

Abstract
Cytochrome c has been shown to play a role in cell-free models of apoptosis. During NGF withdrawal–induced apoptosis of intact rat superior cervical ganglion (SCG) neurons, we observe the redistribution of cytochrome c from the mitochondria to the cytoplasm. This redistribution is not inhibited by the caspase inhibitor Z-Val-Ala-Asp-fluoromethylketone (ZVADfmk) but is blocked by either of the neuronal survival agents 8-(4-chlorophenylthio)adenosine 3′:5′-cyclic monophosphate (CPT-cAMP) or cycloheximide. Moreover, microinjection of SCG neurons with antibody to cytochrome c blocks NGF withdrawal–induced apoptosis. However, microinjection of SCG neurons with cytochrome c does not alter the rate of apoptosis in either the presence or absence of NGF. These data suggest that cytochrome c is an intrinsic but not limiting component of the neuronal apoptotic pathway.
Keywords: cytochrome c, mitochondria, neuron, apoptosis, cAMP
Apoptosis is a morphologically and biochemically distinct form of cell death, activated in multiple tissues during development and homeostasis (Ellis et al., 1991; Jacobson et al., 1997). It is thought to play an important role in the development and shaping of the nervous system (Oppenheim, 1991; Johnson and Deckwerth, 1993; Burek and Oppenheim, 1996). Many neurons are dependent upon neurotrophic factors, which stimulate survival pathways within the cell, and their removal can lead to apoptosis (Raff et al., 1993; Silos-Santiago et al., 1995; Segal and Greenberg, 1996). In addition, there is accumulating evidence that apoptosis is involved in neurodegenerative diseases (Bredesen, 1995; Thompson, 1995; Johnson et al., 1996), such as Parkinson's disease (Mochizuki et al., 1996), Alzheimer's disease (Cotman and Anderson, 1995), amylotrophic lateral sclerosis (Yoshiyama et al., 1994; Ghadge et al., 1997), and stroke (Linnik et al., 1993; Choi, 1996).
Many of the genes involved in apoptosis have first been identified in the nematode Caenorhabditis elegans (Hengartner and Horvitz, 1994 a). These include the prototype gene for the caspase family of proteases, ced-3 (Yuan et al., 1993), ced-9, which is homologous to the bcl-2 gene family (Hengartner and Horvitz, 1994b ), and most recently Apaf-1, which is homologous to both ced-4 and ced-3 (Zou et al., 1997). Caspases are activated by cleavage after aspartate residues, and it has been shown in vitro that they can be activated by other active caspases (Fernandes-Alnemri et al., 1996; Liu et al., 1996a ; Srinivasula et al., 1996a ,b). Caspases are now thought to form a proteolytic network within the cell, resulting in the breakdown of key enzymes and cellular structures (Nicholson and Thornberry, 1997; Salvesen and Dixit, 1997). Their target proteins include poly(ADP-ribose) polymerase (Nicholson et al., 1995; Tewari et al., 1995), nuclear lamins (Lazebnik et al., 1995; Takahashi et al., 1996), retinoblastoma protein (Janicke et al., 1996), DNA-dependent protein kinase (Song et al., 1996), and Bcl-2 family members (Cheng et al., 1997; Clem et al., 1998). In addition, caspase 3 has been shown to cause activation of DNase activity thought to be responsible for the chromatin degradation seen in apoptosis (Bortner et al., 1995; Liu et al., 1997; Enari et al., 1998; Sakahira et al., 1998).
Bcl-2 family members encode proteins that either inhibit or accelerate apoptosis in response to a wide range of death stimuli (Nunez and Clarke, 1994; Kroemer, 1997). The mechanism for this is unclear, but they are known to form dimers, which, while located predominantly in the outer mitochondrial membrane, are also found in the nuclear envelope and endoplasmic reticulum (Krajewski et al., 1993). They may function as channels to regulate permeability across these membranes (Antonsson et al., 1997; Minn et al., 1997; Reed, 1997; Schendel et al., 1997). A further insight into the function of Bcl-2–like proteins has come about after the surprising discovery that cytochrome c plays a role in apoptosis. Cytochrome c, originally termed Apaf-2,1 was isolated as one of the factors that mediated caspase activation when dATP was added to normal cell extracts (Liu et al., 1996b ). It has also been reported that cytochrome c partitioned into the cytoplasmic fraction of cells undergoing apoptosis but partitioned into the mitochondrial fraction of normal cells (Liu et al., 1996b ; Kharbanda et al., 1997; Kluck et al., 1997). This apparent redistribution could be prevented by overexpression of bcl-2 (Kluck et al., 1997; Yang et al., 1997). Cytochrome c was found to require other cytoplasmic partners to activate caspases. Recently, Apaf-1 (Zou et al., 1997) and caspase 9 (Apaf-3) have been isolated and shown to be sufficient, with cytochrome c and dATP, to activate caspase 3 (P. Li et al., 1997). These experiments suggest that Bcl-2–like proteins may act by preventing the release of cytochrome c from the mitochondria, but the possibility that they have additional functions is not excluded (Murphy et al., 1996; F. Li et al., 1997; Reed, 1997). From studies in C. elegans, we know that ced-4 lies downstream of ced-9 but upstream of ced-3 (Shaham and Horvitz, 1996). In addition, CED-4 has been shown to directly interact with CED-9, CED-3, and Bcl-2 (Chinnaiyan et al., 1997; Wu et al., 1997; Huang et al., 1998). Caspase 9 and Apaf-1 association has been demonstrated in vitro (P. Li et al., 1997), so by analogy with C. elegans, a bcl-2–like protein may interact directly with Apaf-1. There have also been reports of cytochrome c interacting directly with Bcl-xL (Kharbanda et al., 1997).
To examine whether cytochrome c plays a role in neuronal cell death, we have first determined that after removal of NGF, cytochrome c redistributes from the mitochondria to the cytoplasm. This event was antagonized by two different neuroprotective agents, 8-(4-chlorophenylthio)adenosine 3′:5′-cyclic monophosphate (CPT-cAMP) and cycloheximide, placing their action upstream of cytochrome c, but not by a caspase inhibitor. We have also demonstrated that an antibody to cytochrome c, when microinjected into superior cervical ganglion (SCG) neurons, was able to prevent apoptosis after NGF withdrawal. Finally, cytochrome c injected into neurons did not increase the amount of death either in the presence or absence of NGF, suggesting that redistribution of cytochrome c is not the only regulated step during neuronal cell death.
Materials and Methods
Cell Culture and Apoptosis Induction
Rat SCG neurons were isolated and maintained as described previously (Philpott et al., 1996). Cells were routinely maintained in 100 ng/ml NGF (Promega Corp., Madison, WI). 5–7-d-old cells were deprived of NGF by rinsing twice in media without NGF and incubating the cells with 100 ng/ml anti-NGF antibody (GIBCO BRL; Life Technologies, Rockville, MD). CPT-cAMP, cycloheximide, and actinomycin D were obtained from Sigma Chemical Co. (St. Louis, MO). Z-Val-Ala-Asp-fluoromethylketone (ZVADfmk) was obtained from Enzyme Systems Products (Dublin, CA). Stock solutions of CPT-cAMP were in water, and the others were in DMSO.
Jurkat cells were grown in DME (4.5 mg/ml glucose)/10% FCS and were cultured at 37°C in a 10% CO2 atmosphere.
Immunofluorescence
Cells were fixed with 3% paraformaldehyde in PBS for 15 min, blocked with 10 mM glycine in PBS for 10 min, and then rinsed in PBS. The cells were permeabilized in binding buffer (0.5% Triton X-100, 0.2% gelatine, 0.5% BSA, PBS) for 5 min before incubation in this solution with 20 μg/ml of the 2G8.B6 anti–cytochrome c antibody (a kind gift from Dr. R. Jemmerson, University of Minnesota, Minneapolis, MN; Mueller and Jemmerson, 1996) for 1–2 h. After a 20-min wash in fresh binding buffer, the cells were incubated in 1:100 FITC-conjugated anti–mouse antibody (Jackson Laboratories, Bar Harbor, ME) for an additional 1 h. The cells were finally washed in fresh binding buffer for up to 1 h and costained with 1 μg/ml Hoechst 33342 in water before mounting in 0.25% n-propylgalate and 90% glycerol in PBS.
For labeling of functional mitochondria, cells were incubated in the presence of 450 nM Mitotracker (Molecular Probes, Eugene, OR) for 30– 40 min followed by a 30–60-min incubation in fresh medium and fixation as described above.
Fluorescence Assay of Caspase Activation
Jurkat cytosol preparation and caspase activation were according to a modification of Liu et al. (1996b). Cells were harvested by centrifugation, washed in ice-cold PBS, and incubated on ice for 15 min in a fivefold volume of buffer A (10 mM KCl, 20 mM Hepes, pH 7.4, 1.5 mM MgCl2, 1 mM DTT, 1 μg/ml each of pepstatin and leupeptin, 5 μg/ml antipain, 10 μg/ml chymostatin, and 100 μM PMSF). The cells were disrupted by 20 strokes of a glass/Teflon Dounce homogenizer, and the nuclei and debris were removed by centrifugation for 15 min at 1,000 g, followed by 1 h at 100,000 g. Cytosol was frozen in aliquots in liquid N2. Caspase 3 activation reactions consisted of cytosol (50 μg protein) with 0.002 mg/ml cytochrome C (Sigma Chemical Co.), 0.25 mM dATP (Ultrapure; Pharmacia Biotech, Piscataway, NJ) made to a final volume of 20 μl with buffer A. Reactions were incubated for 1 h at 30°C. For fluorescence assay of active caspase, 25 μg protein (in 10 μl) was mixed with 200 μl of 100 mM Hepes, pH 7.4, 10% sucrose, 0.1% CHAPS, 10 μg/ml DEVD-AMC (Enzyme Systems), of which 100 μl was placed in duplicate wells of a 96-well plate. The plates were incubated for 60 min at 37°C and measured in a fluorimeter (model LS 50B; Perkin-Elmer Corp., Norwalk, CT).
Immunoblot Analysis
For immunoblot analysis, 25 μg protein from the activation reactions (for caspase 3) or 8–16 × 103 cells (for cytochrome c/ERK1/2) were used. Samples were run under reducing conditions on a 15% SDS–polyacrylamide gel and electroblotted onto nitrocellulose membrane. Membranes were blocked in TBST (10 mM Tris-HCl, pH 8, 150 mM NaCl, 0.1% Tween 20) supplemented with 1% Tween 20 and 5% dried low-fat milk for 20 min and incubated in the same buffer with 250 ng/ml anti–caspase 3 mAb (Transduction Laboratories, Lexington, KY) or 1:1,000 anti–cytochrome c antibody, 7H8.2C12, ascites (a kind gift from Dr. R. Jemmerson; Jemmerson et al., 1991). This was followed by up to 1 h each with 1:1,000 dilution of biotin anti–mouse IgG and HRP-streptavidin (Amersham Corp., Arlington Heights, IL). Filters were washed for up to 1 h with TBST between each reagent and before development of signal by enhanced chemiluminescence (Amersham Corp.). For analysis of ERK1/2, blots were reprobed with mAb E16220 or M12320 (Affiniti Research Products, Nottingham, UK) at 1:500 and treated as above. Blots were quantified using a scanner (model GS670; Bio-Rad Labs, Hercules, CA) and NIH Image software.
Microinjection of Neurons
Neurons were microinjected with 0.5× PBS, 5 mg/ml 70-kD Texas red dextran (Molecular Probes), plus either bovine cytochrome c (Sigma Chemical Co.), microperoxidase (Sigma Chemical Co.), 2G8.B6, or 6H2.B4 (PharMingen, San Diego, CA) anti–cytochrome c antibody or mouse (Pierce and Warriner, Rockford, IL) IgG at 20 mg/ml. Cells were counted 2–4 h after injection (time zero) and again 48 or 72 h later. Cells were evaluated by microscopic assessment of several parameters. Initially, only cells that were stained with Texas red, indicating that these were injected cells and that they possessed an uncompromised plasma membrane, were counted. These cells were further assessed under phase illumination for normal nuclear morphology and phase-bright cell body. Cells that possessed all of these characteristics were counted as having survived. Apoptotic cells possessed a condensed and/or fragmented nucleus and phase dark, deformed, and shrunken cell body. Approximately 90–95% of injected cells survive the injection process itself.
Results
Cytochrome c Redistributes from the Mitochondria to the Cytoplasm after NGF Withdrawal
Previous studies in numerous model systems have shown that upon physical disruption and fractionation of cells, cytochrome c partitions with the mitochondria of normal cells but with the cytosol of cells induced to undergo apoptosis (Liu et al., 1996b ; Kharbanda et al., 1997; Kluck et al., 1997). To determine if this differential partitioning indeed represents a redistribution in intact cells, we have examined by immunofluorescence the cellular location of cytochrome c in SCG neurons. SCG neurons cultured in the presence of NGF display a normal morphology with plump nuclei (Fig. 1 A). Approximately 45% of these neurons cultured for 24 h in the absence of NGF had shrunken cytoplasm (data not shown) and pyknotic nuclei (Fig. 1 B), which is consistent with apoptosis. Immunofluorescent staining showed that healthy cells had a bright, punctate cytoplasmic cytochrome c distribution, clearly excluded from the nuclear space and consistent with a mitochondrial location. However in cells with pyknotic nuclei, the staining was diffuse and uniform throughout the cytoplasm and nuclear space, indicating release from the mitochondria. Of note was the small proportion of cells found in both conditions that displayed a normal nuclear morphology but the diffuse cytochrome c pattern (Fig. 1, A and B). This suggests that cytochrome c is released from the mitochondria during NGF withdrawal–induced apoptosis before nuclear condensation.
Figure 1.
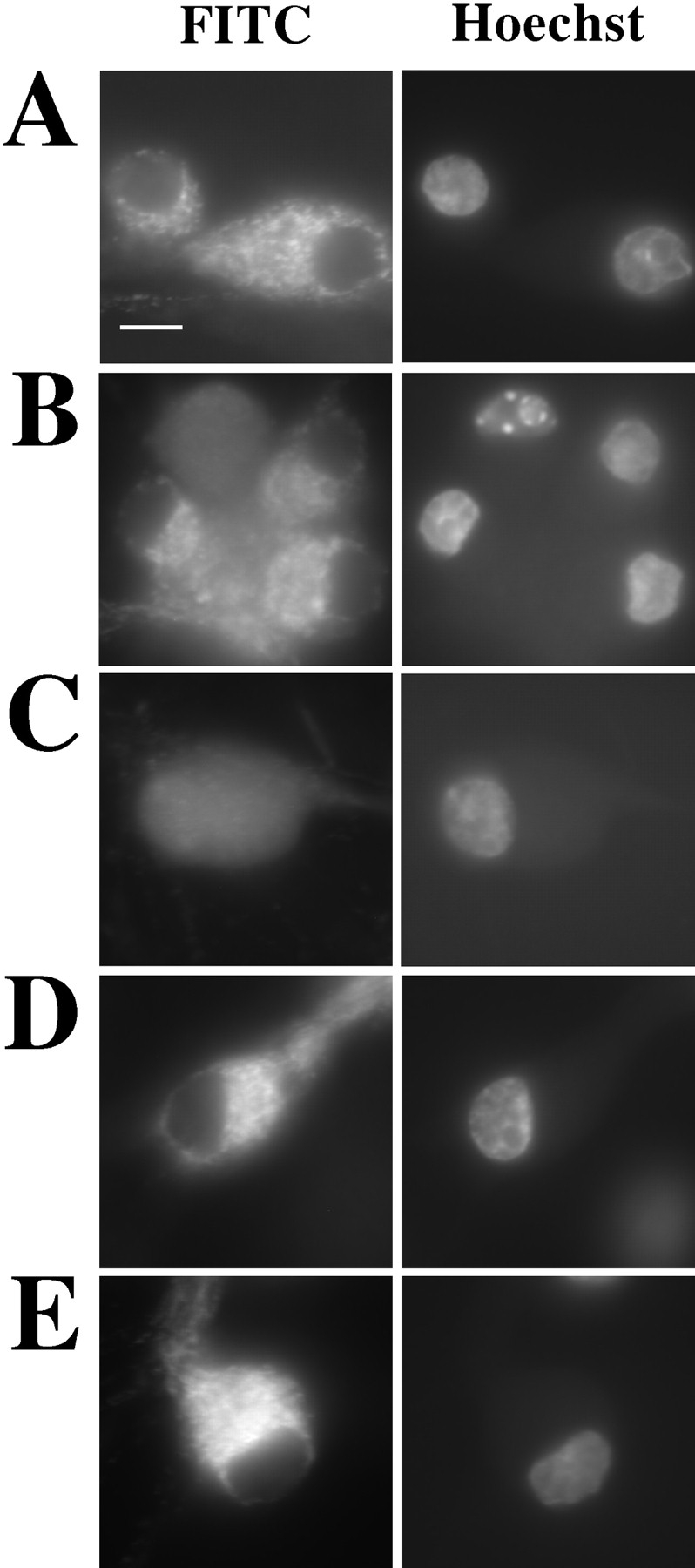
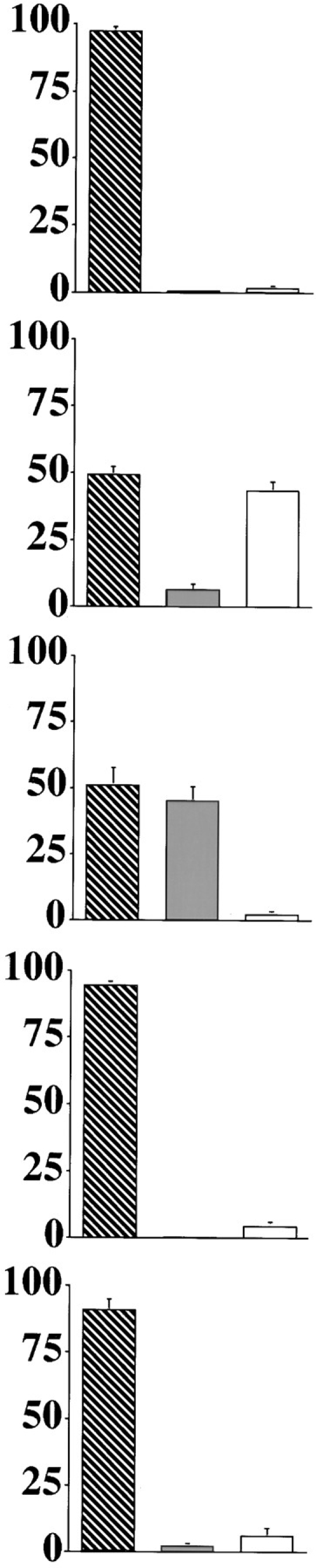
Cytochrome c redistributes from the mitochondria to the cytoplasm before nuclear condensation. SCG neurons were cultured on coverslips for 24 h in medium with NGF (A), or medium lacking NGF supplemented with carrier alone (B), 100 μM ZVADfmk (C), 1 μg/ml cycloheximide (D), or 100 μM CPT-cAMP (E). The cells were then fixed and stained for cytochrome c (FITC) and chromatin (Hoechst). Representative examples were imaged using a Xillix Microimager (Improvision-Image Processing and Vision Co. Ltd., Coventry, UK). Coverslips were mixed in a blind manner, and 10 random fields were counted per condition. Cells were evaluated as being either normal in cytochrome c distribution and nuclear morphology (hatched bars), diffuse in cytochrome c distribution and normal in nuclear morphology (shaded bars), or diffuse in cytochrome c and having pyknotic nuclei (open bars). The graphs show mean percentage of cells in each category and are the result of between four to seven experiments for each condition. The error bars represent SEM. Bar, 10 μm.
Cytochrome c Release from the Mitochondria Is Inhibited by the Neuroprotective Agents CPT-cAMP and Cycloheximide but Not by the Caspase Inhibitor ZVADfmk
We have previously shown that the cell-permeable caspase inhibitor ZVADfmk can inhibit apoptosis in SCG neurons, as determined by nuclear morphology and plasma membrane integrity (McCarthy et al., 1997). We therefore examined the effect of ZVADfmk on cytochrome c redistribution. Addition of 100 μM ZVADfmk to the medium at the time of NGF withdrawal had no effect on the number of cells in which cytochrome c was released from the mitochondria (Fig. 1 C). The addition of ZVADfmk did, however, inhibit nuclear pyknosis, indicating that it was effectively inhibiting cellular caspases.
There are a number of other agents, including cycloheximide and CPT-cAMP, known to provide protection against NGF withdrawal–induced apoptosis in SCG neurons. Cycloheximide may act by inhibiting translation of proteins necessary for apoptosis (Martin et al., 1988; Rydel and Greene, 1988; Edwards et al., 1991; Buckmaster and Tolkovsky, 1994). Alternatively, the reduction in protein synthesis may result in additional cysteine being available for the formation of the antioxidant glutathione (Ratan et al., 1994). cAMP might function via activation of protein-dependent kinase A (PKA) (Martin et al., 1988, 1992; Rydel and Greene, 1988; Edwards et al., 1991; Buckmaster and Tolkovsky, 1994). We therefore studied the cytochrome c distribution in SCG neurons deprived of NGF and treated with these agents. Both cycloheximide and CPT-cAMP prevented nuclear condensation and cytochrome c redistribution (Fig. 1, D and E). Similar experiments with 100 ng/ml actinomycin D, another neuronal survival agent thought to act through its inhibition of transcription (Martin et al., 1988), also resulted in blockade of NGF withdrawal–induced cytochrome c redistribution (data not shown). These results demonstrate that the redistribution of cytochrome c is dependent upon transcription/translation and is regulated by systems that are influenced by CPT-cAMP. In addition, they suggest that the activation of caspases, leading to nuclear condensation, occurs downstream or parallel to cytochrome c redistribution.
Cytochrome c Is Degraded after Release from the Mitochondria
We observed in our immunofluorescence experiments that there was some variation in the intensity of cytochrome c stain in cells that had lost the normal punctate pattern. A small number of bright, diffusely stained cells were always observed, but the majority of the cells were fainter. While the stain in all of these cells was uniformly spread throughout the cytoplasm and nucleus, there was a wide range of intensity from cell to cell, with some appearing barely above background. It seemed that this reduction of intensity must start soon after release of cytochrome c from the mitochondria since the number of very bright cells was always small. To determine whether this was due to a conformational change or veiling of the epitope recognized by the 2G8.B6, or more simply due to protein degradation, we analyzed cell lysates by immunoblot. Cells were cultured in NGF or withdrawn from NGF in the presence or absence of ZVADfmk for 24 h. The cells were harvested and analyzed by immunoblotting using an alternative anti– cytochrome c antibody, 7H8.2C12. We saw a clear reduction in the amount of cytochrome c present when NGF was removed, either in the presence or absence of ZVADfmk (Fig. 2). After 24 h, there was only 24–27% of the cytochrome c remaining, while the levels of ERK1 and ERK2 changed little as described by Deshmukh et al. (1996). This therefore suggests that in SCG neurons deprived of NGF, cytochrome c is initially released from the mitochondria and is then rapidly degraded. Furthermore, ZVADfmk was unable to prevent this decay, suggesting that caspases are not involved in the degradation.
Figure 2.
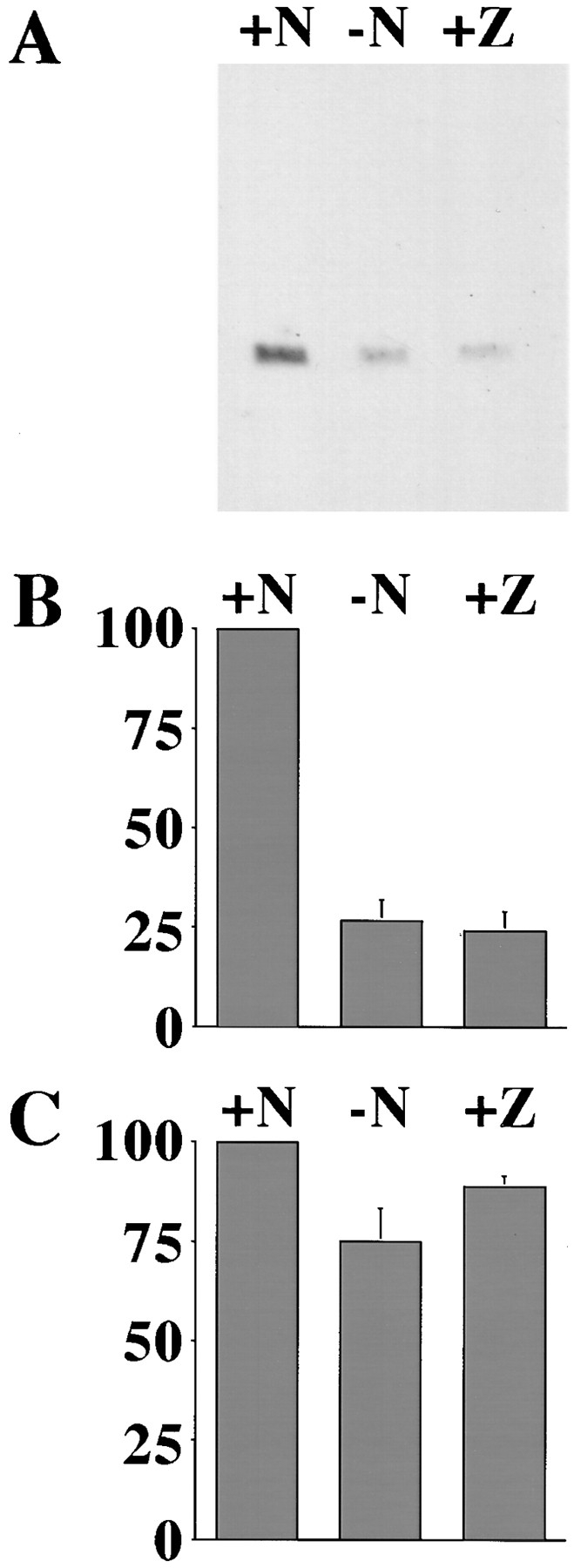
Cytochrome c degrades after release from the mitochondria. (A) SCG cultures were maintained in NGF (+N) or withdrawn from NGF in the absence (−N) or presence (+Z) of 100 μM ZVADfmk. Cells were lysed after 24 h, and protein from an equivalent number of cells was analyzed with an anti–cytochrome c antibody, 7H8.2C12. (B) Three representative blots were scanned on a densitometer, and the bars show the mean + SEM of the values obtained for cells maintained in NGF (+N) or withdrawn from NGF without (−N) or with (+Z) ZVADfmk. (C) These blots were later reanalyzed by immunoblot for ERK1/2, and the results are expressed as for the cytochrome c blots in B.
Cytochrome c Release from the Mitochondria Occurs before Breakdown of Mitochondrial Inner Membrane Potential
We were interested to discover if the observed apoptotic release of cytochrome c from the mitochondria occurred in conjunction with or independently of a generalized disruption of the mitochondrial integrity. Mitotracker, a fluorescent dye that is accumulated by functional mitochondria, was used to stain SCG neurons that were costained for cytochrome c and chromatin. Cells in the presence of NGF with normal nuclei displayed bright, punctate, and overlapping cytochrome c and Mitotracker staining patterns (Fig. 3 A). However, in the absence of NGF, cells with condensed nuclei and diffuse cytochrome c stain frequently had bright Mitotracker staining that was clearly still arranged in a punctate pattern excluded from the nucleus (Fig. 3 B, right-hand cell), indicating that cytochrome c release from the mitochondria could occur without disruption of the inner membrane. Cells in which apoptosis was well advanced (as indicated by low or complete loss of cytochrome c and chromatin stain) did, however, show reduced and diffuse Mitotracker stain, suggesting that mitochondrial inner membrane potential (ΔΨm) had decayed (Fig. 3 B, left-hand cell). This terminal ΔΨm decay was apparently not inhibited by treatment with ZVADfmk in this system (data not shown).
Figure 3.

Redistribution of cytochrome c precedes the loss of mitochondrial inner membrane potential. SCG neurons were cultured on coverslips for 23 h in medium with NGF (A) or medium lacking NGF (B). Mitotracker was added to the medium at 450 nM and incubated for 30 min, and the medium was changed. The cells were incubated for a further 30 min and then fixed and stained as in Fig. 1. Representative cells are shown displaying Mitotracker (MT), cytochrome c (FITC), and chromatin (Hoechst) localization. Bar, 10 μm.
An Anti–cytochrome c Antibody Blocks Caspase Activation in Extracts and Apoptosis in Neurons
The above data show a correlation between cytochrome c distribution and survival in intact neurons. However, we wanted to determine whether mitochondrial loss of cytochrome c was essential for neuronal apoptosis. We therefore used the 2G8.B6 anti–cytochrome c mAb in an in vitro system to ascertain whether it could prevent the activation of caspases. Normal Jurkat cytosol was incubated either in the absence of cytochrome c and dATP or in the presence of cytochrome c and dATP, with or without the anti–cytochrome c or control antibody. The extracts were then analyzed for activation of caspase 3 by immunoblot. This demonstrated that the activation of caspase 3 leading to the loss of the p32 precursor, induced by the addition of cytochrome c and dATP, was clearly inhibited by the anti– cytochrome c mAb but not by the control mouse IgG (Fig. 4 A). The extracts were further tested by a fluorescence assay based upon hydrolysis of DEVD-AMC, a substrate for caspase 3. The extracts incubated with no antibody or control antibody generated a significant amount of free AMC, whereas the extract incubated with the 2G8.B6 mAb did not (Fig. 4 B). These experiments therefore indicate that the 2G8.B6 mAb is able to prevent the cytochrome c–mediated activation of caspase 3 in vitro.
Figure 4.
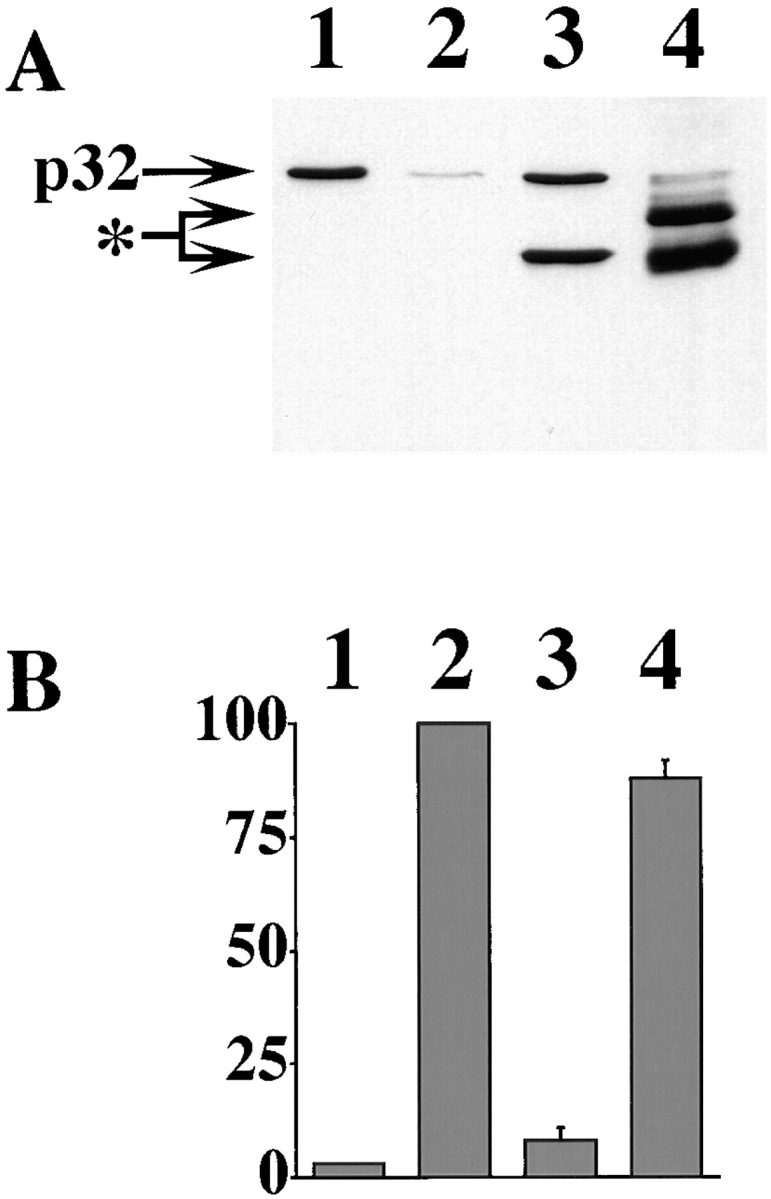
Anti–cytochrome c mAb 2G8.B6 inhibits the activation of caspase 3 in a cell-free assay. 50 μg Jurkat cytosol was incubated for 1 h without cytochrome c and dATP (lane 1) or with cytochrome c and dATP and with no additional components (lane 2), with 2 μg 2G8.B6 anti–cytochrome c mAb (lane 3), or with 2 μg mouse Ig (lane 4). The reaction mix was split into two and analyzed by (A) PAGE and immunoblot for the proteolytic processing of caspase 3. p32 indicates the pro-caspase 3 band and the asterisk indicates the added antibody bands. (B) DEVD-AMC cleavage, for generation of active caspase. Results are expressed as the percentage fluorescence generated over an additional 1-h incubation and are the average of three to six experiments. The error bars represent SEM.
The effect of this antibody was then examined in intact neurons. 2G8.B6 mAb or control mouse IgG was injected into the cytoplasm of SCG neurons, and the cells were withdrawn from NGF 2–4 h later. Cells that survived injection were counted at this time and 72 h later. On average, 86% of cells injected with 2G8.B6 mAb, but only 16% of the control IgG–injected cells, displayed a normal morphology after 72 h of NGF deprivation (Fig. 5). This indicates that blocking the action of cytochrome c is sufficient to halt neuronal apoptosis as defined by morphological criteria.
Figure 5.

Microinjection of anti–cytochrome c mAb 2G8.B6 inhibits SCG apoptosis. SCG neurons were microinjected with either 20 mg/ml 2G8.B6 mAb (lane 1) or an equivalent concentration of mouse Ig (lane 2). After 2–4 h, the injected cells were counted, and the cultures were withdrawn from NGF. At 48 h, the number of surviving injected cells were counted and expressed as a percentage of the cells initially surviving injection. 150–200 cells were injected per coverslip, and the results shown are the average of five experiments. The error bars represent SEM.
We have recently obtained another anti–cytochrome c mAb, 6H2.B4, which also inhibits caspase 3 activation in extracts. Microinjection of this mAb into SCG neurons (three experiments) leads to survival of 65% (SEM 19%) of injected cells (control Ig injection: mean survival = 15%, SEM 5%).
Microinjection of Cytochrome c Does Not Kill SCG Neurons
Since the presence of cytochrome c in the cytoplasm did appear to play a crucial role in neuronal apoptosis, we wanted to determine whether microinjection of cytochrome c was in itself sufficient to induce apoptosis in neurons. We therefore microinjected SCG neurons with a wide range of cytochrome c concentrations and maintained them in the presence of NGF for 48 h. By comparison with known amounts of cytochrome c on Western blots, we estimate that SCG neurons contain between 100– 500 fg of cytochrome c per cell (data not shown). The volume of an SCG neuron is ∼10–20 pl, and we injected a maximum of 1/10th volume into each cell. From these estimates, we concluded that to introduce a single cell equivalent of cytochrome c, we should inject a solution in the range of 70 μg/ml. The range of concentrations of cytochrome c injected cover this value and several log10 concentrations higher and lower than this.
Measuring cell survival as described above, we detected no significant increase in death at any cytochrome c concentration (Fig. 6 A). The small fall in viability at the greatest cytochrome c concentrations was also seen with similar molar concentrations of microperoxidase, a control heme containing fragment of cytochrome c (Fig. 6 B). Thus, cytochrome c alone is not able to induce apoptosis in these cells.
Figure 6.
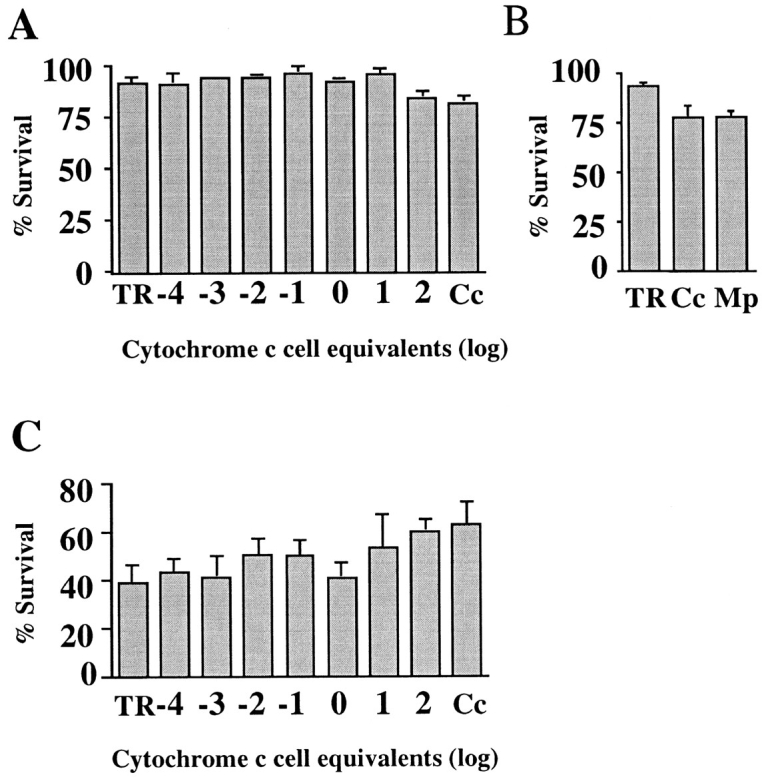
Microinjection of cytochrome c does not induce or accelerate apoptosis in SCG neurons. SCG neurons were microinjected with cytochrome c and counted 2–4 h later. (A) Cells were maintained in NGF for 48 h before counting the surviving cells. The amount of cytochrome c injected is shown as log10 multiples of 1 cell equivalent (70 μg/ml in needle), except for lane TR, which contained no cytochrome c, and lane Cc, in which 17.5 mg/ml of cytochrome c was used. (B) Cells were maintained in NGF for 72 h before counting surviving cells. The microinjection mix in lane TR contained no cytochrome c, 1.45 mM cytochrome c (17.5 mg/ml) in lane Cc, and 1.45 mM microperoxidase in lane Mp. (C) Cells were withdrawn from NGF for 48 h before counting the surviving cells. The amount of cytochrome c injected is shown as log10 multiples of 1 cell equivalent (70 μg/ml in needle), except for lane TR, which contained no cytochrome c, and lane Cc, in which 17.5 mg/ml of cytochrome c was used. The results are expressed as a percentage of the cells initially surviving injection. 150–200 cells were injected per coverslip, and the results shown are the average of three to four experiments. The error bars represent SEM.
If the cytoplasmic presence of cytochrome c were a limiting factor in neuronal apoptosis, then we might expect its microinjection to enhance the rate of death in SCG neurons deprived of NGF. We therefore repeated the above experiment but withdrew the cells from NGF for 48 h after microinjection (Fig. 6 C). Again, no clear enhancement of death was detected under these conditions, suggesting that cytoplasmic cytochrome c is not a rate-limiting factor in neuronal apoptosis.
Microinjection of Cytochrome c with dATP Does Not Kill SCG Neurons
In cell-free apoptotic cell extract systems, dATP significantly increased the rate of cytochrome c–induced caspase activation (Liu et al., 1996b ). We therefore examined whether dATP was a limiting factor in neuronal apoptosis induced by cytochrome c. We chose a concentration of cytochrome c, which we estimated was between 1–10× the cytochrome c cell content, and coinjected dATP in the range 100 μM–10 mM (in the needle). This would give an approximate dATP concentration of 10 μM–1 mM within the cell (assuming that 10% of the cell volume was injected), which is in a similar range to that used in in vitro systems. At the lower concentrations of dATP, no apoptotic effect could be seen (Fig. 7). However, when 10 mM dATP was used, the cells showed a small decrease in viability in the presence or absence of coinjected cytochrome c. No further decrease in viability was detected when higher concentrations of dATP were used (data not shown). Hence, we conclude that dATP, alone or in conjunction with additional cytochrome c, does not induce apoptosis in SCG neurons but may itself have some effect on survival (Wakade et al., 1995).
Figure 7.
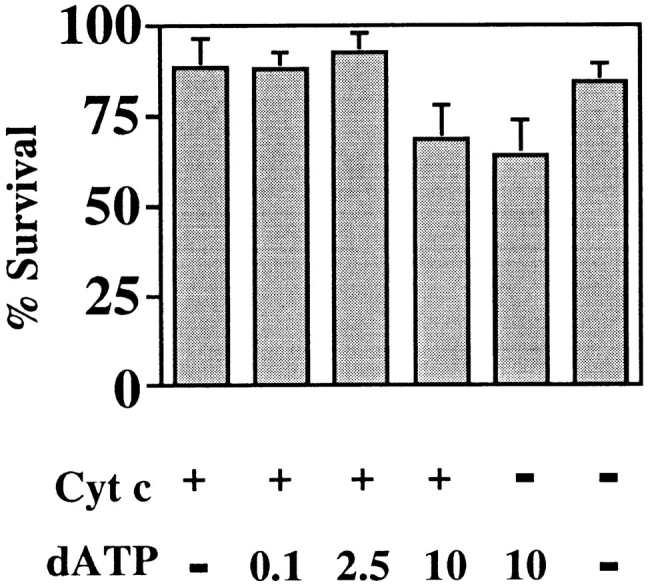
Coinjection of dATP does not enable cytochrome c to initiate apoptosis in SCG neurons. SCG neurons were microinjected with cytochrome c and dATP, counted 2–4 h later, and maintained in NGF for a further 72 h. The microinjection mix in the first four bars contained 0.7 mg/ml cytochrome c. dATP was added in the second bar at 100 μM, in the third bar at 2.5 mM, and in the fourth bar at 10 mM. The fifth bar contained 10 mM dATP alone, and the sixth bar contained no cytochrome c or dATP. The results are expressed as a percentage of the cells initially surviving injection. 150–200 cells were injected per coverslip, and the results shown are the average of three experiments. The error bars represent SEM.
Discussion
We have examined the role of cytochrome c during apoptosis in a model of physiological neuronal cell death, NGF withdrawal–induced death of SCG neurons (Martin et al., 1988; Edwards et al., 1991). Initially, we studied the location of cytochrome c during apoptosis by fluorescence microscopy. We observed that in healthy neurons, cytochrome c was found in a punctate pattern, in keeping with its normal mitochondrial location, but that in neurons with pyknotic nuclei it had assumed a diffuse distribution, implying release from the mitochondria. This first observation is in agreement with the findings of groups who have compared the partition of cytochrome c between the cytosol and mitochondria of normal and apoptotic populations of cells using disruption and fractionation techniques (Liu et al., 1996b ; Ellerby et al., 1997; Kharbanda et al., 1997; Kim et al., 1997; Kluck et al., 1997; Yang et al., 1997) and more recent observations in intact nonneuronal cells (Bossy-Wetzel et al., 1998).
After NGF withdrawal, we consistently observed a small number of cells, which although displaying normal nuclear morphology, had a diffuse cytochrome c staining pattern. This would suggest that the process of nuclear condensation occurred after the release of cytochrome c from the mitochondria. The number of cells with this intermediate morphology was greatly increased by treatment with the broad range caspase inhibitor ZVADfmk. Hence caspase activation, thought to be necessary to induce pyknosis (Liu et al., 1997), is downstream or independent of cytochrome c release from the mitochondria. These data are also in keeping with groups who have reported that caspase inhibitors do not affect the partition of cytochrome c between the cytosolic and mitochondrial cellular fractions during apoptosis (Kharbanda et al., 1997; Kluck et al., 1997; Bossy-Wetzel et al., 1998). We found that many neurons deprived of NGF showed weak cytochrome c staining and that the level of staining was not maintained by the addition of ZVADfmk. This, together with our immunoblotting data, suggests that cytochrome c is degraded after release from the mitochondria and that this is a caspase-independent proteolysis. A previous report has shown that anti-Fas induction of apoptosis in Jurkat cells involves a down regulation of cytochrome c oxidase activity indirectly mediated by the inactivation of cytochrome c (Krippner et al., 1996). They detected no degradation of cytochrome c during this process. The cells in their study were treated for 2 h, so this difference, compared with our results, may be simply due to cytochrome c degradation only being detectable over a longer period. However, in their model ZVADfmk inhibited both the nuclear condensation and the cytochrome oxidase inactivation, suggesting an entirely different sequence of events from that in SCG neurons undergoing NGF withdrawal.
The neuroprotective agents cycloheximide and CPT-cAMP were also examined for their effect on cytochrome c redistribution. Both agents prevented cytochrome c release from the mitochondria, suggesting they act upstream of cytochrome c. The points of action of these compounds are thought to be distinct (Edwards et al., 1991); NGF and cAMP can rescue SCG neurons from NGF withdrawal at a later time than cycloheximide. This suggests that while cells can be rescued by preventing the production of proteins required for apoptosis (Martin et al., 1988, 1992; Edwards et al., 1991), there is a period in which cells can be rescued by posttranslational mechanisms by NGF or cAMP (Edwards et al., 1991; Deckwerth and Johnson, 1993). How might these agents be inhibiting cytochrome c release? One possible explanation is that NGF and cAMP addition may result in the phosphorylation of Bad, a proapoptotic member of the Bcl-2 family (Yang et al., 1995). Phosphorylated Bad is unable to displace Bax from Bax:BclxL heterodimers, which would result in the formation of Bax homodimers and apoptosis (Yang et al., 1995; Zha et al., 1996). One kinase that phosphorylates Bad in cerebellar granule neurons is Akt kinase (Datta et al., 1997). We, and others, have previously demonstrated that Akt kinase can inhibit apoptosis in primary neurons (Dudek et al., 1997; Philpott et al., 1997), and Zha and colleagues (1996) demonstrated that a form of PKA could phosphorylate Bad in vitro. In addition, in cells microinjected with Akt kinase and withdrawn from NGF, we find that the cytochrome c distribution is normal in surviving cells (data not shown), indicating that Akt kinase is upstream of cytochrome c. Thus, we can speculate a survival pathway activated by the NGF receptor, stimulating phosphatidylinositol 3-kinase and Akt kinase, leading to the phosphorylation of Bad, which in turn antagonizes the formation of an apoptotic Bcl-2 family member complex in the mitochondrial membrane (Fig. 8). How such a complex would lead to cytochrome c release is not clear, but Bcl-2–like proteins have a structure resembling pore-forming proteins and could function in this manner (Reed, 1997).
Figure 8.

A model for the mode of action of NGF, cAMP, and cycloheximide in neuronal cell death. In this model, NGF is shown to stimulate phosphatidylinositol 3-kinase, hence Akt kinase activity. Akt kinase can phosphorylate Bad, resulting in its sequestration in the cytosol and survival. However, in the absence of NGF, unphosphorylated Bad disrupts Bax:BclxL dimers in the mitochondrial membrane with the subsequent freeing of Bax to form Bax homodimers. These might be involved in the release of cytochrome c from the mitochondria. Cytochrome c forms a complex with Apaf-1, dATP, and pro-caspase 9, activating caspase 9. cAMP may act to phosphorylate Bad, and the possible sites of cycloheximide intervention are indicated by asterisks.
Additional sites of action of cAMP could be put forward. cAMP is known to modulate gene expression via the phosphorylation of transcription factors by PKA (Lalli and Sassone-Corsi, 1994) and may be acting at the transcriptional level. Indeed modulation of transcription factors, such as c-Jun, can prevent apoptosis in SCG neurons (Ham et al., 1995). Cycloheximide could act at multiple sites by inhibiting the translation of many proteins, including proapoptotic members of the bcl-2 family and proteins necessary for the activation of caspase 9 (Fig. 8). We are currently examining some of these issues.
Having demonstrated that cytochrome c was indeed released from SCG neurons after NGF withdrawal, we wished to determine whether this release was associated with a loss of ΔΨm. This has been reported to be the cause of cytochrome c release (Susin et al., 1996, 1997) and conversely to be the result of caspase activation (Bossy-Wetzel et al., 1998). Thus, for some models of apoptotic cell death it is unclear if ΔΨm loss is crucial to cytochrome c release from the mitochondria. However, in the case of NGF-deprived sympathetic neurons, it was clear that complete cytochrome c dispersal and nuclear pyknosis preceded loss of ΔΨm.
In vitro experiments by several groups have shown that addition of cytochrome c to normal cytosolic extracts causes activation of endogenous caspase 3 (Liu et al., 1996b ; Ellerby et al., 1997; Kharbanda et al., 1997; Kluck et al., 1997; Neame, S.J., unpublished data). Our mAb microinjection results show that cytochrome c activity is important for regulating neuronal apoptosis. This demonstrates for the first time that cytochrome c is an essential component of NGF withdrawal–induced apoptosis of sympathetic neurons. Since the anti–cytochrome c mAbs were injected into the cell cytoplasm, we assume that the inhibition of cytochrome c occurs post efflux from the mitochondria. We infer that the cytochrome c mode of action is by activation of a caspase in a similar manner to that previously described in vitro (Liu et al., 1996b ; P. Li et al., 1997; Zou et al., 1997), involving a CED-4 homologous protein and a caspase with a CED-3 homologous prodomain. We are presently investigating the identity of the protein partners of cytochrome c in SCG neurons.
While in several reports the optimal in vitro activation of caspase 3 was achieved upon the addition of both cytochrome c and dATP, some sources have found that cytochrome c alone is sufficient (Ellerby et al., 1997; Kluck et al., 1997; Hampton et al., 1998; Neame, S.J., unpublished data). This suggests that all the necessary partners of cytochrome c are already present in the cells from which the cytoplasmic extracts are made. If this was true in SCG neurons, microinjection of cytochrome c would induce apoptosis, as has been found in human kidney 293 cells (F. Li et al., 1997) and NRK cells (Zhivotovsky et al., 1998). To ensure that the cytochrome c we were using was, although clearly active in extracts (Fig. 4), not defective in intact cells, we injected Rat-1 cells with cytochrome c at 5 mg/ml. We found that >90% of injected cells displayed apoptotic morphology within 2 h of injection, while microperoxidase-injected cells appeared normal. Not withstanding the efficacy of the cytochrome c, we observed no apoptosis in SCG neurons injected with a wide range of cytochrome c concentrations, suggesting some other limiting factor. Coinjection of dATP with cytochrome c did not induce apoptosis if introduced at below 1 mM. While there was a small induction of death at 10 mM dATP (with or without cytochrome c), this was not the rapid death seen in other cell types upon injection of cytochrome c (F. Li et al., 1997; Zhivotovsky et al., 1998; data not shown) and so may be caused by nonapoptotic processes. Estimates of normal cellular dATP concentration suggest a range of 5–10 μM (Hunting and Henderson, 1981; Sherman and Fyfe, 1989), implying that we are supplying sufficient dATP. Wakade and colleagues (1995) described a 40-fold increase in dATP concentration upon induction of apoptosis in chick SCG neurons. However, this death was induced by treatment with 2-deoxyadenosine, a precursor of dATP, so the increase in its concentration may not be physiologically relevant to apoptosis. Certainly, dATP levels are not increased in all cases of apoptosis and actually fall during apoptosis induced by IL-3 withdrawal from BAF3 cells (Oliver et al., 1996). The primary physiological enzyme responsible for dATP synthesis is ribonucleotide reductase, an enzyme that has a free radical at its active site, obligatory for function. At low O2 concentrations, this radical is abolished (Brischwein et al., 1997), yet cells can undergo apoptosis under similar conditions (Jacobson and Raff, 1995), implying that increases in dATP are not intrinsic to apoptosis.
That we see no induction of apoptosis upon cytochrome c injection suggests that some partner to cytochrome c in the apoptotic process may be regulated. Clearly, the recently identified Apaf-1 (Zou et al., 1997) and Apaf-3 (caspase 9; P. Li et al., 1997) are possible candidates for this regulation. While we have demonstrated that cycloheximide prevents cytochrome c release, perhaps it also functions to inhibit the translation of these proteins; however, the regulation could equally involve other mechanisms. We find that the accumulation of cytochrome c in the cytoplasm does not appear to be the rate-limiting step in induction of nuclear pyknosis. This implies that the release of cytochrome c, which appears rapid since we very rarely see cells displaying intermediate punctate/diffuse cytochrome c distribution, is differentially regulated compared to its apoptotic partners. It is also possible that the immediate apoptotic partners of cytochrome c may be unregulated, while the downstream mediators are. In this context, it is interesting that F. Li et al. (1997) report a parental MCF7 cell line that was not induced to undergo apoptosis upon injection of cytochrome c, while a sibling cell line in which pro-caspase 3 was expressed, could be. In some neuronal cells, caspase 3 mRNA has been shown to increase in level after an apoptotic insult (Miller et al., 1997). However, while caspase 3 is present in SCG neurons at detectable levels by Western blotting (Deshmukh et al., 1996; McCarthy et al., 1997), it does not appear to increase in level after NGF withdrawal (Deshmukh et al., 1996; data not shown). Thus, it is unclear to date which component is limiting apoptosis in SCG neurons and whether this can vary with cell type.
In conclusion, we have shown that the release of cytochrome c from the mitochondria is of crucial importance in the NGF withdrawal–induced apoptosis of sympathetic neurons. We have also shown that cytochrome c release is independent of loss of ΔΨm and so must be regulated by other means. These in turn are dependent upon transcription/translation and can be modulated by Akt kinase or a cAMP-mediated kinase. We have also implied that the partners in cytochrome c activation of caspases are regulated, since the introduction of cytochrome c into the cytoplasm is not in itself sufficient in SCG neurons to activate or accelerate cell death.
Acknowledgments
We would like to thank Dr. C. Gatchalian and Dr. J. Taylor for critical reading of this manuscript, Ms. K. Ferguson for editorial assistance, and Dr. R. Jemmerson for generously supplying anti–cytochrome c antibodies.
Abbreviations used in this paper
- Apaf
apoptotic protease activating factor
- CPT-cAMP
8-(4-chlorophenylthio)adenosine 3′:5′-cyclic monophosphate
- DEVD-AMC
Asp-Glu-Val-Asp-amino methyl coumarin
- PKA
protein-dependent kinase A
- SCG
superior cervical ganglia
- ZVADfmk
Z-Val-Ala-Asp-fluoromethylketone
- ΔΨm
mitochondrial inner membrane potential
Footnotes
Address all correspondence to Stephen J. Neame, Eisai London Research Laboratories, Bernard Katz Building, University College London, Gower Street, London WC1E 6BT, UK. Tel.: 0171 413 1130. Fax: 0171 413 1121. E-mail: sjneame@elrl.co.uk
References
- Antonsson B, Conti F, Ciavatta A, Montessuit S, Lewis S, Martinou I, Bernasconi L, Bernard A, Mermod J-J, Mazzei G, et al. Inhibition of Bax channel-forming activity by Bcl-2. Science. 1997;277:370–372. doi: 10.1126/science.277.5324.370. [DOI] [PubMed] [Google Scholar]
- Bortner CD, Oldenburg NBE, Cidlowski JA. The role of DNA fragmentation in apoptosis. Trends Cell Biol. 1995;5:21–26. doi: 10.1016/s0962-8924(00)88932-1. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Newmeyer DD, Green DR. Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO (Eur Mol Biol Organ) J. 1998;17:37–49. doi: 10.1093/emboj/17.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredesen DE. Neural apoptosis. Ann Neurol. 1995;38:839–851. doi: 10.1002/ana.410380604. [DOI] [PubMed] [Google Scholar]
- Brischwein K, Engelcke M, Riedinger HJ, Probst H. Role of ribonucleotide reductase and deoxynucleotide pools in the oxygen-dependent control of DNA replication in Ehrlich ascites cells. Eur J Biochem. 1997;244:286–293. doi: 10.1111/j.1432-1033.1997.00286.x. [DOI] [PubMed] [Google Scholar]
- Buckmaster EA, Tolkovsky AM. Expression of the cyclic AMP-dependent protein kinase (PKA) catalytic subunit from a herpes simplex virus vector extends the survival of rat sympathetic neurons in the absence of NGF. Eur J Neurosci. 1994;6:1316–1327. doi: 10.1111/j.1460-9568.1994.tb00322.x. [DOI] [PubMed] [Google Scholar]
- Burek MJ, Oppenheim RW. Programmed cell death in the developing nervous system. Brain Pathol. 1996;6:427–446. doi: 10.1111/j.1750-3639.1996.tb00874.x. [DOI] [PubMed] [Google Scholar]
- Cheng EH, Kirsch DG, Clem RJ, Ravi R, Kastan MB, Bedi A, Ueno K, Hardwick JM. Conversion of Bcl-2 to a Bax-like death effector by caspases. Science. 1997;278:1966–1968. doi: 10.1126/science.278.5345.1966. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan AM, Chaudhary D, O'Rourke K, Koonin EV, Dixit VM. Role of CED-4 in the activation of CED-3. Nature. 1997;388:728–729. doi: 10.1038/41913. [DOI] [PubMed] [Google Scholar]
- Choi DW. Ischemia-induced neuronal apoptosis. Curr Opin Neurobiol. 1996;6:667–672. doi: 10.1016/s0959-4388(96)80101-2. [DOI] [PubMed] [Google Scholar]
- Clem RJ, Cheng EH, Karp CL, Kirsch DG, Ueno K, Takahashi A, Kastan MB, Griffin DE, Earnshaw WC, Veliuona MA, Hardwick JM. Modulation of cell death by Bcl-XL through caspase interaction. Proc Natl Acad Sci USA. 1998;95:554–559. doi: 10.1073/pnas.95.2.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotman CW, Anderson AJ. A potential role for apoptosis in neurodegeneration and Alzheimer's disease. Mol Neurobiol. 1995;10:19–45. doi: 10.1007/BF02740836. [DOI] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- Deckwerth TL, Johnson EM., Jr Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor. J Cell Biol. 1993;123:1207–1222. doi: 10.1083/jcb.123.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh M, Vasilakos J, Deckwerth TL, Lampe PA, Shivers BD, Johnson EM., Jr Genetic and metabolic status of NGF-deprived sympathetic neurons saved by an inhibitor of ICE family proteases. J Cell Biol. 1996;135:1341–1354. doi: 10.1083/jcb.135.5.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- Edwards SN, Buckmaster AE, Tolkovsky AM. The death programme in cultured sympathetic neurones can be suppressed at the posttranslational level by nerve growth factor, cyclic AMP, and depolarization. J Neurochem. 1991;57:2140–2143. doi: 10.1111/j.1471-4159.1991.tb06434.x. [DOI] [PubMed] [Google Scholar]
- Ellerby HM, Martin SJ, Ellerby LM, Naiem SS, Rabizadeh S, Salvesen GS, Casiano CA, Cashman NR, Green DR, Bredesen DE. Establishment of a cell-free system of neuronal apoptosis: comparison of premitochondrial, mitochondrial, and postmitochondrial phases. J Neurosci. 1997;17:6165–6178. [PMC free article] [PubMed] [Google Scholar]
- Ellis RE, Yuan JY, Horvitz HR. Mechanisms and functions of cell death. Annu Rev Cell Biol. 1991;7:663–698. doi: 10.1146/annurev.cb.07.110191.003311. [DOI] [PubMed] [Google Scholar]
- Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;391:43–50. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Armstrong RC, Krebs J, Srinivasula SM, Wang L, Bullrich F, Fritz LC, Trapani JA, Tomaselli KJ, Litwack G, Alnemri ES. In vitro activation of CPP32 and Mch3 by Mch4, a novel human apoptotic cysteine protease containing two FADD-like domains. Proc Natl Acad Sci USA. 1996;93:7464–7469. doi: 10.1073/pnas.93.15.7464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghadge GD, Lee JP, Bindokas VP, Jordan J, Ma L, Miller RJ, Roos RP. Mutant superoxide dismutase-1-linked familial amyotrophic lateral sclerosis: molecular mechanisms of neuronal death and protection. J Neurosci. 1997;17:8756–8766. doi: 10.1523/JNEUROSCI.17-22-08756.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ham J, Babij C, Whitfield J, Pfarr CM, Lallemand D, Yaniv M, Rubin LL. A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 1995;14:927–939. doi: 10.1016/0896-6273(95)90331-3. [DOI] [PubMed] [Google Scholar]
- Hampton MB, Zhivotovsky B, Slater AFG, Burgess DH, Orrenius S. Importance of the redox state of cytochrome c during caspase activation in cytosolic extracts. Biochem J. 1998;329:95–99. doi: 10.1042/bj3290095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengartner MO, Horvitz HR. a. C. eleganscell survival gene ced-9 encodes a functional homolog of the mammalian proto-oncogene bcl-2. Cell. 1994;76:665–676. doi: 10.1016/0092-8674(94)90506-1. [DOI] [PubMed] [Google Scholar]
- Hengartner MO, Horvitz HR. The ins and outs of programmed cell death during C. elegansdevelopment. Phil Trans R Soc Lond Ser B Biol Sci. 1994b;345:243–246. doi: 10.1098/rstb.1994.0100. [DOI] [PubMed] [Google Scholar]
- Huang DCS, Adams JM, Cory S. The conserved N-terminal BH4 domain of Bcl-2 homologues is essential for inhibition of apoptosis and interaction with CED-4. EMBO (Eur Mol Biol Organ) J. 1998;17:1029–1039. doi: 10.1093/emboj/17.4.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunting D, Henderson JF. Determination of deoxyribonucleoside triphosphates using DNA polymerase: a critical evaluation. Can J Biochem. 1981;59:723–727. doi: 10.1139/o81-100. [DOI] [PubMed] [Google Scholar]
- Jacobson MD, Raff MC. Programmed cell death and Bcl-2 protection in very low oxygen. Nature. 1995;374:814–816. doi: 10.1038/374814a0. [DOI] [PubMed] [Google Scholar]
- Jacobson MD, Weil M, Raff MC. Programmed cell death in animal development. Cell. 1997;88:347–354. doi: 10.1016/s0092-8674(00)81873-5. [DOI] [PubMed] [Google Scholar]
- Janicke RU, Walker PA, Lin XY, Porter AG. Specific cleavage of the retinoblastoma protein by an ICE-like protease in apoptosis. EMBO (Eur Mol Biol Organ) J. 1996;15:6969–6978. [PMC free article] [PubMed] [Google Scholar]
- Jemmerson R, Johnson JG, Burrell E, Taylor PS, Jenkins MK. A monoclonal antibody specific for a cytochrome c T cell stimulatory peptide inhibits T cell responses and affects the way the peptide associates with antigen-presenting cells. Eur J Immunol. 1991;21:143–151. doi: 10.1002/eji.1830210122. [DOI] [PubMed] [Google Scholar]
- Johnson EM, Jr, Deckwerth TJ. Molecular mechanisms of developmental neuronal death. Annu Rev Neurosci. 1993;16:31–46. doi: 10.1146/annurev.ne.16.030193.000335. [DOI] [PubMed] [Google Scholar]
- Johnson EM, Jr, Deckwerth TL, Deshmukh M. Neuronal death in developmental models: possible implications in neuropathology. Brain Pathol. 1996;6:397–409. doi: 10.1111/j.1750-3639.1996.tb00872.x. [DOI] [PubMed] [Google Scholar]
- Kharbanda S, Pandey P, Schofield L, Israels S, Roncinske R, Yoshida K, Bharti A, Yuan Z-M, Saxena S, Weichselbaum R, et al. Role for Bcl-xL as an inhibitor of cytosolic cytochrome C accumulation in DNA damage-induced apoptosis. Proc Natl Acad Sci USA. 1997;94:6939–6942. doi: 10.1073/pnas.94.13.6939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CN, Wang X, Huang Y, Ibrado AM, Liu L, Fang G, Bhalla K. Overexpression of Bcl-X(L) inhibits Ara-C-induced mitochondrial loss of cytochrome c and other perturbations that activate the molecular cascade of apoptosis. Cancer Res. 1997;57:3115–3120. [PubMed] [Google Scholar]
- Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- Krajewski S, Tanaka S, Takayama S, Schibler MJ, Fenton W, Reed JC. Investigation of the subcellular distribution of the bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res. 1993;53:4701–4714. [PubMed] [Google Scholar]
- Krippner A, Matsuno-Yagi A, Gottlieb RA, Babior BM. Loss of function of cytochrome c in Jurkat cells undergoing fas-mediated apoptosis. J Biol Chem. 1996;271:21629–21636. doi: 10.1074/jbc.271.35.21629. [DOI] [PubMed] [Google Scholar]
- Kroemer G. The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat Med. 1997;31:614–620. doi: 10.1038/nm0697-614. [DOI] [PubMed] [Google Scholar]
- Lalli E, Sassone-Corsi P. Signal transduction and gene regulation: the nuclear response to cAMP. J Biol Chem. 1994;269:17359–17362. [PubMed] [Google Scholar]
- Lazebnik YA, Takahashi A, Moir RD, Goldman RD, Poirier GG, Kaufmann SH, Earnshaw WC. Studies of the lamin proteinase reveal multiple parallel biochemical pathways during apoptotic execution. Proc Natl Acad Sci USA. 1995;92:9042–9046. doi: 10.1073/pnas.92.20.9042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Srinivasan A, Wang Y, Armstrong RC, Tomaselli KJ, Fritz LC. Cell-specific induction of apoptosis by microinjection of cytochrome c. Bcl-xL has activity independent of cytochrome c release. J Biol Chem. 1997;272:30299–30305. doi: 10.1074/jbc.272.48.30299. [DOI] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Linnik MD, Zobrist RH, Hatfield MD. Evidence supporting a role for programmed cell death in focal cerebral ischemia in rats. Stroke. 1993;24:2002–2008. doi: 10.1161/01.str.24.12.2002. [DOI] [PubMed] [Google Scholar]
- Liu X, Kim CN, Pohl J, Wang X. Purification and characterization of an interleukin-1β-converting enzyme family protease that activates cysteine protease P32 (CPP32) J Biol Chem. 1996a;271:13371–13376. [PubMed] [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996b;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Liu X, Zou H, Slaughter C, Wang X. DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell. 1997;89:175–184. doi: 10.1016/s0092-8674(00)80197-x. [DOI] [PubMed] [Google Scholar]
- Martin DP, Schmidt RE, DiStefano PS, Lowry OH, Carter JG, Johnson EM., Jr Inhibitors of protein synthesis and RNA synthesis prevent neuronal death caused by nerve growth factor deprivation. J Cell Biol. 1988;106:829–844. doi: 10.1083/jcb.106.3.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DP, Ito A, Horigome K, Lampe PA, Johnson EM., Jr Biochemical characterization of programmed cell death in NGF-deprived sympathetic neurons. J Neurobiol. 1992;23:1205–1220. doi: 10.1002/neu.480230911. [DOI] [PubMed] [Google Scholar]
- McCarthy MJ, Rubin LL, Philpott KL. Involvement of caspases in sympathetic neuron apoptosis. J Cell Sci. 1997;110:2165–2173. doi: 10.1242/jcs.110.18.2165. [DOI] [PubMed] [Google Scholar]
- Miller TM, Moulder KL, Knudson CM, Creedon DJ, Deshmukh M, Korsmeyer SJ, Johnson EM., Jr Bax deletion further orders the cell death pathway in cerebellar granule cells and suggests a caspase-independent pathway to cell death. J Cell Biol. 1997;139:205–217. doi: 10.1083/jcb.139.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn AJ, Velez P, Schendel SL, Liang H, Muchmore SW, Fesik SW, Fill SW, Thompson CB. Bcl-x(L) forms an ion channel in synthetic lipid membranes. Nature. 1997;385:353–357. doi: 10.1038/385353a0. [DOI] [PubMed] [Google Scholar]
- Mochizuki H, Goto K, Mori H, Mizuno Y. Histochemical detection of apoptosis in Parkinson's disease. J Neurol Sci. 1996;137:120–123. doi: 10.1016/0022-510x(95)00336-z. [DOI] [PubMed] [Google Scholar]
- Mueller CM, Jemmerson R. Maturation of the antibody response to the major epitope on the self antigen mouse cytochrome c. Restricted V gene usage, selected mutations, and increased affinity. J Immunol. 1996;157:5329–5338. [PubMed] [Google Scholar]
- Murphy AN, Bredesen DE, Cortopassi G, Wang E, Fiskum G. Bcl-2 potentiates the maximal calcium uptake capacity of neural cell mitochondria. Proc Natl Acad Sci USA. 1996;93:9893–9898. doi: 10.1073/pnas.93.18.9893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson DW, Thornberry NA. Caspases: killer proteases. Trends Biochem Sci. 1997;22:299–306. doi: 10.1016/s0968-0004(97)01085-2. [DOI] [PubMed] [Google Scholar]
- Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- Nunez G, Clarke MF. The Bcl-2 family of proteins: regulators of cell death and survival. Trends Cell Biol. 1994;4:399–403. doi: 10.1016/0962-8924(94)90053-1. [DOI] [PubMed] [Google Scholar]
- Oliver FJ, Collins MK, Lopez-Rivas A. Regulation of the salvage pathway of deoxynucleotide synthesis in apoptosis induced by growth factor deprivation. Biochem J. 1996;316:421–425. doi: 10.1042/bj3160421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- Philpott KL, McCarthy MJ, Becker D, Gatchalian C, Rubin LL. Morphological and biochemical changes in neurons: apoptosis versus mitosis. Eur J Neurosci. 1996;8:1906–1915. doi: 10.1111/j.1460-9568.1996.tb01334.x. [DOI] [PubMed] [Google Scholar]
- Philpott KL, McCarthy MJ, Klippel A, Rubin LL. Activated phosphatidylinositol 3-kinase and Akt kinase promote survival of superior cervical neurons. J Cell Biol. 1997;139:809–815. doi: 10.1083/jcb.139.3.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff MC, Barres BA, Burne JF, Coles HS, Ishizaki Y, Jacobson MD. Programmed cell death and the control of cell survival: lessons from the nervous system. Science. 1993;262:695–700. doi: 10.1126/science.8235590. [DOI] [PubMed] [Google Scholar]
- Ratan RR, Murphy TH, Baraban JM. Macromolecular synthesis inhibitors prevent oxidative stress-induced apoptosis in embryonic cortical neurons by shunting cysteine from protein synthesis to glutathione. J Neurosci. 1994;14:4385–4392. doi: 10.1523/JNEUROSCI.14-07-04385.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed JC. Double identity for proteins of the Bcl-2 family. Nature. 1997;387:773–776. doi: 10.1038/42867. [DOI] [PubMed] [Google Scholar]
- Rydel RE, Greene LA. cAMP analogs promote survival and neurite outgrowth in cultures of rat sympathetic and sensory neurons independently of nerve growth factor. Proc Natl Acad Sci USA. 1988;85:1257–1261. doi: 10.1073/pnas.85.4.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakahira H, Enari M, Nagata S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature. 1998;391:96–99. doi: 10.1038/34214. [DOI] [PubMed] [Google Scholar]
- Salvesen GS, Dixit VM. Caspases: intracellular signaling by proteolysis. Cell. 1997;91:443–446. doi: 10.1016/s0092-8674(00)80430-4. [DOI] [PubMed] [Google Scholar]
- Schendel SL, Xie Z, Montal MO, Matsuyama S, Montal M, Reed JC. Channel formation by antiapoptotic protein Bcl-2. Proc Natl Acad Sci USA. 1997;94:5113–5118. doi: 10.1073/pnas.94.10.5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal RA, Greenberg ME. Intracellular signaling pathways activated by neurotrophic factors. Annu Rev Neurosci. 1996;19:463–489. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- Shaham S, Horvitz HR. Developing Caenorhabditis elegansneurons may contain both cell-death protective and killer activities. Genes Dev. 1996;10:578–591. doi: 10.1101/gad.10.5.578. [DOI] [PubMed] [Google Scholar]
- Sherman PA, Fyfe JA. Enzymatic assay for deoxyribonucleoside triphosphates using synthetic oligonucleotides as template primers. Anal Biochem. 1989;180:222–226. doi: 10.1016/0003-2697(89)90420-x. [DOI] [PubMed] [Google Scholar]
- Silos-Santiago I, Greenlund LJ, Johnson EM, Jr, Snider WD. Molecular genetics of neuronal survival. Curr Opin Neurobiol. 1995;5:42–49. doi: 10.1016/0959-4388(95)80085-9. [DOI] [PubMed] [Google Scholar]
- Song Q, Lees-Miller SP, Kumar S, Zhang Z, Chan DW, Smith GC, Jackson SP, Alnemri ES, Litwack G, Khanna KK, Lavin MF. DNA-dependent protein kinase catalytic subunit: a target for an ICE-like protease in apoptosis. EMBO (Eur Mol Biol Organ) J. 1996;15:3238–3246. [PMC free article] [PubMed] [Google Scholar]
- Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Litwack G, Alnemri ES. Molecular ordering of the Fas-apoptotic pathway: the Fas/ APO-1 protease Mch5 is a CrmA-inhibitable protease that activates multiple Ced-3/ICE-like cysteine proteases. Proc Natl Acad Sci USA. 1996a;93:14486–14491. doi: 10.1073/pnas.93.25.14486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasula SM, Fernandes-Alnemri T, Zangrilli J, Robertson N, Armstrong RC, Wang L, Trapani JA, Tomaselli KJ, Litwack G, Alnemri ES. The Ced-3/interleukin 1β-converting enzyme-like homolog Mch6 and the lamin-cleaving enzyme Mch2α are substrates for the apoptotic mediator CPP32. J Biol Chem. 1996b;271:27099–27106. doi: 10.1074/jbc.271.43.27099. [DOI] [PubMed] [Google Scholar]
- Susin SA, Zamzami N, Castedo M, Hirsch T, Marchetti P, Macho A, Daugas E, Geuskens M, Kroemer G. Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J Exp Med. 1996;184:1331–1341. doi: 10.1084/jem.184.4.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin SA, Zamzami N, Castedo M, Daugas E, Wang HG, Geley S, Fassy F, Reed JC, Kroemer G. The central executioner of apoptosis: multiple connections between protease activation and mitochondria in Fas/APO-1/CD95- and ceramide-induced apoptosis. J Exp Med. 1997;186:25–37. doi: 10.1084/jem.186.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi A, Alnemri ES, Lazebnik YA, Fernandes-Alnemri T, Litwack G, Moir RD, Goldman RD, Poirier GG, Kaufmann SH, Earnshaw WC. Cleavage of lamin A by Mch2 α but not CPP32: multiple interleukin 1 β-converting enzyme-related proteases with distinct substrate recognition properties are active in apoptosis. Proc Natl Acad Sci USA. 1996;93:8395–8400. doi: 10.1073/pnas.93.16.8395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewari M, Quan LT, Orourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS, Dixit VM. Yama/CPP32 β, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995;81:801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- Wakade AR, Przywara DA, Palmer KC, Kulkarni JS, Wakade TD. Deoxynucleoside induces neuronal apoptosis independent of neurotrophic factors. J Biol Chem. 1995;270:17986–17992. doi: 10.1074/jbc.270.30.17986. [DOI] [PubMed] [Google Scholar]
- Wu D, Wallen HD, Inohara N, Nunez G. Interaction and regulation of the Caenorhabditis elegansdeath protease CED-3 by CED-4 and CED-9. J Biol Chem. 1997;272:21449–21454. doi: 10.1074/jbc.272.34.21449. [DOI] [PubMed] [Google Scholar]
- Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell. 1995;80:285–291. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]
- Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng T-I, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- Yoshiyama Y, Yamada T, Asanuma K, Asahi T. Apoptosis related antigen, Le(Y) and nick-end labeling are positive in spinal motor neurons in amyotrophic lateral sclerosis. Acta Neuropathol. 1994;88:207–211. doi: 10.1007/BF00293395. [DOI] [PubMed] [Google Scholar]
- Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. The C. eleganscell death gene ced-3 encodes a protein similar to mammalian interleukin-1 β-converting enzyme. Cell. 1993;75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]
- Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L) Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- Zhivotovsky B, Orrenius S, Brustugun OT, Doskeland SO. Injected cytochrome c induces apoptosis. Nature. 1998;391:449–450. doi: 10.1038/35060. [DOI] [PubMed] [Google Scholar]
- Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegansCED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]