

Abstract
Calnexin and calreticulin are homologous molecular chaperones that promote proper folding, oligomeric assembly, and quality control of newly synthesized glycoproteins in the endoplasmic reticulum (ER). Both are lectins that bind to substrate glycoproteins that have monoglucosylated N-linked oligosaccharides. Their binding to newly translated influenza virus hemagglutinin (HA), and various mutants thereof, was analyzed in microsomes after in vitro translation and expression in live CHO cells. A large fraction of the HA molecules was found to occur in ternary HA– calnexin–calreticulin complexes. In contrast to calnexin, calreticulin was found to bind primarily to early folding intermediates. Analysis of HA mutants with different numbers and locations of N-linked glycans showed that although the two chaperones share the same carbohydrate specificity, they display distinct binding properties; calreticulin binding depends on the oligosaccharides in the more rapidly folding top/hinge domain of HA whereas calnexin is less discriminating. Calnexin's binding was reduced if the HA was expressed as a soluble anchor-free protein rather than membrane bound. When the co- and posttranslational folding and trimerization of glycosylation mutants was analyzed, it was observed that removal of stem domain glycans caused accelerated folding whereas removal of the top domain glycans (especially the oligosaccharide attached to Asn81) inhibited folding. In summary, the data established that individual N-linked glycans in HA have distinct roles in calnexin/calreticulin binding and in co- and posttranslational folding.
The ER of most eukaryotic cells contains two homologous lectin-like chaperones called calnexin and calreticulin. Calnexin is a membrane protein and calreticulin a soluble lumenal protein. They interact transiently with a variety of newly synthesized glycoproteins by attaching to partially trimmed N-linked oligosaccharide moieties carrying a single glucose residue in the α 1–3 antenna (Ou et al., 1993; Hammond and Helenius, 1994; Hammond et al., 1994; Hebert et al., 1995; Peterson et al., 1995; Tector and Salter, 1995; Ware et al., 1995; Spiro et al., 1996). The association of these chaperones with their substrate glycoproteins promotes correct folding and oligomeric assembly, prevents degradation, and supports quality control (Rajagopalan and Brenner, 1994; Hebert et al., 1996; Vassilakos et al., 1996).
When transferred to growing nascent chains, the N-linked core oligosaccharides carry three glucoses. ER glucosidases I and II rapidly remove two of them, thus generating the monoglucosylated forms (Glc1Man7-9NAcGlc2) that serve as ligands for calnexin and calreticulin binding. The remaining single glucose residue is subsequently removed by glucosidase II, resulting in the dissociation of the chaperone complex (Hebert et al., 1995, 1996; Rodan et al., 1996; Van Leeuwen and Kearse, 1996). The monoglucosylated form of the oligosaccharides is also generated in the ER by the action of UDP-glucose/glycoprotein glucosyltransferase. This lumenal enzyme selectively reglucosylates glycoproteins that possess high mannose glycans only if the proteins are incompletely folded (Sousa et al., 1992; Trombetta and Parodi, 1992). Thus, by adding and removing glucoses, glucosidase II and the glucosyltransferase drive their substrates through a cycle of calnexin/calreticulin binding and release (Hammond and Helenius, 1993; Hebert et al., 1995; Van Leeuwen and Kearse, 1997). Glycoproteins stay in the cycle as long as they have a nonnative conformation, with the glucosyltransferase serving as a folding sensor (Suh et al., 1989; Hammond et al., 1994).
Although identical in their oligosaccharide specificity (Hammond et al., 1994; Peterson et al., 1995; Ware et al., 1995; Spiro et al., 1996), recent studies suggest that calnexin and calreticulin may differ in their substrate selection. Vesicular stomatitis virus G protein binds to calnexin but not to calreticulin (Hammond and Helenius, 1994; Peterson et al., 1995). The bound proteins observed by coimmunoprecipitation from pulse labeled cells are not identical (Peterson et al., 1995; Wada et al., 1995). The two chaperones bind to major histocompatibility complex (MHC) class I antigens at different stages of maturation (Sadasivan et al., 1996; Van Leeuwen and Kearse, 1996), and in the case of influenza virus hemagglutinin (HA),1 calreticulin dissociates more rapidly than calnexin as folding proceeds (Hebert et al., 1996). It is possible that the two chaperones have distinct functions during glycoprotein biosynthesis.
In this paper, we have addressed the functional differences between calnexin and calreticulin using the well characterized influenza HA as a model substrate. After mutating the seven N-linked glycosylation consensus sequences in different combinations, we observed that effects on calnexin and calreticulin binding were distinct. Calreticulin binding depended on the glycans located in the more rapidly folding, globular top domain whereas calnexin appeared to be able to bind to glycans all over the molecule. Evidence was obtained for the presence of ternary HA– calnexin–calreticulin complexes. We also analyzed the role of individual oligosaccharides in the folding process, and found that some of them affected both rate and efficiency.
Materials and Methods
Reagents
Components for the cell-free translation, translocation, and folding system (rabbit reticulocyte lysate, amino acids, DTT, and RNasin) were purchased from Promega Corp. (Madison, WI). The canine pancreas microsomes were a generous gift of R. Gilmore (University of Massachusetts Medical Center, Worcester, MA). Radiolabeled [35S]methionine/cysteine, oxidized glutathione (GSSG), and CHAPS (3-[3-cholamidopropyl]-dimethylammonino-1-propanesulfate), were purchased from Amersham Corp. (Arlington Heights, IL), Fluka Chemical Corp. (Ronkonkoma, NY), and Pierce Chemical Co. (Rockford, IL), respectively. All other chemicals were purchased from Sigma Chemical Co. (St. Louis, MO).
HA Mutants
The various HA glycosylation mutants were created by changing the Asn in the consensus glycosylation sequence to a Gln by standard molecular biological techniques (Sambrook et al., 1989). This included the use of a site-directed mutagenesis kit (QuikChange; Stratagene, La Jolla, CA). The truncated, soluble form of HA was generated by substituting the codon for the first transmembrane residue (T514) with the stop codon, TGA. Wild-type and mutant HA were cloned into a pBluescript expression system (Stratagene), and mRNA was transcribed (after linearization at the SalI or KpnI sites) with T7 RNA polymerase (Boehringer Mannheim, Indianapolis, IN).
Translation, Translocation, and Folding of HA
35S-Labeled HA was translated and translocated into canine pancreas microsomes as previously described (Hebert et al., 1995). HA was translated in the presence of 4.0 mM GSSG at 32°C for cotranslational folding studies. For posttranslational folding, HA was translated in the absence of GSSG. After 1 h of translation, protein synthesis was inhibited with 50 mM cycloheximide and 4.5 mM GSSG was added to initiate synchronous oxidation. Samples were treated at the indicated time points with 20 mM N-ethylmaleimide (NEM) to block free thiols by alkylation before immunoprecipitation.
Infection of CHO Cells and Metabolic Labeling
CHO cells were infected with influenza virus (X31) and labeled as described (Chen et al., 1995). The recombinant vaccinia virus with the T7 polymerase and the cowpox hr gene was gift of R. Drillien (CisINSERM, Strasbourg, France). The presence of cowpox hr gene allows the virus to infect CHO cells. The virus was propagated in BHK cells on 15-cm plates for 3 d after infection with 0.1 plaque formation units (pfu)/cell. The infected cells were homogenized and centrifuged at 2,000 g for 10 min. The supernatant was titered for plaque formation units.
For the infection of CHO cells with vaccinia virus, 6-cm dishes of nearly confluent cells were incubated at 37°C with 1-ml serum-free DME containing 2.5 × 107 virus. 30 min after incubation, the infection medium was aspirated, and 3 ml of transfection medium containing 20 μl lipofectamine and 4 μg HA DNA (plasmid carrying the HA mutant genes) were added and incubated at 37°C for 4 h. The cells were then starved in the cysteine-/ methionine-free medium for 30 min and then labeled with 200–250 μCi [35S]cysteine/methionine for 2 min. Immediately after labeling, the cells were washed twice with cold PBS containing 20 mM NEM, and lysed with 2.0% CHAPS in 50 mM Hepes, 200 mM NaCl, pH 7.5, containing 10 μg/ml each of chymostatin, leupeptin, antipain, and pepstatin (CLAP), 1 mM PMSF, 20 mM NEM, and 1 mM EDTA.
Immunoprecipitation and SDS-PAGE
Immunoprecipitation of 35S-labeled HA with anti-HA, -trimer specific HA, -calnexin, or -calreticulin antibodies was performed as described previously (Hebert et al., 1995, 1996). HA antibodies were a mixture of polyclonals raised to whole influenza virus and an anti-peptide polyclonal generated against a peptide corresponding to an NH2-terminal peptide of HA (NHA). Calreticulin antibodies were obtained from Affinity BioReagents, Inc. (Golden, CO), and calnexin antisera was raised against a peptide corresponding to the COOH-terminal cytosolic tail of calnexin. Immunoprecipitated HA was resolved on nonreducing and reducing (7.5%) SDS-PAGE.
Precipitation for cotranslational studies used the anti-peptide antibody corresponding to the NHA only. The sequential precipitations and the two-dimensional (2-D)-gel system used with the cotranslational studies were described previously (Chen et al., 1995). HA bands were quantified by densitometry with a digital gel scanner or phoshorimager (Visage 200; Molecular Dynamics, Inc., Sunnyvale, CA).
Results
Binding of Calnexin and Calreticulin to Partially Folded Forms of HA
To analyze how calnexin and calreticulin bind to partially folded forms of HA, we translated HA in vitro in the presence of canine pancreas microsomes and [35S]methionine/ cysteine. The redox conditions were adjusted so that the translocated HA molecules would undergo disulfide bond formation and folding (Hebert et al., 1995, 1996). After synthesis, the microsomes were treated with NEM to alkylate-free sulfhydryl groups, and solubilized with nonionic detergent. After immunoprecipitating with antibodies to HA, calnexin, or calreticulin, the samples were analyzed without reduction by SDS-PAGE and autoradiography.
Our previous studies have shown that the newly synthesized, nonreduced, full-length HA runs as three bands (Braakman et al., 1991; Marquardt et al., 1993; Braakman, I., and A. Helenius, manuscript in preparation). The band called IT1 corresponds to an ensemble of incompletely folded HA molecules that lack disulfide bonds C52–C277 and C14–C466. These two disulfide bonds form large loops in the HA molecule, and their presence considerably increases the electrophoretic mobility of the SDS complexes. The IT2 band contains molecules that have disulfide C52– C277 but do not contain C14–C466. The fastest migrating band (NT, for native) is composed of monomeric molecules that have both of these disulfide bonds.
The coimmunoprecipitations in Fig. 1 (A–C, lanes 1–3) indicate that calreticulin bound preferentially (but not exclusively) to the less oxidized folding intermediates IT1 and IT2. In contrast, calnexin bound to all three forms, IT1, IT2, and NT. When quantified by gel scanning, the average IT1/IT2/NT binding ratio in 10 experiments was 1.0: 0.65:0.63 for calnexin and 1.0:0.57:0.16 for calreticulin, with normalization with anti-HA precipitations. Since folding of HA proceeds from IT1 and IT2 to NT (Braakman et al., 1991; Marquardt et al., 1993), the results indicate that calreticulin participates mainly in early stages of folding whereas calnexin association occurs throughout the folding of HA monomers.
Figure 1.

Calnexin and calreticulin differ in their association with HA in microsomes. 35S-HA was translated in vitro in the presence of canine pancreas microsomes at 32°C under oxidizing conditions (A–C, Co, lanes 1–3), or under reducing conditions. At time zero (lane 4), GSSG was added to initiate posttranslational oxidation (lanes 5–9). Alkylated samples were divided into three fractions for immunoprecipitation with anti-HA (A, HA), anti-calnexin (B, CNX), or anti-calreticulin antibodies (C, CRT), and then subjected to nonreducing SDS-PAGE and visualized by autoradiography. Partially oxidized intermediates IT1 and IT2 and the fully oxidized native HA (NT) were resolved.
Differential binding of calnexin and calreticulin was also observed when HA folding occurred posttranslationally. In this case translation was performed in the presence of a high concentration of the reducing agent DTT, and folding began posttranslationally with the addition of GSSG (Fig. 1, lanes 4–9) (Marquardt et al., 1993). The presence of DTT not only inhibits formation of disulfide bonds in HA but also prevents efficient binding of calnexin and calreticulin (Fig. 1, lane 4; Hebert et al., 1995). When GSSG was added, calnexin and calreticulin associated with HA. The same preference of calreticulin for IT1 and IT2 was seen again (Fig. 1, lanes 5–9), indicating that it was determined by the intrinsic properties of the HA molecule. It did not depend on the vectorial translocation of the growing chain into the ER, or on limited accessibility of the HA polypeptide within the translocon complex.
To determine whether the differences in calnexin and calreticulin binding could be observed in live cells, we used a 2-D SDS-PAGE system recently developed in our lab to simultaneously monitor folding of nascent and full-length HA chains. CHO cells infected with influenza virus were pulse labeled for 1.5 min with [35S]methionine/cysteine. Some experiments were performed in the presence of castanospermine, a competitive inhibitor of the ER α-glucosidases, to inhibit calnexin and calreticulin binding to HA (Hammond et al., 1994; Chen et al., 1995; Hebert et al., 1995; Peterson et al., 1995). The cells were alkylated, solubilized, and the lysates subjected to immunoprecipitation with antibodies against the NH2-terminal peptide of HA, to calnexin or to calreticulin. To separate the chaperone-bound HA molecules from other substrates, the anti-calnexin and -calreticulin precipitates were dissolved and reprecipitated with the anti-HA antibodies. The samples were then subjected to 2-D SDS-PAGE in which the first dimension was without reduction and the second with reduction (Chen et al., 1995). Proteins that lack disulfide bonds run on the diagonal, proteins with intermolecular disulfides migrate above the diagonal, and proteins with intramolecular disulfides, as a general rule, run below the diagonal.
The three spots at the extreme right of Fig. 2 A represent the labeled IT1, IT2, and NT forms of the full-length HA. The spurs on the left of the spots emanating diagonally correspond to the labeled nascent chains of variable length (Chen et al., 1995). The spur connected to the IT1 spot contains nascent chains that lack disulfide bonds C52– C277 and C14–C466. They may contain one or more of the small intramolecular disulfide loops. The spur connecting to the IT2 spot corresponds to nascent chains that had disulfide C52–C277 but not C14–C466.
Figure 2.

Cotranslational association of HA with calnexin and calreticulin. Influenza-infected CHO cells were pulsed for 1.5 min with [35S]methionine/cysteine, and then the lysate was divided into three, immunoprecipitated, and analyzed by 2-D SDS-PAGE (nonreducing in the first dimension and reducing in the second dimension). (A) HA immunoprecipitated with an antibody against the NH2 terminus of HA (α-NHA). (B) HA immunoprecipitated sequentially with antibody against calnexin and antibody against HA NH2 terminus (α-CNX/α-NHA). (C) HA immunoprecipitated sequentially with antibody against calreticulin and antibody against HA NH2 terminus (α-CRT/α-NHA). The circles above the 2-D gels denote molecular markers of 66 and 45 kD. Exposure time for B and C was five times longer than A.
It is apparent from the gel patterns in Fig. 2, A–C that both nascent and full-length HA molecules associate with calnexin and calreticulin in living cells. Association was dependent on the trimming of glucoses from the N-linked oligosaccharides because it was blocked by castanospermine (data not shown) (Hammond et al., 1994; Chen et al., 1995; Peterson et al., 1995). From the length of the main spur, it could be estimated that binding occurred by the time the length of the nascent chains was ∼40 kD. Among the full-length forms, calreticulin bound preferentially to the least folded molecules (IT1) whereas calnexin bound to all forms (IT1, IT2, and NT).
Here for the first time, we found that the soluble and lumenal calreticulin has access to the growing nascent chains in the ER of living cells. These results also confirmed that calreticulin participates mainly in the early stages of folding whereas calnexin is involved throughout. The capacity of the chaperones to bind to nascent chains indicated, moreover, that trimming of the two outermost glucoses from the N-linked glycans can occur on the growing nascent chain in <1 min after addition of the glycan by the oligosaccharyl transferase.
HA Forms Ternary Complexes with Calnexin and Calreticulin
The HA monomer has seven N-linked glycans. Four are located in the stem domain attached to asparagines 8, 22, 38, and 483. One is in the hinge region (Asn 285), and two (Asn 81 and Asn 165) are in the top domain (Fig. 9). With so many glycans distributed widely over the surface, the protein has the potential of binding more than one calnexin or calreticulin molecule at a time.
Figure 9.
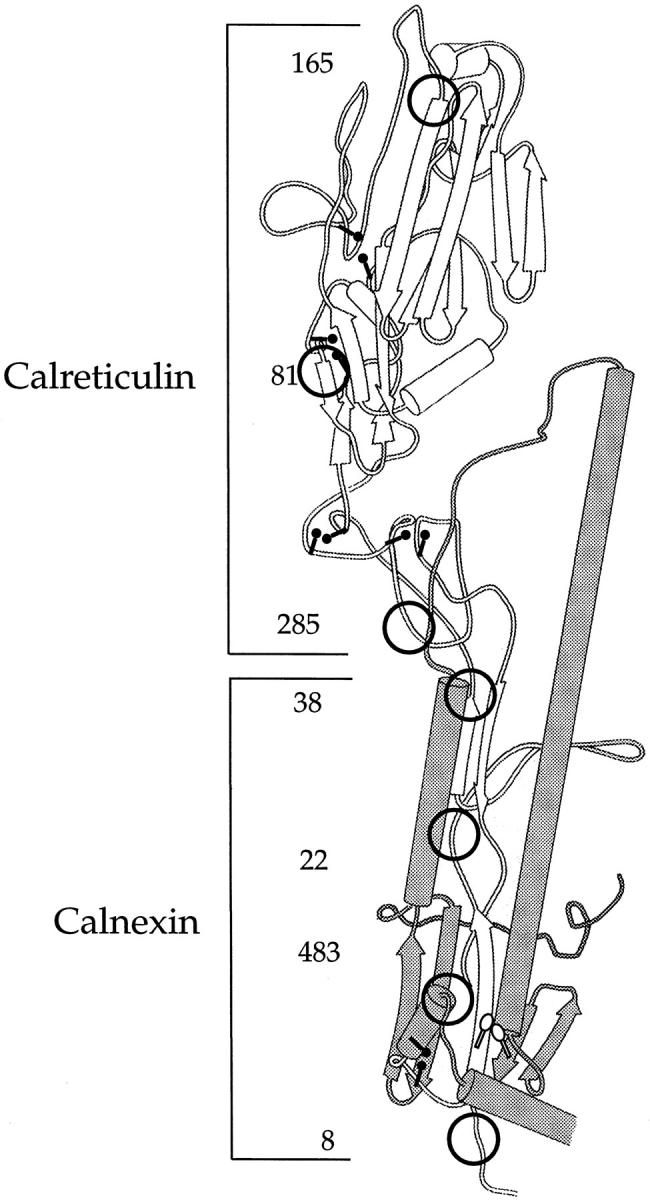
Calnexin and calreticulin binding regions on HA. The structure of the ectodomain of HA, modified from Wiley and co-workers (Wilson et al., 1981) is depicted with N-linked glycans and the disulfide bonds designated by the circles and the ball-and-sticks, respectively. The large disulfide loop, Cys 14-466, is represented by the unfilled ball-and-sticks. The numbers refer to the Asn residue that contain the N-linked glycosylations.
To test whether ternary complexes containing HA and both of the chaperones occurred in the ER-derived microsomes, sequential double immunoprecipitations were performed (Fig. 3, A and B). Primary precipitations with α-HA, -calnexin, or -calreticulin (Hebert et al., 1996) showed that 53% of the 35S-labeled HA was associated with calnexin, and 45% with calreticulin (Fig. 3, lanes 3 and 6, respectively). When the supernatants from these precipitations were reprecipitated with the same antibodies, no additional HA was brought down (Fig. 3, lanes 2, 4, and 7), indicating that the primary precipitations were quantitative. In contrast, when aliquots of the supernatants were reprecipitated after the first precipitation with antibodies against the second chaperone, additional HA was precipitated (Fig. 3, lanes 5 and 8). This HA corresponded to molecules that were associated only with one of the chaperones. The relatively small amount of HA in these complexes suggested that most of the complexes contained both chaperones.
Figure 3.

HA is found in calnexin–calreticulin ternary complexes. The presence of ternary calnexin–calreticulin–HA complexes was analyzed using double immunoprecipitations. (A) HA was translated as in Fig. 1 for 1 h at 32°C under oxidizing conditions. HA was first immunoprecipitated with anti-HA (1°, HA, lanes 1 and 2), -calnexin (1°, CNX, lanes 3–5), and -calreticulin (1°, CRT, lanes 6–8) antisera. The supernatants were cleared with protein A and reprecipitated with the indicated antisera (2°). (B) Bands from the autoradiogram in A were quantified by a digital densitometer and plotted as the fraction of total HA (IT1 + IT2 + NT from lane 1).
When the radioactivity in the various bands was quantified using a phosphorimager (Fig. 3 B), it was found that 10% of the HA was present in calreticulin–HA complexes and 8% in calnexin–HA complexes. As much as 40% of the HA occurred in ternary calnexin–calreticulin–HA complexes. A large fraction of the HA was thus associated with both chaperones simultaneously. Consistent with calreticulin's preference for IT1 and IT2, the ternary complexes and the binary calreticulin–HA complexes were enriched in IT1 and IT2 (Fig. 3, lanes 6 and 5, respectively). The binary calnexin–HA complexes contained more HA of the fully oxidized NT type (Fig. 3, lane 8). It could be concluded that the majority of the chaperone-bound HA occurred in complexes containing both calnexin and calreticulin.
Maturation and Calnexin/Calreticulin Binding of Single Glycosylation Site Mutants
Since a large amount of HA was associated with both chaperones, it was of interest to determine whether these chaperones bound to different regions of the HA molecule. A series of mutants were constructed in which glycosylation sites were eliminated by point mutations in the consensus glycosylation sites. Typically, the Asn in the consensus sequence was replaced with Gln. In specific cases, other changes were also made to confirm that the changes were due to the loss of the oligosaccharide and not to the amino acid change.
In the first series of mutants, glycosylation sequences were eliminated one by one. The mutant proteins were translated in the presence of microsomes and analyzed for folding and oligomerization as well as calnexin/calreticulin binding. We found that six out of seven mutants were able to fold to the NT form (Fig. 4 A). The same mutant proteins also assembled into trimers, judging by immunoprecipitation, with a trimer-specific anti-HA monoclonal antibody (Fig. 4 B). Clearly, the oligosaccharide moieties in positions 8, 22, 38, 165, 285, and 483 were dispensable when eliminated singly.
Figure 4.

Folding, oligomerization, and calnexin/calreticulin binding of single glycan deletion mutants. (A) Wild-type and single glycosylation mutants of HA were translated under oxidizing conditions in the presence of canine pancreas microsomes at 32°C for 1 h. At the indicated times, samples were removed, alkylated, lysed, and immunoprecipitated with anti-HA antibodies. HA was resolved by nonreducing SDS-PAGE and autoradiography. (B) Translations were performed as above with oxidation carried out for 8 h at 32°C. The alkylated HA was immunoprecipitated with anti-trimer specific HA antibodies and resolved by reducing SDS-PAGE and autoradiography. (C) Translation of 35S-labeled HA were performed as in A for 1 h at 32°C. After alkylation, HA was immunoprecipitated with anti-HA, -calnexin, and -calreticulin antibodies. HA was resolved upon nonreducing and reducing SDS-PAGE and autoradiography. The fraction HA coprecipitating with calnexin or calreticulin was calculated as the amount of anti-calnexin or -calreticulin precipitable HA divided by the total HA precipitated with anti-HA antibodies as described in Fig. 3.
However, the folding of some of these six mutant molecules was not identical to the wild type in every respect. For example, whereas the rate of NT formation for mutants Δ8, Δ22 and Δ38 was similar to wild type, only minor amounts of IT2 was seen as intermediates. Evidently, the transition from IT2 to NT (i.e., the formation of the disulfide C14–C466) was accelerated.
Deletion of the COOH-terminal stem glycan in Δ483, on the other hand, resulted in oxidation to NT at a rate faster than the wild type (Fig. 4 A, below in Fig. 6 B, and data not shown). Such differences indicated that although changes in these six glycosylation sites did not affect the final outcome of the folding process, they did affect the kinetics of folding and the expression of intermediate forms.
Figure 6.

Determination of the number of glycans required for calnexin or calreticulin binding. Consensus glycosylation sequences were deleted by increasing, in single increments, from the NH2 to the COOH terminus (lanes 2–8), and from the COOH to the NH2 terminus (lanes 9–15). 35S-HA was translated in vitro for 1 h at 32°C, alkylated, lysed, and precipitated with anti-HA, -calnexin (CNX), or -calreticulin (CRT) antisera. The labeled HA was resolved by nonreducing (A, NR) and reducing (B–D, RD) SDS-PAGE and autoradiography. The band designated as UT corresponds to the untranslocated and unglycosylated HA.
Mutant Δ81, which lacks one of the two oligosaccharides present in the top domain, was an exception. It was greatly inhibited in its ability to fold beyond a form that was either fully reduced or equivalent to IT1 with some of the small disulfide bonded loops in place (Fig. 4 A). Surprisingly, this protein seemed unable to form interchain disulfide bonds which are common for misfolded forms of HA (Hurtley et al., 1989; Braakman et al., 1992b ). Abolishing the glycosylation site by a T83A mutation rather than the N81Q mutation resulted in the same folding phenotype (data not shown) indicating that the effect was not due to a specific amino acid change. The inability of Δ81 to oxidize was likely due to disruption in the formation of disulfide C67– C76, which is very close to this glycan (see Fig. 9), and known to be crucial for formation of other intrachain disulfides in the HA monomer (Braakman, I., and A. Helenius, manuscript in preparation).
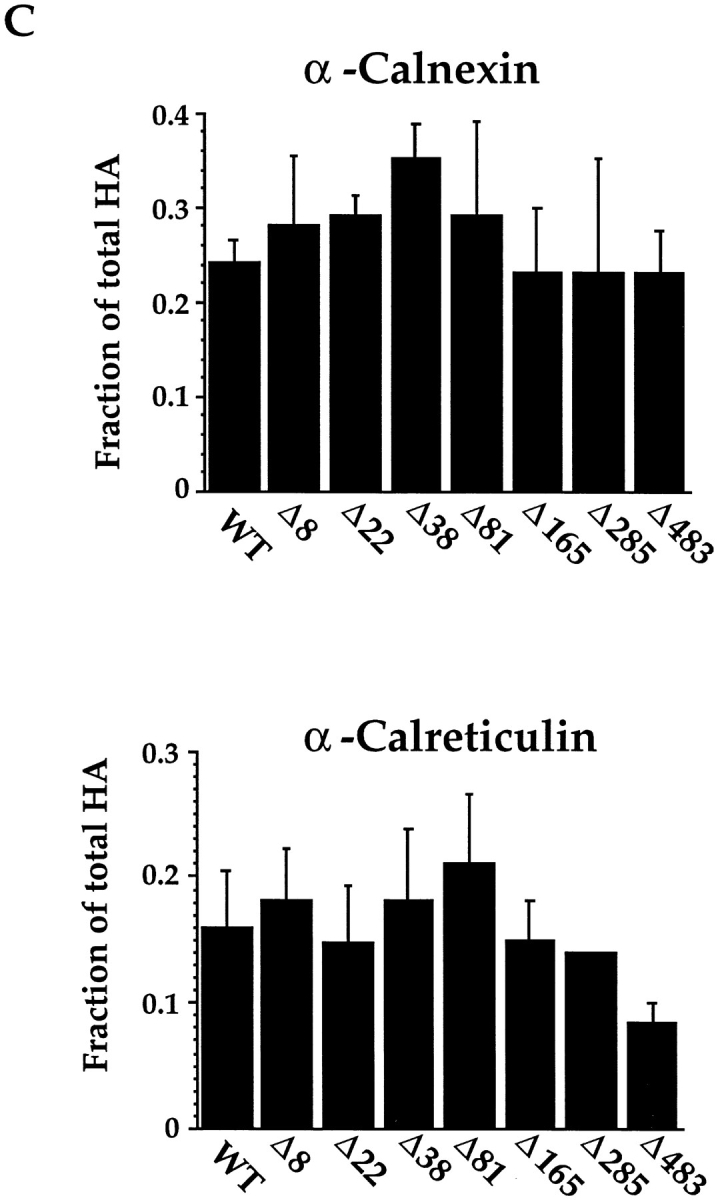
When the calnexin and calreticulin binding to the mutant proteins was measured by coimmunoprecipitation, no significant differences compared to wild-type HA were seen for any of them, including Δ81 (Fig. 4 C). With exception of Δ483, they also bound normal levels of calreticulin. The 40% reduction in calreticulin binding observed for Δ483 was likely caused by the rapid folding of this mutant HA, resulting in a shorter exposure of the preferred calreticulin binding conformers, IT1 and IT2 (see below). We concluded that no single glycan in HA served as the exclusive binding site for either calnexin or calreticulin.
Calreticulin Binds Preferentially to Top and Hinge Glycans
Since we were unable to disrupt binding of calnexin or calreticulin with single glycan deletions, combinations of multiple deletions were tested. Depending on the combination, differences in chaperone binding now emerged. Elimination of the three sites in the top/hinge domain (Δ81,165,285) resulted in almost complete loss of calreticulin binding (Fig. 5 A). In contrast, the mutant protein bound almost normal amounts of calnexin. The top/hinge glycans were evidently needed for calreticulin but not for calnexin binding.
Figure 5.

Mapping of calnexin and calreticulin binding sites by mutating consensus glycosylation sequences. The consensus sequences for glycosylation were eliminated by changing the Asn to Gln. The HA was translated under oxidizing conditions for 1 h at 32°C. (A) The fraction of total HA coprecipitating with the various antisera is plotted. (B) Binding to calnexin and calreticulin was monitored as described above for a set of HA mutants which had, in addition to mutations in the designated consensus glycosylation sites, an additional mutation, C305S, which arrested HA oxidation in the IT1 conformation. The disruption of disulfide 281– 305 by changing Cys305 to a Ser results in the accumulation of nonaggregated IT1 (not shown). The values given are the average of four experiments. (C) HA binding to calnexin and calreticulin in CHO cells was quantified as in A and B. 35S-HA was expressed in CHO cells by use of the T7-based vaccinia expression system. 4 h after infection, cells were pulsed with [35S]methionine/cysteine for 2 min at 37°C. Alkylated and lysed samples were then processed as above.
Surprisingly, the reverse experiment in which three stem glycans (Δ8,22,38) were eliminated also showed reduced binding of calreticulin (Fig. 5 A), whereas calnexin binding was only marginally reduced. Further studies showed that the apparent inability of calreticulin to bind to this particular mutant was caused by its accelerated folding (see below). The early oxidative intermediates IT1 and IT2 that normally bind calreticulin were so short-lived in this mutant that the calreticulin-binding phase during folding could not be experimentally observed.
To correct for this effect, an additional mutation (C305S) was introduced. This mutation prevents the formation of disulfide bond C281–C305 which is needed to convert IT1 and IT2 to NT (Braakman, I., and A. Helenius, manuscript in preparation). It effectively traps the protein in a form equivalent to early folding intermediates. With this mutation in place, the protein stayed as IT1 (data not shown). In this mutant background, the loss of the three-stem domain glycans in positions 8, 22, and 38 had little or no effect on calreticulin binding (Fig. 5 B). However, removal of the three top/hinge domain glycans resulted in a 64% drop, confirming the preferential binding of calreticulin to the top/hinge glycans. Removal of all three glycans was required for calreticulin binding to be affected. Calnexin binding was again virtually unaffected by the loss of glycans, suggesting that it can bind both to top/hinge and the stem domain glycans.
Calnexin and Calreticulin Binding to HA Mutants in Living Cells
To test whether elimination of multiple glycans would have similar effects on calreticulin and calnexin binding in live cells, wild-type HA and two mutants were expressed in CHO cells using the T7 vaccinia virus system. In addition to including the C305S mutation to prevent full oxidation, the mutant proteins tested were devoid of top and hinge oligosaccharides (Δ81,165,285/C305S) and stem glycans (Δ8,22,38/C305S). 3 h after transfection, the cells were pulse labeled for 2 min. After alkylation and solubilization (Braakman et al., 1991), lysates were immunoprecipitated. SDS-PAGE was performed and the HA bands quantified using the phosphorimager.
Calnexin binding to Δ8,22,38/C305S and Δ81,165,285/ C305S mutants was found to be 33% lower than to wild-type HA (Fig. 5 C). A similar decrease was observed in the binding of calreticulin to Δ8,22,38/C305S, but its binding to Δ81,165,285/C305S was found to be dramatically decreased. Taken together, the results suggest that in live cells, as well as in microsomes, calreticulin binds preferentially, but not exclusively, to the top domain glycans. Calnexin, on the other hand, can use glycans in any part of the protein. Since the stem disulfides are the last to form during folding (Braakman et al., 1991), the results are consistent with the observation that calnexin binding continues for a longer period during HA folding than calreticulin binding (Fig. 1).
Calnexin and Calreticulin Binding to Mutants with Decreasing Numbers of Oligosaccharide Moieties
That calnexin can bind to many parts of the HA molecule was confirmed by a series of mutants in which glycans were progressively removed one at a time from the NH2 terminus towards the COOH terminus (Fig. 6, lanes 2–8), or conversely, from the COOH terminus to the NH2 terminus (Fig. 6, lanes 9–15). The stepwise loss of glycans resulted in a ladder of HA molecules with faster electrophoretic mobilities the fewer the oligosaccharides. Thus, with about 2-kD steps for each glycan removed, the HA band approached the mobility of the untranslocated HA (denoted as UT, Fig. 6 A) present in the lysates. In the nonreduced gels (Fig. 6, B–D), UT could not be seen because it formed disulfide cross-linked aggregates under the oxidizing conditions used.
Immunoprecipitation with anti-calnexin antibodies showed that calnexin binding to the various mutant HA molecules did not diminish significantly with decreasing number of glycans until a molecule was reached that had only a single glycan (Fig. 6 C, compare lanes 1–6 with 7 and 8, and lanes 9–13 with 14 and 15). In other words, calnexin formed stable immunoprecipitable complexes with all HA molecules except those that had less than two glycans. This finding was consistent with observations using other substrate glycoproteins (Cannon et al., 1996; Rodan et al., 1996).
In the case of calreticulin (Fig. 6 D), binding already started to drop significantly when three or four out of the seven glycans were lost (Fig. 6, compare lanes 1–5 with 6–8, and lanes 9–11 with 12–15). In this case interpretation was, however, more complicated because some of the mutants folded faster than others. If the early folding events are rapid, less binding of calreticulin is expected to occur. However, the results were consistent with the multiple glycan deletion mutants discussed above (Fig. 5, B and C) that showed that the presence of at least one of the top domain glycans is required for calreticulin binding.
Folding of Glycosylation Mutants in Microsomes and Live Cells
It was evident from Fig. 6 B, which shows the nonreduced anti-HA precipitates, that the progressive loss of oligosaccharides not only affects calnexin and calreticulin binding, but also influences how HA folds. The accelerated folding of mutants lacking glycans in positions N8, 22, and 38 is clearly visible (Fig. 6 B, compare lanes 3 and 4 with 1). When more than half of the wild-type HA was still in the form of IT1 and IT2 (Fig. 6 B, lane 1), most of mutants Δ8, 22 (Fig. 6, lane 3) and Δ8,22,38 (Fig. 6, lane 4) had been converted to NT. Folding was also accelerated in mutants in which the most COOH-terminal glycan (Δ483; Fig. 6 B, lane 9 or Fig. 4 A), or the two most COOH-terminal glycans (Δ285,483, Fig. 6 B, lane 10), had been eliminated.
It was also evident that folding was severely suppressed when four or more glycans were removed starting from either the NH2 or the COOH terminus (Fig. 6 B, lanes 5–8 and 12–15). This was not unexpected since the fourth glycosylation site from either terminus is N81, which, when removed, alone caused inhibition of NT formation and HA trimerization (Fig. 4, A and B).
To test whether folding of glycosylation mutants would also be abnormal in live cells, we expressed the wild-type HA and two mutants (Δ8,22,38 and Δ81) in CHO cells using the T7-based vaccinia virus expression. The 2-D SDS-PAGE system already described above was used to analyze the folding status of immunoprecipitated nascent and full-length HA molecules after a 2-min pulse of [35S]methionine/cysteine.
The Δ8,22,38 mutant and the wild type displayed quite different 2-D gel patterns (Fig. 7, A and B). Whereas the IT1 spur was visible both in the wild type and in the mutant, the IT2 spur in the mutant was longer and more prominent. This indicated that cotranslational formation of disulfide C52–C277 occurred earlier and more efficiently. The NT spot was also more prominent, suggesting more rapid formation of the C14–C466 disulfide. We concluded that co- and posttranslational folding of HA was accelerated when the three glycans in the NH2-terminal part of the sequence were eliminated.
Figure 7.

Cotranslational folding of wild-type and mutant HA, Δ8,22,38, and Δ81. CHO cells were infected with recombinant vaccinia virus and then transfected with the various HA genes. 4 h after transfection, the cells were starved in cysteine-/methionine-free medium for 30 min and then pulse labeled with [35S]cysteine/ methionine for 2 min. Immediately after labeling, the cells were lysed and precipitated with antibodies raised against a peptide corresponding to the NH2 terminus of HA. The precipitates were subjected to 2-D SDS-PAGE as in Fig. 2.
Such acceleration of HA folding has been previously observed in microsomes when calnexin and calreticulin binding is inhibited with castanospermine (Hebert et al., 1996). Taken together with the observed acceleration of folding of mutants Δ8,22, Δ8,22,38, Δ483 and Δ285,483 in microsomes (Figs. 4 A and 6 B), the data suggest that calnexin binding to the oligosaccharides present in the stem domain in positions 8, 22, 38, and 483 serves to slow down HA folding and the formation of NT. Removal of some of these glycans, or inhibition of calnexin binding to them, results in faster folding.
As expected, the Δ81 mutant folded inefficiently to the NT form in the live cells (Fig. 7 C). Essentially no protein reached the IT2 or NT forms after a 2-min pulse. A similar result was obtained whether N81Q mutation or the T83A mutation was used to eliminate the consensus glycosylation sequence (data not shown). The folding defect observed for these mutants confirmed that the oligosaccharide in position N81 is essential for proper folding of the HA in the X31 strain of virus.
Calnexin Binds to Membrane-bound Forms of HA More Efficiently
An important difference between calnexin and calreticulin is that one is membrane bound and the other is soluble in the ER lumen. Although both are known to interact with membrane glycoproteins and soluble proteins (Helenius et al., 1997), it was of interest to determine whether the division of labor observed in their interaction with the membrane-bound wild-type HA would apply for a soluble anchor-free form of HA.
We translated wild-type HA and its anchor-free truncated counterpart by in vitro translation in microsomes, and assayed for folding and calnexin and calreticulin binding. The anchor− HA was found to undergo normal folding to NT, but as reported previously for live cells (Singh et al., 1990); it was unable to trimerize (data not shown). Whereas anti-calreticulin antibodies coprecipitated equal amounts wild-type and anchor− HA (50%), calnexin precipitated 82% of the wild type, but only 54% of anchor− HA (Fig. 8). The soluble HA thus associated somewhat less efficiently with the membrane-bound chaperone. Presumably, the oligosaccharides in the stem region of the anchor− HA were not as easily accessed by calnexin as in the membrane-bound wild-type HA.
Figure 8.

Binding of anchor− HA to calnexin and calreticulin. A soluble anchor-free form of HA was expressed truncated at the first transmembrane residue by replacing the codon for T514 with the stop codon, TGA. Wild-type and truncated (Anchor −) forms of HA were translated for 1 h at 32°C under oxidizing conditions in the presence of microsomes. After 1h, HA was alkylated, lysed, and precipitated with anti-HA, -calreticulin, and -calnexin antisera. HA was resolved, visualized, and quantified as in Fig. 3. Note the levels of chaperone binding in this experiment were higher throughout, likely due to the use of different antibody bleeds. This experiment was repeated three times with similar levels observed.
Discussion
Our analysis of wild-type and mutant forms of HA confirmed the importance of the N-linked oligosaccharides in the general process of glycoprotein folding calnexin and calreticulin binding. Not only is the presence of N-linked glycans necessary for HA folding as previously shown (Hurtley et al., 1989; Gallagher et al., 1992), but their number and location in the molecule are also crucial. The results showed that the glycans affect the structural maturation of the protein locally and globally. They influence the rate of folding and the formation of disulfide bonds. By determining the interactions with chaperones such as calnexin and calreticulin, they help to define the orderly progression of folding from one domain to the next. Calnexin and calreticulin, although working according to the same overall principles, were found to display distinct interactions with folding intermediates of HA.
When growing nascent chains of wild-type HA were analyzed, it could be shown that both calnexin and calreticulin already begin to associate during translation and translocation. Association was first detected in cells when the HA chain was about half finished, i.e., at a time when it was still ∼1 min from termination (Braakman et al., 1991). At this time, five of the seven oligosaccharide chains (N8, 22, 38, 81, and 165) had been added. Since calnexin and calreticulin binding was castanospermine sensitive, at least some of them must have been trimmed to the monoglucosylated form by glucosidase I and II. It is known that these ER glucosidases have access to nascent chains, and that glucose trimming is rapidly initiated (Hubbard and Ivatt, 1981). No indication of disulfide bond formation was detectable at the time of initial calnexin and calreticulin association.
Removal of NH2-terminal glycans in positions N8,22, and 38 resulted in HA molecules that reached the fully oxidized form more rapidly than wild-type HA. When assayed in living cells, cotranslational folding of the Δ8,22, and 38 mutant protein was also found to fold faster. We have previously reported that inhibition of calnexin and calreticulin binding by addition of castanospermine has a similar effect; the rate of NT formation is accelerated and aberrant nonnative disulfide bonds are formed cotranslationally (Chen et al., 1995; Hebert et al., 1996). Based on these observations, we hypothesize that by binding to the NH2-terminal glycans, the chaperones sequester the NH2-terminal portion of the growing chain. This is likely to be important because the sequence comprising residues 1–50 is predestined to be an integral part of the COOH-terminal stem domain (Fig. 9), and, therefore, has to wait before it can begin to fold. For example, Cys 14 emerges from the translocon in cells 1.5 min or more before its partner, Cys 466. By binding to the NH2-terminal glycans, the chaperones may serve to postpone the folding of the NH2-terminal segment and prevent Cys 14 from oxidizing with the eight intervening cysteines in the polypeptide chain. This could, in fact, explain why three glycans are located in the first 38 residues of the X31 HA used in this study.
Our results indicate that as the folding of the HA chain progresses, calreticulin associated preferentially with the three top/hinge domain glycans, N81, 165, and 285 (Fig. 9). The preferential binding to these glycans may have several reasons. First, the oligosaccharides in the top part of the molecule may be more easily accessed from the lumen of the ER, thus attracting the soluble calreticulin more efficiently than the membrane-bound calnexin. Second, being a membrane protein, calnexin may bind more efficiently to the membrane-proximal stem domain glycans, with the result that only the top domain glycans are left for calreticulin to bind to. Support for this notion comes from the observed reduction in calnexin binding to anchor-free HA compared to wild-type HA. Mere membrane binding that affects substrate specificity is shown by the observations of Wada and co-workers (Wada et al., 1995), who found that the pattern of calreticulin-bound proteins in HepG2 cells became similar to that of calnexin when the calreticulin was anchored to the membrane by calnexin's transmembrane region.
It is also possible that the steric arrangement of the top domain glycans may find a better fit in calreticulin's binding site. The glycan binding site(s) are thought to be located in the P-domains of calnexin and calreticulin. Although highly homologous to calnexin's, the P-domain of calreticulin is smaller and has three double sets of sequence repeats instead of four (Wada et al., 1991; Michalak et al., 1992; David et al., 1993). These differences in the proteins may contribute to selectivity in binding.
Finally, the distinct binding specificities could be determined by protein–protein contacts. Such interactions have been postulated to occur between MHC class I and II heavy chains and calnexin (Arunachalam and Cresswell, 1995; Williams, 1995; Zhang et al., 1995). Although protein–protein contacts are not required for the formation of stable complexes between substrate glycoproteins and the two chaperones (Rodan et al., 1996; Zapun et al., 1997), they cannot be ruled out as additional factors contributing to the specificity of binding.
As a consequence of binding to the top/hinge glycans, calreticulin dissociates sooner from the folding HA chain than calnexin. This is consistent with results indicating that folding of HA begins from the top domain and proceeds from there towards the stem (Braakman et al., 1992a ; Braakman, I., and A. Helenius, manuscript in preparation). The progression of folding is best illustrated by the orderly, hierarchical formation of disulfide bonds that starts with the formation of either C67–C76 and C97–C139 in the top domain, and proceeds to the hinge domain disulfide C281–C305, then disulfide C52–C277 (Fig. 9)(Braakman, I., and A. Helenius, manuscript in preparation). We have, in this study, taken advantage of this stringent oxidation program by using the mutation C305S, which arrests oxidation at IT1, to study calreticulin binding to glycosylation mutants that would otherwise fold too rapidly.
One possible reason why calreticulin dissociates before the HA molecule is fully folded is that the top domain fails to be reglucosylated by the UDP-glucose:glycoprotein glucosyltransferase. It may no longer be recognized as unfolded by this folding sensor. This would imply that the transferase senses unfold locally rather than globally. In other words, in a protein with several domains, only the misfolded ones may serve as substrates. Further studies are needed to define how close to a misfolded domain a glycan has to be to serve as a substrate for reglucosylation.
The elimination of most of the glycans singly did not affect the outcome of the folding process. It did, however, affect the folding rate by either increasing (glycans 8, 22, 38, and 483) or decreasing it (glycans 81, 165, and 285). Of the seven oligosaccharides only glycan N81 proved to be essential for HA folding in microsomes. Mutant HA molecules devoid of this glycan were virtually incapable of acquiring intrachain disulfide bonds. They also failed to form the interchain disulfides typically observed when HA misfolds (Hurtley et al., 1989; Marquardt and Helenius, 1992). The protein remained essentially unoxidized for a long time. The explanation for the poor folding may be the close vicinity of N81 to a top domain disulfide bond, C67– C76 (Fig. 9). Interestingly, when the cysteines of this disulfide are mutated to serines a similar oxidation-deficient phenotype is observed (Braakman, I., and A. Helenius, manuscript in preparation). Thus, the most likely explanation for the folding defect in Δ81 is that the formation of a disulfide bond C67–C76 is inhibited when the glycan is missing. This disulfide is important as it serves as an early obligatory link in a chain of downstream oxidation events.
It is somewhat surprising that glycan N81 should prove to be so essential because it is not conserved among influenza strains. The conserved glycans are all in the stem domain and correspond to N22, 38, and 483 in the X31 HA. However, Gallagher and co-workers (1992) have suggested that the oligosaccahrides in positions 8 or 81 of the X31 HA may, in fact, be essential for maturation. They found that the HA of the Japan strain, which does not have these glycans, reaches a transport-competent conformation after tunicamycin treatment, whereas the HA of X31 HA that has them is completely tunicamycin sensitive (Gallagher et al., 1992). However, when they analyzed single-site glycosylation mutants of X31 HA, they found that no single glycan was essential for the acquisition of a transport-competent conformation in live cells. In their study, transport ability was analyzed after extended times of chase, whereas here we have monitored the folding or oxidation process. In live cells the Δ81 mutant displayed a clear-cut cotranslational folding defect, but after a time some of it was able to reach a folded conformation (data not shown) and therefore, would be expected to be competent for transport.
The differences observed among strains of influenza are further illustrated by studies performed by Roberts and co-workers, who have found that two out of three of the conserved stem domain glycans are required for the proper maturation and transport of HA in the strain of fowl plague virus (A/FPV/Rostock/34[H7N1]) (Roberts et al., 1993). However, the location of the top glycans in this strain are very different from that of the X31 strain glycans. Furthermore, most HAs from avian and porcine influenza strains have no glycans in the top or hinge domain. Depending on the strain, glycans can, therefore, be dispensable in the formation of the top domain disulfide bonds.
In summary, we find that N-linked glycosylation on HA influences the folding process in many complex ways. One thing is clear, however; the N-linked glycans allow the newly synthesized protein to interact with the calnexin– calreticulin cycle. Though not totally essential for the folding of HA, this serves to decrease the rate of folding, prevent premature trimerization, inhibit degradation, increase the efficiency of folding, and expose the protein to efficient quality control (Hammond et al., 1994; Hebert et al., 1996). HA folding involves calnexin and calreticulin in an elaborate series of interactions starting on the nascent chain. Binding of calnexin and calreticulin to distinct oligosaccharides and different domains is apparently used by HA to organize the processes of folding. For example, it seems likely that calnexin and calreticulin are used to temporarily sequester the NH2-terminal segment of HA, thus promoting more efficient cotranslational folding of NH2-terminal and COOH-terminal sequences to form the stem domain.
It is also clear that glycans have effects on HA folding that are independent of calnexin and calreticulin. For the X31 HA, this is illustrated by the observation that the protein is completely misfolded in the presence of tunicamycin but only partially affected by castanospermine (Hurtley et al., 1989; Hammond et al., 1994; Hebert et al., 1996). The requirement for a glycan in position N81 provides one example of a direct effect. The importance of this oligosaccharide seems to be based on a local influence on the formation of a nearby disulfide bond. In addition to local effects, the presence of oligosaccharides is likely to have more global influences on folding by increasing overall polarity of folding intermediates, thus counteracting their tendency to aggregate (Kern et al., 1992; Marquardt and Helenius, 1992; Kern et al., 1993).
Acknowledgments
This work was supported by the Patrick and Catherine Weldon Donaghue Medical Research Foundation to D.N. Hebert, and by National Institutes of Health grants to A. Helenius (5-RO1-GM38346 and 1-RO1-GM52972) and J.-X. Zhang (1-532-DKO9309).
Abbreviations used in this paper
- GSSG
oxidized glutathione
- HA
hemagglutinin
- NEM
N-ethylmaleimide
- NHA
NH2-terminal peptide of HA
- 2-D
two-dimensional
Footnotes
We would like to acknowledge R. Gilmore (University of Massachusetts Medical Center), I. Braakman (University of Amsterdam, Amsterdam, The Netherlands), and T. Marquardt (University of Meunster, Muenster, Germany) for providing the canine pancreas microsomes and assistance with some of the glycosylation and truncation mutants, respectively. We would also like to thank N. Ayad (Yale University, New Haven, CT) and E.S. Trombetta (Yale University) for helpful discussions.
Address all correspondence to Ari Helenius, Department of Cell Biology, Yale Univeristy School of Medicine, P.O. Box 208002, New Haven, CT 06520-8002. Tel.: (203) 785-4313. Fax: (203) 737-1756. E-mail: ari_helenius @qm.yale.edu
References
- Arunachalam B, Cresswell P. Molecular requirements for the interaction of class II major histocompatibility complex molecules and invariant chain with calnexin. J Biol Chem. 1995;270:2784–2790. doi: 10.1074/jbc.270.6.2784. [DOI] [PubMed] [Google Scholar]
- Braakman I, Hoover-Litty H, Wagner KR, Helenius A. Folding of influenza hemagglutinin in the endoplasmic reticulum. J Cell Biol. 1991;114:401–411. doi: 10.1083/jcb.114.3.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braakman I, Helenius J, Helenius A. Manipulating disulfide bond formation and protein folding in the endoplasmic reticulum. EMBO (Eur Mol Biol Organ) J. 1992a;11:1717–1722. doi: 10.1002/j.1460-2075.1992.tb05223.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braakman I, Helenius J, Helenius A. Role of ATP and disulphide bonds during protein folding in the endoplasmic reticulum. Nature (Lond) 1992b;356:260–262. doi: 10.1038/356260a0. [DOI] [PubMed] [Google Scholar]
- Cannon KS, Hebert DN, Helenius A. Glycan-dependent and -independent association of vesicular stomatitis virus G protein with calnexin. J Biol Chem. 1996;271:14280–14284. doi: 10.1074/jbc.271.24.14280. [DOI] [PubMed] [Google Scholar]
- Chen W, Helenius J, Braakman I, Helenius A. Cotranslational folding and calnexin binding of influenza hemagglutinin in the endoplasmic reticulum. Proc Natl Acad Sci USA. 1995;92:6229–6233. doi: 10.1073/pnas.92.14.6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David V, Hochstenbach F, Rajagopalan S, Brenner MB. Interaction with newly synthesized and retained proteins in the endoplasmic reticulum suggests a chaperone function for human integral membrane protein IP90 (calnexin) J Biol Chem. 1993;268:9585–9592. [PubMed] [Google Scholar]
- Gallagher PJ, Henneberry JM, Sambrook JF, Gething M-J. Glycosylation requirements for intracellular transport and function of the hemagglutinin of influenza virus. J Virol. 1992;66:7136–7145. doi: 10.1128/jvi.66.12.7136-7145.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond C, Helenius A. A chaperone with a sweet tooth. Curr Biol. 1993;3:884–885. doi: 10.1016/0960-9822(93)90226-e. [DOI] [PubMed] [Google Scholar]
- Hammond C, Helenius A. Folding of VSV G protein: sequential interaction with BiP and calnexin. Science (Wash DC) 1994;266:456–458. doi: 10.1126/science.7939687. [DOI] [PubMed] [Google Scholar]
- Hammond C, Braakman I, Helenius A. Role of N-linked oligosaccharides, glucose trimming, and calnexin during glycoprotein folding in the endoplasmic reticulum. Proc Natl Acad Sci USA. 1994;91:913–917. doi: 10.1073/pnas.91.3.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert DN, Foellmer B, Helenius A. Glucose trimming and reglucosylation determines glycoprotein association with calnexin. Cell. 1995;81:425–433. doi: 10.1016/0092-8674(95)90395-x. [DOI] [PubMed] [Google Scholar]
- Hebert DN, Foellmer B, Helenius A. Calnexin and calreticulin promote folding, delay oligomerization, and suppress degradation of influenza hemagglutinin in microsomes. EMBO (Eur Mol Biol Organ) J. 1996;15:2961–2968. [PMC free article] [PubMed] [Google Scholar]
- Helenius A, Trombetta ES, Hebert DN, Simons JF. Calnexin, calreticulin, and the folding of glycoproteins. Trends Cell Biol. 1997;7:193–200. doi: 10.1016/S0962-8924(97)01032-5. [DOI] [PubMed] [Google Scholar]
- Hubbard SC, Ivatt RJ. Synthesis and processing of asparagine-linked oligosaccharides. Annu Rev Biochem. 1981;50:555–584. doi: 10.1146/annurev.bi.50.070181.003011. [DOI] [PubMed] [Google Scholar]
- Hurtley SM, Bole DG, Hoover-Litty H, Helenius A, Copeland CS. Interactions of misfolded influenzahemagglutinin with binding protein (BiP) J Cell Biol. 1989;108:2117–2126. doi: 10.1083/jcb.108.6.2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern G, Schülke N, Schmid FZ, Jaenicke R. Stability, quaternary structure, and folding of internal, external, and core-glucosylated invertase form yeast. Protein Sci. 1992;1:120–131. doi: 10.1002/pro.5560010112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern G, Kern D, Jaenicke R, Seckler R. Kinetics of folding and association of differently glycosylated variants of invertase from Saccharomyces cerevisiae. . Protein Sci. 1993;2:1862–1868. doi: 10.1002/pro.5560021108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquardt T, Helenius A. Misfolding and aggregation of newly synthesized proteins in the endoplasmic reticulum. J Cell Biol. 1992;117:505–513. doi: 10.1083/jcb.117.3.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquardt T, Hebert DN, Helenius A. Posttranslational folding of influenza hemagglutinin in isolated endoplasmic reticulum-derived microsomes. J Biol Chem. 1993;268:19618–19625. [PubMed] [Google Scholar]
- Michalak M, Milner RE, Burns K, Opas M. Calreticulin. Biochem J. 1992;285:681–692. doi: 10.1042/bj2850681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou W-J, Cameron PH, Thomas DY, Bergeron JJM. Association of folding intermediates of glycoproteins with calnexin during protein maturation. Nature (Lond) 1993;364:771–776. doi: 10.1038/364771a0. [DOI] [PubMed] [Google Scholar]
- Peterson JR, Ora A, Nguyen P, Van, Helenius A. Transient, lectin-like association of calreticulin with folding intermediates of cellular and viral glycoproteins. Mol Biol Cell. 1995;6:1173–1184. doi: 10.1091/mbc.6.9.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan S, Brenner MB. Calnexin retains unassembled major histocompatibility complex class I free heavy chains in the endoplasmic reticulum. J Exp Med. 1994;180:407–412. doi: 10.1084/jem.180.1.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts PC, Garten W, Klenk H-D. Role of conserved glycosylation sites in maturation and transport of influenza A virus hemagglutinin. J Virol. 1993;67:3048–3060. doi: 10.1128/jvi.67.6.3048-3060.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodan AR, Simons JF, Trombetta ES, Helenius A. N-linked oligosaccharides are necessary and sufficient for association of RNase B with calnexin and calreticulin. EMBO (Eur Mol Biol Organ) J. 1996;15:6921–6930. [PMC free article] [PubMed] [Google Scholar]
- Sadasivan B, Lehner PJ, Ortmann B, Spies T, Cresswell P. Roles for calreticulin and a novel glycoprotein, tapasin, in the interaction of MHC class I molecules with TAP. Immunity. 1996;5:103–114. doi: 10.1016/s1074-7613(00)80487-2. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning: A Laboratory Manual. 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 545 pp.
- Singh I, Doms RW, Wagner KR, Helenius A. Intracellular transport of soluble and membrane-bound glycoproteins: folding, assembly, and secretion of anchor-free influenza hemagglutinin. EMBO (Eur Mol Biol Organ) J. 1990;9:631–639. doi: 10.1002/j.1460-2075.1990.tb08155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa MC, Ferrero-Garcia MA, Parodi AJ. Recognition of the oligosaccharide and protein moieties of glycoproteins by the UDP-Glc:glycoprotein glucosyltransferase. Biochemistry. 1992;31:97–105. doi: 10.1021/bi00116a015. [DOI] [PubMed] [Google Scholar]
- Spiro RG, Zhu Q, Bhoyroo V, Söling H-D. Definition of the lectin-like properties of the molecular chaperone, calreticulin, and demonstration of its copurification with endomannosidase from rat liver Golgi. J Biol Chem. 1996;271:11588–11594. doi: 10.1074/jbc.271.19.11588. [DOI] [PubMed] [Google Scholar]
- Suh P, Bergmann JE, Gabel CA. Selective retention of monoglycosylated high mannose oligosaccahrides by a class of mutant vesicular stomatitis virus G proteins. J Cell Biol. 1989;108:811–819. doi: 10.1083/jcb.108.3.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tector M, Salter RD. Calnexin influences folding of human class I histocompatibility proteins but not their assembly with β2-microglobulin. J Biol Chem. 1995;270:19638–19642. doi: 10.1074/jbc.270.33.19638. [DOI] [PubMed] [Google Scholar]
- Trombetta SE, Parodi AJ. Purification to apparent homogeneity and partial characterization of rat liver UDP-glucose:glycoprotein glucosyltransferase. J Biol Chem. 1992;267:9236–9240. [PubMed] [Google Scholar]
- Van Leeuwen JEM, Kearse KP. Deglucosylation of N-linked glycans is an important step in the dissociation of calreticulin-class I-TAP complexes. Proc Natl Acad Sci USA. 1996;93:13997–14001. doi: 10.1073/pnas.93.24.13997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Leeuwen JEM, Kearse KP. Reglucosylation of N-linked glycans is critical for calnexin assembly with T cell receptor (TCR) α proteins but not TCR β proteins. J Biol Chem. 1997;272:4179–4186. doi: 10.1074/jbc.272.7.4179. [DOI] [PubMed] [Google Scholar]
- Vassilakos A, Cohen-Doyle MF, Peterson PA, Jackson MR, Williams DB. The molecular chaperone calnexin facilitates folding and assembly of class I histocompatibility molecules. EMBO (Eur Mol Biol Organ) J. 1996;15:1495–1506. [PMC free article] [PubMed] [Google Scholar]
- Wada I, Rindress D, Cameron PH, Ou W-J, Doherty JJ, II, Louvard D, Bell AW, Dignard D, Thomas DY, Bergeron JJM. SSRα and associated calnexin are major calcium binding proteins of the endoplasmic reticulum membrane. J Biol Chem. 1991;266:19599–19610. [PubMed] [Google Scholar]
- Wada I, Imai S-I, Kai M, Sakane F, Kanoh H. Chaperone function of calreticulin when expressed in the endoplasmic reticulum as the membrane-anchored and soluble forms. J Biol Chem. 1995;270:20298–20304. doi: 10.1074/jbc.270.35.20298. [DOI] [PubMed] [Google Scholar]
- Ware FE, Vassilakos A, Peterson PA, Jackson MR, Lehrman MA, Williams DB. The molecular chaperone calnexin binds Glc1Man9GlcNAc2oligosaccharides as an initial step in recognizing unfolded glycoproteins. J Biol Chem. 1995;270:4697–4704. doi: 10.1074/jbc.270.9.4697. [DOI] [PubMed] [Google Scholar]
- Williams DB. Calnexin: a molecular chaperone with a taste for carbohydrate. Biochem Cell Biol. 1995;73:123–132. doi: 10.1139/o95-015. [DOI] [PubMed] [Google Scholar]
- Wilson IA, Skehel JJ, Wiley DC. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature (Lond) 1981;289:366–373. doi: 10.1038/289366a0. [DOI] [PubMed] [Google Scholar]
- Zapun A, Petrescu SM, Rudd PM, Dwek RA, Thomas DY, Bergeron JJM. Conformation independent binding of monoglucosylated ribonuclease B to calnexin. Cell. 1997;88:29–38. doi: 10.1016/s0092-8674(00)81855-3. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Tector M, Salter RD. Calnexin recognizes carbohydrate and protein determinants of class I major histocompatibility complex molecules. J Biol Chem. 1995;270:3944–3948. doi: 10.1074/jbc.270.8.3944. [DOI] [PubMed] [Google Scholar]