

Abstract
Abstract. CCS embryonic stem (ES) cells possessing two mutant alleles (ry1r−/ry1r−) for the skeletal muscle ryanodine receptor (RyR) have been produced and injected subcutaneously into severely compromised immunodeficient mice to produce teratocarcinomas in which Ry1R expression is absent. Several primary fibroblast cell lines were isolated and subcloned from one of these tumors that contain the knockout mutation in both alleles and exhibit a doubling time of 18–24 h, are not contact growth inhibited, do not exhibit drastic morphological change upon serum reduction, and possess the normal complement of chromosomes. Four of these fibroblast clones were infected with a retrovirus containing the cDNA encoding myoD and a puromycin selection marker. Several (1–2 μg/ml) puromycin-resistant subclones from each initial cell line were expanded and examined for their ability to express myoD and to form multinucleated myotubes that express desmin and myosin upon removal of mitogens. One of these clones (1B5 cells) was selected on this basis for further study. These cells, upon withdrawal of mitogens for 5–7 d, were shown by Western blot analysis to express key triadic proteins, including skeletal triadin, calsequestrin, FK506-binding protein, 12 kD, sarco(endo)plasmic reticulum calcium–ATPase1, and dihydropyridine receptors. Neither RyR isoform protein, Ry1R (skeletal), Ry2R (cardiac), nor Ry3R (brain), were detected in differentiated 1B5 cells. Measurements of intracellular Ca2+ by ratio fluorescence imaging of fura-2–loaded cells revealed that differentiated 1B5 cells exhibited no responses to K+ (40 mM) depolarization, ryanodine (50–500 μM), or caffeine (20–100 mM). Transient transfection of the 1B5 cells with the full-length rabbit Ry1R cDNA restored the expected responses to K+ depolarization, caffeine, and ryanodine. Depolarization-induced Ca2+ release was independent of extracellular Ca2+, consistent with skeletal-type excitation–contraction coupling. Wild-type Ry1R expressed in 1B5 cells were reconstituted into bilayer lipid membranes and found to be indistinguishable from channels reconstituted from rabbit sarcoplasmic reticulum with respect to unitary conductance, open dwell times, and responses to ryanodine and ruthenium red. The 1B5 cell line provides a powerful and easily managed homologous expression system in which to study how Ry1R structure relates to function.
Elucidation of the molecular details of excitation– contraction (e–c)1 coupling is a major area of research in muscle biology. The process by which depolarization of the sarcolemma causes the release of calcium from the sarcoplasmic reticulum (SR) to initiate crossbridge formation and, ultimately, muscle contraction is defined as e–c coupling (36). The ryanodine receptor (RyR) is the site of calcium release from SR in skeletal and cardiac muscle (28). Studies that have addressed the molecular details of how ryanodine receptor structure relates to function have thus far been limited to heterologous expression systems that lack key accessory proteins known to be essential for e–c coupling. The full-length cDNA encoding for the skeletal isoform of ryanodine receptor (Ry1R) has been expressed in CHO cells (32), Xenopus oocytes (31), COS-1 cells (9), and Sf9 insect cells (5). Expression of the Ry1R cDNA in these cells has resulted in channels whose properties are only similar to those of native Ca2+ channels isolated from skeletal SR. We believe that this is because studies that use heterologous expression systems to reconstitute Ry1R function do so in cells that do not express the other important proteins involved in e–c coupling that are normally targeted to the skeletal muscle triad and that have been shown to interact with and modulate the function of Ry1R. These proteins include the α-1 subunit of the L-type voltage-dependent Ca2+ channel dihydropyridine receptor (α1-DHPR) (11, 24, 27, 31, 39), FK506 binding protein, 12 kD (FKBP12) (26), triadin (4, 8, 16, 19, 20), calsequestrin (15–17), and calmodulin (29).
The present paper introduces a myogenic cell line (the 1B5 cell line), which expresses the major proteins known to exist at the skeletal triad, but has been genetically engineered to delete expression of Ry1R. The 1B5 cell line provides a unique and powerful system with which to answer questions concerning how the structure of ryanodine receptors relate to Ca2+ channel function and the relationship among triadic proteins involved in e–c coupling by providing a homologous expression system that lacks constitutive expression of known RyR isoforms.
Materials and Methods
Creation of a Myogenic Cell Line Lacking Expression of RyR Proteins
The procedures used to create a myogenic cell line lacking expression of RyR proteins, the ry1r−/ry1r− 1B5 cell line, are outlined in Fig. 1 and explained in detail below.
Figure 1.
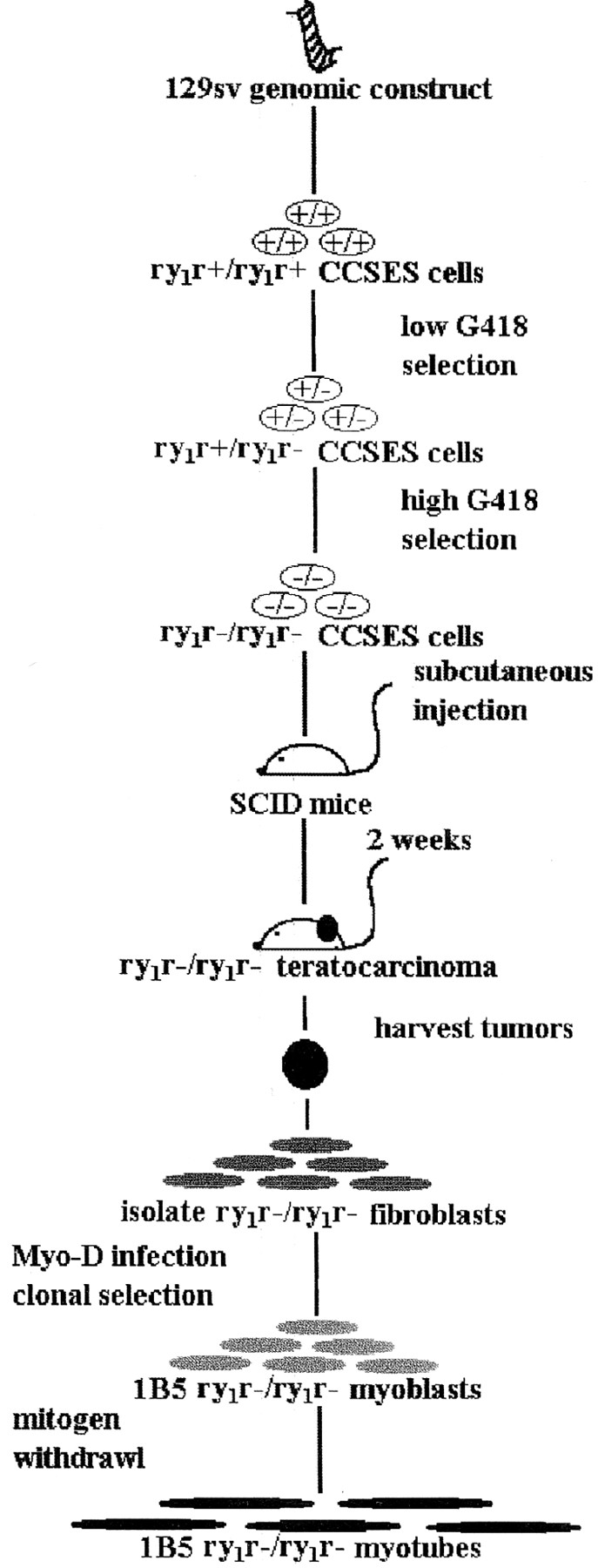
Development of the 1B5 ry1r−/ry1r− cell line. Diagram illustrating the major steps undertaken to create a myogenic cell line that lacks ryanodine receptor expression (the 1B5 ry1r−/ry1r− cell line). Details are given in Materials and Methods.
Creation of an Embryonic Stem Cell Line Possessing Two Disrupted ry1r Alleles.
A 9.0-kb EcoRI–Tth111I mouse genomic restriction fragment, characterized as part of the skeletal ryanodine receptor (ry1r) was isolated from a 129sv mouse genomic library. A neomycin cassette driven by phosphoglycerate kinase (PGK) promoter was inserted into exon 10 at nucleotide (nt) 840 of the mRNA (Fig. 2 A). This disrupted the transcription reading frame and added an additional stop codon. A PGK-driven thymidine kinase gene cassette was attached to the 3′ end as a second selection marker to help in the efficiency of screening for homologous events. The linearized construct was electroporated into CCS embryonic stem (ES) cells, which were in turn grown on neomyocin antibiotic analogue (G418)- resistant mouse embryonic fibroblast feeder cells in a medium consisting of DME supplemented with 20% FBS, with added 2.5 mM glutamine, penicillin, and streptomycin. The ES cells were selected in low G418 (0.4 mg/ml) for 2 d, and then G418 (0.4 mg/ml) and gangcyclovir (250 μM) for 8 d to improve the likelihood of eliminating clones where G418 resistance occurred as a result of random integration (10). The resistant cell clones were then examined for a homologous event by genomic Southern blot analysis.
Figure 2.


Targeting construct and creation of ry1r−/ry1r− cell lines. (A) The targeting construct used to disrupt the ry1r gene in CCS ES cells was composed of a 9-kb fragment of mouse 129sv genomic DNA with an insertion vector containing a phosphoglycerate kinase (pgk) promoter driven neomycin (Neo) cassette inserted at nt 840 disrupting the transcription as described in Materials and Methods. A PGK-driven thymidine kinase (pgk-TK) gene cassette was attached to the 3′ end as a second selection marker to help in the efficiency of screening for a homologous recombination event. N, NcoI; R, EcoRI; K, KpnI; M, MunI; and T, Tth111I.) (B) Southern blot analysis of genomic DNA. Lane 1, size markers obtained from bands 3–6 of BsteI-digested λ DNA. Lane 2, wild-type CCS cells that possess only the wild-type ry1r allele. Lanes 3 and 4, CCS ES cell DNA after low (0.4 mg/ml; lane 3) and high (3.2 mg/ml; lane 4) selection with G418. Lanes 5–7 three independent representative fibroblast cell lines isolated from a teratocarcinoma produced in SCID mice by injection of the ES cells homozygous for the knockout allele. Lanes 8–10, three independent representative myoblast cell lines that were derived by transforming the fibroblast cell lines with a myoD-containing retrovirus. The latter cell lines were selected by their proven expression of myosin and desmin, and for their ability to form multinucleated “myotubes” after withdrawal of mitogens. Cell genomic DNA was digested with EcoRI–MunI, and then hybridized with a 600-bp EcoRI–Tth111I probe. The location of the probe is shown in (A).
A second round of selection using high G418 (3.2 mg/ml) has been shown to select for the rare homologous recombination of the disrupted allele that will yield ES cells where both alleles are disrupted (30). This technique was successfully used to produce ES cells in which both Ry1R alleles were disrupted by the neomycin cassette.
Creation of the 1B5 ry1r−/ry1r− Myogenic Cell Line.
ES cells that were homozygous for the disrupted ry1r allele were injected subcutaneously into the hind quarters of severely compromised immunodeficient (SCID) mice at a concentration of 2–5 × 106 ES cells/ml in a volume of 1 ml. The injections resulted in ry1r−/ry1r− teratocarcinomas composed of several cell types, including skeletal muscle (5–10%). Although we were unable to isolate myoblast cell lines using this procedure, we were able to isolate several ry1r−/ry1r− fibroblast clonal lines that were proven to possess the same ry1r−/ry1r− mutation of the parent ES cells by both Southern blot and PCR analyses. Each of the fibroblast cell lines shared the following characteristics: (a) they exhibited a doubling time of 18–24 h, (b) they were not contact growth inhibited, and (c) they did not change morphology when mitogens were withdrawn from the culture medium. These cells have been tested for chromosome number on several occasions and have maintained a normal complement of chromosomes after several months in culture. Four of the ry1r−/ry1r− fibroblast lines were infected with a retrovirus encoding myoD and bearing a puromycin resistance gene as a selectable marker (a gift of Dr. A. Lassar, Harvard School of Medicine, Cambridge, MA). The virus was packaged in Bosc23 cells (a gift of Dr. W. Pear, Massachusetts Institute of Technology, Cambridge, MA), an ecotropic envelope-expressing packaging cell line. The Bosc23 cells were transfected with the myoD retroviral plasmid construct using CaCl2 shock protocol. The retroviral supernatant was harvested 48 h posttransfection by gently removing the supernatant and filtering it through a 0.45 μM filter. The retroviral supernatant was kept on ice until it was used, and the unused supernatant was quickly frozen and kept at −80°C. Before infection, the adherent ry1r−/ry1r− fibroblasts had been plated at a density of 5 × 105 cells in growth medium per 100-mm plate.
For infection of cells with retrovirus, the standard growth medium was replaced with 3 ml of a cocktail consisting of: (a) retroviral-containing supernatant, (b) Polybrene at a final concentration of 4 μg/ml, and (c) growth media, and allowed to incubate at 37°C for 3 h. Growth media (7 ml) supplemented with bFGF (fibroblast growth factor) (25 ng/ml) was then added to the cells. 48 h after infection, selection was begun with puromycin (1 and 2 μg/ml) and clones were selected after 7–10 d. Puromycin- resistant clones were subcloned in growth media consisting of DME and 20% FBS (Gibco Laboratories, Grand Island, NY). Several of the independent subclones that were shown to express myoD by immunocytochemistry, exhibited contact growth inhibition at high density in growth medium and could be induced to differentiate into noncontractile multinucleated myotubes when 5% heat-inactivated horse serum (HIHS) was substituted for 20% FBS in DME medium (see Results). These myotubes, but not the undifferentiated cells were shown by immunocytochemistry to express skeletal-specific myosin heavy chain and desmin. One of the subclones, the 1B5 cell line, was chosen for more complete study.
Southern Blot Analysis of ES Cells, Tumor Fibroblast Cells, and Converted “Myoblast” Cells.
The resistant CCS ES cells were expanded on gelatin for genomic Southern blot analysis. Analysis of the targeted ry1r alleles in ES cells was performed after digesting the genomic DNA with both EcoRI and MunI, size selecting it on a 1% agarose gel, and then transferring the DNA onto nitrocellulose paper. The DNA digest yielded a 3.0-kb fragment in the wild-type allele and a 4.8-kb fragment in the disrupted allele using a 600-bp Tth111I–EcoRI randomly labeled fragment as probe. The Tth111I–EcoRI probe was from a region outside but adjacent to the transfected region of homology. Results from Southern blot analysis showed the presence of one disrupted and one wild-type allele (i.e., ry1r+/ ry1r−) in ES cells selected with low G418, whereas ES cells subjected to high G418 possessed only disrupted alleles (i.e., ry1r−/ry1r−). All of the fibroblast cell lines and the converted fibroblast cell lines possessed only the disrupted alleles (i.e., ry1r−/ry1r−) derived from the ES precursor cells.
Cell Culture.
All cell lines including the 1B5 cells were cultured in growth medium consisting of DME containing 100 units/ml penicillin-G and 100 μg/ml streptomycin sulfate, 250 nM Fungizone®, 2 mM glutamine, and 20% FBS (vol/vol) (Gibco Laboratories) at 37°C in 7% CO2. Cells were passaged at 50–80% confluence (two to three times weekly). For ratio fluorescence imaging measurements, cells were grown in 2 ml 20% FBS-DME on 25-mm glass coverslips, and then differentiated by replacing the growth medium with 5% HIHS (differentiation medium). The cells were allowed to differentiate 5–7 d and then studied by immunohistochemistry, Western blot analysis, radioligand receptor binding analysis, and ratio fluorescence imaging microscopy.
Immunohistochemistry.
Differentiated 1B5 cells were fixed in buffered zinc-formalin for 15 min and washed with PBS/Tween-20 three times before blocking with 3% H2O2 for 10 min. Cells were washed again and then blocked with 10% normal horse serum in PBS/Tween-20 for 30 min. Blocking buffer was drained and anti-Ry1R 34C monoclonal (a gift of Dr. J. Sutko, University of Nevada, Reno, NV) was applied at a concentration of 1:20 in PBS for 1.5 h at 25°C. Cells were washed with PBS/Tween-20 three times and then incubated with CY-3-linked goat anti–mouse IgG (1: 250) for 30 min. The cells were rinsed with Tris buffer and epifluorescence visualized at 570 nm (excitation at 550 nm).
Microsomal Membrane Preparations.
Microsomal membranes were isolated from 1B5 cells that had been allowed to differentiate for 5–7 d. Cells were scraped from 75-cm2 flasks or 100-mm plates, and then centrifuged for 10 min at 250 g. The pellet was resuspended in buffer consisting of 250 mM sucrose, 10 mM Hepes, 1 mM EDTA, 1 mM DTT, 1 mM benzamidine, 1 μg/ml leupeptin, 0.7 μg/ml pepstatin A, and 0.1 mM PMSF, pH 7.4, and then homogenized using a PowerGen 700D (Fisher Scientific, Pittsburgh, PA). The homogenate was centrifuged for 5 min at 250 g. The pellet was discarded and the 250 g supernatant was centrifuged at 10,000 g for 20 min at 4°C. The 10,000 g supernatant was centrifuged at 100,000 g for 1 h at 4°C, and the microsomal pellet was resuspended at 6–15 μg/ml (23) in 10% sucrose, 20 mM Hepes, pH 7.2, frozen in liquid nitrogen, and stored at −80°C.
For reconstitution studies in bilayer lipid membranes, differentiated 1B5 cells transiently transfected with rabbit wild-type Ry1R cDNA were rinsed twice with ice-cold PBS, and then harvested in 137 mM NaCl, 3 mM KCl, 8 mM Na2HPO4, 1.5 mM KH2PO4, 0.5 mM EDTA, pH 7.4. The collected cells were pelleted at 600 g for 5 min. The cell pellet was then resuspended in ice-cold hypotonic lysis buffer (1 mM EDTA, 10 μg/ml leupeptin, 10 μg/ml pepstatin A, 10 mg/ml benzamidine, 10 mM Hepes, pH 7.4) and homogenized by 10 strokes in a tight-fitting Dounce homogenizer followed by 15 strokes after addition of an equal volume of sucrose buffer (500 mM sucrose, 10 mM Hepes, pH 7.4). The homogenate was then run in a Sorvall centrifuge at 10,000 g for 15 min. Microsomes were collected by centrifugation of supernatant at 110,000 g using a Beckman Ti80 for 45 min at 4°C. The resulting pellet was then resuspended in 300 mM sucrose, 10 mM Hepes-Tris, pH 7.4. The microsomal vesicles were then frozen in N2 and stored at −80°C.
Membranes enriched in markers of junctional SR were prepared from rabbit fast skeletal SR using the method of Saito and coworkers (37) in the presence of protease inhibitors PMSF (100 μM) and leupeptin (10 μg/ml). Purified heavy SR from the 38–45% interface of sucrose density gradients was pelleted, resuspended at 3–6 mg/ml (23), frozen in liquid nitrogen, and stored at −80°C until needed. Membranes enriched in cardiac junctional SR were prepared from rabbit heart using the method of Feher and coworkers (12). Avian junctional SR membranes were isolated from avian pectoralis muscle according to the method of Airey and coworkers (2).
Radioligand Binding Assay.
High affinity binding of [3H]ryanodine (84 Ci/mmol; New England Nuclear, Boston, MA) to membrane homogenates (0.40–1 mg/ml) prepared from differentiated 1B5 (and their fibroblast precursor) cell lines was performed in the presence of 250 mM or 1 M KCl, 15 mM NaCl, 20 mM Hepes, pH 7.1, 50 μM Ca2+, and 1–60 nM [3H]ryanodine. The binding reaction was initiated by addition of cell membranes to the medium and the mixture was permitted to equilibrate at 25°C for 16 h or 37°C for 3 h. Non-specific binding was assessed in the presence of 0.5 or 1 μM unlabeled ryanodine. Under these assay conditions, the K D for Ry1R in skeletal SR preparations has been found to be 1–5 nM (29). Separation of bound and free ligand was performed by rapid filtration through Whatman GF/B glass fiber filters using a Brandel (Gaithersburg, MD) cell harvester. Filters were washed with three volumes of 0.5 ml ice-cold wash buffer containing 20 mM Tris-HCl, 250 mM KCl, 15 mM NaCl, 50 μM CaCl2, pH 7.1, and placed into vials with 5 ml scintillation cocktail (Ready Safe; Beckman Instruments, Inc., Fullerton, CA). The [3H]ryanodine remaining on the filters was quantified by liquid scintillation spectrometry.
The binding of [3H]PN200-110 to membrane-associated DHPR-α1 was measured in 0.5 ml of buffer consisting of 5 mM KCl, 140 mM NaCl, 20 mM Hepes, pH 7.4, and 0.1–8 nM [3H]PN200-110 (specific activity 80–87 Ci/ mmol). Cell membranes (50 μg protein per assay) were incubated for 1 h at 25°C in the dark. Nonspecific binding was determined in the presence of 100 μM nifedipine. Reactions were quenched by filtration as described above and washed four times with 5 ml of wash buffer.
Western Blot Analysis.
Proteins were denatured in reducing sample buffer consisting of 48 mM NaH2PO4, 170 mM Na2HPO4, pH 7.4, 0.02% bromophenol blue, 1% SDS (wt/vol), and 5% β-mercaptoethanol (final concentrations). The samples were placed in boiling water for 6 min and 1–50 μg of protein was loaded onto 3–10% or 4–20% gradient SDS-PAGE gels (21) and electrophoresed at constant voltage (200 V). The size-separated proteins were transferred onto polyvinylidene difluoride (PVDF) microporous membranes by electroblotter (“Mini Trans-Blot; ” Bio-Rad Laboratories, Hercules, CA) overnight at 30 V (4°C) and 1 h at 100 V. PVDF transfers were incubated with TTBS (137 mM NaCl, 20 mM Tris-HCl, pH 7.6, 0.05% Tween-20) containing 5% (wt/vol) non-fat milk for 1 h to block nonspecific reactions. The blots were incubated with the selected antibody in TTBS, pH 7.6, with 1% BSA overnight at 4°C. The immunoblots were incubated with HRP-conjugated anti–mouse or anti– rabbit IgG (Sigma Chemical Co., St. Louis, MO) for 1 h. Enhanced chemiluminescent techniques (Pierce Chemical Co., Piscataway, NJ) were used to visualize the immunoblots. The antibodies used for Western analysis were: anti-Ry1R monoclonal 34C (2), anti-Ry2R monoclonal C3-33 (22), anti-Ry3R receptor polyclonal (14), human anti-FKBP12 monoclonal (Vertex Pharmaceuticals, Cambridge, MA), anti-triadin monoclonal IIG12A8 (19), anti-calsequestrin, anti–α1-DHPR, anti–sarco(endo)plasmic reticulum Ca2+-ATPase pump (SERCA)1, and skeletal muscle–specific anti–myosin heavy chain monoclonal antibodies (Affinity BioReagents, Neshanic, NJ).
Ratio Fluorescence Imaging.
After plating in growth media for 24 h, cells were grown on collagen-coated coverslips in differentiation medium for 5–7 d. The differentiated cells were loaded with 5 μM fura-2AM (Molecular Probes, Inc., Eugene, OR) and 0.05% BSA (fraction V) at 37°C in 5% CO2 for 20 min in loading buffer consisting of 125 mM NaCl, 5 mM KCl, 2 mM KH2PO4, 2 mM CaCl2, 25 mM Hepes, 6 mM glucose, and 1.2 mM MgSO4, pH 7.4. The coverslips were washed twice with 1 ml imaging buffer (loading buffer with 250 μM sulfinpyrazone), placed in a clamp chamber (Medical Systems, Chicago, IL), and then 1 ml of imaging buffer placed on the cells. The cells were transferred to an inverted stage microscope (Nikon Diaphot, Melville, NY) equipped with a thermostat controlled plate set at 37°C. Fura-2 fluorescence emission was measured from cells visible with a × 20 quartz objective. Fura-2 was excited alternately at 340 and 380 nm using dual automated monochrometers interfaced with a reflective chopper and bifurcated fiber optic cable (Deltascan 1000; Photon Technology International, Princeton, NJ). Fluorescence emission was measured at 510 nm. Data were collected from 25–50 individual cells with imaging equipment (intensified CCD camera; Hamamatsu Photonics, Bridgewater, NJ) using ImageMaster software (Photon Technology International) in Free Run mode. The data were presented as the ratio of the emissions obtained at 340 and 380 nm (340/380).
Transient Transfection of the 1B5 Cell Line.
In a six-well tissue culture plate or 35-mm dish, 1–3 × 105 cells per dish were seeded in 2 ml of 20% FBS DME on 12-mm glass coverslips. Cells were incubated at 37°C in a CO2 incubator until 80% confluent. Growth medium was then replaced with differentiation medium consisting of 5% HIHS DME and allowed to differentiate for 4 d. For each well to be transfected, solution A was composed of 0.75 μg of Ry1R cDNA (pCMVRy1R) diluted into 100 μl Opti-MEM I reduced serum medium (GIBCO BRL, Gaithersburg, MD) and solution B was composed of 18 μl of Lipofectamine reagent (GIBCO BRL) diluted into 100 μl Opti-MEM I reduced serum medium. For mock transfections Ry1R cDNA (pCMVRy1R) was replaced with 1.5 μg of β-galactosidase cDNA or all cDNA was completely removed. Solutions A and B were prepared and mixed gently by vortex for about 5 s. Solutions A and B were combined, mixed gently, and incubated at room temperature for 45 min to permit DNA–liposome complexes to form. Transfection medium composed of 0.8 ml of serum-free medium was added to the tube containing the DNA–liposome complexes and mixed gently. Each well was rinsed once with 2 ml of serum-free medium and 1 ml of transfection medium was then added. Transfection proceeded for 5 h at 37°C in a CO2 incubator and was terminated by addition of 0.5 ml 40% FBS DME. The transfected cells were permitted to recover for 19 h, and differentiation medium was introduced (5% HIHS DME). The cells were monitored for functional responses using ratio fluorescence imaging microscopy 1 d after transfection with Ry1R cDNA. pCMVRy1R was constructed by cloning the full-length Ry1R cDNA into the HindIII site of pCMV5. The full-length Ry1R cDNA was obtained by cloning several smaller fragments from a rabbit muscle cDNA library (a gift of Dr. A.R. Marks, Columbia College of Physicians and Surgeons, New York) and assembled in the same manner as has been previously described (4).
Electrophysiological Planar Lipid Bilayer Experiments.
Planar lipid membrane (BLM) reconstitution experiments were carried out with SR vesicles prepared as described above from 1B5 cells transfected with Ry1R cDNA and differentiated as described above. SR vesicles were fused into a BLM made from a 5:3:2 mixture of phosphatidylethanolamine, phosphatidylserine, and phosphatidylcholine suspended in decane at 50 mg/ml. The BLM was formed across a 250-μm hole in a polystyrene cup separating two chambers of 0.7 ml each. SR vesicles (3–15 μg protein) were added to the cis chamber in the presence of 200 μM Ca2+. The cis chamber contained 500 mM CsCl, 20 mM Hepes, pH 7.4. The trans chamber contained 100 mM CsCl, 20 mM Hepes, pH 7.4. After fusion of a single vesicle, 300 μM EGTA was added to the cis chamber of the BLM to prevent any additional fusion events. In some experiments the cis and trans chambers were perfused with a solution composed of 20 mM Hepes, pH 7.4, and 100 mM Cs+ as either the chloride or methane sulfonate salt (symmetrical conditions). In other experiments the cis chamber was perfused with 500 mM CsCl (asymmetrical 5:1 cis/trans conditions). A patch clamp amplifier (model 3900; DAGAN, Minneapolis, MN) was used to measure currents through a single channel. The single-channel data was filtered at 2–5 kHz (Digidata 1200; Axon Instruments, Inc., Foster City, CA) and stored on computer. Depending on the experimental conditions, calcium, ryanodine, or ruthenium red was added to the cis chamber and stirred. Subsequent channel gating behavior was recorded using Axotape software (Axon Instruments, Inc.) for 1–5 min. At the end of each experiment, 10 μM ruthenium red was added to block channel function and confirm the source of the gating currents. Single-channel data from BLM experiments were analyzed using the pCLAMP software (pCLAMP version 6.0; Axon Instruments, Inc.). Open and closed time constants were determined by least squares fit to double exponentials and steady-state channel open probability determined.
Results and Discussion
Creation of ry1r−/ry1r− ES cells
Fig. 2 A shows the targeting construct used to disrupt the ry1r gene in CCS ES cells. Analysis of the targeted ry1r alleles was performed after digesting genomic DNA with both EcoRI and MunI. The DNA digest yielded a 3.0-kb fragment in the wild-type allele and a 4.8-kb fragment in the disrupted allele using a 600-bp Tth111I–EcoRI randomly labeled fragment as probe. This probe was from a region outside but adjacent to the transfected region of homology. Southern blot analysis indicated one disrupted and one wild-type allele (i.e., ry1r+/ry1r−; Fig. 2 B) in the heterozygous ES cells selected with low G418, whereas some ES cells subjected to high G418 possessed only disrupted alleles (i.e., ry1r−/ry1r−) (Fig. 2 B). Expansion of one of these clones resulted in an ES cell line homozygous for the disrupted allele.
ES cells injected subcutaneously into SCID mice produce teratocarcinomas in which expression of Ry1R protein is absent. Several primary fibroblast cell lines were isolated and subcloned from one of these tumors that contained the knockout mutation in both alleles. These cell lines exhibited a doubling time of 18–24 h were not contact growth inhibited, did not exhibit drastic morphological change upon serum reduction, and possessed the normal complement of chromosomes. Four of these fibroblast clones were infected with a retrovirus containing the cDNA encoding myoD and a puromycin selection marker. Several (1–2 μg/ml) puromycin-resistant subclones from each initial cell line were expanded. Upon removal of mitogens these cell lines were examined for their ability to express myoD and to form multinucleated myotubes that express desmin and myosin. One of these clones (1B5 cell line) was selected for further study. All of the tumor fibroblast cell lines derived from these ES cells and the myoD-converted “myoblasts” derived from the fibroblasts showed the identical Southern pattern, thereby verifying their origin. Fig. 2 B shows Southern blot results from each of three representative fibroblast and myoblast cell lines.
Morphology of 1B5 Cells
The typical 1B5 (ry1r−/ry1r−) cell, passaged in growth medium at low density, exhibits a triangular morphology and a single nucleus (Fig. 3). These cells expand rapidly, having a doubling time of 18–24 h under the culture conditions used. They can grow to near confluence without spontaneously differentiating but do show growth inhibition at high density. Once mitogens are withdrawn from the growth medium, cell division slows and the cells undergo myogenic differentiation. After 5–7 d in differentiation medium, most of the cells become very long and multinucleated (Fig. 3). Immunohistochemical staining of all of the many (>12) independent converted clonal lines demonstrated the presence of two muscle-specific proteins, skeletal muscle myosin heavy chain and desmin, in all of the multinucleated cells (data not shown), whereas Ry1R was not detected (Fig. 4). In consonance with immunohistochemical results, the 1B5 cells have not been observed to contract either spontaneously or in response to K+ depolarization or a pharmacological agent (e.g., caffeine).
Figure 3.


Morphology of 1B5 myogenic cell lines. The top panel is a representative phase-contrast images of ry1r−/ry1r− 1B5 cells undifferentiated and differentiated. Cells were cultured in growth medium and photographed at a density of 50–70% (see Materials and Methods). The 1B5 cell line undergoes significant morphological change when cultured in 5% HIHS (differentiation medium) for 5–7 d, exhibiting long myotubes with multiple nuclei (arrows). The bottom panel shows immunocytochemical staining of differentiated 1B5 cells with anti-Ry1R antibody. 1B5 cells were permitted to differentiate after serum withdrawal as described in Materials and Methods. The cells were fixed and stained with 34C monoclonal antibody selective for Ry1R before and after transient transfection with wild-type Ry1R cDNA. Bars: (Nondifferentiated) 40 μm; (Differentiated) 20 μm.
Figure 4.

1B5 cells lack detectable levels of the known RyR isoforms by Western analysis. Western blot analysis was performed as described in Materials and Methods and visualized by the ECL method. Ry1R protein is not detected in 1B5 cells. 1B5 cells do not possess detectable levels of Ry2R or Ry3R immunoreactive protein. The lanes were loaded as follows: Avian protein (avi; 10 μg), rabbit cardiac (crd; 15 μg), rabbit skeletal junctional sarcoplasmic reticulum (jsr; 5 μg), and 1B5 protein (1B5; 50 μg). Gels were 3–10% SDS–polyacrylamide. These experiments were repeated at least twice with each primary antibody. ECL exposure time of up to 24 h showed no detectable levels of the known RyR isoforms.
1B5 ry1r−/ry1r− Myogenic Cells Express Major Elements of the Skeletal Triad
Although 1B5 cells do not express detectable levels of Ry1R by immunohistochemical methods, it is important to ascertain if the alternate isoforms Ry2R or Ry3R are expressed. Western blot analysis was performed with isoform-selective antibodies and visualized using the highly sensitive enhanced chemiluminescence (ECL) method. The sensitivity of this method was verified by titrating standard SR membrane preparations (skeletal [Ry1R], cardiac [Ry2R], or avian [Ry3R]) in the linear range of 0.1–5 μg protein per lane. The threshold for detecting RyR protein by laser densitometry was determined to be <0.1 μg protein/lane for each of the RyR antibodies used in the present study. The absence of detectable Ry1R protein in 1B5 cells was confirmed by Western analysis of their microsomal fraction, whereas an immunoreactive band at M r ∼565,000 was observed with rabbit junctional SR (Fig. 4). Neither Ry2R nor Ry3R immunoreactive protein was detected using antibodies selective for their respective isoforms in preparations from the 1B5 cell line (Fig. 4). Independent support for the absence of functional Ry1R complexes in 1B5 cell lines comes from radioligand receptor binding studies with [3H]ryanodine. Microsomal membranes isolated from 1B5 cells as well as their precursor fibroblast line do not possess detectable specific binding in the range of 1–30 nM [3H]ryanodine. The highly sensitive ECL Western blot protocol fails to detect any of the known RyR isoforms in 1B5 cells implying that either they are not expressed or expressed at extremely low levels.
The functional importance of dihydropyridine receptors (DHPRs) in e–c coupling has been amply demonstrated in studies of the dysgenic mouse, which lacks expression of α1-DHPR subunit and DHPR function (1). Elements of the cytoplasmic loop between repeats 2 and 3 of DHPR-α1 interact with and modulate the function of Ry1R (25). A homologous expression system would therefore require expression of α1-DHPR to reconstitute the tetradic alignment α1-DHPR (13) and wild-type e–c coupling. Western blot analysis of membranes isolated from differentiated 1B5 cells revealed an immunoreactive band at M r ∼170,000 that coincides with α1-DHPR found in rabbit junctional membranes (Fig. 5). The presence of α1-DHPR in 1B5 cells was confirmed with radioligand binding studies with [3H]PN200 (0.1–5 nM) and crude membrane homogenates that possess a B max of 122 ± 13 fmol/mg of specific binding sites having a K D of 0.5 ± 0.03 nM.
Figure 5.

1B5 cells express key triadic proteins. Western blots were developed by ECL technique for the length of time indicated as follows: α1-DHPR, 30 s; FKBP12, 1 min; triadin, 1 min jsr and 10 min 1B5; and calsequestrin, 1 min jsr and 10 min 1B5. Two additional skeletal muscle proteins detected in 1B5 cells are SERCA1 and skeletal myosin heavy chain (ECL exposure time of 5 and 1 min, respectively). The lanes were loaded as follows: rabbit skeletal junctional sarcoplasmic reticulum (jsr) 5 μg except for myosin and SERCA1; 1 μg) and 1B5 protein (50 μg). Gels were 3–10% or 4–20% (for FKBP12) SDS–polyacrylamide. This experiment was repeated at least twice with each primary antibody.
Interestingly these results with ry1r−/ry1 r− cell lines parallel those recently found in neonate dyspedic and wild-type skeletal muscle (7). Dyspedic muscle possesses a less-developed t-system and the DHPR fails to properly align into tetrads (38), as is normally seen in skeletal muscle. L-type Ca2+ inward current is significantly (∼30-fold) reduced in cultured dyspedic myotubes. However it is unlikely that this can be accounted for by reduced density of DHPRs since immobilization-resistant charge movement is not significantly diminished (31). Likewise, 1B5 cells also exhibit specific [3H]PN200-binding sites but lack detectable high affinity [3H]ryanodine-binding sites (7). Further, differentiated 1B5 myotubes possess numerous clusters of large intramembrane particles of α1-DHPR which lack tetradic arrangement. However, differentiated 1B5 cells transiently transfected with wild-type Ry1R cDNA have restored arrays of tetrads in the 1B5 homologous expression system (35).
Several other proteins have been either proposed or demonstrated to be important for the normal function of Ry1R. These include (a) FKBP12, which has been shown to stabilize the full conductance gating behavior of the channel; (b) triadin, which has been proposed either to assist in coupling the DHPR and Ry1R or to couple Ry1R and calsequestrin; and (c) calsequestrin, which has been proposed to receive its signal to release its bound calcium from the Ry1R. As was found in dyspedic skeletal muscle, all three of these proteins were detected by Western analysis of differentiated 1B5 cell membranes (Fig. 5). Immunolocalization studies with antibodies directed against triadin and α1 and α2 subunits of DHPR have revealed a similar punctate pattern of labeling of these proteins in differentiated 1B5 cells thus confirming expression and targeting of these important triadic proteins (35). Two additional skeletal muscle–specific proteins were also detected in differentiated 1B5 cells by Western analysis: Ca2+-(Mg2+) ATPase (SERCA1), the pump protein responsible for Ca2+ uptake into SR, and myosin heavy chain (Fig. 5).
Taken together, these biochemical findings reveal that the ry1r−/ry1r− 1B5 cell line expresses the major elements of the triad junction involved in e–c coupling with the exception of RyR proteins. The 1B5 cell line therefore provides a unique opportunity to study how RyR structure relates to function within the context of a skeletal muscle phenotype.
Transient Transfection of 1B5 Cells with Ry1R cDNA Restores Function
1B5 cells grown in differentiation medium were loaded with fura-2 and examined for functional responses in an extracellular medium that contained 10 nM free Ca2+ and responses were measured from individual cells using ratio fluorescence imaging. 1B5 cells mock transfected with Lipofectamine showed robust responses to ionophore 4-BrA23187 (not shown) demonstrating the integrity of the Ca2+ stores in these cells. The magnitude of this response to ionophore was similar in sham-transfected cells and cells transfected with Ry1R cDNA, suggesting a similar amount of Ca2+ within SR stores. Sham-transfected 1B5 myotubes possessed no detectable responses to K+ (⩽80 mM) depolarization, ryanodine (⩽500 μM), or caffeine (⩽100 mM) (Fig. 6).
Figure 6.

Transient transfection of 1B5 cells with Ry1R cDNA restores responses to K+ depolarization, ryanodine, and caffeine. Ratio fluorescence imaging of individual differentiated myotubes was performed as described in Materials and Methods. All experiments were performed in the presence of an extracellular medium buffered to 10 nM free Ca2+ to eliminate Ca2+ influx. The top panel shows a typical experiment demonstrating lack of response to K+ depolarization (40 mM) in mock-transfected 1B5 cells (of 48 cells examined, 0 responded), and restoration of the response after transient transfection with wild-type Ry1R cDNA (of 163 cells examined, 50 responded). The middle panel shows a typical result with ryanodine (200 μM) on differentiated 1B5 cells before and after transfection with wild-type Ry1R cDNA. In n = 260 total measurements, no Ry1R null cell was ever found to respond to ryanodine. After transfection using the wild-type Ry1R cDNA, 106 1B5 cells responded to 200 μM ryanodine out of 111 cells examined in calcium-free media. The bottom panel shows a typical response to 50 mM caffeine from mock transfected (of 34 cells examined, 0 responded) and Ry1R cDNA–transfected (of 82 cells examined, 51 responded) 1B5 cells. In 109 total measurements, no Ry1R null cell was ever found to respond to caffeine (10–100 mM).
Differentiated 1B5 cells were transiently transfected with Lipofectamine reagent and a plasmid containing the wild-type Ry1R cDNA under the control of a CMV promoter as described in Materials and Methods. The transfection protocol results in efficient reconstitution of detectable Ry1R protein in 1B5 cells, and immunofluorescence reveals punctate rather than diffuse labeling (Fig. 3). The functional responses of fura-2–loaded 1B5 cells transfected with wild-type Ry1R cDNA were analyzed using ratio fluorescence imaging microscopy. Transfection of 1B5 myotubes with the full-length Ry1R cDNA restored responses to K+ (40 mM) depolarization that was independent of extracellular Ca2+ (Fig. 6), suggesting a restoration of skeletal-type e–c coupling in the 1B5 cell. In this respect, 1B5 cells expressing functional Ry1R protein and skeletal-type e–c coupling would be expected to regain an ordered tetradic array of α1-DHPR subunits. Initial results obtained from freeze fractures performed with 1B5 cells transfected with Ry1R cDNA has confirmed this hypothesis (34). The efficiency of recovering depolarization induced Ca2+ release using transient transfection with Lipofectamine ranged from 20 to 50% within any given field in 14 different experiments. Transfection of 1B5 cells also restored responses to ryanodine in 10 out of 10 independent transfections, with at least six coverslips examined per transfection. The typical response to ryanodine was also independent of extracellular Ca2+, although the rate of change in intracellular Ca2+ was significantly slower than the response seen with K+ (Fig. 6). Caffeine as high as 100 mM failed to elicit a response in sham-transfected 1B5 cells whereas transfection with Ry1R cDNA resulted in the anticipated response. Myotube contractions were observed during experiments using ryanodine and caffeine on 1B5 cells transiently transfected with Ry1R cDNA (data not shown).
Additional evidence that Ry1R protein was targeted to SR membranes of 1B5 cells transiently transfected with Ry1R cDNA was obtained from imaging studies performed in Ca2+ replete medium and 1 μM thapsigargin. Thapsigargin, a known selective cell permeant blocker of SERCA pumps was used to assess the nature of the ryanodine-sensitive store. Ryanodine (200 μM) produced a transient elevation in intracellular Ca2+ which was similar to that seen in Ca2+-free medium (Fig. 7 A). Subsequent addition of thapsigargin revealed that the SR store reaccumulated Ca2+. These results are consistent with the known sequential effects of micromolar ryanodine on SR Ca2+ channel function, first activating and then inhibiting (33). Importantly, if thapsigargin is introduced into the cell medium first to completely deplete the microsomal stores, subsequent addition of ryanodine fails to produce a detectable response (Fig. 7 B). These results provide independent verification that Ry1R protein is targeted correctly to the SR membrane.
Figure 7.
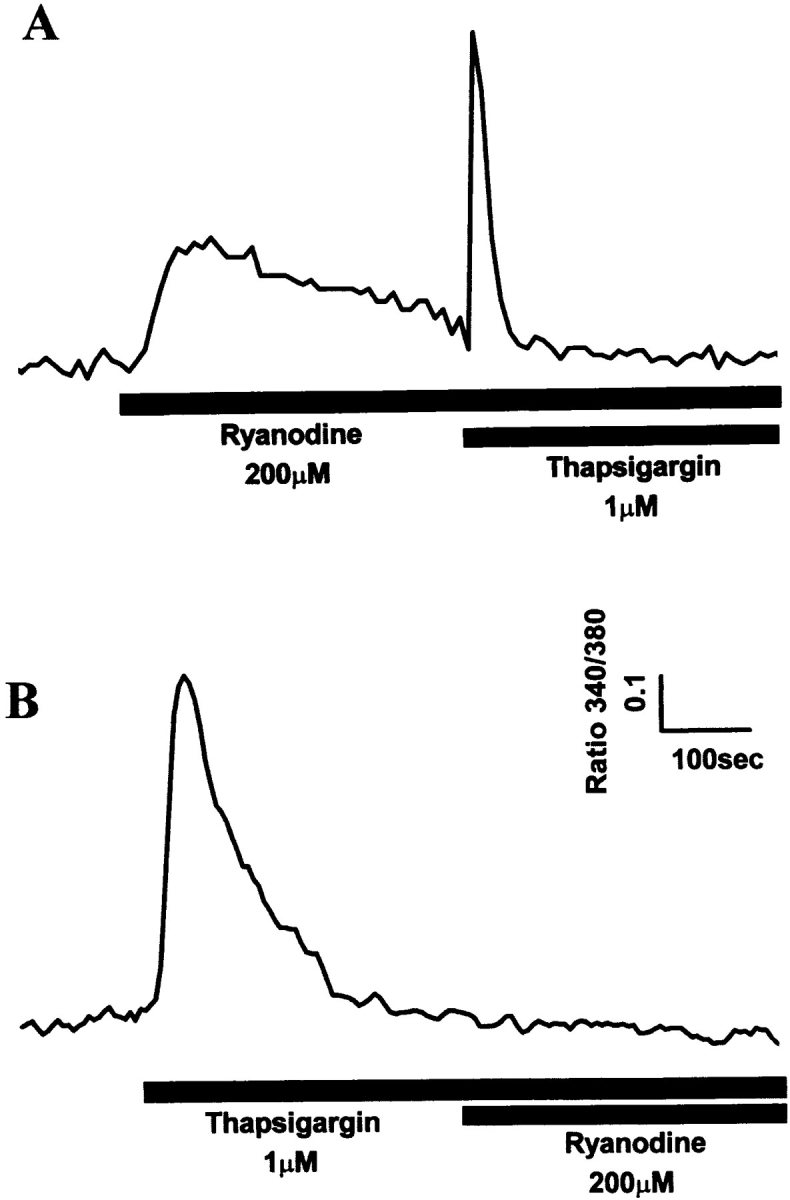
Targeting of Ry1R protein to SR membranes of 1B5 cells transiently transfected with Ry1R cDNA. Ratio fluorescence imaging of individual differentiated myotubes in 2 mM extracellular Ca2+ was performed as described in Materials and Methods. (A) Typical experiment demonstrating the response to ryanodine (200 μM) in 1B5 myotubes transiently transfected with wild-type Ry1R cDNA followed by the addition of thapsigargin (1 μM) (n = 63). (B) Typical response of 1B5 cells transiently transfected with wild-type Ry1R cDNA to thapsigargin (1 μM) followed by ryanodine (200 μM) (n = 61).
Reconstitution of Wild-Type Single Channels from 1B5 Cells Expressing Ry1R cDNA
Several (>30) attempts to reconstitute ryanodine-sensitive Ca2+ channels from nontransfected 1B5 microsomes into planar bilayer membranes have failed to produce the large, ryanodine-sensitive current fluctuations that are consistent with Ry1R. SR membranes prepared from 1B5 cells transfected with wild-type rabbit Ry1R cDNA produce successful channel fusions with gating transitions; these gating transitions are predominately of the full-conductance type, characteristic of those seen with reconstituted channels from rabbit junctional SR (Fig. 8 A). The current/voltage relationship from channels isolated from 1B5 cells yielded a linear current/voltage relationship from −40 to +40 mV that are indistinguishable from channels reconstituted from rabbit SR (Fig. 8 B). Further, the unitary conductance of n = 8 expressed channels averaged 451 ± 77 pS, which is not statistically different from that of channels obtained from native rabbit skeletal muscle SR (6). Channel fluctuations from the recombinant channels are very rapid with τ1 and τ2 averaging 0.6 and 2.9 ms, as expected for a wild-type channel. The addition of 4 μM ryanodine to the cis chamber produced a long-lived subconductance ∼50% of the native channel, whereas 10 μM ruthenium red completely blocked the Cs+ current (Fig. 8 C). The steady-state open probability was highly dependent on the concentration of Ca2+ in the cis chamber (not shown) with the open probability (P o) ranging from 0.1 to 0.4 at 50 μM Ca2+ cis (n = 8 channels).
Figure 8.



Reconstitution of single channels from 1B5 cells expressing wild-type rabbit skeletal Ry1R. (A) Single channel records (450 ms/trace) obtained from 1B5 cell SR expressing Ry1R. Channel currents were measured with 50 μM Ca2+ in the cis chamber and symmetric 100 mM Cs2+. (B) Current–voltage relationship from a single expressed ryanodine receptor and a control ryanodine receptor. (C) The effect of 4 μM ryanodine and 10 μM ruthenium red on expressed ryanodine receptor activated with 100 μM Ca2+ in the cis chamber at +30 mV in asymmetric Cs2+ (5:1). Similar gating behavior was obtained from n = 8 channels (see text for details).
Conclusion
The 1B5 myogenic cell line provides a truly homologous expression system to investigate the relationships between structure and function of ryanodine receptors. Several benefits are realized from the 1B5 cell model over other cell lines used to express ryanodine receptors. First, key triadic proteins normally present at the skeletal muscle triad are also present in 1B5 cells including α1-DHPR, FKBP12, triadin, calsequestrin, and SERCA1. Second, by virtue of being a stable line, the 1B5 cells permit efficient transient transfection of cDNAs encoding for RyR proteins. Transient transfection of 1B5 cells with pCMVRy1R using Lipofectamine reagent reveals that Ry1R function can be efficiently restored at the level of the cell (Figs. 6 and 7) and single channel (Fig. 8). Further, the 1B5 line will permit creation of subclones permanently expressing RyR proteins, which possess defined mutations that should prove invaluable for structure/function studies at both the cellular and single channel levels. This approach should circumvent all of the major problems currently confronting transient transfection studies in heterologous systems. Finally, the biochemical and functional studies presented here indicate that 1B5 cells provide a truly null background for ryanodine receptors, which will permit unambiguous interpretations of results from structure function studies of this protein.
Footnotes
We gratefully acknowledge Drs. E. Buck and W. Feng (University of California, Davis, CA) for assistance with BLM experiments. We also thank T. Meloy, J. Abenojar, T. Taras, E. Kuehnis, and W. Brackney for their technical assistance.
This work was supported by a Public Health Service grant AR 413140 (P.D. Allen and I.N. Pessah), Muscular Dystrophy Association (P.D. Allen), American Heart Association, California Affiliate (I.N. Pessah), and National Institutes of Health Training Grant in Cardiovascular and Neurophysiology HL07682 (R.A. Moore), and American Heart Association, California Affiliate 97-403 (R.A. Moore).
Address all correspondence to Dr. Isaac N. Pessah, Department of Molecular Biosciences, School of Veterinary Medicine, University of California, Davis, CA 95616. Tel.: (530) 752-6696. Fax: (530) 752-4698. E-mail: inpessah@ucdavis.edu
Dr. Hanh Nguyen's present address is Department of Medicine, Division of Cardiology, Albert Einstein College of Medicine, Bronx, NY.
Joan Galceran's present address is Department of Microbiology and Immunology, University of California, San Francisco, CA.
1. Abbreviations used in this paper: BLM, bilayer lipid membrane; α1-DHPR, α-1 subunit of L-type voltage-dependent Ca2+ channel dihydropyridine receptor; e–c, excitation–contraction; ES, embryonic stem; FKBP12, FK506 binding protein, 12 kD; G418, neomyocin antibiotic analogue; HIHS, heat-inactivated horse serum; P o, open probability; Ry1R, Ry2R, and Ry3R, skeletal isoform of ryanodine receptor, cardiac isoform of ryanodine receptor, and brain isoform of ryanodine receptor; SCID, severely compromised immunodeficient; SERCA, sarco(endo)plasmic reticulum Ca2+-ATPase; SR, sarcoplasmic reticulum.
References
- 1.Adams BA, Tanabe T, Mikami A, Numa S, Beam KG. Intramembrane charge movement restored in dysgenic skeletal muscle by injection of dihydropyridine receptor cDNAs. Nature. 1990;346:569–572. doi: 10.1038/346569a0. [DOI] [PubMed] [Google Scholar]
- 2.Airey JA, Beck CF, Murakami K, Tanksley SJ, Deerinck TJ, Ellisman MH, Sutko JL. Identification and localization of two triad junctional foot protein isoforms in mature avian fast twitch skeletal muscle [published erratum appears in J. Biol. Chem. 1990. 265:22057.] . J Biol Chem. 1990;265:14187–14194. [PubMed] [Google Scholar]
- 3.Bhat MB, Zhao J, Hayek S, Freeman EC, Takeshima H, Ma J. Deletion of amino acids 1641-2437 from the foot region of skeletal muscle ryanodine receptor alters the conduction properties of the Ca release channel. Biophys J. 1997;73:1320–1328. doi: 10.1016/S0006-3495(97)78165-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brandt, N.R., A.H. Caswell, J.P. Brunschwig, J.J. Kang, B. Antoniu, and N. Ikemoto. Effects of anti-triadin antibody on Ca2+ release from sarcoplasmic reticulum. FEBS (Fed. Eur. Biochem. Soc.) Lett. 299:57–59. [DOI] [PubMed]
- 5.Brillantes AB, Ondrias K, Scott A, Kobrinsky E, Ondriasova E, Moschella MC, Jayaraman T, Landers M, Ehrlich BE, Marks AR. Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell. 1994;77:513–523. doi: 10.1016/0092-8674(94)90214-3. [DOI] [PubMed] [Google Scholar]
- 6.Buck E, Zimanyi I, Abramson JJ, Pessah IN. Ryanodine stabilizes multiple conformational states of the skeletal muscle calcium release channel. J Biol Chem. 1992;267:23560–23567. [PubMed] [Google Scholar]
- 7.Buck ED, Nguyen HT, Pessah IN, Allen PD. Dyspedic mouse skeletal muscle express major elements of the triadin junction but lacks detectable ryanodine receptor protein and function. J Biol Chem. 1997;272:7360–7367. doi: 10.1074/jbc.272.11.7360. [DOI] [PubMed] [Google Scholar]
- 8.Caswell AH, Brandt NR, Brunschwig JP, Purkerson S. Localization and partial characterization of the oligomeric disulfide-linked molecular weight 95,000 protein (triadin) which binds the ryanodine and dihydropyridine receptors in skeletal muscle triadic vesicles. Biochemistry. 1991;30:7507–7513. doi: 10.1021/bi00244a020. [DOI] [PubMed] [Google Scholar]
- 9.Chen SR, Vaughan DM, Airey JA, Coronado R, MacLennan DH. Functional expression of cDNA encoding the Ca2+ release channel (ryanodine receptor) of rabbit skeletal muscle sarcoplasmic reticulum in COS-1 cells. Biochemistry. 1993;32:3743–3753. doi: 10.1021/bi00065a029. [DOI] [PubMed] [Google Scholar]
- 10.Doetschman TC, Eistetter H, Katz M, Schmidt W, Kemler R. The in vitrodevelopment of blastocyst-derived embryonic stem cell lines; formation of visceral yolk sac, blood islands and myocardium. J Embryol Exp Morph. 1985;82:27–45. [PubMed] [Google Scholar]
- 11.el-Hayek R, Antoniu B, Wang J, Hamilton SL, Ikemoto N. Identification of calcium release-triggering and blocking regions of the II-III loop of the skeletal muscle dihydropyridine receptor. J Biol Chem. 1995;270:22116–22118. doi: 10.1074/jbc.270.38.22116. [DOI] [PubMed] [Google Scholar]
- 12.Feher JJ, Davis MD. Isolation of rat cardiac sarcoplasmic reticulum with improved Ca2+uptake and ryanodine binding. J Mol Cell Cardiol. 1991;23:249–258. doi: 10.1016/0022-2828(91)90061-p. [DOI] [PubMed] [Google Scholar]
- 13.Franzini-Armstrong C, Pincon-Raymond M, Rieger F. Muscle fibers from dysgenic mouse in vivo lack a surface component of peripheral couplings. Dev Biol. 1991;146:364–736. doi: 10.1016/0012-1606(91)90238-x. [DOI] [PubMed] [Google Scholar]
- 14.Giannini G, Conti A, Mammarella S, Scrobogna M, Sorrentino V. The ryanodine receptor/calcium channel genes are widely and differentially expressed in murine brain and peripheral tissues. J Cell Biol. 1995;128:893–904. doi: 10.1083/jcb.128.5.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gilchrist JS, Belcastro AN, Katz S. Intraluminal Ca2+ dependence of Ca2+ and ryanodine-mediated regulation of skeletal muscle sarcoplasmic reticulum Ca2+release. J Biol Chem. 1992;267:20850–20856. [PubMed] [Google Scholar]
- 16.Guo W, Campbell KP. Association of triadin with the ryanodine receptor and calsequestrin in the lumen of the sarcoplasmic reticulum. J Biol Chem. 1995;270:9027–9030. doi: 10.1074/jbc.270.16.9027. [DOI] [PubMed] [Google Scholar]
- 17.Ikemoto N, Antoniu B, Kang JJ, Meszaros LG, Ronjat M. Intravesicular calcium transient during calcium release from sarcoplasmic reticulum. Biochemistry. 1991;30:5230–5237. doi: 10.1021/bi00235a017. [DOI] [PubMed] [Google Scholar]
- 18.Inesi G, Sagara Y. Specific inhibitors of intracellular Ca2+transport ATPase. J Membr Biol. 1994;141:1–6. doi: 10.1007/BF00232868. [DOI] [PubMed] [Google Scholar]
- 19.Knudson CM, Stang KK, Jorgensen AO, Campbell KP. Biochemical characterization of ultrastructural localization of a major junctional sarcoplasmic reticulum glycoprotein (triadin) J Biol Chem. 1993;268:12637–12645. [PubMed] [Google Scholar]
- 20.Knudson CM, Stang KK, Moomaw CR, Slaughter CA, Campbell KP. Primary structure and topological analysis of a skeletal muscle-specific junctional sarcoplasmic reticulum glycoprotein (triadin) J Biol Chem. 1993;268:12646–12654. [PubMed] [Google Scholar]
- 21.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 22.Lai FA, Dent M, Wickenden C, Xu L, Kumari G, Misra M, Lee HB, Sar M, Meissner G. Expression of cardiac Ca2+-release channel isoform in mammalian brain. Biochem J. 1992;288:553–564. doi: 10.1042/bj2880553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lowry OH, Rosenbrough NJ, Farr AL, Randall RJ. Protein measurement with folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 24.Lu X, Xu L, Meissner G. Activation of the skeletal muscle calcium release channel by a cytoplasmic loop of the dihydropyridine receptor. J Biol Chem. 1994;269:6511–6516. [PubMed] [Google Scholar]
- 25.Lu X, Xu L, Meissner G. Phosphorylation of dihydropyridine receptor II-III loop peptide regulates skeletal muscle calcium release channel function. Evidence for an essential role of the β-OH group of Ser687. J Biol Chem. 1995;270:18459–18464. doi: 10.1074/jbc.270.31.18459. [DOI] [PubMed] [Google Scholar]
- 26.Marks A, Fleisher S, Nadal-Ginard B, Tempst P. Surface topography analysis of the ryanodine receptor/junctional channel complex based on proteolysis sensitivity mapping. J Biol Chem. 1990;265:13143–13149. [PubMed] [Google Scholar]
- 27.Marty I, Robert M, Villaz M, Catterall WA, Ronjat M. Biochemical evidence for a complex involving dihydropyridine receptor and ryanodine receptor in triad junction of skeletal muscle. Proc Natl Acad Sci USA. 1994;91:2270–2274. doi: 10.1073/pnas.91.6.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McPherson PS, Campbell KP. The ryanodine receptor/Ca2+release channel. J Biol Chem. 1993;268:13765–13768. [PubMed] [Google Scholar]
- 29.Meissner G. Annu Rev Physiol. 1994;56:485–508. doi: 10.1146/annurev.ph.56.030194.002413. [DOI] [PubMed] [Google Scholar]
- 30.Mortensen RM, Conner DA, Chao S, Geisterfer-Lowrance AA, Seidman JG. production of homozygous mutant ES cells with a single targeting construct. Mol Cell Biol. 1992;12:2391–2395. doi: 10.1128/mcb.12.5.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakai J, Dirksen RT, Nguyen HT, Pessah IN, Beam KG, Allen PD. Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature. 1996;380:72–75. doi: 10.1038/380072a0. [DOI] [PubMed] [Google Scholar]
- 32.Penner R, Neher E, Takeshima H, Nishimura S, Numa S. Functional expression of the calcium release channel from skeletal muscle ryanodiine receptor cDNA. FEBS (Fed Eur Biochem Soc) Lett. 1989;259:217–221. doi: 10.1016/0014-5793(89)81532-7. [DOI] [PubMed] [Google Scholar]
- 33.Pessah IN, Zimanyi I. Characterization of multiple [3H]ryanodine binding sites on the Ca2+release channel of sarcoplasmic reticulum from skelet al and cardiac muscle: evidence for a sequential mechanism in ryanodine action. Mol Pharmacol. 1991;39:679–689. [PubMed] [Google Scholar]
- 34.Philips MS, Fujii J, Khanna VK, DeLeon S, Yokobata K, de Jong PJ, MacLennan DH. The structure organization of the human skeletal muscle ryanodine (RyR1) gene. Genomics. 1996;34:24–41. doi: 10.1006/geno.1996.0238. [DOI] [PubMed] [Google Scholar]
- 35.Protasi F, Franzini-Armstrong C, Allen PD. Role of ryanodine receptors in the assembly of calcium release units in skeletal muscle. J Cell Biol. 1998;140:831–842. doi: 10.1083/jcb.140.4.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rios E, Ma JJ, Gonzalez A. The mechanical hypothesis of excitation-contraction (EC) coupling in skeletal muscle. J Muscle Res Cell Motil. 1991;12:127–135. doi: 10.1007/BF01774031. [DOI] [PubMed] [Google Scholar]
- 37.Saito A, Seiler S, Chu A, Fleischer S. Preparation and morphology of sarcoplasmic reticulum terminal cisternae from rabbit skeletal muscle. J Cell Biol. 1984;99:875–885. doi: 10.1083/jcb.99.3.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takekura H, Nishi M, Noda T, Takeshima H, Franzini-Armstrong C. Abnormal junctions between surface membrane and sarcoplasmic reticulum in skeletal muscle with a mutation targeted to the ryanodine receptor. Proc Natl Acad Sci USA. 1995;92:3381–3385. doi: 10.1073/pnas.92.8.3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tanabe T, Beam KG, Adams BA, Niidome T, Numa S. Regions of the skeletal muscle dihydropyridine receptor critical for excitation-contraction coupling. Nature. 1990;346:567–569. doi: 10.1038/346567a0. [DOI] [PubMed] [Google Scholar]