

Abstract
Cell death by apoptosis is a tightly regulated process that requires coordinated modification in cellular architecture. The caspase protease family has been shown to play a key role in apoptosis. Here we report that specific and ordered changes in the actin cytoskeleton take place during apoptosis.
In this context, we have dissected one of the first hallmarks in cell death, represented by the severing of contacts among neighboring cells. More specifically, we provide demonstration for the mechanism that could contribute to the disassembly of cytoskeletal organization at cell–cell adhesion. In fact, β-catenin, a known regulator of cell–cell adhesion, is proteolytically processed in different cell types after induction of apoptosis. Caspase-3 (cpp32/apopain/yama) cleaves in vitro translated β-catenin into a form which is similar in size to that observed in cells undergoing apoptosis. β-Catenin cleavage, during apoptosis in vivo and after caspase-3 treatment in vitro, removes the amino- and carboxy-terminal regions of the protein. The resulting β-catenin product is unable to bind α-catenin that is responsible for actin filament binding and organization. This evidence indicates that connection with actin filaments organized at cell–cell contacts could be dismantled during apoptosis. Our observations suggest that caspases orchestrate the specific and sequential changes in the actin cytoskeleton occurring during cell death via cleavage of different regulators of the microfilament system.
Apoptosis is a fundamental cellular process in the development and homeostasis of all multicellular organisms (Raff, 1992; Wyllie, 1995). Genetic analysis in Caenorhabditis elegans has identified both positive and negative regulators of apoptosis (Ellis et al., 1991).
The ced-3 gene is essential for developmentally programmed cell death in C. elegans: it encodes a protein which shares homology with mammalian interleukin- 1β–convertase enzyme (ICE; Yuan et al., 1993).
ICE is a cystein protease that cleaves interleukin-1β precursor at two aspartic residues and is activated by cleavage after specific aspartic residues of a 45-kD proenzyme, thus forming two subunits that together form the active site of the enzyme (Thornberry et al., 1992). The regulatory mechanisms of the ICE remain to be clarified (Chinnaiyan et al., 1997). In general, it exhibits full activity only after proteolytic processing, which occurs either by self-cleavage or through cleavage by other proteases (Nicholson, 1996).
Nine additional members of ICE-related proteases have been identified in mammals (Fernandes-Alnemri et al., 1994; Kumar et al., 1994; Wang et al., 1994; Faucheau et al., 1995; Fernandes-Alnemri et al., 1995a ,b; Boldin et al., 1996; Duan et al., 1996a ,b; Fernandes-Alnemri et al., 1996; Muzio et al., 1996). Based on amino acid (aa)1 sequences and recognized substrates, they have been grouped in different sub-families, and named according to the recently proposed caspase nomenclature (Alnemri et al., 1996). These proteases may overlap in their functions and/or regulate one another's activities.
Indeed, recent observations suggest the existence of a hierarchically ordered proteolytic cascade during apoptosis. In the case of CD95/FAS/APO-1-induced apoptosis, an initial activity related to ICE itself (caspase-1), and a later activity related to CPP32 (caspase-3), have been described using specific inhibitor substrates (Enari et al., 1996). Moreover, CD95/FAS/APO-1 receptors use the adaptor molecule MORT1/FADD to physically engage a cytosolic ICE-related cystein protease termed FLICE/ MACH (caspase-8) (Boldin et al., 1996; Muzio et al., 1996). Caspase-8 might represent the most upstream protease involved in generating a death signal.
The central role of the caspase proteases in mammalian apoptosis is supported by the existence of specific inhibitors. The viral proteins p35 and CrmA (Bump et al., 1995; Xue and Horvitz, 1995), or aldehyde and fluormethylketone derivatives of the target cleavage sequences, suppress mammalian apoptosis as triggered by different stimuli (Miura et al., 1993; Gagliardini et al., 1994; Beidler et al., 1995; Nicholson et al., 1995).
If cystein proteases play a crucial role in apoptosis, specific target proteins or death substrates become critically relevant for the execution phase of the apoptotic program.
Specific cleavage by ICE-related cystein proteases of the DNA repair enzymes poly(ADP-ribose) polymerase (PARP) and the catalytic subunit of the DNA-dependent protein kinase (DNA-PK), is possibly related to the inactivation of the DNA repair pathway as a choice of economy (Lazebnik et al., 1994; Song et al., 1996). Cleavage of the nuclear lamins is dependent on Mch2 (caspase-6) and seems to be essential for disassembling the nuclear structure (Lazebnik et al., 1995; Takahashi et al., 1996). Other substrates, whose function is still unclear during apoptosis, are the 70-kD protein of the U1 small nuclear ribonucleoprotein (Casciola-Rosen et al., 1994), the sterol regulatory element-binding proteins SREBP-1 and SRBP-2 (Wang et al., 1996), the huntingtin protein (Goldberg et al., 1996), D4 GDP dissociation inhibitor (Na et al., 1996), and pRb tumor suppressor (Janicke et al., 1996).
Two interesting substrates for caspases are the protein kinase Cδ and Gas2, the last being a component of the microfilament system (Brancolini et al., 1992). In both cases, proteolytic processing results in a gain of function which relates to increased kinase activity in the case of PKCδ (Emoto et al., 1995), or to an activity on the microfilament system and on cell morphology in the case of Gas2 (Brancolini et al., 1995). Recently, gain of function after caspase-3 processing has also been demonstrated for the p21-activated kinase 2 (Rudel and Bokoch, 1997), and the DNA fragmentation factor (Liu et al., 1997).
Apoptotic morphological features are generally similar in all systems where they have been studied: the nucleus condenses and the cell shrinks and often fragments into apoptotic bodies that are rapidly phagocytosed by neighboring cells (Kerr et al., 1972; Wyllie et al., 1980).
The present study addresses the roles and changes of the microfilament system during apoptosis. By a detailed confocal analysis on apoptotic NIH3T3 cells, we have identified specific sequential changes in the microfilament system. Coordinately with actin filament reorganization in the perinuclear area, the cell retracts from the adhesion substrate and severs contacts with neighboring cells.
Germane to the above mentioned changes, we also observed a specific modulation of the cell–cell contacts during apoptosis through proteolytic processing of β-catenin thereby losing the ability to bind α-catenin. Such proteolytic cleavage in vitro is mediated by the cystein-protease cpp32/ apopain/yama (caspase-3).
In summary, the complex reorganization of cell morphology during apoptosis seems to be achieved by specific proteolytic processing of different regulators of actin cytoskeleton.
Materials and Methods
Cell Lines and Culture Conditions
NIH3T3 and MDCK cells were grown in DME supplemented with 10% FCS, penicillin (100 U/ml), and streptomycin (100 μg/ml). In each experiment 2.5 × 104 cells/ml were seeded in 35-mm Petri dishes.
For serum starvation, medium was changed to 0.5% FCS when cells were subconfluent and left in this medium for 48 h. For density-dependent inhibition, cells were plated at 104/cm2 in 10% FCS. 24 h after plating the medium was changed every 2 d. For induction of apoptosis the culture medium of arrested cells was replaced with serum-free DME. Nonadherent and adherent cells were harvested, washed in serum-free medium, and solubilized in SDS-PAGE buffer. In the case of genotoxic-dependent apoptosis, density-inhibited cells were treated with 20 μg/ml of cisplatin for 4 h; nonadherent and adherent cells were harvested separately 24 h later. For UV treatment, culture medium was removed, dishes were washed once with PBS, UVC irradiated (180 J/m2 in PBS), and fresh medium, containing 10% FCS, was added to the cells. Adherent and nonadherent cells were harvested separately 24 h later. NIH3T3 cells were transfected as previously described (Brancolini et al., 1995).
Plasmid Construction
Mouse β-catenin cDNA (Hoschuetzky et al., 1994) was subcloned in-frame with a COOH-terminal VSV tag (β-catenin–VSV) in pMT2SM-tag (Bardelli et al., 1996) and pGDSV7S-tag (Brancolini et al., 1995). The β-catenin cDNA was amplified by PCR using a sense primer corresponding to the T7 promoter (5′-AATACGACTCACTATAGGGC-3′) and a reverse primer (VSV1) containing a SmaI site (5′-CCAAACTATGACTGGAGGGCCCATT-3′). The carboxy-terminal deleted derivative of β-catenin was produced by endonuclease digestion at the EcoRI site (nucleotides 1361/aa 421) and a COOH-terminal VSV tag was introduced by PCR using a sense primer corresponding to the T7 promoter, and a reverse primer (VSV2) containing a NotI site (5′-GATTGCGGCCGCAAGTGAGG-3′).
A site-directed mutagenesis method by overlap extension through PCR as previously described (Brancolini et al., 1995) was used to produce the point mutants D→ A at different positions of the amino-terminal region of β-catenin. As external primers, the olT7 and olVSV2 were used. The pairs of complementary inverse oligos were the following: β-catenin 162–164D/A: ol162–164u:5′-CTAAACGCTGAGGCCCAGGTGGTA-3′ ol162–164d: 5′-TACCACCTGGGCCTCAGCGTTTAG-3′; β-catenin 144–145D/A: ol144–145u:5′-TATCAGGCTGCCGCGGAACTT-3′ ol144–145d:5′-AAGTTCCGCGGCAGCCTGAT A-3′.
All constructs generated were sequenced using an automated (ALF) system to check for the respective introduced mutations, deletions, and translating fidelity of the inserted PCR fragments.
Immunofluorescence Microscopy
NIH3T3 cells were grown under the described conditions and then fixed with 3% paraformaldehyde in PBS for 20 min at room temperature. Fixed cells were washed with PBS/0.1 M glycine, pH 7.5, and then permeabilized with 0.1% Triton-X100 in PBS for 5 min. The coverslips were treated with the anti–β-catenin monoclonal antibody directed against aa 571–781 (Signal Transduction Laboratories, Lexington, KY) for 1 h in a moist chamber at 37°C. TRITC anti–mouse (Southern Biotechnologies, Birmingham, AL) was used as secondary antibody. Actin filaments were detected using FITC-phalloidin (Sigma Chemical Co., St. Louis, MO) and nuclei were labeled with propidium iodide 5 mM (Sigma Chemical Co.).
Cells were examined with a laser scan microscope (LSM 410; Carl Zeiss, Inc., Thornwood, NY) equipped with a 488 λ argon laser and a 543 I helium neon laser. The following sets of filters were used: rhodamine (BP546, FT580, and LP 590); and fluoresceine (450–490, FT 510, and LP520).
Two-dimensional Gel Electrophoresis
NIH3T3 cells grown for 36 h in 0.5% FCS were labeled for 12 h in 1 ml of DME methionine-free 0.5% FCS, containing 400 mCi/ml [35S]methionine (Amersham Int'l., Little Chalfont, UK). Serum-free medium containing a cold methionine chase, (0.5 mM final concentration) was then added for 6 h.
Samples of total cellular proteins were prepared from adherent and nonadherent NIH3T3 cells after washing with cold PBS. Cells were lysed in hot (100°C) lysis solution (0.3% SDS, 200 mM dithiothreitol, 50 mM Tris-HCl, pH 8). Samples were then treated with 1:10 vol of nuclease solution (50 mM MgCl2, 50 mM Tris-HCl, pH 7, 1 mg/ml DNaseI, 0.25 mg/ml RNaseA) for 10 min on ice, precipitated by adding ice-cold acetone to 80% vol/vol, and centrifuged at 14,000 g for 10 min. Air-dried pellets were resuspended in a mixture of 1 vol lysis solution and 4 vol of sample buffer (9.9 M urea, 4% NP-40, 2.2% Millipore ampholytes, 100 mM dithiothreitol). Two-dimensional gels were run using the investigator two-dimensional electrophoresis system (Millipore Corp., Waters Chromatography, Milford, MA) with pH 3–10 and pH 4–8 Millipore ampholytes in the first dimension and 10% acrylamide in the second dimension.
Immunoblotting
For Western blotting, proteins were transferred to 0.2-μm pore-size nitrocellulose (Schleicher & Schuell Inc., Keene, NH) using a semidry blotting apparatus (Bio-Rad Laboratories, Hercules, CA) (transfer buffer: 20% methanol, 48 mM Tris, 39 mM glycine, and 0.0375% SDS). After staining with Ponceau S, the nitrocellulose sheets were saturated for 2 h in Blotto-Tween 20 (50 mM Tris-HCl, pH 7.5, 500 mM NaCl, 5% nonfat dry milk, and 0.1% Tween 20; Sigma Chemical Co.) and incubated overnight at room temperature with a specific antibody: anti-Gas2, anti-actin, anti–β-catenin and anti–α-catenin (Transduction Laboratories, Lexington, KY), anti– β-catenin amino-terminal (McCrea et al., 1993), anti–PKC-δ (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), and anti-VSV (Sigma Chemical Co.). Blots were then rinsed three times with Blotto-Tween 20 and reacted with peroxidase-conjugated goat anti–rabbit (Sigma Chemical Co.) or goat anti–mouse (Sigma Chemical Co.) for 1 h at room temperature. The blots were then washed four times in Blotto-Tween 20, rinsed in PBS, and developed with an enhanced chemiluminescent kit, as recommended by the vendor (Amersham Int'l.).
Expression of Caspase-3 (CPP32) in Bacteria and In Vitro Protease Assay
Caspase-3 cDNA was subcloned in-frame into the BamH1 site of the bacterial expression vector pQE-12 (QIAGEN Inc., Santa Clarita, CA). Cells were grown to an A600 of 0.6 and expression of caspase-3 was induced by adding isopropyl-β-d-thiogalactopyranoside to a final concentration of 1 mM. After 2 h, cells were collected by centrifugation at 3,000 g for 5 min, and then resuspended in 5 vol of caspase-3 buffer (Goldberg et al., 1996) with protease inhibitors (1 mM PMSF and 10 μg/ml each aprotinin, leupeptin, and pepstatin). Cells were lysed by sonication and debris was sedimented by centrifugation at 14,000 g for 20 min. The resulting supernatants were used for in vitro protease assays.
Caspase-3 and β-catenin were labeled with 35S using the TNT-coupled reticulocyte lysate system (Promega Corp., Madison, WI). 1 μl of translated reticulocyte lysate was incubated with the appropiate dilution of the bacterial lysates in caspase-3 buffer (final volume of 10 μl) for 1 h at 37°C. Reactions were terminated by adding 1 vol of SDS gel loading buffer and boiling for 3 min. The specific caspase-3/cpp32/apopain inhibitor Ac-Asp-Glu-Val-Asp-CHO was obtained from Bachem Bioscience (Bubendorf, Switzerland).
Immunoprecipitation
Analysis of β-catenin–α-catenin complexes in apoptotic and nonapoptotic cells was performed as previously described (Hulsken et al., 1994) with some modifications. Cells were extracted with 140 mM NaCl, 4.7 mM KCl, 0.7 mM MgSO4, 1.2 CaCl2, 20 mM Tris-HCl, pH 7.5, 5% glycerol, containing 1% Triton X-100 and 1 mM PMSF, and 10 μg/ml each of aprotinin, leupeptin, antipain, and pepstatin. After centrifugation at 14,000 g for 15 min monoclonal antibodies, anti–β-catenin (Transduction Laboratories), or anti-pan cadherin (Sigma Chemical Co.) were added. After 3 h on ice, protein A–Sepharose (Pharmacia Biotech., Inc., Piscataway, NJ) was added and incubation was prolonged for 1 h on ice. After a brief centrifugation in Eppendorf centrifuge, immunoprecipitates were washed with 100 mM NaCl, 5 mM EDTA, 20 mM Tris-HCl, pH 7.5, 1% Triton X-100 containing 1 mM PMSF, and 10 μg/ml each of aprotinin, leupeptin, antipain, and pepstatin.
Immunocomplexes were released by boiling 5 min in SDS sample buffer, separated in a 10% SDS-PAGE, and Western blots were performed as described.
Results
Microfilament Reorganization during Apoptosis in NIH3T3 Cells
NIH3T3 fibroblasts efficiently organize stress fibers and adhesion plaques and form cell–cell junctional complexes and membrane ruffles. They represent an ideal system to study microfilament organization in response to different environmental conditions. Furthermore, growth-arrested NIH3T3 cells readily respond to complete absence of serum in the culture medium by undergoing apoptosis.
To study the changes in microfilament organization in cells undergoing apoptosis, immunofluorescence analysis was performed on growth-arrested NIH3T3 cells cultured for a further 3 h in serum-free medium.
As shown in Fig. 1 a, after 3 h of incubation in serum-free medium, changes in the microfilament organization become clearly detectable in some cells as shown by phalloidin staining (arrows). As a response to the absence of survival factors, cells apparently sever contacts with neighboring cells, retract from the adhesion substrate, and dismantle stress fibers, extensively reorganizing their actin cytoskeleton.
Figure 1.

Confocal analysis of the organization of actin in NIH3T3 cells induced to enter apoptosis by deprivation of survival factors. Growth-arrested NIH3T3 cells were cultured for 4 h in serum-free medium, fixed, and stained for actin filaments using phalloidin-FITC (a), or double stained with propidium iodide, to visualize nuclei (A, C, E, and G) and phalloidin-FITC (B, D, F, and H). Bars: (a) 5 μm; (A–H) 1 μm.
Apoptosis is always accompanied by alterations in the nuclear morphology, finally leading to nuclear fragmentation and formation of the apoptotic bodies. We, therefore, decided to follow such nuclear alterations in relation to the changes in actin organization during apoptosis.
When an initial alteration in nuclear architecture was first detected (arrow, Fig. 1 A) changes in the actin cytoskeleton were also evident (Fig. 1 B). As described above, the peculiar features at this stage included the severing of contacts with neighboring cells, retraction of fibers, accumulation of actin in the perinuclear region and loss of stress fibers.
At further stages, nuclear alterations become more evident leading to nuclear fragmentation (Fig. 1 C). At this stage, the actin cytoskeleton is clearly organized in a ring-like structure (Fig. 1 D, arrow) with membrane blebbing also clearly evident (data not shown). It is interesting to note that nuclear fragments are external with respect to the observed actin ring. This observation suggests that contraction of actin filaments present in the perinuclear region might be important in the sprouting of the nuclear bodies. This hypothesis was supported by the most altered phenotypes, showing the actin reorganization as summarized in Fig. 1, E–H. At these stages the actin ring appeared collapsed, with the nuclear fragments tending to drop away from the actin filaments. This detachment probably leads to the appearance of the phenotype represented in Fig. 1, G and H, where only remnants of cytoplasm containing a collapsed actin ring structure were detected. Such a phenotype could represent a final stage of the microfilament changes taking place during apoptosis.
From this preliminary analysis, it could be appreciated that apoptosis, as triggered by serum deprivation in NIH3T3 fibroblasts, is coupled to well-defined and ordered changes of the microfilament system. Such changes are concomitant with membrane blebbing and alteration of the nuclear architecture.
Analysis of Actin Cleavage during Apoptosis in NIH3T3
Since it has been reported that actin can be cleaved in an in vitro apoptotic system using purified caspase-1 (Mashima et al., 1995; Kayalar et al., 1996), we analyzed as if in vivo, during apoptosis, actin was cleaved in our defined cellular system. In fact, this could possibly explain the dramatic reorganization of the microfilament system occurring during cell death.
A time course analysis was performed on growth-arrested NIH3T3 cells incubated in serum-free medium. Cell survival was markedly decreased after few hours in serum-free medium (Fig. 2 a) and apoptotic cells could be easily observed floating in the medium (data not shown).
Figure 2.


(a) NIH3T3 cells grown for 48 h in 0.5% FCS were incubated in serum-free medium for the indicated times (relative numbers and percentages of viable cells were determined by trypan blue dye exclusion). Data represent arithmetic means ± SD for three independent experiments. (b) Growth-arrested NIH3T3 cells were incubated in serum-free medium for the indicated times. Lysates from both adherent viable cells and nonadherent apoptotic cells were combined and Western blot analysis was performed. (c) Two-dimensional gel electrophoresis analysis showing actin patterns in adherent and nonadherent apoptotic cells.
To study actin cleavage during apoptosis, both adherent as well as nonadherent (floating dead cells) were combined and Western analysis was performed.
As shown in Fig. 2 b, Gas2 was proteolytically processed during apoptosis as previously reported (Brancolini et al., 1995). Appearance of a band with increased mobility was faintly detectable after 3 h from survival signal deprivation. The intensity of the apoptotic band increased after 6 h, concomitant with the accumulation of apoptotic cells in the culture medium.
Under identical culture conditions, actin failed to show any appreciable variation in size or expression levels as observed in the different lanes (Fig. 2 b). As a further control, we analyzed the proteolytic cleavage of PKC-δ, another identified substrate of caspase proteases (Emoto et al., 1995). PKC-δ showed a similar kinetics of proteolysis during apoptosis as previously described for Gas2 (Fig. 2 b).
The behavior of actins during apoptosis in vivo was also analyzed by two-dimensional electrophoresis. NIH3T3 cells grown for 36 h in 0.5% FCS were labeled for 12 h with [35S]methionine, and then simultaneously chased with cold methionine and cultured in serum-free medium. After 6 h, adherent and nonadherent cells were harvested separately and two-dimensional gel electrophoresis was performed as described in Materials and Methods. The two-dimensional electrophoretic pattern of the different actin isoforms (Garrels and Franza, 1989) did not show appreciable differences between normal and apoptotic cells.
Interestingly, a new band, which might represent a posttranslational modification of an unknown product, became detectable in the lysates from apoptotic cells (Fig. 3 C, arrowhead). These results clearly demonstrate that during apoptosis actin fails to be proteolytically processed, as triggered by survival signal deprivation in NIH3T3 cells. Indeed, resistance of actin to cleavage during apoptosis has been recently reported (Song et al., 1997). Nevertheless, we can not exclude that a minor fraction of actin is proteolytically processed during apoptosis, or that this processing does not occur in NIH3T3 cells.
Figure 3.
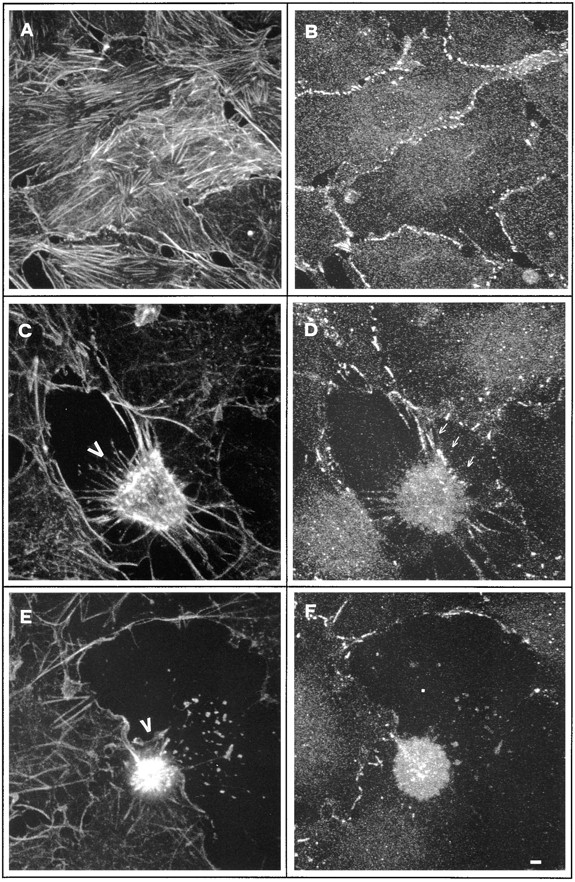
Confocal analysis of cell–cell contacts changes during apoptosis in NIH3T3 cells. Apoptosis was induced by culturing growth-arrested confluent NIH3T3 cells for 4 h in serum-free medium. Confluent growth-arrested NIH3T3 cells (A and B) and apoptotic NIH3T3 cells (C–F) were fixed and double stained for actin filaments using phalloidin-FITC (A, C, and E) and for β-catenin distribution (B, D, and E), using anti–mouse TRITC as secondary antibody. Bar, 1.5 μm.
Modulation of Cell–Cell Contacts during Apoptosis in NIH3T3 Fibroblasts
Having demonstrated that actin is not cleaved in vivo during apoptosis in NIH3T3, we decided to search for other regulators of the microfilament system that could be processed during apoptosis and possibly involved in the dramatic reorganization occurring during cell death.
The previously reported analysis of microfilament changes during apoptosis clearly suggested that a first hallmark of cell death is the severing of contacts among neighboring cells.
Cell–cell contacts have been intensively studied in epithelial cells, where specialized domains of the plasma membrane and the adherens junctions play a critical role in cellular adhesiveness by providing a link between cell surface adhesion molecules and the cytoskeleton (Geiger and Ayalon, 1992). Such an adhesive role is dependent on members of the cadherin superfamily of adhesive receptors and the cytoplasmic adaptor proteins α-, β-, and γ-catenin/plakoglobin, which connect the cadherins to the actin filaments (for review see Gumbiner, 1996).
Fibroblasts in culture express functional cadherins (Itoh et al., 1991), interact with one another, and assemble cadherins–catenin complexes (Knudsen et al., 1995).
To analyze the dynamics of cell–cell contacts during apoptosis in NIH3T3, as a first step we analyzed for the presence of cadherin–catenin complexes in this cell line. NIH3T3 cell lysates were immunoprecipitated using anti-pan cadherin antibody. After electrophoretic separation, the immunoblots were revealed with anti–α-catenin or –β-catenin antibody. α-Catenin, β-catenin, and γ-catenin/plakoglobin were all detected in complexes with cadherin in NIH3T3 cells as previously demonstrated in other fibroblasts (data not shown). As a next step we performed double immunofluorescences to compare β-catenin distribution in relation to actin cytoskeleton in apoptotic and nonapoptotic confluent NIH3T3 cells. Intense staining for β-catenin was observed at the cell–cell contacts in nonapoptotic NIH3T3 cells (Fig. 3, A and B). At an early apoptotic stage (Fig. 3 C, arrowhead), when a retraction response was observed, β-catenin staining was rather diffuse, suggesting that disassembly of cell–cell contacts occurred. However, some localization at the cell–cell junction could still be detected, especially where the apoptotic cell still maintains contacts with its neighbors (Fig. 3 D, arrows). In the more pronounced apoptotic phenotype observed in Fig. 3 E (arrowhead), β-catenin staining was mainly diffuse in the cytoplasm with no evidence for its presence at the cell border (Fig. 3 F).
In summary, this analysis suggested that β-catenin localization at the cell periphery was temporally and spatially regulated during apoptosis in NIH3T3 cells.
Apoptosis Triggered by Both Survival Signal Deprivation and Genotoxic Stress Induces Proteolytic Cleavage of β-Catenin
We, therefore, asked whether the cell–cell adhesion dynamic could be regulated by proteolytic processing of the cytosolic adaptor proteins during apoptosis. To test this hypothesis, Western analysis of β-catenin on both adherent as well as nonadherent (floating dead cells) NIH3T3 cells induced to enter apoptosis following serum-free culture condition was performed.
Deprivation of survival signals induced proteolytic cleavage of β-catenin giving rise to an ∼65-kD stable form clearly evident at 6 h after treatment (Fig. 4 a).
Figure 4.

Analysis of β-catenin cleavage during apoptosis in NIH3T3 and MDCK cells. (a) Growth-arrested NIH3T3 cells were incubated in serum-free medium for the indicated times. Lysates from both adherent viable cells and nonadherent apoptotic cells were combined and Western blot analysis was performed. (b) Density-arrested NIH3T3 cells were treated with 20 μg/ml of cisplatin or UV irradiated as described in Materials and Methods. After 24 h adherent (A) and nonadherent (NA) cells were processed separately and Western analysis was performed. (c) MDCK cells grown for 4 d in 10% FCS were incubated in serum-free medium for 12 h. Adherent (A) and nonadherent (NA) cells were processed separately and Western analysis was performed. (d) MDCK cells grown for 4 d in 10% FCS were treated for 4 h with the indicated amount of MMS. After 24 h, lysates from both adherent viable cells and nonadherent apoptotic cells were combined and Western blot analysis was performed.
The temporal pattern of such β-catenin processing was closely related to the above described Gas2 and PKC-δ apoptotic processing. In fact, processing of β-catenin paralleled the appearance of apoptotic cells in the culture medium (data not shown).
Since β-catenin is known to associate with α-catenin, to mediate the formation of cell–cell adhesion we also followed α-catenin behavior during apoptosis. In this case there was no evidence for proteolytic degradation of α-catenin in NIH3T3 cells induced to enter apoptosis under the same conditions (Fig. 4 a).
β-Catenin proteolytic processing was next analyzed using different apoptotic stimuli, such as genotoxic stress as mediated by DNA-damaging agents. NIH3T3 cells were treated with the chemotherapeutic agent cisplatin or irradiated with UV, and after 24 h adherent and nonadherent (floating dead cells) were separately harvested. Western blot analysis revealed that β-catenin was cleaved to a ∼65-kD form exclusively in the apoptotic cells and, on the contrary, α-catenin failed to be processed under the same conditions (Fig. 4 b).
Adherens junctions are found in many cell types, including cardiac myocytes and fibroblasts, but they have been extensively studied in epithelial cells. Therefore, we similarly investigated proteolytic processing of β-catenin during apoptosis in MDCK cells. Apoptosis was induced by both serum deprivation and DNA damage using increasing amounts of the alkylating agent methylmethanesulfonate (MMS). Western analysis was performed on adherent and nonadherent cells harvested separately, in the case of serum starvation, or jointly, in the case of MMS treatment.
Proteolytic processing of β-catenin in MDCK cells was detected when apoptosis was induced either by serum starvation (Fig. 4 c) or by DNA damage (Fig. 4 d). In the last instance, the relative amount of the processed form paralleled the increased level of genotoxic insult and the appearance of apoptotic cells in the culture medium (data not shown). Here again, both α-catenin and actin failed to be processed.
Since it has been demonstrated that Bcl-2 oncoprotein prevents/delays apoptosis and inhibits activation of the caspase proteases (Shimizu et al., 1996; Chinnaiyan et al., 1997), we investigated the fate of β-catenin during apoptotic stimuli in the presence of increased Bcl-2 expression.
When NIH3T3 overexpressing Bcl-2 were analysed for proteolytic processing of β-catenin under serum-free conditions, apoptosis and proteolytic cleavage of Gas2 and β-catenin were dramatically suppressed (Fig. 5).
Figure 5.
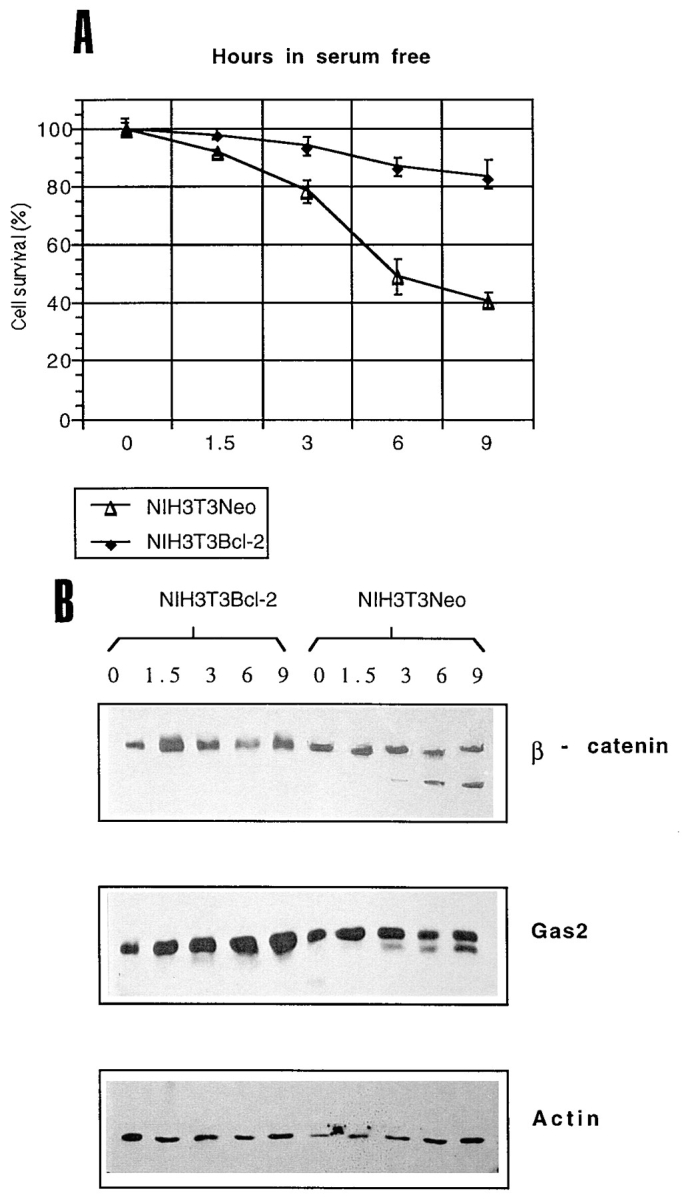
(A) NIH3T3-NEO or NIH3T3 cell lines transfected with bcl-2 were grown for 48 h in 0.5% FCS and incubated in serum-free medium (relative numbers and percentages of viable cells were determined by trypan blue dye exclusion). Data represent arithmetic means ± SD for three independent experiments. (B) Growth-arrested NIH3T3-NEO and NIH3T3–bcl-2 cells were incubated in serum-free medium for the indicated times. Lysates from both adherent viable cells and nonadherent apoptotic cells were combined and Western blot analysis was performed.
β-Catenin cleavage during apoptosis was also observed in the presence of the proteosome inhibitor N-acetyl-leucinyl-leucinyl-norleucinal-H, an inhibitor of the chymotryptic site on the proteosome (Pagano et al., 1995), thus suggesting that it is not mediated by the ubiquitin/proteosome system (data not shown).
We can, therefore, conclude that proteolytic cleavage of β-catenin correlates with activation of a cell death program in both fibroblast and epithelial cells, all the β-catenin pool is cleaved during apoptosis and this processing is suppressed by increased expression of Bcl-2.
β-Catenin Is Cleaved by Caspase-3 In Vitro
We next asked whether enzymes of the caspase family could be responsible for the proteolytic cleavage of β-catenin during apoptosis. We analyzed whether caspase-3, one of the late-activated proteases in the apoptotic proteolytic cascade, was able to cleave β-catenin in vitro.
Full-length β-catenin cDNA was translated in vitro and treated with caspase-3 from a bacterial lysate (Fernandes-Alnemri et al., 1995b ; Song et al., 1996; Xue et al., 1996).
Treatment with caspase-3 specifically cleaved β-catenin, producing a fragment of ∼65 kD, whereas lysates from bacteria not expressing caspase-3, grown and extracted under identical conditions, failed to cleave β-catenin (Fig. 6 a). Moreover, caspase-3–dependent proteolytic cleavage of β-catenin could be prevented by addition of the specific caspase inhibitor, the acidic tetrapeptide aldehyde Ac-DEVD-CHO (Fig. 6 b). It is worth noting that when Ac-DEVD-CHO was used at a final concentration of 20 nM, only a partial inhibition of the β-catenin cleavage occurred, and in parallel an intermediate proteolytic product was detected.
Figure 6.
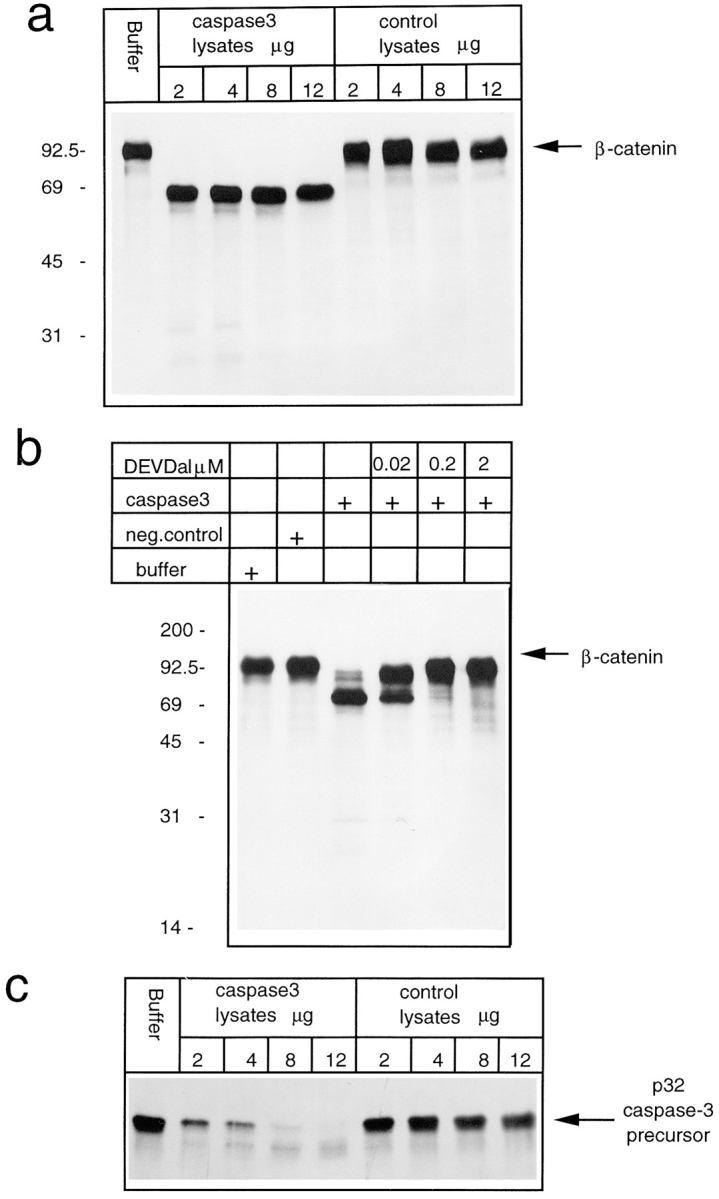
In vitro protease assays. (a) 35S labeled in vitro translated β-catenin was incubated for 1 h at 37°C with caspase-3 buffer alone or with the indicated amounts of caspase-3–expressing bacteria lysates or control bacteria lysates. (b) 35S labeled in vitro translated β-catenin was incubated for 1 h at 37°C with caspase-3 buffer alone, with control bacteria lysates, with caspase-3–expressing bacteria lysates, or in the presence of the indicated amount of caspase-3–specific inhibitor Ac-DEVD-CHO. (c) 35S labeled in vitro translated caspase-3 precursor was incubated for 1 h at 37°C with caspase-3 buffer alone, or with the indicated amount of caspase-3–expressing bacterial lysates or control bacterial lysates.
Having demonstrated that caspase-3 cleaves β-catenin, we tried to define whether β-catenin is a relevant substrate for this protease. Caspase-3, like other proteases of this family, can undergo autocatalytic cleavage, thus producing an intermediate form (p21) and two active forms, p17 and p12 (Fernandes-Alnemri et al., 1995b ; Nicholson et al., 1995; Martin et al., 1996; Wang et al., 1996; Xue et al., 1996).
Caspase-3 autoprocessing can be easily monitored by following the disappearance of the p32 precursor form using an in vitro protease assay (Fernandes-Alnemri et al., 1995b ; Martin et al., 1996; Wang et al., 1996; Xue et al., 1996). In vitro translated caspase-3 was incubated with increasing amounts of bacterial lysates expressing active caspase-3 and compared to the efficiency of β-catenin cleavage over the same time course.
The same caspase-3 activity that is responsible for full β-catenin processing could only partially self-cleave the in vitro translated caspase-3 precursor form (Fig. 6 c). In fact, 12 μg, the equivalent of total proteins from bacterial extracts expressing caspase-3, were required for full processing of caspase-3 precursor with respect to the 2 μg sufficient for full β-catenin processing. Caspase-3–dependent processing specificity of β-catenin is also strengthened by the observation that an amount of caspase-2 (ICH-1), enough for full autoprocessing, was unable to cleave β-catenin in vitro (data not shown).
In summary, these data indicate that β-catenin is a better substrate for caspase-3 than itself, thus strongly suggesting that β-catenin is a relevant caspase-3 substrate.
β-Catenin Is Cleaved at Different Sites by Caspase-3
Experiments using the caspase-3 inhibitor Ac-DEVD-CHO suggested that β-catenin could be cleaved at different sites. To study the overall pattern of β-catenin processing, we performed a time course analysis of β-catenin processing in vitro using bacterial lysates expressing caspase-3. In vitro translated β-catenin was incubated for the indicated times at 37°C with caspase-3–expressing or control bacterial lysates.
After 2.5 min of incubation, partial proteolytic processing of β-catenin could be detected (Fig. 7 a). A major cleavage product of ∼90 kD, in addition to the common but minor ∼65-kD form, was, in fact, revealed at this time. After a 10-min incubation, only the ∼65-kD form was present, thus suggesting that the ∼90-kD form could represent an intermediate degradation product of β-catenin. This result, however, suggested that caspase-3 has greater affinity for full-length β-catenin to generate the ∼90 kD than for the full-length or the ∼90 kD to generate the ∼65-kD form. Incubation with control bacterial lysates did not induce such proteolytic processing.
Figure 7.

(a) 35S labeled in vitro translated β-catenin was incubated for 1 h at 37°C with caspase-3 buffer (lane B), or with 2 μg of caspase-3–expressing bacterial lysates or control bacterial lysates for the indicated times. (b) Western immunoblots were performed on in vitro translated β-catenin and cellular lysates from (A) adherent nonapoptotic, and (NA) nonadherent apoptotic MDCK cells using antibodies against β-catenin or against VSV-tag. In vitro translated β-catenin was incubated for 1 h at 37°C with caspase-3 buffer (lane B) or with 2 μg of caspase-3–expressing bacterial lysates or control bacterial lysates.
As a first step to identify the sites of β-catenin that are cleaved by caspase-3, we constructed a tagged β-catenin with the in-frame VSV epitope at its carboxy terminus.
In vitro translated β-catenin–VSV was incubated for 2.5 or 60 min at 37°C with caspase-3-expressing or control bacterial lysates and then Western blot was performed using antibodies against β-catenin or VSV.
After 2.5 min of incubation with caspase-3, a prominent ∼90-kD form and a minor ∼65-kD band were revealed by antibodies against β-catenin. After 60 min of incubation, only the ∼65-kD form was detectable (Fig. 7 b).
Since antibodies against VSV failed to detect both the ∼90- and ∼65-kD cleaved forms of β-catenin, we can suggest that the carboxy tail is the first one to be proteolytically attacked by caspase-3.
Moreover, comparison between β-catenin in extracts from MDCK cells undergoing apoptosis revealed a similar electrophoretic mobility with respect to the in vitro, caspase-3 processed, ∼65-kD form of β-catenin.
From this analysis we can conclude that β-catenin, as synthesized in vitro, is cleaved by caspase-3 at at least two different sites generating the final ∼65-kD form that closely resembles the processed form previously identified in vivo during apoptosis.
To explain the observed processing dynamics, we can hypothesize an initial caspase-3–dependent cleavage event that removes a fragment of ∼2 kD located at the carboxy-terminal of β-catenin.
Cleavage of β-Catenin during Apoptosis Abolishes Its Ability to Bind α-Catenin
We next assessed whether β-catenin was cleaved at the carboxy-terminal domain during apoptosis in vivo. NIH3T3 were transfected with the β-catenin–VSV construct, apoptosis was induced by serum deprivation, and adherent and nonadherent cells were harvested separately for Western analysis.
The apoptotic cleaved form of β-catenin (65 kD) was detected in apoptotic cells (nonadherent) using the antibody specific for aa 571–781, but it was undetectable using antibodies against the VSV tag (Fig. 8 a). To exclude the possibility that during apoptosis the VSV tag was cleaved instead of the β-catenin carboxy-terminal region, we transfected the cytoplasmic adenylate kinase containing the VSV tag at its carboxy-terminus as a control. Apoptosis was similarly induced by serum deprivation, and nonadherent cells were harvested separately for Western analysis. Adenylate kinase VSV was similarly detected in both apoptotic and nonapoptotic cells (Fig. 8 a). These results indicate that the carboxy terminal domain of β-catenin is specifically processed during apoptosis at a site further upstream of the tag in vivo.
Figure 8.

(a) NIH3T3 cells transfected with β-catenin–VSV or with adenylate kinase were incubated in serum-free medium for 12 h. Adherent (A) and nonadherent (NA) cells were processed separately and Western analysis was performed using anti–β-catenin (aa 571–781) or anti–VSV-tag antibodies. (b) Growth-arrested NIH3T3 cells were incubated in serum-free medium for 12 h. Adherent (A) and nonadherent (NA) cells were processed separately and Western analysis was performed using anti–β-catenin (aa 571–781), or anti–amino-terminal β-catenin (aa 6–138) (McCrea et al., 1993). (c) NIH3T3 and MDCK cells were incubated for 12 h in serum-free medium. Adherent (A) and nonadherent (NA) cells were processed separately. Immunoprecipitation using anti–β-catenin was performed as described in Materials and Methods. The immunocomplexes were resolved in SDS-PAGE and processed for Western blot analysis with anti–α-catenin or anti–β-catenin as indicated.
To analyze whether the amino-terminal region of β-catenin was also cleaved during apoptosis in vivo, we used an antibody against the amino-terminal region of β-catenin (aa 6–138) (McCrea et al., 1993). Apoptosis was induced in NIH3T3 cells by serum deprivation and in MDCK by MMS treatment, and adherent and nonadherent cells were harvested separately for Western analysis.
As shown in Fig. 8 b, antibodies specific for the amino-terminal region recognize β-catenin only in nonapoptotic cells, thus indicating that the amino-terminal region of β-catenin is also proteolytically cleaved both in MDCK and NIH3T3 apoptotic cells.
Since the amino-terminal region of β-catenin is critically involved in binding α-catenin, we decided to analyze whether, during apoptosis, β-catenin was able to maintain the complex with α-catenin or, due to proteolytic cleavage at the amino-terminal, such a complex was compromised.
Apoptosis was induced in both NIH3T3 and MDCK cells by serum deprivation, and nonapoptotic and apoptotic floating cells were harvested separately. The same amount of proteins derived from apoptotic and nonapoptotic cell lysates were immunoprecipitated using anti–β-catenin antibody. After electrophoretic separation, the immunoblots were revealed with anti–α-catenin antibody.
Fig. 8 c shows that the same amount of α-catenin could be detected in the lysates of apoptotic and nonapoptotic cells used for immunoprecipitation. However, α-catenin was not found in complexes with the apoptotic form of β-catenin, whereas it was detected in complexes with β-catenin in the viable cells.
The same blots were also analyzed with antibodies against β-catenin to demonstrate that the two different forms of β-catenin were similarly immunoprecipitated under the different experimental conditions (Fig. 8 c).
These results clearly demonstrate that the proteolytic processing at the amino-terminal of β-catenin during apoptosis is the major determinant for the loss of interaction with α-catenin, which should, therefore, contribute to dismantling actin anchorage from the cell adhesion receptors.
Caspase-3–Dependent Proteolytic Processing of the Amino-Terminal Region of β-Catenin
To further confirm that the amino-terminal region of β-catenin was cleaved during apoptosis by a caspase, a deleted carboxy-terminal version of β-catenin (β-cateninΔCT), missing aa 422–781 was produced, and a VSV tag was introduced at the carboxy-terminal. Digestion of the in vitro translated β-cateninΔCT with caspase-3 produced a major band of ∼32 kD, which was completely suppressed by addition of the specific inhibitor Ac-DEVD-CHO (Fig. 9 a). Using decreasing amounts of caspase-3, a complex pattern of digestion products was observed, probably representing partial proteolytic products (Fig. 9 a). This evidence suggests the existence of multiple target sites for caspase proteases within the amino-terminal region of β-catenin.
Figure 9.

(a) 35S labeled in vitro translated β-cateninΔCT was incubated for 1 h at 37°C with control bacterial lysates or with increasing amount of caspase-3–expressing bacterial lysates. (b) 35S labeled in vitro translated β-cateninΔCTwtVSV, β-cateninΔCT162–164D/AVSV, and the β-cateninΔCT144–145D/AVSV were incubated with caspase-3–expressing bacterial lysates or with caspase-3–expressing bacterial lysates in the presence of 200 nM Ac-DEVD-CHO. Immunoprecipitations were performed using anti-VSV antibody.
As a first attempt to map the amino-terminal cleavage sites and to further confirm that caspase-3 cleaves the amino-terminal domain of β-catenin, we decided to perform a mutagenesis analysis. Different mutants of the β-catenin amino-terminal region were constructed by introducing point mutations that substitute the aspartic residue with alanine within the amino-terminal region. More specifically, the double-point mutants β-cateninΔCT144– 145D/A and β-cateninΔCT162–164D/A were created, and the resulting constructs were in vitro transcribed/translated. The in vitro translated products were then incubated with caspase-3–expressing bacterial lysates and immunoprecipitated using the antibody against the VSV tag.
As predicted by the existence of multiple cleavage sites, the different point mutants tested were cleaved by caspase-3, and Ac-DEVD-CHO completely abolished this processing (Fig. 9 b). The cleaved products were efficiently immunoprecipitated with the anti-VSV antibody, thus indicating that the multiple cleavage sites of β-cateninΔCT lie in the amino-terminal part of the protein (Fig. 9 b).
Most interestingly, different electrophoretic shifts were observed, distinguishing the proteolytic products of the double mutants 144–145D/A and 162–164D/A from the one generated from the wtβ-cateninΔCT (Fig. 9 b).
The small electrophoretic shift of the digested β-cateninΔCT162–164D/A suggests that at least one or both of the two aspartic residues represent cleavage sites for caspase-3. Since caspase-3 digestion of β-cateninΔCT144– 145D/A double mutants produced a larger electrophoretic shift, we can infer that the mutated 144–145 aspartic residues could represent caspase-3 cleavage sites.
A possible explanation for the described proteolytic pattern is that in vitro cleavage at residues 162 and/or 164 requires previous cleavage at position 144 and/or 145. Substitution of aspartic residues 144 and/or 145 should also interfere with proteolytic processing at position 162–164, thus leading to an incomplete digestion product with a more evident reduced electrophoretic mobility.
In summary, these results demonstrate that within the amino-terminal domain of β-catenin, different aspartic residues are targets for caspase-3 cleavage. More specifically, aspartic residues at position 162 and/or 164 and 144 and/or 145 of β-catenin serve as caspase-3 targets within the amino-terminal region.
Discussion
It is becoming evident that the caspase family of cellular proteases plays an important role in the execution phase of apoptosis (Kumar, 1995; Fraser and Evan, 1996; Nicholson, 1996). The activity of a protease results in the cleavage of various substrates. Some specific substrates for caspase proteases have been identified and in some cases a relationship with the apoptotic process could be clarified (Casciola-Rosen et al., 1994; Lazebnik et al., 1994; Brancolini et al., 1995; Emoto et al., 1995; Lazebnik et al., 1995; Goldberg et al., 1996; Janicke et al., 1996; Na et al., 1996; Song et al., 1996; Wang et al., 1996; Liu et al., 1997; Rutel and Bokoch, 1997). However, further identification of critical substrates for caspase proteases is fundamental to unveil the execution phase of apoptosis.
In this report we show that when apoptosis was induced either by complete growth factor removal or genotoxic stress, β-catenin becomes efficiently cleaved to produce a stable ∼65-kD form in both epithelial and fibroblast cells. β-Catenin is a ∼92-kD protein component of cell–cell contacts and adherens junctions having both structural and signaling functions (Miller and Moon, 1996). The reported cleavage of β-catenin seems to be (a) specific, since α-catenin, another component of adherens junctions, is not apparently processed during apoptosis, (b) coupled to the onset of apoptosis, since it was coincident with Gas2 activation, and (c) suppressed by the antiapoptotic protein Bcl-2 and proportional to the level of apoptotic insult.
The cleavage product generated after in vitro treatment of translated β-catenin protein with active caspase-3 (cpp32/ apopain/yama) migrates with the same apparent size as the β-catenin cleavage product of apoptotic MDCK cells, supporting the idea that in vivo such cleavage is also mediated by a caspase protease. Kinetics studies in vitro and functional studies in vivo indicate that β-catenin is cleaved at different sites, thus trimming both the amino- and carboxy-terminal regions of the protein.
The caspase family of proteases contains a QACXG pentapeptide in which the cysteine participates in catalysis and is characterized by the absolute requirement for an aspartic acid residue in the substrate P1 position (Thornberry et al., 1992). In addition to the requirement for a P1 Asp, caspase-3 shows preference for an anionic Asp residue in the P4 residue (DXXD) (Nicholson, 1996). A DXXD consensus site is located in the carboxy-terminal part of β-catenin (aa 760–764: DLMDG), thus possibly explaining the rapid cleavage of the carboxy-terminal site of the protein as observed in vitro after incubation with caspase-3. This sequence is similar to the DQMDG site in p35, a baculovirus antiapoptotic protein that is efficiently cleaved by caspase-3 (Xue and Horvitz, 1995).
We have demonstrated that aspartic residues 144 and/or 145, 162 and/or 164 are target sites for caspase-3 cleavage in vitro and that upstream, different aspartic residues within the amino-terminal region of β-catenin are also cleaved by caspase-3, none of them showing a canonical DXXD sequence. These observations raised the question whether caspase-3 is the protease involved in processing of the amino-terminal domain in vivo, or other caspases show higher affinity for such amino-terminal sites. Indeed, the recent generation of caspase-3 null mice (Kuida et al., 1996), clearly demonstrates that caspase-3 play a critical role during morphogenetic cell death in the mammalian brain but raises the possibility that other caspase proteases may have an important role during apoptosis in other tissues or in response to cell death-trigger.
Therefore, we can hypothesize that β-catenin could also be a target for other caspases; further studies are required to understand if other caspases are involved in β-catenin processing, and to identify the different aspartic residues cleaved during apoptosis.
Adherens junctions link cells together by organizing actin filaments to the plasmamembrane. The transmembrane receptors are the cadherins, a family of homophilic Ca2+-dependent cell–cell adhesion molecules (Takeichi, 1995). Their crystal structures suggest that cadherin-mediated cell adhesion is not based solely on the stability of association among individual molecules, but rather by the generation of a cell adhesion zipper which provides a mechanism to form strong intercellular bonds (Shapiro et al., 1995). In vivo strong intercellular bonds are dependent upon the association of the cadherin carboxy-terminal cytoplasmic domain to the central region of β-catenin and γ-catenin/plakoglobin (Nagafuchi and Takeichi, 1988; Ozawa et al., 1989; Hulsken et al., 1995; Sacco et al., 1995). Deletion of the β-catenin–binding site from cadherin cytoplasmic domain renders the cadherin nonfunctional in a cell aggregation assay (Nagafuchi and Takeichi, 1988; Ozawa et al., 1989).
The central function of β-catenin as a regulator of adherens junctions has been further demonstrated in different organisms using different experimental approaches (Peifer et al., 1993; Oyama et al., 1994; Haegel et al., 1995). It has been extensively demonstrated that β-catenin serves as an adapter to link cadherins to α-catenin and thereby to the actin cytoskeleton, since α-catenin seems to interact with both actin and α-actinin (Knudsen et al., 1995; Rimm et al., 1995). In this context, β-catenin seems to be the critical component for cadherin–catenin complexes in the regulation of cellular adhesiveness (Hamaguchi et al., 1993; Cowin and Bruke, 1996).
The fundamental roles of α-catenin in cadherin cytoskeleton interactions have also been clearly established in various cell types. Nonadhesive epithelial tumor cells lacking α-catenin can be induced to form tightly adherent epithelia after reintroduction of α-catenin (Watabe et al., 1994). Moreover, expression of E-cadherin–α-catenin chimeras has been reported to induce a strong and inflexible adhesive phenotype (Nagafuchi et al., 1994).
We have demonstrated in vivo that proteolytic processing of the β-catenin at its amino-terminal domain impairs its ability to bind α-catenin. Amino acid residues 120–151 of β-catenin, embedded in the amino-terminal domain and containing the first ARM repeat, are required for interaction with α-catenin (Aberle et al., 1994). This is confirmed in the human gastric cell line HSC-39 where a truncated β-catenin originated from a homozygous in-frame deletion that removes aa 28–134, cannot interact with α-catenin (Oyama et al., 1994; Kawanishi et al., 1995). The observations that: (a) an antibody raised against aa 6–138 (McCrea et al., 1993) of the amino-terminal region of β-catenin fails to detect the apoptotic form of β-catenin, and (b) aspartic residues 144 and/or 145, 162 and/or 164 are targets for caspase-3 cleavage in vitro, further confirming that proteolytic processing of β-catenin during apoptosis impairs interaction with α-catenin. Removal of the β-catenin domain responsible for interaction with α-catenin should contribute to disassemble the connections among the adhesion receptors, cadherins, and the actin cytoskeleton, thereby impairing a sustained cell–cell adhesive interaction (Fig. 10). An early marker of apoptosis in confluent cells is the retraction from neighboring cells, which is probably coincident with disassembly of cell–cell contacts. This process seems to be both temporally and spatially regulated and probably not exclusively dependent on β-catenin cleavage. In fact, β-catenin–related member, γ-catenin/plakoglobin, also plays a similar role in regulating cadherin adhesive activity in adherens junctions and in the specialized desmosomal junctions. The aspartic residues 144 and/or 145 and 162 and/or 164 possible target sites for caspase activity in β-catenin, are conserved among β-catenin, γ-catenin, and armadillo (the Drosophila form of β-catenin). Indeed, we also noticed that γ-catenin/plakoglobin was proteolytically processed both in vitro after incubation with caspase-3, and in vivo during apoptosis in fibroblast and epithelial cells (Brancolini, C., manuscript in preparation).
Figure 10.

Schematic representation of β-catenin cleavage during apoptosis. Apoptotic stimuli lead to activation of the caspase proteases, removing both a segment of β-catenin from the carboxy-terminal domain and from the amino-terminal domain including the first armadillo repeat (black arrows). Cleaved β-catenin is released from α-catenin, thereby losing the connections to the cytoskeleton. Unmapped caspase cleavage sites are indicated by hatched arrows.
It is interesting to note that E-cadherin complexes may promote cell survival and suppress cell death in epithelial cells (Hermiston and Gordon, 1995). In this context, cleavage of β-catenin might also suppress transduction of survival signals, consequently contributing to the one-way proceeding of the apoptotic process. The evidence that some components of microfilament system are substrates for the caspase proteases (Brancolini et al., 1995; Martin et al., 1996; Na et al., 1996), suggests that these proteases coordinate actin architectural changes during apoptosis and that such changes are important for a normal apoptotic phenotype (Cotter et al., 1992).
In conclusion, a complex reorganization of the microfilament system was observed during cell death: cells retract from adhesion substrate, sever the contacts with neighbors, and actin filaments are concentrated in the perinuclear area in a ring-like organization. Collapsing of the ring-like structure becomes evident with the proceeding of the morphological changes accompanied by nuclear fragmentation, membrane blebbing, and formation of the apoptotic bodies.
In this context, β-catenin and Gas2 processing seem to have different roles. Cleavage of Gas2 seems to be important to switch on the ability to rearrange the microfilament system, which may be relevant for the organization of the ring-like structure (Brancolini et al., 1995). Cleavage of β-catenin seems to be responsible for switching off its function as a regulator of adherens junctions, and is possibly instrumental for severing contacts with neighboring cells.
β-Catenin is also found in the nucleus and cytoplasm, where its function is not restricted to modulation of adherens junctions. Association of β-catenin with the tumor-suppressor gene product APC (adenomatous polyposis colon) (Rubinfeld et al., 1993) and the transcription factors LEF-1/XTcf-3 (Behrens et al., 1996; Molenaar et al., 1996) have, in fact, been reported. Moreover, β-catenin, together with the related protein γ-catenin/plakoglobin and Armadillo, is implicated in the Wnt/wingless signaling pathway, which determines the dorsoventral axis in Xenopus and segment polarity in Drosophila (for review see Miller and Moon, 1996).
To unveil the relationships among the specific roles of β-catenin in the regulation of cell growth/differentiation and its proteolytic processing during apoptosis will be an exciting challenge for the future.
Acknowledgments
We would like to thank A. Sgorbissa for help in some experiments. We are grateful to B.M. Gumbiner for providing anti–amino-terminal β-catenin antibody. D. Lazarevic is a European Association Cancer Research fellow. J. Rodriguez is a Howard Hughes Medical Institute Predoctoral Fellow.
This work was supported by Associazione Italiana Ricerca sul Cancro, Consorzio Nazionale delle Ricerche–Applicazioni Cliniche della Ricerca Oncologica, Ministero dell' “UNIVERSITA” and della Ricerca Scientifica and Tecnologica. 40% to C. Schneider, and CNR Progetto Strategico Ciclo cellulare e Apoptosi to C. Brancolini.
Abbreviations used in this paper
- aa
amino acid
- Gas
growth arrest specific protein
- ICE
interleukin-1β–convertase enzyme
- MMS
methylmethanesulfonate
Footnotes
Address all correspondence to Claudio Schneider, Laboratorio Nazionale, Consorzio Interuniversitario Biotechnologie, AREA Science Park, Padriciano 99, 34142 Trieste, Italy. Tel.: 39.40.398.985. Fax: 39.40.398.990. E-mail: sch@icgeb.trieste.it
References
- Aberle H, Butz S, Stappert J, Weissig H, Kemler R, Hoschuetzky H. Assembling of the cadherin catenin complex in vitro with recombinant proteins. J Cell Sci. 1994;107:3655–3663. doi: 10.1242/jcs.107.12.3655. [DOI] [PubMed] [Google Scholar]
- Alnemri ES, Livingston DJ, Nicholson DW, Salvesen G, Thornberry NA, Wong WW, Yuan J. Human ICE/CED3 protease nomenclature. Cell. 1996;87:171. doi: 10.1016/s0092-8674(00)81334-3. [DOI] [PubMed] [Google Scholar]
- Bardelli A, Longati P, Albero D, Goruppi S, Schneider C, Ponzetto C, Comoglio PM. HGF receptor associates with the antiapoptotic protein BAG-1 and prevents cell death. EMBO (Eur Mol Biol Organ) J. 1996;15:6205–6212. [PMC free article] [PubMed] [Google Scholar]
- Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. Functional interaction of β-catenin with the transcription factor LEF-1 . Nature (Lond) 1996;382:225–230. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- Beidler D, Tewari M, Friesen P, Poirier GG, Dixit V. The baculovirus p35 protein inhibits Fas- and tumor necrosis factor-induced apoptosis. J Biol Chem. 1995;270:16526–16528. doi: 10.1074/jbc.270.28.16526. [DOI] [PubMed] [Google Scholar]
- Boldin MK, Goncharov TM, Goltsev Y, Wallach D. Involvement of MACH a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell. 1996;85:803–815. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- Brancolini C, Bottega S, Schneider C. Gas2, a growth arrest-specific protein, is a component of the microfilament network system. J Cell Biol. 1992;117:1251–1261. doi: 10.1083/jcb.117.6.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brancolini C, Benedetti M, Schneider C. Microfilament reorganization during apoptosis: the role of Gas2, a possible substrate for ICE-like proteases. EMBO (Eur Mol Biol Organ) J. 1995;14:5179–5190. doi: 10.1002/j.1460-2075.1995.tb00202.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bump NJ, Hackett M, Hugunin M, Seshagiri S, Brady K, Chen P, Ferenz C, Franklin S, Ghayur T, Li P, et al. Inhibition of ICE family protease by baculovirus antiapoptotic protein p35. Science (Wash DC) 1995;269:1885–1888. doi: 10.1126/science.7569933. [DOI] [PubMed] [Google Scholar]
- Casciola-Rosen LA, Miller DK, Anhalt GJ, Rosen A. Specific cleavage of the 70-kD protein component of small nuclear ribonucleoprotein is a characteristic biochemical feature of apoptotic cell death. J Biol Chem. 1994;269:30757–30760. [PubMed] [Google Scholar]
- Chinnaiyan AM, O'Rouke K, Lane BR, Dixit VM. Interaction of CED-4 with CED-3 and CED-9: a molecular framework for cell death. Science (Wash DC) 1997;275:1122–1126. doi: 10.1126/science.275.5303.1122. [DOI] [PubMed] [Google Scholar]
- Cotter TG, Lennon SV, Glynn JM, Green DR. Microfilament disrupting agents prevent the formation of apoptotic bodies in tumor cells undergoing apoptosis. Cancer Res. 1992;52:997–1005. [PubMed] [Google Scholar]
- Cowin P, Bruke B. Cytoskeleton-membrane interactions . Curr Opin Cell Biol. 1996;7:56–65. doi: 10.1016/s0955-0674(96)80049-4. [DOI] [PubMed] [Google Scholar]
- Duan H, Chinnaiyan AM, Hudson PL, Wing JP, He W-W, Dixit VM. ICE-LAP3, a novel mammalian homologue of the Caenorhabditis eleganscell death protein Ced-3 is activated during fas- and tumor necrosis factor-induced apoptosis. J Biol Chem. 1996a;271:1621–1625. doi: 10.1074/jbc.271.3.1621. [DOI] [PubMed] [Google Scholar]
- Duan H, Orth K, Chinnaiyan AM, Poirier GG, Froelich CJ, He W-W, Dixit VM. ICE-LAP6, a novel member of the ICE/CED3 gene family, is activated by the cytotoxic T cell protease granzyme B. J Biol Chem. 1996b;271:16720–16724. doi: 10.1074/jbc.271.28.16720. [DOI] [PubMed] [Google Scholar]
- Ellis RE, Yuan J, Horvitz HR. Mechanisms and functions of cell death. Annu Rev Cell Biol. 1991;7:663–698. doi: 10.1146/annurev.cb.07.110191.003311. [DOI] [PubMed] [Google Scholar]
- Emoto Y, Manome Y, Meinhardt G, Kisaki H, Kharbanda S, Roberston M, Ghayur T, Wong WW, Kamen R, Weichselbaum R, Kufe D. Proteolytic activation of protein kinase Cδ by an ICE-like protease in apoptotic cells. EMBO (Eur Mol Biol Organ) J. 1995;14:6148–6156. doi: 10.1002/j.1460-2075.1995.tb00305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enari M, Talanian R, Wong W, Nagata S. Sequential activation of ICE-like and CPP32-like proteases during Fas-mediated apoptosis. Nature (Lond) 1996;380:723–726. doi: 10.1038/380723a0. [DOI] [PubMed] [Google Scholar]
- Faucheau C, Diu A, Chan AWE, Blanchet A-M, Miossec C, Herve F, Collard-Dutilleul V, Gu Y, Aldape RA, Lippke JA, et al. A novel human protease similar to the interleukin-1β converting enzyme induces apoptosis in transfected cells. EMBO (Eur Mol Biol Organ) J. 1995;14:1914–1922. doi: 10.1002/j.1460-2075.1995.tb07183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Litwack G, Alnemri ES. CPP32, a novel human apoptotic protein with homology to Caenorhabditis eleganscell death protein ced-3 and mammalian interleukin-1β–convertase enzyme. J Biol Chem. 1994;269:30761–30764. [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Litwack G, Alnemri ES. Mch2, a new member of the apoptotic CED3/ICE cysteine protease gene family. Cancer Res. 1995a;55:2737–2742. [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Takahashi A, Armstrong R, Krebs J, Fritz L, Tomaselli KJ, Wang L, Yu Z, Croce CM, Salveson G, et al. Mch3, a novel human apoptotic cystein protease highly related to CPP32. Cancer Res. 1995b;55:6045–6052. [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Armstrong C, Krebs J, Srinivasula SM, Wang L, Bullrich F, Fritz LC, Trapani JA, Tomaselli KJ, Litwack G, Alnemri ES. In vitro activation of CPP32 and Mch3 by Mch4, a novel human apoptotic cystein protease containing two FADD-like domains. Proc Natl Acad Sci USA. 1996;93:7464–7469. doi: 10.1073/pnas.93.15.7464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser A, Evan G. A license to kill. Cell. 1996;85:781–784. doi: 10.1016/s0092-8674(00)81005-3. [DOI] [PubMed] [Google Scholar]
- Gagliardini V, Fernandez PA, Lee RK, Drexler HC, Rotello RJ, Fishman MC, Yuan J. Prevention of vertebrate neuronal cell death by the crmA gene. Science (Wash DC) 1994;263:826–828. doi: 10.1126/science.8303301. [DOI] [PubMed] [Google Scholar]
- Garrels JI, Franza RB., Jr The REF52 protein database. J Biol Chem. 1989;264:5283–5298. [PubMed] [Google Scholar]
- Geiger B, Ayalon O. Cadherins. Annu Rev Cell Biol. 1992;8:307–332. doi: 10.1146/annurev.cb.08.110192.001515. [DOI] [PubMed] [Google Scholar]
- Goldberg YP, Nicholson DW, Rasper DM, Kalchmen MA, Koide HB, Graham RK, Bromm M, Kazemi-Esfarjani P, Thornberry NA, Vaillancourt JP, Hayden MR. Cleavage of huntingtin by apopain, a proapoptotic cysteine protease, is modulated by the polyglutamine tract. Nat Genet. 1996;13:442–449. doi: 10.1038/ng0896-442. [DOI] [PubMed] [Google Scholar]
- Gumbiner BM. Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell. 1996;84:345–357. doi: 10.1016/s0092-8674(00)81279-9. [DOI] [PubMed] [Google Scholar]
- Haegel H, Larue M, Ohsugi M, Fedorov L, Herrenknecht K, Kemler R. Lack of β-catenin affects mouse development at gastrulation. Development (Camb) 1995;121:3529–3537. doi: 10.1242/dev.121.11.3529. [DOI] [PubMed] [Google Scholar]
- Hamaguchi M, Matsuyoshi N, Ohnishi Y, Gotoh B, Takeichi M, Nagai Y. p60v-src causes tyrosine phosphorylation and inactivation of the N-cadherin-catenin cell adhesion system. EMBO (Eur Mol Biol Organ) J. 1993;12:307–314. doi: 10.1002/j.1460-2075.1993.tb05658.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermiston ML, Gordon JI. In vivo analysis of cadherin function in the mouse intestinal epithelium: essential roles in adhesion, maintenance of differentiation, and regulation of programmed cell death. J Cell Biol. 1995;129:489–506. doi: 10.1083/jcb.129.2.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoschuetzky H, Aberle H, Kemler R. β-Catenin mediates the interaction of the cadherin–catenin complex with epidermal growth factor receptor. J Cell Biol. 1994;127:1375–1380. doi: 10.1083/jcb.127.5.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulsken J, Birchmeier W, Behrens J. E-Cadherin and APC compete for the interaction with β-catenin and the cytoskeleton. J Cell Biol. 1994;127:2061–2069. doi: 10.1083/jcb.127.6.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh M, Yonemura S, Nagafuchi A, Tsukita S, Tsukita S. A 220-kD undercoat-constitutive protein: its specific localization at cadherin-based cell–cell adhesion sites. J Cell Biol. 1991;115:1449–1462. doi: 10.1083/jcb.115.5.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janicke RU, Walker PA, Lin XY, Porter AG. Specific cleavage of the retinoblastoma protein by an ICE-like protease in apoptosis. EMBO (Eur Mol Biol Organ) J. 1996;15:6969–6978. [PMC free article] [PubMed] [Google Scholar]
- Kawanishi J, Kato J, Sasaki K, Fujii S, Watanabe N, Niitsu Y. Loss of E-cadherin-dependent cell adhesion due to mutation of the β-catenin gene in a human cancer cell line, HSC-39. Mol Cell Biol. 1995;15:1175–1181. doi: 10.1128/mcb.15.3.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayalar C, Ord T, Testa MP, Zhong L-T, Bredesen DE. Cleavage of actin by interleukin 1β–converting enzyme to reverse DNase I inhibition . Proc Natl Acad Sci USA. 1996;93:2234–2238. doi: 10.1073/pnas.93.5.2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JFR, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuida K, Zheng TS, Na S, Kuan C-y, Yang D, Karasuyama H, Rakic P, Flavell RA. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature (Lond) 1996;384:368–375. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- Kumar S. ICE-like proteases in apoptosis. Trends Biochem Sci. 1995;20:198–202. doi: 10.1016/s0968-0004(00)89007-6. [DOI] [PubMed] [Google Scholar]
- Kumar S, Kinoshita M, Noda M, Copeland NG, Jenkins NA. Induction of apoptosis by the mouse Nedd2 gene, which encodes a protein similar to the product of the Caenorhabditis eleganscell death gene ced-3 and the mammalian IL-1β-converting enzyme. Genes Dev. 1994;8:1613–1626. doi: 10.1101/gad.8.14.1613. [DOI] [PubMed] [Google Scholar]
- Kundsen KA, Soler AP, Johnson KR, Wheelock MJ. Interaction of α-actinin with the cadherin/catenin cell–cell adhesion complex via α-catenin. J Cell Biol. 1995;130:67–77. doi: 10.1083/jcb.130.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature (Lond) 1994;371:346–347. doi: 10.1038/371346a0. [DOI] [PubMed] [Google Scholar]
- Lazebnik Y, Takahashi A, Moir RD, Goldman RD, Poirier GG, Kaufmann SH, Earnshaw WC. Studies of the lamin proteinase reveal multiple parallel biochemical pathways during apoptotic execution. Proc Natl Acad Sci USA. 1995;92:9042–9046. doi: 10.1073/pnas.92.20.9042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zou H, Slaughter C, Wang X. DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell. 1997;89:175–184. doi: 10.1016/s0092-8674(00)80197-x. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Amarante-Mendes GP, Shi L, Chuang T-H, Casiano CA, O'Brien GA, Fitzgerald P, Tan EM, Bokoch GM, Greenberg AH, Green DR. The cytotoxic cell protease granzyme B initiates apoptosis in a cell-free system by proteolytic processing and activation of the ICE/CED-3 family protease, CPP32, via a novel two-step mechanism. EMBO (Eur Mol Biol Organ) J. 1996;15:2407–2416. [PMC free article] [PubMed] [Google Scholar]
- Mashima T, Naito M, Kataoka S, Kawai H, Tsuruo T. Identification of Actin as substrate of ICE and an ICE-like protease and involvement of an ICE-LIKE protease but not ICE in VP-16-induced U937 apoptosis. Biochem Biophys Res Commun. 1995;217:1185–1192. doi: 10.1006/bbrc.1995.2894. [DOI] [PubMed] [Google Scholar]
- McCrea PD, Brieher WM, Gumbiner BM. Induction of a secondary body axis in Xenopusby antibodies to β-catenin. J Cell Biol. 1993;123:477–484. doi: 10.1083/jcb.123.2.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JR, Moon RT. Signal transduction through β-catenin and specification of cell fate during embryogenesis. Genes Dev. 1996;10:2527–2539. doi: 10.1101/gad.10.20.2527. [DOI] [PubMed] [Google Scholar]
- Miura M, Zhu H, Rotello R, Hartweig EA, Yuan J. Induction of apoptosis in fibroblasts by IL-1 β-converting enzymes, a mammalian homologue of the C. eleganscell death gene ced-3. Cell. 1993;75:653–660. doi: 10.1016/0092-8674(93)90486-a. [DOI] [PubMed] [Google Scholar]
- Molenaar M, van de Wetering M, Oosterwegel M, Peterson-Maduro J, Godsave S, Korinek V, Roose J, Destree O, Clevers H. XTcf-3 transcription factor mediates β-catenin–induced axis formation in Xenopus embryos. Cell. 1996;86:391–399. doi: 10.1016/s0092-8674(00)80112-9. [DOI] [PubMed] [Google Scholar]
- Muzio M, Chinnaiyan AM, Kischkel FC, O'Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz JD, Zhang M, Gentz R, et al. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- Na S, Chuang TH, Cunningham A, Turi TG, Hanke JH, Bokoch GM, Danley DE. D4-GDI, a substrate of CPP32, is proteolyzed during Fas-induced apoptosis. J Biol Chem. 1996;271:11209–11213. doi: 10.1074/jbc.271.19.11209. [DOI] [PubMed] [Google Scholar]
- Nagafuchi A, Takeichi A. Cell binding function of E-cadherin is regulated by the cytoplasmic domain. EMBO (Eur Mol Biol Organ) J. 1988;7:3679–3684. doi: 10.1002/j.1460-2075.1988.tb03249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagafuchi A, Ishihara S, Tsukita S. The roles of catenins in the cadherin-mediated cell adhesion: functional analysis of E-cadherin–α-catenin fusion molecules. J Cell Biol. 1994;127:235–245. doi: 10.1083/jcb.127.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson DW. ICE/CED3-like protease as therapeutic targets for the control of inappropriate apoptosis. Nat Biotechnol. 1996;14:297–301. doi: 10.1038/nbt0396-297. [DOI] [PubMed] [Google Scholar]
- Nicholson DW, Ambereen A, Thornberry N, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik Y, et al. Identification and inhibition of the ICE/CED3 protease necessary for mammalian apoptosis. Nature (Lond) 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- Oyama T, Kanai Y, Ochiai A, Akimoto S, Oda T, Yanagihara K, Nagafuchi A, Tuskita S, Shibamoto S, Ito F, et al. A truncated β-catenin disrupts the interaction between E-cadherin and α-catenin: a cause of loss of intercellular adhesiveness in human cancer cell lines. Cancer Res. 1994;54:6282–6287. [PubMed] [Google Scholar]
- Ozawa M, Baribault H, Kemler R. The cytoplasmic domain of the cell adhesion molecule uvomorulin associates with three independent proteins structurally related in different species. EMBO (Eur Mol Biol Organ) J. 1989;8:1711–1717. doi: 10.1002/j.1460-2075.1989.tb03563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano M, Tam SW, Theodoras AM, Beer-Romero P, Del Sal G, Chau V, Yew PR, Draetta GF, Rolfe M. Role of the ubiquitin-proteosome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science (Wash DC) 1995;269:682–685. doi: 10.1126/science.7624798. [DOI] [PubMed] [Google Scholar]
- Peifer M, Orsulic S, Sweeton D, Wieschaus E. A role for the Drosophila segment polarity gene armadilloin cell adhesion and cytoskeletal integrity during oogenesis. Development. 1993;111:1029–1043. doi: 10.1242/dev.118.4.1191. [DOI] [PubMed] [Google Scholar]
- Raff MC. Social controls on cell survival and death: an extreme view. Nature (Lond) 1992;356:397–400. doi: 10.1038/356397a0. [DOI] [PubMed] [Google Scholar]
- Rimm DL, Koslov ER, Kebriaei P, Cianci CD, Morrow JS. α 1(E)-catenin is an actin binding and bundling protein mediating the attachment of F-actin to the membrane adhesion complex. Proc Natl Acad Sci USA. 1995;92:8813–8817. doi: 10.1073/pnas.92.19.8813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinfeld B, Souza B, Albert I, Muller O, Chamberlain SH, Masiarz FR, Munemitsu S, Polakis P. Association of the APC gene product with β-catenin. Science (Wash DC) 1993;262:1731–1734. doi: 10.1126/science.8259518. [DOI] [PubMed] [Google Scholar]
- Rudel T, Bokoch GM. Membrane and morphological changes in apoptotic cells regulated by caspase-mediated activation of PAK2. Science (Wash DC) 1997;276:1571–1574. doi: 10.1126/science.276.5318.1571. [DOI] [PubMed] [Google Scholar]
- Sacco PA, McGranahan M, Wheelock MJ, Johnson KR. Identification of plakoglobin domains required for association with N-cadherin and α-catenin. J Biol Chem. 1995;270:20201–20206. doi: 10.1074/jbc.270.34.20201. [DOI] [PubMed] [Google Scholar]
- Shapiro L, Fannon AM, Kwong PD, Thompson A, Lehmann MS, Grubel G, Legrand JF, Als-Neilson A, Colman J, Hendrickson WA. Structural basis of cell-cell adhesion by cadherins. Nature (Lond) 1995;374:327–336. doi: 10.1038/374327a0. [DOI] [PubMed] [Google Scholar]
- Shimizu S, Eguchi Y, Kamiike W, Matsuda H, Tsujimoto Y. Bcl-2 expression prevents activation of the ICE protease cascade. Oncogene. 1996;12:2251–2257. [PubMed] [Google Scholar]
- Song Q, Less-Miller SP, Kumar S, Zhang N, Chan DW, Smith GCM, Jackson SP, Alnemri ES, Litwack G, Khanna KK, Lavin MF. DNA-dependent protein kinase catalytic subunit: a target for an ICE-like protease in apoptosis. EMBO (Eur Mol Biol Organ) J. 1996;15:3238–3246. [PMC free article] [PubMed] [Google Scholar]
- Song Q, Wei T, Lees-Miller S, Alnemri E, Watters D, Lavin MF. Resistance of actin to cleavage during apoptosis. Proc Natl Acad Sci USA. 1997;94:157–162. doi: 10.1073/pnas.94.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi A, Alnemri ES, Lazebnik Y, Fernandes-Alnemri T, Litwack G, Moir RD, Goldman RD, Poirier GG, Kaufmann S, Earnshaw WC. Cleavage of lamin A by Mch2a but not CPP32: multiple interleukin 1β–converting enzyme-related proteases with distinct substrate recognition properties are active in apoptosis. Proc Natl Acad Sci USA. 1996;93:8395–8400. doi: 10.1073/pnas.93.16.8395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeichi M. Morphogenetic role of classical cadherins. Curr Opin Cell Biol. 1995;7:619–627. doi: 10.1016/0955-0674(95)80102-2. [DOI] [PubMed] [Google Scholar]
- Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, Miller DK, Molineaux SM, Weidner JR, Aunins J, et al. A novel heterodimeric cysteine protease is required for interleukin-1β processing in monocytes. Nature (Lond) 1992;356:768–774. doi: 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- Wang L, Miura M, Bergeron L, Zhu H, Yuan J. Ich-1, an Ice/ ced3-related gene, encodes both positive and negative regulators of programmed cell death. Cell. 1994;78:739–750. doi: 10.1016/s0092-8674(94)90422-7. [DOI] [PubMed] [Google Scholar]
- Wang X, Zelenski NG, Yang J, Sakai J, Brown MS, Goldstein J. Cleavage of sterol regulatory element binding proteins (SREBPs) by CPP32 during apoptosis. EMBO (Eur Mol Biol Organ) J. 1996;15:1012–1020. [PMC free article] [PubMed] [Google Scholar]
- Watabe M, Nagafuchi A, Tsukita S, Takeichi M. Induction of polarized cell–cell association and retardation of growth by activation of the E-cadherin-catenin adhesion system in a disperse carcinoma line. J Cell Biol. 1994;127:247–256. doi: 10.1083/jcb.127.1.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyllie AH. The genetic regulation of apoptosis. Curr Opin Genet Dev. 1995;5:97–104. doi: 10.1016/s0959-437x(95)90060-8. [DOI] [PubMed] [Google Scholar]
- Wyllie AH, Kerr JFR, Currie AR. Cell death: the significance of apoptosis. Int Rev Cytol. 1980;68:251–306. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- Xue D, Horvitz HR. Inhibition of the Caenorhabditis eleganscell-death protease CED-3 by a CED-3 cleavage site in baculovirus p35 protein. Nature (Lond) 1995;377:248–251. doi: 10.1038/377248a0. [DOI] [PubMed] [Google Scholar]
- Xue D, Shaham S, Horvitz HR. The Caenorhabditis eleganscell-death protein CED-3 is a cystein protease with substrate specificities similar to those of the human CPP32 protease. Genes Dev. 1996;10:1073–1083. doi: 10.1101/gad.10.9.1073. [DOI] [PubMed] [Google Scholar]
- Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. The Caenorhabditis eleganscell death gene ced-3 encodes a protein similar to mammalian interleukin-1β–converting enzyme. Cell. 1993;75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]