

Abstract
Breakdown of microvilli is a common early event in various types of apoptosis, but its molecular mechanism and implications remain unclear. ERM (ezrin/radixin/moesin) proteins are ubiquitously expressed microvillar proteins that are activated in the cytoplasm, translocate to the plasma membrane, and function as general actin filament/plasma membrane cross-linkers to form microvilli. Immunofluorescence microscopic and biochemical analyses revealed that, at the early phase of Fas ligand (FasL)–induced apoptosis in L cells expressing Fas (LHF), ERM proteins translocate from the plasma membranes of microvilli to the cytoplasm concomitant with dephosphorylation. When the FasL-induced dephosphorylation of ERM proteins was suppressed by calyculin A, a serine/threonine protein phosphatase inhibitor, the cytoplasmic translocation of ERM proteins was blocked. The interleukin-1β–converting enzyme (ICE) protease inhibitors suppressed the dephosphorylation as well as the cytoplasmic translocation of ERM proteins. These findings indicate that during FasL-induced apoptosis, the ICE protease cascade was first activated, and then ERM proteins were dephosphorylated followed by their cytoplasmic translocation, i.e., microvillar breakdown. Next, to examine the subsequent events in microvillar breakdown, we prepared DiO-labeled single-layered plasma membranes with the cytoplasmic surface freely exposed from FasL-treated or nontreated LHF cells. On single-layered plasma membranes from nontreated cells, ERM proteins and actin filaments were densely detected, whereas those from FasL-treated cells were free from ERM proteins or actin filaments. We thus concluded that the cytoplasmic translocation of ERM proteins is responsible for the microvillar breakdown at an early phase of apoptosis and that the depletion of ERM proteins from plasma membranes results in the gross dissociation of actin-based cytoskeleton from plasma membranes. The physiological relevance of this ERM protein–based microvillar breakdown in apoptosis will be discussed.
Apoptotic cell death is an active process, which is a critical feature of the regulated development of multicellular organisms (Wyllie et al., 1980; Nagata and Goldstein, 1995; Chinnaiyan and Dixit, 1996; Fraser and Evans, 1996; Jacobson, 1997; Nagata, 1997). Apoptosis is characterized by marked morphological alterations of the nucleus, such as chromatin condensation. Various types of stimuli are known to cause apoptosis, and irrespective of stimuli, apoptotic cell death is usually accompanied by the activation of interleukin-1β–converting enzyme (ICE)1 family members of cysteine proteases followed by the fragmentation of nuclear DNA into oligonucleosomal-sized units (Enari et al., 1995; Los et al., 1995; Alnemri et al., 1996). Relatively few apoptosis-related substrates for the ICE family members have been reported, and our knowledge of the roles of these substrates in DNA fragmentation is still limited (Chinnaiyan and Dixit, 1996; Fraser and Evans, 1996; Jacobson, 1997; Nagata, 1997). Plasma membranes and the cytoskeleton also undergo marked morphological changes during apoptosis. Among these changes, the disappearance of microvilli has been recognized as one of the common early events of apoptosis, although its molecular mechanism and physiological implications in apoptosis are unknown.
Microvilli are specific sites of actin filament/plasma membrane interaction and are composed of core actin filaments and several actin-binding proteins, such as villin, fimbrin, and ERM proteins (Bretscher, 1991; Sato et al., 1992; Arpin et al., 1994; Tsukita et al., 1997a ,b). At least one member of the ERM family is found in microvilli in all types of cells, whereas the expression and distribution of villin and fimbrin are restricted to some specific types of cell. When the expression of ERM proteins was suppressed by antisense oligonucleotides, microvillar structures completely disappeared from the cell surface, indicating that these proteins play a key role in microvillar formation in general (Takeuchi et al., 1994b ).
The ERM family consists of three closely related proteins: ezrin, radixin, and moesin (Arpin et al., 1994; Tsukita et al., 1997a ,b). Sequence analyses of cDNAs have revealed that the amino acid sequence identity among ERM proteins is 70–80% (Gould et al., 1989; Funayama et al., 1991; Lankes and Furthmayr, 1991; Sato et al., 1992; Tsukita et al., 1997a ,b). The sequences of their amino-terminal halves are highly conserved (∼85% identity) and homologous to the amino-terminal ends of some membrane-associated proteins such as band 4.1 protein, talin, and merlin/schwannomin (a tumor suppressor molecule for neurofibromatosis type II) (Rouleau et al., 1993; Trofatter et al., 1993), indicating that the ERM family is included in the band 4.1 superfamily (Takeuchi et al., 1994a ).
ERM proteins function as general cross-linkers between actin filaments and specific groups of integral membrane proteins such as CD44, CD43, ICAM-2, etc. (Turunen et al., 1989; Yonemura et al., 1993; Tsukita et al., 1994; Helander et al., 1996; Tsukita et al., 1997a ,b). Their carboxy- and amino-terminal halves bind to actin filaments and the cytoplasmic domains of integral membrane proteins, respectively. Several lines of evidence indicate that the amino- and carboxy-terminal halves of ERM proteins suppress the functions of their carboxy- and amino-terminal halves, respectively, by binding to each other in a head- to-tail manner (Henry et al., 1995; Magendantz et al., 1995; Martin et al., 1995; Hirao et al., 1996; Tsukita et al., 1997a ,b). In the cytoplasm, this head-to-tail association results in folded monomers or homotypic/heterotypic oligomers of ERM proteins, which are inactivated as plasma membrane/actin filament cross-linkers. Some signals have been suggested to disrupt this intramolecular or intermolecular head-to-tail interaction to expose both membrane- and actin filament–binding domains, activating ERM proteins to function as cross-linkers. Recently, the Rho signaling pathway has been implicated in this activation process of ERM proteins (Hirao et al., 1996; Tsukita et al., 1997a ,b). Serine/ threonine as well as tyrosine phosphorylation have also been suggested to be involved in the regulation of ERM protein function (Urushidani et al., 1989; Berryman et al., 1995; Jiang et al., 1995; Nakamura et al., 1995).
In the present study, to clarify the molecular mechanism of microvillar disappearance during apoptosis, we examined the behavior of ERM proteins at the early phase of apoptosis. It is well known that Fas ligand (FasL), a death factor, binds to Fas, a cell surface receptor, to induce apoptosis (Suda et al., 1993; Nagata and Goldstein, 1995). Here, we induced apoptosis by adding human FasL to mouse L fibroblasts expressing human Fas (LHF cells). In this system, within 1 h of incubation FasL induced not only the translocation of ERM proteins from the plasma membrane (microvilli) to the cytoplasm but also the dephosphorylation of ERM proteins. Close analyses using phosphatase inhibitors or ICE protease inhibitors revealed that the ICE protease cascade is first activated by FasL stimulation, and then ERM proteins are dephosphorylated, followed by the cytoplasmic translocation of ERM proteins. Furthermore, we found that the cytoplasmic translocation of ERM proteins resulted in the gross dissociation of the actin-based cytoskeletal components from the plasma membrane. These findings clarified the molecular mechanism of microvilli disappearance at the early phase of apoptosis, providing a clue to understanding its physiological relevance.
Materials and Methods
Cells and Antibodies
Mouse L929 cells expressing recombinant human Fas antigen (LHF) and mouse MTD-1A cells were cultured in DME supplemented with 10% FCS. HL-60 cells were cultured in RPMI1640 supplemented with 10% FCS.
Ezrin-, radixin-, and moesin-specific mAbs (M11, R21, and M22) were raised in rats using recombinant ezrin, radixin, and moesin, respectively, as antigens (Hirao et al., 1996). Two pAbs, TK89 and TK90, were raised in rabbits against synthesized peptides corresponding to the mouse radixin and ezrin sequences (amino acids 551–570 and 480–489), respectively. TK89 detected all ERM proteins, whereas T90 was specific for ezrin.
Induction of Apoptosis
LHF cells were detached from the dishes by trypsinization and washed with culture medium. They were then cultured on 2% agar-coated 12-well plates in 1 ml of culture medium at the concentration of 5 × 105 cells/ml. To induce apoptosis, FasL was added to the culture medium at the final concentration of 1 μg/ml (Suda et al., 1993). In some experiments, calyculin A (Research Biochemicals International, Natick, MA) was added to the culture medium with FasL at the final concentration of 300 nM (Morana et al., 1996).
To induce apoptosis, MTD-1A cells were incubated with 0.5 μM staurosporine for 16 h, and HL-60 cells were cultured for 16 h in the presence of 50 μM C2 ceramide, 0.5 μM staurosporine, or 5 μg/ml actinomycin D.
Immunofluorescence Microscopy
LHF cells were collected by centrifugation, washed with PBS, and then placed on poly-l-lysine–coated coverslips for 15 min for attachment. LHF cells on coverslips or cultured MTD-1A cells were fixed with 3% formaldehyde in PBS for 10 min at room temperature and treated with 0.1% Triton X-100 in PBS for 5 min. After soaking in PBS containing 1% BSA for 10 min, samples were incubated with the first antibody, washed with PBS, and then incubated with the second antibody (FITC-conjugated goat anti– rat IgG or FITC-conjugated goat anti–rabbit IgG [Tago, Inc., Burlingame, CA]) in the presence of 4′,6′-diamidino-2-phenylindole (DAPI). Samples were then washed with PBS and examined using a fluorescence microscope (Axiophoto photomicroscope; Carl Zeiss, Oberkochen, Germany).
Fractionation of Soluble and Insoluble ERM Proteins
Collected LHF cells or HL-60 cells were resuspended and homogenized by weak sonication in a solution containing 130 mM KCl, 20 mM NaCl, 2 mM EDTA, 1 mM EGTA, 50 mM Tris-HCl, pH 7.4, 1 mM p-amidino PMSF, 10 μg/ml leupeptin, and 10 μg/ml aprotinin at 4°C. The homogenates were centrifuged at 100,000 g for 10 min at 4°C to recover the soluble and insoluble fractions in the supernatant and pellet, respectively. Equivalent amounts of supernatant and pellet were applied to SDS–polyacrylamide gels, electrophoresed, and subsequently subjected to immunoblotting with pAb TK89, mAb M11, mAb R21, or mAb M22.
Labeling of Cellular Phosphoproteins and Immunoprecipitation
LHF cells were placed in 2% agar-coated 12-well plates in phosphate-free DME with 10% FCS. Phosphate-free FCS was prepared by dialyzing against 0.9% NaCl in 10 mM Hepes buffer, pH 7.4. The cells were cultured for 4 h in the same medium containing 0.5 mCi/ml [32P]orthophosphate (Phosphorous-32; NEN Life Science Products, Boston, MA).
The labeled cells were lysed and incubated for 5 min in 0.1 ml of solubilization buffer (1% SDS, 10 mM Tris-HCl, pH 7.4, 10 mM Na3VO4, 1 mM Na2MoO4, and 10 mM p-amidinoPMSF). After addition of 0.9 ml of IP buffer (10 mM Tris-HCl, pH 7.4, 1% Triton X-100, 1% NaDOC, 150 mM NaCl, 2 mM EDTA, 1 mM EGTA, 5 mM NaF, 1 mM Na3VO4, 0.1 mM Na2MoO4, 1 mM p-amidinoPMSF, and 1 μg/ml leupeptin), the lysate stood on ice for an additional 10 min and was centrifuged at 50,000 g for 20 min. The supernatant was immunoprecipitated with 20 μl of protein G–Sepharose 4B (Zymed Laboratories, Inc., South San Francisco, CA) conjugated with pAb TK89, pAb T90, or mAb M22. Sepharose 4B–bound immune complexes were washed five times with IP buffer containing 0.1% SDS. Immune complexes were then eluted by boiling in SDS-PAGE sample buffer and resolved by SDS-PAGE. After transferring from gels onto nitrocellulose sheets, the 32P signals were analyzed by autoradiography (Fujix Bioimage Analyzer Bas 200 System; Fuji Photofilm Corp., Tokyo, Japan), and ERM proteins were detected by immunoblotting with pAb TK89.
Phosphoamino Acid Analysis
Antiezrin pAb (TK90) or antimoesin mAb (M22) immunoprecipitates from 32P-labeled LHF cells with or without 1-h FasL treatment were resolved by SDS-PAGE and transferred onto polyvinyl difluoride membranes (Immobilon; Millipore Corp., Bedford, MA). Phosphoamino acids were analyzed based on the method of Boyle et al. (1991) with minor modifications. 32P-labeled phosphorylated ezrin or moesin bands were excised from membranes and hydrolyzed in 200 μl of 6 M HCl at 110°C for 60 min. The hydrolysates were lyophilized using a Speed Vac Concentrator (Savant Instruments, Hicksville, NY) and resuspended in 6 μl of pH 3.5 buffer (5% glacial acetic acid, 0.5% pyridine) containing cold phosphoamino acid standards. The samples were then spotted onto silica gel– coated thin-layer chromatography plates and resolved by electrophoresis at 1,300 V for 1.3 h using pH 3.5 buffer at 4°C (model Multiphor II; Pharmacia Biotech AB, Uppsala, Sweden). The positions of 32P-labeled phosphoamino acids were determined by autoradiography, and cold phosphoamino acid standards were visualized by ninhydrin staining.
ICE-like or CPP32-like Protease Inhibitor Treatment
LHF cells were labeled with [32P]orthophosphate for 3 h, and then Ac-YVAD-cho or Ac-DEVD-cho (Takara Shuzo Co., Ltd., Kyoto, Japan) was added at the final concentration of 300 μM. After 1 h incubation, 1 μg/ml of FasL was added. At 3 h after the FasL stimulation, the phosphorylation level of ERM proteins was examined as described above.
Gel Electrophoresis and Immunoblotting
One-dimensional SDS-PAGE (7.5%) was performed, based on the method of Laemmli (1970). After electrophoresis, proteins were electrophoretically transferred from gels onto nitrocellulose membranes, which were then incubated with the first antibody. Bound antibodies were visualized with alkaline phosphatase–conjugated goat anti–rabbit IgG and the appropriate substrates as described by the manufacturer (Amersham International, Buckinghamshire, UK).
Analysis of Chromosomal DNA
Cells (1 × 106) were washed with PBS and incubated in 15 μl of 10 mM Tris-HCl, pH 8.0, 150 mM NaCl, 10 mM EDTA, 0.5% SDS, and 1 μg/ml proteinase K at 50°C for 30 min. Then, 3 μl of 1 mg/ml RNase A was added and incubated for an additional 1 h. The digested samples were added to the wells of a 2% agarose gel. After electrophoresis, the DNA was visualized by UV illumination after ethidium bromide staining.
Single-layered Plasma Membrane Preparation
LHF cells were cultured in 2% agar-coat plates and vitally labeled for 1.5 h with DiO (170 μg/ml) (Hong and Hume, 1986). The FasL-treated (1 h) or nontreated cells were collected by centrifugation, washed with PBS, and placed on poly-l-lysine–coated coverslips for 15 min for attachment. Then, cells were subjected to jet streams of 5–10 ml of buffer containing 10 mM Hepes, pH 7.4, 1 mM MgCl2, and 50 mM KCl using a 10-ml syringe with a 23-gauge needle (Tsukita et al., 1984). The isolated plasma membranes on the glass were fixed and stained with rhodamine phalloidin or with pAb TK89 followed by FITC-conjugated goat anti–rat IgG (Tago, Inc., Burlingame, CA). Samples were then washed with PBS and examined using a fluorescence microscope (Axiophoto photomicroscope; Carl Zeiss).
Results
Translocation of ERM Proteins from Plasma Membranes to Cytoplasm at an Early Phase of Fas-mediated Apoptosis
To examine whether ERM proteins are involved in the microvillar disappearance commonly observed in various types of apoptosis, mouse L929 fibroblast transfectants expressing human Fas (LHF) were cultured on agar-coated dishes (Yonehara et al., 1989; Itoh et al., 1991; Nagata and Golstein, 1995), and the apoptosis was initiated by addition of recombinant human FasL (Suda et al., 1993; Nagata and Golstein, 1995). On agar-coated dishes, LHF cells were not spread out but rounded, and they were killed by apoptosis within 3 h as determined by DNA fragmentation ladder formation, retaining their spherical shape (Fig. 1). The fairly quick and synchronized induction of apoptosis in this system allowed us to analyze the cellular events at the early stage of apoptosis in detail. Furthermore, since LHF cells retained their spherical shape without attachment to the dish, this system was advantageous for morphological analysis of the apoptotic cellular changes as well as for biochemical analyses.
Figure 1.

FasL-induced apoptosis in LHF cells. After incubation of LHF cells with FasL for 0, 1, 2, and 3 h, the chromosomal DNA was extracted and analyzed by gel electrophoresis. Judging from the DNA fragmentation ladder formation, apoptosis was induced within 2–3 h.
Using this system, we first examined the behavior of radixin at the early stage of Fas-mediated apoptosis by immunofluorescence microscopy. In LHF cells before FasL stimulation, radixin was concentrated at the plasma membranes, especially in microvilli, and were also distributed rather diffusely in the cytoplasm (Fig. 2). Within 1 h after FasL stimulation, marked deformation of plasma membranes such as bleb formation was observed. Concomitantly, the concentration of radixin on the plasma membranes faded. At 2–3 h after FasL stimulation, radixin was distributed evenly in the cytoplasm, and DAPI staining identified condensed and fragmented nuclei. As shown in Fig. 3, ezrin and moesin showed the same behavior as radixin at the early stage of Fas-mediated apoptosis of LHF cells.
Figure 2.
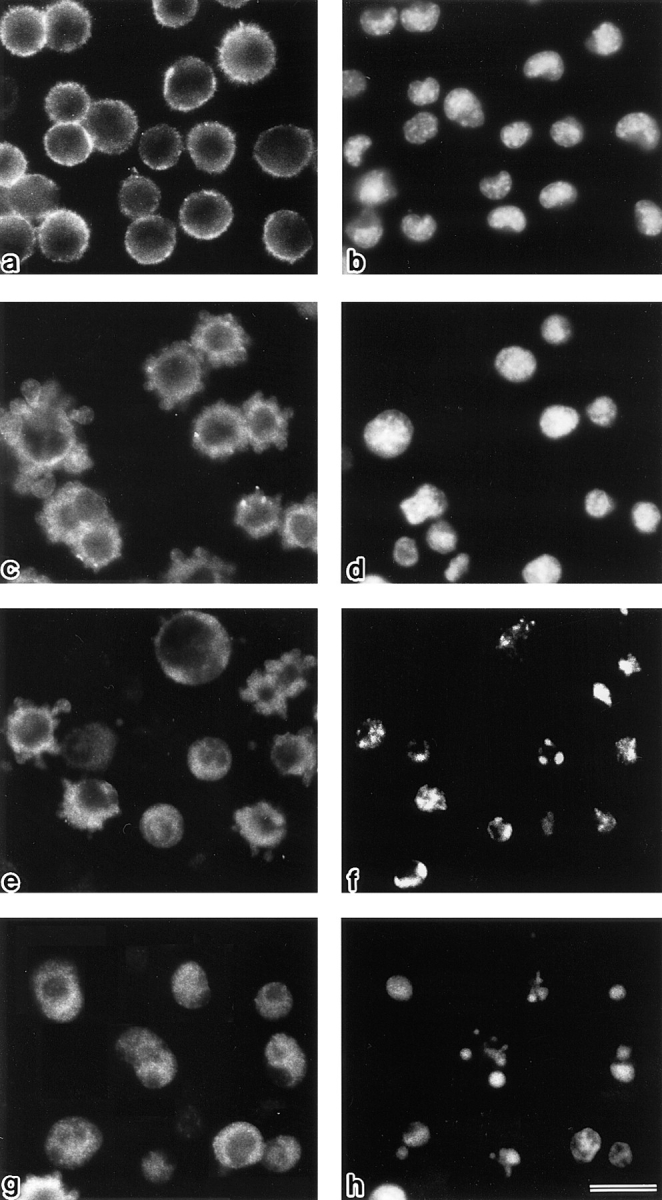
Behavior of radixin in LHF cells at 0 h (a and b), 1 h (c and d), 2 h (e and f), and 3 h (g and h) after FasL stimulation. LHF cells were doubly stained with antiradixin mAb, R21 (a, c, e, and g) and DAPI (b, d, f, and h). After 1 h of incubation, radixin translocated from the plasma membrane to the cytoplasm, and the nuclear change, such as chromatin condensation, began to be observed after 2 h of incubation. Bar, 20 μm.
Figure 3.

Behavior of ezrin and moesin in LHF cells at 0 h (a and c) and 2 h (b and d) after FasL stimulation. LHF cells were stained with ezrin-specific pAb TK90 (a and b) or moesin-specific mAb M22 (c and d). Behavior of ezrin and moesin during apoptosis was the same as that of radixin (see Fig. 2). Bar, 20 μm.
The cytoplasmic translocation of ERM proteins shown by immunofluorescence microscopy was also detected biochemically (Fig. 4). At various times after FasL stimulation, LHF cells were homogenized in physiological solution and divided into soluble and insoluble fractions by centrifugation. Equivalent amounts of the soluble and insoluble fractions were resolved by SDS-PAGE, and the amount of ERM proteins in each fraction was estimated by immunoblotting with pAb TK89 that recognized all ERM proteins. As shown in Fig. 4 a, in the absence of FasL, ∼50% of ERM proteins were recovered in the insoluble fraction. FasL stimulation gradually decreased the amount of insoluble ERM proteins, and at 1, 2, and 3 h after FasL stimulation, ∼35, 15, and 5% of total ERM proteins were recovered in the insoluble fraction, respectively. The cytoplasmic translocation of ERM proteins was also detected by immunoblotting with ezrin-, radixin-, or moesin-specific antibodies (Fig. 4 b). We then concluded that the microvillar ERM proteins translocate from the plasma membrane to the cytoplasm at an early stage of Fas-mediated apoptosis.
Figure 4.

Solubility of ERM proteins in LHF cells at 0, 1, 2, and 3 h after addition of FasL. LHF cells were homogenized in physiological saline and centrifuged to separate the soluble and insoluble fractions into the supernatant (S) and pellet (P), respectively. Equivalent amounts of supernatant and pellet were applied to SDS-PAGE and subsequently subjected to immunoblotting. To detect all members of ERM proteins, pAb TK89 was used (a). To detect ezrin, radixin, and moesin separately, mAb M11, mAb R21, and mAb M22 were used, respectively (b). ERM proteins translocated from the insoluble (P) to the soluble fraction (S) as apoptosis proceeded. Arrows in a indicate ezrin, radixin, and moesin, respectively, from the top.
The apoptosis-associated cytoplasmic translocation of ERM proteins was also observed in cultured mouse epithelial cells (MTD-1A) and human promyelocytic leukemic cells (HL-60), although the fairly slow and nonsynchronized induction of apoptosis in these cells prevented analysis of the early events of apoptosis in detail. As shown in Fig. 5 A, when apoptosis was induced in MTD-1A cells by 16 h incubation with staurosporine, radixin as well as ezrin and moesin were translocated from apical microvilli and lateral cell–cell contact sites to the cytoplasm. The 16-h incubation with C2 ceramide, staurosporine, or actinomycin D induced apoptosis in HL-60 cells, and in all cases the cytoplasmic translocation of ERM proteins was clearly detected (Fig. 5 B).
Figure 5.


Behavior of ERM proteins during apoptosis in mouse epithelial cells (MTD-1A) and human promyelocytic leukemic cells (HL-60). (A) Immunofluorescence micrographs of MTD-1A cells at 0 h (a and b) and 16 h (c and d) incubation with staurosporine. MTD-1A cells were doubly stained with antiradixin mAb R21 (a and c) and DAPI (b and d). After 16 h of incubation, radixin translocated from apical microvilli and lateral cell–cell contact sites to the cytoplasm, and nuclear changes such as chromatin condensation were observed. Ezrin and moesin showed the same behavior as radixin (data not shown). (B) Solubility of ERM proteins in HL-60 cells at 0 (control) and 16 h incubation with C2 ceramide, staurosporine, or actinomycin D. HL-60 cells were homogenized in physiological saline and centrifuged to separate the soluble and insoluble fractions into the supernatant (S) and pellet (P), respectively. Equivalent amounts of supernatant and pellet were applied to SDS-PAGE and subsequently subjected to immunoblotting with pAb TK89 capable of recognizing all ERM proteins. ERM proteins translocated from the insoluble (P) to the soluble fraction (S) as apoptosis proceeded. Arrows indicate ezrin, radixin, and moesin, respectively, from the top. Bar, 20 μm.
FasL-induced Dephosphorylation of ERM Proteins Coupled with Their Cytoplasmic Translocation
We next examined what types of modification of ERM proteins occur at the time of their cytoplasmic translocation at the early stage of the Fas-mediated apoptosis. The possible degradation of ERM proteins was first postulated since ICE proteases are known to be commonly activated as an early event of apoptosis. As shown in Fig. 4, however, ERM translocation was not associated with ERM degradation. We next examined the phosphorylation levels of ERM proteins since some reports suggested that phosphorylated ERM proteins are concentrated at the plasma membrane. LHF cells were metabolically labeled with [32P]orthophosphate and stimulated with FasL. At various times after FasL stimulation, ERM proteins were recovered by immunoprecipitation with pAb TK89 that recognized all ERM proteins and analyzed by autoradiography (Fig. 6 a). Before stimulation, ERM proteins were fairly highly phosphorylated, and FasL markedly decreased their phosphorylation level within 1 h. This downregulation continued up to the completion of apoptosis.
Figure 6.

Dephosphorylation of ERM proteins in LHF cells during Fas-mediated apoptosis. (a) Time course of apoptosis-associated dephosphorylation of ERM proteins. LHF cells were metabolically labeled with [32P]orthophosphate. At 0, 1, 2, and 3 h after FasL stimulation, the cell lysate was immunoprecipitated with anti-ERM pAb, TK89, and analyzed by autoradiography ([32P]) and immunoblotting ([Blotting]). Equal amounts of protein were applied to each lane as revealed in accompanying immunoblots with anti-ERM pAb, TK89. The phosphorylation level of ERM proteins markedly decreased after a 1-h incubation. Arrows indicate ezrin, radixin, and moesin, respectively, from the top. (b) Autoradiography of antiezrin pAb (TK90) or antimoesin mAb (M22) immunoprecipitates from 32P-labeled LHF cells. Immunoprecipitated ezrin and moesin were phosphorylated and dephosphorylated before and 1 h after FasL stimulation, respectively (arrows). (c) Phosphoamino acid analysis. 32P-labeled phosphorylated ezrin or moesin bands (arrows in b) were excised from membranes and processed for phosphoamino acid analysis. The positions of phosphoserine (p-S), phosphothreonine (p-T), and phosphotyrosine (p-Y) were determined by autoradiography through comparison with the ninhydrin staining profiles on unlabeled phosphoamino acid standards. In both ezrin and moesin, phosphothreonine appeared to be preferentially dephosphorylated after 1 h of incubation with FasL, although the levels of phosphoserine and phosphotyrosine were also decreased.
To determine which types of phosphoamino acid residues were dephosphorylated, we analyzed phosphoamino acids of ezrin and moesin after 0 and 1 h of incubation with FasL using LHF cells that were metabolically labeled with [32P]orthophosphate. Ezrin and moesin were recovered by immunoprecipitation with their specific antibodies, pAb TK90 and mAb M22, respectively. Radixin, however, was not purified from LHF cells by immunoprecipitation, partly because the available radixin-specific mAb R21 was not potent for immunoprecipitation, and partly because the expression level of radixin was fairly low in LHF cells. Recovered ezrin and moesin were phosphorylated and dephosphorylated before and 1 h after FasL stimulation, respectively (Fig. 6 b). As shown in Fig. 6 c, without FasL stimulation, serine/threonine as well as tyrosine residues of ezrin and moesin were phosphorylated. Among these phosphoamino acids, phosphothreonine appeared to be preferentially dephosphorylated after 1 h of incubation with FasL, although the levels of phosphorylation of serine and tyrosine residues were also partially decreased. These observations suggested that dephosphorylation on threonine residues of ERM proteins is primarily accompanied by their cytoplasmic translocation during Fas-mediated apoptosis.
To clarify whether the dephosphorylation of ERM proteins is coupled with their cytoplasmic translocation, and, if so, which is located upstream in the FasL-triggered signaling pathway, we stimulated LHF cells with FasL in the presence of 300 nM calyculin A, a potent serine/threonine phosphatase inhibitor (Morana et al., 1996). As shown in Fig. 7 a, calyculin A suppressed the FasL-induced dephosphorylation of ERM proteins at 1 h of incubation, conversely elevating the phosphorylation level of ERM proteins. This suppression continued up to 3 h after FasL stimulation, and the phosphorylation level of ERM proteins remained at the high level. Then, we examined the effects of calyculin A on the FasL-induced cytoplasmic translocation of ERM proteins. Immunoblotting of soluble and insoluble fractions from FasL/calyculin A–incubated LHF cells revealed that ∼90% of ERM proteins were recovered in the insoluble fraction at 1 h of incubation, which continued for at least 4 h, indicating that the FasL-induced cytoplasmic translocation of ERM proteins was suppressed by calyculin A (Fig. 7 b). In agreement with these observations, when these FasL/calyculin A–incubated cells were examined by immunofluorescence microscopy using anti-ERM antibodies, ERM proteins were shown to be concentrated at the plasma membrane (Fig. 7 c). These findings indicated that FasL-induced dephosphorylation of ERM proteins is coupled with their cytoplasmic translocation and that the former is located upstream of the latter.
Figure 7.
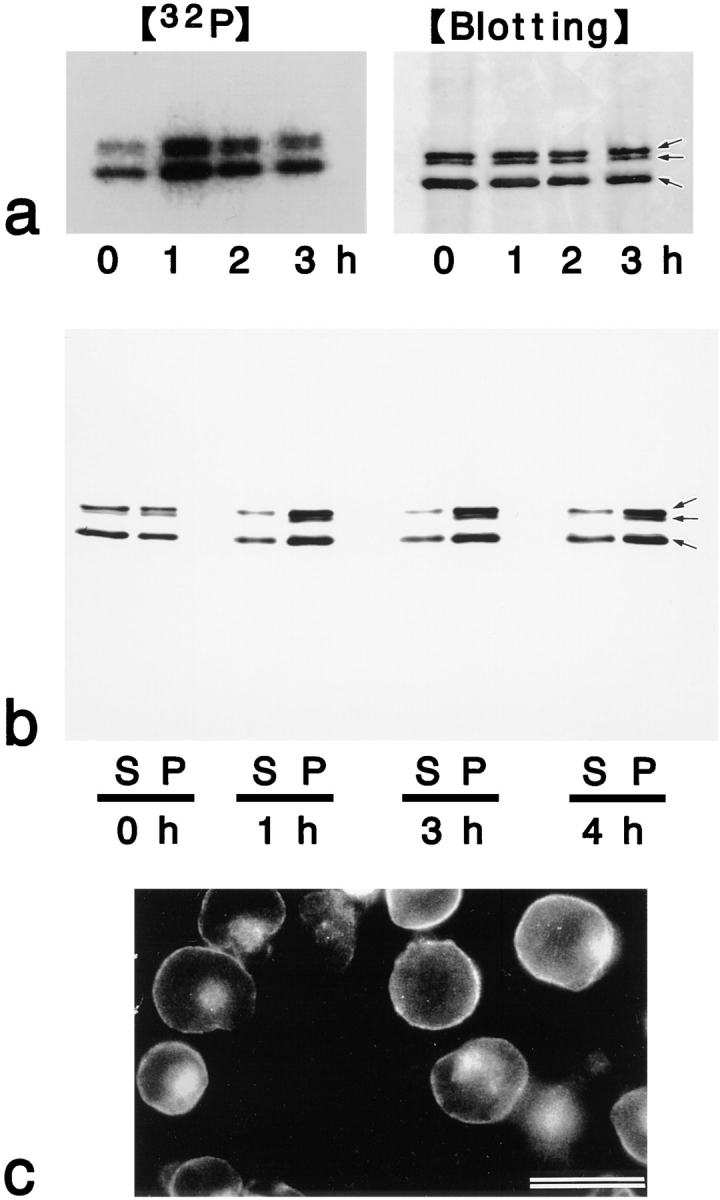
Effects of calyculin A on FasL-induced changes of ERM proteins in LHF cells. (a) Suppression of FasL-induced dephosphorylation of ERM proteins by calyculin A. LHF cells were metabolically labeled with [32P]orthophosphate. At 0, 1, 2, and 3 h after FasL stimulation in the presence of calyculin A, the cell lysate was immunoprecipitated with anti-ERM pAb, TK89, and then analyzed by autoradiography ([32P]) and immunoblotting ([Blotting]). Equal amounts of protein were applied to each lane as revealed in accompanying immunoblots with anti-ERM pAb, TK89. The FasL-induced dephosphorylation of ERM proteins (see Fig. 6 a) was suppressed, while conversely the phosphorylation level was elevated. Arrows indicate ezrin, radixin, and moesin, respectively, from the top. (b) Suppression of the FasL-induced cytoplasmic translocation of ERM proteins by calyculin A. Calyculin A was added to LHF cells together with FasL, and, after incubations for 1, 2, 3, and 4 h, ERM proteins were partitioned into soluble (S) and insoluble (P) fractions as described in Fig. 4 a. Immunoblots with anti-ERM pAb, TK89, revealed that FasL-induced cytoplasmic translocation of ERM proteins was suppressed and that conversely calyculin A increased the amounts of insoluble ERM proteins. Arrows indicate ezrin, radixin, moesin, respectively, from the top. (c) Antiradixin mAb immunofluorescence micrograph of LHF cells after 2 h of incubation of FasL and calyculin A. Radixin still remained on plasma membranes. Ezrin and moesin showed the same changes as radixin (data not shown). Bar, 20 μm.
Relationship between Dephosphorylation of ERM Proteins and Activation of ICE Protease Cascade
We next examined whether the dephosphorylation of ERM proteins is located upstream or downstream of the ICE protease cascade. As previously reported, in the presence of 300 μM Ac-YVAD-cho or Ac-DEVD-cho (ICE-like or CPP32-like protease-specific inhibitors, respectively), LHF cells resisted FasL-induced apoptosis (data not shown) (Nicholson et al., 1995). Under these conditions, i.e., when the ICE protease cascade was completely suppressed, the phosphorylation level of ERM proteins was analyzed after FasL stimulation. LHF cells were metabolically labeled with [32P]orthophosphate and then stimulated with FasL in the presence of 300 μM Ac-YVAD-cho or Ac-DEVD-cho. After incubation for 3 h, ERM proteins were recovered by immunoprecipitation, and their phosphorylation level was examined by autoradiography. As shown in Fig. 8, the FasL-induced dephosphorylation of ERM proteins was mostly suppressed by either Ac-YVAD-cho or Ac-DEVD-cho, indicating that the dephosphorylation of ERM proteins is located downstream of the FasL-induced activation of ICE proteases.
Figure 8.

Suppression of FasL-induced dephosphorylation of ERM proteins by ICE protease inhibitors. LHF cells were metabolically labeled with [32P]orthophosphate for 3 h, and then ICE protease inhibitors (Ac-YVAD-cho or Ac-DEVD-cho) were added at 300 μM. After 1 h incubation, FasL was added. At 3 h of incubation, the cell lysate was immunoprecipitated with anti-ERM pAb, TK89, and analyzed by autoradiography ([32P]) and immunoblotting ([Blotting]). Equal amounts of protein were applied to each lane as revealed in accompanying immunoblots with anti-ERM pAb, TK89. The FasL-induced dephosphorylation of ERM proteins was suppressed by both inhibitors. Arrows indicate ezrin, radixin, and moesin, respectively, from the top.
Subsequent Events in the Cytoplasmic Translocation of ERM Proteins during FasL-induced Apoptosis
The question then arose as to what type of cellular event is evoked by the cytoplasmic translocation of ERM proteins, i.e., the disappearance of microvilli, in apoptosis. Considering that ERM proteins function as general cross-linkers between plasma membranes and actin filaments, it was expected that gross dissociation of the actin-based cytoskeleton and plasma membranes occur as a result of the cytoplasmic translocation of ERM proteins. To evaluate this expectation, we prepared DiO-labeled single-layered plasma membranes from FasL-treated and nontreated LHF cells (Tsukita et al., 1984; Hong and Hume, 1986). Briefly, free-floating LHF cells were incubated with DiO to vitally label their plasma membrane proper, stimulated with FasL, and then attached to poly-l-lysine–coated coverslips. In control experiments, without FasL stimulation, cells were attached to poly-l-lysine–coated coverslips. At 1 h of incubation, cells were ruptured with a jet stream of hypotonic solution, and the remnant single-layered plasma membranes with the cytoplasmic side upward on the glass were stained with anti-ERM antibodies. As shown in Fig. 9, a and b, when the single-layered plasma membranes were prepared from nontreated cells, they were double positive with DiO and anti-ERM antibodies. In contrast, FasL-treated cells left various sizes of DiO-positive plasma membranes on the cover glasses, almost free of ERM proteins (Fig. 9, c and d). This finding confirmed the FasL-induced cytoplasmic translocation of ERM proteins. Also, in this system the suppression of the cytoplasmic translocation of ERM proteins by calyculin A was observed (Fig. 9, e and f).
Figure 9.

Detection of ERM proteins on single-layered plasma membranes isolated on cover glasses. After vital labeling of plasma membranes proper with DiO, single-layered plasma membranes with the cytoplasmic surface freely exposed were prepared from nontreated (a and b), FasL–1 h–treated (c and d), and FasL/calyculin A–1 h–treated (e and f) LHF cells as described in Materials and Methods. These preparations were immunofluorescently labeled with anti-ERM pAb, TK89. Each single-layered plasma membrane was detected by DiO signal (a, c, and e). ERM signal was intense from plasma membranes of nontreated cells (b), whereas it was undetectable from those of FasL-treated cells (d). Calyculin A suppressed the FasL-induced depletion of ERM proteins from plasma membranes (f). Bar, 20 μm.
Next, the single-layered plasma membranes were stained with rhodamine phalloidin to visualize actin filaments. As shown in Fig. 10, the membranes prepared from the nonstimulated cells were associated with dense actin filaments, whereas those from the FasL-stimulated cells were almost free of actin filaments. Furthermore, calyculin A suppressed this FasL-induced dissociation of actin filaments from plasma membranes. We then concluded that FasL stimulation induced the gross dissociation of the actin-based cytoskeleton from plasma membranes through the cytoplasmic translocation of ERM proteins.
Figure 10.
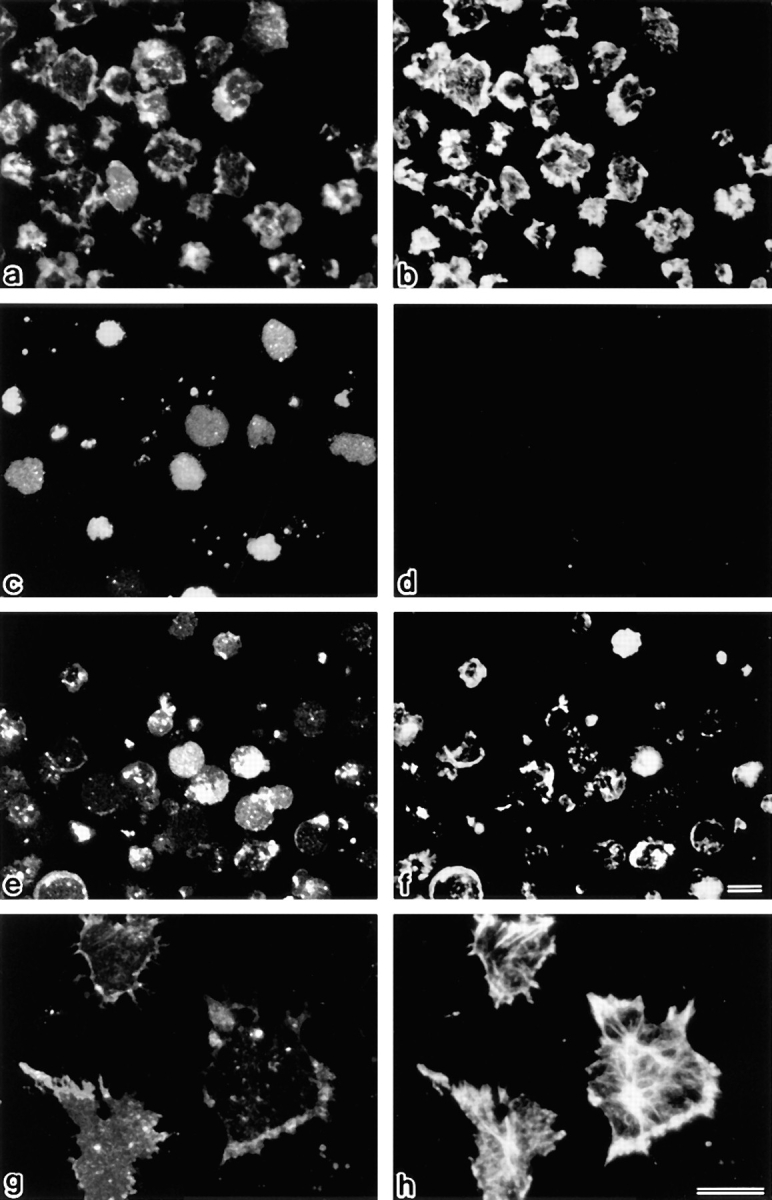
Detection of actin filaments on the single-layered plasma membranes isolated on cover glasses. After vital labeling of plasma membranes proper with DiO, single-layered plasma membranes with the cytoplasmic surface freely exposed were prepared from nontreated (a, b, g, and h), FasL–1 h–treated (c and d), and FasL/calyculin A–1 h–treated (e and f) LHF cells as described in Materials and Methods. These preparations were labeled with rhodamine phalloidin to visualize actin filaments (b, d, f, and h). Each single-layered plasma membrane was detected by DiO signal (a, c, e, and g). Actin filaments were densely associated with plasma membranes of nontreated cells (b and h), whereas they were undetectable from those of FasL-treated cells (d). Calyculin A suppressed the FasL-induced dissociation of actin filaments from plasma membranes (f). Bars: (a–f) 10 μm; (g and h) 15 μm.
Discussion
Microvillar disappearance is one of the early common events in various types of apoptosis. In this study, using FasL-induced apoptosis in LHF cells, we found that plasma membrane–bound ERM proteins, ubiquitously expressed microvillar proteins, are translocated to the cytoplasm concomitantly with microvillar disappearance (within 1 h of incubation with FasL). Considering that the depletion of ERM proteins from living cells by antisense oglionucleotide treatment resulted in the complete disappearance of microvilli from the cell surface (Takeuchi et al., 1994b ), we conclude that in FasL-induced apoptosis the cytoplasmic translocation of ERM proteins, i.e., the depletion of ERM proteins from plasma membranes, is directly responsible for microvillar disappearance. Furthermore, in this study we analyzed the upstream and downstream events of the microvillar disappearance, i.e., the cytoplasmic translocation of ERM proteins, in the FasL-triggered death signaling pathway, and our conclusions were as follows. FasL stimulation upregulates the ICE protease cascade as recently clarified, which facilitated the dephosphorylation of ERM proteins. This dephosphorylation event is required for ERM proteins to translocate from the plasma membrane to the cytoplasm, which induces gross dissociation of actin filaments from the plasma membrane.
The first question is how activated proteases induce the dephosphorylation of ERM proteins and their subsequent cytoplasmic translocation. In most types of cells, approximately half of the ERM protein is distributed in the cytoplasm (Sato et al., 1992). As described in the introduction, these cytoplasmic soluble ERM proteins take an inactive form, in which the amino- and carboxy-terminal halves mutually suppress their functions through head-to-tail association (Henry et al., 1995; Magendantz et al., 1995; Martin et al., 1995). Some kinds of activation signals may release this mutual suppression by disrupting the intramolecular or intermolecular head-to-tail interaction, allowing the activated ERM proteins to bind to the plasma membranes and actin filaments simultaneously (Arpin et al., 1994; Tsukita et al., 1997a ,b). The serine/threonine phosphorylation of ERM proteins has been reported to cause their translocation from the cytoplasm to the plasma membrane (Urushidani et al., 1989; Chen 1995; Nakamura et al., 1995). These observations are consistent with our present finding that the dephosphorylation of ERM proteins is coupled with their cytoplasmic translocation during apoptosis, although dephosphorylation was detected not only at serine/threonine but also at tyrosine residues. Ezrin has been reported to be a good in vivo substrate for tyrosine kinases, but its physiological relevance remains unclear (Bretscher, 1989; Hunter and Cooper, 1981). As during apoptosis, phosphothreonine of ERM proteins appeared to be preferentially dephosphorylated as compared to phosphoserine and phosphotyrosine, and as calyculin A, a potent serine/threonine phosphatase inhibitor, suppressed the apoptosis-associated dephosphorylation of ERM proteins, it is likely that the FasL-activated ICE protease cascade directly or indirectly downregulates some serine/threonine kinase or activates some serine/threonine phosphatase, resulting in the dephosphorylation of ERM proteins. On the other hand, Rho, a small GTP-binding protein, was reported to regulate the activation of ERM proteins through its downstream signaling molecules (Hirao et al., 1996). It is thus also possible that the FasL-activated ICE protease cascade affects the activity of ERM proteins through the Rho-dependent signaling pathway. In this context, it is interesting that D4-GDI, a hematopoietic cell-specific homologue of Rho-GDI, is cleaved by ICE proteases in FasL- and staurosporine-induced apoptosis of T-cell lines (Songqing et al., 1996).
The second question was the subsequent event of the cytoplasmic translocation of ERM proteins, i.e., the breakdown of microvilli at the early phase of apoptosis. ERM proteins are general cross-linkers between actin filaments and the plasma membranes. In this sense, microvilli can be regarded as membrane domains that are specialized for the ERM protein–based interaction of actin filaments with the plasma membrane. Thus the depletion of ERM proteins from the plasma membrane, i.e., the breakdown of microvilli, is expected to result in the gross dissociation of actin filaments from the plasma membrane. The DiO-stained single-layered membrane preparations developed in this study allowed us to evaluate this expectation and led us to conclude that at an early phase of FasL-induced apoptosis, the actin-based cytoskeleton and plasma membrane are completely dissociated, leaving actin filament–free plasma membranes. Many actin filament–plasma membrane cross-linkers other than ERM proteins have been identified, but such gross dissociation of actin filaments and plasma membranes has not been observed by the downregulation of their expression, suggesting the predominant role of ERM proteins in the regulation of actin filament/plasma membrane interaction. Furthermore, this gross dissociation is consistent with our previous results obtained with antisense oligonucleotides that the suppression of ERM protein expression affected several independent cellular events, such as cell–cell adhesion, cell–matrix adhesion, and microvillar formation, where actin filament/plasma membrane interactions may play important roles (Takeuchi et al., 1994b ).
During apoptosis, breakdown of microvilli has been reported to be followed by marked changes in cytoskeletal organization. Several cytoskeletal proteins and related signaling molecules, such as actin, fodrin, Gas2, and protein kinase Cδ, have been identified as potential targets for ICE proteases (Brancolini et al., 1995; Chinnaiyan and Dixit, 1996; Fraser and Evans, 1996). These targets may play synchronized roles together with the dephosphorylation of ERM proteins, resulting in marked changes in cytoskeletal organization.
Considering that microvillar breakdown is a common early event in apoptosis, it is likely that the gross dissociation between actin filaments and plasma membranes occurs commonly at an early phase of various types of apoptosis. Actually, the apoptosis-associated cytoplasmic translocation of ERM proteins was observed not only in LHF cells but also in cultured epithelial cells (MTD-1A) and promyelocytic leukemic cells (HL-60). Furthermore, this gross dissociation is located downstream of activation of the ICE protease cascade, which is commonly activated in apoptosis (Enari et al., 1996). This raises questions regarding the physiological relevance of this gross dissociation in apoptosis in general and whether this apoptosis-associated cytoplasmic event is independent from the nuclear events such as chromatin condensation and DNA fragmentation. It is noteworthy that calyculin A significantly suppressed the FasL-induced apoptosis of LHF cells under the conditions used in this study (data not shown). Calyculin A has also been reported to delay the apoptotic process in lymphoma cells induced by gamma-irradiation, tetrandrine, bistratene A, cisplatin, and etoposide (Song and Lavin, 1993; Morana et al., 1996). Considering that calyculin A retained ERM proteins on plasma membranes, it is tempting to speculate that the ERM-based tight association of the actin-based cytoskeleton with the plasma membranes makes cells resistant to death signals, i.e., that destruction of ERM-based actin filament/plasma membrane interactions is required for the apoptotic changes in the nucleus. This speculation is similar to the recent observation that mutated lamin, a nuclear skeletal protein that is resistant to ICE protease–dependent proteolysis, delayed apoptosis (Rao et al., 1996). Of course, it is possible that calyculin A does not suppress the nuclear apoptosis through ERM proteins, but through other proteins directly involved in the nuclear apoptosis. Further analysis of the physiological relevance of the gross dissociation of actin filaments from plasma membranes, i.e., the disappearance of microvilli, in apoptosis will lead to a better understanding of the total picture of the molecular mechanism of apoptosis.
Acknowledgments
We thank all the members of our laboratory (Department of Cell Biology, Faculty of Medicine, Kyoto University) for their helpful discussions. T. Kondo thanks Prof. H. Saito (Nagoya University) for providing him with the opportunity to work in the Department of Cell Biology, Faculty of Medicine, Kyoto University.
This work was supported in part by a Grant-in-Aid for Scientific Research (B) (to Sa. Tsukita), a Grant-in-Aid for Cancer Research and a Grant-in-Aid for Scientific Research (A) from the Ministry of Education, Science, and Culture of Japan (to Sh. Tsukita).
Abbreviations used in this paper
- DAPI
4′,6′-diamidino-2-phenylindole
- ERM
ezrin/radixin/moesin
- FasL
Fas ligand
- ICE
interleukin-1β-converting enzyme
Footnotes
Address all correspondence to Sachiko Tsukita, Ph.D., Department of Cell Biology, Kyoto University Faculty of Medicine, Konoe-Yoshida, Sakyo-ku, Kyoto 606, Japan. Tel.: 81-75-753-4373. Fax: 81-75-753-4660. E-mail: atsukita@mfour.med.kyoto-u.ac.jp
References
- Alnemri ES, Livingston DJ, Nicholoson DW, Salvensen G, Thornberry NA, Wong WW, Yuan J. Human ICE-CED-3 protease nomenclature. Cell. 1996;87:171. doi: 10.1016/s0092-8674(00)81334-3. [DOI] [PubMed] [Google Scholar]
- Arpin M, Algrain M, Louvard D. Membrane-actin microfilament connections: an incresing diversity of players related to band 4.1. Curr Opin Cell Biol. 1994;6:136–141. doi: 10.1016/0955-0674(94)90127-9. [DOI] [PubMed] [Google Scholar]
- Berryman M, Gary R, Bretscher A. Ezrin oligomers are major cytoskeletal components of placental microvilli: a proposal for their involvement in cortical morphogenesis. J Cell Biol. 1995;131:1231–1242. doi: 10.1083/jcb.131.5.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle WJ, van der Geer P, Hunter T. Phosphopeptide mapping and phosphoamino acid analysis by two-dimensional separation on thin-layer cellulose plates. Methods Enzymol. 1991;201:110–148. doi: 10.1016/0076-6879(91)01013-r. [DOI] [PubMed] [Google Scholar]
- Brancolini C, Benedetti M, Schneider C. Microfilament reorganization during apoptosis: the role of Gas2, a possible substrate for ICE-like proteases. EMBO (Eur Mol Biol Organ) J. 1995;14:5179–5190. doi: 10.1002/j.1460-2075.1995.tb00202.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretscher A. Rapid phosphorylation and reorganization of ezrin and spectrin accompany morphological changes induced in A-431 cells by epidermal growth factor. J Cell Biol. 1989;108:921–930. doi: 10.1083/jcb.108.3.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretscher A. Microfilament structure and function in the cortical cytoskeleton. Annu Rev Cell Biol. 1991;7:337–374. doi: 10.1146/annurev.cb.07.110191.002005. [DOI] [PubMed] [Google Scholar]
- Chen J, Cohn A, Mandel LJ. Dephosphorylation of ezrin as an early event in renal microvillar breakdown and anoxic injury. Proc Natl Acad Sci USA. 1995;92:7495–7499. doi: 10.1073/pnas.92.16.7495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnaiyan KM, Dixit VM. The cell-death machine. Curr Biol. 1996;6:555–562. doi: 10.1016/s0960-9822(02)00541-9. [DOI] [PubMed] [Google Scholar]
- Enari M, Hug H, Nagata S. Involvement of an ICE-like protease in Fas-mediated apoptosis. Nature (Lond) 1995;375:78–81. doi: 10.1038/375078a0. [DOI] [PubMed] [Google Scholar]
- Enari M, Talanian RV, Wong WW, Nagata S. Sequential activation of ICE-like and CPP32-like proteases during Fas-mediated apoptosis. Nature (Lond) 1996;380:723–726. doi: 10.1038/380723a0. [DOI] [PubMed] [Google Scholar]
- Fraser A, Evans G. A license to kill. Cell. 1996;85:781–784. doi: 10.1016/s0092-8674(00)81005-3. [DOI] [PubMed] [Google Scholar]
- Funayama N, Nagafuchi A, Sato N, Sa, Tsukita, Sh, Tsukita Radixin is a novel member of the band 4.1 family. J Cell Biol. 1991;115:1039–1048. doi: 10.1083/jcb.115.4.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould KL, Bretscher A, Esch FS, Hunter T. cDNA cloning and sequencing of the protein-tyrosine kinase substrate, ezrin, reveals homology to band 4.1. EMBO (Eur Mol Biol Organ) J. 1989;8:4133–4142. doi: 10.1002/j.1460-2075.1989.tb08598.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helander TS, Carpen O, Turunen O, Kovanen PE, Vaheri A, Timonen T. ICAM-2 redistributed by ezrin as a target for killer cells. Nature (Lond) 1996;382:265–268. doi: 10.1038/382265a0. [DOI] [PubMed] [Google Scholar]
- Henry MD, Gonzalez C, Agosti, Solomon F. Molecular dissection of radixin: distinct and interdependent functions of the amino- and carboxy-terminal domains. J Cell Biol. 1995;4:1007–1022. doi: 10.1083/jcb.129.4.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirao M, Sato N, Kondo T, Yonemura S, Monden M, Sasaki T, Takai Y, Sh, Tsukita, Sa, Tsukita Regulation mechanism of ERM (ezrin/radixin/moesin) protein/plasma membrane association: possible involvement of phosphatidylinositol turnover and Rho-dependent signaling pathway. J Cell Biol. 1996;135:37–51. doi: 10.1083/jcb.135.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong MB, Hume RI. Fluorescent carbocyanine dyes allow living neurons of identified origin to be studied in long-term cultures. J Cell Biol. 1986;103:171–187. doi: 10.1083/jcb.103.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter T, Cooper JA. Epidermal growth factor induces tyrosine phosphorylation of proteins in A431 human tumor cells. Cell. 1981;24:741–752. doi: 10.1016/0092-8674(81)90100-8. [DOI] [PubMed] [Google Scholar]
- Itoh N, Yonehara S, Ishii A, Yonehara M, Mizushima S, Sameshima M, Hase A, Seto Y, Nagata S. The polypeptide encoded by the cDNA for human cell surface antigen Fas can medite apoptosis. Cell. 1991;66:233–243. doi: 10.1016/0092-8674(91)90614-5. [DOI] [PubMed] [Google Scholar]
- Jacobson MD. Programmed cell death in animal development. Cell. 1997;88:347–354. doi: 10.1016/s0092-8674(00)81873-5. [DOI] [PubMed] [Google Scholar]
- Jiang WG, Hiscox S, Singhrao SK, Puntis MC, Nakamura T, Mansel RE, Hallett MB. Induction of tyrosine phosphorylation and translocation of ezrin by hepatocyte growth factor/scatter factor. Biochem Biophys Res Commun. 1995;217:1062–1069. doi: 10.1006/bbrc.1995.2877. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage. J Cell Biol. 1970;105:1395–1404. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lankes WT, Furthmayr H. Moesin: a member of the protein 4.1-talin-ezrin family of proteins. Proc Natl Acad Sci USA. 1991;88:8297–8301. doi: 10.1073/pnas.88.19.8297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Los M, Van de Craen M, Penning LC, Schenk H, Westendorp M, Baeuerle PA, Droge W, Krammer PH, Fiers W, Schulze-Osthoff K. Requirement of an ICE/CED-3 protease for Fas/APO-1-mediated apoptosis. Nature (Lond) 1995;375:81–83. doi: 10.1038/375081a0. [DOI] [PubMed] [Google Scholar]
- Magendantz M, Henry MD, Lander A, Solomon F. Interdomain interactions of radixin in vitro. J Biol Chem. 1995;270:25324–25327. doi: 10.1074/jbc.270.43.25324. [DOI] [PubMed] [Google Scholar]
- Martin SJ, O'Brien GA, Nishioka WK, McGahon AJ, Mahboubi A, Saido TC, Green DR. Proteolysis of fodrin (non-erythroid spectrin) during apoptosis. J Biol Chem. 1995;270:6425–6428. doi: 10.1074/jbc.270.12.6425. [DOI] [PubMed] [Google Scholar]
- Morana SJ, Wolf CM, Li J, Reynolds JE, Brown MK, Eastman A. The involvement of protein phosphatase in the activation of ICE/ CED-3 protease, intercellular acidification, DNA digestion, and apoptosis. J Biol Chem. 1996;271:18263–18271. doi: 10.1074/jbc.271.30.18263. [DOI] [PubMed] [Google Scholar]
- Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- Nagata S, Goldstein P. The Fas death factor. Science (Wash DC) 1995;267:1449–1456. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- Nakamura F, Amieva M, Furthmayr H. Phosphorylation of threonine 558 in the carboxyl-terminal actin-binding domain of moesin by thrombin activation of human platelets. J Biol Chem. 1995;270:31377–31385. doi: 10.1074/jbc.270.52.31377. [DOI] [PubMed] [Google Scholar]
- Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature (Lond) 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- Rao L, Perez D, White E. Lamin proteolysis facilitates nuclear events during apoptosis. J Cell Biol. 1996;135:1441–1455. doi: 10.1083/jcb.135.6.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouleau GA, Merel P, Lutchman M, Sanson M, Zucman J, Marineau C, Hoang-Xuan K, Demczuk S, Desmaze C, Plougastel B, et al. Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature (Lond) 1993;363:515–521. doi: 10.1038/363515a0. [DOI] [PubMed] [Google Scholar]
- Sato N, Funayama N, Nagafuchi A, Yonemura S, Sa, Tsukita, Sh, Tsukita A gene family consisting of ezrin, radixin, and moesin. Its specific localization at actin filament/plasma membrane association sites. J Cell Sci. 1992;103:131–143. doi: 10.1242/jcs.103.1.131. [DOI] [PubMed] [Google Scholar]
- Song Q, Lavin MF. Calyculin A, a potent inhibitor of phosphatase-1 and -2A, prevents apoptosis. Biochem Biophys Res Commun. 1993;190:47–55. doi: 10.1006/bbrc.1993.1009. [DOI] [PubMed] [Google Scholar]
- Songqing N, Chuangs TH, Cunningham A, Turi TG, Hanke JH, Bokch GM, Danley DE. D4-GDI, a substrate of CPP32, is proteolyzed during Fas-induced apoptosis. J Biol Chem. 1996;271:11209–11213. doi: 10.1074/jbc.271.19.11209. [DOI] [PubMed] [Google Scholar]
- Suda T, Takahashi T, Goldstein P, Nagata S. Molecular cloning and expression of the Fas ligand, a novel member of the tumor necrosis factor family. Cell. 1993;75:1169–1178. doi: 10.1016/0092-8674(93)90326-l. [DOI] [PubMed] [Google Scholar]
- Takeuchi K, Kawashima A, Nagafuchi A, Sh, Tsukita Structural diversity of band 4.1 superfamily members. J Cell Sci. 1994a;107:1921–1928. doi: 10.1242/jcs.107.7.1921. [DOI] [PubMed] [Google Scholar]
- Takeuchi K, Sato N, Kasahara H, Funayama N, Nagafuchi A, Yonemura S, Sa, Tsukita, Sh, Tsukita Perturbation of cell adhesion and microvilli formation by antisense oligonucleotides to ERM family members. J Cell Biol. 1994b;125:1371–1384. doi: 10.1083/jcb.125.6.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trofatter JA, MacCollin MM, Rutter JL, Murrell JR, Duyao MP, Parry DM, Eldridge R, Kley N, Menon AG, et al. A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell. 1993;72:791–800. doi: 10.1016/0092-8674(93)90406-g. [DOI] [PubMed] [Google Scholar]
- Tsukita Sa, Tsukita Sh, Ishikawa H. Bidirectional polymerization of G-actin on the human erythrocyte membrane. J Cell Biol. 1984;98:1102–1110. doi: 10.1083/jcb.98.3.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukita Sa, Oishi K, Sato N, Sagara J, Kawai A, Tsukita Sh. ERM family members as molecular linkers between the cell surface glycoprotein CD44 and actin-based cytoskeletons. J Cell Biol. 1994;126:391–401. doi: 10.1083/jcb.126.2.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukita Sa, Yonemura S, Tsukita Sh. ERM proteins: head-to-head regulation of actin-plasma membrane interaction. Trends Biochem Sci. 1997a;22:53–58. doi: 10.1016/s0968-0004(96)10071-2. [DOI] [PubMed] [Google Scholar]
- Tsukita Sa, Yonemura S, Tsukita Sh. ERM(ezrin/radixin/moesin) family: from cytoskeleton to signal transduction. Curr Opin Cell Biol. 1997b;9:70–75. doi: 10.1016/s0955-0674(97)80154-8. [DOI] [PubMed] [Google Scholar]
- Turunen O, Winqvist R, Pakkanen R, Grzeschik KH, Wahlstrom T, Vaheri A. Cytovillin, a microvillar Mr 75,000 protein. cDNA sequence, prokaryotic expression, and chromosomal localization. J Biol Chem. 1989;264:16727–16732. [PubMed] [Google Scholar]
- Urushidani T, Hanzel DK, Forte JG. Characterization of an 80 kD phosphoprotein involved in parietal cell stimulation. Am J Physiol. 1989;256:G1070–G1081. doi: 10.1152/ajpgi.1989.256.6.G1070. [DOI] [PubMed] [Google Scholar]
- Wyllie AH, Kerr JFR, Currie AR. Cell death: the significance of apoptosis. Int Rev Cytol. 1980;68:251–300. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- Yonehara S, Ishii A, Yonehara M. A cell-killing monoclonal antibody (anti-Fas) to a cell surface antigen co-downregulated with the receptor of tumor necrosis factor. J Exp Med. 1989;169:1747–1756. doi: 10.1084/jem.169.5.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonemura S, Nagafuchi A, Sato N, Sh, Tsukita Concentration of an integral membrane protein, CD43 (leukosialin, sialophorin), in the cleavage furrow through the ineraction of its cytoplasmic domain with actin-based cytoskeletons. J Cell Biol. 1993;120:437–449. doi: 10.1083/jcb.120.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]