

Abstract
Abstract. Carcinoma cells selected for their ability to migrate in vitro showed enhanced invasive properties in vivo. Associated with this induction of migration was the anchorage-dependent phosphorylation of p130CAS (Crk-associated substrate), leading to its coupling to the adaptor protein c-CrkII (Crk). In fact, expression of CAS or its adaptor protein partner Crk was sufficient to promote cell migration, and this depended on CAS tyrosine phosphorylation facilitating an SH2-mediated complex with Crk. Cytokine-stimulated cell migration was blocked by CAS lacking the Crk binding site or Crk containing a mutant SH2 domain. This migration response was characterized by CAS/Crk localization to membrane ruffles and blocked by the dominant-negative GTPase, Rac, but not Ras. Thus, CAS/Crk assembly serves as a “molecular switch” for the induction of cell migration and appears to contribute to the invasive property of tumors.
During wound repair and inflammation, cells can be induced to migrate by cytokines or adhesive proteins within the provisional extracellular matrix (ECM)1 (Clark et al., 1996). Recent evidence indicates that cytokine receptors as well as integrins transmit signals in response to extracellular cues (for reviews see Schwartz et al., 1995; Parsons, 1996). While these biochemical signals are likely to be involved in the induction of cell migration, little is known about the actual signal transduction events responsible for this process or how they impact the cells' migration machinery.
Ultimately, the biochemical signals responsible for controlling cell migration must impact adhesion and deadhesion of integrin receptors as well as organization of the actin and myosin cytoskeleton since these events are critical for movement (Huttenlocher et al., 1996). However, the complexity of cell motility suggests that multiple signaling mechanisms exist to regulate this process. For example, Ras/MAP kinase (ERK1 and 2) signaling has been shown to promote phosphorylation of myosin light chain kinase leading to activation of the actin/myosin motor and cell migration (Klemke et al., 1997), whereas the Rho family of small GTPases (i.e., Rac, Rho, Cdc42) control actin organization associated with cell motility (Ridley et al., 1992; Takaishi et al., 1993; Hotchin and Hall, 1995; Michiels et al., 1995). Furthermore, the adhesion-dependent signaling molecules focal adhesion kinase (FAK) and c-src may be involved in focal contact disassembly during cell migration (Ilic et al., 1995; Rodier et al., 1995; Cary et al., 1996; Gilmore and Romer, 1996; Parsons, 1996; Hanks and Polte, 1997). Phosphatidylinositol 3-kinase (Rodriguez- Viciana et al., 1997) and PLCγ (Chen et al., 1994) are also thought to regulate cell migration. Therefore, it is likely that cell migration depends on the coordinate regulation of a number of these signaling events.
Ligation of integrin or cytokine receptors promote a cascade of biochemical signals, including the activation of tyrosine kinases leading to phosphorylation of multiple cellular substrates. A family of adaptor proteins (Nck, Crk, Grb2), which consist primarily of src homology 2 (SH2) and 3 (SH3) domains, coordinate these biochemical events by assembling signal-generating complexes. For example, the SH2 domains of these adaptor proteins bind specifically to phosphotyrosine-binding sites in many signaling molecules, while the SH3 domains are critical for coupling to effector molecules and targeting of signaling complexes to discrete sites within the cell (Bar-Sagi et al., 1993).
The adaptor protein p130CAS (Crk-associated substrate) is a member of a family of structurally related proteins (i.e., Efs/Sin and Hef 1) that was originally identified as a prominent tyrosine-phosphorylated protein in v-crk– and v-src–transformed cells (Sakai et al., 1994; Greulich and Hanafusa, 1996; Matsuda and Kurata, 1996). CAS contains an SH3 domain, two proline-rich regions, and a substrate domain consisting of 15 potential SH2-binding motifs (Sakai et al., 1994). C-Crk II (Crk) is an adaptor protein that contains an SH2 and two SH3 domains capable of interacting with numerous effector molecules, including tyrosine-phosphorylated CAS (for reviews see Feller et al., 1994a ; Matsuda and Kurata, 1996). In fact, 9 of the 15 tyrosine phosphorylation sites present in the substrate domain of CAS conform to the SH2-binding motif for Crk (YD(V/T)P, suggesting that Crk is the primary “docking protein” for CAS. However, Nck also binds to CAS and may play a role in regulation of CAS activity (Schlaepfer et al., 1997). Recently, it was reported that cell adhesion to ECM proteins promotes FAK and c-src kinase activity leading to tyrosine phosphorylation of CAS and its association with Crk or Nck (Vuori et al., 1996; Schlaepfer et al., 1997). While this suggests a role for these molecules in mediating cellular responses to the ECM, the biological consequences of this signaling event are not known.
In this report, we investigate the role of CAS and Crk in regulation of cell migration on the ECM. Evidence is provided that tyrosine phosphorylation of CAS is associated with induction of migration and enhanced invasive potential of carcinoma cells in vivo. This migration response was shown to depend on the assembly of a CAS/Crk adaptor protein complex, and the resulting migration was disrupted by a dominant-negative form of Rac, but not Ras. These findings suggest that formation of a CAS/Crk adaptor protein complex serves as a “molecular switch” facilitating a Rac-dependent cell migration response on the extracellular matrix.
Materials and Methods
Cells and Cell Culture
Previous reports have described the FG human pancreatic carcinoma cell adhesive and migratory properties and integrin profile (Klemke et al., 1994). FG-M is a cell line derived from FG cells that are migration competent on vitronectin. To obtain these cells, FG cells were serum starved for 48 h before being removed from the culture dish with Hanks' balanced salt solution containing 5 mM EDTA and 25 mM Hepes, pH 7.2, and allowed to migrate for 24 h on vitronectin-coated (10 μg/ml in PBS, pH 7.4) migration wells as previously reported (Klemke et al., 1994). The migratory cells on the underside of the membrane were removed with trypsin/ EDTA solution (GIBCO BRL, Gaithersburg, MD) and cultured in RPMI 10% FCS until enough cells were obtained to repeat the process. After 10 rounds of selection, we obtained a stable cell line (FG-M cells) that spontaneously migrates on vitronectin to levels comparable to that of parental FG cells on collagen. FG and FG-M cells were grown in RPMI 1640 (GIBCO BRL) supplemented with 10% FBS and 50 μg/ml gentamicin and were free from mycoplasma during these studies. COS-7 cells were grown in DME with 10% FBS and 50 μg/ml gentamicin. Before testing, all cells were starved for 24–48 h by replacing serum-containing culture media with FBS-free media.
Expression Vectors and Constructs
The expression plasmid pUCCAGGS containing full-length CrkII as well as c-CrkII cDNA in which either the amino-terminal SH3 domain (tryptophan 109 to cysteine) or the SH2 domain (arginine 38 to valine) were mutated have been described previously (Matsuda et al., 1993, 1994; Tanaka et al., 1993). The pEBG expression plasmid containing glutathione-S-transferase (gst)-tagged or untagged wild-type p130CAS or CAS with an in frame deletion of its substrate domain (CAS-SD, amino acids [aa] 213–514) has been described previously (Mayer et al., 1995). Myc-tagged dominant-positive Rac1 (Q61L) or dominant-negative Rac 1 (N17) in pcDNA3, and dominant-negative Ras (N17) in pCMV5 vectors have been described previously (Minden et al., 1995).
Antibodies and Reagents
Anti-Crk was from Transduction Laboratories (Lexington, KY). Anti-CAS used for Western blotting and immunofluorescent staining was from Transduction Laboratories, and anti-CAS antibodies for immunoprecipitation experiments were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Anti-myc antibody 9E10 and anti-Ras (259) was from Santa Cruz Biotechnology. Anti-gst and antiphosphotyrosine monoclonal antibody (4G10) was from Upstate Biotechnology, Inc. (Lake Placid, NY). Goat anti–rabbit and anti–mouse antibodies were from Bio-Rad Labs (Hercules, CA). Anti-FAK antibody BC3 was kindly provided by Dr. J. Thomas Parsons (University of Virginia Medical Center, Charlottesville, VA). Human recombinant EGF, IGF-1, and insulin were obtained from Genzyme (Cambridge, MA).
Adhesive Ligands
Vitronectin was prepared as described by Yatohgo et al. (1988). Fibronectin and collagen type I were obtained from Upstate Biotechnology, Inc.
Transfection of COS-7 and FG -M Cells
Transient transfection of COS cells was performed as previously described (Klemke et al., 1997). Briefly, to investigate the role of CAS or Crk in cell migration, COS-7 cells (0.75 × 106 cells/10-cm plate) were cotransfected with lipofectamine (20 μl/10-cm plate; GIBCO BRL) and 1.5 μg of the expression vector containing the cDNA encoding wild-type and/or mutant forms of c-Crk or CAS, along with 0.5 μg of a reporter construct encoding β-galactosidase (pSV–β-galactosidase; Promega Corp., Madison, WI). In some cases, cells were transfected with the expression vectors encoding wild-type CAS and Crk (1.5 μg of each construct per plate) along with dominant-negative Rac construct (1 μg/plate) or Ras (0.25 μg/plate) as described above. Mock cells were transfected as described above with the appropriate amount of the empty expression vectors. Cells were allowed to incorporate the cDNA constructs for 6–8 h, washed, and then allowed to incubate for 40 h, which provides optimal transient expression. FG-M cells (1 ×106 cells/10-cm dish) were cotransfected with lipofectamine (40 μl/10-cm plate) and 10 μg of the expression vector containing CAS without its substrate domain (CAS-SD) or the empty expression vector along with the β-galalctosidase reporter construct (10 μg/10-cm plate). Cells were allowed to incorporate the cDNAs for 16 h, washed, and then incubated for 36 h. COS and FG-M cells were then prepared for cell migration or analyzed for expression of specific proteins as described below. Cells cotransfected with β-galactosidase were developed using X-gal as a substrate according to the manufacturer's recommendation (Promega Corp.). In typical transfection experiments of FG-M cells, we achieved 10–15% expression efficiency, and in COS-7 cells we obtained 40–60% efficiency providing a 10–30-fold increase in the level of Crk, 10–15-fold increase in CAS, or 10–30-fold increase in Rac and Ras compared with endogenous levels of these proteins as determined by Western blotting.
Haptotaxis Migration Assays
Cell adhesion and migration assays were performed as previously described with minor modifications (Klemke et al., 1997). Briefly, cell migration assays were performed using modified Boyden chambers (tissue culture–treated, 6.5-mm diameter, 10-μm thickness, 8-μm pores, Transwell®; Costar Corp., Cambridge, MA) containing polycarbonate membranes coated on the underside of the membrane with 10 μg/ml vitronectin, fibronectin, or collagen type I in PBS for 2 h at 37°C, rinsed once with PBS, and then placed into the lower chamber containing 500 μl migration buffer (fibroblast basal medium [FBM] with 0.5% BSA; Clonetics, San Diego, CA). Serum-starved cells were removed from culture dishes with Hanks' balanced salt solution containing 5 mM EDTA and 25 mM Hepes, pH 7.2, and 0.01% trypsin, washed twice with migration buffer, and then resuspended in FBM 0.5% BSA (106 cells/ml). 50,000–100,000 cells were then added to the top of each migration chamber and allowed to migrate to the underside of the top chamber for various times in the presence or absence of either insulin (25 μg/ml), insulin-like growth factor 1 (IGF-1; 20 ng/ml), or epidermal growth factor (50 ng/ml), which had been added to the lower chamber. The nonmigratory cells on the upper membrane surface were removed with a cotton swab, and the migratory cells attached to the bottom surface of the membrane stained with X-gal substrate as described above (for β-galactosidase–expressing cells) or 0.1% crystal violet in 0.1 M borate, pH 9.0, and 2% ethanol for 20 min at room temperature. The number of migratory cells per membrane were either counted with an inverted microscope using a 40× objective, or the stain was eluted with 10% acetic acid, the absorbance was determined at 600 nm, and migration was enumerated from a standard curve. Each determination represents the average of three individual wells, and error bars represent the standard deviation (SD). All values have had background subtracted, which represents cell migration on membranes coated on the bottom with BSA (1%). In control experiments, cell migration on BSA was less than 0.01% of the total cell population.
Chick Embryo Metastasis Assay
The chick embryo metastasis assay was performed as previously described (Brooks et al., 1997). Briefly, FG or FG-M carcinoma cells (5–10 × 106) were inoculated on the back of the chorioallantoic membrane of a 9-d-old chick embryo and allowed to incubate for 7–9 d. Embryos were then killed, tumors were excised, and the weights were determined. Pulmonary metastasis was quantified by determining the percentage of human cells present in a single cell suspension of whole lung tissue by flow cytometry as described by Brooks et al. (1997).
Cell Adhesion and Transfection Efficiency
Cell adhesion was performed according to Klemke et al. (1994) with minor modifications. Briefly, 48-well cluster plates (polystyrene, non-tissue culture–treated; Costar, Cambridge, MA) were coated with either 10 μg/ml vitronectin, fibronectin, or collagen 1 in PBS, pH 7.4. Proteins were allowed to bind for 2 h at 37°C before the wells were rinsed and blocked for 1 h with 1% heat-denatured BSA (RIA grade; Sigma Chemical Co., St. Louis, MO) in PBS. Cells were treated and harvested as for the migration assay and added to the wells at a concentration of 100,000 cells/0.1 ml FBM-BSA. Nonadherent cells were removed by washing, and the adherent cells were stained with crystal violet and enumerated from dye uptake or stained with X-gal substrate and counted as described above. Each data point was calculated from triplicate wells and was expressed as the mean ± SD. Nonspecific cell adhesion as measured on BSA-coated wells has been subtracted.
Immunoprecipitation and Immunoblotting of Proteins
Immunoprecipitation and immunoblotting of proteins was performed as previously described (Klemke et al., 1997). Briefly, cells were rinsed twice with PBS and then lysed with either RIPA buffer for Western blotting of total cellular proteins or NP-40 buffer (50 mM Tris, pH 7.5, containing 150 mM NaCl, 5 mM EDTA, 1 mM sodium orthovanadate, a cocktail of protease inhibitors [Boehringer Mannheim cocktail tablet; Boehringer Mannheim Corp., Indianapolis, IN], and 1% NP-40) for immunoprecipitation of cellular proteins for 1 h at 4°C. The protein concentration of the samples were normalized before either direct immunoblotting of total cell proteins or immunoprecipitation of equivalent amounts of proteins with Crk, CAS, or gst antibodies bound to protein A– or G–Sepharose beads. Samples were analyzed by immunoblot analysis using antibodies to Crk, CAS, gst, or antiphosphotyrosine monoclonal antibody (4G10) and horseradish peroxidase–conjugated goat anti–mouse or rabbit secondary antibodies and the ECL system. In some cases, the immunoblots were stripped and reprobed with antibodies according to the manufacturer's recommendations (ECL; Amersham Corp., Arlington Heights, IL).
Immunofluorescent Staining and Confocal Microscopy
COS-7 cells either transfected with CAS and Crk or mock-transfected cells were removed from the culture dish and resuspended in FBM (0.5% BSA) as described above and allowed to attach to glass coverslips coated with 10 μg/ml vitronectin for 2 h at 37 °C. In some cases, cells grown on glass coverslips were serum-starved for 24 h and then treated with or without insulin (25 μg/ml) or IGF-1 (20 ng/ml) for 15 min. Cells were fixed in 4% paraformaldehyde, permeablized with 0.2% Triton X-100, and stained with rhodamine-conjugated phalloidin and anti-Crk or anti-CAS antibodies followed by FITC-conjugated goat anti–mouse or rabbit secondary antibodies. Cell fluorescence was analyzed with a laser confocal microscope (model 1024; Bio-Rad Labs) and a Zeiss Axiovert microscope (Thornwood, NY) focused at the cell–substratum interface.
Results
Selection of FG Pancreatic Carcinoma Cells with Increased Migration In Vitro and Metastatic Properties In Vivo
While cell adhesion to the extracellular matrix is required for cell migration, it is not sufficient. For example, FG pancreatic carcinoma cells attach to vitronectin but fail to migrate on this adhesive ligand, whereas, they readily migrate on collagen (Klemke et al., 1994). This may be explained by differences in adhesion-dependent signaling events that ultimately facilitate cell movement. To identify putative signaling molecules that are associated with the migratory phenotype, FG cells were allowed to attach to either collagen or vitronectin and examined for differences in their phosphoprotein profile. As shown in Fig. 1, cell attachment to collagen, a migration-competent ligand, resulted in significantly enhanced tyrosine phosphorylation of a 70- and 125–130-kD protein(s), whereas, cell adhesion to vitronectin, which does not support FG cell migration, showed only little tyrosine phosphorylation of these proteins.
Figure 1.

FG cell migration on collagen is associated with increased tyrosine phosphorylation of p125–130 proteins. Serum-starved FG cells were held in suspension (0 time point) or allowed to attach to Petri dishes coated with collagen (COLL) or vitronectin (VN) for various times before being lysed and examined for changes in tyrosine phosphorylation of total cellular proteins by phosphotyrosine immunoblotting as described in Materials and Methods. Cell migration was determined in modified Boyden chambers (Transwells) coated with either collagen or vitronectin as described in Materials and Methods. Cell migration on collagen (+) was greater than 50% of the cell population in 24 h, while on vitronectin less than 1.0% of the cells migrated.
To determine whether the phosphorylation status of the 125–130-kD phosphoprotein(s) was associated with the migratory phenotype of these cells, we first selected a stable population of FG cells that were migration competent on vitronectin since a small percentage of the parental FG cells (<0.05%) were capable of movement on this substrate. After 10 rounds of selection, a stable population was obtained (FG-M cells) that showed spontaneous motility on vitronectin that was comparable to FG cell migration on collagen (Fig. 2 A). While FG-M cells gained the ability to migrate on vitronectin, there was no difference in their integrin profile or their capacity to adhere to vitronectin (Fig. 2 A) or a number of other extracellular matrix proteins (data not shown). Therefore, FG-M cells represent a stable migratory subpopulation of the parental FG cells.
Figure 2.
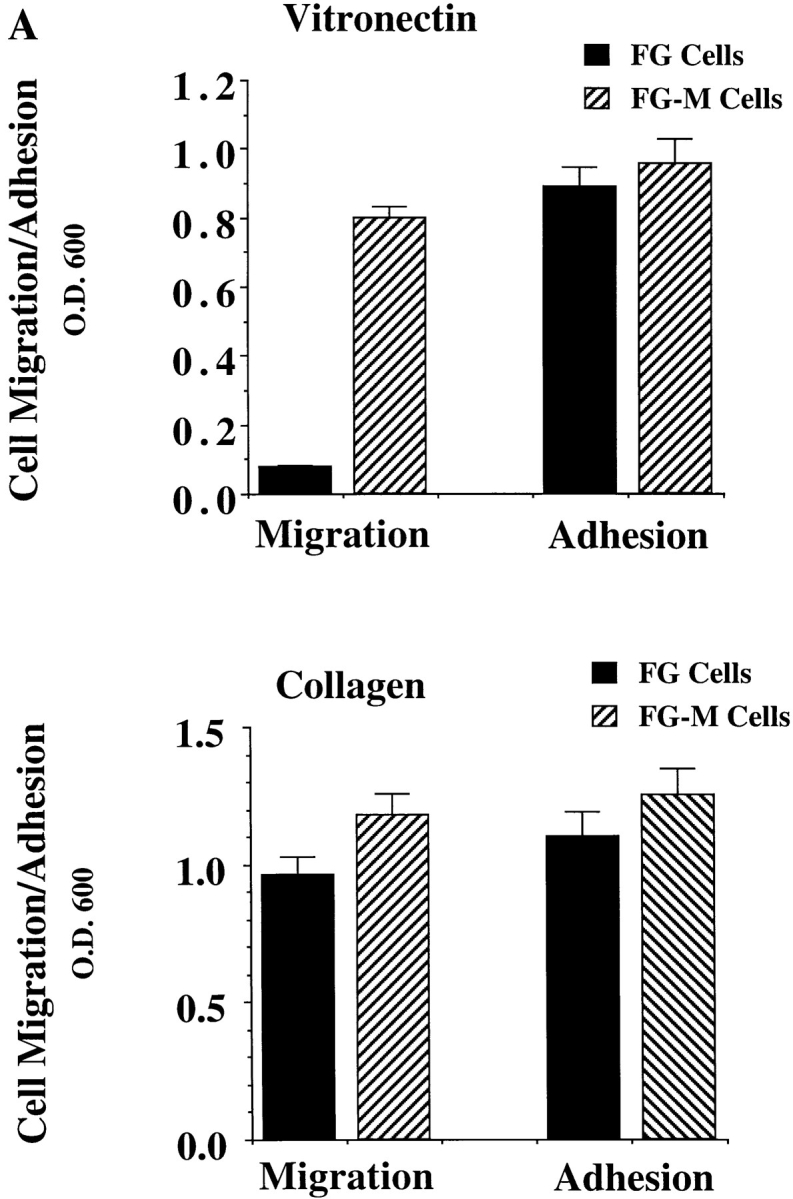
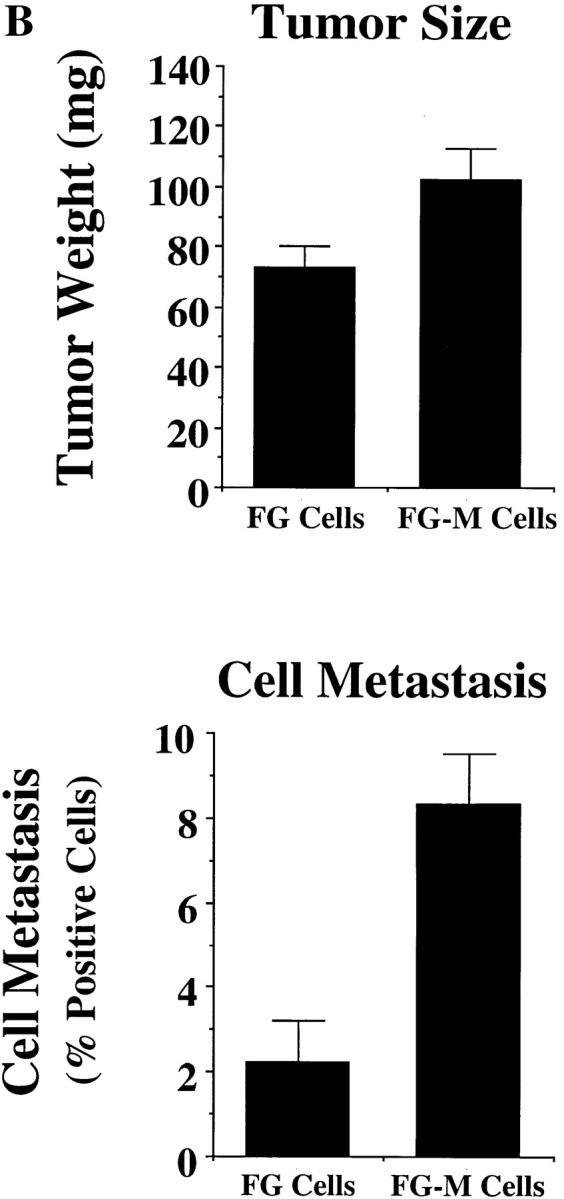
Comparison of FG and FG-M cell migration in vitro and metastasis in vivo. (A) FG-M cells are a stable cell line derived from FG cells selected for their ability to migrate on a vitronectin substrate as described in Materials and Methods. Cells were allowed to adhere for 30 min to Petri dishes or migrate for 4 h on Transwell migration chambers coated with vitronectin (top) or collagen (bottom) and quantified as described in Materials and Methods. Each bar represents the mean ± SD of triplicate migration wells of one of three representative experiments. (B) FG or FG-M carcinoma cells (5 × 106) inoculated onto chorioallantoic membrane of 9-d-old chick embryos were allowed to form tumors for 9–10 d, at which time the tumors were resected and weighed (top). Pulmonary metastasis (bottom) was measured as described in Materials and Methods. Tumor weight is the mean ± SEM. Percent positive metastasis is the mean ± SEM of the relative percentage of tumor cells in the lungs of the chick embryos. FG-M cells showed a statistically significant increase in metastasis over FG cells (P < 0.01) as determined by Student's t test.
Cell migration on vitronectin in vitro has been linked to the metastatic capacity of tumor cells in vivo (Nip et al., 1992; Brooks et al., 1997). Therefore, we compared FG and FG-M cells for their ability to metastasize spontaneously from the chick chorioallantoic membrane to the lungs of 10-d-old chick embryos. As shown in Fig. 2 B (bottom), FG-M cells showed a fourfold increase in spontaneous metastasis compared with FG cells. However, there was only a slight increase in the average FG-M primary tumor weight (1.4-fold) compared with primary tumors formed by FG cells (Fig. 2 B, top). Furthermore, FG and FG-M cells had identical growth rates in vitro (data not shown). These findings suggest that the enhanced migratory properties of FG-M cells in vitro are primarily associated with their increased metastatic properties in vivo.
FG-M Cells Show Increased CAS Tyrosine Phosphorylation
To establish a possible role of the p125–130 protein(s) in the migratory capacity of FG-M cells, we compared the phosphoprotein profile of FG and FG-M cells during their attachment to vitronectin. As shown in Fig. 3 A, FG-M, but not FG cell attachment to vitronectin, resulted in the tyrosine phosphorylation of proteins with a molecular mass of 125–130 kD. This phosphorylation pattern appeared similar to what was observed between FG cells attached to collagen vs. vitronectin and thus appeared to be associated with FG cell migration in general (Fig. 1). Together, these data demonstrate that cells selected for spontaneous migration in vitro show enhanced invasive properties in vivo as well as adhesion-dependent phosphorylation of proteins with a molecular mass of 125–130 kD.
Figure 3.



FG-M, but not FG cells, show increased tyrosine phosphorylation of p130CAS upon attachment to vitronectin. (A) Serum-starved FG or FG-M cells were held in suspension or allowed to attach to vitronectin-coated Petri dishes for various times before being lysed in detergent and analyzed for changes in tyrosine phosphorylation of total cellular proteins as described in Materials and Methods. The bracket indicates p125–130 proteins that show increased tyrosine phosphorylation in FG-M but not FG cell protein extracts. (B, top) Phosphotyrosine immunoblot of FAK immunoprecipitated from FG or FG-M cells either held in suspension (S) or allowed to attach (A) to vitronectin-coated Petri dishes for 15 min. (B, bottom) Blot was stripped and reprobed with anti-FAK antibodies to confirm equal amounts of protein were precipitated in this experiment. (C) Phosphotyrosine immunoblot of CAS immunoprecipitated from FG or FG-M cells held in suspension (S) or allowed to attach (A) to vitronectin-coated Petri dishes for 15 min. (C, bottom) The blot was reprobed with anti-CAS antibodies to confirm equal amounts of protein were precipitated in this experiment. The result shown is representative of at least three independent experiments.
Previous studies have demonstrated that p125 FAK and p130CAS are phosphorylated upon cell adhesion to matrix proteins (Ilic et al., 1995; Nojima et al., 1995; Vuori and Ruoslahti, 1995; Hanks and Polte, 1997). Recently, FAK has been linked to cell migration since cells lacking this protein show reduced cell migration in vitro (Ilic et al., 1995). Therefore, we investigated the possibility that FAK was a component of the p125–130-kD proteins phosphorylated in the FG-M cells. FAK was immunoprecipitated from vitronectin-attached FG or FG-M cells. The immunoprecipitated proteins were then analyzed for the presence of phosphotyrosine by immunoblotting. Interestingly, both FG and FG-M cells showed a similar increase in FAK tyrosine phosphorylation upon adhesion to vitronectin (Fig. 3 B). Importantly, FAK phosphorylation was not readily detectable in total cell lysates prepared from these cells since they express low levels of this protein (data not shown). While tyrosine phosphorylation of FAK appeared identical in these cells, CAS, immunoprecipitated from FG-M cells attached to vitronectin, was highly tyrosine-phosphorylated compared with CAS precipitated from FG cells (Fig. 3 C). However, CAS immunoprecipitated from FG cells attached to collagen was highly tyrosine phosphorylated, indicating that these cells have the capacity to phosphorylate this protein (data not shown). In either case, the enhanced phosphorylation was not the result of an increase in CAS expression since similar amounts of this protein were immunoprecipitated from both cell types (Fig. 3 C). These findings suggest that phosphorylation of CAS may be associated with the migratory properties of these cells.
Expression of CAS in Cells Is Sufficient to Promote Cell Migration on the ECM
To establish whether CAS played a direct role in cell motility, we expressed the wild-type form of this molecule in COS cells since these cells efficiently express proteins encoded by eukaryotic expression vectors and show negligible migration on several ECM proteins after serum starvation (Klemke et al., 1997). Transient expression of CAS in these serum-starved cells promoted a fourfold increase in their migration on vitronectin (Fig. 4 A, top) as well as on collagen and fibronectin (data not shown). Associated with this induction of migration was increased tyrosine phosphorylation of both the expressed CAS (Fig. 4 A, bottom) and endogenous CAS protein (data not shown). Importantly, expression of CAS did not alter the ability of these cells to attach to any of the substrates tested (data not shown), suggesting that CAS expression and its associated phosphorylation is sufficient to promote cell migration.
Figure 4.


Expression of CAS in cells is sufficient to induce cell migration. (A, top) Serum-starved COS cells were allowed to migrate for 3 h on vitronectin-coated membranes after transient transfection with either the empty expression vector or the expression vector containing gst-tagged wild-type CAS (gstCAS-WT) or gst-tagged CAS lacking a substrate domain (i.e., aa 213–514 and tyr-377 to tyr-414; gstCAS-SD). The number of transfected cells migrating were enumerated by counting cells on the underside of the membrane that coexpressed a β-galactosidase vector as described in Materials and Methods. Each bar represents the mean ± SD of triplicate migration wells of one of three representative experiments. Identical results were obtained with cells transfected with wild-type CAS or CAS-SD without the gst tag (data not shown). (A, middle) Phosphotyrosine immunoblot of gstCAS-WT and gstCAS-SD immunoprecipitated from COS cells treated as for the migration assay above and attached to the culture dish. (A, bottom) The blot was reprobed with anti-gst antibodies to confirm equal loading of gstCAS-WT and gstCAS-SD. (B) Serum-starved FG-M cells were allowed to migrate on vitronectin- or collagen-coated Transwell membranes after being transiently transfected with either the empty expression vector or the vector containing CAS-SD along with the β-galactosidase reporter construct as described in the Materials and Methods. Migration was enumerated as described in A above. The results shown are representative of at least three independent experiments.
The substrate domain of CAS contains most of the putative tyrosine phosphorylation sites in this molecule (tyr-377 to tyr-414) and accounts for a significant portion of its tyrosine phosphorylation (Sakai et al., 1994). To examine the role of the CAS substrate domain in CAS-mediated migration, cells were transfected with either wild-type CAS or CAS containing an in-frame deletion of its substrate domain (aa 213–514; CAS-SD). As shown in Fig. 4 A (top), cells expressing CAS-SD failed to migrate and did not show CAS tyrosine phosphorylation in response to cell attachment (Fig. 4 A, middle). In addition, transfection of FG-M cells with CAS-SD reduced their migration on vitronectin and collagen substrates by >50%. (Fig. 4 B). These findings provide further evidence that CAS signaling is involved in cell migration and suggests that CAS tyrosine phosphorylation of its substrate domain is required for CAS-induced migration.
Crk Binding to CAS Is Required for the Induction of Cell Migration
Tyrosine phosphorylation of CAS within its substrate domain is known to promote coupling to the adaptor protein Crk (Matsuda et al., 1993; Feller et al., 1994b ). In fact, 9 of the 15 tyrosine phosphorylation sites in the CAS substrate domain conform to the Crk SH2–binding motif (YD (V/T)P. To investigate the possibility that CAS binding to Crk was involved in promoting cell migration, COS cells were transfected with Crk and either wild-type CAS or CAS-SD and then allowed to migrate on a vitronectin substrate after serum starvation. As shown in Fig. 5 A (top), expression of either wild-type CAS or Crk promoted a three- to fourfold increase in cell migration compared with cells expressing an empty vector. Interestingly, coexpression of both CAS and Crk in these cells further enhanced their migratory properties resulting in a seven- to eightfold increase in cell migration (Fig. 5 A, top). However, when Crk was expressed together with CAS-SD, motility was blocked (Fig. 5 A). Similar results were obtained when cells were allowed to migrate on collagen and fibronectin, and expression of these cDNAs did not influence the ability of these cells to attach to any of these matrix proteins (data not shown). In addition, stable expression of Crk in FG cells was sufficient to promote a 10-fold increase in cell migration on vitronectin compared with mock-transfected cells (data not shown). Together, these results suggest that the interaction between Crk and CAS is critical for migration on multiple ECM proteins but not attachment of these cells and that CAS-SD serves as a dominant-negative regulator of cell migration.
Figure 5.




CAS and Crk promote cell migration. (A) Serum-starved COS cells were allowed to migrate for 3 h on vitronectin-coated membranes (top) or lysed on the culture dish and analyzed for expression of CAS (middle) and Crk (bottom) by immunoblotting after transfection with either the empty expression vector or the expression vector containing wild-type Crk (Crk-WT) and/or gstCAS-WT or gstCAS-SD. The number of transfected migratory cells that coexpressed β-galactosidase were enumerated by counting cells on the underside of the membrane as described in Materials and Methods. Each bar represents the mean ± SD of triplicate migration wells of one of three representative experiments. (B) Crk immunoprecipitated from detergent lysates prepared from COS cells transfected as for the migration assay above were analyzed by blotting with anti-gst (top) or anti-Crk antibodies (bottom). (C) gstCAS-WT or gstCAS-SD immunoprecipitated from detergent lysates prepared from COS cells treated as described in B was analyzed by immunoblotting with anti-gst (top) or anti-Crk antibodies (bottom). (D) Crk immunoprecipitated from detergent lysates prepared from FG or FG-M cells either held in suspension (S) or allowed to attach (A) to vitronectin for 15 min were analyzed by immunoblotting with anti-CAS (top) or anti-Crk antibodies (bottom). The results shown are representative of at least three independent experiments.
To establish whether the molecular interaction between CAS and Crk within these migratory cells depended on the CAS-SD domain, Crk was immunoprecipitated from an aliquot of the cells used for the migration experiments above. This immunoprecipitated material was then analyzed for the presence of tyrosine-phosphorylated CAS. As shown in Fig. 5 B, wild-type CAS, but not the CAS-SD, coprecipitated with Crk. As expected, the wild-type CAS, but not the CAS-SD, was highly tyrosine phosphorylated (data not shown). These same results were obtained when CAS was immunoprecipitated and probed for Crk association (Fig. 5 C). Endogenous Crk could also be coprecipitated with wild-type CAS but not the CAS-SD (data not shown).
To determine whether coupling of endogenous CAS/ Crk proteins was associated with the migratory phenotype, FG or FG-M cells were either held in suspension or allowed to attach to vitronectin for 15 min and examined for changes in CAS/Crk binding. As shown in Fig. 5 D, FG-M cell adhesion to vitronectin resulted in significantly increased CAS/Crk coupling compared with FG cells. These findings reveal that both endogenous and exogenously expressed CAS and Crk interact with one another through tyrosine residues present in the substrate domain of CAS and that this interaction directly influences the migratory properties of these cells.
The CRK SH2 Domain Is Required for Binding to CAS and Induction of Cell Migration
To further establish the role of Crk/CAS binding in cell migration, cells were cotransfected with wild-type CAS and Crk with a point mutation in the SH2 domain (Crk-SH2) that prevents its binding to CAS (Matsuda et al., 1993; Tanaka et al., 1993). These cells were then tested for migration and their ability to assemble a CAS/Crk complex. Expression of this mutant Crk with wild-type CAS not only failed to induce cell migration but was also unable to couple to CAS (Fig. 6, A and B), indicating that Crk-SH2 served as dominant-negative in this case. Importantly, while these cells were deficient in migration, they showed no differences in their ability to attach or spread on various ECM proteins (data not shown). Together, these results support the notion that Crk binding to CAS is critical for the migratory properties of these cells and that the substrate domain of CAS as well as the SH2 domain of Crk contribute to this response.
Figure 6.


Crk binding to CAS is required for cell migration. (A) Serum- starved COS cells were allowed to migrate for 3 h on vitronectin-coated membranes (top) or lysed on the culture dish and analyzed for expression of Crk and CAS by immunoblotting (middle and bottom) after transfection with either empty expression vector or the expression vector containing gstCAS-WT and either Crk-WT or CRK with a mutated SH2 domain (Crk-SH2). The number of transfected cells migrating were enumerated by counting β-galactosidase– positive cells on the underside of the membrane. Each bar represents the mean ± SD of triplicate migration wells of one of three representative experiments. (B) Crk immunoprecipitated from detergent lysates prepared from COS cells transfected as described for the migration experiment in A and analyzed by immunoblotting with anti-gst (top) or anti-Crk antibodies (bottom). The results shown are representative of at least three independent experiments.
Cell Migration Requires the NH2-terminal SH3 Domain of Crk
Crk-dependent signaling events likely depend on the ability of this protein to couple via its two SH3 domains to one or more downstream effector molecules (Matsuda et al., 1994; Hasegawa et al., 1996). Therefore, we investigated the role of these SH3 domains in CAS/Crk migration. We first investigated the role of the NH2-terminal SH3 domain in the cell migration response by expressing a Crk construct (Crk-SH3N) with a point mutation in this portion of the molecule, rendering it incapable of binding to effector molecules (Matsuda et al., 1994). Cells were transfected with wild-type CAS and either Crk or Crk-SH3N. While Crk-SH3N readily bound to wild-type CAS protein in these cells (Fig. 7, bottom), it did not promote cell migration, whereas the wild-type form of Crk did (Fig. 7, top). In contrast, expression of Crk with its COOH-terminal SH3 domain truncated readily bound to wild-type CAS and promoted migration that was comparable to that of cells expressing wild-type CAS and Crk (data not shown). These findings reveal that the NH2-terminal, but not the COOH-terminal, SH3 domain of Crk is critical for cell migration. Therefore, the commitment to migrate not only involves assembly of a CAS/Crk complex but depends on the NH2-terminal SH3 domain of Crk, which is known to bind various effector molecules and promote downstream signaling events.
Figure 7.

The NH2-terminal SH3 domain of Crk is required for CAS/Crk-induced cell migration. (Top) Serum-starved COS cells were allowed to migrate for 3 h on vitronectin-coated membranes after transfection with either empty expression vector or the expression vector containing gstCAS-WT and either Crk-WT or CRK with a mutation in the NH2-terminal SH3 domain (c-Crk-SH3(mut)). The number of transfected cells migrating were enumerated by counting β-galactosidase–positive cells on the underside of the membrane. Each bar represents the mean ± SD of triplicate migration wells of one of three representative experiments. (Bottom) Crk-WT or Crk-SH3N immunoprecipitated from detergent extracts from COS cells transfected as described for the migration experiment above and immunoblotted for gstCAS or Crk. The results shown are representative of at least three independent experiments.
Insulin Promotes Migration in a CAS/Crk-dependent Manner
Previous work has established that cytokines or growth factors such as insulin are physiological activators of the molecular switch leading to cell migration/invasion. This is important during development, angiogenesis, and wound repair. In fact, various tumor cell lines stimulated with insulin or insulin-like growth factor not only show increased migration in vitro but gain the ability to metastasize in vivo (Klemke et al., 1994, 1997; Brooks et al., 1997). Serum-starved COS cells can be stimulated to migrate after treatment with various cytokines. Therefore, experiments were designed to investigate whether cytokine-stimulated cell migration was also dependent on CAS and Crk. To investigate this possibility, serum-starved COS cells expressing wild-type CAS or CAS-SD were allowed to migrate in the presence or absence of insulin. As shown in Fig. 8, expression of CAS-SD, but not wild-type CAS, in COS cells blocked insulin-induced cell migration without impacting the adhesion of these cells. Furthermore, expression of Crk, lacking a functional SH2 or an NH2-terminal SH3 domain, also blocked this response (data not shown). Similar findings were obtained when cells were stimulated with IGF-1 and EGF (data not shown). Together, these findings demonstrate that CAS and Crk play a critical role in cytokine/growth factor–mediated cell migration.
Figure 8.

Requirement of CAS for cytokine-induced cell migration. Serum-starved COS cells were allowed to migrate for 3 h on vitronectin-coated membranes in the presence or absence of insulin after transfection with either empty expression vector or the vector containing gstCAS-WT or gstCAS-SD as described in Materials and Methods. The number of transfected cells migrating were enumerated by counting β-galactosidase–positive cells on the underside of the membrane. Each bar represents the mean ± SD of triplicate wells. Similar results were obtained after stimulation with IGF-1 and EGF (not shown).
Crk and CAS Localize to Membrane Ruffles in Migratory Cells
Targeting of signaling protein complexes to specific regions of the cell is important for the control of various cellular functions (Bar-Sagi et al., 1993). To investigate the intracellular localization of Crk and CAS, cells expressing wild-type Crk or CAS were allowed to migrate on vitronectin-coated coverslips for 2–4 h and then examined by immunofluorescent staining and confocal microscopy. As shown in Fig. 9 A, cells expressing either Crk or CAS had a classical motile phenotype with a leading lamellae containing membrane ruffles rich in F-actin. Interestingly, both Crk and CAS proteins were localized to these membrane ruffles (Fig. 9 A, arrowhead). Importantly, cells expressing wild-type CAS together with Crk showed a similar motile phenotype and colocalization of these proteins in membrane ruffles (data not shown). In contrast, cells within the field of view not expressing the Crk or CAS cDNAs contained little or no membrane ruffling and appeared nonmotile (Fig. 9 A).
Figure 9.
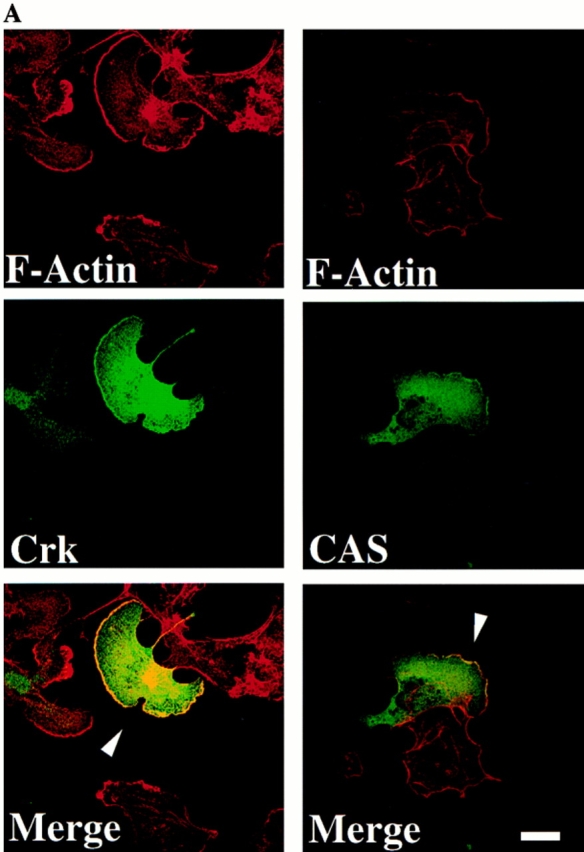
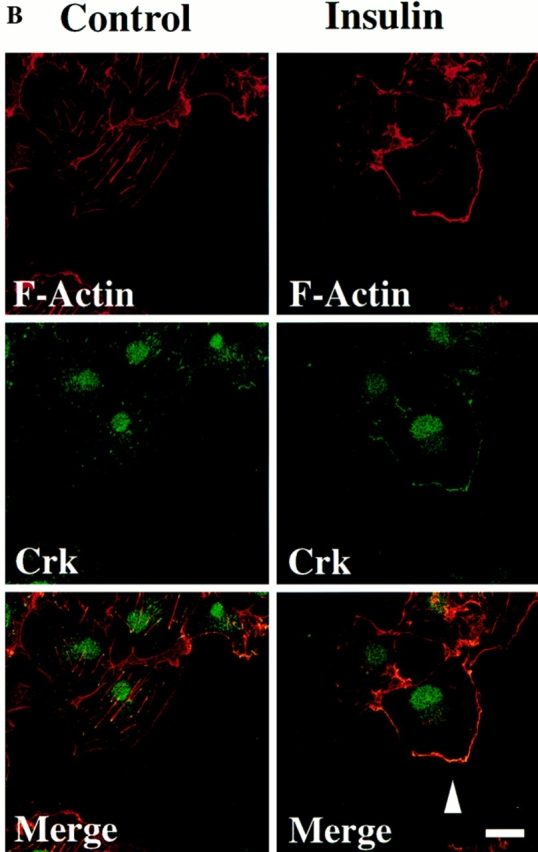
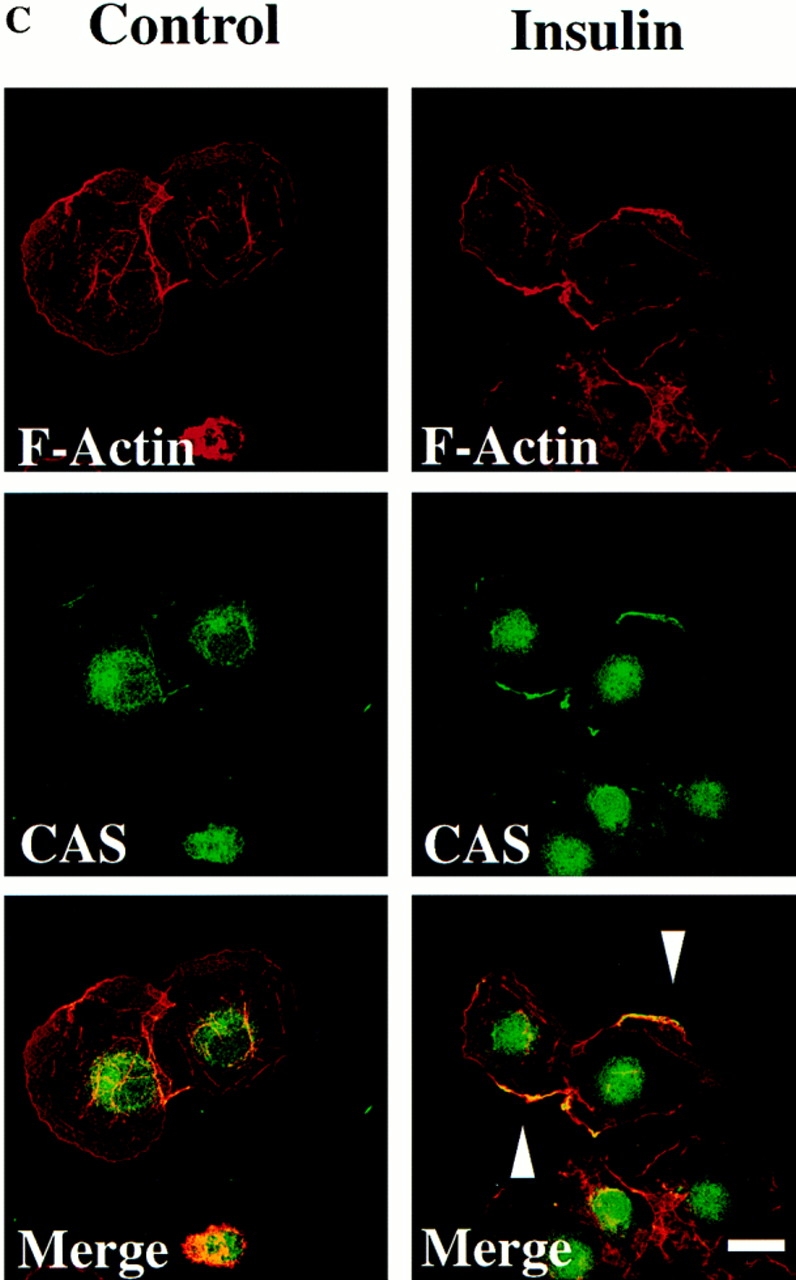
Localization of CAS and Crk to membrane ruffles in migratory cells. (A) Serum-starved COS cells were allowed to attach to vitronectin-coated glass coverslips after transfection with the expression vector containing either wild-type Crk or CAS and examined by confocal immunofluorescent imaging as described in Materials and Methods. Rhodamine-conjugated phalloidin was used to visualize F-actin (red), whereas FITC-conjugated secondary antibody was used to visualize primary antibodies directed to Crk or CAS (green). Yellow staining represents the colocalization of red and green staining in the merged image. Control cells in which the primary antibody was omitted showed no immunofluorescent staining (data not shown). Photomicrographs were taken with a Bio-Rad Labs 1024 laser and Zeiss Axiovert microscope focused at the cell–substratum interface (630×). The arrowheads indicate colocalization of Crk or CAS with F-actin and represent membrane ruffling of cells showing motile phenotype. (B) Serum-starved COS cells attached to vitronectin-coated coverslips in the presence (15 min) or absence of insulin were analyzed by immunofluorescent confocal imaging for intracellular localization of endogenous Crk with F-actin as described above. Photomicrographs were taken at the cell–substratum interface (630×). The arrowhead indicates colocalization of Crk with F-actin containing membrane ruffles in cells stimulated to migrate with insulin. (C) Confocal immunofluorescent imaging of endogenous CAS in COS cells treated as described in B above. Photomicrographs were taken at the cell–substratum interface (630×). The arrowheads indicate colocalization of CAS with F-actin containing membrane ruffles in cells stimulated to migrate with insulin. Bars, 10 μm.
To establish the subcellular localization of endogenous Crk and CAS, serum-starved cells were treated with or without insulin and stained for the presence of Crk and CAS. Nonmigratory serum-starved cells showed prominent Crk staining of focal contacts associated with the end of actin filaments at the cell–substrate interface (Fig. 9 B). However, after insulin treatment cells showed prominent membrane ruffles that contained both Crk and CAS (Fig. 9, B and C). Within the insulin treated cell population, few focal contacts or stress fibers were observed, which is characteristic of motile cells (data not shown). Together, these findings demonstrate that both Crk and CAS localize to the membrane ruffles of migratory cells.
Rac but Not Ras Activity Is Required for Crk/CAS-induced Cell Migration
The small G proteins Ras and Rac have been associated with cell movement (Ridely et al., 1992; Takaishi et al., 1993; Michiels et al., 1995). In fact, membrane ruffling has been linked to the activation state of each of these small G proteins (Ridely et al., 1992). Therefore, we investigated the role of Rac and Ras in the Crk/CAS-induced migration. In these experiments, COS cells cotransfected with wild-type Crk and CAS together with either dominant-negative Rac (Rac−) or dominant-negative Ras (Ras−) were tested for their ability to migrate on ECM proteins. Expression of Rac− but not Ras− in these cells blocked Crk/CAS-induced cell migration (Fig. 10) and membrane ruffling (data not shown). In fact, expression of Ras− was greater than that of Rac− in these cells (Fig. 10), yet it still failed to suppress the Cas/Crk-mediated cell motility response. In addition, expression of a dominant-active form of Rac in these cells failed to promote migration, suggesting that Rac activity was not sufficient to facilitate this event (data not shown). Neither Rac nor Ras influenced the adhesive or spreading capacity of these cells, indicating expression of these constructs was not toxic (data not shown). Together these results indicate that Rac activity, but not Ras activity, is required for Crk/CAS-induced cell migration on the ECM.
Figure 10.

Requirement of Rac, but not Ras, for CAS/Crk-mediated cell migration. (Top) Serum-starved COS cells were allowed to migrate for 3 h on vitronectin-coated membranes after transfection with either the empty expression vector or the expression vector containing gstCAS-WT and Crk-WT along with dominant-negative Rac (−) or dominant-negative Ras (−) . The number of transfected cells migrating were enumerated by counting cells on the underside of the membrane as described above. Each bar represents the mean ± SD of triplicate migration wells of one of three representative experiments. (Bottom) An aliquot of those cells used for the migration assay above were lysed in detergent and analyzed for myc-tagged Rac or Ras expression as described in Materials and Methods.
Discussion
During wound repair and inflammation, cells gain the ability to migrate once they are stimulated with growth factors/cytokines. However, cell migration also depends on adhesive proteins present within the ECM (Clark et al., 1996; Palecek et al., 1997). In fact, both growth factor receptors and integrins promote intracellular signals that are associated with the migratory phenotype of cells (Ridley et al., 1992; Chen et al., 1994; Klemke et al., 1994, 1997; Brooks et al., 1997). However, little is known regarding how these signals activate the cells' migration machinery, thereby converting a stationary cell to a migratory cell.
While adhesion is necessary for cell motility, it is not sufficient. For example, FG cell attachment to collagen is permissive for migration, while attachment to vitronectin is not. This prompted us to examine whether FG cell attachment to collagen or vitronectin produced distinct signaling events. As we show in this study, the increased phosphorylation of CAS was associated with FG cell adhesion to collagen but not to vitronectin. In addition, selection of FG-M cells for their ability to spontaneously migrate on vitronectin resulted in a cell population that showed increased CAS phosphorylation and CAS/Crk coupling upon adhesion to vitronectin. It is this CAS/Crk complex that has been associated with downstream signaling events, yet little is known how these signals influence the biology of cells.
In this report, we demonstrate that assembly of the CAS/Crk adaptor protein complex serves as a molecular switch committing a cell to migrate on the extracellular matrix. This was investigated using cDNAs encoding alleles of wild-type Crk and CAS or cDNAs that had been mutationally altered in critical effector domains. Several lines of evidence are presented showing that CAS and Crk play a central role in cell migration. First, expression of wild-type CAS or Crk in cells was sufficient to induce cell movement without affecting cell adhesion on various matrix proteins. Second, this migration event requires the assembly of a CAS/Crk complex involving the binding of Crk via its SH2 domain to the substrate domain of CAS. Third, cell migration induced by known regulators of motility including insulin, EGF, and IGF-1 required functional Crk and CAS, which were driven to the membrane ruffles of migratory cells. Finally, the small G protein Rac, which is involved in regulating signaling events important for membrane ruffling and cell migration, is required for migration induced by Crk/CAS. Surprisingly, a dominant-negative form of Ras failed to block CAS/Crk-induced motility (Fig. 10), even though it blocked cytokine-mediated cell migration (data not shown). These data suggest that Rac and Ras promote somewhat distinct pathways of cell migration. In fact, evidence from our laboratory demonstrates that the Ras/Map kinase (Erk 1 and 2) promotes cell motility based on the ability of ERK to phosphorylate myosin light chain kinase (Klemke et al., 1997). While a dominant-negative form of myosin light chain kinase blocked Ras-induced motility (Klemke et al., 1997), it failed to inhibit CAS/Crk-induced migration. Furthermore, the MEK inhibitor PD98059 failed to block CAS/Crk-induced cell migration (Klemke, R., unpublished data). Therefore, it appears that CAS/Crk-dependent cell migration may represent a distinct pathway from that initiated by Ras/MAP kinase.
CAS phosphorylation has been linked to cell adhesion and depends on kinases such as c-Src and FAK (Harte et al., 1996; Vuori et al., 1996; Schlaepfer et al., 1997); however, its susceptibility to phosphatases plays a key role in its level of phosphorylation (Garton et al., 1996). Therefore, exogenous expression of CAS could serve as a competitive substrate for such phosphatases, thereby increasing the overall level of phosphorylated CAS in the cell. In fact, we observed that exogenous expression of wild-type CAS not only caused increased migration but also led to increased phosphorylation of endogenous as well as expressed CAS protein (Fig. 4). Likewise, exogenous expression of Crk is known to protect CAS from dephosphorylation by phosphatases (Birge et al., 1992), providing a second mechanism for enhancing the level of phosphorylated CAS in these cells (data not shown). Therefore, exogenous expression of either wild-type CAS or Crk would be expected to enhance the level of CAS phosphorylation, resulting in the generation of CAS/Crk complexes.
This study demonstrates that CAS without a substrate domain and Crk without a functional SH2 or NH2-terminal SH3 domain serve as dominant-negative proteins specifically blocking cell migration without affecting attachment or spreading of these cells on vitronectin, collagen, or fibronectin. CAS-SD may block migration by preventing CAS/Crk complexes from localizing to the appropriate subcellular location, such as focal adhesion sites or membrane ruffles. Both wild-type and CAS-SD have been shown to localize to focal contacts in adherent cells, an event that required the SH3 domain of CAS (Nakamoto et al., 1997). Expression of Crk-SH2 in cells may block CAS-mediated migration by competing with endogenous Crk for important effector molecules that associate with the NH2-terminal SH3 domain. That expression of Crk-SH3 (N) in cells also blocks CAS-mediated migration supports this notion. In fact, several signaling molecules are known to interact with the NH2-terminal SH3 domain of CRK, including Abl kinase (Feller et al., 1994a ), DOCK 180 (Hasegawa et al., 1996), GTP exchange proteins SOS and C3G (Matsuda et al., 1994; Feller et al., 1995), and the EGF receptor substrate protein Eps15 (Schumacher et al., 1995). Alternatively, this SH3 domain may also be involved in targeting the CAS/Crk complex to a specific cellular location (Bar-Sagi et al., 1993). To this end, Crk and CAS were found to colocalize in membrane ruffles of migratory cells (Fig. 9).
While cell adhesion events are required for cell migration, not all adherent cells move. In fact, cell migration often depends on signals generated from both growth factor and adhesion receptors. Our findings suggest that CAS/Crk coupling provides the adhesion-dependent component of this signaling cascade and, thereby, serves as a molecular switch promoting cell migration on the extracellular matrix. However, in transformed cells, CAS/Crk coupling may be constitutive. In fact, v-Crk, a viral oncogene, has been linked to cell transformation and the malignant phenotype of tumors (for review see Matsuda and Kurata, 1996). We provide evidence that CAS/Crk signaling may also influence tumor cell metastasis. Specifically, FG-M carcinoma cells selected for spontaneous migration on vitronectin not only showed enhanced CAS phosphorylation and CAS/Crk coupling upon adhesion to vitronectin in vitro but also had an increased metastatic capacity in vivo compared with parental cells that showed no adhesion-dependent CAS phosphorylation.
The data presented here provide a mechanism to link adhesion-dependent signaling events and the formation of a CAS/Crk complex to the activation of a Rac-dependent cell migration response. The phosphorylation of CAS and its coupling to Crk serves as a fundamental component of this signaling cascade. These events may be critical for growth factor– or cytokine-induced cell migration associated with development, wound repair, and inflammation. However, in transformed cells, CAS/Crk complexes may form spontaneously, leading to invasion events critical for the dissemination of tumors.
Footnotes
1. Abbreviations used in this paper: aa, amino acid(s); CAS, Crk-associated substrate; ECM, extracellular matrix; FAK, focal adhesion kinase; FBM, fibroblast basal medium; IGF-1, insulin-like growth factor 1; SH, src homology.
The authors would like to thank Drs. A. Minden and M. Karin (University of California, San Diego, CA) for providing Rac and Ras constructs. We are also grateful to Dr. M. Matsuda (International Medical Center of Japan, Tokyo, Japan) for original Crk constructs, and Drs. H. Hirai (University of Tokyo, Tokyo, Japan) and B. Mayer (Howard Hughes Medical Institute, The Children's Hospital, Boston, MA) for CAS cDNA constructs and expression vectors.
D.A. Cheresh was supported by grants HL-54444, CA-50286, and CA-45726 from the National Institutes of Health (NIH). R.L. Klemke was supported by a fellowship award from the Joseph Drown Foundation. J. Leng was supported by the U.S. Army Medical Research and Material Command under DAMD17-96-1-6104. K. Vuori was supported by grant CA-71560 from the NIH. K. Vuori is a PEW scholar in Biomedical Sciences. This manuscript is number 11052-IMM from The Scripps Research Institute.
Address all correspondence to Richard L. Klemke, Departments of Immunology and Vascular Biology, IMM-24, The Scripps Research Institute, 10550 N. Torrey Pines Rd., La Jolla, CA 92037. Tel.: (619) 784-8164. Fax: 619-784-8926. E-mail: klemke@scripps.edu
References
- Bar-Sagi D, Rotin D, Batzer A, Mandiyan V, Schlessinger J. SH3 domains direct cellular localization of signaling molecules. Cell. 1993;74:83–91. doi: 10.1016/0092-8674(93)90296-3. [DOI] [PubMed] [Google Scholar]
- Birge B, Fajardo J, Mayer B, Hanafusa H. Tyrosine-phosphorylated epidermal growth factor receptor and cellular p130 provide high affinity binding substrates to analyze Crk-phosphotyrosine-dependent interactions in vitro. J Biol Chem. 1992;267:10588–10595. [PubMed] [Google Scholar]
- Brooks PC, Klemke RL, Schön S, Lewis JM, Schwartz MA, Cheresh DA. Insulin-like growth factor receptor cooperates with integrin αvβ5 to promote tumor cell dissemination in vivo. J Clin Invest. 1997;99:1390–1398. doi: 10.1172/JCI119298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cary LA, Chang JF, Guan J. Stimulation of cell migration by overexpression of focal adhesion kinase and its association with Src and Fyn. J Cell Sci. 1996;109:1787–1794. doi: 10.1242/jcs.109.7.1787. [DOI] [PubMed] [Google Scholar]
- Chen P, Xie H, Sekar M, Gupta K, Wells A. Epidermal growth factor receptor–mediated cell motility: phospholipase C activity is required but mitogen-activated protein kinase is not sufficient for induced cell movement. J Cell Biol. 1994;127:847–857. doi: 10.1083/jcb.127.3.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RAF, Tonnesen MG, Gailit J, Cheresh DA. Transient functional expression of αvβ3 on vascular cells during wound repair. Am J Pathol. 1996;148:1407–1421. [PMC free article] [PubMed] [Google Scholar]
- Feller S, Ren R, Hanafusa H, Baltimore D. SH2 and SH3 domains as molecular adhesives: the interactions of Crk and Abl. Trends Biochem Sci. 1994a;19:453–458. doi: 10.1016/0968-0004(94)90129-5. [DOI] [PubMed] [Google Scholar]
- Feller S, Knudsen B, Hanafusa H. C-Abl kinase regulates the protein binding activity of c-Crk. EMBO (Eur Mol Biol Organ) J. 1994b;13:2341–2351. doi: 10.1002/j.1460-2075.1994.tb06518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller S, Knudsen B, Hanafusa H. Cellular proteins binding to the first Src homology 3 (SH3) domain of the proto-oncogene product c-Crk indicate Crk-specific signaling pathways. Oncogene. 1995;10:1465–1473. [PubMed] [Google Scholar]
- Garton A, Flint A, Tonks N. Identification of p130CAS as a substrate for the cytosolic protein tyrosine phosphatase PTP-PEST. Mol Cell Biol. 1996;271:6408–6418. doi: 10.1128/mcb.16.11.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore AP, Romer LH. Inhibition of focal adhesion kinase (FAK) signaling in focal adhesions decreases cell motility and proliferation. Mol Biol Cell. 1996;7:1209–1224. doi: 10.1091/mbc.7.8.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greulich H, Hanafusa H. A role for Ras in v-Crk transformation. Cell Growth Diff. 1996;7:1443–1451. [PubMed] [Google Scholar]
- Hanks SK, Polte TR. Signaling through focal adhesion kinase. BioEssays. 1997;19:137–145. doi: 10.1002/bies.950190208. [DOI] [PubMed] [Google Scholar]
- Harte MT, Hildebrand JD, Burnham MR, Bouton AH, Parsons JT. p130CAS, a substrate associated with v-Src and v-Crk, localizes to focal adhesions and binds to focal adhesion kinase. J Biol Chem. 1996;271:13649–13655. doi: 10.1074/jbc.271.23.13649. [DOI] [PubMed] [Google Scholar]
- Hasegawa H, Kiyokawa E, Tanaka S, Nagashima K, Gotoh N, Shibuya M, Kurata T, Matsuda M. DOCK180, a major CRK-binding protein, alters cell morphology upon translocation to the cell membrane. Mol Cell Biol. 1996;16:1770–1776. doi: 10.1128/mcb.16.4.1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotchin NA, Hall A. The assembly of integrin complexes requires both extracellular matrix and intracellular rho/rac GTPases. J Cell Biol. 1995;131:1857–1865. doi: 10.1083/jcb.131.6.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenlocher A, Ginsberg M, Horwitz A. Modulation of cell migration by integrin-mediated cytoskeletal linkages and ligand-binding affinity. J Cell Biol. 1996;134:1551–1562. doi: 10.1083/jcb.134.6.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilic D, Furuat Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T, Aizawa S. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377:539–544. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- Klemke R, Yebra M, Bayna EM, Cheresh DA. Receptor tyrosine kinase signaling required for integrin αvβ5-directed cell motility but not adhesion on vitronectin. J Cell Biol. 1994;127:859–866. doi: 10.1083/jcb.127.3.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemke R, Cai S, Giannini A, Gallagher P, de Lanerolle P, Cheresh D. Regulation of cell motility by mitogen-activated protein kinase. J Cell Biol. 1997;137:481–492. doi: 10.1083/jcb.137.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda M, Kurata T. Emerging components of the Crk oncogene product: the first identified adaptor protein. Cell Signalling. 1996;8:335–340. doi: 10.1016/0898-6568(96)00067-8. [DOI] [PubMed] [Google Scholar]
- Matsuda M, Nagata S, Tanaka S, Nagashima K, Kurata T. Structural requirement of CRK SH2 region for binding to phosphotyrosine-containing proteins. J Biol Chem. 1993;268:4441–4446. [PubMed] [Google Scholar]
- Matsuda Mi, Hashimoto Y, Muroya K, Hasegawa H, Kurata T, Tanaka S, Nakamura S, Hattori S. CRK protein binds to two guanine nucleotide-releasing proteins for the Ras family and modulates nerve growth factor-induced activation of ras in PC12 cells. Mol Cell Biol. 1994;14:5495–5500. doi: 10.1128/mcb.14.8.5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer BJ, Hirai H, Sakai R. Evidence that SH2 domains promote processive phosphorylation by protein-tyrosine kinases. Curr Biol. 1995;5:296–305. doi: 10.1016/s0960-9822(95)00060-1. [DOI] [PubMed] [Google Scholar]
- Michiels F, Habets GGM, Stam JC, van der Kammen RA, Collard JG. A role for Rac in Tiam1-induced membrane ruffling and invasion. Nature. 1995;375:338–340. doi: 10.1038/375338a0. [DOI] [PubMed] [Google Scholar]
- Minden A, Lin A, Claret F, Abo A, Karin M. Selective activation of the JNK signaling cascade and c-jun transcriptional activity by the small GTPases Rac and Cdc42HS. Cell. 1995;81:1147–1157. doi: 10.1016/s0092-8674(05)80019-4. [DOI] [PubMed] [Google Scholar]
- Nakamoto T, Sakai R, Honda H, Ogawa S, Ueno H, Suzuki T, Aizawa S, Yazaki Y, Hirai H. Requirements for localization of p130CAS to focal adhesions. Mol Cell Biol. 1997;17:3884–3887. doi: 10.1128/mcb.17.7.3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nip J, Shibata H, Loskutoff D, Cheresh D, Brodt P. Human melanoma cells derived from lymphatic metastasis use integrin αvβ3 to adhere to lymph node vitronectin. J Clin Invest. 1992;90:1406–1413. doi: 10.1172/JCI116007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nojima Y, Morino N, Mimura T, Hamasaki K, Furuya H, Sakai R, Sato T, Tachibana K, Morimoto C, Yazaki Y, Hirai H. Integrin-mediated cell adhesion promotes tyrosine phosphorylation of p130Cas, a Src homology 3-containing molecule having multiple Src homology 2-binding motifs. J Biol Chem. 1995;270:15398–15402. doi: 10.1074/jbc.270.25.15398. [DOI] [PubMed] [Google Scholar]
- Palecek S, Loftus J, Ginsberg M, Lauffenburger D, Horwitz A. Integrin-ligand binding properties govern speed through cell-substratum adhesiveness. Nature. 1997;385:537–540. doi: 10.1038/385537a0. [DOI] [PubMed] [Google Scholar]
- Parsons T. Integrin-mediated signaling: regulation by protein tyrosine kinases and small GTP-binding proteins. Curr Opin Cell Biol. 1996;8:146–152. doi: 10.1016/s0955-0674(96)80059-7. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- Rodier J, Valles AM, Denoyelle M, Thiery JP, Boyer B. pp60c-srcis a positive regulator of growth factor-induced cell scattering in a rat bladder carcinoma cell line. J Cell Biol. 1995;131:761–773. doi: 10.1083/jcb.131.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Warne PH, Khwaja A, Marte BM, Pappin D, Das P, Waterfield MD, Ridley A, Downward J. Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell. 1997;89:457–467. doi: 10.1016/s0092-8674(00)80226-3. [DOI] [PubMed] [Google Scholar]
- Sakai R, Iwamatsu A, Hirano N, Ogawa S, Tanaka T, Mano H, Yazaki Y, Hirai H. A novel signaling molecule, p130, forms stable complexes in vivo with v-Crk and v-Src in a tyrosine phosphorylation-dependent manner. EMBO (Eur Mol Biol Organ) J. 1994;13:3748–3756. doi: 10.1002/j.1460-2075.1994.tb06684.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer D, Broome M, Hunter T. Fibronectin-stimulated signaling from a focal adhesion kinase-c-Src complex: involvement of the Grb2, p130 CAS and Nck adaptor proteins. Mol Cell Biol. 1997;17:1702–1713. doi: 10.1128/mcb.17.3.1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher C, Knudsen B, Ohuchi T, Di Fiore P, Glassman R, Hanafusa H. The SH3 domain of Crk binds specifically to a conserved proline-rich motif in Eps 15 and Eps 15R. J Biol Chem. 1995;270(25):15341–15347. doi: 10.1074/jbc.270.25.15341. [DOI] [PubMed] [Google Scholar]
- Schwartz M, Schaller M, Ginsberg M. Integrins: emerging paradigms of signal transduction. Annu Rev Cell Dev Biol. 1995;11:549–599. doi: 10.1146/annurev.cb.11.110195.003001. [DOI] [PubMed] [Google Scholar]
- Takaishi K, Kiluchi A, Kuroda S, Kotani K, Sasaki T, Takai Y. Involvement of rho p21 and its inhibitory GDP/GTP exchange protein (rho GDI) in cell motility. Mol Cell Biol. 1993;13:72–79. doi: 10.1128/mcb.13.1.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Hattori S, Kurata T, Nagashima K, Fukui Y, Nakamura S, Matsuda M. Both the SH2 and SH3 domains of human Crk protein are required for neuronal differentiation of PC12 cells. Mol Cell Biol. 1993;13:4409–4415. doi: 10.1128/mcb.13.7.4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuori K, Ruoslahti E. Tyrosine phosphorylation of p130Cas and cortactin accompanies integrin-mediated cell adhesion to the extracellular matrix. J Biol Chem. 1995;270:22259–22262. doi: 10.1074/jbc.270.38.22259. [DOI] [PubMed] [Google Scholar]
- Vuori K, Hirai H, Aizawa S, Ruoslahti E. Induction of p130Cas signaling complex formation upon integrin-mediated cell adhesion: a role for Src family kinases. Mol Cell Biol. 1996;16:2606–2613. doi: 10.1128/mcb.16.6.2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatohgo T, Izumi M, Kashiwagi H, Hiyashi M. Novel purification of vitronectin from human plasma by heparin affinity chromatography. Cell Struct Funct. 1988;13:281–292. doi: 10.1247/csf.13.281. [DOI] [PubMed] [Google Scholar]