

Abstract
Analysis of complex biological samples by MALDI-TOF mass spectrometry has been generally limited to the detection of low-mass protein (or protein fragment) peaks. We have extended the mass range of MALDI-TOF high-sensitivity detection by an order of magnitude through the combined optimization of instrument parameters, data processing, and sample preparation procedures for affinity capture. WCX, C3, and IMAC magnetic beads were determined to be complementary and most favorable for broad mass range protein profiling. Key instrument parameters for extending mass range included adjustment of the ADC offset and preamplifier filter values of the TOF detector. Data processing was improved by a combination of constant and quadratic down-sampling, preceded by exponential baseline subtraction, to increase sensitivity of signal peaks. This enhancement in broad mass range detection of protein signals will be of direct benefit in MS expression profiling studies requiring full linear range mass detection.
Keywords: mass spectrometry, proteomics, magnetic beads, affinity capture, broad mass range, enhanced sensitivity, protein profiling
Introduction
Proteomic pattern analysis by mass spectrometry (MS) is an established approach, especially for the identification of potential biomarkers for disease diagnosis.1,2 Within this approach, the landscape of sample preparation procedures, MS, and computational methods being employed is quite diverse. For MS, ionization methods available for generating protein patterns include surface-enhanced laser desorption/ionization (SELDI),3–5 matrix-assisted laser desorption/ionization (MAL-DI),6 and electrospray (ES).7 Of these, MALDI-TOF MS has emerged as the preferred strategy for MS detection of intact proteins. MALDI-TOF in linear mode has a small mass preference due to the ionization efficiency of intact protein molecules and the focusing of flight of ions toward the detector. Many applications would benefit from increased mass linear detection. For biofluids, MALDI-TOF profiling is often preceded by fractionation of serum or plasma proteins using various techniques including chemical affinity magnetic beads.6,8–12 These prefractionation and MS analysis steps can be automated for high reproducibility and throughput, while consuming small amounts of sample.6,9,10 Despite the advancements in MS-derived protein patterns, interpretation is complicated by difficulties in differentiating biological variation (in vivo) from analytical variation (in vitro). Thus, there is critical need for technical improvements in all steps of sample preparation, MS data collection, and data processing.
To date, virtually all biomarkers reported from SELDI- or MALDI-TOF MS analysis of biofluids are small proteins or peptides with masses less than 20 kDa.8,11–13 This is mainly a factor of the limited sensitivity of TOF analyzers for masses above 20 kDa. To address the need for advancements in MS profiling, our objective is to extend the mass range for protein analysis beyond the customary 20 000 m/z limit for typical state-of-the-art linear TOF–MS instrumentation. We report on the optimization of procedures for broad mass range coverage in high-throughput experiments using derivatized magnetic beads for robotic sample extraction and Ultraflex MALDI-TOF analysis. Furthermore, we optimized detector ADC settings and applied down-sampling techniques for enhancing sensitivity of high masses.
Experimental Procedures
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless noted otherwise. Protein standards, α-cyano-4-hydroxycinnamic acid (CHCA), sinapinic acid (SA), 2,5-dihy-droxybenzoic acid (DHB), and CLINPROT magnetic beads (MB) kits (C3, C8, WCX, WAX, LAC Con A, IMAC-Fe) were obtained from Bruker Daltonik (Leipzig, Germany). Overall sensitivity enhancement was measured by comparing detection SNR (signal-to-noise ratio) of signals after preprocessing with default versus optimized instrumental settings for the same amount of samples.
Serum Collection
Serum samples were obtained from the Virginia Prostate Center Tissue and Body Fluid Bank. Serum samples were collected from consented patients under a study protocol approved by the Eastern Virginia Medical School Institutional Review Board. Briefly, after obtaining informed consent from a patient, a blood sample was collected into a 10-cc Serum Separator Vacutainer Tube and centrifuged 30 min later at 3000 rpm for 5 min. The serum was distributed into 0.5 mL aliquots and stored frozen at −80 °C. A quality control (QC) sample was prepared by pooling serum from a controlled normal group as previously described.14 The QC sample was used to determine reproducibility and gauge high mass sensitivity in MALDI-TOF analysis.
Sample Processing
A protein standard (PS) consisting of 7 proteins was made by mixing equal volumes of Bruker’s MALDI Mass Calibration Protein Standards I and II. Purification and fractionation of serum samples were performed with CLIN-PROT magnetic beads. Bead functionalities of C3, C8, WCX (weak cation exchange), WAX (weak anion exchange), IMAC-Fe (immobilized metal-ion affinity chromatography), and Con A (Concanavalin A) were utilized, and the samples were purified on a ClinProt robotic platform (Bruker Daltonik) as per manufacturer’s instructions. The only deviation was a 20 μL starting material for serum with subsequent reagent volumes adjusted accordingly. For optimization of robotic preparation, different matrices (SA, DHB, and CHCA), dilutions (1:2, 1:5, and 1:10), and plates (stainless steel or AnchorChip) were evaluated based on reproducibility and higher SNR of resulting spectra. Determination of the optimized matrix, dilution, and surface is discussed below. Samples were purified using three steps: binding, washing, and elution. Unbound proteins were discarded, and bound proteins were eluted and mixed 1:5 with α-cyano-4-hydroxy-cinnamic acid (CHCA) in an acetone/ ethanol mixture (1:2). Samples were spotted robotically by depositing 0.8 μL of eluate onto an AnchorChip target (Bruker Daltonik, 600 μm diameter anchors). A serum sample was also fractionated on a SELDI-IMAC3 ProteinChip (Ciphergen Bio-systems, Fremont, CA) as previously described.15 The protein chip array was then analyzed using the SELDI ProteinChip System (PBS-II, Ciphergen) as described elsewhere,14 and also spotted onto an AnchorChip and analyzed by MALDI-TOF MS. For each sample, 5–10 replicate spots were analyzed.
MALDI-TOF MS Analysis
Mass spectra were acquired in linear positive ion mode in the m/z range of 2000–100 000 using an Ultraflex III MALDI-TOF/TOF mass spectrometer controlled by FlexControl software v. 2.0 (Bruker Daltonik). The instrument was equipped with a smartbeam laser (Bruker Daltonik), and acquisition laser power was optimized using the PS calibration mixture before the collection of sample data. Default operating conditions were as follows: ion source 1, 25.0 kV; ion source 2, 23.7 kV; lens voltage, 6.0 kV; pulsed ion extraction time, 200 ns; gain factor, 24×; matrix suppression mass cutoff, 1500; ADC offset, 50; preamplifier filter bandwidth, high; digitizer sampling frequency, 500 MHz. Instrument settings optimized for broad mass range include the following changes from default: ion source 2, 23.0; ADC offset, 40; preamplifier filter bandwidth, medium. All acquisitions were generated automatically in the instrument software and based on averaging 1000 shots from 10 non-overlapping positions (100 shots/position). Data was calibrated externally with the PS calibration mixture consisting of masses in the range of 5734–66431 Da.
Data Processing
For baseline subtraction, the baseline trend was modeled as a decaying exponential (starting after the mass deflection region) plus a constant offset: A exp(−t/B) + C. The model coefficients for the data shown in this paper were (A, B, C) ) = (1.8 × 104, 9.5 × 103, 24.83 × 103) for IMAC bead spectra, (1.3 × 103, 5.3 × 103, 2.25 × 103) for SELDI IMAC chip, (1.8 × 104, 1.1 × 104, 24.9 × 103) for Con A beads, (4.6 ×103, 1.0 ×104, 24.5 × 103) for WAX beads, (9.7 × 103, 1.12 × 104, 24.6 × 103) for WCX beads, (9.5 × 103, 1.25 × 104, 24.5 × 103) for C8 beads, and (4.2 × 104, 8 × 103, 25.1 × 103) for C3 beads. The constant offset coefficient, C, was calculated as a mean of the late TOF portion of the spectra that did not contain peaks. The exponential decay, B, and amplitude, A, were obtained by a linear least-squares fit to the natural log of the data without signals in the early TOF region after subtraction of the constant offset.
To enhance sensitivity over the full length of the TOF record, constant-width integrative down-sampling of the signal was conducted, followed by quadratic down-sampling as described previously.16 Integrative quadratic down-sampling16 is an automated procedure that down-samples the data with a time step, Tr, according to the quadratic dependence on time: Tr ) C1t2 + C2t + C3, and assigns an integrated intensity of the original time sample to a new time sample in the middle of the down-sampling interval. For even intervals, half-intensity of the edge points is added to the new neighboring samples. The quadratic dependence of the down-sampling step on time follows the same trend as the experimentally observed TOF peak width. The model coefficients were obtained by measuring the half-width of peaks in the original data as a function of TOF and by performing last-squares fit to a quadratic function. For the spectra shown in this work, (C1, C2, C3) = (3.96 × 10−8, −0.25× 10−3, 27.26) for Bruker Ultraflex spectra and (1.27 × 10−7, −1.97 × 10−3, 31.15) for the PBS II spectrum.
TOF signals for heavy masses (above the optimal mass focusing region) correspond to broader peaks due to the approximately linear dependence of peak width on mass (quadratic in time17). This property of TOF detection leads to quadratic over-sampling (higher point density per peak) of late TOF signals compared to those before the optimum, which have approximately constant peak width in time.16 Under the acquisition and mass focusing conditions used in this work, the TOF optimum was below 40 μs (~6.5 kDa). To achieve the broadest mass coverage with the highest possible sampling rate, we used a 500 MHz sampling frequency in 2–100 kDa range. The half-width of peaks in the optimization range (m/z 2000–6500) was about 20 time steps (40 ns). Half-widths were constant in time although growing in m/z.18
The model wavelet describing the signal for an Ultraflex instrument was asymmetric Gaussian,16 with a right skew of 5 time steps, and producing a full peak width at half-maximum (fwhm) of 45 time steps. In the case of noiseless signal, three time points within the peak shape were sufficient to completely describe the signal wavelet model parameters (maximum, half-width, and skew). With the addition of experimental noise, 8–10 time points per fwhm of a peak should be adequate. Our experimental data provided five times this point density. This allowed performing constant-width integrative down-sampling of rate 5 for the original signal, prior to quadratic down-sampling as previously described.16
The described preprocessing procedure is generally applicable to data produced by different TOF instruments employing flash ADC in their detector circuit. Quadratic down-sampling has been previously demonstrated for the PBS series16 and is extended with constant down-sampling for the Ultraflex in this work. The constant down-sampling allowed about a 5-fold gain in preprocessing speed and an additional 3-fold enhancement of SNR. The Matlab scripts for integrative down-sampling are available for academic users upon request from the authors.
Results and Discussion
Optimization of MS Instrument Parameters for Broad Mass Range
The mass range of high-sensitivity detection for TOF instruments in linear mode is generally in the 2–20 kDa range.5 In essence, mass spectrometric profiling from complex biological samples has been limited to the analysis of peptides, low-mass proteins, or protein fragments. This limitation is due to lower desorption and m/z analysis efficiency at higher mass, and is exacerbated by suboptimal sample preparation, data acquisition, and data processing procedures. For data acquisition, we have enhanced detection sensitivity in an extended mass range up to 100 kDa for Bruker MALDI-TOF/TOF mass spectrometers by optimization of specific instrument parameters
A time-of-flight tube is the most common analyzer geometry used for MS protein profiling due to the high reproducibility and mass accuracy of resulting spectral data sets. There are numerous parameters for manipulating ion flights through a TOF system, and operators usually adjust parameters to collect data in either the low-mass or high-mass range. For instance, higher laser intensity and longer time-lag focusing (delayed extraction)19,20 are needed to enhance detection at heavy mass. The broader m/z focusing requires a compromise with optimal instrumental resolution.21 However, for example, for an Ultra-flex TOF instrument, a 2-fold compromise in resolution helps achieve more than a 20-fold gain in sensitivity (detector SNR) for masses above 8 kDa.22 Furthermore, lower mass focusing voltage helps to preserve peak width over the broader mass range, although at the expense of the lower “optimal” resolution. For instance, at the default setting of ion source 2 voltage (see Experimental Procedures) the optimal resolution of 850 is achieved at about m/z 5000, but there is a fast increase in the signal width outside mass focusing range (e.g., fwhm doubles at m/z 9000). With the slightly lower setting of this voltage that we used to optimize the broad mass range acquisition, the optimal resolution is lower (~500); however, the signal width changes slower (e.g., doubles at m/z 25 000). As a result, compared to default focusing voltage, the peaks are broader for lower masses (below m/z 10 000), nominally the same width between m/z 20 000 and 50 000 (Figure 1 range), and narrower for heavy masses. This is a compromise setting, which does not have a major impact on overall sensitivity for signals below 50 000 m/z, although the heavy mass signals may exhibit up to 2-fold enhancement of SNR per point due to partial narrowing.
Figure 1.
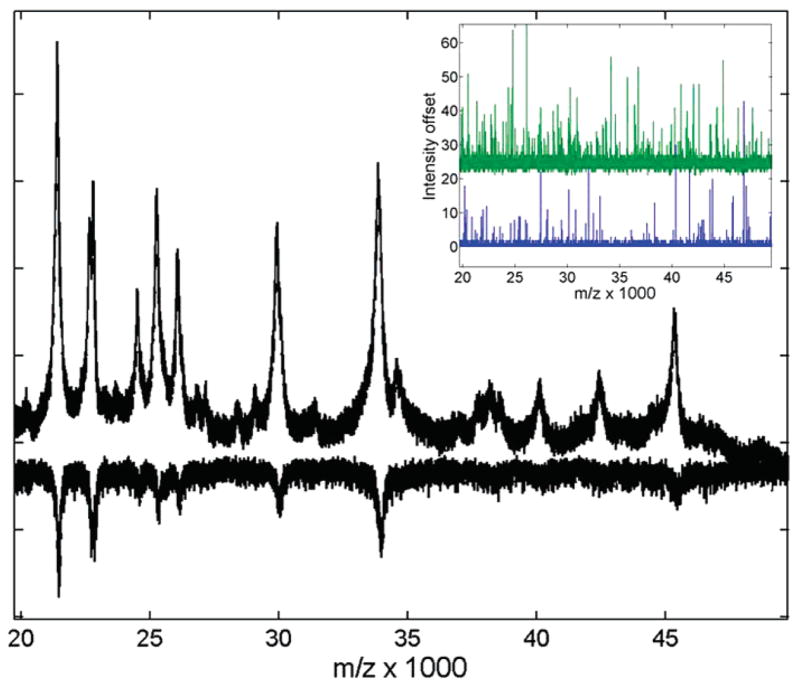
Average of 5 protein standard spectra acquired with optimized and default (inverted) ADC offset parameters. The raw spectra are scaled to the same noise level to illustrate improvement in detection sensitivity (SNR) for low-intensity signals. Inset shows single shot records in the same m/z range with optimized (top trace, 40%) and default (bottom trace, 50%) ADC offset settings. The upward one-point-wide spikes are due to dark current electronic noise. Note the clipping of “negative” bit noise in the lower (default) trace, and its recovery in the upper (optimized) trace. A 10% change in the ADC offset causes a constant (vertical) intensity offset centered at 24 (~10% of the full 8-bit range of 256).
More critical improvement of detection sensitivity can be achieved in the broad mass range by changing the default settings for the ADC offset and preamplifier bandwidth. A MALDI-TOF mass spectrum is usually the summation of several hundred laser shots taken at one or more positions from a sample spot. The dynamic range of intensities that can be detected by a TOF instrument after a single laser shot is limited by the detector ADC precision (8 bits in our study, Acquiris DP240). The default offset is typically set at a half of the ADC scale, which effectively uses all 8 bits of ADC range (256 intensity values) for detection of positive MS signals. If no signal is present, only the positive half of bit-noise is collected (typically, the 2 lower bits are lost, Figure 1 inset). However, due to intrinsic fluctuations of the ADC base level, low-intensity signals may fall below the detection cutoff for some of the laser shots, especially when superimposed on negative noise signals. As a result, in the average spectrum of multiple shots, these low signals get preferentially clipped at their base under such settings. This is especially detrimental to the detection of naturally broad signals from overlapping peaks or intermediate and heavy masses. Figure 1 shows raw spectra of the protein standard acquired under default and optimized conditions for the same m/z range. Note, for example, how in Figure 1 the shoulder peak and triplet between 35 and 40 kDa get completely lost in the noise with the default ADC offset (inverted spectrum). Changing the offset to about 40% of the scale value helps recover “negative” bits of the baseline noise and minimizes peak base clipping for low-intensity signals (Figure 1 inset). The recovery of “negative” bits happens abruptly at any offset value lower than 49%, since negative noise is about 1% of the full ADC range for a single shot (Figure 1 inset). However, due to (vertical) offset fluctuations, some residual clipping may still be sporadically observed for the single shots even at 44–46% offset (data not shown). Any setting below 40% avoids clipping completely. However, lowering the offset by 10% from default value causes a constant vertical shift in baseline intensity (Figure 1 inset) by about 10% of the dynamic range (~24 of 256). Thus, especially large signals may get clipped on the top for some single shots, causing signal misshaping. This was not observed in the studied samples. Hence, the compromise value of 40% was optimal to allow the broadest dynamic range of ADC (~225 for a single shot) without substantial clipping of signal base.
Adjustment of the preamplifier filter bandwidth is another hardware setting that can enhance the sensitivity of low-intensity signals. Without this filter, the “flash” Ultraflex ADC (Acqiris DP240) samples the signals with a defined frequency without integration of the signals between sample points. This leads to lost signal per sample point for lower sampling rates. Because of limitations on the memory buffer length, data collection in a broad mass range requires using lower sampling rates. Switching on the preamplifier filter allows integration of the signal within the bandwidth and, thus, recovery of lost sensitivity per sample point. However, if the filter bandwidth is higher than the signal width, this may cause undesirable over-broadening and lost resolution. After studying the effects of three allowed bandwidth settings on the broadening of dark current noise from a single shot, we determined that a reasonable compromise setting of the preamplifier filter is at “medium” bandwidth in the acquisition software which corresponds to 200 MHz (Acqiris DP240 ADC specifications). Such a setting does not over-broaden true signal peaks and potentially helps recover some under-sampled intensities in broad mass range acquisition through integration of under-sampled signals.
Although the specific acquisition parameter optimization discussed above was performed for the Bruker Ultraflex instrument, it would be generally applicable to any TOF instrument whose detection system is based on the flash ADC and includes a preamplifier. For instance, the AB 4800 MALDI-TOF/TOF allows software adjustment of the ADC scale and percent-offset as well as preamplifier filter bandwidths. The PBS instruments do not allow such adjustments. The ADC offset is set in the hardware to 20% of scale for better broad mass coverage, however, at the expense of smaller dynamic range.
TOF–MS Data Processing
Since the described down-sampling procedure (in Experimental Procedures) involves signal integration, it had to be preceded by proper baseline subtraction to avoid contributions of a slowly varying baseline trend, while still preserving peak shapes and intensities. The manufacturer’s (Bruker) baseline subtraction algorithm (convex hull) was not adequate for heavy masses (above 20 kDa), since it apparently subtracted signals from the base of broader peaks. Such subtraction reduced sensitivity to broad heavy masses or overlapping signals. A de-trending model that we found to be more appropriate in the broad mass range was a decaying exponential, which is concave along the TOF record.
Baseline subtraction followed by integrative down-sampling processing is essentially doing the job of an “integrative ADC”. Flash ADC, set at a lower sampling rate, loses the under-sampled signal compared to higher sampling rates. Integration adds contributions from the under-sampled signal between sampling points and suppresses random noise. Similarly, integrative down-sampling postacquisition enhances the SNR per sample point proportionally to the square root of the down-sampling window length in the presence of random Gaussian noise. In the high-mass range (above 10 kDa), where both constant and quadratic down-sampling can be used, the SNR enhancement is a product of the gains from each procedure separately. In contrast to, for example, moving average integrative filtering,23 there is no signal broadening by this procedure, because each intensity point is counted once in non-overlapping windows. This procedure does not change the signal shape and width. The peak width in mass is the same before and after processing; only the point density per signal peak is smaller compared to raw data.16
After down-sampling, the intensity per point is more representative of the total signal, since the constant point density per peak is recovered.16 Hence, relative intensities of signal maxima are brought on comparable scale for visual inspection over the whole recorded mass range. This is different for the raw data, where the heavy mass peaks may appear lower just due to their broadness. This down-sampling is also a prerequisite for flawless peak detection, which assumes nominally constant peak-width. The precision of signal location and intensity, determined by peak detection algorithm, is limited by SNR. Thus, SNR enhancement also leads to improved precision of signal detection. The overall results of processing by constant and quadratic down-sampling preceded by baseline subtraction are (1) a constant point density of about 9 time steps for fwhm is recovered over the 100 kDa mass range; (2) the processed data is compressed by a factor of 20 compared to the original 1 Mb of raw data per spectrum, and (3) the resulting MS spectra have SNR values enhanced 10-fold and more above 10 kDa.
The improved sensitivity achieved through integrative down-sampling is illustrated in Figure 2, which shows the MALDI-TOF spectrum for serum purified with C3 magnetic beads. The bottom trace (inverted) is the raw spectrum, and the top trace is the processed spectrum after down-sampling. The spectra are scaled to the same noise level to illustrate enhanced SNR per signal point achieved by down-sampling. Note that the noise level remains constant after processing in the full m/z range. This allows the introduction of a global threshold for peak detection. The low-mass range (Figure 2a) exhibits about 3-time SNR enhancement ( ) mainly due to constant-rate integrating down-sampling (Tr = 5) that we applied. The high-mass range (Figure 2b) is improved by a combination of constant and quadratic down-sampling (10-fold and more above 10 kDa). Note also that the relative intensities of peak maxima in the processed spectra are now more representative of total relative intensities of signals (e.g., higher 1+with respect to 2+ and 3+ charge states, as seen for albumin: 1+ m/z = 66 433). For unprocessed data, peaks are broader for higher m/z and can obscure relative visual estimates from peak maxima.
Figure 2.
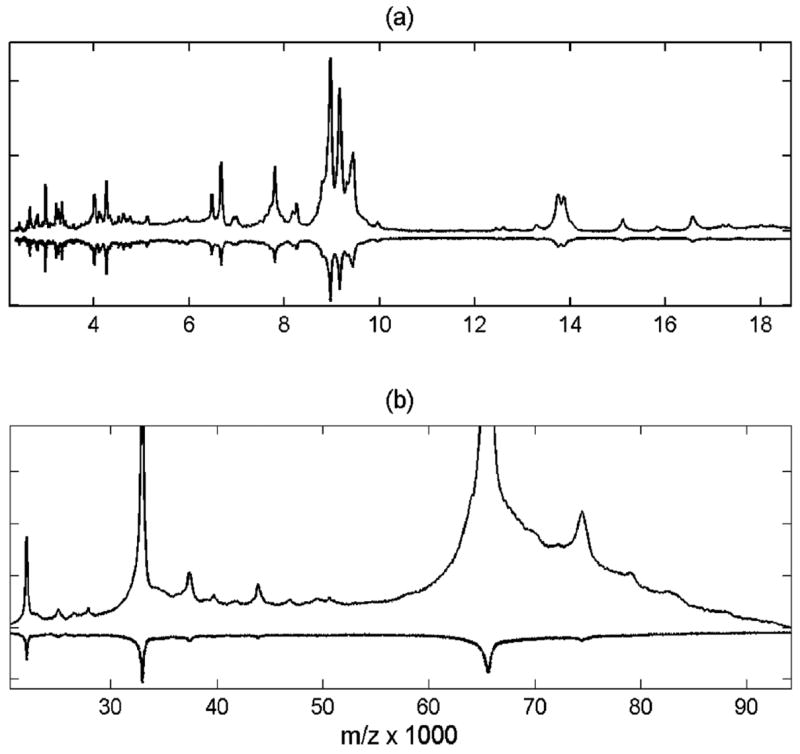
Pooled serum spectrum was processed by integrative down-sampling and is shown in low (a) and high (b) mass range compared to raw data (inverted). Processed data is scaled to the same noise level as in the raw spectrum.
Enhancement of sensitivity to heavy mass signals above 20 kDa (shown in Figure 2b) allows detection of about 15 more nonredundant signals (not counting ionization satellites) in addition to about 40 abundant peaks visible below 20 kDa. For instance, in Figure 2, the only dominant signals above 20 kDa in the raw spectrum are the probable 1+ to 3+ charge states of albumin. After processing, many other features are now visible at higher m/z. The SNR of early mass signals is also enhanced. This allows lowering of the thresholds for peak detection and obtaining higher precision on peak locations and intensities of less abundant signals over the full MS record range of 100 kDa. Many of these low-abundance signals may be related to ionization satellites and chemical adducts, which then can be deconvoluted from parent peaks. Without the achieved sensitivity enhancement over the broad mass range, it would be impossible to discriminate between many ionization satellites and parent protein signals. For instance, if only data below 20 kDa is analyzed without enhancement of heavy mass signals, assignment of the peak cluster near 17 kDa to a particular serum protein moiety is not straightforward (e.g., myoglobin is at 16.9 kDa). After broad-mass enhancement, this cluster is easily identified as the +4 charge state of the peak cluster surrounding parent albumin peak near 70 kDa.
Enrichment and Purification of Proteins
The transition from chromatographic separation to solid phase via derivatized paramagnetic beads facilitates high-throughput, robotics-based applications and is a preferred up-front step for reduction in sample complexity. Predictably, we established that robotic sample preparation was more reproducible than manual procedures and also gave higher quality spectra. Thus, all samples were processed robotically in this study. To ascertain the preferred sample preparation conditions for broad mass coverage, we evaluated a variety of parameters (bead functionalities, MALDI matrices, dilutions, and plate surfaces) to determine reproducibility, number of detected peaks, and overall SNR in spectra spanning 100 000 m/z. We experimented with solution conditions for MALDI spotting using SA, DHB, and CHCA matrices. Although DHB and SA are known to enhance heavy mass signals more effectively, we found this enhancement to be quite minimal (~10% more heavy m/z signals) and at the expense of considerable loss of low m/z signals compared to CHCA (~30% less). More importantly, DHB and SA crystal formation was less uniform on MALDI plates, which severely compromised reproducibility (>50% variability in replicate signals). Likewise, the robotic spotting on an AnchorChip plate was much more uniform compared to stainless steel. Thus, with CHCA matrix on an AnchorChip, the changes of intensities in the spectra resulting from (up to 10) replicate spotting of the same sample were within 5–10%. Therefore, CHCA matrix and the AnchorChip surface were found to be more reliable for high-throughput MS protein profiling. The sample-to-matrix dilution of 1:5 was also helpful both for better reproducibility and higher SNR in the higher mass range. Sample replicates (aliquots) were found to be the major source of spectral intensity variation, producing 20–40% variability in peak signals. This is observed to a higher degree for peaks below m/z 20 000.
We tested a number of different bead functionalities including C3, C8, WCX, WAX, IMAC, and Con A. Beads functionalized with C3 and C8 capture proteins and peptides from biological samples based on hydrophobic interaction; WCX and WAX are weak ion exchangers; IMAC preferentially captures phosphorylated proteins; and Con A enriches specific glycoproteins. Samples were processed robotically using AnchorChips with CHCA matrix (1:5 dilution). Data was collected with optimized MS acquisition settings in the 2000–100 000 m/z range. Replicate spectra from 6 spots for each affinity bead prep were averaged, baseline-subtracted, and preprocessed by down-sampling before signal detection. For quantitative comparison of processed spectra, we performed signal detection using a trivial peak detection algorithm provided by MD Anderson freeware in Matlab (Cromwell package).24 No wavelet smoothing was used from the Cromwell package, only a trivial peak detection routine. The algorithm is based on detection of the change in sign for the first difference (local maxima) at each point in a spectrum and does not include any thresholds. Although more elaborate peak detection algorithms are available, their comparison is outside of the scope of this study. We found this simple trivial detection procedure to be sufficient for quantitative analysis of our data after SNR was enhanced by optimized acquisition and preprocessing. To avoid detection of noise peaks, we added a condition of having the difference between the peak and the closest minimum on either the left or right of a peak to be greater than SNR = 1. SNR is defined as the ratio of measured signal intensity to the mean noise amplitude within the 10· fwhm of the peaks (100 time points for the data in this study). At least three noise measurements from peakless regions of each spectrum were averaged to estimate the noise. The error of the measured SNR for detected peaks was within ±0.5, with the minimum SNR for detected signals of about 0.5. The adequate performance of this automated signal detection procedure was ensured by visual overlay of detected peaks with the corresponding spectrum. The results of signal detection and SNR statistics are summarized in Table 1 for the six studied bead functionalities.
Table 1.
Signal Detection Statistics for TOF–MS Spectra of Pooled Serum on C3, C8, IMAC, WCX, WAX, and Con A ClinProt Beads
| bead type | total peaks | max(SNR) total | median(SNR) total | no. of peaks>15 kDa | max(SNR) >15 kDa | median(SNR) >15 kDa |
|---|---|---|---|---|---|---|
| C3 | 141 | 458 | 21 | 37 | 458 | 22 |
| C8 | 131 | 330 | 21 | 46 | 330 | 29 |
| IMAC | 165 | 849 | 11 | 48 | 849 | 21 |
| WCX | 161 | 777 | 39 | 48 | 364 | 36 |
| WAX | 109 | 154 | 8 | 26 | 29 | 8 |
| Con A | 80 | 844 | 11 | 19 | 53 | 16 |
About a quarter of detected signals for all bead functionalities were above 15 kDa. The median SNR for these >15 kDa masses was higher than the overall median. This illustrates an effective extension of the TOF detection range to heavy masses. Con A and WAX beads show lower efficiency in binding higher MW proteins compared to low MW species. This may be related to a higher selectivity of these bead types. WCX shows the best performance both in terms of median SNR and peak numbers for low mass, and comparatively lower intensities of high-mass signals (max(SNR)) with respect to low-mass intensities in the same spectra). IMAC also provides good overall mass coverage, but shows an opposite behavior with higher affinity to heavy mass proteins with respect to lower mass species (total median-(SNR) < heavy m/z median(SNR)). C3 and C8 beads produced highly similar spectra with less than 10% difference both from m/z peak lists and relative intensity patterns. These beads offer the best overall compromise in detection of low- versus high-mass signals (note similar maximum and median SNR values independent of mass range).
Figures 3 and 4 show processed MALDI spectra for the 6 different affinity beads. WAX (Figure 3, green) exhibits the lowest SNR and weakest affinity to heavy masses. In the high-mass range, Con A beads apparently cause attachment of their chemical component to albumin, producing broadened flat-top features (Figure 3, magenta). There are very few features in the Con A spectrum due to the more specific protein enrichment. The most abundant feature in Con A spectra was a cluster centered around m/z 7800 and its doubly charged reflection. This peak cluster was also common for all other beads that we studied. Our results show that, although valuable for selective enrichment studies, the Con A and WAX beads do not provide the best choice for broad range m/z survey scanning. C8 spectra (Figure 3, blue) are highly similar to C3 (Figure 4, blue) with a slightly lower overall peak number and SNR for C8, so C3 is a slightly better choice of the two.
Figure 3.
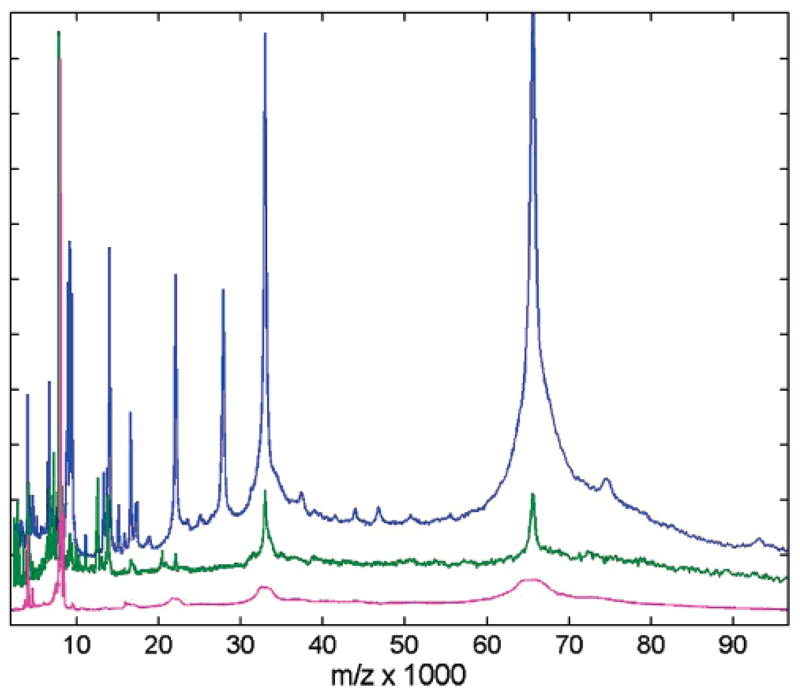
Comparison of mass spectral content for three affinity capture magnetic bead surfaces using CHCA matrix: C8 (blue), WAX (green), Con A (magenta). Spectra are shown after integrative down-sampling, and are normalized to maximum intensity and vertically offset for clarity.
Figure 4.
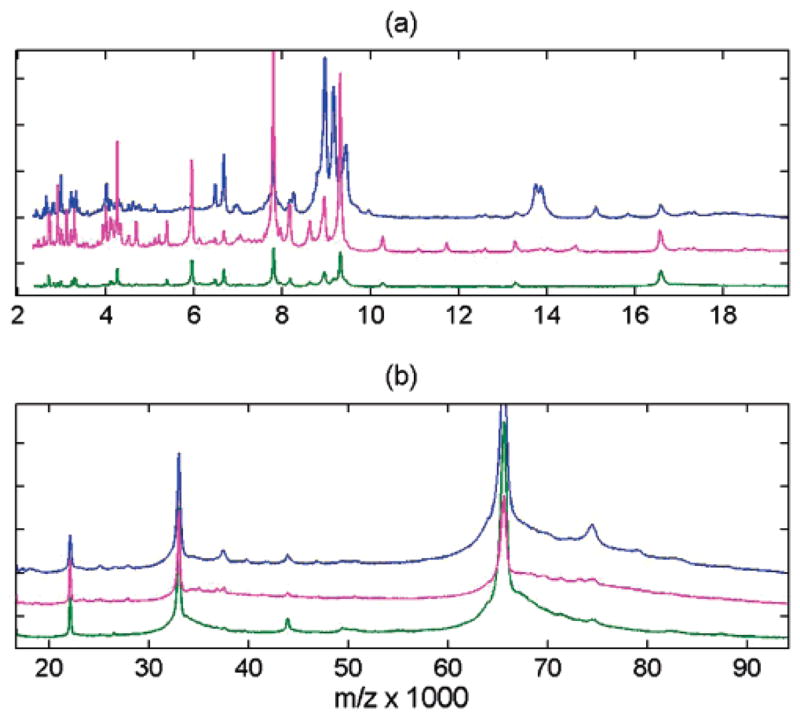
Comparison of mass spectral content for three affinity capture magnetic bead surfaces using CHCA matrix: C3 (blue), IMAC (green), WCX (magenta). Spectra are shown after integrative down-sampling. The view is subdivided in two mass ranges (a) and (b), and spectra are normalized to maximum intensity and vertically offset for clarity.
WCX (Figure 4, magenta) has reduced efficiency at high m/z compared to low m/z, yet the spectral quality for lower masses is visibly the best (as the numbers in Table 1 clearly indicate). There are more peak differences observed between C3 and IMAC or WCX in the low-mass range (~30% signals, Figure 4a) then in the high-mass range (Figure 4b). Some examples of highly abundant peaks observed for C3, but suppressed for IMAC and WCX, include the singly charged ions at m/z 8200 and 15 100 and a doublet at 13750/13870 with their corresponding doubly charged reflections. Conversely, the peak at m/z 5940 is 10-fold more abundant for WCX and IMAC compared to C3. The most prominent common peaks for the three surfaces (in addition to 1+ through 4+ charges of albumin (m/z = 66 400, 33 200, 22 100, 16 600)) include a peak doublet at m/z 6500/6700, a cluster centered around 7800, and triplet at 8950/9160/9310. Detailed results on the common and different subsets of peaks can be found in a table in Supporting Information. We also noticed that robotic C3 preparation resulted in greater reproducibility in MS signals versus IMAC or WCX preparation. This is most likely due to the manufacturer’s protocol for IMAC and WCX which have smaller elution volumes and increased number of steps. Of the beads tested, we concluded that C3, IMAC, and WCX gave the most favorable and complementary protein profiles for a large mass window (WCX being more efficient in lower m/z range, while IMAC is slightly better for higher m/z). We recommend their complementary use for high-throughput MALDI–TOF profiling of complex protein mixtures.
As expected, biofluid sample preparation protocols have a major effect on the spectral quality, as illustrated in Figure 5. IMAC affinity capture of serum was compared in 3 different systems: Ciphergen SELDI chips with sinapinic acid (SA) matrix recorded on a Ciphergen PBS II instrument, SELDI chips with SA matrix analyzed on a Bruker Ultraflex III, and ClinProt magnetic beads with CHCA matrix analyzed on an Ultraflex III. The top two traces in Figure 5 show that affinity capture on IMAC chip surfaces produces similar mass records independent of changes in detector systems (PBS II versus Ultraflex III spectrometer). Similarity is measured by the correlation of intensity patterns and peak positions, and is more than 0.95 (on 0 to 1 scale) for the example shown. In contrast, the spectrum produced after affinity capture using magnetic beads is significantly different (lower trace in Figure 5, correlation of 0.75), especially in intermediate- and low-mass range (<5 kDa). This is notwithstanding the fact that both chip and beads possess IMAC chemical affinity. Different protocols were used for preparation, however, which accounts for some of the observed differences. In particular, urea is used in the binding step for the IMAC chip. For instance, the peak at 12 kDa is present in the SELDI chip spectra in Figure 5, but absent in the bead spectrum. In fact, we found that the 12 kDa protein (or protein fragment) is absent in the bead spectrum whether urea is used or not (data not shown). This may be due to different requirements for binding or elution of protein/peptide when utilizing bead-based approaches.
Figure 5.
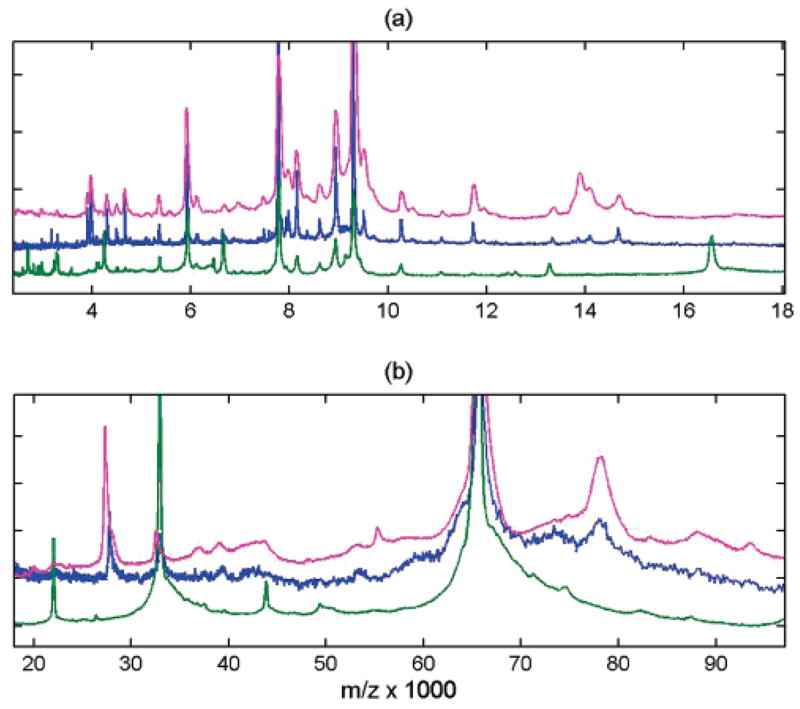
Comparison of mass spectral content for IMAC affinity capture by SELDI chip with SA matrix recorded on Ciphergen PBS II instrument (magenta), by SELDI chip recorded on Bruker Ultraflex III (blue), and by ClinProt magnetic beads with CHCA matrix recorded on Bruker Ultraflex III (green). Robotic preparation was used for all samples. Middle (blue) record is at about twice the resolution of the top spectrum (magenta). Spectra are processed by averaging 7 spots, subtracting exponential baseline, and performing constant and quadratic integrative down-sampling. The view is subdivided in two mass ranges (a) and (b), and spectra are normalized to maximum intensity and vertically offset for clarity.
Other noticeable differences for IMAC beads include, for example, suppression of registered intensities for peak clusters around 14, 29, and 79 kDa. These m/z peak values are consistent with literature references of known abundant serum protein molecules.25–31 Also, IMAC beads show enhancement of peptide signals below 4 kDa, as well as observation of the 1+ to 5+ charge states of albumin. The observed differences in ionization satellite content can be largely attributed to the change of matrix. For instance, higher charge states and less matrix adducts and neutral loss is characteristic of CHCA compared to SA. In general, we observed that the albumin cluster dominates the heavy mass spectra from magnetic beads, while some abundant serum proteins in the intermediate- and high-mass range are captured with lower efficiency. Thus, binding to IMAC beads is more selective compared to the IMAC chip.
In summary, we have extended the mass range of MALDI-TOF high-sensitivity detection for profiling of complex protein mixtures. This has been accomplished through the combined optimization of sample preparation procedures, MS instrument parameters, and data processing. C3, IMAC, and WCX beads were determined to be complementary and favorable for broad mass range protein detection. Key instrument parameters for enhancing sensitivity in an extended mass range included the adjustment of the ADC offset and preamplifier filter values. Data processing utilized a combination of exponential baseline subtraction, and constant and quadratic down-sampling, to increase SNR and precision of signal peak detection. Overall, this “broad range” enhancement will extend the utility of MS-based analysis of biological samples through the improvement of protein/peptides detection sensitivity. The improvement in the relative linear range of detection will also complement efforts to enrich for lower abundant proteins.32 Our developed methods are relevant to applications where broad mass range detection is advantageous, such as in clinical biomarker discovery, microorganism identification,33,34 and MS imaging of biosamples.35–37
Acknowledgments
This research was supported by grant CA126118 from the Advanced Proteomics Platforms and Computational Sciences Program within Clinical Proteomic Technologies Initiative of the National Cancer Institute. We wish to thank Sergei Dikler of Bruker Daltonics for his advice on ADC settings, and Professor Gene Tracy of the College of William and Mary for his critical review and contributions to this work.
Footnotes
Supporting Information Available: Tables with detailed results on the common and different subsets of peaks. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Gillette MA, Mani DR, Carr SA. Place of pattern in proteomic biomarker discovery. J Proteome Res. 2005;4(4):1143–54. doi: 10.1021/pr0500962. [DOI] [PubMed] [Google Scholar]
- 2.Conrads TP, Hood BL, Issaq HJ, Veenstra TD. Proteomic patterns as a diagnostic tool for early-stage cancer: a review of its progress to a clinically relevant tool. Mol Diagn. 2004;8(2):77–85. doi: 10.1007/BF03260049. [DOI] [PubMed] [Google Scholar]
- 3.Petricoin EF, Ardekani AM, Hitt BA, Levine PJ, Fusaro VA, Steinberg SM, Mills GB, Simone C, Fishman DA, Kohn EC, Liotta LA. Use of proteomic patterns in serum to identify ovarian cancer. Lancet. 2002;359(9306):572–7. doi: 10.1016/S0140-6736(02)07746-2. [DOI] [PubMed] [Google Scholar]
- 4.Cazares LH, Adam BL, Ward MD, Nasim S, Schellhammer PF, Semmes OJ, Wright GL., Jr Normal, benign, preneoplastic, and malignant prostate cells have distinct protein expression profiles resolved by surface enhanced laser desorption/ ionization mass spectrometry. Clin Cancer Res. 2002;8(8):2541–52. [PubMed] [Google Scholar]
- 5.Issaq HJ, Conrads TP, Prieto DA, Tirumalai R, Veenstra TD. SELDI-TOF MS for diagnostic proteomics. Anal Chem. 2003;75(7):148A–155A. [PubMed] [Google Scholar]
- 6.Villanueva J, Philip J, Entenberg D, Chaparro CA, Tanwar MK, Holland EC, Tempst P. Serum peptide profiling by magnetic particle-assisted, automated sample processing and MALDI-TOF mass spectrometry. Anal Chem. 2004;76(6):1560–70. doi: 10.1021/ac0352171. [DOI] [PubMed] [Google Scholar]
- 7.VerBerkmoes NC, Bundy JL, Hauser L, Asano KG, Razumovskaya J, Larimer F, Hettich RL, Stephenson JL., Jr Integrating “top-down” and “bottom-up” mass spectrometric approaches for proteomic analysis of Shewanella oneidensis. J Proteome Res. 2002;1(3):239–52. doi: 10.1021/pr025508a. [DOI] [PubMed] [Google Scholar]
- 8.Shin S, Cazares L, Schneider H, Mitchell S, Laronga C, Semmes OJ, Perry RR, Drake RR. Serum biomarkers to differentiate benign and malignant mammographic lesions. J Am Coll Surg. 2007;204(5):1065–71. doi: 10.1016/j.jamcollsurg.2007.01.036. [DOI] [PubMed] [Google Scholar]
- 9.Baumann S, Ceglarek U, Fiedler GM, Lembcke J, Leichtle A, Thiery J. Standardized approach to proteome profiling of human serum based on magnetic bead separation and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Clin Chem. 2005;51(6):973–80. doi: 10.1373/clinchem.2004.047308. [DOI] [PubMed] [Google Scholar]
- 10.West-Norager M, Kelstrup CD, Schou C, Hogdall EV, Hogdall CK, Heegaard NH. Unravelling in vitro variables of major importance for the outcome of mass spectrometry-based serum proteomics. J Chromatogr, B: Anal Technol Biomed Life Sci. 2007;847(1):30–7. doi: 10.1016/j.jchromb.2006.09.048. [DOI] [PubMed] [Google Scholar]
- 11.Zhang X, Leung SM, Morris CR, Shigenaga MK. Evaluation of a novel, integrated approach using functionalized magnetic beads, bench-top MALDI-TOF-MS with prestructured sample supports, and pattern recognition software for profiling potential biomarkers in human plasma. J Biomol Tech. 2004;15(3):167–75. [PMC free article] [PubMed] [Google Scholar]
- 12.Sparbier K, Wenzel T, Kostrzewa M. Exploring the binding profiles of ConA, boronic acid and WGA by MALDI-TOF/TOF MS and magnetic particles. J Chromatogr, B: Anal Technol Biomed Life Sci. 2006;840(1):29–36. doi: 10.1016/j.jchromb.2006.06.028. [DOI] [PubMed] [Google Scholar]
- 13.Adam BL, Qu Y, Davis JW, Ward MD, Clements MA, Cazares LH, Semmes OJ, Schellhammer PF, Yasui Y, Feng Z, Wright GL., Jr Serum protein fingerprinting coupled with a pattern-matching algorithm distinguishes prostate cancer from benign prostate hyperplasia and healthy men. Cancer Res. 2002;62(13):3609–14. [PubMed] [Google Scholar]
- 14.Semmes OJ, Feng Z, Adam BL, Banez LL, Bigbee WL, Campos D, Cazares LH, Chan DW, Grizzle WE, Izbicka E, Kagan J, Malik G, McLerran D, Moul JW, Partin A, Prasanna P, Rosenzweig J, Sokoll LJ, Srivastava S, Srivastava S, Thompson I, Welsh MJ, White N, Winget M, Yasui Y, Zhang Z, Zhu L. Evaluation of serum protein profiling by surface-enhanced laser desorption/ionization time-of-flight mass spectrometry for the detection of prostate cancer: I. Assessment of platform reproducibility. Clin Chem. 2005;51(1):102–12. doi: 10.1373/clinchem.2004.038950. [DOI] [PubMed] [Google Scholar]
- 15.Wadsworth JT, Somers KD, Cazares LH, Malik G, Adam BL, Stack BC, Jr, Wright GL, Jr, Semmes OJ. Serum protein profiles to identify head and neck cancer. Clin Cancer Res. 2004;10(5):1625–32. doi: 10.1158/1078-0432.ccr-0297-3. [DOI] [PubMed] [Google Scholar]
- 16.Malyarenko DI, Cooke WE, Tracy ER, Drake RR, Shin S, Semmes OJ, Sasinowski M, Manos DM. Resampling and deconvolution of linear time-of-flight records for enhanced protein profiling. Rapid Commun Mass Spectrom. 2006;20(11):1670–8. doi: 10.1002/rcm.2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cotter RJ. Time-of-Flight Mass Spectrometry. American Chemical Society; Washington, DC: 1997. p. 326. [Google Scholar]
- 18.Malyarenko DI, Cooke WE, Adam BL, Malik G, Chen H, Tracy ER, Trosset MW, Sasinowski M, Semmes OJ, Manos DM. Enhancement of sensitivity and resolution of surface-enhanced laser desorption/ionization time-of-flight mass spectrometric records for serum peptides using time-series analysis techniques. Clin Chem. 2005;51(1):65–74. doi: 10.1373/clinchem.2004.037283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bahr U, Stahl-Zeng J, Gleitsmann E, Karas M. Delayed extraction time-of-flight MALDI mass spectrometry of proteins above 25,000 Da. J Mass Spectrom. 1997;32(10):1111–6. doi: 10.1002/(SICI)1096-9888(199711)32:10<1111::AID-JMS567>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 20.Vestal ML, Jahasz P, Martin SA. Delayed extraction matrix-assisted laser desorption time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. 1995;9:1044–50. [Google Scholar]
- 21.Vestal ML, Juhasz P. Resolution and mass accuracy in matrix-assisted laser desorption ionization-time-of-flight. J Am Soc Mass Spectom. 1998;9:892–911. [Google Scholar]
- 22.Malyarenko DI, Cooke WE, Tracy ER, Drake RR, Shin S, Semmes OJ, Sasinowski M, Manos DM. Time Series Filtering of Linear MALDI-TOF Records for Full Mass Range Enhancement of Protein Survey; Proceedings of the 54th Annual American Society of Mass Spectrometry Conference; Seattle, WA. May 2006. [Google Scholar]
- 23.Fung ET, Enderwick C. ProteinChip clinical proteomics: computational challenges and solutions. BioTechniques. 2002;(Suppl):34–8. 40–1. [PubMed] [Google Scholar]
- 24.Cromwell Package homepage: http://bioinformatics.mdanderson.org/cromwell.html
- 25.Marchi N, Fazio V, Cucullo L, Kight K, Masaryk T, Barnett G, Vogelbaum M, Kinter M, Rasmussen P, Mayberg MR, Janigro D. Serum transthyretin monomer as a possible marker of blood-to-CSF barrier disruption. J Neurosci. 2003;23(5):1949–55. doi: 10.1523/JNEUROSCI.23-05-01949.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moshkovskii SA, Serebryakova MV, Kuteykin-Teplyakov KB, Tikhonova OV, Goufman EI, Zgoda VG, Taranets IN, Makarov OV, Archakov AI. Ovarian cancer marker of 11.7 kDa detected by proteomics is a serum amyloid A1. Proteomics. 2005;5(14):3790–7. doi: 10.1002/pmic.200401205. [DOI] [PubMed] [Google Scholar]
- 27.Semmes OJ, Cazares LH, Ward MD, Qi L, Moody M, Maloney E, Morris J, Trosset MW, Hisada M, Gygi S, Jacobson S. Discrete serum protein signatures discriminate between human retrovirus-associated hematologic and neurologic disease. Leukemia. 2005;19(7):1229–38. doi: 10.1038/sj.leu.2403781. [DOI] [PubMed] [Google Scholar]
- 28.Timms JF, Arslan-Low E, Gentry-Maharaj A, Luo Z, T’Jampens D, Podust VN, Ford J, Fung ET, Gammerman A, Jacobs I, Menon U. Preanalytic influence of sample handling on SELDI-TOF serum protein profiles. Clin Chem. 2007;53(4):645–56. doi: 10.1373/clinchem.2006.080101. [DOI] [PubMed] [Google Scholar]
- 29.Tirumalai RS, Chan KC, Prieto DA, Issaq HJ, Conrads TP, Veenstra TD. Characterization of the low molecular weight human serum proteome. Mol Cell Proteomics. 2003;2(10):1096–103. doi: 10.1074/mcp.M300031-MCP200. [DOI] [PubMed] [Google Scholar]
- 30.Watkins LK, Bondarenko PV, Barbacci DC, Song S, Cockrill SL, Russell DH, Macfarlane RD. Fast C18 solid-phase desalting/delipidation of the human serum apolipoproteins for matrix-assisted laser desorption ionization and electrospray ionization mass spectrometric analysis. J Chromatogr, A. 1999;840(2):183–93. doi: 10.1016/s0021-9673(99)00205-8. [DOI] [PubMed] [Google Scholar]
- 31.Ye B, Cramer DW, Skates SJ, Gygi SP, Pratomo V, Fu L, Horick NK, Licklider LJ, Schorge JO, Berkowitz RS, Mok SC. Haptoglobin-alpha subunit as potential serum biomarker in ovarian cancer: identification and characterization using proteomic profiling and mass spectrometry. Clin Cancer Res. 2003;9(8):2904–11. [PubMed] [Google Scholar]
- 32.Pieper R, Su Q, Gatlin CL, Huang ST, Anderson NL, Steiner S. Multi-component immunoaffinity subtraction chromatography: an innovative step towards a comprehensive survey of the human plasma proteome. Proteomics. 2003;3(4):422–32. doi: 10.1002/pmic.200390057. [DOI] [PubMed] [Google Scholar]
- 33.Demirev PA, Feldman AB, Kowalski P, Lin JS. Top-down proteomics for rapid identification of intact microorganisms. Anal Chem. 2005;77(22):7455–61. doi: 10.1021/ac051419g. [DOI] [PubMed] [Google Scholar]
- 34.Fenselau C, Demirev PA. Characterization of intact micro-organisms by MALDI mass spectrometry. Mass Spectrom Rev. 2001;20(4):157–71. doi: 10.1002/mas.10004. [DOI] [PubMed] [Google Scholar]
- 35.Pacholski ML, Winograd N. Imaging with mass spectrometry. Chem Rev. 1999;99(10):2977–3006. doi: 10.1021/cr980137w. [DOI] [PubMed] [Google Scholar]
- 36.Stoeckli M, Chaurand P, Hallahan DE, Caprioli RM. Imaging mass spectrometry: a new technology for the analysis of protein expression in mammalian tissues. Nat Med. 2001;7(4):493–6. doi: 10.1038/86573. [DOI] [PubMed] [Google Scholar]
- 37.Chaurand P, Schwartz SA, Caprioli RM. Profiling and imaging proteins in tissue sections by MS. Anal Chem. 2004;76(5):86A–93A. [PubMed] [Google Scholar]