

Abstract
Abstract. Pelizaeus-Merzbacher disease (PMD) is a dysmyelinating disease resulting from mutations, deletions, or duplications of the proteolipid protein (PLP) gene. Distinguishing features of PMD include pleiotropy and a range of disease severities among patients. Previously, we demonstrated that, when expressed in transfected fibroblasts, many naturally occurring mutant PLP alleles encode proteins that accumulate in the endoplasmic reticulum and are not transported to the cell surface. In the present communication, we show that oligodendrocytes in an animal model of PMD, the msd mouse, accumulate Plp gene products in the perinuclear region and are unable to transport them to the cell surface. Another important aspect of disease in msd mice is oligodendrocyte cell death, which is increased by two- to threefold. We demonstrate in msd mice that this death occurs by apoptosis and show that at the time oligodendrocytes die, they have differentiated, extended processes that frequently contact axons and are expressing myelin structural proteins. Finally, we define a hypothesis that accounts for pathogenesis in most PMD patients and animal models of this disease and, moreover, can be used to develop potential therapeutic strategies for ameliorating the disease phenotype.
Pelizaeus-merzbacher disease (PMD)1 is an X-linked leukodystrophy (McKusick, 1994) primarily associated with the duplication, deletion, or mutation of the proteolipid protein locus (PLP), and a connection to several other diseases with overlapping clinical profiles has recently become apparent from extensive genetic screening (Hodes et al., 1993; Boespflug-Tanguy et al., 1994; Kobayashi et al., 1994; Cambi et al., 1996). Yet despite these advances, there has been little progress in defining and characterizing the molecular mechanisms underlying pathogenesis. From our analyses of PLP trafficking in transfected fibroblasts (Gow et al., 1994a ,b; Gow and Lazzarini, 1996; Tosic et al., 1996, 1997), we have developed a hypothesis that posits that the principal effect of many mutations in the coding region of PLP is to disrupt the higher-ordered structure of the resulting protein isoforms, DM-20 and PLP, causing their accumulation in the ER of oligodendrocytes and ultimately leading to the diminished biosynthetic capacity or survival of these cells. This accumulation can be substantial since PLP mRNA constitutes 10% of the total mRNA in myelinating oligodendrocytes (Milner et al., 1985), a level that is 100 times greater than the average abundant message. Here, we provide in vivo evidence to further support this hypothesis, which we have broadened to account for the majority of phenotypes in transgenic mice, Plp mutant mice, and patients with X-linked PMD.
Previous studies have reported dramatic reductions in the number of mature oligodendrocytes in biopsies from patients with PMD (Watanabe et al., 1973; Koeppen, 1992) and in several animal models of this disease, including jimpy and msd mice (Skoff and Knapp, 1992). Moreover, the absence of these cells may be a major factor contributing to the severe disease phenotype (Schneider et al., 1992). A point mutation that inactivates the splice-acceptor site of exon 5 in jimpy mice results in a dramatic alteration to the primary structure of the PLP gene products with the absence of exon 5–encoded amino acids and a concomitant shift in the reading frame in exons 6 and 7. On the other hand, the missense mutation in msd mice causes a conservative substitution of valine for alanine at amino acid 242 of PLP (A242V). In considering these alleles, it is surprising that the disease phenotypes for jimpy and msd are virtually indistinguishable (Skoff and Knapp, 1992), although we cannot rule out the possibility that subtle differences in pathogenesis may exist. In detailed electron microscopic and light microscopic investigations of cellular pathology in the central nervous system (CNS) of jimpy mice, a model for severe PMD, Knapp et al. (1986) have convincingly demonstrated that oligodendrocytes are the principal cell type that degenerate in white matter tracts, although apoptosis was ruled out as the mode of cell death. More recent studies have yielded mixed conclusions; increased levels of apoptotic oligodendrocyte death is absent (Owen and Skoff, 1995; Williams and Gard, 1995) or a feature of (Skoff, 1995) pathogenesis in the CNS of jimpy mice. Two missense alleles causing relatively mild forms of disease have been characterized in the rsh mouse (I186T) and the pt rabbit (H36Q), where oligodendrocyte death does not appear to be elevated above controls, and the mutants have a normal life span (Taraszewska and Zelman, 1987; Fanarraga et al., 1992, 1993; Schneider et al., 1992). In addition to PLP coding region mutations, overexpression of the wild-type Plp gene (Kagawa et al., 1994; Readhead et al., 1994) or a cDNA encoding DM-20 in transgenic mice (Mastronardi et al., 1993; Nadon et al., 1994; Johnson et al., 1995) is associated with hypomyelination, and the severity of disease is proportional to the level of overexpression (Ikenaka and Kagawa, 1995). Partial X chromosomal duplications encompassing the PLP locus also give rise to PMD (for review see Hodes and Dlouhy, 1996) and may account for a substantial proportion of patients that do not harbor coding region mutations (Carango et al., 1995; Harding et al., 1995; Inoue et al., 1996a ). Finally, the minimal behavioral phenotype in mice with targeted disruptions of the Plp gene apparently in the absence of significant compensation by other gene products (Boison and Stoffel, 1994; Rosenbluth et al., 1996; Klugmann et al., 1997) indicates that the hypomyelinating phenotypes observed in jimpy and msd mice, as well as other allelic mutants, do not result from the loss of a functional protein. In man, complete deletions of the PLP gene or mutations that generate in-frame stop codons near the initiating methionine give rise to clinical disease, albeit generally a less severe form when compared with patients with duplications or coding region mutations (Hodes et al., 1993; Garbern et al., 1997).
In this report, we have characterized oligodendrocyte cell death in the CNS of jimpy and msd mice, and we have shown biochemically and histologically using terminal transferase labeling of fragmented DNA that apoptosis in white matter tracts of these mutants is increased several-fold compared with normal littermates. In addition, we demonstrate using five oligodendrocyte-specific antibody markers that the majority of cells undergoing apoptosis in white matter tracts are mature oligodendrocytes that have extended cellular processes and engaged nearby axons, and we show by in situ hybridization that abundant DM-20/PLP–positive oligodendrocytes are present in the brains of msd mice. Furthermore, we show that despite the dramatic differences in disease severity between msd and rsh mice, DM-20/PLP are largely confined to the perinuclear region of oligodendrocytes in the brains of both of these mutants. Thus, the in vivo immunocytochemical results are entirely consistent with our hypothesis that mutant PLP gene products misfold and accumulate in the ER (Gow et al., 1994b ; Gow and Lazzarini, 1996; Gow et al., 1997). These data were presented in part at the American Neurochemistry Society annual meeting (Gow et al., 1995).
Materials and Methods
Purification of Genomic DNA
Optic nerves and sections of trigeminal nerves were rapidly dissected from p18 jimpy, msd, and wild-type mice and frozen on dry ice. The tissue samples were digested overnight at 50°C in 200 μl of 50 mM Tris, pH 8.0, containing 0.5% SDS, 100 mM EDTA, and 240 μg/ml proteinase K (Sigma Chemical Co., St. Louis, MO) and then extracted with phenol/chloroform (1:1 by volume), pH 8.0. Finally, the genomic DNA was precipitated with sodium acetate/ethanol and redissolved in 20 μl of TE. To identify msd/Y male and msd/X carrier female mice of all ages by genotyping, 3–5-mm tail tips were digested overnight at 55°C in 100 μl of TE buffer, pH 8, containing 0.1% Triton X-100 and 100 μg/ml proteinase K and boiled for 10 min. 1 or 2 μl of each digest was amplified in 50 μl PCRs containing 50 ng each of sense-PLP (5′-TTGTG CTTGC TTTTC TGTTC TAAGA AATAA-3′) and antisense-PLP (5′-TTACC AGGGA AACTA GTGTG GCCTC AGCAC-3′) primers for 40 cycles: 1 min at 94°C, 1 min at 55°C, 1 min at 72°C. Aliquots of the PCRs were restricted with 10 U of BsoFI (New England Biolabs, Beverly, MA) for 2 h and then electrophoresed on 4% NuSieve agarose (FMC BioProducts, Rockland, ME) gels containing ethidium bromide. PCR products from wild-type Plp alleles were cut into 88- and 37-bp fragments, while those from msd alleles remained intact (125 bp). A deliberate base change (G-to-T) in the antisense primer (underlined) eliminates in the PCR fragments a BsoFI site that is present in the genomic DNA to optimize size discrimination between wild-type and msd alleles on agarose gels.
Terminal Transferase Labeling
All reactions for labeling tissue sections contained terminal transferase (TdT) at a concentration of 0.25 U/μl and 2.5 mM CoCl2 in buffer provided with the enzyme (Boehringer Mannheim Corp., Indianapolis, IN) according to the method of Gold et al. (1993). Tetramethylrhodamine- 6-dTTP (Boehringer Mannheim Corp.) was included at a final concentration of 10 μM to label vibratome and cryostat sections, which were overlaid with 30 μl of the reaction mixture and incubated in a humidified chamber for 1 h at 37°C. Alternatively, to label genomic DNA in solution ∼1 μg of purified DNA (with or without prior RNase A treatment) and 50 μCi of [α-32P]ddATP (Amersham Corp., Arlington Heights, IL) were added to the buffer/enzyme mixture in a total volume of 20 μl, and the reaction was incubated for 1 h at 37°C. The labeled DNA was separated from free nucleotides using a spin column (Stratagene, La Jolla, CA) and precipitated with sodium acetate/ethanol/glycogen, and 5 × 104 dpm were loaded onto 4% NuSieve agarose gels. After electrophoresis, the DNA samples were transferred to Nytran Plus membranes (Schleicher and Schuell, Inc., Keene, NH) using a standard Southern blotting protocol (Sambrook et al., 1989) and exposed to x-ray film at −80°C.
Immunocytochemistry
Mice were anesthetized with tribromoethanol and fixed by vascular perfusion with 100 ml of freshly prepared 4% paraformaldehyde in 0.1 M sodium phosphate, pH 7.2. Brains and spinal cords were dissected and stored for up to 1 wk at 4°C in phosphate buffer. Some tissue blocks were infiltrated with 20% sucrose in phosphate buffer at 4°C and embedded in OCT for cryostat sectioning. Frozen sections (10 μm) were thaw-mounted on Superfrost Plus slides (Fisher Scientific, Pittsburgh, PA) and stored at −20°C. For labeling with the O4 and Ranscht antibodies (M. Schachner, Eidgenössische Techische Hochschule, Zurich, Switzerland), sections remained unpermeabilized until after incubation with the secondary antibody and fixation in 4% paraformaldehyde/phosphate buffer for 10 min. For antibodies against myelin basic protein (MBP) (130–137; Boehringer Mannheim Corp.; No. 644, D.R. Colman, Mount Sinai School of Medicine), DM-20/PLP (AA3 [Yamamura et al., 1991]), platelet-derived growth factor α–receptor (PDGFαR) (Upstate Biotechnology, Inc., Lake Placid, NY; R7, C. Heldin, Ludwig Institute, Uppsala, Sweden), neurofilaments (2H3 [Guthrie and Pini, 1995], Developmental Hybridoma Bank, University of Iowa, Iowa City, IA; SMI31/32, Sternberger Monoclonals, Inc., MD), and histones (MAB052, Chemicon International, Temecula, CA), the sections were permeabilized for 15 min in methanol and then incubated for 30 min in blocking solution (Gow et al., 1994a ). The sections were incubated in primary antibodies at room temperature for 2 d or overnight, and subsequent antibody steps were for 3 h duration. Intervening wash steps were carried out for 2× 10 min in TBS. Finally, the sections were incubated in 2 μg/ml 4′,6-diamidino-2-phenylindole (DAPI) to stain nuclei and mounted in 2.5% 1,4-diazabicyclo-(2,2,2)-octane (DABCO) (Sigma Chemical Co.) and 50 mM Tris, pH 8.6, in 90% glycerol (Johnson et al., 1982). Alternatively, 50-μm vibratome sections of transverse spinal cord were cut and then processed while free-floating. These sections were permeabilized in TBS containing 0.5% Triton X-100 before being labeled with antibodies using the same protocol described for cryostat sections.
In Situ Hybridization
Digoxigenin-labeled sense and antisense RNA probes were synthesized by in vitro transcription (Genius 4 labeling kit; Boehringer Mannheim Corp.) of a linearized plasmid comprising a 1.4-kb EcoRI human PLP cDNA (Puckett et al., 1987) in pBS using T3 or T7 RNA polymerases. The probes were not hydrolyzed, and their concentrations were determined after agarose/formaldehyde electrophoresis and Northern blotting using anti–digoxigenin-alkaline phosphatase (Genius 3 detection kit) by comparison of signal intensities with known digoxigenin-labeled standards. 10-μm cryostat sections were permeabilized in 0.1% Triton X-100/ TBS for 30 min and overlaid with 25 μl of a hybridization solution (Strable et al., 1994) containing 50% formamide, 2× sodium chloride/sodium phosphate/ethylmaleimide-tetra-acetate (SSPE), 10% dextran-sulfate, 1× Denhardt's (Sambrook et al., 1989), and 1 mg/ml tRNA and prehybridized for 2 h at 55°C under a parafilm “coverslip.” Probes were boiled for 5 min and added to fresh prehybridization solution at a final concentration of 0.2 ng/μl, overlaid onto the sections, and coverslipped for overnight hybridization at 55°C. Thereafter, the slides were washed 2× 30 min at 65°C in 5× SSPE/50% formamide, 2× 30 min at 65°C in 1× SSPE/25% formamide/0.5× PBS, and finally at 25°C in 0.1% Tween-20/PBS for 30 min. Sections were processed for alkaline phosphatase immunohistochemistry using the Genius 3 detection kit. The enzyme reactions with NBT/BCIP were carried out at 20°C for ∼2 h on wild-type sections and 20 h on jimpy and msd sections.
Results
Elevated Levels of Apoptosis in jimpy and msd Mice
Studies by many investigators over the last few years have established that cell death can occur by at least two distinct mechanisms: necrosis, which is often the result of physical tissue damage, and apoptosis, which can be initiated by a variety of noxious stimuli. Three analytical techniques have been used to identify cells undergoing apoptosis in vivo. Electron microscopy reveals the condensation of chromatin along the inner nuclear membrane, while cytoplasmic organelles remain relatively unaffected in apoptotic cells. Gel electrophoresis of DNA from apoptotic cells reveals a characteristic fragmentation into small segments that are multiples of 180 bp. This DNA fragmentation is exploited in the third method of analysis, terminal transferase (TdT) labeling of the fragments with labeled deoxynucleotide triphosphates.
Previous studies (Barres et al., 1992) have showed that a low level of oligodendrocyte apoptosis occurs normally in developing brain. This level of cell death is reflected as a faint 180-bp ladder in the electrophoretogram of DNA from the optic nerves of a wild-type mouse (Fig. 1, second lane). The intensity of the DNA ladder obtained with DNA from the optic nerve of jimpy mice (fourth lane), however, is several-fold more intense. The amounts of the high molecular weight DNA in the second and fourth lanes are similar, indicating that similar amounts of DNA for both samples were loaded onto the gel and that the greater intensity of the lower bands in jimpy mice results from an increase in DNA fragmentation. Prominent DNA ladders from the optic nerves of msd mice (data not shown) are indistinguishable from that shown for jimpy optic nerve. In contrast to CNS, the level of apoptosis in peripheral nervous system trigeminal nerves (Fig. 1, Trig.) of wild-type and jimpy mice is not detectable (third and fifth lanes). These lanes are included as controls, showing that the laddering is not a product of our DNA isolation procedure.
Figure 1.

Autoradiogram showing terminal transferase–labeling of 180-bp DNA ladders in optic nerve (Opt.) samples from p18 wild-type (second lane) and jimpy (fourth lane) mice. DNA ladders are not detectable in trigeminal nerve (Trig.) samples (third and fifth lanes). A radiolabeled 123-bp ladder (first lane) was used to calculate M r of DNA fragments in the tissue samples.
Transverse sections of thoracic spinal cord from p18 wild-type, jimpy, and msd mice show terminal transferase labeling with a fluorescent substrate (Fig. 2, red) in the region of the lateral ventral white matter tracts (Fig. 2, a–c). While labeled nuclei are present in wild-type tissue, they are much more abundant in jimpy and msd tissue, consistent with the data in Fig. 1. The condensation of chromatin is apparent in two terminal transferase–labeled nuclei from msd spinal cord shown at high power (Fig. 2 d). Margination of the chromatin in four foci along the inner nuclear membrane is visible in the nucleus on the right as shown by a DAPI stain of the same field (Fig. 2 e). The nuclei not labeled by terminal transferase in Fig. 2 d have a normal appearance and stain relatively evenly with DAPI. The transverse spinal cord sections in Fig. 2, a–c, are counterstained (green) with an antiserum against MBP that reveals many heavily myelinated axons as bright rings of fluorescence in wild-type specimens (Fig. 2 a). Sections from the mutant mice (Fig. 2, b and c) are also stained, albeit at dramatically reduced levels, where oligodendrocyte-derived MBP-positive membranes have enveloped some axons. This feature is most obvious in Fig. 2 b, where the oblique plane of the section clearly demonstrates that oligodendrocytes in these animals are able to fully differentiate, contact nearby axons, and synthesize myelin membrane components.
Figure 2.

Immunofluorescence images of lateral ventral spinal cord white matter tracts in transverse sections from p18 wild-type (a), msd (b), and jimpy (c) mice. MBP-positive membrane (green) surrounds a few axons in the mutants and terminal transferase–labeled nuclei (red) are present in all three sections. (d) High magnification of two terminal transferase– labeled nuclei in a longitudinal section of spinal cord from an msd mouse. Many normal-appearing nuclei are present in this field and are revealed by the DAPI stain (e). The terminal transferase reaction product colocalizes with the DAPI staining of the condensed marginated chromatin. Bars: (a–c) 50 μm; (d and e) 10 μm.
Using the terminal transferase detection of apoptotic cells, coupled with DAPI staining to independently assess the condensation of chromatin and abnormal appearance of the nuclei, we quantified cell death in cryostat sections from four regions in the spinal cords of 10-d-old (p10) msd mice and wild-type male littermates (Table I). Counts were obtained for 4–12 transverse serial sections from each of the cervical, thoracic, lumbar, and sacral regions, and these data show that apoptosis is increased by two- to threefold along the length of the spinal cords of msd mice compared with littermate controls. These results accord with the elevated levels of cell death quantified in the cervical spinal cords of jimpy mice, which were found to be 2.7- and 4.8-fold above that in wild-type littermates at ages p7 and p12, respectively (Knapp et al., 1986). Surprisingly, the absolute numbers of degenerating cells reported for cervical spinal cord in that study, which were identified on the basis of morphological changes in 1-μm plastic sections, are ∼10-fold greater than our data for an equivalent thickness of section. While a portion of this difference likely reflects the greater severity of disease caused by the jimpy mutation compared with msd and differences in genetic background between the two alleles, we surmise that the TdT assay detects cells in the final stages of degeneration at a point when the cellular dissolution may well be proceeding at its maximal rate. On the other hand, the early morphological changes catalogued by Knapp et al. (1986) may proceed at a slow rate, thereby rendering individual degenerating cells visible for a greater length of time.
Table I.
Quantitation of Apoptotic Nuclei in the Spinal Cords of Wild-Type and msd Mice*
| Region | Wild-type | msd | Ratio (msd/wt) | |||
|---|---|---|---|---|---|---|
| Cervical | 12.8 ± 2.0 | 43.9 ± 5.9 | 3.4 | |||
| Thoracic | 9.4 ± 0.3 | 24.5 ± 1.5 | 2.6 | |||
| Lumbar | 9.7 ± 1.4 | 22.2 ± 2.1 | 2.3 | |||
| Sacral | 6.4 ± 0.3 | 10.6 ± 2.6 | 1.7 |
Apoptotic nuclei were counted in 4–12 serial transverse sections in each region of spinal cord from all animals at age p10. The data are presented as mean ± SEM from three wild-type and two msd mice.
Mature Oligodendrocytes Undergo Apoptosis in msd Mice
Previous electron microscopic analyses of degenerating cells in jimpy mouse CNS identified oligodendrocytes by characteristic morphological criteria (Knapp et al., 1986); however, the stage in the oligodendrocyte lineage that these cells had attained was not easily discernible. Thus, to assess the degree to which oligodendrocytes of the mutant animals differentiate before they undergo apoptosis, we double labeled parasagittal sections from msd mice using terminal transferase (Fig. 3, red) and antibodies to several oligodendrocyte-specific markers (green): O4, which is synthesized by progenitor cells as well as mature oligodendrocytes (Fig. 3 a); Ranscht antigen, which is synthesized by postmitotic and mature oligodendrocytes (Fig. 3 b); and DM-20/PLP and MBP, which are synthesized by mature myelinating oligodendrocytes (Fig. 3, c and d, respectively). Apoptotic nuclei are shown in Fig. 3 a labeled with O4 antibody and in Fig. 3 b labeled with Ranscht antibody. A considerable amount of myelin-like membrane debris is apparent in these micrographs that presumably resulted from oligodendrocyte degeneration. The presence of DM-20/PLP–positive membrane surrounding many apoptotic nuclei (Fig. 3 c) indicates that oligodendrocytes in the mutant animals fully differentiate to the point at which they are synthesizing myelin membrane components. Fig. 3 d shows an MBP-positive oligodendrocyte at high power with several processes extending away from the cell body. Condensed, marginated chromatin in the nucleus of this cell is clearly revealed by the terminal transferase labeling (red). Together, these data substantiate the view that oligodendrocytes differentiate and begin to synthesize myelin membrane components before they undergo apoptosis.
Figure 3.

Immunofluorescence staining of cells undergoing apoptosis (red) in the midbrains of p18 msd mice. These cells are stained by antibodies to different oligodendrocyte-specific markers: O4 (a), Ranscht (b), PLP (c), and MBP (d). Bars: (a–c) 10 μm; (d) 2 μm.
The proportions of TdT-positive apoptotic cells that were labeled with one of five oligodendrocyte-specific markers were quantified in the hypothalamus, pons, medulla, and cervical spinal cords of msd mice (Table II). Several studies have shown that the expression of these markers, once initiated, is maintained during subsequent stages of cellular differentiation through to myelinating oligodendrocytes with the exception of PDGFαR. The gene encoding this protein is expressed in O-2A progenitor cells but downregulated in immature oligodendrocytes before the appearance of mature markers such as MBP and PLP by immunocytochemistry (Ellison and de Vellis, 1994; Nishiyama et al., 1996). Approximately 75% of apoptotic cells in the msd mice are stained with antibodies against DM-20/PLP, a percentage that is corroborated by the O4 and Ranscht antigen markers, each of which stain 83% of apoptotic cells. On the other hand, we found that only a small proportion of TdT-labeled cells were also labeled by either of two independently derived polyclonal antibodies directed against PDGFαR. Although we cannot rule out the possibility that epitopes on the PDGFαR are unstable or masked in apoptotic cells, the results obtained for this antigen are consistent with the DM-20/PLP data. Reasons for the comparatively low proportion of MBP-positive apoptotic nuclei are not certain; however, MBP is a soluble protein that is normally very susceptible to attack by proteases, and our data may reflect the instability of the antibody epitope in apoptotic cells. Finally, if we assume that the O4 antibody is able to detect essentially all of the oligodendrocyte lineage cells that undergo apoptosis in msd mice, then our data show that 90 ± 5% of those cells are mature oligodendrocytes.
Table II.
Proportion (%) of Apoptotic Cells That Stain Positive for Oligodendrocyte-specific Markers by Immunofluorescence
| Oligodendrocyte- specific antigen | No. of mice | No. of apoptotic cells counted | Antigen-positive cells (%) | |||
|---|---|---|---|---|---|---|
| PLP | 3 | 183 | 75 ± 0.5 | |||
| O4 | 3 | 110 | 83 ± 4 | |||
| Ranscht | 3 | 176 | 83 ± 3 | |||
| MBP | 3 | 134 | 43 ± 8 | |||
| PDGFαR | 2 | 353 | 8 ± 0.5 |
Oligodendrocyte-specific antigen-positive/TdT-positive/DAPI-positive cells were located in the hypothalamus, pons, medulla, and cervical spinal cord. Counts are presented as mean ± SEM and were obtained from 10-μm sagittal cryostat sections of brain from two or three p18 msd mice.
Abundant DM-20/PLP mRNA–positive Oligodendrocytes in msd Mice
While the majority of apoptotic nuclei are surrounded by membrane that is strongly DM-20/PLP–positive, the micrographs in Fig. 3 do not impart any appreciation for the number of viable oligodendrocytes in the CNS of mutant animals. To this end, we performed in situ hybridization on parasagittal sections of brain from p18 wild-type and msd mice using a digoxigenin-labeled PLP cRNA probe. In Fig. 4 a, the corpus callosum (cc) and subcortical white matter tract extending to the right are heavily populated by DM-20/PLP–positive oligodendrocytes and many mature oligodendrocytes also line the periventricular zone. In Fig. 4 b, DM-20/PLP–positive oligodendrocytes are present in the same regions of white matter as for the wild-type animal, again supporting the notion that many msd oligodendrocytes fully differentiate and express genes encoding myelin-specific proteins before degenerating. Similar results were obtained for jimpy mice. The development times for alkaline phosphatase histochemistry were 5–10 times longer for sections from the mutants than for controls suggesting that the levels of DM-20/PLP mRNA in msd oligodendrocytes is lower than in wild-type cells.
Figure 4.

In situ hybridization in brain sections of p18 wild-type (a) and msd (b) mice using an antisense-PLP cRNA probe. Adjacent sections from each animal hybridized with sense-PLP probes were unstained. cc, corpus callosum; hc, hippocampus. Bar, 100 μm.
DM-20/PLP Remain Perinuclear in msd and rsh Mouse Oligodendrocytes
With the strong indication that the majority of oligodendrocytes in jimpy and msd mice express myelin-specific proteins at the time they undergo apoptosis (Figs. 2–4; Table II), we investigated the intracellular distributions of two of these proteins, MBP and PLP. Immunofluorescence staining of spinal cords from the mutant animals (Fig. 2) shows that oligodendrocyte processes recognize and envelop axons with MBP-positive membrane. On the other hand, our hypothesis predicts that DM-20/PLP in jimpy and msd mice should accumulate in the ER of oligodendrocytes. To test this prediction, we labeled sagittal brain sections of p18 wild-type msd and rsh mice with a monoclonal antibody raised to the carboxyl terminus of DM-20/PLP and one of two antibodies against MBP to locate myelinating oligodendrocytes. A spatial and temporal wave of myelination spreads through the developing CNS with a well-defined course, and the region of the cerebral cortex shown in Fig. 5 was chosen to illustrate an area that is relatively sparsely myelinated. Oligodendrocyte cell bodies in wild-type brain (Fig. 5, a–c, arrowheads) contain both MBP (Fig. 5 a) and DM-20/PLP (Fig. 5 b). Myelination is evident in this field from the numerous axons that have been enveloped in MBP- and DM-20/PLP–positive membrane, thereby rendering them yellow in the overlay (Fig. 5 c). A comparable region of cortex from an msd littermate is shown in Fig. 5, d–f, and oligodendrocyte cell bodies in this field are positive for both MBP and DM-20/ PLP (arrowheads). However, there is little evidence of DM-20/PLP in oligodendrocyte-derived membranes surrounding nearby axons. Rather, these data clearly demonstrate that mutant Plp gene products are essentially confined to the perinuclear region of oligodendrocytes. The fact that msd oligodendrocytes are able to extend processes that contact and invest axons is evident from the MBP staining surrounding axons (Fig. 5 d) in a similar fashion to that in wild-type cortex (Fig. 5 a).
Figure 5.
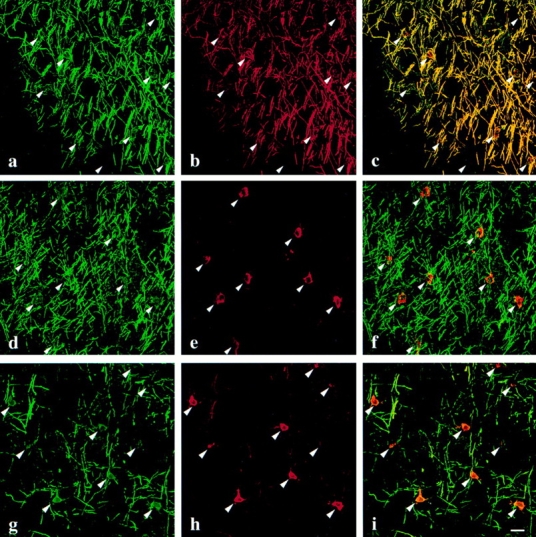
Immunofluorescence staining for MBP (a, d, and g) and DM-20/PLP (b, e, and h) in p18 wild-type (a–c), msd (d–f), and rsh (g–i) mouse neocortex. In wild-type mice, MBP and DM-20/ PLP staining are extensively colocalized in the cell bodies (arrowheads) as well as in oligodendrocyte processes that are in contact with nearby axons (c). In msd mice, the presence of oligodendrocyte processes wrapped around axons is inferred from the MBP staining (d), which has a similar morphology to that in wild-type mice (a). However, DM-20/PLP staining is limited to the perinuclear region of oligodendrocytes, suggesting that the intracellular trafficking of this mutant protein is disrupted (e). In rsh mice, the trafficking of DM-20/PLP (h) is disrupted in a similar fashion to msd mice (e) except that low levels of DM-20/ PLP are visible in myelin sheaths. Bar, 20 μm.
In contrast to the severe dysmyelination in the CNS of msd mice, which live only 3–4 weeks after birth, the disease in rsh mice on the same genetic background is relatively mild and is characterized by hypomyelination. Furthermore, hemizygous males and females homozygous for the rsh allele can breed and have a normal life span. Nonetheless, the intracellular trafficking defect revealed for DM-20/PLP in msd mice is also apparent in oligodendrocytes in the cortex of p21 rsh mice (Fig. 5, g–i). Similar to the immunofluorescence staining for msd mice, oligodendrocytes in rsh mice invest nearby axons in MBP-positive membrane (Fig. 5 g). However, the majority of the myelin sheaths have low levels of DM-20/PLP staining despite the tremendous amount of these proteins in the perinuclear region of oligodendrocytes (Fig. 5 h, arrowheads). Thus, these data are consistent with previous immunoelectron microscopic studies in jimpy mice (Roussel et al., 1987) and strongly support our hypothesis (Gow et al., 1994a ,b; Gow and Lazzarini, 1996; Gow et al., 1997) that many missense PLP mutations cause the encoded gene products to accumulate in the ER. This accumulation probably stems from misfolding of the mutant proteins, a notion which is consistent with our immunocytochemical data in transfected cells using three conformationally sensitive anti– DM-20/PLP antibodies, 1A9 and O10 (Gow et al., 1997) and 81-11 (unpublished data).
An exciting aspect of the data in Fig. 5 is the differential effect of the rsh allele on the intracellular trafficking of the mutant DM-20/PLP proteins. Previous immunocytochemical investigations of rsh CNS with PLP-specific antibodies indicated that this isoform (i.e., PLPrsh) is present at low concentrations in myelin sheaths (Fanarraga et al., 1992). Moreover, western blots from these mutant mice showed that in purified myelin fractions, the amount of PLPrsh is sharply reduced compared with littermates, while DM-20rsh is present at levels that are close to normal (Mitchell et al., 1992). These observations bear a striking resemblance to our previous analyses of the intracellular trafficking of mutant DM-20/PLPs in transiently transfected fibroblasts where DM-20rsh was found to traverse the secretory pathway to the cell surface while PLPrsh accumulated in the ER (Gow et al., 1994b ; Gow and Lazzarini, 1996). We also showed that when coexpressed, DM-20rsh mediated the trafficking of some of the PLPrsh to the cell surface in a concentration-dependent manner, perhaps by forming transport-competent heteromeric complexes and thereby reducing the amount of mutant PLP in the ER. On the other hand, the presence of DM-20msd, which by itself accumulates in the ER, had no effect on the intracellular localization of PLPmsd, and both proteins remained perinuclear. The coexpression of several other mutant DM-20/PLPs in fibroblasts yielded identical results (Gow and Lazzarini, 1996; Tosic et al., 1997), suggesting that an “assisted transport” mechanism may be of particular importance to the pathogenesis of PMD. Thus, in the light of previous biochemical analyses in rsh mice (Fanarraga et al., 1992; Mitchell et al., 1992), the data shown in Fig. 5, d–i, provide strong in vivo evidence for an inverse correlation between the proficiency of mutant PLP gene products to traverse the secretory pathway and the severity of disease elicited by the mutation, as predicted from our transfection studies (Gow and Lazzarini, 1996).
Pathogenesis in Plp-overexpressing Transgenic Mice
Transgenic mice bearing supernumerary autosomal copies of a wild-type murine Plp gene develop a dose-dependent (Ikenaka and Kagawa, 1995) hypomyelinating disease with a number of similarities to msd and rsh mice. Three independent lines of Plp-overexpresser transgenic mice, No. 72, No. 66, and No. 4e, have been characterized (Kagawa et al., 1994; Readhead et al., 1994), and each exhibits an increase in overall DM-20/PLP expression from 1.3- to 5.2-fold above littermate controls that is further increased in the homozygous animals to as much as 7.4-fold (Inoue et al., 1996b ). These mice develop a nonimmune-mediated demyelinating disease, the severity of which is proportional to the level of Plp overexpression (Ikenaka and Kagawa, 1995). A demyelinating disease stemming from the overexpression of DM-20 cDNA in oligodendrocytes has also been reported (Mastronardi et al., 1993; Johnson et al., 1995).
To determine whether the intracellular trafficking of DM-20/PLP is disrupted in Plp-overexpresser mice in a manner similar to that shown in Fig. 5 for the msd and rsh mutants, we examined tissue sections from line No. 72 transgenic mice using immunocytochemistry. Fig. 6 a shows a parasagittal section of neocortex from a 3-mo-old wild-type mouse using an antibody raised against neurofilaments (red). Many radiating and longitudinal axons tracking along the superior surface of the corpus callosum are ensheathed in DM-20/PLP–positive myelin (green) and are continuous for considerable distances through the section. Immunofluorescence staining in the neocortex of an age-matched homozygous Plp-overexpresser (line No. 72) is shown in Fig. 6 b. Disease in this mouse was at an advanced stage at the time of perfusion. While the neuronal architecture in this animal appears relatively intact, the amount of myelin revealed by DM-20/PLP staining in the cortex is drastically reduced compared with the wild-type, and abnormalities are apparent at the cellular level. Arrowheads point to several neurofilament-rich lesions that were observed throughout the brain as previously noted in many myelin mutants (Seitelberger, 1970; Dentinger et al., 1982; Rosenfeld and Freidrich, 1983; Readhead et al., 1994). Small arrows show large swellings in myelin sheaths that are abundant throughout the CNS of these and other myelin mutants (Rickman, D., and R.A. Lazzarini, manuscript in preparation; Gravel et al., 1996). DM-20/PLP– positive oligodendrocyte cell bodies are visible in Fig. 6 b (thick arrows); however, these cells appear to have lost their processes. While this aspect of pathology accounts for the paucity of myelinated axons, it precludes us from determining whether or not the trafficking of DM-20/PLP has been disrupted. Immunofluorescence staining of adjacent brain sections with antibodies against 2′,3′-cyclic nucleotide 3′-phosphodiesterase, myelin-associated glycoprotein, MBP, and myelin oligodendrocyte glycoprotein yielded staining patterns that were extensively colocalized with the DM-20/PLP staining and confirm the lack of processes extending from the majority of these cells (data not shown). The inset in Fig. 6 b shows an oligodendrocyte cell body at high magnification stained with antibodies against DM-20/PLP (green), MBP (red), and histones (blue) to highlight the nucleus. Previous studies have suggested that oligodendrocytes fail to differentiate in the Plp-overexpressers (Kagawa et al., 1994; Readhead et al., 1994); however, our data clearly show an abundance of cells expressing multiple markers of mature oligodendrocytes.
Figure 6.

Immunofluorescence staining for DM-20/PLP (green) and neurofilaments (red) in 3-mo-old wild-type (a) and No. 72 homozygous Plp-overexpresser mice (b) showing comparable regions of the cerebral cortex superior to the corpus callosum (bottom of panels). (Inset) High-power image showing a typical oligodendrocyte cell body from a No. 72 mouse stained with antibodies against DM-20/PLP (green), MBP (red), and histones (blue). Bar, 50 μm.
Discussion
The cellular and molecular defects responsible for the pleiotrophic phenotypes of the PLP mutants have been subjects of intense investigation. An underlying theme of many earlier studies has been the suggestion that dysmyelination in the CNS of these mutants stems from an absence of functional DM-20/PLP, which arrests the differentiation of virtually all oligodendrocyte progenitors at an immature stage, resulting in the accumulation of premyelinating cells that subsequently die (Webster and Sternberger, 1980; Skoff and Knapp, 1992; Nadon and Duncan, 1995). However, our analyses of jimpy and msd mice affords an alternative explanation of pathogenesis—that precursors differentiate into mature oligodendrocytes that extend processes, make contact with nearby axons, and initiate the synthesis of proteins such as MBP and PLP (see Table II; Figs. 2–5) before undergoing apoptosis. The presence of oligodendrocyte-derived membrane around the axons of jimpy and msd mice (Fig. 5; Ghandour and Skoff, 1988) and the virtual absence of compact myelin in these mutants (Billings-Gagliardi et al., 1980) further substantiate this notion. Thus, our data strongly support a view that the primary pathologic effect of many PLP mutations is the perinuclear accumulation of mutant proteins in mature oligodendrocytes, which frequently induces apoptotic death. Studies by Kagawa et al. (1994) and Inoue et al. (1996a ,b) suggest that the overexpression of wild-type PLP, if sufficiently robust, also causes a dysmyelinating disease with a phenotype very similar to that observed in jimpy and msd mice. Our analysis of the homozygous No. 72 Plp-overexpressers clearly indicates that mature oligodendrocytes are abundant in the brain and that demyelination likely stems from an inability of these cells to maintain contact with and/or remyelinate nearby axons rather than an impaired differentiation of oligodendrocyte lineage cells as previously proposed (Kagawa et al., 1994; Readhead et al., 1994).
In light of our evidence that oligodendrocyte differentiation is not impaired in the Plp mutants, what kind of mechanism might elicit disease in these animals? We hypothesize that the accumulation of nascent gene products in the secretory pathway governs the pathogenesis. If mutant protein is synthesized at a slow rate, the oligodendrocyte can degrade the misfolded nascent polypeptides in the ER without any lasting damage to the cell. On the other hand, if the rate of mutant protein synthesis exceeds the capacity of the cell to degrade it, the protein may accumulate to levels that interfere with cellular function and eventually trigger apoptosis. This concept has considerable merit and enables us to account for the phenotypes observed in most patients with PMD. Our previous studies in transfected cells (Gow et al., 1994a ,b; Gow and Lazzarini, 1996; Tosic et al., 1996, 1997) demonstrated that eleven disease-causing missense mutations in extracellular, transmembrane, and cytoplasmic regions cause DM-20 and/or PLP to accumulate in the ER of COS-7 cells, suggesting that some form of disruption in the secretory pathway may cause disease. This conclusion is consistent with the immunocytochemical data in Fig. 5 showing disrupted DM-20/PLP trafficking in the msd and rsh mice, previous immunoelectron microscopic analyses showing that DM-20/PLP is confined to the ER of jimpy oligodendrocytes in vivo (Nussbaum and Roussel, 1983; Schwob et al., 1985; Roussel et al., 1987), and by the jimpy-like phenotype that is observed in transgenic mice (Turnley et al., 1991; Yoshioka et al., 1991) in which the integral membrane protein, MHC-Kb, accumulates in the ER of oligodendrocytes (Power et al., 1996). Furthermore, elevated expression of the gene encoding the ER luminal chaperone BiP in the brains of jimpy and msd mice (Hudson and Nadon, 1992) is consistent with metabolic stress (Shamu et al., 1994).
Our hypothesis also accommodates the apparent continuum of disease severities observed among patients with coding region mutations by taking into account epistatic effects such as those that influence the pathogenesis of other diseases. For example, previous studies have shown that the rate of degradation of mutant protein in the ER is variable within the population and that susceptibility to liver disease in patients harboring PiZZ mutations in the gene encoding α1-antitrypsin is inversely correlated with the protein degradation rate (Wu et al., 1994). Thus, for PMD we hypothesize that some alleles at secondary sites that slightly reduce the operating efficiency of the ER degradation pathway in oligodendrocytes and other cell types should confer a greater susceptibility to the effects of mutant DM-20/PLP accumulation than others, thereby giving rise to a spectrum of disease severities both within and between pedigrees. Other mechanisms may diminish the rate of accumulation of mutant PLP gene products. In this regard, we have previously demonstrated that facilitated trafficking through the secretory pathway of mutant PLPs by their cognate DM-20s is correlated with less severe forms of disease such as classical PMD and spastic paraplegia (Gow and Lazzarini, 1996).
Finally, protein accumulation in the secretory pathway may also account for disease associated with the overexpression of wild-type PLP gene products (Carango et al., 1995; Inoue et al., 1996a ). Although we have not established whether DM-20/PLP accumulates in the perinuclear region of oligodendrocytes in the No. 72 strain of Plp-overexpressing transgenic mice, these data do not preclude the possibility that disruptions to DM-20/PLP trafficking may underlie pathogenesis. Kagawa et al. (1994) and Inoue et al. (1996b) have reported that 4e homozygous mice develop a robust behavioral phenotype and exhibit pathological changes in the CNS that are strikingly similar to jimpy and msd mice, including the accumulation of DM-20/PLP in the perinuclear region of oligodendrocytes (Macklin et al., 1995) and markedly increased levels of oligodendrocyte apoptosis. With regard to pathogenesis in the No. 72 Plp-overexpressers, the perinuclear accumulation of DM-20/PLP in 4e oligodendrocytes suggests that the oligodendrocyte pathology illustrated in Fig. 6 may reflect a DM-20/PLP trafficking disturbance that is insufficient to trigger apoptosis but nevertheless impairs the biosynthetic capacity of oligodendrocytes and decreases their ability to maintain their processes and myelin sheaths. This view is consistent with a dependence of disease severity on the level of Plp overexpression, starting with less than a twofold increase in steady-state mRNA levels, and with early pathological changes that have been observed in oligodendrocytes by electron microscopy, including distention and vesiculation of the ER and/or Golgi apparatus (Kagawa et al., 1994; Readhead et al., 1994; Inoue et al., 1996b ).
Apart from defining the molecular mechanism of pathogenesis in PMD, our hypothesis makes strong predictions about strategies that may be used to ameliorate the severity of this disease. In general, the synthesis of mutant PLP gene products or overexpression of the wild-type gene is of greater detriment to oligodendrocytes than the absence of these proteins (Hodes et al., 1993; Boison and Stoffel, 1994; Sistermans et al., 1996; Garbern et al., 1997; Klugmann et al., 1997; Yang and Skoff, 1997), which suggests that reducing PLP expression should benefit most PMD patients. In this regard, antisense phosphorothioate oligonucleotides have been shown to dramatically improved the survival of jimpy oligodendrocytes in culture (Yang and Skoff, 1997) and suggest that this strategy might also succeed in vivo. Furthermore, our hypothesis predicts that therapies based on gene complementation strategies would be largely unsuccessful in most DM-20/PLP–linked diseases because increased levels of PLP gene products may further increase the accumulation of protein in the ER and exacerbate pathology. Indeed, studies from three independent groups have failed to rescue jimpy mice with supernumerary copies of the Plp gene (Kagawa et al., 1994; Nadon et al., 1994; Readhead et al., 1994; Schneider et al., 1995). As shown in Fig. 6 and studies by other groups, the overexpression of Plp transgenes in wild-type mice causes a hypomyelinating phenotype (Kagawa et al., 1994; Readhead et al., 1994). On the other hand, PMD patients in which PLP is deleted, or is functionally null, may well benefit from gene replacement, provided the level of transgene expression does not exceed that of the wild-type gene.
Footnotes
1. Abbreviations used in this paper: CNS, central nervous system; DAPI, 4′,6-diamidino-2-phenylindole; MBP, myelin basic protein; PDGFαR, platelet derived growth factor α–receptor; PLP, proteolipid protein; PMD, Pelizaeus-Merzbacher disease; TdT, terminal transferase.
We thank: V. Friedrich, Jr and Susan Morgello for many helpful discussions involving neuroanatomy, morphometry, and statistics; Kazu Ikenaka and Marjorie Lees for providing us with the PLP antibodies; and Hans Lassmann and Helena Breitschopf for sharing their unpublished methods on terminal transferase labeling and in situ hybridization.
This work was supported by research grants awarded to A. Gow from the National Multiple Sclerosis Society (RG2891A1/1) and to R.A. Lazzarini from the National Multiple Sclerosis Society (RG2734-A3) and the National Institutes of Health (3P01NS33165-O1A1S1). This is manuscript number 248 from the Brookdale Center for Developmental and Molecular Biology, Mount Sinai School of Medicine.
Address all correspondence to R.A. Lazzarini, Brookdale Center for Developmental and Molecular Biology, Box 1126, Mount Sinai School of Medicine, One Gustave L. Levy Place, New York, NY 10029-6574. Tel.: (212) 241-4272. Fax: (212) 860-9279. E-mail: rlazzar@smtplink.mssm.edu
References
- Barres BA, Hart IK, Coles HS, Burne JF, Voyvodic JT, Richardson WD, Raff MC. Cell death and control of cell survival in the oligodendrocyte lineage. Cell. 1992;70:31–46. doi: 10.1016/0092-8674(92)90531-g. [DOI] [PubMed] [Google Scholar]
- Billings-Gagliardi S, Adcock LH, Wolf MK. Hypomyelinated mutant mice: description of jpmsd and comparison with jp and qkon their present genetic backgrounds. Brain Res. 1980;194:325–338. doi: 10.1016/0006-8993(80)91215-9. [DOI] [PubMed] [Google Scholar]
- Boespflug-Tanguy O, Mimault C, Melki J, Cavagna A, Giraud G, Pham D, Dinh, Dastugue B, Dautigny A, Group PC. Genetic homogeneity of Pelizaeus-Merzbacher disease: tight linkage to the proteolipoprotein locus in 16 affected families. Am J Hum Genet. 1994;55:461–467. [PMC free article] [PubMed] [Google Scholar]
- Boison D, Stoffel W. Disruption of the compacted myelin sheath of axons of the central nervous system in proteolipid protein-deficient mice. Proc Natl Acad Sci USA. 1994;91:11709–11713. doi: 10.1073/pnas.91.24.11709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambi F, Tang XM, Cordray P, Fain PR, Keppen LD, Barker DF. Refined genetic mapping and proteolipid protein mutation analysis in X-linked pure hereditary spastic paraplegia. Neurology. 1996;46:1112–1117. doi: 10.1212/wnl.46.4.1112. [DOI] [PubMed] [Google Scholar]
- Carango P, Funanage VL, Ruben RE, Debruyn CS, Marks HG. Overexpression of DM20 messenger RNA in two brothers with Pelizaeus-Merzbacher disease. Ann Neurol. 1995;38:610–617. doi: 10.1002/ana.410380409. [DOI] [PubMed] [Google Scholar]
- Dentinger MP, Barron KD, Csiza CK. Ultrastructure of the central nervous system in a myelin deficient rat. J Neurocytol. 1982;11:671–691. doi: 10.1007/BF01262431. [DOI] [PubMed] [Google Scholar]
- Ellison JA, de Vellis J. Platelet-derived growth factor receptor is expressed by cells in the early oligodendrocyte lineage. J Neurosci Res. 1994;37:116–128. doi: 10.1002/jnr.490370116. [DOI] [PubMed] [Google Scholar]
- Fanarraga ML, Griffiths IR, McCulloch MC, Barrie JA, Kennedy PG, Brophy PJ. Rumpshaker: an X-linked mutation causing hypomyelination: developmental differences in myelination and glial cells between the optic nerve and spinal cord. Glia. 1992;5:161–170. doi: 10.1002/glia.440050302. [DOI] [PubMed] [Google Scholar]
- Fanarraga ML, Sommer IU, Griffiths IR, Montague P, Groome NP, Nave K-A, Schneider A, Brophy PJ, Kennedy PG. Oligodendrocyte development and differentiation in the rumpshaker mutation. Glia. 1993;9:146–156. doi: 10.1002/glia.440090208. [DOI] [PubMed] [Google Scholar]
- Garbern JY, Cambi F, Tang X-M, Sima AAF, Vallet JM, Bosch EP, Lewis R, Shy M, Sohi J, Kraft G, et al. Proteolipid protein is necessary in peripheral as well as central myelin. Neuron. 1997;19:205–218. doi: 10.1016/s0896-6273(00)80360-8. [DOI] [PubMed] [Google Scholar]
- Ghandour M, Skoff R. Expression of galactocerebroside in developing normal and jimpy oligodendrocytes in situ. . J Neurocytol. 1988;17:485–498. doi: 10.1007/BF01189804. [DOI] [PubMed] [Google Scholar]
- Gold R, Schmied M, Rothe G, Zischler H, Breitschopf H, Wekerle H, Lassmann H. Detection of DNA fragmentation in apoptosis: application of in situ nick translation to cell culture systems and tissue sections. J Histochem Cytochem. 1993;41:1023–1030. doi: 10.1177/41.7.8515045. [DOI] [PubMed] [Google Scholar]
- Gow A, Lazzarini RA. A cellular mechanism governing the severity of Pelizaeus-Merzbacher disease. Nat Genet. 1996;13:422–428. doi: 10.1038/ng0896-422. [DOI] [PubMed] [Google Scholar]
- Gow A, Friedrich VL, Jr, Lazzarini RA. Intracellular transport and sorting of the oligodendrocyte transmembrane proteolipid protein. J Neurosci Res. 1994a;37:563–573. doi: 10.1002/jnr.490370503. [DOI] [PubMed] [Google Scholar]
- Gow A, Friedrich VL, Jr, Lazzarini RA. Many naturally occurring mutations of myelin proteolipid protein impair its intracellular transport. J Neurosci Res. 1994b;37:574–583. doi: 10.1002/jnr.490370504. [DOI] [PubMed] [Google Scholar]
- Gow A, Friedrich VL, Jr, Lazzarini RA. Apoptosis of myelin basic protein-positive oligodendrocytes in JIMPYMSDmice. J Neurochem. 1995;64:S44. [Google Scholar]
- Gow A, Gragerov A, Gard A, Colman DR, Lazzarini RA. Conservation of topology, but not conformation, of the proteolipid proteins of the myelin sheath. J Neurosci. 1997;17:181–189. doi: 10.1523/JNEUROSCI.17-01-00181.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravel M, Peterson J, Yong VW, Kottis V, Trapp B, Braun PE. Overexpression of 2′,3′-cyclic nucleotide 3′-phosphodiesterase in transgenic mice alters oligodendrocyte development and produces aberrant myelination. Mol Cell Neurosci. 1996;7:453–466. doi: 10.1006/mcne.1996.0033. [DOI] [PubMed] [Google Scholar]
- Guthrie S, Pini A. Chemorepulsion of developing motor axons by the floor plate. Neuron. 1995;14:1117–1130. doi: 10.1016/0896-6273(95)90260-0. [DOI] [PubMed] [Google Scholar]
- Harding B, Ellis D, Malcolm S. A case of Pelizaeus-Merzbacher disease showing increased dosage of the proteolipid protein gene. Neuropathol Appl Neurobiol. 1995;21:111–115. doi: 10.1111/j.1365-2990.1995.tb01036.x. [DOI] [PubMed] [Google Scholar]
- Hodes M, Pratt V, Dlouhy S. Genetics of Pelizaeus-Merzbacher disease. Dev Neurosci. 1993;15:383–394. doi: 10.1159/000111361. [DOI] [PubMed] [Google Scholar]
- Hodes ME, Dlouhy SR. The proteolipid protein gene: double, double, ... and trouble. Am J Genet. 1996;59:12–15. [PMC free article] [PubMed] [Google Scholar]
- Hudson, L.D., and N.L. Nadon. 1992. Amino acid substitutions in proteolipid protein that cause dysmyelination. In Myelin: Biology and Chemistry. R.E. Martenson, editor. CRC Press, Boca Raton, FL. 677–702.
- Ikenaka K, Kagawa T. Transgenic systems in studying myelin gene expression. Dev Neurosci. 1995;17:127–136. doi: 10.1159/000111281. [DOI] [PubMed] [Google Scholar]
- Inoue K, Osaka H, Sugiyama N, Kawanishi C, Onishi H, Nezu A, Kimura K, Kimura S, Yamada Y, Kosaka K. A duplicated PLPgene causing Pelizaeus-Merzbacher disease detected by comparative multiplex PCR. Am J Genet. 1996a;59:32–39. [PMC free article] [PubMed] [Google Scholar]
- Inoue Y, Kagawa T, Matsumura Y, Ikenaka K, Mikoshiba K. Cell death of oligodendrocytes or demyelination induced by overexpression of proteolipid protein depending on expressed gene dosage. Neurosci Res. 1996b;25:161–172. doi: 10.1016/0168-0102(96)01039-5. [DOI] [PubMed] [Google Scholar]
- Johnson GD, Davidson RS, McNamee KC, Russell G, Goodwin D, Holborow EJ. Fading of immunofluorescence during microscopy: a study of the phenomenon and its remedy. J Immunol Methods. 1982;55:231–242. doi: 10.1016/0022-1759(82)90035-7. [DOI] [PubMed] [Google Scholar]
- Johnson RS, Roder JC, Riordan JR. Over-expression of the DM-20 myelin proteolipid causes central nervous system demyelination in transgenic mice. J Neurochem. 1995;64:967–976. doi: 10.1046/j.1471-4159.1995.64030967.x. [DOI] [PubMed] [Google Scholar]
- Kagawa T, Ikenaka K, Inoue Y, Kuriyama S, Tsujii T, Nakao J, Nakajima K, Aruga J, Okano H, Mikoshiba K. Glial cell degeneration and hypomyelination caused by overexpression of myelin proteolipid protein gene. Neuron. 1994;13:427–442. doi: 10.1016/0896-6273(94)90358-1. [DOI] [PubMed] [Google Scholar]
- Klugmann M, Schwab MH, Pullhofer A, Schneider A, Zimmermann F, Griffiths IR, Nave K-A. Assembly of CNS myelin in the absence of proteolipid protein. Neuron. 1997;18:59–70. doi: 10.1016/s0896-6273(01)80046-5. [DOI] [PubMed] [Google Scholar]
- Knapp P, Skoff R, Redstone D. Oligodendroglial cell death in jimpymice: an explanation for the myelin deficit. J Neurosci. 1986;6:2813–2822. doi: 10.1523/JNEUROSCI.06-10-02813.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H, Hoffman E, Marks H. The rumpshakermutation in spastic paraplegia. Nat Genet. 1994;7:351–352. doi: 10.1038/ng0794-351. [DOI] [PubMed] [Google Scholar]
- Koeppen, A. 1992. Pelizaeus-Merzbacher disease: X-linked proteolipid protein deficiency in the human central nervous system. In Myelin: Biology and Chemistry. R. Martenson, editor. CRC Press, Boca Raton, FL. 703–721.
- Macklin WB, Trapp BD, Kagawa T, Ikenaka K. Hypomyelination in transgenic mice with multiple copies of the myelin proteolipid protein gene. J Neurochem. 1995;64:S86. [Google Scholar]
- Mastronardi FG, Ackerley CA, Arsenault L, Roots BI, Moscarello MA. Demyelination in a transgenic mouse: a model for multiple sclerosis. J Neurosci Res. 1993;36:315–324. doi: 10.1002/jnr.490360309. [DOI] [PubMed] [Google Scholar]
- McKusick, V.A. 1994. Mendelian Inheritance in Man. (11th edition). Johns Hopkins University Press, Baltimore, MD. 3009 pp.
- Milner R, Lai C, Nave K, Lenoir D, Ogata J, Sutcliffe G. Nucleotide sequences of two mRNAs for rat brain myelin proteolipid protein. Cell. 1985;42:931–939. doi: 10.1016/0092-8674(85)90289-2. [DOI] [PubMed] [Google Scholar]
- Mitchell LS, Gillespie SC, McAllister F, Fanarraga ML, Kirkham D, Kelly B, Brophy PJ, Griffiths IR, Montague P, Kennedy PG. Developmental expression of major myelin protein genes in the CNS of X-linked hypomyelinating mutant rumpshaker. J Neurosci Res. 1992;33:205–217. doi: 10.1002/jnr.490330204. [DOI] [PubMed] [Google Scholar]
- Nadon NL, Duncan ID. Gene expression and oligodendrocyte development in the myelin deficient rat. J Neurosci Res. 1995;41:96–104. doi: 10.1002/jnr.490410111. [DOI] [PubMed] [Google Scholar]
- Nadon NL, Arnheiter H, Hudson LD. A combination of PLP and DM20 transgenes promotes partial myelination in the jimpymouse. J Neurochem. 1994;63:822–833. doi: 10.1046/j.1471-4159.1994.63030822.x. [DOI] [PubMed] [Google Scholar]
- Nishiyama A, Lin XH, Giese N, Heldin CH, Stallcup WB. Co-localization of NG2 proteoglycan and PDGF α-receptor on O-2A progenitor cells in the developing rat brain. J Neurosci Res. 1996;43:299–314. doi: 10.1002/(SICI)1097-4547(19960201)43:3<299::AID-JNR5>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Nussbaum JL, Roussel G. Immunocytochemical demonstration of the transport of myelin proteins through the Golgi apparatus. Cell Tiss Res. 1983;234:547–559. doi: 10.1007/BF00218650. [DOI] [PubMed] [Google Scholar]
- Owen C, Skoff RP. Glial cells in myelin deficient jimpy mice in situdo not show increased DNA fragmentation compared with normals. J Neurochem. 1995;64:S63C. [Google Scholar]
- Power C, Kong P, Trapp B. Major histocompatibility class I expression in oligodendrocytes induces hypomyelination in transgenic mice. J Neurosci Res. 1996;44:165–173. doi: 10.1002/(SICI)1097-4547(19960415)44:2<165::AID-JNR9>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Puckett C, Hudson L, Ono K, Friedrich V, Benecke J, Dubois-Dalcq M, Lazzarini R. Myelin-specific proteolipid protein is expressed in myelinating Schwann cells but is not incorporated into myelin sheaths. J Neurosci Res. 1987;18:511–518. doi: 10.1002/jnr.490180402. [DOI] [PubMed] [Google Scholar]
- Readhead C, Schneider A, Griffiths I, Nave K-A. Premature arrest of myelin formation in transgenic mice with increased proteolipid protein gene dosage. Neuron. 1994;12:583–595. doi: 10.1016/0896-6273(94)90214-3. [DOI] [PubMed] [Google Scholar]
- Rosenbluth J, Stoffel W, Schiff R. Myelin structure in proteolipid protein (PLP)-null mouse spinal cord. J Comp Neurol. 1996;371:336–344. doi: 10.1002/(SICI)1096-9861(19960722)371:2<336::AID-CNE12>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Rosenfeld J, Freidrich VL., Jr Axonal swellings in jimpy mice: does lack of myelin cause neuronal abnormalities? . Neuroscience. 1983;10:959–966. doi: 10.1016/0306-4522(83)90233-6. [DOI] [PubMed] [Google Scholar]
- Roussel G, Meskovic N, Trifilieff E, Artault J-C, Nussbaum J-L. Arrest of proteolipid transport through the Golgi apparatus in jimpybrain. J Neurocytol. 1987;16:195–204. doi: 10.1007/BF01795303. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning. A Laboratory Manual. Cold Spring Harbor Press, Cold Spring Harbor, NY.
- Schneider A, Montague P, Griffiths I, Fanarraga M, Kennedy P, Brophy P, Nave K-A. Uncoupling of hypomyelination and glial cell death by a mutation in the proteolipid protein gene. Nature. 1992;358:758–761. doi: 10.1038/358758a0. [DOI] [PubMed] [Google Scholar]
- Schneider A, Griffiths I, Readhead C, Nave K-A. Dominant-negative action of the jimpymutation in mice complemented with an autosomal transgene for myelin proteolipid protein. Proc Natl Acad Sci USA. 1995;92:4447–4451. doi: 10.1073/pnas.92.10.4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwob VS, Clark HB, Agrawal D, Agrawal HC. Electron microscopic immunocytochemical localization of myelin proteolipid protein and myelin basic protein to oligodendrocytes in rat brain during myelination. J Neurochem. 1985;45:559–571. doi: 10.1111/j.1471-4159.1985.tb04024.x. [DOI] [PubMed] [Google Scholar]
- Seitelberger, F. 1970. Pelizaeus-Merzbacher disease. In Handbook of Clinical Neurology: Leukodystrophies and Polydystrophies. Vol. 10. P. Vinken and G. Bruyn, editors. North-Holland, Amsterdam. 150–202.
- Shamu CE, Cox JS, Walter P. The unfolded-protein-response pathway in yeast. Trends Cell Biol. 1994;4:56–60. doi: 10.1016/0962-8924(94)90011-6. [DOI] [PubMed] [Google Scholar]
- Sistermans EA, de Wijs IJ, de Coo RFM, Smit LME, Menko FH, van Oost BA. A (G-to-A) mutation in the initiation codon of the proteolipid protein gene causing a relatively mild form of Pelizaeus-Merzbacher disease in a Dutch family. Hum Genet. 1996;97:337–339. doi: 10.1007/BF02185767. [DOI] [PubMed] [Google Scholar]
- Skoff, R., and P. Knapp. 1992. Phenotypic expression of X-linked genetic defects affecting myelination. In Myelin: Biology and Chemistry. R. Martenson, editor. CRC Press, Boca Raton, FL. 653–676.
- Skoff RP. Programmed cell death in the dysmyelinating mutants. Brain Pathol. 1995;5:283–288. doi: 10.1111/j.1750-3639.1995.tb00605.x. [DOI] [PubMed] [Google Scholar]
- Strable U, Blader P, Adam J, Ingham PW. A simple and efficient procedure for non-isotopic in situ hybridization to sectioned material. Trends Genet. 1994;10:75–76. doi: 10.1016/0168-9525(94)90221-6. [DOI] [PubMed] [Google Scholar]
- Taraszewska A, Zelman IB. Electron microscopic study of glia in ptrabbit during myelination. Neuropatol Pol. 1987;25:351–368. [PubMed] [Google Scholar]
- Tosic M, Gow A, Dolivo M, Domanska-Janik K, Lazzarini RA, Matthieu J-M. Proteolipid/DM20 proteins bearing the paralytic tremor mutation in peripheral nerves and transfected COS-7 cells. Neurochem Res. 1996;21:423–430. doi: 10.1007/BF02527706. [DOI] [PubMed] [Google Scholar]
- Tosic M, Matthey B, Gow A, Lazzarini RA, Matthieu J-M. Intracellular transport of the DM-20 bearing shaking pup (shp)mutation and its possible phenotypic consequences. J Neurosci Res. 1997;50:844–852. doi: 10.1002/(SICI)1097-4547(19971201)50:5<844::AID-JNR20>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Turnley AM, Morahan G, Okano H, Bernard O, Mikoshiba K, Allison J, Bartlett PF, Miller JAFP. Dysmyelination in transgenic mice resulting from expression of class I histocompatibility molecules in oligodendrocytes. Nature. 1991;353:566–569. doi: 10.1038/353566a0. [DOI] [PubMed] [Google Scholar]
- Watanabe I, Patel V, Goebel HH, Siakotos AN, Zeman W, DeMyer W, Dyer JS. Early lesion of Pelizaeus-Merzbacher disease: electron microscopic and biochemical study. J Neuropath Exp Neurol. 1973;32:313–333. doi: 10.1097/00005072-197304000-00010. [DOI] [PubMed] [Google Scholar]
- Webster, H., deF, and N.H. Sternberger. 1980. Morphological features of myelin formation. In Neurological Mutations Affecting Myelination. N. Baumann, editor. Elsevier, Amsterdam. 73–86.
- Williams WC, II, Gard AL. DNA fragmentation is not a prominent feature of oligodendrocyte death in the jimpy mutant. J Neurochem. 1995;64:S98C. [Google Scholar]
- Wu Y, Whitman I, Molmenti E, Moore K, Hippenmeyer P, Perlmutter D. A lag in intracellular degradation of mutant α1-antitrypsin correlates with the liver disease phenotype in homozygous PiZZ α1-antitrypsin deficiency. Proc Natl Acad Sci USA. 1994;91:9014–9018. doi: 10.1073/pnas.91.19.9014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamura T, Konola JT, Wekerle H, Lees MB. Monoclonal antibodies against myelin proteolipid protein: identification and characterization of two major determinants. J Neurochem. 1991;57:1671–1680. doi: 10.1111/j.1471-4159.1991.tb06367.x. [DOI] [PubMed] [Google Scholar]
- Yang X, Skoff RP. Proteolipid protein regulates the survival and differentiation of oligodendrocytes. J Neurosci. 1997;17:2056–2070. doi: 10.1523/JNEUROSCI.17-06-02056.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka T, Feigenbaum L, Jay G. Transgenic mouse model for central nervous system demyelination. Mol Cell Biol. 1991;11:5479–5486. doi: 10.1128/mcb.11.11.5479. [DOI] [PMC free article] [PubMed] [Google Scholar]