

Abstract
We have generated F9 murine embryonal carcinoma cells in which either the retinoid X receptor (RXR)α and retinoic acid receptor (RAR)α genes or the RXRα and RARγ genes are knocked out, and compared their phenotypes with those of wild-type (WT), RXRα−/−, RARα−/−, and RARγ−/− cells. RXRα−/−/ RARα−/− cells were resistant to retinoic acid treatment for the induction of primitive and parietal endodermal differentiation, as well as for antiproliferative and apoptotic responses, whereas they could differentiate into visceral endodermlike cells, as previously observed for RXRα−/− cells. In contrast, RXRα−/−/RARγ−/− cells were defective for all three types of differentiation, as well as antiproliferative and apoptotic responses, indicating that RXRα and RARγ represent an essential receptor pair for these responses. Taken together with results obtained by treatment of WT and mutant F9 cells with RAR isotype– and panRXR-selective retinoids, our observations support the conclusion that RXR/ RAR heterodimers are the functional units mediating the retinoid signal in vivo. Our results also indicate that the various heterodimers can exert both specific and redundant functions in differentiation, proliferation, and apoptosis. We also show that the functional redundancy exhibited between RXR isotypes and between RAR isotypes in cellular processes can be artifactually generated by gene knockouts. The present approach for multiple gene targeting should allow inactivation of any set of genes in a given cell.
Retinoids exert their pleiotropic effects on vertebrate development, cellular differentiation, proliferation, and homeostasis through two classes of ligand-dependent transactivators: the retinoic acid receptors (RARs)1, and the retinoid X receptors (RXRs) (for reviews see De Luca, 1991; Blomhoff, 1994; Chambon, 1994, 1996; Kastner et al., 1995; Mangelsdorf and Evans, 1995). RARs are activated by all-trans retinoic acid (tRA) and by 9-cis retinoic acid (9C-RA), whereas RXRs are activated by 9C-RA only. The various RAR (RARα, β, and γ) and RXR (RXRα, β, and γ) isotypes are encoded by different genes, and their isoforms, which differ in their NH2-terminal regions, are generated by differential promoter usage and alternative splicing. The multiple RAR and RXR isotypes and isoforms are conserved in vertebrate evolution, and display distinct spatiotemporal expression patterns in developing embryos and adult tissues, suggesting that each receptor performs some unique functions (for reviews see Leid et al., 1992a ; Chambon, 1994; Kastner et al., 1994). RXR/RAR heterodimers bind much more efficiently to retinoic acid response elements (RAREs) than their respective homodimers in vitro (for reviews see Leid et al., 1992a ; Chambon, 1994, 1996; Giguère, 1994; Glass, 1994; Kastner et al., 1994; Mangelsdorf et al., 1994; Gronemeyer and Laudet, 1995; Keaveney and Stunnenberg, 1995; Mangelsdorf and Evans, 1995), and several lines of evidence support the idea that these heterodimers represent the functional units transducing the retinoid signal in vivo (Kastner et al., 1995; Chambon, 1996).
The F9 murine embryonal carcinoma (EC) cell line expresses all types of RARs and RXRs (Zelent et al., 1989; Martin et al., 1990; Wan et al., 1994), and upon retinoic acid treatment, it differentiates into cells resembling three distinct extraembryonic endoderm (primitive, parietal, and visceral), depending on the culture conditions (for reviews see Strickland, 1981; Hogan et al., 1983; Gudas et al., 1994). Retinoid-induced differentiation is accompanied by an apoptotic response and a dramatic decrease in the rate of proliferation (Sleigh, 1992; Atencia et al., 1994; Clifford et al., 1996). Thus, F9 cells provide an attractive system for the analysis of retinoid signaling in vivo.
To further investigate the roles of RXRs and RARs in differentiation, proliferation, and apoptosis, we have now generated F9 cells lacking either RXRα and RARα, or RXRα and RARγ, and then compared their phenotypes with those of wild-type (WT), RXRα−/− (Clifford et al., 1996), RARα−/− (Boylan et al., 1995), and RARγ−/− (Boylan et al., 1993; Taneja et al., 1995) F9 cells. Multiple gene targeting in a given cell has been achieved by using a Cre/loxP system (Sauer and Henderson, 1990; Metzger et al., 1995), which allows removal of the antibiotic resistance gene from a targeted locus, and therefore subsequent mutagenesis of the second allele of a given gene with the same targeting construct, as well as the targeting of additional genes. We demonstrate that tRA-treated RXRα−/−/ RARα−/− cells differentiate poorly into primitive and parietal endodermlike cells and are impaired in both antiproliferative and apoptotic responses, whereas they fully differentiate into visceral endoderm (VE)-like cells, as previously observed for RXRα−/− cells (Clifford et al., 1996). In contrast, RXRα−/−/RARγ−/− cells are defective for all three types of endodermal differentiation, as well as for the antiproliferative and apoptotic responses, indicating that the absence of both RXRα and RARγ cannot be functionally compensated by the other retinoid receptors in these cells. Taken together with results obtained by treatment of WT and mutant F9 cells with panRXR- and RAR isotype– selective retinoids, our findings support the conclusion that RXR/RAR heterodimers are the functional units mediating the retinoid signal in vivo. Furthermore our results indicate that RXR/RAR heterodimers can exert both specific and redundant functions in differentiation, proliferation, and apoptosis. We also show that functional redundancy between RXR isotypes and between RAR isotypes can be artifactually generated by gene knockouts.
Materials and Methods
Cell Culture
F9 cells were cultured and induced to differentiate into primitive, parietal, and visceral endodermlike cells as previously described (Boylan et al., 1993; Clifford et al., 1996). The retinoids (tRA, Am80, BMS753, BMS453, BMS961, and BMS649) were dissolved in ethanol.
Targeting of the RARα or RARγ Genes in RXRα-null F9 Cells.
The RARα targeting vector, pRARα(LNL), was previously described (Metzger et al., 1995). The RARγ targeting vector, pRARγ(LPL), was derived from pDγ6.5A (a gift from D. Lohnes, IGBMC, CNRS/INSERM/ ULP, Illkirch, France), which contains a 6-kb genomic fragment including exons 5 and 8. A unique SmaI site, followed by stop codons in all three reading frames, was introduced into pDγ6.5A at the KpnI site located in exon 8 of RARγ by inserting the oligonucleotides 5′-CCCCGGGTAGGTAGATAGCGTAC-3′ and 5′-GCTATCTACCTACCCGGGGGTAC-3′, yielding the pRARγT4 construct. An XhoI-BamHI fragment containing the phosphoglycerate kinase (PGK) promoter-driven, puromycin- resistance (puro) gene, flanked by loxP sites, was isolated from pHRLpuro1, and blunt ended with T4 DNA polymerase, followed by ligation into the SmaI site of pRARγT4. pHRLpuro1 was constructed from VS-1, a plasmid containing a loxP site-flanked PGKpuroA+ cassette, by mutating the SalI site. The PGKpuroA+ cassette was obtained by ligating the PGK promoter (a 500-bp EcoRI-PstI fragment isolated from pKJ-1 [Adra et al., 1987]) to the coding sequence of the puro gene (a 600-bp HindIII-ClaI fragment isolated from pLXPB [Imler et al., 1996]), and by inserting the SV-40 polyadenylation signal (a 160-bp BglII-XbaI fragment isolated from pSG5 [Green et al., 1988]) using synthetic oligonucleotides. This cloning resulted in the loss of the PstI, HindIII, and ClaI restriction sites, and the introduction of HindIII and EcoRI sites at 5′ and 3′ ends of the PGK promoter, respectively. KpnI, ApaI, XhoI, and BglII restriction sites, and BamHI and SacI sites are located at 5′ and 3′ ends of the loxP-flanked cassette, respectively.
Electroporation, selection of neomycin-resistant clones, Cre-mediated excision of the resistance genes, and Southern blot analysis were performed as previously described (Metzger et al., 1995; Clifford et al., 1996). Puromycin selection (500 ng/ml) was carried out for 10 d.
Western Blotting and Electrophoretic Mobility Shift Assays (EMSA)
Isolation of whole cell extracts from F9 cells and transfected COS-1 cells, Western blot analysis, and electrophoretic mobility shift assays were performed as previously described (Rochette-Egly et al., 1991; Gaub et al., 1992; Boylan et al., 1993).
Reverse Transcription (RT)-PCR
RNA preparation, RT-PCR, and Southern blotting were performed as previously described (Bouillet et al., 1995; Roy et al., 1995). The PCR primers and end-labeled oligonucleotide probes for collagen IVα1, laminin B1, α-fetoprotein (AFP), and 36B4, were described previously (Clifford et al., 1996). Transcript levels were quantified using a BAS 2000 bio-imaging analyzer (Fuji Ltd., Tokyo, Japan), and were normalized to the corresponding 36B4 signals.
Analysis of Cellular Growth
Cells were plated in triplicate 3-cm culture wells (5 × 102 cells per well), and cell counting experiments were performed as previously described (Clifford et al., 1996). [3H]Thymidine incorporation assays were performed essentially as described (Clifford et al., 1996), with the following modifications. Cells were cultured for 4 d in six replicate 3-cm wells, in the presence or absence of 1 μM tRA, and three of the six wells were treated with 8 μCi/well [3H]methylthymidine (20.0 Ci/mmol; Dupont-NEN, Boston, MA) for 2 h before harvesting. The cell cycle profile of WT and mutant cells was determined as previously described (Clifford et al., 1996).
Analysis of Apoptosis
Apoptosis was analyzed by both Hoechst staining of nuclei and FACS® analysis, as previously described (Clifford et al., 1996).
Results
Targeted Disruption of the RARα or RARγ Genes in RXRα Null F9 Cells
The RXRα-null F9 cell line, C2RXRα−(L)/−(L), which constitutively expresses a ligand-dependent chimeric Cre- recombinase (Cre-ER; Metzger et al., 1995), was electroporated with the targeting constructs pRARα(LNL) (Fig. 1 A) or pRARγ(LPL) (Fig. 2 A) to generate F9 cells disrupted in either the RXRα and RARα genes or the RXRα and RARγ genes. These targeting constructs contain translation stop codons and loxP site–flanked neomycin- or puromycin-resistance genes in exon 9 of the RARα gene or exon 8 of the RARγ gene, respectively (Materials and Methods; Figs. 1 A and 2 A). Since these exons encode the B region, which is common to all isoforms of a given RAR isotype (Kastner et al., 1990; Leroy et al., 1991; Chambon, 1994), the expression of RARα and RARγ proteins is suppressed by these mutations. Homologous recombination (HR) and Cre-mediated excision of the resistance genes were verified by Southern blotting (Figs. 1 B and 2 B).
Figure 1.

Disruption of both alleles of the RARα gene by HR in a RXRα−(L)/−(L) cell line. (A) Schematic diagram of the pRARα(LNL) targeting construct, the WT RARα locus, and the recombined locus after integration (HR[I]) and after Cre-mediated excision (HR[E]). Dark boxes indicate exons. The exons 4–8 encoding the NH2-terminal part of minor isoforms (RARα3-7) (Leroy et al., 1991) are not represented. Restriction enzyme sites and the location of probes are indicated. The neo and a1 probes have been previously described (Metzger et al., 1995). The numbers in the lower part of diagram are in kb. K, KpnI; L, loxP recombination site; S, SalI; ST, two translation stop codons; Xb, XbaI; Xh, XhoI. (B) Southern blot analysis indicating the targeting of the RARα gene in a RXRα−(L)/−(L) cell line. The genotypes of different cell lines (e.g., 9) and their subclones (9a, etc.) are indicated at the top of each lane, and correspond to all three panels. (C) Western blot analysis indicating the absence of RARα protein in RXRα−(L)/−(L)/ RARα−(L)/−(LNL) cell lines. Lanes 1 and 2 contain 2 μg of whole cell extracts from COS cells transfected with either the pSG5 (Green et al., 1988) or mRARαø expression construct (Zelent et al., 1989), and lanes 3–6 contain 60 μg of whole cell extracts from WT and mutant F9 cells, as indicated. RARα protein was detected using the rabbit polyclonal antibody RPα(F), followed by chemiluminescence detection. Mol wt is shown in kD.
Figure 2.

Disruption of both alleles of the RARγ gene by HR in a RXRα−(L)/−(L) cell line. (A) Schematic diagram of the pRARγ(LPL) targeting construct, the WT RARγ locus, and the recombined locus after integration (HR[I]) and after Cre-mediated excision (HR[E]). Dark boxes indicate exons. The exons 6 and 7 encoding the NH2-terminal part of minor isoforms (RARγ4 and 6; Kastner et al., 1990) are not represented. Restriction enzyme sites and the location of probes are indicated. The puro probe corresponds to a 0.7-kb EcoRI-XbaI fragment derived from pHRLpuro1. The r1 probe corresponds to a 1.5-kb BamHI–EcoRI fragment derived from the RARγ genomic clone λG1mRARγ (Lohnes et al., 1993). The r2 probe corresponds to a 1.6-kb EcoRI–PstI fragment derived from pRARγ(LPL). The numbers in the lower part of the diagram are in kb. B, BamHI; Bg, BglII; E, EcoRI; H, HindIII; L, loxP recombination site; S, SalI; ST, three translation stop codons inserted in all reading frames. Asterisk indicates that these sites are not present in the WT gene, and dashed line represents vector sequence. (B) Southern blot analysis indicating the disruption of the RARγ gene in a RXRα−(L)/−(L) cell line. The genotypes of different cell lines (e.g., 25) and their subclones (25a, etc.) are indicated at the top of each lane and correspond to all four panels. (C) EMSA indicating the absence of RARγ protein in RXRα−(L)/−(L)/ RARγ−(L)/−(LNL) cells. A radiolabeled oligonucleotide corresponding to the Hoxa-1/RARβ RARE was incubated with 20 μg of whole cell extracts from WT cells (lanes 1 and 4), RXRα−(L)/−(L)/ RARγ−(L)/−(LNL) cells (lanes 2 and 5) and RARγ−/− cells (lanes 3 and 6), or with 2 μg of whole cell extracts from COS cells transfected with either the pSG5 (lanes 7 and 9; Green et al., 1988) or mRARγø expression construct (lanes 8 and 10; Zelent et al., 1989), together with 0.5 μg of whole cell extracts from COS cells transfected with mRXRαø expression construct (Leid et al., 1992b ). The arrows indicate the shifted complex formed in the presence of mouse monoclonal antibodies Ab2γ(F) and Ab10γ (A2).
Southern blot analysis using a 3′ probe (a1) located outside of the pRARα(LNL) targeting construct (Fig. 1 A), indicated that, after electroporation, 5 out of 96 C2RXRα−(L)/−(L) neomycin-resistant clones had one targeted RARα allele (Fig. 1, A, HR[I]; and B, compare lanes 3 and 4 with lanes 1 and 2; data not shown). One RXRα−(L)/−(L)/RARα+/−(LNL) cell line (clone nine) was treated with estradiol (E2) to excise the loxP-flanked cassette (−[LNL] and −[L]) designate the targeted allele before and after Cre-mediated excision, respectively). Southern blot analysis using a1 and “neo” probes revealed that excision of the resistance gene occurred in two out of six subclones treated with E2 (Fig. 1, A, HR[E]; and B, compare lane 5 with lanes 3 and 4; see also Metzger et al., 1995). The second allele of the RARα gene was targeted in the RXRα−(L)/−(L)/RARα+/−(L) cell line (Fig. 1 B, clone 9a) using the same targeting construct and strategy. 2 of 96 neomycin-resistant clones were positive for the desired recombination event, resulting in RXRα−(L)/−(L)/RARα−(L)/−(LNL) cell lines (Fig. 1 B, clones 9a-10 and 9a-26; compare lanes 6 and 7 with lanes 1–5). No wild-type RARα transcripts were detected in RXRα−(L)/−(L)/ RARα−(L)/−(LNL) cells by semi-quantitative RT-PCRs (data not shown). Similarly, no RARα protein could be detected in RXRα−(L)/−(L)/RARα−(L)/−(LNL) cells (called hereafter RXRα−/−/RARα−/−) by Western blotting using the polyclonal antibody RPα(F) (Gaub et al., 1992), directed against the F region of the RARα protein (Fig. 1 C, compare lanes 3 and 4 with lanes 2, 5, and 6).
To establish F9 cells in which both RXRα and RARγ genes are inactivated, C2RXRα−(L)/−(L) cells were electroporated with the pRARγ(LPL) targeting construct (Fig. 2 A). Southern blot analysis using the r1 probe, located 5′ to the pRARγ(LPL) sequence (Fig. 2 A), revealed that 3 out of 96 puromycin-resistant clones had one targeted RARγ allele (Fig. 2, A, HR[I]; and B, compare lanes 3 and 4 with lanes 1 and 2; and data not shown). One RXRα−(L)/−(L)/ RARγ+/−(LPL) cell line (clone 25) was transiently transfected with a Cre recombinase expression construct (pPGK-Cre) (Clifford et al., 1996), since for unknown reasons the loxP-flanked, puromycin-resistance gene was not excised by treatment of the cells with E2 (data not shown; −[LPL] and −[L] designate the targeted allele before and after Cre-mediated excision, respectively). The pattern obtained by Southern blot analysis, using r1, r2, and puro probes, clearly indicated that 2 out of 96 subclones had lost the puromycin-resistance cassette (Fig. 2, A, HR[E]; and B, compare lane 5 with lane 4; and data not shown). The second allele of the RARγ gene was inactivated in one RXRα−(L)/−(L)/RARγ+/−(L) cell line (Fig. 2 B, clone 25a) using the same targeting construct and strategy, yielding a RXRα−(L)/−(L)/RARγ−(L)/−(LPL) cell line (Fig. 2 B, clone 25a-3, compare lane 6 with lanes 1–5). No wild-type RARγ RNA was detected in RXRα−(L)/−(L)/RARγ−(L)/−(LPL) cells (data not shown). The absence of RARγ protein was verified by EMSA using the monoclonal antibodies Ab2γ(F) and Ab10γ(A2) (Rochette-Egly et al., 1991), directed against the F and A2 regions of the RARγ protein, respectively. No antibody-shifted complex was observed in RXRα−(L)/−(L)/ RARγ−(L)/−(LPL) cells (called hereafter RXRα−/−/RARγ−/−) (Fig. 2 C, compare lane 5 with lanes 4, 6, and 10). Note that the RXRα loci, which contain loxP sites, were not rearranged during excision of the resistance genes at the RARα or RARγ loci (data not shown). Note also that the knockout of a given receptor(s) did not result in major variations of those remaining (Chiba et al., 1997).
Function of RXRs and RARs in the Retinoid-induced Differentiation of F9 Cells into Primitive and Parietal Endodermlike Cells
WT F9 cells differentiate into primitive and parietal endodermlike cells, when grown in monolayer culture in the presence of tRA alone and tRA in combination with dibutyryl cAMP (bt2cAMP), respectively (Strickland, 1981; Hogan et al., 1983). Previous studies have shown that these two types of differentiation are severely impaired in RARγ−/− and RXRα−/− cells (Boylan et al., 1993; Clifford et al., 1996). The differentiation patterns of RXRα−/−/ RARα−/− and RXRα−/−/RARγ−/− cells were compared with those of WT and single knockout cell lines. After 4 d of treatment, <10% of RXRα−/−/RARα−/− cells became flatter and irregular in shape, or rounded with long cell processes, which are the morphological characteristics of primitive or parietal endodermal differentiation of WT F9 cells, respectively. The same results were obtained with 9a-10 and 9a-26 RXRα−/−/RARα−/− cell lines (Fig. 3 A, compare g–i with a–f, and data not shown). No morphological differentiation at all was observed in RXRα−/−/ RARγ−/− cells after 4 d of treatment, and <0.1% of the cells exhibited a differentiated morphology after 6 d of treatment (Fig. 3 A, j–l, and data not shown). This undifferentiated phenotype persisted after 10 d of treatment.
Figure 3.

RXRα−/−/RARγ−/− F9 cells do not differentiate into primitive and parietal endodermlike cells. (A) WT (a– c), RXRα−/− (d–f), RXRα−/−/ RARα−/− (g–i) and RXRα−/−/ RARγ−/− (j–l) cells were treated with control vehicle (a, d, g, and j), 1 μM tRA alone (b, e, h, and k) or 1 μM tRA and 250 μM bt2cAMP (c, f, i, and l) for 4 d. Cells were photographed under a phase–contrast microscope at ×125 magnification. (B) Total RNA from WT and mutant cells, treated with control vehicle or 1 μM tRA for 48 h, was analyzed by RT-PCR analysis for collagen type IVα1, laminin B1, and 36B4. (C) RT-PCR analysis was performed as in B, for three separate experiments. The levels of RNA transcripts were expressed relative to the amount present in tRA-treated WT cells, which was taken as 100. The white and black bars correspond to transcript levels expressed in vehicle- and tRA-treated cells, respectively. Bar, 100 μm.
The extent of differentiation of WT and mutant F9 cells was further investigated biochemically by determining the mRNA levels of collagen type IVα1 and laminin B1, two markers of endodermal differentiation (Fig. 3, B and C). After 48 h of 1 μM tRA treatment, the induction of collagen type IVα1 and laminin B1 was reduced in RARγ−/− cells (10-fold and 6-fold lower levels, respectively) and in RXRα−/− cells (3-fold and 6-fold lower levels, respectively) when compared to WT cells, whereas these inductions were not altered in RARα−/− cells (Boylan et al., 1993, 1995; Clifford et al., 1996; Taneja et al., 1996). The induction of both transcripts was also impaired in RXRα−/−/ RARα−/− cells (fivefold and sevenfold lower levels, respectively), whereas it was fully abrogated in RXRα−/−/ RARγ−/− cells. Thus, RA-induced primitive and parietal endodermal differentiation of WT F9 cells appears to be mainly mediated by the RXRα/RARγ pair, whereas it cannot be mediated by combinations of RXR(β+γ) with either RARα or RARβ (see Table IV).
Table IV.
Summary of the Involvement of the Various RARs and RXRs in the Transduction of the Retinoid Signal in F9 Cells, as Deduced from the Present and Previous Studies of RAR and RXR Mutant Cells and the Use of Receptor-specific Retinoids
| Retinoid-induced events and RXR/RAR pairs capable of transducing the signal | Role of RXRs | Role of RARs | ||
|---|---|---|---|---|
| Primitive endodermal differentiation | RARγ | |||
| RARγ ligand active on its own at saturating | ||||
| RXR ligand inactive on its own (Roy et al., 1995) | concentration (Taneja et al., 1996) | |||
| RARγ−/− cells differentiate very poorly (Boylan et al., 1993) | ||||
| RXRα/RARγ in all instances | RXRα−/− cells differentiate very poorly (Clifford et al., 1996) | synergizes with RXRα at suboptimal ligand concentration (Clifford et al., 1996) | ||
| weakly hindered by RARα in WT cells (Taneja et al., 1996) | ||||
| RXR ligand is required at suboptimal concentration | RARα | |||
| of RARγ ligand (Roy et al., 1995; Taneja et al., 1996) | RARα ligand inactive on its own (Taneja et al., 1996) | |||
| RARα−/− cells differentiate normally (Boylan et al., 1995) | ||||
| can poorly replace RARγ provided RXRα is activated | ||||
| RXRα can be poorly replaced by RXR(β+γ) | RARβ | |||
| provided that RARγ is present | inactive or very poorly active (Taneja et al., 1996) | |||
| Visceral endodermal differentiation | RARγ | |||
| RXR ligand inactive on its own RXR ligand is required at suboptimal concentration of a RAR ligand RXRα can be efficiently replaced by RXR(β+γ) provided that RARγ is present RXRα prevents efficient synergism between RXR(β + γ) and RARγ | indispensable in WT cells | |||
| RARγ ligand active on its own at saturating concentration | ||||
| RXRα/RARγ in WT cells | synergizes with RXRs at suboptimal ligand concentration | |||
| hindered by RARα in WT cells | ||||
| RARα | ||||
| RXR(β+γ)/RARγ in absence | RARα ligand inactive on its own or very poorly active | |||
| of RXRα (efficiently) | RARα−/− cells differentiate normally | |||
| synergizes with RXRα in the absence of RARγ | ||||
| RXRα/RARα or RARβ in | hindered by RARγ in WT cells | |||
| absence of RARγ (efficiently) | RARβ | |||
| RARβ ligand inactive on its own or very poorly active | ||||
| synergizes with RXRα in the absence of RARγ | ||||
| blocked by RARγ in WT cells | ||||
| Inhibition of proliferation | RARγ | |||
| not indispensable in WT cells | ||||
| RXRα/RARγ or RARα in WT cells | RARγ ligand active on its own at saturating concentration | |||
| RXR ligand inactive on its own | synergizes with RXRs at suboptimal ligand concentration | |||
| partially hindered by RARα in WT cells | ||||
| RXRα/RARβ in absence of RARγ | RXR ligand is required at limiting concentration of RAR ligand | RARα | ||
| RARα ligand inactive on its own | ||||
| synergizes with RXRα in WT cells | ||||
| RXR(β+γ)/RARγ in absence of RXRα | RXRα can be partially replaced by RXR(β+γ) provided that RARγ is present | partially hindered by RARγ in WT cellsRARβ | ||
| RARβ ligand inactive on its own | ||||
| synergizes with RXRα in the absence of RARγ | ||||
| blocked by RARγ in WT cells | ||||
| Induction of proliferation in | RARα | |||
| RXRα−/−/RARγ−/−cells | RARα ligand active on its own | |||
| RXR ligand inactive on its own | RARβ | |||
| RXR(β+γ)/RARα | RARβ ligand active on its own | |||
| RXR(β+γ)/RARβ | synergizes with RXR(β+γ) | |||
| Apoptosis | RARγ | |||
| indispensable in WT cells | ||||
| RXRα/RARγ in WT cells | RXR ligand inactive on its own | RARα ligand active on its own at saturating ligand concentration | ||
| RXR(β+γ)/RARγ in absence of RXRα, but very inefficiently | RXRα ligand is required at suboptimal ligand concentration of RAR ligand | synergizes with RXRα at suboptimal ligand concentration | ||
| hindered by RARα in WT cells | ||||
| RARα | ||||
| RXRα/RARα or RARβ in absence of RARγ, but inefficiently | RXRα can be poorly replaced by RXR(β+γ) provided that RARγ is present | RARα ligand inactive on its own weakly synergizes with RXRα in the absence of RARγ blocked by RARγ in WT cells | ||
| RARβ | ||||
| RARβ ligand inactive on its own | ||||
| weakly synergizes with RXRα in the absence of RARγ | ||||
| blocked by RARγ in WT cells |
Function of RXRs and RARs in the Retinoid-induced Differentiation of F9 Cells into VE-like Cells
When F9 cells are grown in suspension as aggregates, low levels of tRA induce a VE phenotype in the outermost layer of cells, which display an irregular surface (Strickland, 1981; Hogan et al., 1983; see also Fig. 4 A, WT, a and b, brackets). We have previously shown that, in contrast to primitive and parietal endodermal differentiation, VE differentiation can be induced by tRA in RXRα−/− F9 cells (Clifford et al., 1996). Similarly, after 10 d of treatment, >80% of the outer layer of RARα−/− and RXRα−/−/ RARα−/− cell aggregates differentiated into VE-like cells, which were indistinguishable from those of WT and RXRα−/− cells (Fig. 4 A, compare f with panels b and d; and Table I). In contrast, <10% of the outer layer of RARγ−/− cells exhibited VE conversion after 10 d of treatment, whereas full VE differentiation was eventually achieved after 14 d of treatment (Fig. 4 A, compare h with g and b; and Table I). In RXRα−/−/RARγ−/− cells, the surface of the aggregates was as smooth after 10 d of treatment as in untreated controls (Fig. 4 A, compare j with a and i), and <10% of the aggregates displayed only a spotty VE conversion after 12 or 18 d of treatment (Table I). To exclude the possibility that this very poor differentiation of the RXRα−/−/RARγ−/− cells could be due to some clonal variation, rather than the presence of the RXRα-null mutation in the RARγ-null background, we expressed the RXRα cDNA in RXRα−/−/RARγ−/− cells. As expected, cells expressing the RXRα cDNA exhibited a phenotype identical to that of RARγ−/− cells, i.e., the RA- induced VE differentiation was restored at late time (14 d) of RA treatment (data not shown).
Figure 4.


RXRα−/−/RARγ−/− F9 cells are defective for tRA-induced differentiation into VE-like cells. (A) WT (a and b), RXRα−/− (c and d), RXRα−/−/RARα−/− (e and f), RARγ−/− (g and h), and RXRα−/−/RARγ−/− (i and j) cells were grown in suspension in the absence (a, c, e, g, and i) or presence (b, d, f, h, and j) of 50 nM tRA for 10 d. The aggregates were photographed under a phase–contrast microscope at ×125 magnification. The arrows and brackets indicate VE morphology. (B) Total RNA from WT and mutant aggregates, treated with control vehicle or 50 nM tRA for 10 d, was subjected to RT-PCR analysis for collagen IVα1, laminin B1, AFP, and 36B4. Similar results were obtained for three independent experiments. Bar, 100 μm.
Table I.
Effect of Various Retinoids on Morphological Differentiation of WT and Mutant F9 Cells into Visceral Endoderm (VE)-like Cells
| Treatment | WT and mutant F9 cells | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | RXRα−/− | RARα−/− | RXRα−/− RARα−/− | RARγ−/− | RXRα−/− RARγ−/− | |||||||
| Ethanol | (−) | (−) | (−) | (−) | (−) | (−) | ||||||
| 50 nM tRA | +++ | +++ | +++ | +++ | ±(+++*) | (−) (−‡) | ||||||
| 1 μM panRXR agonist | (−) | (−) | (−) | (−) | (−) | (−) | ||||||
| 10 nM RARα agonist | (−) | (−) | ND | ND | (−) | ND | ||||||
| 100 nM RARα agonist | (−) | (−) | (−) | (−) | ± | (−) | ||||||
| 500 nM RARβ agonist | (−) | (−) | (−) | (−) | ± | (−) | ||||||
| 1 nM RARγ agonist | (−) | (−) | (−) | (−) | (−) | ND | ||||||
| 10 nM RARγ agonist | ± | + | +++ | + | (−) | ND | ||||||
| 100 nM RARγ agonist | +++ | +++ | +++ | +++ | (−) | (−) | ||||||
| 10 nM RARα + 1 μM panRXR agonists | (−) | (−) | ND | ND | (−) | ND | ||||||
| 100 nM RARα + 1 μM panRXR agonists | ± | ± | (−) | (−) | ++ | (−) (±§) | ||||||
| 500 nM RARβ + 1 μM panRXR agonists | (−) | (−) | (−) | (−) | ++ | (−) (±§) | ||||||
| 1 nM RARγ + 1 μM panRXR agonists | (−) | (−) | + | (−) | (−) | ND | ||||||
| 10 nM RARγ + 1 μM panRXR agonists | + | +++ | +++ | +++ | (−) | (−) | ||||||
| 100 nM RARγ + 1 μM panRXR agonists | +++ | +++ | +++ | +++ | (−) | (−) | ||||||
WT and mutant F9 cell aggregates were treated for 10 d with the indicated retinoids, and their differentiation was scored according to the proportion of outer layer of cells displaying VE morphology. +++, more than 80%; ++, 50–80%; +, 10–50%; ±, not more than 10%; (−), no visible effect; ND, not determined.
After 14 d of treatment.
After 12 or 18 d of treatment, <10% of the aggregates exhibited a spotty VE morphology only. The RARα, RARβ, RARγ, and panRXR agonists were BMS753, BMS453, BMS961, and BMS649, respectively (see text).
After 12 or 18 d of treatment. Note that this visual scoring correlated well with the determination of the relative level of induction of α-fetoprotein RNA using semi-quantitative RT-PCR (Fig. 4 B).
We also analyzed the mRNA levels of collagen type IVα1, laminin B1 and AFP in WT and mutant F9 cells (Fig. 4 B). After 10 d of aggregate culture in the presence of 50 nM tRA, the three markers were similarly induced in WT, RXRα−/−, RARα−/−, and RXRα−/−/RARα−/−cells. In RARγ−/− cells, the induction of laminin B1 was similar to that of WT cells, whereas the induction of collagen IVα1 was slightly reduced (twofold lower than in WT cells). In contrast, the induction of AFP, a specific marker of VE differentiation, was hardly detectable in RARγ−/− cells after 10 d of RA treatment (Fig. 4 B). There was no induction of either collagen IVα1, laminin B1 or AFP in RXRα−/−/ RARγ−/− cells, in agreement with their lack of morphological differentiation into VE. Thus, RXRα and RARγ play an essential role in VE differentiation of WT F9 cells.
To further investigate the functions of RARs and RXRs in VE differentiation, WT and mutant F9 cells were treated for 10–18 d with tRA or synthetic retinoid agonist selective for RARα (BMS188,753 [BMS753]; Taneja et al., 1996), RARβ (BMS189,453 [BMS453]; Chen et al., 1995), RARγ (BMS188,961 [BMS961]; Taneja et al., 1996) and all three RXRs (panRXR, BMS188,649 [BMS649]; also known as SR11237; Lehmann et al., 1992; see Roy et al., 1995) (Table I). In WT cells, VE differentiation was triggered by 100 nM of the RARγ agonist as effectively as by 50 nM tRA, and it was synergistically induced by a combination of 10 nM RARγ and 1 μM panRXR agonists (Table I). In contrast, VE differentiation of RARα−/− cells was triggered by 10 nM RARγ agonist as efficiently as by 50 nM tRA, and it was even synergistically induced by 1 nM of the RARγ agonist in combination with 1 μM panRXR agonist, indicating that RARα partially hinders the RARγ function in WT cells. The RARγ agonist alone, or together with the panRXR agonist, was more efficient in RXRα−/− and RXRα−/−/RARα−/− cells than in WT cells, but weaker than in RARα−/− cells, demonstrating that RXRα prevents an efficient synergism between RXR(β+γ) and RARγ (Table I; see Table IV). As expected, no VE differentiation was observed in RARγ−/− and RXRα−/−/ RARγ−/− cells treated with the RARγ/panRXR agonist combination.
The RARα agonist, BMS753, on its own did not trigger VE differentiation of WT and RXRα−/− cells, whereas the combination of 100 nM RARα and 1 μM panRXR agonists was much less efficient than the RARγ/panRXR agonist combination. As expected, no VE differentiation was seen in RARα−/− and RXRα−/−/RARα−/− cells upon treatment with the RARα/panRXR agonist combination. In contrast, RARγ−/− cells weakly differentiated into VE-like cells upon treatment with 100 nM RARα agonist alone, and this differentiation was markedly enhanced by addition of 1 μM panRXR agonist. This synergistic stimulation was almost abrogated in RXRα−/−/RARγ−/− cells.
A combination of RARβ (BMS453) and panRXR agonists did not trigger VE differentiation in WT, RXRα−/−, RARα−/−, and RXRα−/−/RARα−/− cells. Interestingly, this combination synergistically induced VE differentiation of RARγ−/− cells, and this effect was almost absent in RXRα−/−/RARγ−/− cells, as in the case of the RARα/panRXR agonist combination (Table I). Thus, RARγ strongly prevents RARα and RARβ to synergize with RXRα, and mutation of RARγ artefactually generates functional redundancy between RARs for VE differentiation of F9 cells (see Table IV).
Function of RXRs and RARs in the Retinoid-induced Antiproliferative Response of F9 Cells and Retinoid-induced Proliferation of RXRα/RARγ Null F9 Cells
The effect of tRA on proliferation of WT, RXRα−/−, RXRα−/−/RARα−/−, and RXRα−/−/RARγ−/− F9 cells was investigated (Fig. 5 A). After 6 d of 1 μM tRA treatment, the inhibition of growth as determined by cell counting, was lower for RXRα−/− than for WT cells (58 and 79% inhibition relative to untreated control cells, respectively) (Clifford et al., 1996). The antiproliferative effect of tRA was decreased to the same extent for RXRα−/−/RARα−/− cells (54% inhibition) and RXRα−/− cells. On the other hand, tRA did not reduce, but slightly increased the number of RXRα−/−/RARγ−/− cells. The rate of DNA synthesis was also compared for WT and mutant cell lines by measuring [3H]thymidine incorporation during the anti-proliferative response to tRA (Fig. 5 B). After 4 d of 1 μM tRA treatment, [3H]thymidine incorporation was reduced by 54% in WT cells relative to vehicle-treated control cells, and only by 20% in RXRα−/− and RXRα−/−/RARα−/− cells. In contrast, there was no inhibition of [3H]thymidine incorporation in RXRα−/−/RARγ−/− cells.
Figure 5.
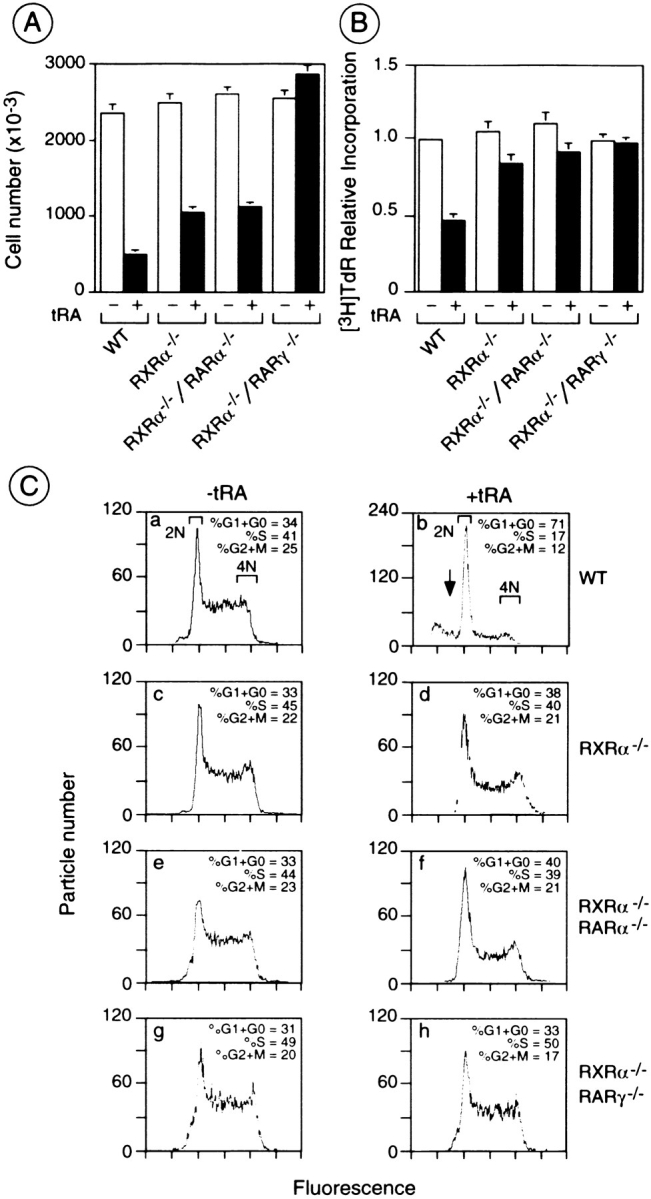
The antiproliferative response to tRA is impaired in RXRα−/− and RXRα−/−/RARα−/− cells, and is abolished in RXRα−/−/RARγ−/− F9 cells. (A) The number of cells after 6 d of culture in the presence (black bars) or absence (white bars) of 1 μM tRA are indicated for WT and mutant cells. The bars represent the mean ± SEM for triplicate cultures within the same experiment. (B) Cells were cultured for 4 d with or without 1 μM tRA, followed by 2 h of [3H]thymidine ([3H]TdR) labeling. The bars represent the mean ± SEM for three different experiments, setting the amount of [3H]TdR incorporation per 1,000 cells equal to one, for WT control cells. (C) Subconfluent cultures of WT (a and b), RXRα−/− (c and d), RXRα−/−/RARα−/− (e and f), and RXRα−/−/RARγ−/− (g and h) cells were grown for 5 d in the presence (b, d, f, and h) or absence (a, c, e, and g) of 1 μM tRA, and analyzed by FACS®. The X axis indicates the integrated fluorescence intensity and the Y axis the particle number. Approximately 20,000 particles are represented in each histogram. The percentage of cells in G1+G0, S, and G2+M phases are indicated. The arrow highlights the sub-2N size, DNA-containing particles corresponding to “apoptotic bodies.”
FACS® analysis has previously shown that tRA treatment of WT F9 cells results in an accumulation of cells in the G0 and G1 phases of the cell cycle, and that this accumulation was decreased in RXRα−/− cells (Clifford et al., 1996) (Fig. 5 C). The cell cycle profile of untreated RXRα−/−/RARα−/− and RXRα−/−/RARγ−/− cells was the same as that of WT and RXRα−/− cells. After 5 d of 1 μM tRA treatment, the proportion of cells in the G0 and G1 phases, was 71% for WT cells, whereas it was lower for RXRα−/− and RXRα−/−/RARα−/− cells (38 and 40%, respectively; Fig. 5 C, compare b, d, and f). Interestingly, the cell cycle profile of RXRα−/−/RARγ−/− cells was not significantly affected by tRA treatment, and was almost identical to that of untreated WT cells (Fig. 5 C, compare h with g and a).
To further dissect the roles of RARs and RXRs in the control of proliferation, WT and mutant F9 cells were treated for 6 d with tRA or receptor-selective retinoids, and cell numbers were counted (Table II). In WT cells, 100 nM RARγ agonist efficiently reduced the proliferation, and 10 nM of the same agonist, which had no effect on its own, synergized with 1 μM panRXR agonist. The effect of these retinoids on proliferation was reduced, while not abolished, in RXRα−/− and RXRα−/−/RARα−/− cells, indicating that RXRα can be partially replaced by RXR(β+γ) for synergizing with RARγ (see Table IV). Interestingly, the RARγ/panRXR agonist combination was more efficient in RARα−/− cells than in WT cells, revealing that RARα partially hinders the antiproliferative effect of RARγ in WT cells (Table II; see Table IV). As expected, this combination did not inhibit the proliferation of RARγ−/− and RXRα−/−/RARγ−/− cells. The combination of 100 nM RARα and 1 μM panRXR agonists, which reduced the proliferation of WT cells less efficiently than the RARγ/panRXR agonist combination, was more efficient in RARγ−/− cells than in WT cells (Table II), indicating that RARγ partially hinders the antiproliferative effect of RARα in WT cells (see Table IV). The RARα/panRXR agonist combination had no effect on the proliferation of RXRα−/− cells, showing that RARα can only synergize with RXRα to inhibit proliferation (see Table IV).
Table II.
Effect of Various Retinoids on Proliferation of WT and Mutant F9 Cells
| Treatment | WT and mutant F9 cells | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | RXRα−/− | RARα−/− | RXRα−/− RARα−/− | RARγ−/− | RXRα−/− RARγ−/− | |||||||
| Ethanol | 100 | 100 | 100 | 100 | 100 | 100 | ||||||
| 1 μM tRA | 21 | 42 | 22 | 46 | 28 | 112 | ||||||
| 1 μM panRXR agonist | 100 | 100 | 100 | 100 | 100 | 100 | ||||||
| 10 nM RARα agonist | 100 | 100 | 100 | 100 | 100 | 124 | ||||||
| 100 nM RARα agonist | 100 | 100 | 100 | 100 | 100 | 143 | ||||||
| 50 nM RARβ agonist | 100 | 100 | ND | ND | 100 | ND | ||||||
| 500 nM RARβ agonist | 100 | 100 | 100 | 100 | 100 | 120 | ||||||
| 10 nM RARγ agonist | 100 | 100 | 100 | 100 | 100 | 100 | ||||||
| 100 nM RARγ agonist | 37 | 51 | 30 | 49 | 100 | 100 | ||||||
| 10 nM RARα + 1 μM panRXR agonists | 100 | 100 | 100 | 100 | 100 | 130 | ||||||
| 100 nM RARα + 1 μM panRXR agonists | 56 | 100 | 100 | 100 | 40 | 146 | ||||||
| 50 nM RARβ + 1 μM panRXR agonists | 100 | 100 | ND | ND | 53 | ND | ||||||
| 500 nM RARβ + 1 μM panRXR agonists | 100 | 100 | 100 | 100 | 47 | 154 | ||||||
| 10 nM RARγ + 1 μM panRXR agonists | 60 | 81 | 42 | 77 | 100 | 100 | ||||||
| 100 nM RARγ + 1 μM panRXR agonists | 31 | 44 | ND | ND | 100 | ND | ||||||
WT and mutant F9 cells were treated for 6 d in monolayer culture with the indicated retinoids. In each case, the number of cells was expressed in percent relative to the number of cells grown in 0.1% ethanol, which was taken as 100%. ND, not determined. These values correspond to the average of at least three experiments (± 10%). RARα, RARβ, RARγ, and panRXR specific agonists were as in Table I.
Surprisingly, a treatment with 10 and 100 nM RARα agonist increased the number of RXRα−/−/RARγ−/− cells, indicating that RARα can mediate a proliferative effect in the absence of both RXRα and RARγ. As expected, the RARα/panRXR agonist combination did not affect proliferation of RARα−/− and RXRα−/−/RARα−/− cells. Neither the RARβ agonist alone nor in combination with the panRXR agonist affected the proliferation of WT, RXRα−/−, RARα−/−, and RXRα−/−/RARα−/− cells, whereas this combination synergistically reduced the proliferation of RARγ−/− cells. Note that 500 nM RARβ agonist alone increased the cell number of RXRα−/−/RARγ−/− cells, and that this effect was enhanced by addition of 1 μM panRXR agonist. Thus, the presence of RARγ not only hinders the antiproliferative effect of the RXRα/RARβ pair, but also the proliferation-promoting effects of the combinations of RXR(β+γ) with RARβ, showing again that knockouts generate artifactual effects not observed under WT conditions, as already seen above in the case of F9 cell differentiation (see Table IV).
Function of RXRs and RARs for the Retinoid-induced Apoptotic Response of F9 Cells
Since retinoids can induce apoptosis, which also contributes to the decrease in cell number in retinoid-treated F9 cells (Atencia et al., 1994), we determined the extent of the tRA-induced apoptotic response of WT and mutant F9 cells by FACS® analysis. Sub-2N–size, DNA-containing particles corresponding to “apoptotic bodies” appeared in WT cells after 5 d of tRA treatment, whereas they were not detected in tRA-treated RXRα−/− (see also Clifford et al., 1996), RXRα−/−/RARα−/−, and RXRα−/−/RARγ−/− cells (Fig. 5 C, compare d, f, and h with b [arrow]). Apoptosis was confirmed by staining with the DNA-binding fluorochrome Hoechst 33258. Apoptotic particles and condensed chromatin were similarly observed in tRA-treated WT and RARα−/− cells (Fig. 6, a, b, e, and f; Table III). In contrast, tRA-induced apoptosis was reduced in RARγ−/− cells, rarely seen in RXRα−/− (Clifford et al., 1996) and RXRα−/−/RARα−/− cells, and abolished in RXRα−/−/ RARγ−/− cells (Fig. 6, c, d, and g–l; Table III). Note that a background level of apoptosis occurred at high cell density even in the absence of tRA, as previously mentioned (Clifford et al., 1996).
Figure 6.

The apoptotic response to tRA is severely impaired in RXRα−/− and RXRα−/−/RARα−/− cells, and is abrogated in RXRα−/−/RARγ−/− F9 cells. WT (a and b), RXRα−/− (c and d), RARα−/− (e and f), RXRα−/−/RARα−/− (g and h), RARγ−/− (i and j), and RXRα−/−/RARγ−/− (k and l) cells were treated for 6 d with control vehicle (a, c, e, g, i, and k) or 1 μM tRA (b, d, f, h, j, and l), followed by fixation and staining with Hoechst dye. Cells were photographed under a fluorescence microscope at ×120 magnification. Arrows indicate condensed chromatin in the nuclei of apoptosing cells or in apoptotic bodies. Arrowheads indicate mitotic cells. Bar, 150 μm.
Table III.
Effect of Various Retinoids on Apoptosis of WT and Mutant F9 Cells
| WT and mutant F9 cells | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Treatment | WT | RXRα−/− | RARα−/− | RXRα−/− RARα−/− | RARγ−/− | RXRα−/− RARγ−/− | ||||||
| Ethanol | (−) | (−) | (−) | (−) | (−) | (−) | ||||||
| 1 μM tRA | ++ | ± | ++ | ± | + | (−) | ||||||
| 1 μM panRXR agonist | (−) | (−) | (−) | (−) | (−) | (−) | ||||||
| 100 nM RARα agonist | (−) | (−) | (−) | (−) | (−) | (−) | ||||||
| 500 nM RARβ agonist | (−) | (−) | (−) | (−) | (−) | (−) | ||||||
| 10 nM RARγ agonist | (−) | ND | (−) | ND | ND | ND | ||||||
| 100 nM RARγ agonist | + | ±* | + | ±* | (−) | (−) | ||||||
| 100 nM panRAR agonist | + | ±* | + | ±* | ± | (−) | ||||||
| 100 nM RARα + 1 μM panRXR agonists | (−) | (−) | (−) | (−) | ± | (−) | ||||||
| 500 nM RARβ + 1 μM panRXR agonists | (−) | (−) | (−) | (−) | ± | (−) | ||||||
| 10 nM RARγ + 1 μM panRXR agonists | ± | (−) | + | (−) | ND | ND | ||||||
| 100 nM RARγ + 1 μM panRXR agonists | ++ | ± | ++ | ± | (−) | (−) | ||||||
| 100 nM panRAR + 1 μM panRXR agonists | ++ | ± | ++ | ± | + | (−) | ||||||
WT and mutant F9 cells were treated for 6 d in monolayer culture with the indicated retinoids, and their apoptosis was scored according to the proportion of apoptotic nuclei and subcellular fragments upon staining of fixed cells with Hoechst 33258, as shown in Fig. 6. ++, >10% ; +, 1–10%; ±, <1%; (−), no visible effect; asterisks, less than that in combination with 1 μM panRXR agonist; ND, not determined. RARα, RARβ, RARγ, and panRXR agonists were as in Table I. The panRAR agonist was AM80.
To further investigate the role played by the different RAR and RXR isotypes in apoptosis, WT and mutant F9 cells were treated for 6 d with receptor-selective retinoids, and stained with Hoechst dye (Table III). In WT cells, 100 nM RARγ agonist triggered apoptosis, and the addition of 1 μM panRXR agonist resulted in a synergistic effect. The effect of these retinoids was markedly reduced in RXRα−/− and RXRα−/−/RARα−/− cells, indicating that RXRα can only poorly be replaced by RXR(β+γ) for this response (Table IV). In contrast, the RARγ/panRXR agonist combination was more efficient in RARα−/− cells than in WT cells, indicating that RARα partially prevents the apoptotic response mediated by RARγ in WT cells (Table IV). As expected, this combination had no effect on the apoptosis of RARγ−/− and RXRα−/−/RARγ−/− cells. Neither the RARα/panRXR nor the RARβ/panRXR agonist combination induced the apoptosis of WT, RXRα−/−, RARα−/−, RXRα−/−/RARα−/−, and RXRα−/−/RARγ−/− cells, whereas they weakly triggered apoptosis of RARγ−/− cells (Table III). 100 nM Am80, which acts as panRAR agonist at this concentration (Hashimoto et al., 1990), was as efficient as 100 nM RARγ agonist for WT, RXRα−/−, RARα−/−, and RXRα−/−/RARα−/− cells. The panRAR/ panRXR combination was more effective than either the RARα/panRXR or the RARβ/panRXR combination in RARγ−/− cells, whereas these retinoids had no effects on the apoptosis of RXRα−/−/RARγ−/− cells (Table III). Thus, RARγ fully prevents the weak apoptotic response that can be mediated by the RXRα/RARα and RXRα/ RARβ pairs, and mutation of RARγ artifactually generates some functional redundancy (Table IV).
Discussion
In vitro studies using either cell-free systems or cultured cells cotransfected with vectors overexpressing the various retinoid receptors and cognate recombinant reporter genes, have suggested that RXR/RAR heterodimers could be the functional units transducing the retinoid signal in vivo. These studies have also indicated that the various RXR/RAR heterodimers, resulting from the combination of either one of three RXRs (α, β, or γ) with either one of the three RARs (α, β, or γ), could be differentially involved in the numerous physiological events that are controlled by retinoids in vivo (Chambon, 1994, 1996). The results of RAR and RXR gene knockout studies in the mouse have supported these suggestions, but their interpretation remains equivocal, in particular because cell- autonomous and non–cell-autonomous effects cannot be distinguished in the intact animal (Kastner et al., 1995, 1997).
The aim of the present study was to determine the actual role of the various RXR/RAR combinations as retinoid transducers in a well-established, cell-autonomous system, namely that provided by the retinoid-responsive F9 EC cells. To this end, differentiation into primitive, parietal, and visceral endoderms, as well as antiproliferative and apoptotic responses have been studied in RXR and RAR single or compound mutant F9 EC cells cultured in the presence of either tRA or panRXR- and/or RAR isotype-selective synthetic retinoids. Our present results are summarized in Table IV with relevant data from previous reports (Roy et al., 1995; Clifford et al., 1996; Taneja et al., 1996), and lead to several important conclusions, which are in keeping with those recently drawn from a study of the expression of RA-responsive genes in the same mutant F9 EC cells (Chiba et al., 1997).
Taken together, our genetic data and those obtained with selective retinoids in WT or mutated cells establish that RXR/RAR pairs are always involved in the transduction of the retinoid signal, irrespective of the nature of the retinoid-induced event examined (differentiation, antiproliferative, or apoptotic effects). This is obvious from both the comparison of single and double mutants, and the combined use of the panRXR ligand with suboptimal concentrations of either one of the RAR isotype–specific, synthetic retinoids. Thus, since the panRXR-specific agonist is never active on its own, all cellular events induced by retinoids in F9 EC cells appear to be mediated by RXR/ RAR heterodimers. Note that the “subordination” of RXRs to RARs (i.e., that a RXR cannot be transcriptionally activated unless its heterodimer RAR partner is liganded), which has been repeatedly observed in different cell systems (Roy et al., 1995; Chen et al., 1996; Horn et al., 1996; Taneja et al., 1996), as well as in some in vitro studies (Durand et al., 1994; Apfel et al., 1995; Forman et al., 1995), may be important to prevent the promiscuous activation of the retinoid and other signaling pathways (e.g., those of thyroid hormones and vitamin D3) by RXR ligands (Mangelsdorf and Evans, 1995; Chambon, 1996). The dispensability of the RXR ligand that can be observed in some instances when a saturating amount of a RAR- selective ligand (notably in the case of RARγ) is used, has been previously noted (Roy et al., 1995). This dispensability most probably reflects the fact that the RAR activation functions of RXR/RAR heterodimers alone are sufficient to trigger the expression of the genes involved in the cellular event considered, whereas the synergistic effect of the activation functions of the RXR heterodimeric partner becomes indispensable at lower concentrations of the RAR ligand (Clifford, J., unpublished results), which are probably closer to physiological retinoic acid concentrations.
The second important conclusion of the present study is that the various RXR/RAR heterodimers that can be formed in F9 EC cells exhibit some functional specificity. Indeed, each of the cellular events that are RA-induced in F9 cells appears to preferentially involve a specific RXR/ RAR isotype combination (or set of combinations) (Table IV). It appears that in all cases the RA signal is transduced by RXRα/RARγ heterodimers in WT F9 cells. However, both the RXRα/RARγ and RXRα/RARα heterodimers can mediate the RA-induced inhibition of WT F9 cell proliferation. Thus, in WT F9 cells, depending on the cellular event considered, different RXR/RAR isotype heterodimers possess both specific functions and redundant functions shared with other heterodimers. Interestingly, additional redundant functions, not seen in WT cells, are revealed when either RXRα or RARγ are not expressed. The presence of RARγ often hinders or blocks the activity of RARα and RARβ, where the presence of RXRα can hinder the activity of RXR(β+γ) (Table IV). In several instances, the retinoid-induced cellular events mediated by RXRα/RARγ in WT cells can be mediated by RXR(β+γ)/RARγ heterodimers in the absence of RXRα, and by RXRα/RAR(α and/or β) heterodimers in the absence of RARγ (Table IV). Again, these redundancies vary according to the cellular event examined, further supporting the conclusion that the different RXR/RAR isotype heterodimers possess some functional specificity.
The third conclusion is that gene knockouts generate artifactual conditions unmasking potential functional redundancies, which actually do not occur in the WT situation (Table IV). For instance, in the case of visceral endoderm differentiation, RXR(β+γ)/RARγ heterodimers can efficiently substitute for RXRα/RARγ heterodimers in the absence of RXRα; in addition, either RXRα/RARα or RXRα/RARβ heterodimers can efficiently substitute for RXRα/RARγ heterodimers in the absence of RARγ, even though RXRα/RARγ heterodimers essentially mediate this differentiation in WT F9 EC cells. How the presence of RXRα/RARγ heterodimers prevents potentially functionally redundant heterodimers from transducing the RA signal is unknown, but it could be related to their differential affinities for the RAREs of the target genes involved in the cellular processes induced by RA in F9 cells. In any event, it is clear that the functional redundancies that are revealed by gene knockout cannot be taken as evidence for a lack of functional specificity of the knockout gene product under WT physiological conditions. It is not unlikely that many of the functional redundancies that have been so far revealed by mouse gene knockouts are similarly artifactually generated.
Interestingly, our present study also reveals that different RXR/RAR heterodimers can have opposite effects on cell proliferation of F9 cells. RXRα/RARγ or RARα heterodimers are involved in the transduction of the antiproliferative effect of RA, but in the absence of RXRα and RARγ, both RXR(β+γ)/RARα and RARβ heterodimers can mediate a proliferative effect of RA (Tables II and IV). Note that induction of proliferation of certain cell types by retinoids has been previously reported (Amos and Lotan, 1990; Koshimizu et al., 1995). Our present observations on retinoid-induced cell antiproliferative and proliferative effects, strengthen the conclusion that different RXR/RAR heterodimers can exert specific functions. These observations also suggest that the actual set of retinoid receptors present in a given cell may have a profound influence on the effects generated by a retinoid treatment.
It is interesting to note that morphological differentiation of WT F9 EC cells can be efficiently triggered by a combination of panRXR/RARγ-specific (BMS961) agonists, but not by a combination of panRXR/RARα-specific (BMS753) agonists, nor by a combination of a panRXR/RARβ-specific (BMS453) agonists. In contrast, P19 EC cell differentiation can be triggered by either a panRXR/RARγ or a panRXR/RARα agonist combination, but not by a panRXR/RARβ agonist combination (Taneja et al., 1996), whereas the differentiation of the NB4 acute promyelocytic leukemia cells, and HL60 myeloblastic leukemia cells can be triggered by a combination of a panRXR/RARα or a panRXR/RARβ agonists, but not by a panRXR/RARγ agonist combination (Chen et al., 1996). Similarly, the apoptosis of NB4 cells can be induced by a panRXR/RARβ agonist combination (Chen et al., 1996), which on the other hand is inefficient in the case of F9 cells (Table III). Thus, different RAR isotype-specific agonists acting synergistically with a panRXR agonist are not only more restricted than tRA in their effects on various cellular events in a given cell type (e.g., differentiation and apoptosis), but also affect differentially these events in a cell-specific manner. These cell type–specific effects of synthetic retinoids may extend their potential for therapeutical use.
Finally, to our best knowledge, this study is the first report of multiple gene targeting (two alleles of two genes) in a mammalian cell–autonomous system. Similar approaches will allow the inactivation of any set of genes in a given cell, which will undoubtedly and particularly useful to elucidate the molecular mechanisms underlying complex biological events.
Acknowledgments
We are grateful to J-M. Bornert and P. Unger for technical assistance; to P.R. Reczek (Bristol-Meyers-Squibb, Pharmaceutical Research Institute, Buffalo, NY) for the gift of synthetic retinoids; K. Shudo (University of Tokyo, Tokyo, Japan) for Am80; to J.L. Imler (Transgène, Société Anonyme, Strasbourg, France) for pLXPB; to J. Brocard, P. Kastner, D. Lohnes, and R. Taneja for various oligonucleotides and plasmids; to C. Ebel, and C. Waltzinger for help with FACS® analysis; and to M.P. Gaub, and C. Rochette-Egly for various antibodies and helpful discussions. We thank H. Gronemeyer, P. Kastner, and all members of the retinoid group for helpful discussions. We also thank the cell culture facility for providing cells, I. Colas, F. Ruffenach, and E. Troech for oligonucleotide synthesis, the secretarial staff for typing, and R. Bucher, S. Metz, C. Werlé, B. Boulay, and J.M. Lafontaine for preparing the figures.
This work was supported by funds from the Centre National de la Recherche Scientifique, the Institut National de la Santé et de la Recherche Médicale, the Collège de France, the Centre Hospitalier Universitaire Régional, the Association pour la Recherche sur le Cancer, the Fondation pour la Recherche Médicale, the Human Frontier Science Program, and Bristol-Myers-Squibb. J. Clifford was supported by a fellowship from the Association pour la Recherche sur le Cancer and the US National Institutes of Health (grant No. IF32GM15857), and H. Chiba by a fellowship from the Centre National de la Recherche Scientifique.
Abbreviations used in this paper
- AFP
α-fetoprotein
- E2
estradiol
- EC
embryonal carcinoma
- EMSA
electrophoretic mobility shift assays
- HR
homologous recombination
- PGK
phosphoglycerate kinase
- RAR
retinoic acid receptor
- RXR
retinoid X receptor
- RARE
retinoic acid response element
- RT
reverse transcription
- tRA
all-trans retinoic acid
- VE
visceral endoderm
- WT
wild-type
Footnotes
Address all correspondence to P. Chambon, Institut de Génétique et de Biologie Moléculaire et Cellulaire, Centre National de la Recherche Scientifique/Institut National de la Santé et de la Recherche Médicale/Université Louis Pasteur, Collège de France, BP 163, 67404 Illkirch-Cedex, France. Tel.: (33) 388-65-32-13. Fax: (33) 388-65-32-03. E-mail: IGBMC@ IGBMC.U-STRASBG.FR
H. Chiba's present address is Department of Pathology, Sapporo Medical University, School of Medicine, South-1, West-17, Chuo-ku, Sapporo 060, Japan.
J. Clifford's present address is M.D. Anderson Cancer Center, Department of Clinical Cancer Center, Department of Clinical Cancer Prevention, Box 236, 1515 Holcombe Boulevard, Houston, TX 77030.
References
- Adra CN, Boer PH, McBurney MW. Cloning and expression of the mouse pgk-1gene and the nucleotide sequence of its promoter. Gene (Amst) 1987;60:65–74. doi: 10.1016/0378-1119(87)90214-9. [DOI] [PubMed] [Google Scholar]
- Amos B, Lotan R. Retinoid-sensitive cells and cell lines. Methods Enzymol. 1990;190:217–225. doi: 10.1016/0076-6879(90)90026-w. [DOI] [PubMed] [Google Scholar]
- Apfel C, Kamber M, Klaus M, Mohr P, Keider S, LeMotte PK. Enhancement of HL-60 differentiation by a new class of retinoids with selective activity on retinoid X receptor. J Biol Chem. 1995;270:30765–30772. doi: 10.1074/jbc.270.51.30765. [DOI] [PubMed] [Google Scholar]
- Atencia R, Garcia-Sanz M, Unda F, Arechaga J. Apoptosis during retinoic acid-induced differentiation of F9 embryonal carcinoma cells. Exp Cell Res. 1994;214:663–667. doi: 10.1006/excr.1994.1304. [DOI] [PubMed] [Google Scholar]
- Blomhoff, R. 1994. Overview of vitamin A metabolism and function. In Vitamin A Health and Disease. R. Blomhoff, editor. Marcel Dekker, New York/ Basel/Hong Kong. 1–35.
- Bouillet P, Oulad-Abdelghani M, Vicaire S, Garnier J-M, Schuhbaur B, Dollé P, Chambon P. Efficient cloning of cDNAs of retinoic acid-responsive genes in P19 embryonal carcinoma cells and characterization of a novel mouse gene, Stra1 (Mouse LERK-2/Eplg2) Dev Biol. 1995;170:420–433. doi: 10.1006/dbio.1995.1226. [DOI] [PubMed] [Google Scholar]
- Boylan J, Lohnes D, Taneja R, Chambon P, Gudas LJ. Loss of retinoic acid receptor γ function in F9 cells by gene disruption results in aberrant Hoxa-1expression and differentiation upon retinoic acid treatment. Proc Natl Acad Sci USA. 1993;90:9601–9605. doi: 10.1073/pnas.90.20.9601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boylan J, Lufkin T, Achkar CC, Taneja R, Chambon P, Gudas LJ. Targeted disruption of retinoic acid receptor α (RARα) and RARγ results in receptor-specific alterations in retinoic acid-mediated differentiation and retinoic acid metabolism. Mol Cell Biol. 1995;15:843–851. doi: 10.1128/mcb.15.2.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambon P. The retinoid signaling pathway: molecular and genetic analyses. Semin Cell Biol. 1994;5:115–125. doi: 10.1006/scel.1994.1015. [DOI] [PubMed] [Google Scholar]
- Chambon P. A decade of molecular biology of retinoic acid receptors. FASEB (Fed Am Soc Exp Biol) J. 1996;10:940–954. [PubMed] [Google Scholar]
- Chen J-Y, Penco S, Ostrowski J, Balaguer P, Pons M, Starrett JE, Reczek PR, Chambon P, Gronemeyer H. RAR-specific agonist/antagonists which dissociate transactivation and AP1 transrepression inhibit anchorage-independent cell proliferation. EMBO (Eur Mol Biol Organ) J. 1995;14:1187–1197. doi: 10.1002/j.1460-2075.1995.tb07102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J-Y, Clifford J, Zusi C, Starrett J, Tortolani D, Ostrowski J, Reczek PR, Chambon P, Gronemeyer H. Two distinct actions of retinoid-receptor ligands. Nature (Lond) 1996;382:819–822. doi: 10.1038/382819a0. [DOI] [PubMed] [Google Scholar]
- Chiba H, Clifford J, Metzger D, Chambon P. Distinct retinoid X receptor heterodimers are differentially involved in the control of expression of retinoid target genes in F9 embryonal carcinoma cells. Mol Cell Biol. 1997;17:3013–3020. doi: 10.1128/mcb.17.6.3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifford J, Chiba H, Sobieszczuk D, Metzger D, Chambon P. RXRα-null F9 embryonal carcinoma cells are resistant to the differentiation, anti-proliferative and apoptotic effects of retinoids. EMBO (Eur Mol Biol Organ) J. 1996;15:4142–4155. [PMC free article] [PubMed] [Google Scholar]
- De Luca LM. Retinoids and their receptors in differentiation, embryogenesis, and neoplasia. FASEB (Fed Am Soc Exp Biol) J. 1991;5:2924–2933. [PubMed] [Google Scholar]
- Durand B, Saunders M, Gaudon C, Roy B, Losson R, Chambon P. Activation function 2 (AF-2) of retinoic acid receptor and 9-cis retinoic acid receptor: presence of a conserved autonomous constitutive activating domain and influence of the nature of the response element on AF-2 activity. EMBO (Eur Mol Biol Organ) J. 1994;13:5370–5382. doi: 10.1002/j.1460-2075.1994.tb06872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman BM, Umesono K, Chen J, Evans RM. Unique response pathways are established by allosteric interactions among nuclear hormone receptors. Cell. 1995;81:541–550. doi: 10.1016/0092-8674(95)90075-6. [DOI] [PubMed] [Google Scholar]
- Gaub M-P, Rochette-Egly C, Lutz Y, Ali S, Matthes H, Scheuer I, Chambon P. Immunodetection of multiple species of retinoic acid receptor α: evidence for phosphorylation. Exp Cell Res. 1992;201:335–346. doi: 10.1016/0014-4827(92)90282-d. [DOI] [PubMed] [Google Scholar]
- Giguère V. Retinoic acid receptors and cellular retinoid binding proteins: complex interplay in retinoid signaling. Endocr Rev. 1994;15:61–79. doi: 10.1210/edrv-15-1-61. [DOI] [PubMed] [Google Scholar]
- Glass CK. Differential recognition of target genes by nuclear receptor monomers, dimers, and heterodimers. Endocr Rev. 1994;15:391–407. doi: 10.1210/edrv-15-3-391. [DOI] [PubMed] [Google Scholar]
- Green S, Issemann I, Scheer E. A versatile in vivo and in vitroeukaryotic expression vector for protein engineering. Nucl Acids Res. 1988;16:369. doi: 10.1093/nar/16.1.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronemeyer H, Laudet V. Transcription factors 3: nuclear receptors. Protein Profile. 1995;2:1173–1308. [PubMed] [Google Scholar]
- Gudas, L.J., M.B. Sporn, and A.B. Roberts. 1994. Cellular biology and biochemistry of the retinoids. In The Retinoids: Biology, Chemistry and Medicine. M.B. Sporn, A.B. Roberts, and D.S. Goodman, editors. Raven Press, New York. 443–520.
- Hashimoto Y, Kagechika H, Shudo K. Expression of retinoic acid receptor genes and the ligand-binding selectivity of retinoic acid receptors (RARs) Biochem Biophys Res Commun. 1990;166:1300–1307. doi: 10.1016/0006-291x(90)91007-f. [DOI] [PubMed] [Google Scholar]
- Hogan BLM, Barlow D, Tilly R. F9 teratocarcinoma cells as a model for the differentiation of parietal and visceral endoderm in the mouse embryo. Cancer Surv. 1983;2:115–140. [Google Scholar]
- Horn V, Minucci S, Ogryzko VV, Adamson ED, Howard BH, Levin AA, Ozato K. RAR and RXR selective ligands cooperatively induce apoptosis and neuronal differentiation in P19 embryonal carcinoma cells. FASEB (Fed Am Soc Exp Biol) J. 1996;10:1071–1077. doi: 10.1096/fasebj.10.9.8801169. [DOI] [PubMed] [Google Scholar]
- Imler JL, Chartier C, Dreyer D, Dieterle A, Sainte M, Maie, Faure T, Pavirani A, Methali M. Novel complementation cell lines derived from human lung carcinoma A549 cells support the growth of E1-deleted adenovirus vectors. Gene Ther. 1996;3:75–84. [PubMed] [Google Scholar]
- Kastner P, Krust A, Mendelsohn C, Garnier JM, Zelent A, Leroy P, Staub A, Chambon P. Murine isoforms of retinoic acid receptor γ with specific patterns of expression. Proc Natl Acad Sci USA. 1990;87:2700–2704. doi: 10.1073/pnas.87.7.2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastner, P., M. Leid, and P. Chambon. 1994. The role of nuclear retinoic acid receptors in the regulation of gene expression. In Vitamin A in Health and Disease. R. Blomhoff, editor. Marcel Dekker, New York. 189–238.
- Kastner P, Mark M, Chambon P. Nonsteroid nuclear receptors: what are genetic studies telling us about their role in real life? . Cell. 1995;83:859–869. doi: 10.1016/0092-8674(95)90202-3. [DOI] [PubMed] [Google Scholar]
- Kastner P, Mark M, Ghyselinck N, Krezel W, Dupé V, Grondona JM, Chambon P. Genetic evidence that the retinoid signal is transduced by heterodimeric RXR/RAR functional unit during mouse development. Development (Camb) 1997;124:313–326. doi: 10.1242/dev.124.2.313. [DOI] [PubMed] [Google Scholar]
- Keaveney, M., and H.G. Stunnenberg. 1995. Retinoic acid receptors. In Inducible Gene Expression. vol. 2. P.A. Bauerle, editor. Birkhäeuser, Boston. 187–242.
- Koshimizu U, Watanabe M, Nakatsuji N. Retinoic acid is potent growth activator of mouse primordial germ cells in vitro. Dev Biol. 1995;168:683–685. doi: 10.1006/dbio.1995.1113. [DOI] [PubMed] [Google Scholar]
- Lehmann JM, Jong L, Fanjul A, Cameron JF, Lu XP, Haefner P, Dawson MI, Pfahl M. Retinoids selective for retinoid X receptor response pathways. Science (Wash DC) 1992;258:1944–1946. doi: 10.1126/science.1335166. [DOI] [PubMed] [Google Scholar]
- Leid M, Kastner P, Chambon P. Multiplicity generates diversity in the retinoic acid signaling pathways. Trends Biochem Sci. 1992a;17:427–433. doi: 10.1016/0968-0004(92)90014-z. [DOI] [PubMed] [Google Scholar]
- Leid M, Kastner P, Lyons R, Nakshatri H, Saunders M, Zacharewski T, Chen J-Y, Staub A, Garnier J-M, Mader S, et al. Purification, cloning, and RXR identity of the HeLa cell factor with which RAR or TR heterodimerizes to bind target sequences efficiently. Cell. 1992b;68:377–395. doi: 10.1016/0092-8674(92)90478-u. [DOI] [PubMed] [Google Scholar]
- Leroy P, Krust A, Zelent A, Mendelsohn C, Garnier J-M, Kastner P, Dierich A, Chambon P. Multiple isoforms of the mouse retinoic acid receptor α are generated by alternative splicing and differential induction by retinoic acid. EMBO (Eur Mol Biol Organ) J. 1991;10:59–69. doi: 10.1002/j.1460-2075.1991.tb07921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohnes D, Kastner P, Dierich A, Mark M, LeMeur M, Chambon P. Function of retinoic acid receptor γ in the mouse. Cell. 1993;73:643–658. doi: 10.1016/0092-8674(93)90246-m. [DOI] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83:841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- Mangelsdorf, D.J., K. Umesono, and R.M. Evans. 1994. The retinoid receptors. In The Retinoids: Biology, Chemistry and Medicine. M.B. Sporn, A.B. Roberts, and D.S. Goodman, editors. Raven Press, New York. 319–349.
- Martin CA, Ziegler LM, Napoli JL. Retinoic acid, dibutyryl-cAMP, and differentiation affect the expression of retinoic acid receptors in F9 cells. Proc Natl Acad Sci USA. 1990;87:4804–4808. doi: 10.1073/pnas.87.12.4804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger D, Clifford J, Chiba H, Chambon P. Conditional site-specific recombination in mammalian cells using a ligand-dependent chimeric Cre-recombinase. Proc Natl Acad Sci USA. 1995;92:6991–6995. doi: 10.1073/pnas.92.15.6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochette-Egly C, Lutz Y, Saunders M, Scheuer I, Gaub M-P, Chambon P. Retinoic acid receptor γ: specific immunodetection and phosphorylation. J Cell Biol. 1991;115:535–545. doi: 10.1083/jcb.115.2.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy B, Taneja R, Chambon P. Synergistic activation of expression of retinoic acid (RA)-responsive genes and induction of embryonal carcinoma cell differentiation by an RA receptor α (RARα)-, RARβ-, or RARγ-selective ligand in combination with a retinoid X receptor-specific ligand. Mol Cell Biol. 1995;15:6481–6487. doi: 10.1128/mcb.15.12.6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer B, Henderson N. Targeted insertion of exogenous DNA into the eukaryotic genome by the Cre recombinase. New Biol. 1990;2:441–449. [PubMed] [Google Scholar]
- Sleigh MJ. Differentiation and proliferation in mouse embryonal carcinoma cells. Bioessays. 1992;14:769–775. doi: 10.1002/bies.950141109. [DOI] [PubMed] [Google Scholar]
- Strickland S. Mouse teratocarcinoma cells: prospects for the study of embryogenesis and neoplasia. Cell. 1981;24:277–278. doi: 10.1016/0092-8674(81)90313-5. [DOI] [PubMed] [Google Scholar]
- Taneja R, Bouillet P, Boylan JF, Gaub M-P, Roy B, Gudas LJ, Chambon P. Reexpression of retinoic acid receptor (RAR)γ or overexpression of RARα or RARβ in RARγ-null F9 cells reveals a partial functional redundancy between the three RAR types. Proc Natl Acad Sci USA. 1995;92:7854–7858. doi: 10.1073/pnas.92.17.7854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneja R, Roy B, Plassat J-L, Zusi CF, Ostrowski J, Reczek PR, Chambon P. Cell-type and promoter-context dependent RAR redundancies for RARβ2 and Hoxa-1 activation in F9 and P19 cells can be artefactually generated by gene knockouts. Proc Natl Acad Sci USA. 1996;93:6197–6202. doi: 10.1073/pnas.93.12.6197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y-J, Wang L, Wu T-C. The expression of retinoid X receptor genes is regulated by all-trans and 9-cis-retinoic acid in F9 teratocarcinoma cells. Exp Cell Res. 1994;210:56–61. doi: 10.1006/excr.1994.1009. [DOI] [PubMed] [Google Scholar]
- Zelent A, Krust A, Petkovich M, Kastner P, Chambon P. Cloning of murine α and β retinoic acid receptors and a novel receptor γ predominantly expressed in skin. Nature (Lond) 1989;339:714–717. doi: 10.1038/339714a0. [DOI] [PubMed] [Google Scholar]