

1. Introduction
A member of the pentose phosphate pathway, ribose 5-phosphate (R5P) exists at higher levels (5–20 μM) in those cells requiring reduced nicotinamide adenine dinucleotide phosphate (NADPH) for reductive biosynthesis or in dividing cells that require the molecule as a precursor for purine and pyrimidine biosynthesis. As a sugar, R5P is subject to spontaneous Maillard reactions with cellular amines, including the N-terminal, lysine (Lys) and arginine (Arg) groups of proteins. In fact, the molecule has been shown to react at much higher rates than most other common sugars (including glucose, ribose, and fructose) and sugar phosphates (including glucose 6-phosphate and fructose 1,6-bisphosphate).1-3 Based on UV absorbance and browning results, R5P reacts at rates that exceed 100-fold that of glucose.1 A reaction with glycine at pH 8 and 37 °C monitored by 1H NMR spectroscopy gave a bimolecular rate constant for the disappearance of R5P from solution of 0.22 M-1 h-1.1 Considering this first step of the Maillard reaction involves the attack of the deprotonated form of the amine, a pH-independent rate equation for this system can be rewritten as:
where [R-NH2] reflects the pH-dependent concentration of the deprotonated amine. For the reported glycine system,1 the rate constant would be recalculated as approximately 7 M-1 h-1. Given an amine of moderately low pKa (e.g., 8.0) and 100 μM concentration, this translates into an in vivo R5P glycation rate equaling approximately 0.05% of available amine per day at physiologic R5P levels. Longer lived proteins would thus experience one percent glycation in a three-week period.
The relatively rapid glycation rate of R5P is clearly related in some way to its attached phosphate group.1 Phosphates have been long employed as catalysts of the Maillard browning reaction via a mechanism speculated to be either general base catalysis4 or covalent catalysis.5 In the case of R5P, a proximity effect of the phosphate group perched over the reactive anomeric center apparently promotes a greater reactivity in comparison to that observed by intermolecular phosphate. (Sugar phosphates such as glucose 6-phosphate contain intramolecular phosphate, but the location of the phosphate in an equatorial position is stereochemically distant from the C-1 carbon of the glucopyranoside.) The proximity/orientation rate enhancements could also be supplemented by steric effects of the phosphate on the furanose ring structure leading to a higher level of the acyclic structure, a form generally acknowledged as the true initial reactant of the Maillard sequence. While large discrepancies have been reported in the acyclic levels of sugars as a result of the technique employed, the acyclic level of R5P appears to be at least an order of magnitude in excess of D-ribose and 2–3 orders of magnitude greater than D-glucose.6-8
The early steps of the reaction of R5P with amines (Scheme 1) appear to be similar to the Maillard reaction of other sugars. The initial attack of the nitrogen to produce an N-glycoside is followed by a rearrangement to generate an Amadori compound.2,9 The formation of a typical α-dicarbonyl compound has then been suggested1 which subsequently proceeds to a variety of highly UV- and visible-absorbing advanced glycation end-products (AGEs). The reaction of R5P in this sequence appears, however, to be more complex than other sugars. It is possible that the presence of the amine encourages isomerization of R5P to ribulose 5-phosphate (Ru5P) (see Scheme 1), and this reaction may or may not be connected to an observed loss of phosphate from the molecule.1,10 As in other Maillard systems, the availability of O2 allows a different series of subsequent reactions (compared to anaerobic conditions) and the formation of reactive oxygen species superoxide and hydrogen peroxide.11
Scheme 1.
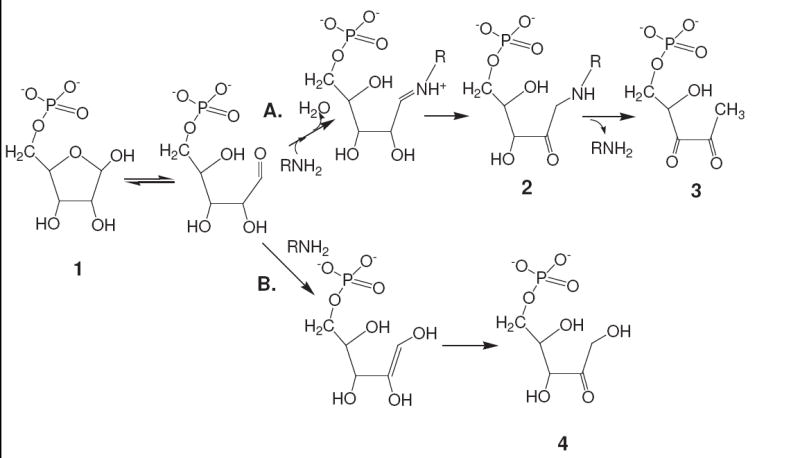
Proposed early reactions of R5P (1) with amines stemming from the acyclic form in equilibrium with the furanose. A traditional series of Maillard reactions (path A) leads through an Amadori product (2) to an α-dicarbonyl product (3), while an isomerization series (path B) leading to ribulose 5-phosphate (4) may be encouraged by a general base catalysis (i.e., removal of the C-2 hydrogen) of the amine.
Like R5P, 2-deoxyribose 5-phosphate (dR5P) has a phosphate moiety perched over a reactive hemiacetal group of a furanose ring. The sugar, however, lacks the hydroxyl group at C-2 necessary for the enaminol tautomerization step that occurs during the Amadori rearrangement process. Thus, while it is likely that dR5P undergoes some initial N-glycoside reaction with amines in solution, it is impossible for dR5P to proceed to melanoidin production and browning via a traditional Maillard series of reactions. However, as previously reported by Wondrak et al.12 and as we report here, dR5P overcomes this “deoxy” barrier to generate a variety of Maillard-like compounds.
We report here the kinetics and major reaction products of R5P and dR5P (along with their corresponding non-phosphorylated counterparts ribose and deoxyribose) with simple amines. The molecules are capable of producing a diverse set of products in a relatively rapid timeframe in simulated physiological conditions and, thus, need to be considered as potentially significant glycation agents in cells.
2. Results
2.1 Rate of sugar consumption
The rate constants reflecting the loss of R5P with time in reactions with glycine (Gly), L-alanine (Ala), glycylglycine (Gly-gly), L-asparagine (Asn), β-alanine (β-Ala), N-acetyl-L-lysine (NAcLys), and N-acetyl-L-arginine (NAcArg) at pH 8.0 and 37 °C are shown in Table 1. (The NAcArg value listed is at best an estimate, given the slow reaction time, i.e., less than 20% reacted over 18 days.) All determinations at this pH were performed by measuring the intensities of the 1H peaks (α and β) of the C-1 hydrogens of R5P. An LC/MS selective ion-monitoring (SIM) method following the m/z 97 fragment of R5P13 was also employed for some amines, and the results (not shown) were in agreement with the given NMR data. The molecular rate constants (k) reflect the kinetics of the reaction between the R5P and amine molecules while the adjusted k* corrects the value for actual deprotonated amine concentrations. The kinetic treatment of the early Maillard sequence is based on the reaction series shown in Scheme 2. For simplicity, the kinetics assumes irreversible steps to generate the Amadori product14 and the α-dicarbonyl compounds, an assumption that does not appear to be completely true.15 (A slow reversal of these steps occurs.)
Table 1.
Kinetic Data for the Rate of Sugar Loss in a Maillard Reaction with Amine at 37 °C
| Sugar‡ | Amine‡ | Technique | pKa* (at 37 °C) |
pH | k
(M-1 h-1) |
k* (M-1 h-1) |
|---|---|---|---|---|---|---|
| R5P | Gly-gly | 1H NMR | 7.8 | 8.0 | 0.6 | 1.0 |
| R5P | L-Asn | 1H NMR | 8.6 | 8.0 | 0.35 | 1.8 |
| R5P | Gly | 1H NMR | 9.46 | 8.0 | 0.26 | 7.8 |
| R5P | L-Ala | 1H NMR | 9.7 | 8.0 | 0.07 | 3.6 |
| R5P | β-Ala | 1H NMR | 9.95 | 8.0 | 0.31 | 28 |
| R5P | L-NAcLys | 1H NMR | 10.25 | 8.0 | 0.20 | 36 |
| R5P | L-NAcArg | 1H NMR | 12.25 | 8.0 | 0.005 | 89 |
| R5P (2X) | L-NAcLys | 1H NMR | 10.25 | 8.0 | 0.63 | 113 |
| R5P | L-NAcLys | 1H NMR | 10.25 | 7.5 | 0.32 | 180 |
| R5P | L-NAcLys (0.5X) | LC/MS | 10.25 | 7.5 | 0.32 | 180 |
| Ribose | L-NAcLys | LC/MS | 10.25 | 7.5 | 0.021 | 11.8 |
| dR5P | L-NAcLys | 1H NMR | 10.25 | 7.5 | 0.015 | 10.7 |
| Deoxyribose | L-NAcLys | LC/MS | 10.25 | 7.5 | ~ 0 | ~ 0 |
Sugar concentrations were 0.050 M and amine concentrations were 0.10 M except where noted.
Gly pKa at 37 °C obtained from a literature value while L-NAcArg was estimated from a literature value at 25 °C. pKa’s for Gly-gly, β-Ala, L-Ala, L-Asn, and L-NAcLys at 37 °C were determined by titration. The ribose and deoxyribose experiments contained equimolar amounts of inorganic phosphate.
Scheme 2.
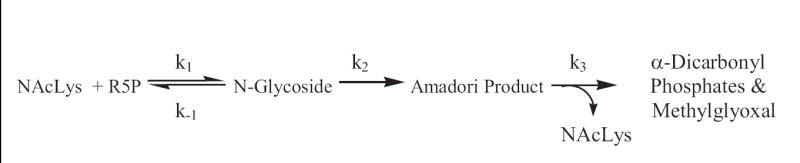
Chemical equations of the early Maillard sequence of R5P and amines defining the rate constants. Methylglyoxal would be generated by a retro-aldolization along with glyceraldehyde phosphate.15
It is important to note that the kinetics of the system is not only impacted by the collision rate of the two reactants, but also by the transition of the N-glycoside to the Amadori rearrangement product and beyond. The initial reaction of R5P and amine (deproto as defined by the equation and Keq below.
Employing 1H NMR spectroscopy to determine the loss of R5P from a NAcLys-containing solution at pH 10.5, we observed rapid equilibrium (within minutes) with a Keq = 7.6 M-1 (37 °C). This value is comparable to Keq = 2.5 M-1 determined by Schendel et al.16 for a R5P/NH3 reaction and to Keq = 5.0 M-1 for a R5P/tris(hydroxymethyl)aminomethane reaction obtained in our laboratory (unpublished data). Given the neutral pH of our reaction systems, only 0.3% of R5P would be converted at equilibrium to the N-glycoside. Thus, while the concentration of deprotonated amine is a component of our rate equation, large scale R5P and NAcLys loss is primarily impacted by the rate of the subsequent step to the Amadori product. The rate constant (k*) listed in Table 1 is essentially a combined rate constant for the first two steps, equivalent to k1k2/k-1 or Keqk2. This translates into a k2 value of 4.7 h-1 for the R5P/NAcLys system.
Confirming the early steps are rate limiting to the overall Maillard series, the trend in the “R5P-loss” rate constants (either k or k*) with the various amino acids correlates well with the corresponding rate constant series calculated for the increase in 280 nm absorbance and browning.1 As can be seen, there is variation in the k* values for R5P loss ranging from the lower Gly-gly value (1.0 M-1 h-1) to higher β-Ala and NAcLys values (~ 27 M-1 h-1). An observed direct relationship between the pKa of the amine and k* suggests that a reaction subsequent to the initial deprotonated amine attack is favored by a protonated form of the amine/imine. Conforming to the general Maillard reaction (see Scheme 3), this likely reflects the need for the Schiff base to be protonated in order to proceed to the enaminol and then on to the Amadori product. (This assumes the pKa of the Schiff base parallels the pKa of the amine.) This reaction would pull the initial equilibrium reaction (most likely favoring the sugar and free amine at neutral pH) towards the Amadori product and beyond. An alternative explanation for the variation observed in k* is that a steric effect may play a role in the initial attack with the long-tailed, less hindered NAcLys and β-Ala having a greater effective collision frequency than the more hindered amino acid reactants. This could be the explanation for why L -Ala, with an α-carbon substituent, has a smaller k* than l -Gly and β-Ala.
Scheme 3.
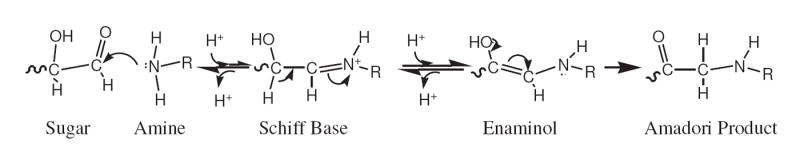
The general reaction mechanism for the formation of the Amadori product from a sugar and an amine.
Additional evidence supporting a second contributor to the initial steps of the Maillard reaction is supplied by pH studies. Given the accepted general pathway states that a deprotonated nitrogen nucleophile is required for attack, the reaction should proceed more rapidly at higher pH. This appears to be the case for Gly (data not shown), but not for NAcLys which causes more R5P loss at pH 7.4 than at pH 8. This supports the suggestion that the initial formation of the Schiff base is an equilibrium reaction favoring the free sugar and amine, and that further progression in the path (leading to the significant losses of sugar observed by 1H NMR spectroscopy or LC/MS) requires a low pH-favoring reaction that drives the reaction through an irreversible step(s).
An interesting effect on the rate constants is observed when R5P concentrations are increased while holding amine levels constant. While altering the initial amine concentration does not result in an apparent change in the rate constant, increasing the initial level of R5P in solution yields significantly higher rate constants for R5P loss (see Table 1). While this evidence may be interpreted as the reaction being of higher order relative to R5P concentration, it is difficult to comprehend how the steps contributing to the rate constant could be anything other than first order (see below and Discussion). One likely contributor to this effect is the higher levels of potentially catalytic phosphate that accompany the increase in R5P concentration. A comparison of the rates of 280 nm absorbance increases for a reaction of glycine in either a glyceraldehyde vs. glyceraldehyde + Pi system or a ribose vs. ribose + Pi system indicates the catalytic effect of phosphate to be approximately 10–12 fold per 1 molar equivalent of soluble phosphate (data not shown). In our system where R5P is doubled (to 100 mM), this would account for only about one-third of the observed rate constant increase. Further explanation may require a full elucidation of the mechanism of the inter- and intramolecular phosphate catalysis.
A similar characterization of a reaction between NAcLys and D-ribose and Pi (equimolar D-ribose and Pi to simulate the concentrations of each in R5P) gave a k = 0.021 M-1 h-1 and a k* = 11.8 M-1 h-1. When interpreted in comparison to the R5P experiment, an approximately fifteen-fold rate enhancement for the disappearance of the sugar occurs when a phosphate group is covalently attached to the ribose sugar. This is of the range typically quoted for catalytic effects due to proximity;17 however, a second explanation may be that the phosphate group encourages higher levels of the acyclic R5P form, the form that is believed to be the true initial reactant (along with the deprotonated amine) in the Maillard reaction. A phosphate group existing on C-5 would increase the level of the acyclic form due to the inability of the phosphorylated sugar to take the more stable pyranose form and possibly to other steric factors (i.e., steric hindrance adding stress to the furanose structure). The cited levels of acyclic forms of D-ribose and R5P are 0.05%7 and 1%,6 respectively, although it should be mentioned two different techniques were used to obtain these values, and discrepancies have frequently been seen among literature values for acyclic form levels. In order to examine this in a different manner, the relative rates of color formation for glyceraldehyde + Pi vs. glyceraldehydes 3-phosphate (G3P) with Gly were determined. Since both glyceraldehyde and G3P exist only in the acyclic forms, any rate enhancement must be solely due to a proximity effect of the intramolecular phosphate. In fact, G3P generated 280-nm products at a six-fold greater rate than glyceraldehyde in the presence of intermolecular phosphate. It is likely that a proximity effect plays a similar role in the intramolecular phosphate catalysis of the R5P Maillard reaction; however, it is still unclear what portion it contributes to the overall increase in rate.
With a k = 0.015 M-1 h-1 in a reaction with NAcLys, the Maillard activity of dR5P is considerably less (factor of 10–20) than that of R5P. This is expected for two reasons. First, replacement of the bulky –OH group with a hydrogen at the C-2 position favors dR5P taking the unreactive cyclic form vs. the reactive acyclic form. Second, the lack of the hydroxyl group on C-2 prevents the N-glycoside from transitioning to an Amadori compound by a normal Maillard reaction scheme. The N-glycoside compound generated from dR5P is unable to convert to the normal enaminol intermediate or the subsequent C-2 carbonyl group, steps which are part of the Maillard reaction sequence. The fact that dR5P does generate Maillard-like reaction products (see below) suggests that the molecule somehow overcomes the C-2 “blockage” to proceed in reactivity.
The loss of deoxyribose from a system containing NAcLys and Pi at pH 7.4 and 37 °C is at least an order of magnitude less rapid than the comparable ribose/NAcLys/Pi system. Over a 150 h incubation period at 37 °C, no statistically significant loss of deoxyribose was noted by analysis using LC/MS. Although not detected, some deoxyribose indeed reacted as evidence of the formation of an N-glycoside/Amadori product was observed by liquid chromatography/mass spectroscopy (LC/MS) (see below). Like dR5P, this slower rate is expected due to the combination of lower acyclic levels (unlike dR5P, deoxyribose also can assume the more stable pyranose form) and the lack of the hydroxyl group on the C-2 needed for normal Maillard reaction progression. The rate is considerable less than that observed for dR5P, suggesting an enhanced reactivity promoted by an attached phosphate group.
2.2 Rate of amine consumption
The disappearance of amine from solution does not stoichiometrically couple with the loss of R5P. Figure 1 shows the loss of R5P vs. the loss of NAcLys in a reaction at 0.10 M NAcLys and 0.050 M R5P at pH 7.5 and 37 °C using peak intensities and a SIMS method of LC/MS13 to determine NAcLys and R5P concentrations, respectively. (The fluorescamine method for determining free NAcLys amine groups yielded similar values.) As can be seen, the loss of NAcLys from solution is approximately three times less than the absolute decrease in R5P concentration. This effect is explained by the reaction mechanism (see Scheme 1) where NAcLys is consumed during the initial attack of the amine onto the sugar but then is released after the Amadori rearrangement as the sugar converts to one of the α-dicarbonyl forms. Under these conditions (pH 7.5; 37 °C), a level of approximately 0.015 M NAcLys is tied up in the N-glycoside/Amadori product as molecules flow through the initial portion of the Maillard reaction. Ultimately, there are some amine products that result in the amine compound being consumed in the reaction; however, it is not at a 1:1 ratio with R5P.
Figure 1.
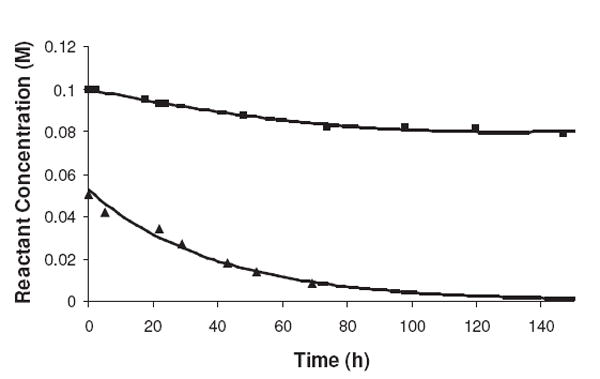
A comparison of the rates of disappearance of NAcLys (■) and R5P (▲) from solution at pH 7.5 and 37 °C. The loss of NAcLys over an approximately 70-h period was 0.015 M while 0.042 M of R5P disappeared from solution during this same timeframe.
The overall amine (NAcLys or β-Ala) loss from solution was related to the initial level of R5P in solution. A doubling of the R5P concentration resulted in an approximately 50% increase in the amine loss. The initial concentration of amine appeared to play less of a factor in the absolute amount of disappearance of amine from solution, e.g., a 0.050 M NAcLys solution vs. a 0.10 M NAcLys solution of the same R5P concentration exhibited approximately equal overall amine losses from solution. Together these results suggest the ultimate loss of amine from solution is related primarily to the synthesis of amine-consuming, post-Amadori R5P products. These products apparently are made in limited amounts in comparison to non-amine consuming, post-Amadori R5P products. The data suggest less than 30% of post-Amadori products involve an amine attachment.
The amine loss in an NAcArg/R5P system as measured by an LC/MS method or 9,10-phenanthrenequinone modification technique was insignificant over a 300 h incubation period at 37 °C. This slow reaction rate corresponds with the negligible loss of R5P over this time frame and indicates a low reactivity of free Arg side groups with initial free sugar. Any observable Arg modification in combined amine systems, therefore, appears to be the result of a reaction between Arg and a product generated by the reaction of R5P and an amine of lower pKa.
The rate of loss of amine from an incubation of NAcLys, D-ribose, and Pi is significantly less than that seen for the R5P system. After 145 hours of reaction, only 0.008 M of NAcLys was observed to be modified (data not shown) in comparison to approximately 0.021 M of the amine when reacted with R5P. Although slower, the relative ratio of D-ribose used to NAcLys modified (1.5: 1) at a point in the reaction where approximately one-third of the sugar consumed is similar to the 2:1 ratio seen for R5P in the R5P/NAcLys reaction. This suggests that the slower initial reaction of ribose to the Amadori product is accompanied by a proportionally slower reaction from the Amadori product (i.e., the step recycling the amine). Thus, the catalytic effect of the intramolecular phosphate in R5P is enhancing both the pre-Amadori and post-Amadori reactions by relatively similar levels.
Over a long period of time (~150 h), amine losses in the dR5P/NAcLys system were approximately the same as the amine consumption in the R5P/NAcLys system, 22% vs. 21%, respectively. As opposed to the R5P reaction, the loss of NAcLys is at a 3:1 stoichiometric ratio to the loss of dR5P, implying that several NAcLys residues ultimately become conjugated to a single dR5P molecule. In a traditional Maillard reaction, the amine is released upon the formation of one of two dicarbonyl compounds via a rearrangement step. Lacking the C-2 hydroxyl group, dR5P cannot undergo this rearrangement and thus appears to stay attached. Additional amine molecules apparently must subsequently attach to this N-glycoside (see below).
The loss of amine from the deoxyribose/Pi/NAcLys system was insignificant over a 165-h incubation period, confirming with the deoxyribose loss studies that the Maillard reaction of deoxyribose is comparably slow. In a manner similar to the ribose/R5P comparison, the relative rates of the deoxyribose vs. dR5P systems point to a large catalytic effect of intramolecular phosphate on this series of reactions.
2.3 Rate of N-glycoside/Amadori product formation
The relative concentration of the combined N-glycoside/Amadori compounds generated by a NAcLys/R5P reaction was determined by LC/MS via an extracted ion method measuring the peak areas under the chromatogram of the m/z 401 ± 0.5 [M+H+] signal. (The N-glycoside and Amadori rearrangement compounds have identical molecular masses. Although it is speculated that the level of N-glycoside is of minor concentrations (see previous section), we do not have conclusive evidence of this and thus will group the two species together for these discussions.) The relative amounts of the N-glycoside/Amadori compounds generated in the reaction with time are shown in Figure 2. As can be seen, the concentration maximum is reached at approximately 30–40 h while the NAcLys levels continue to decline (see Figure 1) until about five days. This signifies that NAcLys molecules continue to attach to R5P-derived compounds even after the maximum level of the N-glycoside/Amadori compounds have been reached. This likely represents the attachment of the amine onto subsequently formed α-dicarbonyls and/or other products (temporary and permanent) later in the sequence. Modeling of the kinetics of the reaction series (see Discussion) gave a k3 value of 0.03 h-1 and a maximum Amadori product level of 0.016 M.
Figure 2.
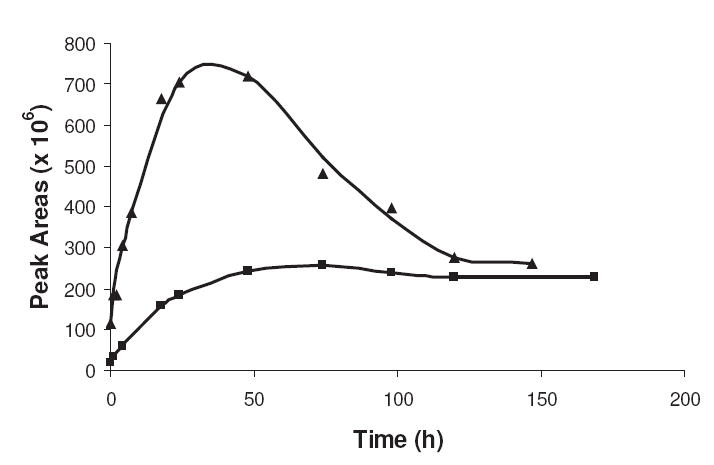
The generation of N-glycoside/Amadori products for R5P (▲) and dR5P (■) in reaction with NAcLys at pH 7.4 and 37 °C. The relative amount of products, represented as peak areas, was obtained by integration of an extracted ion chromatogram performed at the appropriate m/z value. The concentrations of the sugar and NAcLys were 0.05 M and 0.10 M, respectively.
A R5P/NAcLys reaction performed in the absence of oxygen did not significantly alter the N-glycoside/Amadori product ion peak pattern (data not shown) in comparison to the reaction shown in Figure 2 performed in the presence of oxygen. The rates of loss of R5P and NAcLys were also comparable. As expected, this indicates the movement of molecules through the early portion of the Maillard reaction, i.e., up through and including the transition of the Amadori product to the next compound, is a series which proceeds without oxygen involvement.
The formation of N-glycoside/Amadori-like compounds from dR5P is included in Figure 2. As can be seen, there is a slower transition to a maximum level in comparison to the R5P reaction and over the course of the experiment, the ion peak areas of the chromatograms isolated on the selected Amadori/N-glycoside mass (m/z 385 [M + H+] for dR5P) are not as intense. A relatively slower initial reaction rate (with a similar rate of transition away from this form) would account for this rate pattern although it is important to note that the current Maillard reaction mechanism would not allow for an Amadori product to be formed in dR5P where C-2 has lost a necessary hydroxyl participant of the rearrangement. It is possible that the level seen is reflective solely of the N-glycoside only, although it is anticipated that equilibrium levels of the N-glycoside would be established quickly, whereas our data indicates a steady increase of this compound(s) over the first 24 hours.
N-glycoside/Amadori formation in the non-phosphorylated ribose or deoxyribose systems took longer to reach a steady state/maximum level in comparison to R5P or dR5P (see Table 2). N-glycoside/Amadori production in the ribose/Pi system took 105 h to reach 90% of a maximum value while the deoxyribose/Pi system was only at 50% of an estimated peak level at this same time point. The production of an N-glycoside/Amadori-like compound in the deoxyribose system was observed even when no statistically significant loss of deoxyribose or NAcLys from solution was detected. This implies the signals recorded for this compound represent relatively small concentration levels.
Table 2.
Estimated Time to Reach 90 % of Maximum N-Glycoside/Amadori Level
| System | Time (h) |
|---|---|
| R5P and NAcLys | 15 |
| dR5P and NAcLys | 40 |
| Ribose + Pi + NAcLys | 105 |
| Deoxyribose + Pi + NAcLys | > 200 |
The R5P, dR5P, and ribose values were determined graphically while the deoxyribose level was estimated via a calculation from initial rate patterns.
2.4 Hydrolysis of phosphate and formation of new phosphate compounds
The reaction between R5P and amines eventually leads to the cleavage of the phosphate group from the ribose nucleus. Figure 3 shows the rate of phosphate hydrolysis from several amines at 37 °C and pH 8.0 as monitored by 31P NMR. There is less correlation of the rate of Pi formation with the pKa of the attacking amine for this series, although it is likely that the exceptionally slow phosphate cleavage rates of some amines like NAcArg (hydrolysis rate equivalent to the background rate) is likely tied to their high pKa. (The rate of R5P hydrolysis without amine at pH 8 was approximately 1.5% over a 70 h time period.) The rate of phosphate cleavage is directly proportional to both the initial concentration of the amine and the initial concentration of R5P present in the mixture. A lag time of 5–10 hours is typically observed for the first evidence of phosphate cleavage; this suggests an initial transition of R5P to a second compound prior to phosphate removal. The phosphate continues to be cleaved from the pentose nucleus, even after all R5P has disappeared from solution, again indicating that R5P is converted to a second phosphate-containing intermediate. The reaction of poly(lysine) with R5P exhibits a greatly enhanced rate of phosphate cleavage even at a lower molar level of amine. This is not explained solely by its relatively low pKa (9.5) as its phosphate hydrolysis rate is significantly greater than amines of comparable pKa (such as Asn or Gly-gly). The close proximity of several amines around the reaction site must promote a higher rate of the cleavage reaction.
Figure 3.
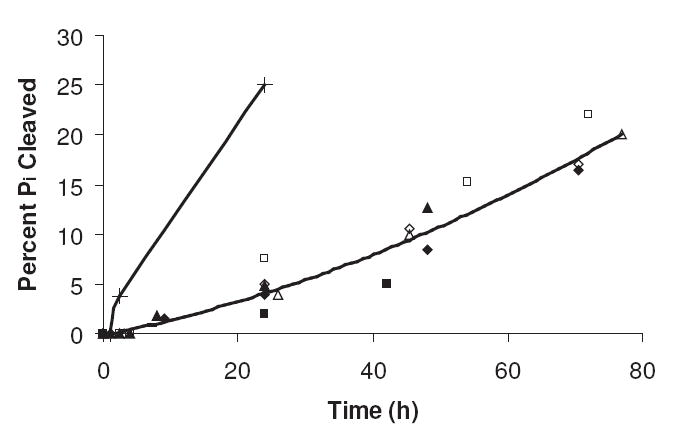
The percent phosphate (Pi) cleaved from various R5P/amine systems at pH 8 and 37 °C as determined by 31P NMR. The R5P concentration was 0.05 M, and the amine concentration was 0.10 M except for poly(lysine) where the amine concentration (of the R groups) was 0.050 M. Symbols: Poly(lysine) (+); Asn (◊); β-Ala (■); l-NAcLys (△); l-Ala (◆); Gly-gly (▲); Gly (□).
The loss of a C-5 phosphate from a ribose-like sugar has been proposed by Hauck et al.10 to be the result of a series of reactions initiated when a sugar intermediate contains a C-3 carbonyl group next to a C-4 hydroxyl group, leading to the ability to form an enediol between these two carbon centers (Scheme 4).
Scheme 4.
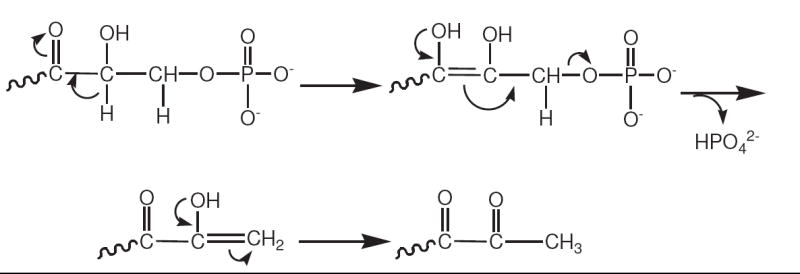
A proposed reaction scheme of Hauck et al.10 for the hydrolysis of an intramolecular 5-phosphate moiety from a pentose sugar containing a carbonyl group at the C-3 position.
Along with the release of Pi from R5P into solution during the Maillard reaction, 31P NMR analysis detected the formation of several new compounds exhibiting peaks 0–0.5 ppm downfield from the phosphate peak of R5P (see Figure 4). It is believed these represent the phosphorylated forms of the Amadori product, the α-dicarbonyl compounds, and other intermediates as the R5P sugar core undergoes the various Maillard alterations. In a reaction between R5P and ammonia, the Maillard product phosphoribosamine exhibits a phosphate peak that is positioned as a shoulder (i.e., less than 0.05 ppm downfield) on the R5P phosphate peak. The phosphate resonance of ribulose 5-phosphate, the C-2 ketose isomer of R5P and a possible product of our reactions, is well differentiated from the R5P resonance at 0.4 ppm downfield.18 It is likely that other compounds existing in a C-2 ketose form (e.g., the phosphorylated forms of the Amadori product and the α-dicarbonyl compounds) would be shifted downfield in a similar fashion.
Figure 4.
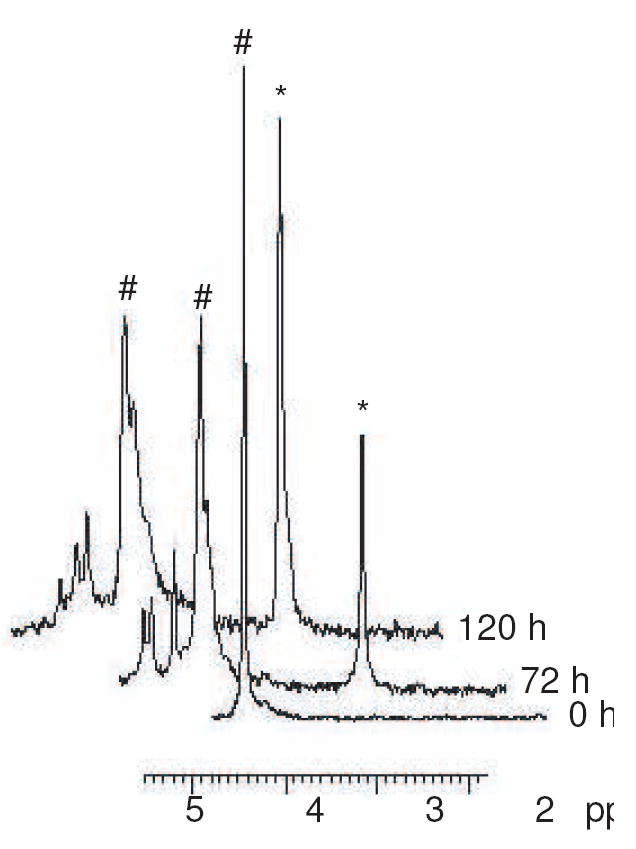
A series of three 31P NMR spectra following the reaction between NAcLys and R5P at pH 7.4 and 37 °C for five days. (Spectra are offset for clarity.) The reaction begins with a single R5P peak [represented by #] at approximately +4.5 ppm and, with time, Pi [represented by *] at approximately +3.2 ppm. Compounds giving downfield signals (+4.6 to +5.0 ppm) were generated.
When the reaction was performed in the absence of oxygen, the rate of phosphate cleavage was approximately 50% more rapid than when performed in the presence of oxygen. This strengthens the evidence that the hydrolysis of phosphate is a non-oxidative process. (The formation of carboxymethyllysine (CML) via an oxidative pathway (see NMR and LC/MS Characterization of Products) would generate 3-phosphoglycerate (3PG) in place of Pi; 3PG yields a phosphate peak in 31P NMR that is superimposable upon the R5P phosphate resonance.18)
A similar analysis was performed using 31P NMR to characterize the loss of phosphate from dR5P during a Maillard-like reaction. Over a 120-h period, a solution of dR5P and NAcLys at pH 7.5 and 37 °C results in the hydrolysis of 18% of its phosphate as Pi. (The comparable solution using R5P shows cleavage of 20% of its intramolecular phosphate.) The loss of dR5P and the formation of Pi appear over time to be at a 1:1 stoichiometric ratio, suggesting that a reaction of dR5P is associated with a simultaneous or nearly simultaneous release of phosphate.
2.5 Absorbance changes
The relative rates of absorbance increases at 280 nm and 420 nm of R5P reacting with amino acids/dipeptides have been previously reported1 to show a partial correlation to the pKa of the attacking amine. The rate of UV absorbance increase (at 280 nm) for the R5P/amine (NAcLys) reaction is faster than other common sugars1,2 and for the comparable dR5P/amine, ribose + Pi/amine, and deoxyribose + Pi/amine systems (see Figure 5A). It is important to note that while the ribose/Pi reaction with NAcLys is less than 15-fold less rapid than the reaction of R5P in terms of sugar loss, the rate of absorbance increase at 280 nm is only approximately three-fold slower. Thus, the comparative color development for these two systems is not based solely on the initial rate of attack of the amine, but more likely to the speed of unique products formed once the reaction progresses beyond the Amadori product.
Figure 5.
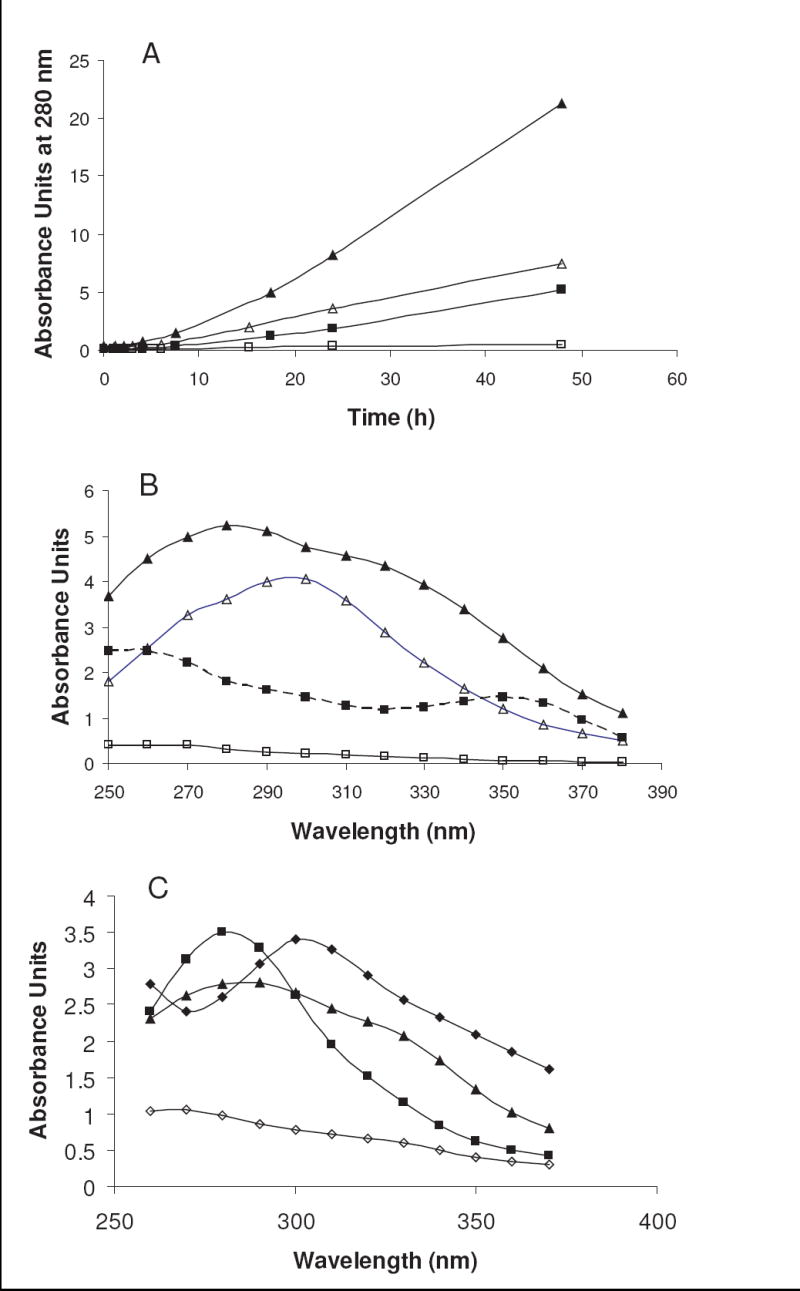
(A) Rate of absorbance increase at 280 nm for R5P (▲), ribose + Pi (△), dR5P (■), and deoxyribose + Pi (□) with NAcLys at pH 7.4 and 37 °C. (B) Absorbance spectra for R5P (▲), ribose + Pi (△), dR5P (■), and deoxyribose + Pi (□) with NAcLys after 24 h (17.5 h for R5P) at pH 7.4 and 37 °C. (C) Absorbance spectra for three amino acids after a reaction with R5P at pH 8.0 and 37 °C. Reaction time was 10 h for Gly (▲) and Glu (◊), 7.3 h for Asn (■), and 6 h for Gly-Gly (◆).
In addition, as can be seen in Figure 5B, R5P is unique among this grouping in its production of an obvious 320 nm product along with a primary UV-absorbing product in the 280–290 nm range. The reaction of R5P with numerous amino acids/dipeptides all show the 320 nm shoulder to some degree, with some (like Gly) having approximately similar absorbances at 320 nm vs. 280 nm, while others (like Asn) have a relatively less intense 320-nm peak. (See Figure 5C.) The 320-nm peak appears to be dependent on the ratio of R5P to amine in solution, a solution containing a R5P-to-NAcLys concentration ratio of 1.5:1 yields a dominant peak at 320 nm with a less intense 290-nm shoulder. Previous reports have indicated that 2,3-α-dicarbonyls (non-phosphorylated) have absorbance maxima at 310 nm.10 In comparison, ribulose 5-phosphate has an absorbance maximum of 280 nm and hydroxymethylfuranone (4-hydroxyl-5-methyl-3(2H)-furanone, HMF), a spontaneous product of ribulose 5-phosphate degradation, has a reported absorbance maximum of 285 nm.10 The lack of significant 310–320 nm absorbance indicates the reaction series is deviating from the normal progression through the α-dicarbonyl forms or that it is rapidly transitioning through these dicarbonyl intermediates. For example, Asn has been reported to react with glucose in an acrylamide-producing, decarboxylation reaction that ultimately results in a fructosamine product.19,20 It is highly possible the same series can happen for the reaction of Asn with R5P, forming a ribulosamine phosphate product with reduced conversion to an α-dicarbonyl form.
The availability of oxygen in the R5P-amine reactions determines the development of the UV-absorbing compounds. As shown in Figure 6, the presence of oxygen in the system reduces the overall UV absorbance, creating a spectrum with broad absorbance maxima of 280 nm and 310 nm. The reaction under anaerobic conditions favors the production of a compound of high absorptivity at 305 nm and a lower absorbing compound at approximately 360 nm.
Figure 6.
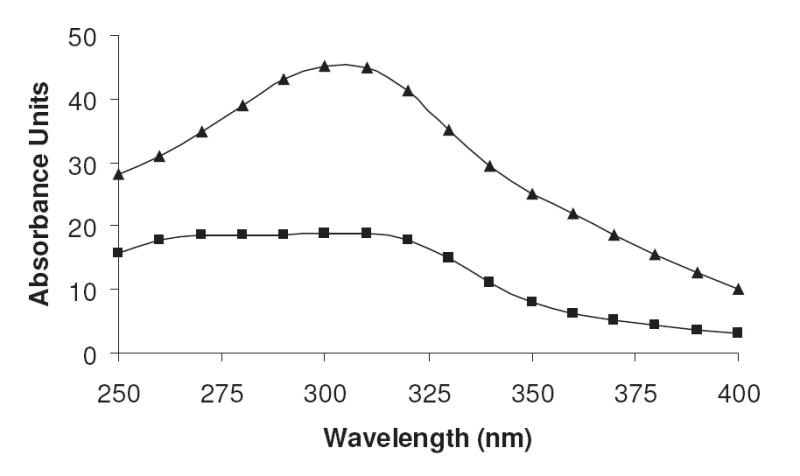
The comparative absorbance spectra of reactions of NAcLys (0.10 M) and R5P (0.050 M) (pH 7.4) performed under the presence (■) and absence (▲) of oxygen. The incubations were held at 37 °C for five days.
The absorbance maximum at 350 nm for dR5P in reaction with NAcLys (see Figure 5B) is interesting. The reaction of this sugar with some amines (e.g., β-Ala and L-Ala) exhibits this absorbance peak, while other amines (e.g., glycine and l-lysine (non-acetylated)) fail to produce significant amounts of the UV-absorbing compound. The difference may be in the ability of some amines to undergo additional rearrangement reactions between the amine group and the α-carbon (such as decarboxylation reactions of the α-carboxylate group21,22) not possible with other amines including NAcLys. This would thus reduce the transition of the intermediate to the 350-nm-absorbing compound. A four-day dR5P/β-Ala reaction performed in the absence of oxygen (data not shown) gave similar 350-nm absorbance to that performed in the presence of oxygen, but a decrease in the 280-nm absorbance was noted.
In a Maillard reaction with NAcLys and a phosphate catalyst, ribose yielded relatively high absorbance in the UV range with A280 rate increases of approximately three-fold less than the corresponding R5P system (see Figure 5). Analysis of the spectra generated by a ribose reaction indicates compounds with absorbance maxima of approximately 280 nm and 300 nm were generated. In a fashion similar to other pentoses, D-ribose undergoes the Maillard reaction to produce α-dicarbonyls23 and thus should also exhibit an absorbance peak at 310 nm. Hauck et al.10 found that a 1-deoxy 2,3-dicarbonyl compound (a potential dicarbonyl formed from ribose) converts through rearrangement to HMF. A relatively rapid conversion of the dicarbonyl to HMF would exhibit a lower 310-nm absorbance and a high 285-nm absorbance (of HMF). The presence of an absorbance maximum for the ribose system in the 280–290 nm region suggests that this may be occurring. (See also Characterization of Major Products by NMR and LC/MS.)
Comparatively, the deoxyribose/Pi/amine (NAcLys) reaction generated a small rate of UV absorbance increase (10-fold less than ribose/Pi/amine) with time over a 150-h incubation period at 37 °C. These data and our results in the visible range are in agreement with the study of Wondrak et al.12 which evaluated the browning (at 420 nm) of ribose vs. deoxyribose solutions in reaction with ω-amino acids. The spectrum at five days indicates the presence of a 260–280 nm absorbing compound along with a 310–330 nm absorbing compound.
2.6 Fluorescence
The general Maillard reaction has been reported to produce glycation products that exhibit fluorescence characteristics and R5P is no exception. As reported previously, a spontaneous reaction of R5P with amines or proteins rapidly produces a total AGEs fluorescence (360 nm excitation; 460 nm emission) (see Figure 7) at a faster rate than most other sugars such as glucose, glucose 6-phosphate, and fructose.2 While the presence of the phosphate bound onto the ribose nucleus appears to greatly speed up the original consumption of the sugar and the production of UV products, the functional group does not add significant advantage over intermolecular inorganic phosphate to the production of total AGE fluorophores (Figure 7). After a brief lag period, mixtures containing amines and d-ribose with equimolar soluble phosphate exhibit a higher fluorescence than R5P. (This greater fluorescence by ribose in comparison to R5P was reported in short-term experiments using bovine serum albumin at 55 °C, but the opposite was observed at 37 °C.2) Total AGE fluorescence with ribose was found to be directly correlated with phosphate concentration added to solution. Since the relatively lower fluorescence shown by R5P vs. ribose/Pi is not due to the original rate of amine attack or to the formation of the Amadori product (both which are more rapid for R5P than D-ribose/Pi), it is likely that R5P is incapable of generating highly fluorescent products until after phosphate hydrolysis.
Figure 7.
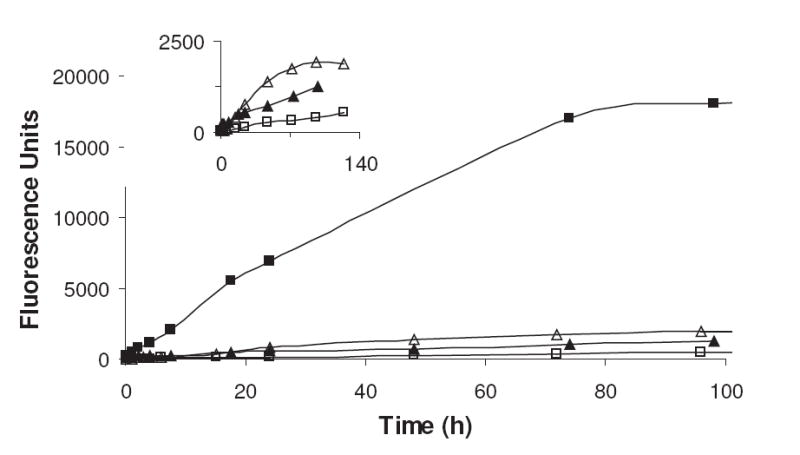
The Total AGEs fluorescence (emission = 360 nm; excitation = 460 nm) generated in a reaction between a pentose sugar and NAcLys at pH 7.5 and 37 °C. The sugars employed are R5P (▲), dR5P (■), ribose and Pi (△), and deoxyribose and Pi (□). Inset: An expanded view of R5P, ribose and Pi, and deoxyribose and Pi.
While R5P is relatively quick in comparison to other sugars to produce fluorescence products, dR5P in a Maillard reaction is a significantly greater fluorescence generator, exceeding the total AGE fluorescence capability of R5P by a factor of approximately fifteen (see Figure 7). When compared to a deoxyribose/Pi system, dR5P generates fluorescent compounds at rates nearly 60-fold higher. A portion of this increased rate vs. that of the deoxyribose/Pi system is likely attributed to the more rapid initial reaction between the sugar and the amine; however, a second contributor could be the presence of the phosphate in directing the molecule to an end-point fluorophore.
2.7 Characterization of Major Products by NMR and LC/MS
The Maillard reaction of amines with sugars is initiated by the attack of the amine nitrogen onto the carbonyl carbon to generate an N-glycoside (Schiff base), which subsequently has the opportunity to undergo rearrangement to form the Amadori product (see Scheme 1). The transition of the free amine and free sugar to the N-glycoside is a reversible process which, as stated previously, slightly favors the N-glycoside for the R5P/amine system. At neutral pH, where deprotonated amine concentrations are relatively low, the amount of N-glycoside in comparison to free sugar and free amine will be comparably low. An 1H NMR investigation of dR5P with β-Ala confirmed this process. Lacking a hydroxyl group on the C-2 carbon, dR5P cannot undergo conversion to the Amadori product via traditional Maillard rearrangement. The overall loss of dR5P from solution was relatively slow. However, as shown in Figure 8, 1H NMR spectroscopy indicates a relatively rapid loss of the resonance of the hydrogen signals of the C-2 carbon during the initial eight-hour period of the reaction (37 °C), a period where no significant loss of C-1 hydrogen intensity was noted. Clearly, the loss of the C-2 hydrogen resonance is due to exchange of these hydrogens with deuteriums of the D2O solvent. The exchange of C-2 hydrogens also occurs without amine12 but at a three-fold slower rate. Thus, either the amine is promoting the proton-to-deuterium exchange in a catalytic process (i.e., the amine is acting as a general base for removal of the proton) or, more likely, the exchange results from a transient binding of the dR5P with the amine and a subsequent temporary rearrangement of the double bond to expel a hydrogen (Scheme 5).12
Figure 8.
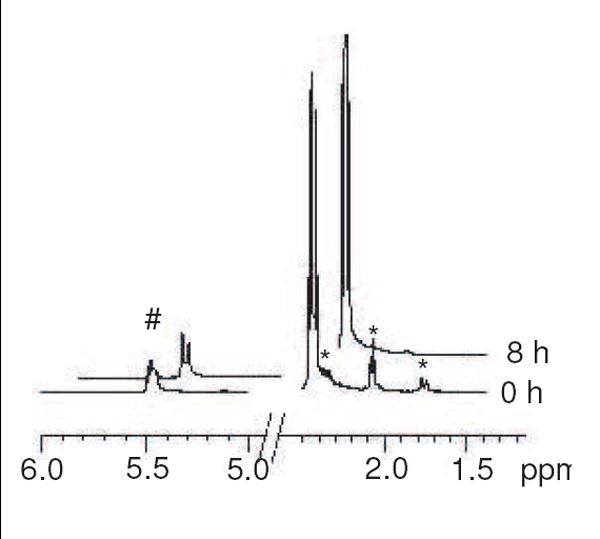
Portions of 1H NMR spectra of 0 h and 8 h reactions of dR5P with β-Ala (pH 7.5, 37 °C) performed in D2O. Peaks at +5.5 ppm [represented by #] indicate the C-1 hydrogen does not lose significant intensity over this time period while peaks at +1.75 ppm, +2.05 ppm, and +2.35 ppm (shoulder) [represented by *] indicate the C-2 hydrogen signals (three sets stemming from the two ring hydrogens and the α/β forms) disappear as exchange for solvent deuterium occurs. Note the splitting pattern for the C-1 hydrogens moves from a multiplet (actually overlapping triplets) to a pair of singlets (for α and β forms).
Scheme 5.
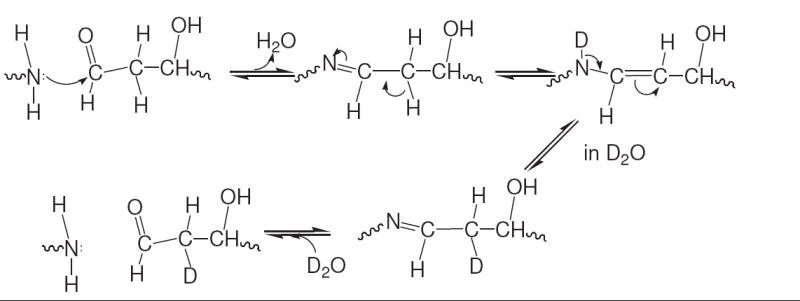
The reaction scheme for the exchange of hydrogen with solvent deuterium at the C-2 carbon of dR5P in a Maillard reaction with an amine. 1H NMR analysis shows full loss of C-2 hydrogens over a period of approximately 10 h at 37 °C, thus indicating the sequence can occur twice to replace both hydrogens with deuterium.
Being a dead-end product for dR5P, the enamine can only reverse in direction, ultimately releasing the free amine and generating the free acyclic sugar. The fact we detected insignificant short-term losses of the C-1 hydrogen (a chemical shift of the resonance would be expected to occur when the nitrogen is attached) while at the same time observing the loss of C-2 hydrogens agrees with our speculations that the equilibrium levels of the N-glycoside at neutral pH are small. Thus, we propose the bulk of the MS ion signal observed for our combination of N-glycoside/Amadori product (see above) represents the Amadori product.
LC/MS analysis of the reaction of D-ribose/Pi and NAcLys at pH 7.4 and 37 °C with extended times showed the development of a compound mass equal to 114 Da which eluted at a longer retention time of 3.6 min in comparison to the majority of reactants and other products. First noted of its 280–290 nm absorbance at 15 h, the compound grew in amount until 96 h, when its level reached a stable value. (At this point the compound accounted for approximately 20% of the overall 280–290 nm absorbance.) Comparison to a pure standard confirmed this product as HMF. This compound is believed to be formed by a reaction series (Scheme 6) involving a 1-deoxy-2,3-dicarbonyl intermediate derived via a traditional Maillard sequence and a subsequent rearrangement that includes cyclization and dehydration.10, 23 After an initial lag period of about 10 h, HMF is generated at a rate of approximately 5–10% of that of d-ribose consumed. Reaching a steady level of 0.35 mM at a point in the reaction sequence (at ~96 h) when less than 20% of the d-ribose has been consumed suggests HMF may be reacting to form a subsequent product (e.g., polymerization).
Scheme 6.
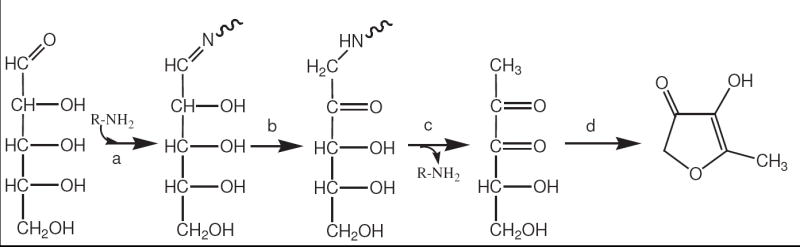
A proposed sequence of reactions between d-ribose and NAcLys leading to a m/z 115 [M + H+] product. The reaction is initiated (step a) by an attachment of the amine to form a Schiff base, followed by Amadori rearrangement (step b) and hydrolysis of the amine to yield a 1-deoxy-2,3-dicarbonyl compound (step c). The dicarbonyl compound can spontaneously rearrange to HMF by a previously reported mechanism.10
LC/MS characterization of the D-ribose/Pi/NAcLys reaction also clearly indicated the oxidative production of a compound with a molecular mass compatible with that of a CML product (actually in this case CM-NAcLys). The compound’s identity was confirmed by an MS2 fragmentation pattern that was in agreement (for the NAcLys version of the molecule) with that reported for CML by Teerlink et al.24 Unlike the formation of HMF, the synthesis of CML appeared to be initiated without any significant lag period. The ion signal for CML continued to rise in intensity throughout the 165 h of the reaction. Also observed was the production of a compound with an appropriate mass for a carboxyethyl (CEL) moiety onto the NAcLys. This occurred with a significant lag time of over 72 h, suggesting its formation was on a slower time scale than the CML product. We did not observe a furfural product (furfural itself or a Schiff base conjugate of furfural) observed of pentoses in Maillard reactions;23,25,26 however, previous studies indicate higher temperatures may be required for its formation.
LC/MS analysis of the reaction of R5P with NAcLys at pH 7.4 and 37 °C with extended time identified the development of several new products, many of which are awaiting further characterization. The appropriate m/z peak representing the production of an N-glycoside/Amadori product was observed immediately in the LC/MS spectrum, and the intensity of this signal rose rapidly to peak levels in a one-day period (for NAcLys and β-Ala). Within relatively short times (10 h), the production of a compound with mass = [R-NH2 + 58] was noted, which matched the molecular mass and fragmentation pattern of the CML product generated in the D-ribose/Pi/NAcLys system. The CML product was detected early in the reaction (at 7.5 h), and it continued to be formed at a linear rate throughout the 150 h of the experiment. Comparison of the intensity of the ion peak areas for this signal in the R5P vs D-ribose/Pi systems indicates CML is generated at approximately the same rate despite R5P loss occurring at a significantly faster pace. Again mirroring the comparative d-ribose reaction, a CEL product was also generated after a noted lag period (> 48 h). It is believed that the synthesis of a carboxymethyl moiety stemming from either ribose or R5P onto NAcLys was via an oxidative mechanism involving a C-2–C-3 enediol as proposed by Smith and Thornalley.14 Confirmation of the mechanism of synthesis of the CEL group awaits further analysis of a methylglyoxal intermediate.
A reaction of a R5P/NAcLys system performed in the absence of oxygen gave some signals observed in the aerobic system including the CML and CEL products. After a five-day incubation, the CML signal was substantially reduced (at only 15%) of that observed during a comparable R5P/NAcLys reaction performed in the presence of oxygen. Representing oxidized products of R5P, the presence of these compounds suggests the conditions of the incubation and analytical system had not fully excluded oxygen. While there may have been other products of minor ion intensity, the major MS signal novel to the oxygen-free reaction was compatible with a compound of a mass equivalent to [R-NH2 + 96]. The compound had an absorbance in the 320 nm range and little absorbance in the 280 nm region. As will be discussed below, analogous compounds were discovered in the dR5P/amine system (under aerobic conditions). The [R-NH2 + 96] compound represents the attachment of a C5H6O2 group onto the deprotonated form of the amine. One candidate for this compound is an imine derivative of HMF. This could result from the initial isomerization of R5P (promoted by the general base action of an amine) to ribulose 5-phosphate (Ru5P), which can undergo spontaneous dephosphorylation, a rearrangement to generate a furanone ring, and then an attack of the amine onto the ring carbonyl. We consider this reaction series unlikely, however, as we fail to see any evidence of HMF itself in a R5P-derived reaction. (Also, a HMF-containing reaction system failed to rapidly react with an amine to generate this compound.) We favor instead an imine conjugate of a triketone previously noted in Maillard reactions.27 The triketone is speculated to result from spontaneous dephosphorylation of a 2,3-α-dicarbonyl compound.
In the dR5P reaction with NAcLys, LC/MS analysis revealed the production of several new compounds, implying that while the loss of dR5P from solution is slow, the lack of the C-2 hydroxyl group does not fully impede some conversion of the molecule to future products. (A practically identical set of products was achieved in a reaction of dR5P with β-Ala; for clarity the masses given will be only for the NAcLys reaction.) This has been reported previously for solutions both with and without amine.12 The likely explanation for how the C-2 barrier is overcome is via a rearrangement of the double bond initially formed between C-1 and C-2 (Scheme 5) to generate an Amadori-like C-3 ketose (Scheme 7). This transition to the C-3 ketose can then, in the manner of R5P, lead to a variety of other reactions including dephosphorylation. Occurring in a manner similar to R5P (Scheme 4) by the establishment of the carbonyl at C-3, phosphate hydrolysis appears to be at a same rate as dR5P loss. Thus, the rate of phosphate cleavage is relatively spontaneous compared to the rate of transition of dR5P to the C-3 ketose form.
Scheme 7.
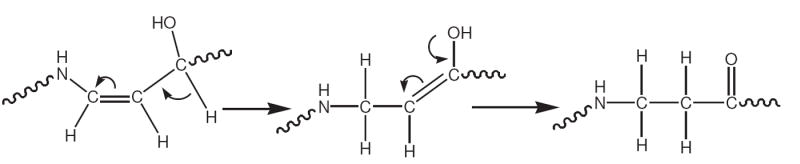
Tautomerization reaction of dR5P originating from an enamine (Scheme 5) leading to an Amadori-like compound.
Most obvious in the LC chromatogram of the dR5P/NAcLys system at extended times was the generation of two unique compounds with extended retention times (tR 4.4 and 6.8 min) on a reversed phase column (Figure 9). Both compounds have an m/z 267 [M + H+] which is compatible with a molecular formula represented by NAcLys attached through a double bond (or two single bonds in a cyclic form) to a C5H4O group. It is important to note that these products are oxidized in respect to the starting reactants. The wavelengths of maximum absorbance for the two compounds are 258 nm and 290 nm for the tR 4.4 min and tR 6.8 min peaks, respectively. The two compounds account for 52% of the 280 nm absorbance and 20% of the 320-nm absorbance, but none of the 350-nm and 420-nm absorbance.
Figure 9.
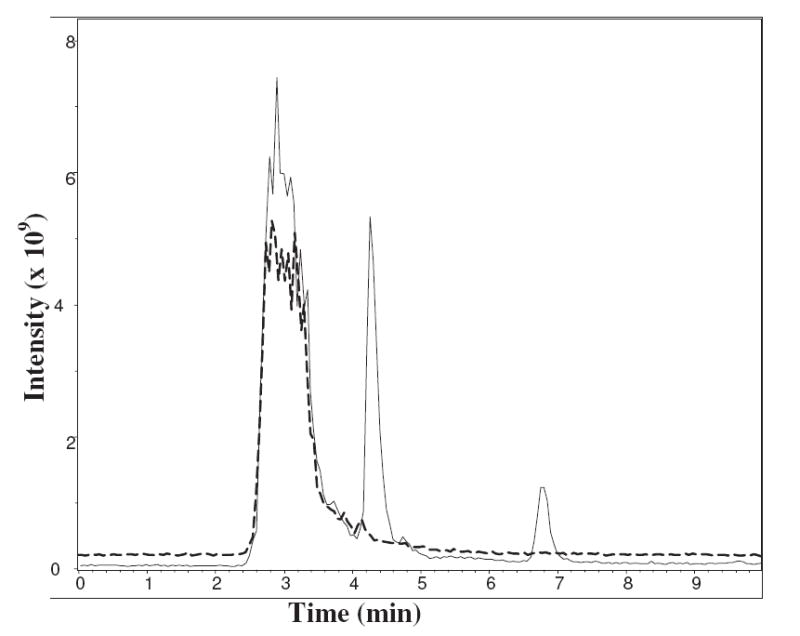
Total ion chromatograms of the seven day reactions (37 °C; pH 7.4) of NAcLys with R5P (– – –) and dR5P (–––). Separation was performed on a 4.6 × 150 mm C8 column using 80:20 H2O:2-PrOH with 0.5% HCO2H at 0.5 mL/min.
Employing β-Ala as the amine component, the two compounds were characterized by 1H NMR spectroscopy, and the structures (see Scheme 8) best fitting the data were an amine derivative of 5-methylene-2-pyrrolone [5-MP, 3-(2-methylene-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)propanoic acid, (tR 4.0 min), and a 2-formylpyrrole compound [3-(2-formyl-1H-pyrrol-1-yl)propanoic acid, (tR 6.0 min)]. Judging from proton spectra of an acetone-extracted solution, the two compounds were synthesized in approximately equal amounts. The reaction scheme in the generation of the compounds requires oxidation, dehydration, and phosphate cleavage steps. A compound similar in structure to the amine derivative of 5-MP is 5-methylene-2-furanone (5-MF). 5-MF, a marker of DNA oxidative damage upon irradiation, has been shown to be formed through an initial hydrogen abstraction from C-1 of a deoxynucleotide (in DNA) by oxygen leading to superoxide.28,29 A scheme for the generation of the amine form of 5-MP would not likely follow a similar path as for the formation of 5-MF29,30 as the nitrogen becomes incorporated in the ring rather than as a double-bonded atom at C-1. It is more likely that the amine ultimately attaches at a newly formed carbonyl at C-4, cyclizes, and then rearranges to the 5-MP form. The reaction mechanism is presently unclear. Another product of DNA oxidative damage (via H2O2 and chromium) is furfural.30 A scheme involving hydrogen abstraction at C-5 leading to furfural generation has been proposed by Mazzer et al.30 While we did not observe furfural in our β-Ala/amine reactions, the pyrrole compound with a longer retention time (at ~six min) is similar in structure to furfural. The mechanism for its formation is speculated to be via a series of reactions generating a pyrrole compound as described by Tressl et al. for deoxyribose.31 Somewhere in the reaction sequence an oxidation of C-5 would be required to generate the aldehydic group during phosphate cleavage; this may occur in an analogous manner as for furfural formation generated from DNA.30
Scheme 8.
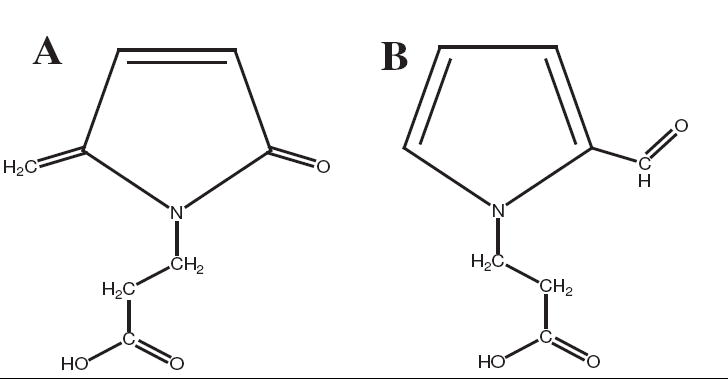
Structures of two products generated from a reaction at 37 °C and pH 7.4 between dR5P and β-Ala. A. Four-min retention-time compound, an amine derivative of 5-MP, with an absorbance maximum at 260 nm. B. Six-min retention-time compound, N-methyl-2-formylpyrrole, with an absorbance maximum at 290 nm.
While these compounds absorb in the 280-nm and 320-nm regions, their absorbance maxima are not at 350 nm, the unique wavelength observed in our UV–vis studies. Instead, a short tR compound (2.8 min) of an m/z 455 [M + H+] is compatible with a compound with a 350-nm wavelength maximum. Similar to the m/z 267 [M + H+] compound, this compound is an oxidized product of the starting dR5P and NAcLys reactants. The fragmentation pattern of the m/z 455 [M + H+] compound reveals a major peak at m/z 268, indicating a loss of the molecular mass (187 amu) of a NAcLys. While the identity of this compound needs further investigation, it is likely that the molecule involves two amine groups attached to a C5H6O moiety.
Additional related compounds observed with time exhibited LC/MS signals at m/z 625 [M + H+] and 689 [M + H+]. These likely correspond to an NAcLys attached to the m/z 455 compound and to an NAcLys bridging two m/z 267 compounds, respectively.
The reaction of dR5P with amine performed in the absence of oxygen for four days at 37 °C generated many of the same products seen with the reaction performed in the presence of oxygen. However, LC/MS analysis indicated significantly lower amounts of signal from the compounds of m/z 267 and 455 (see above) and instead an enhanced signal of compounds two hydrogens greater in mass. The lack of oxygen likely prevents the synthesis of the oxidized versions and instead the non-oxidized forms predominate.
The sugar deoxyribose reacts with an amine in the presence of a phosphate catalyst at extremely slow rates. Over a period of 150 h, no significant deoxyribose or NAcLys loss was detected by LC/MS analysis, and the only major new product observed was a compound consistent with a mass of an N-glycoside (m/z 305 [M + H+] for the NAcLys conjugate.) The intensity of the signal for this compound grew linearly over the length of the incubation, and it accounted for >90% of all new UV–vis absorbance generated. Given that the equilibrium level of N-glycoside is thought to be extremely low at neutral pH, the linear rate of formation of this compound implies its transformation into a secondary, similar mass product. The possibility of a conversion to a pseudo-Amadori product (a 3-ketose) in a similar fashion to dR5P is likely. The lack of significant loss of deoxyribose and NAcLys from solution along with the relatively small intensity of the extracted ion signal suggests the formation of this compound to be relatively small.
3. Discussion
Ribose sugars and sugar phosphates undergo traditional Maillard reactions leading to a diverse set of products. As is common with most Maillard processes, the initial reversible attack of amine onto the carbonyl group yielding an N-glycoside (either in a straight-chain form or in the presumably more stable cyclic form) is likely an equilibrium process connected to the concentration of the deprotonated form. Previous studies on the pentose phosphate pathway metabolite phosphoribosamine have determined an equilibrium constant (2.5 M-1 at 37 °C) that slightly favors the N-glycoside over R5P and ammonia.16 It is probable that other amines reacting with R5P exhibit N-glycosides of approximately the same stability, although steric factors make them to be slightly less favorable. In our attempts to establish equilibrium constants of the R5P N-glycosides of tris(hydroxymethyl)aminomethane (Tris) and NAcLys employing systems of high pH (~10.5), equilibrium constants equaling approximately 5–10 M-1 were achieved (data not shown). dR5P and non-phosphorylated ribose forms would be anticipated to have equilibrium constants of the same range. This implies very little N-glycoside exists at typical reaction concentrations at neutral pH and suggests any significant decrease in observed sugar concentration occurs because of a subsequent Amadori rearrangement step reported to be largely irreversible.14 The lack of observable amounts of deoxyribose or dR5P loss over extended times is consistent with small equilibrium levels of the N-glycoside and with the inability of these molecules to undergo a traditional Amadori rearrangement process.
The kinetics of the early Maillard sequence is complicated by the fact that free amine is consumed and then regenerated in the early portions of the reaction producing α-dicarbonyls. Here, treatment of the rates in a second-order manner for comparative purposes does not depict the true kinetics as shown most clearly by systems where sugars stoichiometrically in excess of amine concentrations continue to readily lose sugar concentrations beyond the expected 1:1 ratio. A closer investigation of the R5P/NAcLys system reveals additional kinetic insights. Making an assumption of comparatively negligible equilibrium levels of the N-glycoside at neutral pH and a recycling of the amine during its release from the Amadori product, kinetics modeling was performed using the reaction sequence of Scheme 2 to fit the pattern of the Amadori product profile (i.e., a maximum at approximately 40 h and a half maximum achieved at 110 h, see Figure 2) with the R5P and NAcLys data (see Figure 1 and kinetic data). The modeling resulted in a k = 0.26 M-1 h-1 and a k3 = 0.03 h-1. Application of our measured Keq = 7.6 M-1 for the deprotonated NAcLys/R5P/N-glycoside equilibrium system yields a k2 = 24.5 h-1 for the system at pH 7.4. This theoretical treatment of the data predicts that at its maximum, the Amadori product at 0.016 M comprises approximately 33% of the sugar and 17% of the NAcLys. This is in comparison to less than 0.3% of each existing as the N-glycoside. In opposition to the predictions of the modeling, the free amine level did not return to original concentrations upon full consumption of the sugar and subsequent movement of the molecules past the Amadori compound. The progressive loss of amine after the maximal level of Amadori product is achieved (see Figures 1 and 2) implies a subsequent amine-consuming reaction occurring post-Amadori rearrangement. It is likely that reaction with the α-dicarbonyl or formation of subsequent Maillard product(s) (like CML) in a stable form accounts for the long-term loss of amine from solution. At 120 h, approximately 12% of the NAcLys in the R5P-containing solution was lost to other compounds with an estimated forty percent of this contained as Amadori product that had yet to be converted to future Maillard compounds.
Treatment of the data in this manner with NAcLys being recycled explains to the discrepancy observed in the rate constants of R5P loss when increasing levels of R5P. When the kinetics are modeled employing the actual observed NAcLys concentrations (involving the recycling of the amine), the solutions containing different starting R5P levels yield rate constants that are more equivalent. (A small increase in rate constant with pKa was still observed.) When treated in this manner, rate constants in the range of 0.3–0.6 M-1 h-1 were obtained.
Despite inaccuracies in the true values, the R5P systems with different amino acids exhibiting k* values varying by more than an order of magnitude shows the early Maillard system to be more complicated than a simple attack of a deprotonated amine onto the sugar. Higher rates at lower pH for some amines indicate a protonation step may be involved, while some steric effects appear to be partially responsible for the differences in rates, e.g., β-alanine vs l-alanine.
The similar kinetics of the early Maillard reaction of amines with R5P in both the presence and absence of oxygen suggests a common pathway to and just beyond the Amadori rearrangement product. The observation of the production of CML in reactions performed in the presence of oxygen was expected as R5P is structurally similar to other sugars at the reducing end of the molecule. In contrast, the reaction of R5P in the absence of oxygen generated little CML but instead produced a compound compatible in mass with a triketopentane conjugate. (The lack of evidence of the formation of HMF suggests this is not a HMF conjugate.) Inorganic phosphate was cleaved from the sugar over time at appreciable rates in both the presence and absence of oxygen. It is unknown whether this is due to a Maillard-related reaction (for example, phosphate cleavage from an α-dicarbonyl) or to the isomerization of R5P to Ru5P, perhaps promoted by the amine in solution.
Previously reported to yield furfural products,25,26 the most widely characterized of the studied group, d-ribose, is believed to react in a traditional manner with amines, generating an Amadori rearrangement product, followed by α-dicarbonyls, and then to cyclization to the furfural/furfurone product. The relatively rapid rate of reaction (in comparison to common aldohexose sugars such as glucose) is speculated to be partially tied to the sugar’s higher level of acyclic form. The primary products observed during the reaction at neutral pH and 37 °C are believed to be an HMF product and CML
While the dR5P system lacks the C-2 hydroxyl group necessary for the transition of the N-glycoside to the traditional Amadori product, the molecule is capable of reacting in a Maillard fashion12 with some products ultimately undergoing hydrolysis of phosphate. Like R5P, the inclusion of a 5-phosphate moiety (which in some cases likely leads to a 5-methyl group) discourages the furfuryl alcohol and 2-(hydroxymethyl)pyrrole products observed for deoxyribose12,31 in favor of unique pyrrole monomeric and polymeric compounds. A pseudo-Amadori product is formed from dR5P with amines at relatively slow rates in comparison to R5P but at a fast pace compared to deoxyribose with Pi. The difference in rates of dR5P and deoxyribose/Pi clearly demonstrates the impact of an intramolecular phosphate group at C-5 on the speed of the reaction. As in the R5P system, this effect may be due to the phosphate causing an increased tendency towards an acyclic form or to a catalytic role it plays in rearrangement steps. The formation of the pseudo-Amadori product apparently is connected to the subsequent loss of the phosphate and ultimately the production of a pair of pyrrole isomers with absorbance in the UV range. The reaction is an oxidative process; the reaction run in the absence of oxygen instead generates a saturated form of this compound. As the O2-exposed reaction progresses, additional amine molecules link to this non-phosphorylated product, generating other UV–vis-absorbing compounds, including (for some amines) a unique 350-nm product. While the exact identity of this product(s) is unknown, the compound(s) bears similar UV–vis, fluorescence, and mass-building patterns to a 2-(2-furoyl)-4(5)-(2-furanyl)-1H-imidazole product formed by the condensation of glucoses with two Lys followed by oxidation and dehydration.32,33 Surprisingly, no carboxyethyl groups (−CH2CH2COOH) were detected as a result of this reaction. These hypothetically could be generated by the reaction of O2 with the dicarbonyl in the dR5P reaction series in a similar manner as it occurs with the Amadori product of any sugar/amine combination. The formation of this compound may indicate the importance of the amine being in a position such that a Schiff base ultimately lies in conjugation with an enediol upon a removal of a hydride by oxygen.
The unique reactivity of pentose and deoxypentose 5-phosphates before and after the hydrolysis of the intramolecular phosphate adds a new variation to a typical Maillard reactions scheme. Further research is needed to determine if these reactions have commercial applications or if they are of consequence physiologically.
4. Experimental
4.1 General methods
d-Ribose 5-phosphate (disodium salt), d-deoxyribose 5-phosphate (sodium salt) (2-deoxy-d-erythropentose 5-phosphate), d-ribose, d-deoxyribose (2-deoxy-d-erythropentose), N-acetyllysine, β-alanine, N-acetylarginine, 4-hydroxyl-5-methyl-3(2H)-furanone (HMF), D2O, and other L-amino acids and reagents were purchased of the highest purity from Sigma–Aldrich Chemical Co. Solvents for liquid chromatography/mass spectrometry (LC/MS) were purchased from Fisher Scientific Co. and were of >99.9% purity.
Unless stated otherwise, all reactions involved the preparation of solutions containing 0.10 M amine and 0.050 M sugar phosphate. For deoxyribose and ribose, a reaction of 0.10 M amine, 0.050 M sugar, and 0.050 M sodium dihydrogen phosphate were prepared. These solutions were brought quickly to the desired pH (usually either pH 7.4 or 8.0) and then incubated in a heating block at 37 °C. Incubations for periods extending to seven days were performed for some experiments. For extended-time experiments of greater than three days, the solutions were sterile-filtered prior to incubation to eliminate the possibility of microbial growth.
For reactions performed in the absence of oxygen, component solutions were brought to the proper pH, bubbled with nitrogen for several minutes, combined, further bubbled with nitrogen, and then sealed tightly. Samples were opened just prior to analysis; for studies requiring a number samples over a period of time, multiple sealed tubes were prepared.
4.2 Rate of sugar consumption
Loss of R5P and dR5P from solution was determined by 1H nuclear magnetic resonance (NMR) spectroscopy using 5-mm tubes and a 400 MHz Bruker Fourier-transform (FT) NMR spectrometer. Typically, each 1H experiment time point involved sixteen scans (scan range equaling 20 ppm), followed by an automatic baseline correction and integration of the proton peaks. All chemical shifts are relative to tetramethylsilane (TMS) hydrogens. The concentrations of R5P or dR5P were determined by comparing the integration of the combined peaks of the α and β hydrogens of the anomeric carbon (at 5.2–5.5 ppm for R5P (two peaks) and at 5.5 ppm for dR5P (one combined peak for both the α and β form) versus either an MeOH hydrogen peak at approximately δ 3.4 or an EtOH hydrogen triplet at approximately δ 1.2. (MeOHG and EtOH are contained in small amounts in the R5P and dR5P purchased from Sigma Chemical Co.) The integration ratio of the initial sample was set as equivalent to the initial R5P or dR5P concentration (usually 0.05 M). This ratio was verified by measuring this integration ratio on the appropriate R5P or dR5P solution that lacked the amine. Measurement of the R5P in solution by this manner was performed until all R5P was consumed or for seven days. The results of the R5P analysis method was shown to be in good agreement with results obtained via an LC–MS technique of Huck et al.13 which monitored the intensity of the ion signal of the m/z 97 collision fragment of the m/z 229 signal of the MS operating in a negative mode. The dR5P results were in good agreement with LC/MS experiments (see below) using an extracted ion method (combined area of the m/z 215, 237, and 259 peaks).
Due to complications of the water peak and multiple sugar forms (pyranose and furanose) in the NMR analysis, the determination of the loss of deoxyribose and ribose from solution was performed by an extracted-ion method using an electrospray injection (ESI) Agilent Ion Trap LC–MS. The combined areas of the extracted ion chromatogram (range = m/z ±0.5) of the [M + H+], [M + Na+], and [M − H+ + 2 Na+] signals were measured for each sugar. This corresponds to the areas of the m/z 135, 157, and 179 signals for deoxyribose and the areas of the m/z 151, 173, and 195 signals for ribose.
Calculations of the kinetics of the sugar loss were performed using a self-designed modeling program employing Microsoft Excel® software. The second-order rate constant was determined by fitting the actual disappearance of the sugar with the best fit relationship using the rate equation
where the sugar and amine concentrations were the actual concentrations prepared. While serving as a good estimate of rate constants for comparative purposes, the rate equation employed assumes a stoichiometric loss of amine with the loss of the sugar, an assumption shown by us and others not to be correct. (The amine apparently is regenerated during the loss of sugar (see Section 3).) Adjusted rate constants (k*) corresponding to the actual deprotonated amine concentrations were determined via the above equation employing deprotonated amine concentrations as calculated using the amine’s pKa. The pKa for all amines except Gly were determined at 37 °C by titration of the amine (0.10 M) with 1.0 M NaOH.
4.3 Rate of amine consumption and rate of N-glycoside/Amadori product formation
The loss of NAcLys from solution was determined by an extracted-ion method measuring the combined MS ion areas (range = m/z ±0.5) of the m/z 189, 211, and 233 signals. (Similar results of other amines (not shown) were performed using the appropriate masses of the mono-protonated and mono- and di-sodiated amine.) The peak area of the extracted ion peaks for the amine at 0 h was set as the initial concentration of the amine (typically 0.10 M). N-glycoside/Amadori product formation was determined in a similar manner by extracting the ion signals corresponding to the masses of the mono-protonated and mono- and di-sodiated forms of the sugar–amine conjugate (mass = sugar plus amine minus water). No direct translation of these values to the corresponding concentrations could be made since pure standards were not available. Comparative concentrations over time are represented by the total MS peak areas of these extracted ions.
In some cases lysine and arginine modifications were assessed by a fluorescamine method34 and a 9,10-phenanthrenequinone method,34 respectively. Measurements were performed with excitation/emission wavelengths (nm) of 380/485 (for Lys modification) and 310/400 (for Arg modification). (Filters employed had 20–40 nm of bandwidth to cover the optimum excitation and emission wavelengths.)
4.4 Hydrolysis of phosphate and formation of new phosphate compounds
The cleavage of inorganic phosphate (Pi) from sugar phosphates was monitored by 31P NMR. All solutions were prepared in D2O rather than H2O at the appropriate concentrations. FT 31P NMR was performed at a frequency of 161.9 MHz with a scan range of 100 ppm and a relaxation delay of 0.25 s. Typically, 200–500 scans were performed for each analysis. The amount of phosphate hydrolyzed was determined by comparison of the integration of the Pi peak at approximately +3.4 ppm (vs. a H3PO4 reference) versus the integration of the total phosphate signals in solution. The phosphate peaks for R5P and dR5P were located at approximately +4.8 ppm. With time, phosphate signals typically developed that either overlapped the R5P or dR5P or were downfield from the R5P or dR5P peak. Our experience with phosphoribosamine (a reaction of R5P with ammonia) suggests the phosphate signals of N-glycosides overlap typically as a downfield shoulder to the R5P peak. It is speculated that Amadori and other rearrangement products (e.g., dicarbonyl phosphates) have chemical shifts slightly downfield from the R5P phosphate signal (i.e., at approximately +5.3 ppm). The signal for ribulose 5-phosphate (Ru5P), a ketose isomer of R5P, is reported to be 0.4 ppm downfield from the R5P signal.18
4.5 Absorbance and fluorescence measurements
All ultraviolet/visible (UV–vis) absorbance spectra, single-wavelength measurements, and fluorescence measurements were performed with a BioTek Synergy HT Microplate Reader using 200-uL samples. For UV–vis characterization, samples were read without dilution unless measurements greater than 2.0 A were obtained, whereupon diluted samples were prepared and measured. All absorbances are adjusted appropriately for any dilution and for a factor that corrects for the pathlength to yield values in the commonly reported cm-1 units.
Fluorescence measurements were performed using black microwell plates, 100 uL of sample combined with 100 uL of water, and a sensitivity of 50. Total Advanced Glycation End-Products (AGEs) fluorescence employed an excitation wavelength of 360 nm (±20 nm) filter with an emission wavelength filter of 460 nm (±20 nm). The values given are not corrected for dilution or for sensitivity.
4.6 Characterization of products by LC–MS and NMR spectroscopy
All LC–MS chromatograms and spectra were obtained using an Agilent Series 1100 LC/MSD Trap XCT Plus Liquid Chromatogram–Mass Spectrometer with electrospray injection. Samples (5–25 μL) were injected, and LC separation of the compounds was performed using a 4.6 × 150 mm C8 Zorbex reversed phase column and an 80:20 H2O–2-PrOH mobile phase (containing 0.5% HCO2H). A photodiode array detector capable of recording full spectra monitored the chromatographic flow. ESI injection conditions employed a drying temperature of 365 °C, a nebulizer pressure of 50 psi, and a dry gas flow of 9 L/min. Typically, a MS experiment was performed in positive mode over a scan range of m/z 50–2200 and a target mass of 250 Da. Use of an automatic MSn mode allowed for fragmentation of prominent MS signals. In some cases (such as for the analysis of R5P loss), a negative-ion mode MS and/or a manual mode was employed for supplemental information. The instrument has the capability of generating chromatograms via an ion-extraction method. This proved useful in identifying unique signals and matching the MS results with the UV–vis results of the LC–MS.
HMF was identified by a combination of 1H NMR and LC/MS. 1H NMR analysis was performed on reaction samples in D2O. Standard solutions (0.0–1.0 mM) of HMF were prepared in water, and 5 μL was injected into the reversed phase column LC–MS system with conditions listed previously. Analysis: UV–vis: λmax 285 nm;35 1H NMR (D2O): δ4.55 (s, 2H), δ2.2 (s, 3H);35 MS: m/z 115 [M + H+] (tR 3.6) with fragmentations of 88 (55) and 87 (100). Compounds in the Maillard reactions which yielded a match of retention time, absorbance characteristics, m/z and fragmentation spectrum, and 1H NMR were identified as HMF. The amount of HMF in the Maillard reaction solution was quantified by a standard relationship generated for A280 peak area vs HMF concentration. (A second method employing the standard relationship between the peak area of the m/z 168 signal vs HMF concentration was not needed for this study as few interfering compounds with UV absorbance exited the reversed phase column at tR 3.6 min.)
The amine form of 5-MP generated by the dR5P/β-Ala (or dR5P/NAcLys) reaction was likewise identified by LC–MS and 1H NMR spectroscopy. (The β-Ala results are presented here.) 1H NMR analysis was performed on samples that were collected at 4.0 min, acidified, extracted with EtOAc, dried and resuspended in CD2Cl2. Analysis: UV–vis: λmax 258 nm; 1H NMR (CD2Cl2): δ 2.7 (t, 2 H), 3.95 (t, 2 H), 4.95 (s, 1 H), 5.05 (s, 1 H), 6.25 (d, 1 H), and 7.1 (d, 1H); MS: m/z 168 [M + H+] (tR 4.0 min) with fragmentations of m/z 150 (100) and 108 (5). COSY NMR analysis indicated the δ 2.7 and 3.95 peaks to be strongly coupled, as were also the δ 6.25 and 7.1 signals. Use of a 15N-labeled amine resulted in a relatively small 15N-1H (ring hydrogens) coupling value (~ 3ppm) suggesting neither hydrogen is directly adjacent to the ring nitrogen.
The 2-formylpyrrole deriviative was characterized on the product of dR5P and β-Ala. 1H NMR analysis was performed on samples that were collected at 6.3 min, acidified, extracted with EtOAc, dried and resuspended in D2O. Analysis: UV–vis: λmax 292 nm; 1H NMR (D2O): δ 2.8 (t, 2 H), 4.1 (t, 2 H; hidden by the water peak but visible in COSY spectrum), 6.18 (m, 1 H), 7.04 (m, 1 H), 7.13 (broad s, 1 H), and 9.35 (s, 1 H, observed in the COSY spectrum); MS: m/z 168 [M + H+] (tR 6.3 min) with fragmentations of m/z 150 (9), 140 (100), 122 (9), and 88 (2). Coupling was observed by COSY analysis to occur between the δ 2.8 and 4.1 signals, between the δ 7.04 and 6.18 signals, and between the δ 6.18 and 7.13 signals. In comparison, the chemical shifts for the aromatic and formyl hydrogens in N-methyl-2-formylpyrrole were reported to be δ 6.19 (m, 1 H), 6.86 (m, 1H), 6.90 (m, 1 H), and 9.53 (d, 1H).31
Acknowledgments
This study was supported by a grant from the Vermont Genetics Network, a five-year IDeA Networks of Biomedical Research Excellence (INBRE) project sponsored by the National Institutes of Health. The LC–MS instrument was funded by a grant from the Major Research Instrumentation program of the National Science Foundation. The authors would like to thank Magdalena Bokiej, Andrew Livermore, and Professor Jeff Byers of Middlebury College for their contributions to this investigation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sandwick R, Johanson M, Breuer E. Ann N Y Acad Sci. 2005;1043:85–96. doi: 10.1196/annals.1333.011. [DOI] [PubMed] [Google Scholar]
- 2.Sharma SD, Pandey BN, Mishra KP, Sivakami S. J Biochem Mol Biol Biophys. 2002;6:233–242. doi: 10.1080/10258140290031554. [DOI] [PubMed] [Google Scholar]
- 3.Farmer LJ, Hagan TDJ, Paraskevaws O. Role of Selected Precursors in Meat Flavor Formation. In: Xiong Y, Ho C-T, Shahidi F, editors. Quality Attributes of Muscle Foods. Kluwer Acad./Plenum Publisher; New York: 1999. pp. 159–172. [Google Scholar]
- 4.Potman RP, van Wijk TA. ACS Symp Ser. Vol. 409. American Chemical Society; Washington: 1989. pp. 182–195. [Google Scholar]
- 5.Rizzi GP. J Agric Food Chem. 2004;52:953–957. doi: 10.1021/jf030691t. [DOI] [PubMed] [Google Scholar]
- 6.Serianni AS, Pierce J, Barker R. Biochemistry. 1979;18:1192–1199. doi: 10.1021/bi00574a012. [DOI] [PubMed] [Google Scholar]
- 7.Hayward LD, Angyal SJ. Carbohydr Res. 1977;53:13–20. [Google Scholar]
- 8.Davies CGA, Kaanane A, Labuza TP, Moscowitz AJ, Guillaume F. Evaluation of the Acyclic State and the Effect of Solvent Type on Mutarotation Kinetics and on Maillard Browning Rate of Glucose and Fructose. In: O’Brien J, Nursten HE, Crabbe MJC, Ames JM, editors. The Maillard Reaction in Foods and Medicine. The Royal Society of Chemistry; London: 1998. pp. 166–171. [Google Scholar]
- 9.Bjergegaard C, Lauridsen L, Mortensen K, Moller P, Sorensen H, Sorensen JC. Abstracts of Papers. Euro Food Chem XIII Symposium; Hamburg, Germany: 2005. Abstr. No. Xxxx. [Google Scholar]
- 10.Hauck T, Hubner Y, Bruhlmann F, Schwab W. Biochim Biophys Acta. 2003;1623:109–119. doi: 10.1016/j.bbagen.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 11.Jiang Z-Y, Woollard ACS, Wolff SP. FEBS Lett. 1990;268:69–71. doi: 10.1016/0014-5793(90)80974-n. [DOI] [PubMed] [Google Scholar]
- 12.Wondrak GT, Tressl R, Rewicki D. J Agric Food Chem. 1997;45:321–327. doi: 10.1021/jf970657c. [DOI] [PubMed] [Google Scholar]
- 13.Huck JHJ, Struys EA, Verhoeven NM, Jakobs C, van der Knaap MS. Clin Chem. 2003;49:1375–1380. doi: 10.1373/49.8.1375. [DOI] [PubMed] [Google Scholar]
- 14.Smith PR, Thornalley PJ. Eur J Biochem. 1992;210:729–739. doi: 10.1111/j.1432-1033.1992.tb17474.x. [DOI] [PubMed] [Google Scholar]
- 15.Martins SIFS, van Boekel MAJS. Carbohydr Res. 2003;338:1665–1678. doi: 10.1016/s0008-6215(03)00174-5. [DOI] [PubMed] [Google Scholar]
- 16.Schendel FJ, Cheng YS, Otvos JD, Wehrli S, Stubbe J. Biochemistry. 1988;27:2614–2623. doi: 10.1021/bi00407a052. [DOI] [PubMed] [Google Scholar]
- 17.Dafforn A, Koshland DE. Biochem Biophys Res Commun. 1973;52:779–785. doi: 10.1016/0006-291x(73)91005-x. [DOI] [PubMed] [Google Scholar]
- 18.Robitaille P-ML, Robitaille PA, Brown GG, Jr, Brown GG. J Magn Reson. 1991;92:73–84. [Google Scholar]
- 19.Mottram DS, Wedzicha BL, Dodson AT. Nature. 2002;419:448–449. doi: 10.1038/419448a. [DOI] [PubMed] [Google Scholar]
- 20.Stadler RH, Blank I, Varga N, Robert F, Hau J, Guy PA, Robert M-C, Riediker S. Nature. 2002;419:449–450. doi: 10.1038/419449a. [DOI] [PubMed] [Google Scholar]
- 21.Schonberg A, Moubacher R, Mostafa A. J Chem Soc. 1948:176–182. doi: 10.1039/jr9480000176. [DOI] [PubMed] [Google Scholar]
- 22.Schonberg A, Moubasher R. Chem Rev. 1952;50:261–277. [Google Scholar]
- 23.Hofmann T. Carbohydr Res. 1998;313:203–213. [Google Scholar]
- 24.Treelink T, Barto R, Ten Brink HJ, Schalkwijk CG. Clin Chem. 2004;50:1222–1228. doi: 10.1373/clinchem.2004.031286. [DOI] [PubMed] [Google Scholar]
- 25.Cuzzoni MT, Stoppini G, Gazzani G, Mazza P. Food Chem Toxicol. 1988;26:815–822. doi: 10.1016/0278-6915(88)90020-8. [DOI] [PubMed] [Google Scholar]
- 26.Cuzzoni MT, Stoppini G, Gazzani G, Mazza P. Food Chem Toxicol. 1989;27:209–213. doi: 10.1016/0278-6915(89)90157-9. [DOI] [PubMed] [Google Scholar]
- 27.Nedvidek W, Ledl F, Fischer P. Z Lebensm-Unters-Forsch. 1992;194:222–228. [Google Scholar]
- 28.Tallman KA, Tronche C, Yoo DJ, Greenberg MM. Nucleic Acids Res. 1999;27:4903–4909. doi: 10.1093/nar/27.19.3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roginskaya M, Bernhard WA, Marion RT, Razskazovskiy Y. Radiat Res. 2005;163:85–89. doi: 10.1667/rr3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mazzer PA, Maurmann L, Bose RN. Inorg Biochem. 2007;101:44–55. doi: 10.1016/j.jinorgbio.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 31.Tressl R, Wondrak GT, Kruger R-P, Rewicki D. J Agric Food Chem. 1998;46:104–110. doi: 10.1021/jf970657c. [DOI] [PubMed] [Google Scholar]
- 32.Pongor S, Ulrich PC, Bencsath FA, Cerami A. Proc Natl Acad Sci USA. 1984;81:2684–2688. doi: 10.1073/pnas.81.9.2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Njoroge FG, Fernandes AA, Monnier VM. J Biol Chem. 1988;263:10646–10652. [PubMed] [Google Scholar]
- 34.Schmitt A, Schmitt J, Munch G, Gasic-Milencovic J. Anal Biochem. 2005;338:201–215. doi: 10.1016/j.ab.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 35.Ames JM, Bailey RG, Mann J. J Agric Food Chem. 1999;47:438–443. doi: 10.1021/jf980528b. [DOI] [PubMed] [Google Scholar]