

Abstract
Abstract. To determine whether the p75 neurotrophin receptor (p75NTR) plays a role in naturally occurring neuronal death, we examined neonatal sympathetic neurons that express both the TrkA tyrosine kinase receptor and p75NTR. When sympathetic neuron survival is maintained with low quantities of NGF or KCl, the neurotrophin brain-derived neurotrophic factor (BDNF), which does not activate Trk receptors on sympathetic neurons, causes neuronal apoptosis and increased phosphorylation of c-jun. Function-blocking antibody studies indicate that this apoptosis is due to BDNF-mediated activation of p75NTR. To determine the physiological relevance of these culture findings, we examined sympathetic neurons in BDNF−/− and p75NTR−/− mice. In BDNF−/− mice, sympathetic neuron number is increased relative to BDNF+/+ littermates, and in p75NTR−/− mice, the normal period of sympathetic neuron death does not occur, with neuronal attrition occurring later in life. This deficit in apoptosis is intrinsic to sympathetic neurons, since cultured p75NTR−/− neurons die more slowly than do their wild-type counterparts. Together, these data indicate that p75NTR can signal to mediate apoptosis, and that this mechanism is essential for naturally occurring sympathetic neuron death.
The p75 neurotrophin receptor (p75NTR)1 (Johnson et al., 1986; Radeke et al., 1987) is the first member discovered of a family of receptors, including fas and TNFR1, which have been shown to mediate cellular differentiation and apoptosis (for review see Chao, 1994). p75NTR can interact with all of the mammalian members of the neurotrophin family (for review see Levi-Montalcini, 1987; Barde, 1989; Snider, 1994), NGF, brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), and neurotrophin-4 (NT-4), with approximately equivalent affinities (Rodriguez-Tebar et al., 1990, 1992). In contrast, the other family of neurotrophin receptors, the Trk tyrosine kinases (for review see Barbacid, 1994), display a specificity for binding individual neurotrophins: NGF and NT-3 for TrkA (Cordon-Cardo et al., 1991; Klein et al., 1991; Kaplan et al., 1991a ,b); BDNF and NT-4 for TrkB (Soppet et al., 1991); and NT-3 for TrkC (Lamballe et al., 1993; Tsoulfas et al., 1993). Whereas much is known about the signaling and biological roles of the Trks in neurons in vivo and in vitro, the functional role of p75NTR in neurons has remained elusive.
p75NTR was originally reported to function as a positive regulator of TrkA activity in a number of neural cell lines (Benedetti et al., 1993; Ip et al., 1993; Barker and Shooter, 1994; Verdi et al., 1994). The initial report of the p75NTR knockout mouse apparently supported this hypothesis, since these mice exhibited a loss of nociceptive and thermosensitive sensory neurons (Lee et al., 1992), phenotypes observed in a more severe form in the NGF (Crowley et al., 1994) and TrkA knockout mice (Smeyne et al., 1994). However, other defects have been observed in the p75NTR knockout mice that are more difficult to explain on the basis of p75NTR positively regulating TrkA; defects such as a selective loss of sympathetic innervation without an apparent loss of sympathetic neurons (Lee et al., 1992, 1994), and an increase in the number of basal forebrain cholinergic neurons (van der Zee et al., 1996) with a coincident increase in cholinergic innervation of the hippocampus (Yeo et al., 1997).
More recent evidence indicates that, in addition to modulating Trk function, p75NTR signals on its own. Specifically, in neural cell lines and glial cells, neurotrophin binding to p75NTR stimulates the generation of ceramide (Dobrowsky et al., 1994, 1995; Casaccia-Bonnefil et al., 1996), activation of nuclear factor (NF)-kB and translocation of this protein to the nucleus (Carter et al., 1996), and enhancement of jun kinase (JNK) activity (Casaccia-Bonnefil et al., 1996). Such signaling likely mediates the effects of NGF on Schwann cells, which do not express TrkA, and which respond to NGF with migration (Anton et al., 1994). Similarly, NGF-mediated activation of p75NTR signaling in cultured oligodendrocytes leads to apoptosis (Casaccia-Bonnefil et al., 1996).
In spite of this progress in our understanding of p75NTR function in cell lines and glial cells, the functional role of this receptor in neurons remains elusive. However, a number of recent studies indicate that p75NTR may play a role in regulating neuronal survival. First, Rabizadeh et al. (1993) reported that p75NTR decreased survival of a neural tumor cell line in a ligand-independent fashion, whereas Barrett and Bartlett (1994) reported that functional ablation of p75NTR enhanced survival of cultured postnatal sensory neurons. However, neither of these reports determined whether p75NTR mediated these responses on its own, or by modulating Trk activity. Second, addition of NGF in vivo to neurons of the developing chick isthmo-optic nucleus, which express p75NTR but not TrkA, led to cell death by an undefined mechanism (von Bartheld et al., 1994). Third, addition of anti-NGF antibodies to the developing chick retina led to decreased death of cells that express p75NTR but not TrkA (Frade et al., 1996). In this latter study, unlike that with the chick isthmo-optic nucleus neurons, addition of excess NGF had no effect. Finally, expression of the intracellular domain of p75NTR as a transgene in mouse neurons resulted in apoptosis of selected populations of peripheral and central neurons, indicating the cell death–inducing potential of p75NTR (Majdan et al., 1997). However, whereas these studies implicate p75NTR in neuronal apoptosis, the mechanism whereby this occurs, and its physiological roles remain unclear.
We have addressed these issues by investigating the role of p75NTR in postnatal rat sympathetic neurons during the developmental period of naturally occurring cell death. These neurons express relatively high levels of TrkA and p75NTR, low levels of functional TrkC (Belliveau et al., 1997), and have an absolute requirement for NGF for survival (Levi-Montalcini et al., 1960a,b). We have recently demonstrated that TrkA activation is necessary for sympathetic neuron survival, and that NGF and NT-3 are both capable of eliciting TrkA activation on these neurons (Belliveau et al., 1997). However, surprisingly, whereas NT-3 is as effective as NGF in mediating neuritogenesis, it is two- to fourfold less effective at mediating neuronal survival at equivalent levels of TrkA activation. Since NT-3 is much less efficient at activating TrkA than NGF (Cordon-Cardo et al., 1991; Belliveau et al., 1997), and approximately equivalent at binding p75NTR (Rodriguez-Tebar et al., 1992), we hypothesized that this difference in survival was a consequence of a balance between differential activation of TrkA versus p75NTR on sympathetic neurons. In studies reported here, we have tested this hypothesis. Our data indicate that p75NTR can signal to mediate neuronal apoptosis, and that this mechanism is essential for naturally occurring sympathetic neuron cell death.
Materials and Methods
Primary Neuronal Cultures
Mass cultures of pure sympathetic neurons from the superior cervical ganglion (SCG) of postnatal day 1 rats were prepared as previously described (Ma et al., 1992). Neurons were plated on rat tail collagen-coated tissue culture dishes; 4-well plates (Falcon Plastics, Cockeysville, MD) for morphological measurements, 6-well plates for biochemistry, and 48-well plates for 3-[4,5-dimethylthio-zol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assays. Low density SCG cultures were used for all of the survival assays; for the MTT assays, neurons were used at a density of 1,500–2,000 neurons per well of a 48-well plate. Similar neuronal densities were used for the TUNEL assays, but the neurons were plated on poly-d-lysine and laminin. For biochemistry, ∼100,000 neurons were plated per well of a 6-well dish. For all experiments, neurons were initially cultured for 5 d in the presence of 10 or 50 ng/ml NGF. At the end of this 5-d selection, neurons were washed several times in neurotrophin-free media before addition of the new neurotrophin- or KCl-containing media.
NGF for these experiments was purified from mouse salivary glands (Cedarlane Labs. Ltd., Hornby, Ontario, Canada). Purified recombinant NT-4 was obtained from Genentech (South San Francisco, CA). Two sources of recombinant human BDNF (Amgen, Thousand Oaks, CA; PreproTech, Inc., Rocky Hill, NJ) were used for these experiments to ensure that the results were not because of unsuspected toxicity associated with one of the sources. Both preparations of BDNF caused TrkB autophosphorylation on TrkB-expressing NIH-3T3 cells (data not shown). Moreover, these sources of BDNF have been used on primary cultures of cortical progenitor cells, cortical neurons, and hippocampal neurons, with no evidence of cytotoxicity (data not shown). The p75NTR function-blocking antibody REX (Weskamp and Reichardt, 1991) (the gift of L. Reichardt, University of California, San Francisco, CA) is directed against the extracellular domain of the receptor, and was used as an antiserum at a dilution of 1:150.
Mouse sympathetic neurons were cultured by a modification of the method used for rat neurons. Specifically, neurons were dissected and triturated as for rat neurons (Ma et al., 1992), except that neurons were dissociated in the presence of media instead of saline solution. Neurons were then plated on collagen-coated culture dishes in Ultraculture media (BioWhittaker, Inc.) containing 2 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 3% FBS (Life Technologies, Inc., Gaithersburg, MD), and 50 ng/ml NGF. 3 d after plating, the neurons were fed with the same media containing 0.5% cytosine arabinoside.
TUNEL and Survival Assays
TUNEL assays were performed as previously described (Slack et al., 1996). For survival assays, analysis was performed 48 h later using nonradioactive cell proliferation (MTT) assays (Celltitre 96; Promega Corp., Madison, WI). 20 μl of the MTT reagent as added to each well and left for 90 min, followed by the addition of 200 μl of solubilization solution to lyse the cells. Each condition was repeated in triplicate. In each assay, baseline (0% survival) was considered to be 0 ng/ml NGF, and 10 ng/ml NGF was considered to be 100% survival. All other conditions were related to these values. For the REX experiment, neurons were washed with neurotrophin-free media for 3 h, and were then preincubated in media + KCl + REX for 2 h, at which point the BDNF was added for 48 h.
Survival assays for p75NTR−/− versus p75NTR+/+ neurons were performed by counting randomly selected fields of cultured neurons. Specifically, after 5 d in culture, neurons were washed with four changes of neurotrophin-free media for a total of 2.5 h, and then maintained in media with no added neurotrophins. The number of phase-bright neurons with neurites in randomly selected, 5.3-mm2 fields was counted immediately after the switch to NGF-free media, and was recounted in the same fields at 24-h intervals for 5 d. In each individual experiment, each condition was repeated in triplicate. As controls for the p75NTR−/− neurons, neurons were cultured from mice of either C129 or CD1 backgrounds. Results for neurons from either background were similar, and were therefore combined. In three of the five experiments analyzed, p75NTR−/− and p75NTR+/+ neurons were cultured at the same time in the same 48-well plates to eliminate potential variability.
Immunoprecipitations and Western Blot Analysis
For Trk assays, sympathetic neurons were lysed in TBS lysis buffer (Knusel et al., 1994) containing 137 mM NaCl, 20 mM Tris, pH 8.0, 1% (vol/ vol) NP-40, 10% (vol/vol) glycerol, 1 mM PMSF, 10 μg/ml aprotinin, 0.2 μg/ml leupeptin, 5 mM phenanthroline, and 1.5 mM sodium vanadate. 1 ml of cold TBS was added to one or two wells of a six-well dish, and cells were collected, resuspended in 100 μl lysis buffer, and rocked for 20 min at 4°C. Each sample was vortexed for 10 s, and cleared by centrifugation. The lysates were normalized for protein concentration using a BCA Protein Assay Reagent (Pierce Chemical Co., Rockford, Ill). Total Trk protein was immunoprecipitated using 3 μl of anti–panTrk 203 (the gift of D. Kaplan, Montreal Neurological Institute, Montreal, Canada), and TrkA using 2 μl of anti–TrkA RTA (the gift of L. Reichardt). The immunoprecipitates were collected with protein A–Sepharose (Pharmacia Biotechnology Inc., Piscataway, NJ) for 1.5 h at 4°C followed by centrifugation.
For Western blot analysis, precipitates were washed three times with cold lysis buffer, boiled in sample buffer (2% SDS, 100 mM dithiothreitol, 10% glycerol, and 0.05% bromophenol blue) for 5 min, and electrophoresed on 8% SDS–polyacrylamide minigels. After electrophoresis, proteins were transferred to 0.2 μm nitrocellulose for 1 h at 0.5 Amps, and the membrane washed twice for 10 min in TBS. For all antibodies except anti-phosphotyrosine, for which membranes were blocked in 2% BSA (Sigma Chemical Co.), membranes were blocked in 5% nonfat dry milk, in TBS for 2.5 h. Membranes were then washed twice for 10 min in TBS, and the primary antibodies were used overnight at 4°C, at a dilution of 1:5,000 for anti-phosphotyrosine 4G10 (Upstate Biotechnology Inc., Lake Placid, NY), and 1:1,000 or 1:2,000 for anti–panTrk 203. Secondary antibodies were incubated for 1.5 h at room temperature, and were used at a dilution of 1:10,000 or 1:20,000 for a goat anti–mouse HRP antibody (Boehringer Mannheim Corp., Indianapolis, IN; used for anti-phosphotyrosine), 1:10,000 for a goat anti–rabbit HRP antibody (Boehringer Mannheim Corp.; used for anti-TrkA), and 1:2,000 for protein A–HRP (Sigma Chemical Co.; used for anti-panTrk). Detection was carried out using enhanced chemiluminescence (Amersham Corp., Arlington Heights, IL) and XAR x-ray film (Eastman Kodak, Rochester, NY). Results were quantitated by image analysis.
For biochemical analysis of c-jun, the collected cells were incubated 20 min at 4°C in lysis buffer plus 0.5% SDS. After protein determination, 50 μg of protein was boiled in sample buffer, and separated on a 10% acrylamide gel, transferred, and then Western blotted with antibodies specific to c-jun at a 1:1,000 dilution, using the protocols described above. For the c-jun experiments, two different antibodies were used, anti–c-jun J3192 (Transduction Labs., Lexington, KY), and anti–c-jun SC-822 (Santa Cruz Biotech, Santa Cruz, CA). Both antibodies produced similar results on Western blots.
Analysis of BDNF−/− and p75NTR−/− Mice
Mice heterozygous for a targeted mutation in the BDNF gene (Ernfors et al., 1994) and homozygous for a targeted mutation in the p75NTR gene (Lee et al., 1992) were obtained from Jackson Labs. (Bar Harbor, ME). The BDNF−/− mice were maintained in a C129/BalbC background. The p75NTR−/− mice were originally generated in a C129/BalbC background (Lee et al., 1992), were crossed back into a C129 background before purchase from Jackson Labs., and were then maintained as homozygotes. Progeny from BDNF heterozygote crosses were screened for the mutant allele(s) using PCR. To amplify the wild-type allele, two PCR oligonucleotide primers were used (5′-ATGAAAGAAGTAAACGTCCAC-3′ and 5′-CCAGCAGAAAGAGTAGAGGAG-3′) to generate a 275-nucleotide product. To amplify the mutant allele, two oligonucleotide primers specific to this allele were used (5′-GGGAACTTCCTGACTAGGGG-3′ and 5′-ATGAAAGAAGTAAACGTCCAC-3′) to generate a 340-nucleotide product.
For morphometric analysis, the SCG were removed and immersion fixed in 4% paraformaldehyde in phosphate buffer (PB) from 1 h to overnight at 4°C. Ganglia were cryoprotected in graded sucrose solutions, 7-μm- thick sections were serially cut on a cryostat, and every section was collected and thaw mounted onto chrom-alum–subbed slides. Slides were stained with cresyl violet and morphometric analyses were performed using a computer-based image analysis system (Biocom, Paris, France). Neuronal sizes were determined by randomly measuring the cross-sectional area of neurons containing a distinct nucleolus. Neuronal numbers were determined by counting all neuronal profiles with nucleoli on every third section for the BDNF−/− mice, and every fourth section for the p75NTR−/− mice, as per Coggeshall (1984). This sampling frequency (every 21 and 28 μm, respectively) ensures that neurons are not double counted, since the average neuronal diameter does not exceed 21 μm in any of the groups examined (see Results section). This method does not correct for split nucleoli. As a second approach, neuronal numbers were estimated using measurements of total SCG volume and neuronal density. To determine ganglion volume, the combined cross-sectional area of every third (BDNF−/−) or every fourth (p75NTR−/−) serial section was measured. This cross-sectional area was normalized for total number of sections, and multiplied by 7 μm (the section thickness) to obtain the total SCG volume. To measure mean neuronal density, the number of neurons in a randomly applied sampling window of 175 μm × 175 μm was counted. For the BDNF−/− and BDNF+/+ animals, the sampling window was 182 μm × 182 μm, and the mean neuronal densities were then normalized to a 175 μm × 175 μm sampling window (a normalization factor of 1.08). To estimate neuronal number from these two parameters, we calculated SCG volume x (mean neuron number/214,375 μm3), where 214,375 μm3 is the sampling window volume (175 μm × 175 μm × 7 μm). This calculation was performed either (a) on an individual ganglion basis or (b) using the mean volume and the mean pooled neuronal density obtained from at least three individual animals. Since the numbers obtained were similar in both cases, only the latter calculation is presented in the results. Statistical results were expressed as the mean ± the standard error of the mean and were tested for significance by a one-tailed Student's t test.
Results
Differential Neurotrophin Binding to TrkA Versus p75NTR Correlates with Sympathetic Neuron Survival Versus Death
To determine whether differences in neurotrophin binding to TrkA versus p75NTR regulate neuronal survival, as suggested by our previous results (Belliveau et al., 1997), we compared NGF and NT-4, both of which bind only TrkA and p75NTR on sympathetic neurons. For these studies, postnatal day 1 sympathetic neurons were first selected for 5 d in 10 ng/ml NGF, washed free of NGF, exposed to varying concentrations of NGF and NT-4, and then analyzed for TrkA activation relative to neuronal survival. For the biochemical measurements, neurons were exposed to these two neurotrophins for 10 min, cell lysates were immunoprecipitated with anti-TrkA (RTA; Clary et al., 1994), and the precipitated TrkA was analyzed for autophosphorylation on Western blots using anti-phosphotyrosine. This biochemical analysis revealed that NT-4 was similar to NT-3 in its ability to activate TrkA on sympathetic neurons (Fig. 1 A; data not shown). NT-4 was, however, ∼10- to 20-fold less efficient at activating TrkA than NGF (Fig. 1 A), with 2.5 ng/ml NGF being equivalent to ∼50 ng/ml NT-4.
Figure 1.

(A) NT-4 activates TrkA on sympathetic neurons. Sympathetic neurons were selected in 10 ng/ml NGF for 5 d, washed free of neurotrophin, and then exposed to various concentrations of NGF (from 2.5 to 10 ng/ml) or NT-4 (50 and 100 ng/ml) for 10 min. Cellular lysates were immunoprecipitated with an antibody specific to TrkA, and probed first with anti-phosphotyrosine (anti-ptyr) to visualize TrkA autophosphorylation, and then reprobed with anti-panTrk to monitor total TrkA levels (anti-pantrk). All samples were normalized for equal amounts of protein. (B and C) BDNF does not activate Trk on sympathetic neurons. (B) To examine short-term Trk autophosphorylation, sympathetic neurons were washed free of NGF and exposed to 2.5, 5, or 10 ng/ml NGF or to 30 or 100 ng/ml BDNF for 10 min. Total Trk protein was immunoprecipitated with anti-panTrk, and Trk autophosphorylation analyzed by blotting with anti-phosphotyrosine (anti-ptyr). In the bottom panel, the same blot was reprobed with panTrk antibody 203 (anti-pantrk) to ensure that similar amounts of total Trk protein were present in all lanes. (C) To examine long-term Trk autophosphorylation, sympathetic neurons were selected in 10 ng/ml NGF, and then switched to 10 ng/ml NGF (10 NGF), 110 ng/ml NGF (110 NGF), or 10 ng/ml NGF + 100 ng/ml BDNF (10 NGF + 100 BDNF). Analysis of Trk autophosphorylation was performed as in B. In all cases, samples were normalized for equal amounts of protein, and blots were reprobed for total Trk protein levels. (D) KCl treatment does not lead to Trk activation on sympathetic neurons. Sympathetic neurons were washed free of neurotrophin and switched to 10 ng/ml NGF for 10 min, or to 25 or 50 mM KCl for 10 min or 24 h. Trk autophosphorylation was analyzed as in B. (E and F) BDNF-mediated activation of p75 leads to increased phosphorylation of c-jun. Sympathetic neurons were selected in 50 ng/ml NGF, washed free of neurotrophin, and then switched to 10 ng/ml NGF, or 12.5 or 25 mM KCl plus or minus BDNF. 18 h later, cellular lysates were analyzed for c-jun on Western blots. (E) As previously reported (Ham et al., 1995), the size of c-jun increases in response to NGF withdrawal, a shift indicative of increased phosphorylation. 200 ng/ml BDNF caused a similar size shift in the presence of 12.5 mM KCl. (F) This size shift was specific to BDNF and not to KCl, as shown here with 25 mM KCl plus or minus 100 ng/ml BDNF.
To compare the survival promoting activity of these two TrkA/p75 ligands, we then cultured the NGF-dependent neurons in 0–10 ng/ml NGF or 10–100 ng/ml NT-4 for 2 d (Fig. 2 A). MTT assays revealed that 2.5 and 5 ng/ml NGF supported ∼55% and 85% sympathetic neuron survival, respectively. In contrast, 50 and 100 ng/ml NT-4, which lead to approximately equal levels of TrkA activation (Fig. 1 A) supported only ∼10% and 25% neuronal survival, respectively. Thus, when compared to NGF, NT-4 was ∼40-fold less potent at mediating neuronal survival, and ∼5-fold less efficient when normalized for similar levels of TrkA autophosphorylation (Figs. 1 A and 2 A). Since NT-4 binds far less well to TrkA than NGF, and more efficiently to p75NTR (Rodriguez-Tebar et al., 1992), we hypothesized that, at low levels of TrkA activity, coincident activation of p75NTR might “override” the TrkA survival signal.
Figure 2.

(A) NT-4 is fivefold less efficient than NGF at supporting sympathetic neuron survival at equivalent levels of TrkA autophosphorylation. A comparison of sympathetic neuron survival in response to 2.5–10 ng/ml NGF versus 10–100 ng/ml NT-4, as monitored using MTT assays that measure mitochondrial function and cell survival. Neonatal sympathetic neurons were cultured in 10 ng/ml NGF for 5 d in 48-well dishes, washed free of neurotrophin-containing medium, and then incubated for 2 d in various concentrations of NGF or NT-4, as indicated on the x-axis. Each point represents the values pooled from three independent sets of survival assays, each of which was performed in quadruplicate. In these assays, absolute values are normalized so that the value obtained without neurotrophin is 0% survival, whereas that obtained with 10 ng/ml NGF is considered 100% survival. Error bars represent the standard error. (B–D) BDNF decreases sympathetic neuron survival in the presence of limiting quantities of NGF (B and C) or KCl (D). Neurons were selected in 50 ng/ml NGF for 5 d, and then switched to 2.5 ng/ml NGF, 5 ng/ml NGF, 10 ng/ml NGF, 12.5 mM KCl, or 25 mM KCL, with or without 100 or 200 ng/ml BDNF for 2 d. Survival was monitored by MTT assays. Results are expressed relative to those obtained with 10 ng/ml NGF, and represent the mean ± standard error. B and D include the combined data from three independent experiments performed in triplicate. C represents the data from one representative experiment performed in quadruplicate (**P < 0.05, ***P < 0.005 relative to NGF [B and C] or KCl [D] alone). (E) BDNF decreases sympathetic neuron survival in a concentration-dependent fashion. Neurons were selected in 50 ng/ml NGF for 5 d, and then switched to 12.5 mM KCl with or without concentrations of BDNF ranging from 30 to 200 ng/ml for 2 d. Survival was monitored by MTT assays. Results are normalized relative to those obtained with 12.5 mM KCl alone, and represent the mean ± standard error from the combined data of four independent experiments (***P < 0.005 relative to 12.5 mM KCl alone).
To test this hypothesis, we cultured sympathetic neurons in limiting amounts of NGF with and without the presence of a p75NTR ligand, BDNF. We chose the neurotrophin BDNF, which binds to p75NTR (Rodriguez-Tebar et al., 1990), since BDNF does not bind to the two Trk receptors present on neonatal sympathetic neurons, TrkA and TrkC (Lamballe et al., 1993). We first confirmed biochemically that BDNF is selective for p75NTR on these neurons, by examining short- and long-term total Trk autophosphorylation. For the short-term experiments, neonatal sympathetic neurons were grown for 5 d in 10 ng/ml NGF, and were then exposed to BDNF for 10 min. Cellular lysates were immunoprecipitated with an antibody that recognizes all members of the Trk receptor family (anti–panTrk 203; Hempstead et al., 1992), and the precipitated Trk examined for BDNF-induced tyrosine autophosphorylation by probing Western blots with anti-phosphotyrosine. These studies demonstrated that concentrations of BDNF ranging from 30 to 100 ng/ml (Fig. 1 B) did not lead to short-term activation of Trk receptors on sympathetic neurons.
For the long-term experiments, sympathetic neurons were selected for 5 d in 10 ng/ml NGF, and then were switched for 2 d either to 10 ng/ml NGF plus 100 ng/ml BDNF, or to 10 ng/ml NGF plus 100 ng/ml NGF; 10 ng/ml NGF was required in these experiments to support neuronal survival. 2 d later, the Trk proteins were analyzed for BDNF or NGF-induced tyrosine phosphorylation (Fig. 1 C). These studies revealed that, as previously reported (Belliveau et al., 1997), 110 ng/ml NGF led to higher levels of long-term Trk autophosphorylation than did 10 ng/ml NGF. However, Trk autophosphorylation in 10 ng/ml NGF plus 100 ng/ml BDNF was unchanged relative to 10 ng/ml NGF alone. Thus, as predicted, BDNF does not activate either TrkA or TrkC on sympathetic neurons, and represents a p75NTR-selective ligand for these experiments.
To test the hypothesis that at low levels of TrkA activity, coincident activation of p75NTR could override the TrkA survival signal, we cultured sympathetic neurons in 50 ng/ml NGF for 5 d, and then switched them into 2.5 ng/ml NGF for 2 d, with or without BDNF. 50 ng/ml NGF was used for the initial selection since we have previously demonstrated (Belliveau et al., 1997) that p75NTR levels are significantly higher in sympathetic neurons cultured in 50 ng/ml NGF versus 10 ng/ml NGF, whereas TrkA levels remain constant. 2.5 ng/ml NGF was chosen for the survival assay since this concentration of NGF is limiting for TrkA activation and neuronal survival under our assay conditions (Fig. 2 A). MTT assays demonstrated that 2.5 ng/ml NGF alone mediated survival of 50–55% of sympathetic neurons, and that the addition of 100 or 200 ng/ml BDNF reduced this survival by 43 and 58%, respectively (Fig. 2 B). In contrast, BDNF had less of an effect when TrkA activation was increased; addition of 100 ng/ml BDNF in the presence of 5 ng/ml NGF reduced survival by 25% (Fig. 2 C), and addition of BDNF to 10 ng/ml NGF (which supports 100% survival) had no effect (Fig. 2 C). Thus, BDNF reduces TrkA-mediated neuronal survival, but only when the survival signal is suboptimal.
BDNF-mediated Activation of p75NTR Causes Sympathetic Neuron Apoptosis in the Absence of TrkA Activation
One explanation for these data is that BDNF causes activation of p75NTR, and that a p75NTR-mediated apoptotic signaling cascade can override survival signals mediated by low levels of TrkA activation. An alternative explanation is that BDNF competes with low levels of NGF for binding to p75NTR, thereby affecting NGF binding to TrkA (Barker and Shooter, 1994). To differentiate these two possibilities, we performed survival assays using KCl instead of NGF to maintain sympathetic neuron survival. We first confirmed, as previously reported (Franklin et al., 1995), that KCl maintained sympathetic neuron survival without activating Trk by measuring Trk autophosphorylation in neurons exposed to KCl for 10 min or 24 h in the absence of neurotrophin (Fig. 1 D). Specifically, after an initial selection for 5 d in NGF, neurons were washed free of NGF, and then were exposed to 25 or 50 mM KCl for 10 min or 24 h. Cellular lysates were immunoprecipitated with anti-panTrk, and the precipitates were analyzed by Western blot analysis with anti-phosphotyrosine (Fig. 1 D). These experiments confirmed that, as previously reported (Franklin et al., 1995), KCl did not lead to an increase in autophosphorylation of Trk either in the short or long term.
We then performed survival assays with KCl as we had done for NGF and BDNF; sympathetic neurons selected in 50 ng/ml NGF for 5 d were switched to KCl with or without BDNF for 2 d. These assays were performed under two conditions. In one case, we used 25 mM KCl, which mediates maximal sympathetic neuron survival (130% relative to 10 ng/ml NGF); and in the second, we used 12.5 mM KCl, which is suboptimal, mediating ∼80– 85% survival (Fig. 2 D). MTT assays revealed that the addition of 100 ng/ml BDNF to 12.5 or 25 mM KCl decreased neuronal survival significantly in both cases (Fig. 2 D). To determine the concentration range of this BDNF-mediated killing, we performed similar experiments using 12.5 mM KCl with the addition of 30–200 ng/ml BDNF. MTT assays revealed a concentration-dependent decrease in neuronal survival commencing at 30 ng/ml BDNF (Fig. 2 E). Thus, BDNF was sufficient to override survival signals mediated by KCl in a concentration-dependent fashion.
To demonstrate that the lack of neuronal survival detected using MTT assays corresponded to neuronal apoptosis, we monitored the neurons morphologically, and by TUNEL assays. Sympathetic neurons were first selected in 50 ng/ml NGF, were washed free of neurotrophin, and then were switched to 12.5 or 25 mM KCl with or without BDNF. As controls, neurons were switched to 50 ng/ml NGF or were maintained without NGF. 18 h after the switch, neurons maintained in 50 ng/ml NGF or 12.5 mM KCl exhibited no TUNEL-positive nuclei, whereas neurons withdrawn from NGF or maintained in 12.5 mM KCl + 100 ng/ml BDNF showed many TUNEL-positive nuclei (Fig. 3, A–D). Similar results were obtained with neurons maintained in 25 mM KCl (data not shown). By 48 h, morphological examination revealed that all neurons maintained in 50 ng/ml NGF were alive (Fig. 3 E), while in 12.5 mM KCl, many neurons were alive, but some were also showing morphological signs of cell death (Fig. 3 F), consistent with the MTT survival assays (Fig. 2 D). In contrast, no neurons were apparently alive after 48 h of culture in 0 NGF (Fig. 3 G) or in 12.5 mM KCl plus 100 ng/ml BDNF (Fig. 3 H).
Figure 3.
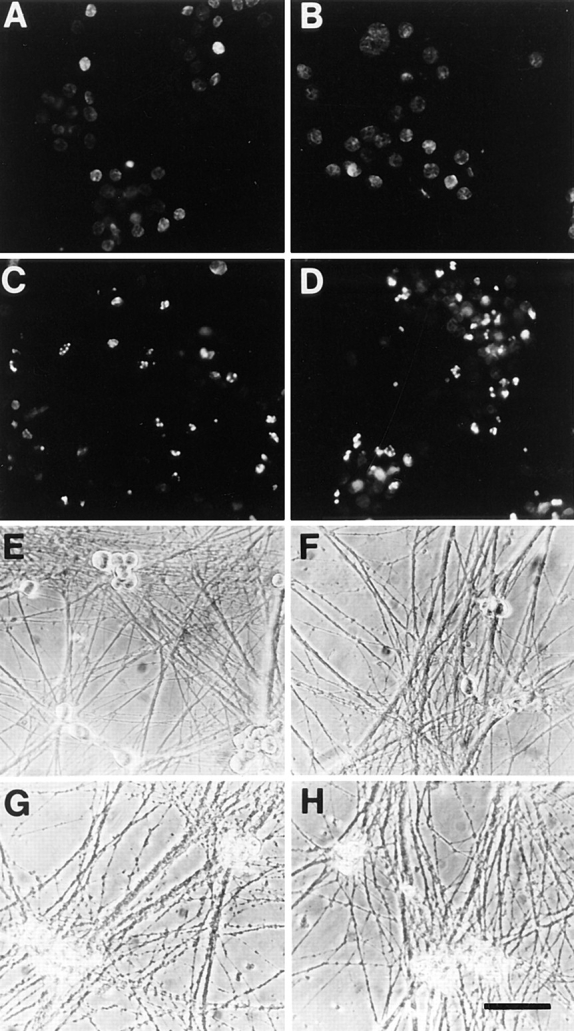
Sympathetic neurons undergo apoptosis in response to BDNF. (A–D) Fluorescence photomicrographs of sympathetic neurons analyzed by TUNEL labeling. Neurons were selected in 50 ng/ml NGF for 5 d, washed free of neurotrophin, and then switched to 12.5 mM KCl with (D) or without (B) 100 ng/ml BDNF for 18 h. As controls, sister cultures were maintained in 50 ng/ml NGF (A) or withdrawn from NGF (C). 18 h later, neurons were analyzed for apoptosis using TUNEL labeling for fragmented nuclear DNA. (E–H) Phase-contrast photomicrographs of cultured sympathetic neurons. Neurons were cultured as above, and then switched to 12.5 mM KCl with (H) or without (F) 100 ng/ml BDNF for 48 h. As controls, sister cultures were maintained in 50 ng/ml NGF (E) or withdrawn from NGF (G). Bar, 74 μm.
We also performed similar TUNEL assays on sympathetic neurons maintained in suboptimal concentrations of NGF and coincidentally exposed to BDNF. Specifically, sympathetic neurons were cultured for 5 d in 50 ng/ml NGF, and switched to 5 ng/ml NGF plus or minus 100 ng/ml BDNF. TUNEL labeling was performed 24 h after this switch; this analysis demonstrated a statistically significant increase in the number of TUNEL-labeled cells in the cultures exposed to BDNF (P < 0.03; data not shown).
Together, these data suggest that BDNF leads to p75NTR activation, and that this activation causes neuronal apoptosis. To directly test this hypothesis, we used the function-blocking p75NTR antibody, REX (Weskamp et al., 1991; Cassaccia-Bonnefil et al., 1996). Specifically, neurons were cultured in 50 ng/ml NGF for 5 d, and then were switched to 25 mM KCl with or without the addition of 100 ng/ml BDNF and/or REX. 2 d later, MTT assays were used to monitor neuronal survival. These experiments (Fig. 4 A) demonstrated that REX was able to inhibit the BDNF-mediated apoptosis of sympathetic neurons, indicating that p75NTR mediates most, if not all, of the apoptotic effects of BDNF.
Figure 4.

(A) The p75NTR antibody, REX, inhibits the BDNF-mediated apoptosis of sympathetic neurons. A comparison of sympathetic neuron survival, as monitored using MTT assays, in response to 25 mM KCl ± BDNF with and without the presence of the function-blocking anti-p75NTR, REX. Sympathetic neurons were cultured in 50 ng/ml NGF for 5 d, washed free of neurotrophin-containing medium, and then incubated for 2 d in 25 mM KCl or 25 mM KCl + 100 ng/ml BDNF with or without REX. Results of two representative experiments, each of which was performed in triplicate, are shown. In all cases, results represent the mean ± standard error, and are normalized so that the survival mediated by 25 mM KCl alone is 100%. REX by itself had no significant effect (P > 0.05) on sympathetic neuron survival as mediated by 25 mM KCl. (***P < 0.004 for the comparison between 25 mM KCl + REX and 25 mM KCL + 100 ng/ml BDNF + REX). (B) Death of cultured p75NTR−/− neurons is delayed after NGF withdrawal. Time course of survival of cultured p75NTR−/− and p75NTR+/+ (Control) neurons following NGF withdrawal. Representative fields of neurons were counted immediately upon NGF withdrawal, and again every 24 h. Results are normalized so that the number of neurons at the time of NGF withdrawal is 100%. Results represent the mean ± standard error of the combined data from five individual experiments each for the p75NTR−/− and control neurons; every individual experiment was performed in triplicate. (***P < 0.002 for the comparison between p75NTR−/− and control neurons at each time point).
BDNF Leads to Increased Phosphorylation of c-jun
Previous work has demonstrated that increased expression and phosphorylation of c-jun is necessary for sympathetic neuron apoptosis caused by NGF withdrawal (Estus et al., 1994; Ham et al., 1995). Moreover, p75NTR has been demonstrated to activate the kinases that phosphorylate c-jun, the jun kinase (JNK) family, in cultured oligodendrocytes (Casaccia-Bonnefil et al., 1996). We therefore examined c-jun in sympathetic neurons induced to undergo apoptosis by p75NTR activation. To perform these experiments, neurons were selected for 5 d in 50 ng/ml NGF, and then were switched to 12.5 or 25 mM KCl plus or minus BDNF. 18 h after this switch, cellular lysates were analyzed by probing Western blots with an antibody to c-jun. As controls, neurons were maintained in 10 ng/ml NGF, or were withdrawn from NGF.
These experiments demonstrated that, as previously reported (Ham et al., 1995), NGF withdrawal led to a shift in c-jun to a larger size, indicative of phosphorylation (Fig. 1 E). A similar shift in c-jun size was detected in neurons cultured in either 12.5 (Fig. 1 E) or 25 (Fig. 1 F) mM KCl plus 100 or 200 ng/ml BDNF, relative to KCl alone. Thus, BDNF induces c-jun phosphorylation in sympathetic neurons coincident with neuronal apoptosis, suggesting that one of the signal transduction pathways leading to neuronal apoptosis in response to NGF withdrawal is similar to that induced by p75NTR activation.
Sympathetic Neuron Number Is Increased and Size Decreased in BDNF−/− Mice
These in vitro experiments indicate that, when neonatal sympathetic neurons are exposed to a suboptimal survival signal, activation of p75NTR by BDNF leads to neuronal apoptosis. If a similar cellular mechanism occurred in vivo during the period of naturally occurring cell death, then one would predict that in the BDNF−/− mice (Ernfors et al., 1994; Jones et al., 1994), there would be an increase in the number of sympathetic neurons. To test this prediction, we analyzed the number of sympathetic neurons of the superior cervical ganglion at postnatal day 15, immediately after the period of target competition and cell death (Hendry and Campbell, 1976; Wright et al., 1983). Specifically, SCG from BDNF+/+ (Fig. 5 B) and −/− (Fig. 5 A) littermates were serially sectioned at 7 μm, and the number of neurons with an identifiable nucleolus were counted on every third section. This analysis demonstrated a statistically significant increase of 36% in the relative number of sympathetic neurons in BDNF−/− mice relative to their BDNF+/+ littermates (BDNF+/+: 15,690 ± 617; BDNF−/−: 21,318 ± 1,627; P = 0.009; n = 6 animals each) (Fig. 6 C; Table I).
Figure 5.

Morphology of sympathetic neurons of the superior cervical ganglion in BDNF−/− and p75NTR−/− mice. Photomicrographs of cresyl violet-stained cross-sections of the SCG from P15 BDNF−/− mice (A), P15 BDNF+/+ littermates (B), P23 p75NTR−/− mice (C), adult C129 mice (D), and adult p75NTR−/− mice (E). Bar, 38.5 μm.
Figure 6.

At P15 to P23, sympathetic neuron number is increased in the SCG of BDNF−/− and p75NTR−/− mice. (A) Cross-sectional area of P15 BDNF−/− neurons relative to control neurons from animals of the same background, plotted as a size distribution histogram, in bins of 50 μm2. Note that the entire population of BDNF−/− sympathetic neurons is shifted to a smaller size relative to BDNF+/+ (Control) neurons of the same age. (B) Mean cross-sectional areas of sympathetic neurons of the SCG in p75NTR−/− mice at various developmental time points relative to control C129 mice. Note that from P1 to P4, and in adulthood, the size of sympathetic neurons is similar in the presence or absence of p75NTR, but that at P23, neurons lacking p75NTR are significantly smaller. Neuron size is expressed as cross-sectional area in μm2, and error bars represent the standard error (**P < 0.05). (C) The number of sympathetic neurons of the SCG is increased in P15 BDNF−/− animals relative to their control littermates, as determined by counting. The number of neurons in each of six control BDNF+/+ and BDNF−/− animals are shown; each bar represents the number of neurons in the SCG of one animal of the indicated genotype. The means of these two groups are significantly different (***P < 0.01). The mean ± the standard error are summarized in Table I as “Total counted neuron number.” (D) Sympathetic neuron number in the SCG of control C129 mice at developmental time points encompassing the period of naturally occurring sympathetic neuron death. The number of neurons counted in the SCG at P1, P4, P23, and adulthood are shown; each bar represents the number of neurons in the SCG of one C129 animal of the indicated age. Five animals were analyzed at P1, three at P4, five at P23, and five at adulthood. The mean ± standard error of these groups are plotted in F. (E) Sympathetic neuron number in the SCG of p75NTR−/− mice at developmental time points encompassing the period of naturally occurring sympathetic neuron death. The number of neurons counted in the SCG at P4, P23, and adulthood are shown; each bar represents the number of neurons counted in the SCG of one p75NTR−/− animal of the indicated age. Four animals were analyzed at P4, five at P23, and five adults. The mean ± standard error of these groups are plotted in F. (F) Comparison of the time course of naturally occurring sympathetic neuron death in the SCG of p75NTR−/− versus control (C129 control) animals. The points represent the mean ± standard error of the counts shown in D and E. There is a highly significant decrease in neuronal number from P4 to P23 in the control C129 mice, and from P23 to adulthood in the p75NTR−/− mice (***P < 0.007). The means for all of these groups are summarized in Table I as “Total counted neuron number”.
Table I.
Morphometric Analysis of the SCG from BDNF−/− and p75NTR−/− Mice
| Group | SCG volume | Neuron number per 214,375 μm3 | Total estimated neuron number | Total counted neuron number | ||||
|---|---|---|---|---|---|---|---|---|
| mm3 | ||||||||
| C129 P1 | 0.106 ± 0.008 | 56 ± 7.2 | 27,698 ± 3,846 | 25,328 ± 927 | ||||
| C129 P4 | 0.121 ± 0.001 | 40 ± 0.3 | 22,577 ± 171 | 23,196 ± 568 | ||||
| C129 P23 | 0.120 ± 0.012 | 32 ± 2.2 | 17,912 ± 1,355 | 13,415 ± 1,683 | ||||
| C129 adult | 0.121 ± 0.009 | 30 ± 1.5 | 16,933 ± 906 | 14,667 ± 799 | ||||
| p75NTR−/− P4 | 0.170 ± 0.021 | 39 ± 2.9 | 30,927 ± 2,576 | 32,468 ± 2,362 | ||||
| p75NTR−/− P23 | 0.202 ± 0.028 | 44 ± 6.7 | 41,460 ± 7,197 | 40,275 ± 1,368 | ||||
| p75NTR−/− adult | 0.172 ± 0.005 | 26 ± 3.9 | 20,860 ± 3,223 | 22,254 ± 1,404 | ||||
| BDNF+/+ P15 | 0.102 ± 0.014 | 30 ± 0.8 | 14,358 ± 437 | 15,690 ± 617 | ||||
| BDNF−/− P15 | 0.083 ± 0.004 | 44 ± 0.8 | 17,150 ± 328 | 21,318 ± 1,627 |
Data are presented for the SCG from two different groups of experimental versus control animals; P15 BDNF+/+ and BDNF−/− littermates in a BalbC/C129 background, and p75NTR−/− mice in a C129 background versus control C129 animals. “SCG volume” and “Neuron number per 214,375 μm3” are expressed as the mean ± SE, and were measured as described in the Materials and Methods (n = 3 animals for each group). The “Total estimated neuron number” is calculated as mean SCG volume × (mean neuron number/214,375 μm3). The “Total counted neuron number” represents the mean ± SE of total SCG neurons determined by counting neurons with a nucleolus in every third (BDNF−/− and BDNF+/+) or fourth (C129 and p75NTR−/−) section, as described in the Materials and Methods. The data obtained from each individual animal is illustrated in Fig. 6. Note that the total estimated neuron numbers and the total counted neuron numbers are statistically similar (P > 0.05 in all cases).
To confirm the increase in number of sympathetic neurons in the absence of BDNF, we estimated neuronal number using a second approach. Specifically, we first determined the volume of the SCG by measuring the total cross-sectional area of every third section throughout the entirety of individual ganglia, and then normalized for sampling frequency and section thickness (Table I). We then determined the mean number of neurons in a randomly selected sampling volume of 214,375 μm3 from three animals of each genotype (Table I). This latter analysis demonstrated that neuronal density was significantly higher in the SCG of BDNF−/− animals versus their BDNF+/+ littermates (P < 0.0001; Table I; Fig. 5, A and B). The volume and density measurements were then used to estimate the total number of neurons within the SCG as 14,358 ± 437 in the BDNF+/+ animals and 17,150 ± 328 in the BDNF−/− animals (Table I), a statistically significant increase (P = 0.003). These numbers are statistically similar to those derived by direct counting (P > 0.05), and support the conclusion that there are increased numbers of sympathetic neurons in the BDNF−/− animals.
Finally, we determined the size of sympathetic neurons from the BDNF+/+ and BDNF−/− animals. This morphological analysis revealed a statistically significant decrease in sympathetic neuron cross-sectional area (Figs. 5, A and B; and 6 A) of ∼1.6-fold in the absence of BDNF (BDNF+/+: 222.24 ± 6.50 μm2; BDNF−/−: 142.49 ± 9.21 μm2; P = 0.002; n = 3 animals each), a finding that is consistent with increased numbers of neurons competing for limiting amounts of NGF. Thus, these findings indicate that BDNF in vivo may well mediate sympathetic neuron apoptosis, as we have documented in vitro.
Sympathetic Neuron Death Is Developmentally Delayed in p75NTR−/− Mice
If, as our results from the BDNF−/− mice suggest, ligand-mediated activation of p75NTR is essential for naturally occurring sympathetic neuron death, then this process should also be perturbed in the absence of p75NTR. To test this prediction, we examined sympathetic neuron numbers in the p75NTR−/− mice during the normal period of developmental death, which occurs during the first few postnatal weeks in rats (Hendry and Campbell, 1976; Hendry, 1977; Wright et al., 1983). Since the p75NTR−/− mice analyzed in this study have been bred into a C129 background and are maintained as homozygotes, we made two types of comparisons: an internal comparison from birth to adulthood in the p75NTR−/− mice, and a comparison of p75NTR−/− mice with control animals of the same C129 background.
For this analysis, we collected serial 7-μm-thick sections of the SCG, and counted the total number of neurons containing a nucleolus in every fourth section (Fig. 5, C–E). Analysis of control C129 mice revealed that, as previously reported in rats, naturally occurring sympathetic neuron death in the SCG occurred between birth and P23. Sympathetic neuron number at P1 was 25,328 ± 927, at P4 was 23,196 ± 568, at P23 was 13,415 ± 1,682 (a number similar to that observed in the BDNF+/+ animals at P15 [Table I]), and by adulthood was 14,667 ± 799 (Fig. 6, D and F; Table I). These counts demonstrated a 42% decrease in number of SCG neurons between birth and P23, after which numbers remained constant (P = .0003, P4 versus P23; P > 0.05, P23 versus adult; n = 5 P1, 3 P4, 5 P23, and 5 adults). In contrast, in p75NTR−/− mice, the neuron number at P4 was 32,468 ± 2,362, at P23 was 40,275 ± 1,368, and by adulthood was 22,254 ± 1,404 (Fig. 6, E and F; Table I). Thus, in the absence of p75NTR, sympathetic neuron number did not decrease between birth and P23, as seen in controls, but instead increased by 24% between birth and P23, and then decreased 45% between P23 and adulthood (P = 0.02, P4 versus P23; P = 0.006, P23 versus adult; n = 4 P4, 5 P23, and 5 adults). This developmental profile of sympathetic neuron number in the p75NTR−/− mice is very similar to that observed when neonatal rats are given NGF systemically from P6 to P21 (Hendry and Campbell, 1976); naturally occurring sympathetic neuron death is eliminated, and the number of sympathetic neurons in the SCG increases 20% between P6 and P21, an increase attributed to the rescue of sympathetic neurons that are born postnatally (Hendry, 1977).
To obtain an independent estimate of sympathetic neuron number over this developmental interval, we measured SCG volumes and neuronal density as we had for the BDNF−/− mice (Table I). Analysis of control C129 mice revealed that SCG volume increased from birth to P23 and then remained relatively constant into adulthood (Table I), as previously observed in rats (Hendry and Campbell, 1976). In contrast, in the p75NTR−/− mice, SCG volume increased from P4 to P23, but then decreased between P23 and adulthood. Moreover, p75NTR−/− SCG volume at P23 was 1.7-fold larger than SCG volume in the corresponding C129 animals (P < 0.05). Similar differences were noted in neuronal density. In the control C129 animals, neuronal density decreased from P4 to P23 (P < 0.05; Table I), and then remained constant into adulthood. In contrast, in the p75 NTR−/− SCG, neuronal density was not significantly altered between P4 and P23 (P = 0.27), but decreased between P23 and adulthood (P < 0.05; Table I).
The neuronal numbers estimated from the measurements of ganglia volume and neuronal density confirmed the conclusions obtained from counting neuronal profiles (Table I). In the control C129 SCG, estimated neuron number decreased between P4 and P23 (P = 0.013), and then remained constant, whereas in the p75 NTR−/− SCG, estimated neuron number increased from P4 to P23, and then decreased from P23 to adulthood (P = 0.030; Table I). Moreover, the absolute numbers obtained using this estimation were statistically similar to those obtained by direct counting (in all cases, P > 0.05; Table I). Thus, in the absence of p75NTR, normal developmental death of sympathetic neurons does not occur, but is instead replaced by a delayed period of neuronal loss between P23 and adulthood.
Since rat sympathetic neurons increase in size over this developmental interval, we also determined neuronal size. As reported in rats (Hendry and Campbell, 1976), neuron size increased gradually from birth to adulthood in control C129 animals (Figs. 5, and 6 B); the average cross-sectional area at P1 to P4 was 114.86 ± 6.24 μm2, at P23 was 240.73 ± 16.43 μm2, and in adults was 317.06 ± 26.60 μm2 (Fig. 6 B). These measurements correspond to diameters of 12.1 μm, 17.5 μm, and 20.1 μm, respectively, numbers very similar to those previously reported in rats (Hendry and Campbell, 1976). Sympathetic neurons from p75NTR−/− mice were similar in size to controls at P4 (cross-sectional area, 121.46 ± 6.44 μm2; 12.4 μm diam; P = 0.25, n = 3 animals each) and in adulthood (cross-sectional area, 318.72 ± 5.17 μm2; 20.1 μm diam; P = 0.47, n = 3 animals each) (Figs. 5, and 6 B). In contrast, at P23, p75NTR−/− sympathetic neurons were significantly smaller than their control counterparts (cross-sectional area, 167.47 ± 11.15 μm2; 14.6 μm diam; P = 0.011, n = 3) (Figs. 5, and 6 B), consistent with increased neurons in the p75NTR−/− mice competing for limiting amounts of trophic support.
Cultured p75NTR−/− Sympathetic Neurons Die More Slowly After NGF Withdrawal
The delay in sympathetic neuron death in the p75NTR−/− mice could be due to alterations either intrinsic or extrinsic to the neurons themselves. To differentiate between these two possibilities, we cultured p75NTR−/− sympathetic neurons and examined their rate of cell death after NGF withdrawal. Specifically, sympathetic neurons of the postnatal day 1 mouse SCG were cultured for 4 or 5 d in 50 ng/ml NGF, washed to remove the NGF, and then maintained in 0 or 10 ng/ml NGF-containing media for 5 d. Fields of neurons were counted immediately upon NGF withdrawal, and at 24-h intervals subsequent to the withdrawal.
This analysis (Fig. 4 B) revealed p75NTR−/− sympathetic neurons died significantly more slowly than their wild-type counterparts in the absence of NGF, indicating that the delay in neuronal loss observed in vivo was intrinsic to the neurons themselves. Specifically, 48 h after NGF withdrawal, 81.6 ± 2.9% p75NTR−/− neurons counted at day 0 were still alive, compared to 55.6 ± 4.8% of control neurons (P < 0.005, n = 5 each). At 72 h, 72.7 ± 3.5% of p75NTR−/− neurons were alive versus 39.8 ± 3.9% of controls (P < 0.0001, n = 5), whereas at 96 h, 63.9 ± 5.8% of p75NTR−/− neurons were still alive, as opposed to 30.14 ± 2.9% of controls (P < 0.005, n = 5). By 120 h, the last time point examined, 59.8 ± 6.4% of p75NTR−/− neurons were still alive, compared to only 19.6 ± 2.3% of control neurons (P < 0.0005, n = 5). Moreover, at this final time point, all of the control neurons were clearly dying, with fragmented neurites, whereas the p75NTR−/− neurons were clearly still healthy with neurites intact.
Thus, the lack of p75NTR in sympathetic neurons themselves leads to a significant delay in apoptosis of these neurons after NGF withdrawal, results that are consistent with the in vivo delay in naturally occurring cell death.
Discussion
Data presented in this paper demonstrate that BDNF- mediated activation of p75NTR is both necessary and sufficient for the developmental apoptosis of sympathetic neurons competing for limiting amounts of survival factors. Specifically, these experiments indicate that, in culture, BDNF-mediated activation of p75NTR is sufficient to override suboptimal survival signals, whether they derive from TrkA activation or chronic depolarization, thereby leading to sympathetic neuron apoptosis. Coincident with this apoptosis, BDNF leads to phosphorylation of the immediate early gene product, c-jun, a biochemical event that is necessary for sympathetic neuron apoptosis after NGF withdrawal (Estus et al., 1994; Ham et al., 1995), presumably via activation of p75NTR signaling. The in vivo relevance of these culture experiments is indicated by two findings. First, in BDNF−/− mice (Ernfors et al., 1994; Jones et al., 1994), sympathetic neuron number is increased relative to BDNF+/+ littermates, an effect consistent with the lack of activation of p75NTR by BDNF. This is the first time such an increase in neuronal number has been reported in neurotrophin knockout mice. Second, naturally occurring sympathetic neuron death does not occur normally in p75NTR−/− animals, but there is instead delayed neuronal loss between P23 and adulthood. This delay in neuronal loss is intrinsic to the neurons themselves, since p75NTR−/− sympathetic neurons display a similar delayed death in culture. Thus, although sympathetic neuron death can occur without p75NTR, this receptor is required for developmental apoptosis to occur rapidly and appropriately.
What is the biological rationale for having two neurotrophin receptors, one of which, TrkA, mediates survival, and one of which, p75NTR, mediates apoptosis? We propose that p75NTR provides a molecular mechanism for ensuring rapid and active apoptosis when a neuron is unsuccessful in competing for adequate amounts of the appropriate neurotrophin. If a sympathetic neuron reaches the appropriate target and sequesters NGF, TrkA is robustly activated, and any coincident activation of p75NTR is insufficient to override this survival signal. Conversely, if a neuron is late-arriving and/or reaches an inappropriate target, then TrkA would be only weakly induced as a consequence of the lack of NGF, and p75NTR would be robustly activated by neurotrophins such as BDNF. Our data indicate that the net outcome of such a scenario would be the rapid apoptotic elimination of that neuron. Such a perturbation in the normal period of naturally occurring cell death could at least partially explain the perturbations in innervation observed previously in the p75NTR−/− mice; there is cholinergic hyperinnervation of the hippocampus in these mice (Yeo et al., 1997), and sympathetic innervation is perturbed, with targets such as the iris being normally innervated, and targets such as the pineal gland completely lacking sympathetic innervation (Lee et al., 1994). Data presented here indicate that this perturbed sympathetic innervation is not because of a deficit in neuronal number, but is instead presumably due to the failure of p75-dependent mechanisms that are essential for matching neurons to their target organs during the period of naturally occurring cell death.
This model does not imply that p75NTR activation is the only means by which sympathetic neurons die in the absence of TrkA activation. In fact, our data demonstrating loss of sympathetic neurons between P23 and adulthood, and the death (albeit slow) of cultured p75NTR−/− sympathetic neurons argue that inadequate levels of TrkA activation do ultimately lead to the death of sympathetic neurons even in the absence of p75NTR. Instead, we propose that p75NTR activation represents a mechanism whereby exposure to an “inappropriate” neurotrophin is sufficient to cause a rapid apoptotic death even in the presence of low levels of TrkA activation. Such a mechanism would ensure that neurotrophins like NT-3 and NT-4, which weakly activate TrkA (as shown here) would not maintain sympathetic neuron survival and circumvent the absolute requirement for NGF, thereby leading to inappropriate target innervation. In fact, our data indicate that BDNF is not the only apoptotic p75NTR ligand in vivo; after naturally occurring cell death, there are only ∼36% more sympathetic neurons in the BDNF−/− mice, but at least twice as many in the p75NTR−/− mice, implying that other ligands, potentially NT-3 and/or NT-4, act through p75NTR during developmental sympathetic neuron death.
If ligand-mediated activation of p75NTR is required for it to play an essential role in sympathetic neuron apoptosis, then why is there a delay in death of cultured p75NTR−/− versus control neurons in the absence of exogenous neurotrophins? We have previously demonstrated (Causing et al., 1997) that BDNF is synthesized in the rodent SCG in vivo. Moreover, cultured sympathetic neurons synthesize BDNF, and BDNF can be detected in media conditioned by sympathetic neurons (Causing, C.G., R. Aloyz, and F.D. Miller, unpublished data). On the basis of these findings, we propose that autocrine BDNF may play a role in cultured sympathetic neuron apoptosis after NGF withdrawal, a hypothesis we are currently testing. Does sympathetic neuron–derived BDNF play a similar role in vivo? Although our data demonstrate an increased number of sympathetic neurons in BDNF−/− mice, it is unclear whether the BDNF that is critical for this effect derives from sympathetic neurons themselves and/or from sympathetic neuron targets. In this regard, we have previously demonstrated that BDNF made by sympathetic neurons in vivo regulates their level of preganglionic input (Causing et al., 1997). One could therefore speculate that BDNF made by sympathetic neurons might serve both to eliminate those sympathetic neurons that did not innervate an appropriate target (and thereby receive adequate TrkA activation), and to ensure adequate preganglionic input for those neurons that did successfully compete for target territory.
The developmental profile of sympathetic neuron number observed in the absence of p75NTR is very similar to that observed when neonates are supplied with exogenous NGF, lending support to the notion that the absence of p75NTR rescues sympathetic neurons from the normal period of naturally occurring cell death. When NGF is given systemically to rats from P6 to P21, not only are the neurons that normally die rescued, but as observed in the p75NTR−/− mice, there is an increase of ∼20% in the number of neurons (Hendry and Campbell, 1976). This increase is presumably because of the fact that, during the early period of naturally occurring cell death, sympathetic neuroblasts continue to proliferate. However, these later-born neurons are preferentially susceptible to programmed cell death, and the increase in number of neurons born is masked by the neuronal death that occurs at the same time (Hendry, 1977). It is therefore likely that the increased number of sympathetic neurons observed at P23 in p75NTR−/− mice is due to the same phenomenon.
What are the biochemical mechanisms that allow p75NTR activation to override a TrkA- or KCl-mediated survival signal? Our results demonstrate that BDNF stimulated a hyperphosphorylation of c-jun concomitant with the appearance of apoptosis. Hyperphosphorylation of c-jun has been previously noted in postnatal rat sympathetic neurons deprived of NGF (Ham et al., 1995). Serum and NGF withdrawal from PC12 cells also leads to sustained activation of JNK, the kinase that phosphorylates c-jun (Xia et al., 1995). The necessity of c-jun phosphorylation in these apoptotic responses was suggested by three types of experiments; (a) expression of dominant-inhibitory c-jun in rat sympathetic neurons protected against apoptosis caused by NGF deprivation (Ham et al., 1995); (b) microinjection of sympathetic neurons with antibodies to c-jun inhibited cell death induced by NGF deprivation (Estus et al., 1995); and (c) expression in PC12 cells of dominant-inhibitory MEKK1 (which inhibits the activity of JNK) reduced apoptosis, whereas expression of activated MEKK1, which indirectly activates JNK, increased apoptosis (Xia et al., 1995). The observation that BDNF also stimulates c-jun phosphorylation suggests that the MEKK1-MKK4-JNK pathway may function in apoptosis induced by both NGF deprivation and by activation of p75NTR. However, we do not know whether the components of the JNK pathway are necessary or sufficient for p75NTR-mediated cell death. p75NTR activates several other signaling proteins, including nuclear factor (NF)-kB and perhaps ceramide-activated kinases, which have been hypothesized to function in the induction of apoptosis (Casaccia-Bonnefil et al., 1996). The role of each of the p75NTR-stimulated signaling proteins in apoptosis awaits experiments in which the activities of these molecules are selectively blocked in BDNF-treated sympathetic neurons.
p75NTR activation could also antagonize a suboptimal TrkA survival signal by a direct action on the TrkA receptor itself. Such a mechanism might involve alterations in p75NTR–TrkA interactions to modify a high affinity signaling complex (Hempstead et al., 1991; Ross et al., 1995; Wolf et al., 1995) and/or a direct effect of p75NTR signaling on the TrkA receptor involving, for example, serine/ threonine phosphorylation by ceramide-activated kinases (MacPhee and Barker, 1997). However, our data indicate that even if p75NTR activation directly regulates TrkA function, there must be further downstream actions of p75NTR that lead to apoptosis. This conclusion derives from our finding that BDNF-mediated activation of p75NTR led to apoptosis in the absence of TrkA activation, when neuronal survival was maintained by chronic depolarization. It is also likely that cross talk between the p75NTR and TrkA is bidirectional. In this regard, Dobrowsky et al. (1994) have demonstrated that a p75NTR-mediated ceramide increase was antagonized by coincident activation of TrkA. Thus, one of the mechanisms whereby TrkA may support neuronal survival is by silencing a neurotrophin-mediated p75NTR death signal.
The finding of an increased number of sympathetic neurons in the BDNF−/− mice was predicted by our culture findings, but was unexpected in light of previous studies on this growth factor. The neurotrophins, including BDNF, are traditionally thought to regulate neuronal survival positively via the Trk family of tyrosine kinase receptors (for review see Snider, 1994). Here, BDNF antagonizes NGF- or KCl-induced survival signals through the activation of p75NTR. Such a mechanism, whereby the precise cohort of neurotrophins to which a neuron is exposed determines life versus death at the time of target innervation provides a level of sophistication that was previously unsuspected. However, this additional level of complexity provides a mechanism whereby neurons can recognize not only whether they are exposed to the “right” neurotrophin, but whether or not they are seeing the “wrong” neurotrophin. It is possible that such functional antagonism between Trk and p75NTR provides not only a mechanism for determining neuronal survival, but also a mechanism for elimination of axonal collaterals that innervate the inappropriate target.
Finally, although our findings indicate that p75NTR can signal to mediate sympathetic neuron apoptosis during the period of naturally occurring cell death, they do not imply that p75NTR-mediated signaling inevitably results in neuronal apoptosis. It is more likely that, like other members of this family, the outcome of p75NTR-mediated signal transduction cascades is a function of cellular context. For example, depending upon the cellular environment, p75NTR regulates cell migration (Anton et al., 1994), gene expression (Itoh et al., 1995), and can positively modulate TrkA signaling (Barker and Shooter, 1994; Verdi et al., 1994). However, our findings indicate that during the period of naturally occurring sympathetic neuron death, the apoptotic actions of p75NTR are essential for the establishment of appropriate neuron–target interactions.
Footnotes
We thank O.-M. Nicolesu (McGill University, Montreal, Canada) for her help with some of these experiments; A. Speelman and R. Varma (both of McGill University) for excellent technical assistance; and D. Kaplan and members of the Miller laboratory for their advice and input throughout the course of this work.
This study was supported by grants from the Canadian Medical Research Council and NeuroSciences Network (to F.D. Miller). During the course of this work, D.J. Belliveau was a Medical Research Council (MRC)/Genentech fellow, R. Aloyz a NeuroSciences Network fellow, and F.D. Miller a Killam Scholar. S.X. Bamji and C.D. Pozniak are funded by MRC studentships, and J. Kohn and C. Causing were supported by studentships from Fonds pour la Formation de Chercheurs et l'Aide à la Recherche and the Savoy Foundation, respectively.
S.X. Bamji and M. Majdan contributed equally to this work.
Address all correspondence to F.D. Miller, Center for Neuronal Survival, Montreal Neurological Institute, McGill University, 3801 rue University, Montreal, Quebec, Canada H3A 2B4. Tel.: (514) 398-4261. Fax: (514) 398-1319. E-mail: mdfm@musica.mcgill.ca
D.J. Belliveau's present address is Department of Anatomy and Cell Biology, University of Western Ontario, London, Canada M6A SC1.
1. Abbreviations used in this paper: BDNF, brain-derived neurotrophic factor; JNK, jun kinase; NT-3 and NT-4, neurotrophin-3 and neurotrophin-4; NTR, neurotrophin receptor; p75NTR, p75 neurotrophin receptor; SCG, superior cervical ganglion.
References
- Anton ES, Weskamp G, Reichardt LF, Matthew WD. Nerve growth factor and its low-affinity receptor promote Schwann cell migration. Proc Natl Acad Sci USA. 1994;91:2795–2799. doi: 10.1073/pnas.91.7.2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbacid M. The trk family of neurotrophin receptors. J Neurobiol. 1994;25:1386–1403. doi: 10.1002/neu.480251107. [DOI] [PubMed] [Google Scholar]
- Barde Y-A. Trophic factors and neuronal survival. Neuron. 1989;2:1525–1534. doi: 10.1016/0896-6273(89)90040-8. [DOI] [PubMed] [Google Scholar]
- Barker PA, Shooter EM. Disruption of NGF binding to the low affinity neurotrophin receptor p75LNTR reduces NGF binding to TrkA on PC12 cells. Neuron. 1994;13:203–215. doi: 10.1016/0896-6273(94)90470-7. [DOI] [PubMed] [Google Scholar]
- Barrett GL, Bartlett PF. The p75 nerve growth factor receptor mediates survival or death depending on the stage of sensory neuron development. Proc Natl Acad Sci USA. 1994;91:6501–6505. doi: 10.1073/pnas.91.14.6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belliveau DJ, Krivko I, Kohn J, Lachance C, Pozniak C, Rusakov D, Kaplan D, Miller FD. NGF and NT-3 both activate TrkA on sympathetic neurons but differentially regulate survival and neuritogenesis. J Cell Biol. 1997;136:374–388. doi: 10.1083/jcb.136.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti M, Levi A, Chao MV. Differential expression of nerve growth factor receptors leads to altered binding affinity and neurotrophin responsiveness. Proc Natl Acad Sci USA. 1993;90:7859–7863. doi: 10.1073/pnas.90.16.7859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter BD, Kaltschmidt C, Kaltschmidt B, Offenhauser N, Bohm-Matthaei R, Baeuerle PA, Barde Y-A. Selective activation of NF-kB by nerve growth factor through the neurotrophin receptor p75. Science. 1996;272:542–545. doi: 10.1126/science.272.5261.542. [DOI] [PubMed] [Google Scholar]
- Casaccia-Bonnefil P, Carter BD, Dobrowsky RT, Chao MV. Death of oligodendrocytes mediated by the interaction of nerve growth factors with its receptor p75. Nature. 1996;383:716–719. doi: 10.1038/383716a0. [DOI] [PubMed] [Google Scholar]
- Causing CG, Gloster A, Aloyz R, Bamji SX, Chang E, Fawcett J, Kuchel G, Miller FD. Synaptic innervation density is regulated by neuron-derived BDNF. Neuron. 1997;18:257–267. doi: 10.1016/s0896-6273(00)80266-4. [DOI] [PubMed] [Google Scholar]
- Chao MV. The p75 neurotrophin receptor. J Neurobiol. 1994;25:1373–1385. doi: 10.1002/neu.480251106. [DOI] [PubMed] [Google Scholar]
- Clary DO, Weskamp G, Austin LR, Reichardt LF. TrkA cross-linking mimics neuronal responses to nerve growth factor. Mol Biol Cell. 1994;5:549–563. doi: 10.1091/mbc.5.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coggeshall RE. An empirical method for converting nucleolar counts to neuronal numbers. J Neurosci Meth. 1984;12:125–132. doi: 10.1016/0165-0270(84)90011-6. [DOI] [PubMed] [Google Scholar]
- Cordon-Cardo C, Tapley P, Jing SQ, Nanduri V, O'Rourke E, Lamballe F, Kovary K, Klein R, Jones KR, Reichardt LF, Barbacid M. The trk tyrosine protein kinase mediates the mitogenic properties of nerve growth factor and neurotrophin-3. Cell. 1991;66:173–183. doi: 10.1016/0092-8674(91)90149-s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley C, Spencer SD, Nishimura MC, Chen KS, Pitts-Meek S, Armanini MP, Ling LH, McMahon SB, Shelton DL, Levinson AD, Phillips HS. Mice lacking nerve growth factor display perinatal loss of sensory and sympathetic neurons yet develop basal forebrain cholinergic neurons. Cell. 1994;76:1001–1011. doi: 10.1016/0092-8674(94)90378-6. [DOI] [PubMed] [Google Scholar]
- Dobrowsky RT, Werner MH, Castellino AM, Chao MV, Hannun YA. Activation of the sphingomyelin cycle through the low-affinity neurotrophin receptor. Science. 1994;265:1596–1599. doi: 10.1126/science.8079174. [DOI] [PubMed] [Google Scholar]
- Dobrowsky RT, Jenkins GM, Hannun YA. Neurotrophins induce sphingomyelin hydrolysis: modulation by co-expression of p75 with Trk receptors. J Biol Chem. 1995;270:22135–22142. doi: 10.1074/jbc.270.38.22135. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Lee KF, Jaenisch R. Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature. 1994;368:147–150. doi: 10.1038/368147a0. [DOI] [PubMed] [Google Scholar]
- Estus S, Zaks WJ, Freeman RS, Gruda M, Bravo R, Johnson EM., Jr Altered gene expression in neurons during programmed cell death: Identification of c-jun as necessary for neuronal apoptosis. J Cell Biol. 1994;127:1717–1727. doi: 10.1083/jcb.127.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frade JM, Rodriguez-Tebar A, Barde Y-A. Induction of cell death by endogenous nerve growth factor through its p75 receptor. Nature. 1996;383:166–168. doi: 10.1038/383166a0. [DOI] [PubMed] [Google Scholar]
- Franklin JL, Sans-Rodriguez C, Juhasz A, Deckwerth TL, Johnson EM., Jr Chronic depolarization prevents programmed death of sympathetic neurons in vitro but does not support growth: requirement for Ca2+influx but not trk activation. J Neurosci. 1995;15:643–664. doi: 10.1523/JNEUROSCI.15-01-00643.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ham J, Babij C, Whitfield J, Pfarr CM, Lallemand D, Yaniv M, Rubin LL. A c-jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 1995;14:927–939. doi: 10.1016/0896-6273(95)90331-3. [DOI] [PubMed] [Google Scholar]
- Hempstead BL, Martin-Zanca D, Kaplan DR, Parada LF, Chao MV. High-affinity NGF binding requires coexpression of the trk proto- oncogene and the low-affinity NGF receptor. Nature. 1991;350:678–683. doi: 10.1038/350678a0. [DOI] [PubMed] [Google Scholar]
- Hempstead BL, Rabin SJ, Kaplan L, Reid S, Parada LF, Kaplan DR. Overexpression of the trk tyrosine kinase rapidly accelerates nerve growth factor-induced differentiation. Neuron. 1992;9:883–896. doi: 10.1016/0896-6273(92)90241-5. [DOI] [PubMed] [Google Scholar]
- Hendry IA. Cell division in the developing sympathetic nervous system. J Neurocytol. 1977;6:299–309. doi: 10.1007/BF01175193. [DOI] [PubMed] [Google Scholar]
- Hendry IA, Campbell J. Morphometric analysis of rat superior cervical ganglion after axotomy and nerve growth factor treatment. J Neurocytol. 1976;5:351–360. doi: 10.1007/BF01175120. [DOI] [PubMed] [Google Scholar]
- Ip NY, Stitt TN, Tapley P, Klein R, Glass DJ, Fandl J, Greene LA, Barbacid M, Yancopoulos GD. Similarities and differences in the way neurotrophins interact with the Trk receptors in neuronal and nonneuronal cells. Neuron. 1993;10:137–149. doi: 10.1016/0896-6273(93)90306-c. [DOI] [PubMed] [Google Scholar]
- Itoh K, Brackenbury R, Akeson RA. Induction of L1 mRNA in PC12 cells by NGF is modulated by cell-cell contact and does not require the high-affinity NGF receptor. J Neurosci. 1995;15:2504–2512. doi: 10.1523/JNEUROSCI.15-03-02504.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson D, Lanahan A, Buck CR, Sehgal A, Morgan C, Mercer E, Bothwell M, Chao M. Expression and structure of the human NGF receptor. Cell. 1986;47:545–554. doi: 10.1016/0092-8674(86)90619-7. [DOI] [PubMed] [Google Scholar]
- Jones KR, Farinas I, Backus C, Reichardt LF. Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor development. Cell. 1994;76:989–999. doi: 10.1016/0092-8674(94)90377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan DR, Hempstead BL, Martin-Zanca D, Chao MV, Parada LF. The trk proto-oncogene product: a signal transducing receptor for nerve growth factor. Science. 1991a;252:554–558. doi: 10.1126/science.1850549. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Martin-Zanca D, Parada LF. Tryosine phosphorylation and tyrosine kinase activity of the trk proto-oncogene product induced by NGF. Nature. 1991b;350:158–160. doi: 10.1038/350158a0. [DOI] [PubMed] [Google Scholar]
- Klein R, Jing S, Nanduri V, O'Rourke E, Barbacid M. The trk proto-oncogene encodes a receptor for nerve growth factor. Cell. 1991;65:189–197. doi: 10.1016/0092-8674(91)90419-y. [DOI] [PubMed] [Google Scholar]
- Knusel B, Rabin SJ, Hefti F, Kaplan DR. Regulated neurotrophin receptor responsiveness during neuronal migration and early differentiation. J Neurosci. 1994;14:1542–1554. doi: 10.1523/JNEUROSCI.14-03-01542.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamballe F, Tapley P, Barbacid M. trkC encodes multiple neurotrophin-3 receptors with distinct biological properties and substrate specificities. EMBO (Eur Mol Biol Organ) J. 1993;12:3083–3094. doi: 10.1002/j.1460-2075.1993.tb05977.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KF, Li E, Huber LJ, Landis SC, Sharpe AH, Chao MV, Jaenisch R. Targeted mutation of the gene encoding the low affinity NGF receptor p75 leads to deficits in the peripheral sensory nervous system. Cell. 1992;69:737–749. doi: 10.1016/0092-8674(92)90286-l. [DOI] [PubMed] [Google Scholar]
- Lee K-F, Bachman K, Landis S, Jaenisch R. Dependence on p75 for innervation of some sympathetic targets. Science. 1994;263:1447–1449. doi: 10.1126/science.8128229. [DOI] [PubMed] [Google Scholar]
- Levi-Montalcini R. The nerve growth factor 35 years later. Science. 1987;237:1154–1162. doi: 10.1126/science.3306916. [DOI] [PubMed] [Google Scholar]
- Levi-Montalcini R, Booker B. Excessive growth of the sympathetic ganglia evoked by a protein isolated from mouse salivary glands. Proc Natl Acad Sci USA. 1960a;46:373–381. doi: 10.1073/pnas.46.3.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi-Montalcini R, Booker B. Destruction of the sympathetic ganglia in mammals by an antiserum to a nerve growth protein. Proc Natl Acad Sci USA. 1960b;46:381–390. doi: 10.1073/pnas.46.3.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Campenot RB, Miller FD. Concentration-dependent regulation of neuronal gene expression by nerve growth factor. J Cell Biol. 1992;117:135–141. doi: 10.1083/jcb.117.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPhee IJ, Barker PA. Brain-derived neurotrophic factor binding to the p75 neurotrophin receptor reduces TrkA signaling while increasing serine phosphorylation in the TrkA intracellular domain. J Biol Chem. 1997;272:23547–23551. doi: 10.1074/jbc.272.38.23547. [DOI] [PubMed] [Google Scholar]
- Majdan M, Lachance C, Gloster A, Aloyz R, Zeindler C, Bamji S, Bhakar A, Belliveau D, Fawcett J, Miller FD, Barker PA. Transgenic mice expressing the intracellular domain of the p75 neurotrophin receptor undergo neuronal apoptosis. J Neurosci. 1997;17:6988–6998. doi: 10.1523/JNEUROSCI.17-18-06988.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabizadeh S, Oh J, Zhong LT, Yang J, Bitler CM, Butcher LL, Bredesen DE. Induction of apoptosis by the low-affinity NGF receptor. Science. 1993;261:345–348. doi: 10.1126/science.8332899. [DOI] [PubMed] [Google Scholar]
- Radeke MJ, Misko TP, Hsu C, Herzenberg LA, Shooter EM. Gene transfer and molecular cloning of the rat nerve growth factor receptor. Nature. 1987;325:593–596. doi: 10.1038/325593a0. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Tébar A, Dechant G, Barde Y-A. Binding of brain- derived neurotrophic factor to the nerve growth factor receptor. Neuron. 1990;4:487–492. doi: 10.1016/0896-6273(90)90107-q. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Tébar A, Dechant G, Götz R, Barde Y-A. Binding of neurotrophin-3 to its neuronal receptors and interactions with nerve growth factor and brain-derived neurotrophic factor. EMBO (Eur Mol Biol Organ) J. 1992;11:917–922. doi: 10.1002/j.1460-2075.1992.tb05130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross AH, Daou M-C, McKinnon CA, Condon PJ, Lachyankar MB, Stephens RM, Kaplan DR, Wolf DE. The neurotrophin receptor, gp75, forms a complex with the receptor tyrosine kinase trkA. J Cell Biol. 1995;132:945–953. doi: 10.1083/jcb.132.5.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack RS, Belliveau DJ, Rosenberg M, Atwal J, Lochmuller H, Aloyz R, Haghighi A, Lach B, Seth P, Cooper E, Miller FD. Adenovirus-mediated gene transfer of the tumor suppressor, p53, induces apoptosis in postmitotic neurons. J Cell Biol. 1996;135:1–12. doi: 10.1083/jcb.135.4.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeyne RJ, Klein R, Schnapp A, Long LK, Bryant S, Lewin A, Lira SA, Barbacid M. Severe sensory and sympathetic neuropathies in mice carrying a disrupted Trk/NGF receptor gene. Nature. 1994;368:246–248. doi: 10.1038/368246a0. [DOI] [PubMed] [Google Scholar]
- Snider WD. Functions of the neurotrophins during nervous system development: what the knockouts are teaching us. Cell. 1994;77:627–638. doi: 10.1016/0092-8674(94)90048-5. [DOI] [PubMed] [Google Scholar]
- Soppet D, Escandon E, Maragos J, Middlemas DS, Reid SW, Blair J, Burton LE, Stanton BR, Kaplan DR, Hunter T, et al. The neurotrophic factors brain-derived neurotrophic factor and neurotrophin-3 are ligands for the trkB tyrosine kinase receptor. Cell. 1991;65:895–903. doi: 10.1016/0092-8674(91)90396-g. [DOI] [PubMed] [Google Scholar]
- Tsoulfas P, Soppet D, Escandon E, Tessarollo L, Mendoza-Ramirez J-L, Rosenthal A, Nikolics K, Parada LF. The rat trkC locus encodes multiple neurogenic receptors that exhibit differential response to neurotrophin-3 in PC12 cells. Neuron. 1993;10:975–990. doi: 10.1016/0896-6273(93)90212-a. [DOI] [PubMed] [Google Scholar]
- Van der Zee CEEM, Ross GM, Riopelle R, Hagg T. Survival of cholinergic forebrain neurons in developing p75(NGFR)-deficient mice. Science. 1996;274:1729–1732. doi: 10.1126/science.274.5293.1729. [DOI] [PubMed] [Google Scholar]
- Verdi JM, Birren SJ, Ibáñez CF, Persson H, Kaplan DR, Benedetti M, Chao MV, Anderson DJ. p75LNGFRregulates Trk signal transduction and NGF-induced neuronal differentiation in MAH cells. Neuron. 1994;12:733–745. doi: 10.1016/0896-6273(94)90327-1. [DOI] [PubMed] [Google Scholar]
- von Bartheld CS, Kiroshita Y, Prevette D, Yin QW, Oppenheim RW, Bothwell M. Positive and negative effects of neurotrophins on the isthmo-optic nucleus in chick embryos. Neuron. 1994;12:639–654. doi: 10.1016/0896-6273(94)90219-4. [DOI] [PubMed] [Google Scholar]
- Weskamp G, Reichardt LF. Evidence that biological activity of NGF is mediated through a novel subclass of high affinity receptors. Neuron. 1991;6:649–663. doi: 10.1016/0896-6273(91)90067-a. [DOI] [PubMed] [Google Scholar]
- Wolf DE, McKinnon CA, Daou MC, Stephens RM, Kaplan DR, Ross AH. Interaction with trkA immobilizes gp75 in the high affinity nerve growth factor receptor complex. J Biol Chem. 1995;270:2133–2138. doi: 10.1074/jbc.270.5.2133. [DOI] [PubMed] [Google Scholar]
- Wright LL, Cunningham TJ, Smolen AJ. Developmental neuron death in the rat superior cervical sympathetic ganglion: cell counts and ultrastructure. J Neurocytol. 1983;12:727–738. doi: 10.1007/BF01258147. [DOI] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- Yeo TT, Chua-Couzens J, Butcher LL, Bredesen DE, Cooper JD, Valletta JS, Mobley WC, Longo FM. Absence of p75NTR causes increased basal forebrain cholinergic neuron size, choline acetyltransferase activity, and target innervation. J Neurosci. 1997;17:7594–7605. doi: 10.1523/JNEUROSCI.17-20-07594.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]