding. Science (Wash DC) 1996;271:1526–1533. doi: 10.1126/science.271.5255.1526. [DOI] [PubMed] [Google Scholar]
- Schonhorn JE, Wessling-Resnick M. Brefeldin A down-regulates the transferrin receptor in K562 cells. Mol Cell Biochem. 1994;135:159–169. doi: 10.1007/BF00926519. [DOI] [PubMed] [Google Scholar]
- Seaman MN, Ball CL, Robinson MS. Targeting and mistargeting of plasma membrane adaptors in vitro. J Cell Biol. 1993;123:1093–1105. doi: 10.1083/jcb.123.5.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafini T, Orci L, Amherdt M, Brunner M, Kahn RA, Rothman JE. ADP-ribosylation factor is a subunit of the coat of Golgi-derived COP-coated vesicles: a novel role for a GTP-binding protein. Cell. 1991;67:239–253. doi: 10.1016/0092-8674(91)90176-y. [DOI] [PubMed] [Google Scholar]
- Shupliakov O, Loew P, Grabs D, Gad H, Chen H, David C, Takei K, De Camilli P, Brodin L. Synaptic vesicle endocytosis impaired by disruption of Dynamin-SH3 domain interactions. Science (Wash DC) 1997;276:259–263. doi: 10.1126/science.276.5310.259. [DOI] [PubMed] [Google Scholar]
- Simon J-P, Ivanov IE, Shopsin B, Hersh D, Adesnik M, Sabatini DD. The in vitro generation of post-Golgi vesicles carrying viral envelope glycoproteins requires an ARF-like GTP-binding protein and a protein kinase C associated with the Golgi apparatus. J Biol Chem. 1996;271:16952–16961. doi: 10.1074/jbc.271.28.16952. [DOI] [PubMed] [Google Scholar]
- Simpson S, Bright NA, West MA, Newman LS, Darnell RB, Robinson MS. A novel adaptor-related protein complex. J Cell Biol. 1996;133:749–760. doi: 10.1083/jcb.133.4.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamnes MA, Rothman JE. The binding of AP-1 clathrin adaptor particles to Golgi membranes requires ADP-ribosylation factor, a small GTP-binding protein. Cell. 1993;73:999–1005. doi: 10.1016/0092-8674(93)90277-w. [DOI] [PubMed] [Google Scholar]
- Takei K, Mundigl O, Daniell L, De Camilli P. The synaptic vesicle cycle: a single vesicle budding step involving clathrin and dynamin. J Cell Biol. 1996;133:1237–1250. doi: 10.1083/jcb.133.6.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takizawa PA, Yucel JK, Veit B, Faulkner DJ, Deerinck T, Soto G, Ellisman M, Malhotra V. Complete vesiculation of Golgi membranes and inhibition of protein transport by a novel sea sponge metabolite, ilimaquinone. Cell. 1993;73:1079–1090. doi: 10.1016/0092-8674(93)90638-7. [DOI] [PubMed] [Google Scholar]
- Tanigawa G, Orci L, Amherdt M, Ravazzola M, Helms JB, Rothman JE. Hydrolysis of bound GTP by ARF protein triggers uncoating of Golgi-derived COP-coated vesicles. J Cell Biol. 1993;123:1365–1371. doi: 10.1083/jcb.123.6.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooze J, Holinshead M. In AtT20 and HeLa cells brefeldin A induces the fusion of tubular endosomes and changes their distribution and some of their endocytic properties. J Cell Biol. 1992;118:813–830. doi: 10.1083/jcb.118.4.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Weert AWM, Dunn KW, Geuze HJ, Maxfield FR, Stoorvogel W. Transport from late endosomes to lysosomes but not sorting of integral membrane proteins in endosomes, depends on the vacuolar proton pump. J Cell Biol. 1995;130:821–834. doi: 10.1083/jcb.130.4.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker MW, Bobak DA, Tsai SC, Moss J, Vaughan M. GTP but not GDP analogues promote association of ADP-ribosylation factors, 21-kDa protein activators of cholera toxin, with phospholipids and PC-12 cell membranes. J Biol Chem. 1992;267:3230–3235. [PubMed] [Google Scholar]
- Waters MG, Serafini T, Rothman JE. ‘Coatomer': a cytosolic protein complex containing subunits of non-clathrin-coated Golgi transport vesicles. Nature (Lond) 1991;349:248–251. doi: 10.1038/349248a0. [DOI] [PubMed] [Google Scholar]
- Weidman PJ, Winter WM. The G protein-activating peptide, mastoparan, and the synthetic NH2-terminal ARF peptide, ARFp13, inhibit in vitro Golgi transport by irreversibly damaging membranes. J Cell Biol. 1994;127:1815–1827. doi: 10.1083/jcb.127.6.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney JA, Gomez M, Sheff D, Kreis TE, Mellman I. Cytoplasmic coat proteins involved in endosome function. Cell. 1995;83:703–713. doi: 10.1016/0092-8674(95)90183-3. [DOI] [PubMed] [Google Scholar]
- Wood SA, Wood WJ. The morphology but not the function of endosomes and lysosomes is altered by brefeldin A. J Cell Biol. 1992;119:273–285. doi: 10.1083/jcb.119.2.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood SA, Park JE, Brown WJ. Brefeldin A causes a microtubule-mediated fusion of the trans-Golgi network and early endosomes. Cell. 1991;67:591–600. doi: 10.1016/0092-8674(91)90533-5. [DOI] [PubMed] [Google Scholar]