

Abstract
A member of the annexin XIII sub-family, annexin XIIIb, has been implicated in the apical exocytosis of epithelial kidney cells. Annexins are phospholipid-binding proteins that have been suggested to be involved in membrane trafficking events although their actual physiological function remains open. Unlike the other annexins, annexin XIIIs are myristoylated. Here, we show by immunoelectron microscopy that annexin XIIIb is localized to the trans-Golgi network (TGN), vesicular carriers and the apical cell surface. Polarized apical sorting involves clustering of apical proteins into dynamic sphingolipid-cholesterol rafts. We now provide evidence for the raft association of annexin XIIIb. Using in vitro assays and either myristoylated or unmyristoylated recombinant annexin XIIIb, we demonstrate that annexin XIIIb in its native myristoylated form stimulates specifically apical transport whereas the unmyristoylated form inhibits this route. Moreover, we show that formation of apical carriers from the TGN is inhibited by an anti-annexin XIIIb antibody whereas it is stimulated by myristoylated recombinant annexin XIIIb. These results suggest that annexin XIIIb directly participates in apical delivery.
Keywords: cholesterol, exocytosis, MDCK, polarity, raft
In the MDCK cell line sorting of apical and basolateral proteins occurs in the TGN (Simons, 1995), from where they are transported to their respective plasma membrane domain (Rodriguez-Boulan and Nelson, 1989; Simons and Ikonen, 1997; Weimbs et al., 1997).
Specific sorting determinants are required for polarized targeting. An increasing body of evidence supports a hierarchical read-out of the determinants.
For membrane proteins, basolateral sorting signals which are dominant over apical ones have been identified (Rodriguez-Boulan and Powell, 1992; Matter and Mellman, 1994). The basolateral signals include tyrosine- or dileucine-based motifs similar to those implicated in clathrin-dependent endocytosis suggesting that these basolateral proteins use similar mechanisms for recruitment into vesicular carriers destined to the cell surface (Scheiffele and Simons, 1998).
In the apical pathway, sorting of proteins can be conferred by glycosylphosphatidylinositol (GPI)1 anchors, signals located in the transmembrane domain (Kundu et al., 1996) and by glycosylation: N-glycosylation for membrane proteins (Gut et al., 1998) and secreted proteins (Scheiffele et al., 1995) as well as O-glycosylation (Yeaman et al., 1997). Not only proteins but also specific lipids e.g., glycosphingolipids are apically targeted in epithelial cells (van Meer, 1989). Analysis of the apical sorting of proteins and lipids led to the raft hypothesis summarized by Simons and Ikonen (1997). It proposes that dynamic lateral assemblies of sphingolipids and cholesterol subcompartimentalize membranes. Apical proteins are recruited into such microdomains from which basolateral proteins are excluded. The exoplasmic sphingolipids possess long saturated fatty acyl chains which allow them to form tightly packed assemblies within the more fluid environment of the kinked cis-unsaturated acyl chain phospholipids (e.g., phosphatidylcholine). Cholesterol, which is present in both leaflets, plays an important role in stabilizing such lipid microdomains (Brown, 1998). Accumulating evidence suggests that the sphingolipid-cholesterol rafts could constitute liquid-ordered phases dispersed in the liquid-crystalline phase formed within phosphatidylcholine enriched regions of cell membranes (Ahmed et al., 1997). The liquid-ordered phase lipids can be isolated as detergent-insoluble glycosphingolipid complexes (DIGs). After detergent extraction, the DIGs float when subjected to density gradient centrifugation, facilitating their separation from the insoluble cytoskeleton-associated fraction (Brown and Rose, 1992). Apical transmembrane and GPI-anchored proteins as well as some double acylated tyrosine kinases have been isolated using this procedure demonstrating their specific interaction with these lipid microdomains (Harder and Simons, 1997; Scheiffele and Simons, 1998). Moreover, by in vitro reconstitution experiments it has been shown that detergent insolubility of the apical GPI-anchored placental alkaline phosphatase depends on a raft lipid environment (Schroeder et al., 1994).
One objection that has been raised to the use of detergent insolubility as a tool to detect lipid domains in cellular membranes is that detergent solubilization might cause aggregation of membrane components that might not have been in proximity in cell membranes. This could be due, for instance, to the intrinsic insolubility of some lipids (Schroeder et al., 1994) or to the coalescence of rafts upon detergent extraction trapping proteins in close vicinity that otherwise are not included into rafts. But, recently, evidence obtained by several laboratories using different approaches excluded such a possibility (Brown and London, 1997). An additional criterion is to show that detergent insolubility depends on the integrity of raft domains by disrupting them with methyl-β-cyclodextrin (MeβCD) which extracts cholesterol from membranes (Klein et al., 1995; Yancey et al., 1996). This treatment renders raft-associated proteins detergent soluble leading to a more stringent definition for raft association, namely that raft association is cholesterol-dependent (Harder and Simons, 1997). Two results consistent with a role of rafts in apical membrane trafficking were recently obtained (Keller and Simons, 1998). First, in cholesterol-depleted cells only apical transport is impaired. Second, apically routed proteins are mistargeted after cholesterol depletion probably due to disorganization of rafts, interfering with apical protein clustering (Cerneus et al., 1993; Hanada et al., 1995; Hannan and Edidin, 1996; Scheiffele et al., 1997). Therefore, although rafts can also be found in the basolateral compartment (Melkonian et al., 1995), it is proposed that apical proteins are recruited into large percolating raft domains before apical carriers bud off the TGN. Another example of such an assembly are caveolae that can be considered as a stable coalescence of rafts (Simons and Ikonen, 1997). The essential components of caveolae are caveolins that can form large oligomers bound to cholesterols as demonstrated for VIP21/caveolin-1 (Murata et al., 1995). De novo formation of caveolae, upon expression of VIP21/caveolin-1 (Fra et al., 1995) suggests that caveolins regulate the dynamics of rafts. But, with the exception of caveolins, proteins modulating raft assembly await to be identified.
After being released from the TGN, basolateral carriers move along microtubules to the basolateral surface (Lafont et al., 1994) where they dock and fuse using the ubiquitous soluble SNARE (N-ethyl maleimide-sensitive factor [NSF] attachment protein [SNAP]) receptors-Rab machinery (Ikonen et al., 1995, for review see Rothman, 1994). In the apical pathway, apical carriers move along microtubules to the apical surface (Lafont and Simons, 1996). After having crossed the subcortical network of actin cytoskeleton (Ojakian and Schwimmer, 1988; Fath and Burgess, 1993; Muallem et al., 1995), the apical carriers dock and fuse with the cell surface by a mechanism remaining to be clarified. Evidence for the involvement of SNAREs in the apical membrane trafficking has been provided from cells overexpressing syntaxin-3 or from permeabilized cells with recombinant proteins and antibodies directed against NSF added (Low et al., 1998; F. Lafont and K. Simons, unpublished results). So far, the apical pathway differs from the basolateral route by the lack of a requirement for NSF (Ikonen et al., 1995; Low et al., 1998).
Annexins belong to a family of proteins that has been implicated in regulating membrane trafficking in exocytosis and endocytosis where the annexins could be involved in membrane-membrane interactions (Creutz, 1992; Moss, 1992; Gruenberg and Emans, 1993; Burgoyne and Clague, 1994; Raynal and Pollard, 1994). Such a role is supported by their calcium-dependent binding to phospholipids due to their endonexin fold repeats forming the annexin core domain. We previously identified a protein in immunoisolated apical carriers that turned out to be a new member of the annexin family, namely annexin XIIIb (Fiedler et al., 1995). Annexin XIIIb is the only annexin found in immunoisolated apical exocytic carriers (Wandinger-Ness et al., 1990) and is an isoform of annexin XIIIa previously described in human intestinal cells (Wice and Gordon, 1992). It is worth mentioning that annexin XIIIs are the only annexins myristoylated in their NH2-terminal part that is the divergent domain among the members of the annexin family. Immunofluorescence experiments showed that annexin XIIIb is located in the apical compartment of MDCK cells (Fiedler et al., 1995). An antibody raised against annexin XIIIb inhibited the apical delivery of vesicular carriers when introduced in streptolysin-O (SLO)- permeabilized cells (Fiedler et al., 1995). The interpretation of these results led us to speculate that annexin XIIIb is involved in apical delivery. In this study, using the anti-annexin XIIIb antibody and recombinant annexin XIIIb, we provide morphological and functional evidence for a direct role of annexin XIIIb in apical transport. Our results suggest that annexin XIIIb binds to apical rafts that bud off the TGN and acts both at the TGN level and at the cell surface.
Materials and Methods
Media and reagents for cell culture were purchased from GIBCO Biocult (Eggestein, Germany). Transwell™ polycarbonate filters (0.4-μm pore size) for cell culture were from Costar (Cambridge, MA). Propidium iodide was from Molecular Probes (Eugene, OR). Restriction endonucleases were from New England Biolabs (Schwalbach/Taunus, Germany), polymerases from Boehringer Mannheim (Mannheim, Germany) and DNA modifying enzymes from USB (Cleveland, OH). Unless otherwise indicated chemicals were from Sigma (Deisenhofen, Germany). Monoclonal anti-annexin II antibody (HH7) was kindly provided by V. Gerke (University of Münster, Münster, Germany). Affinity-purified rabbit anti– vesicular stomatitis virus (VSV) G and anti-Rab 5 antibodies were provided by T. Nilsson and M. Zerial (EMBL, Heidelberg, Germany) respectively. Polyclonal and monoclonal anti-hemagglutinin (HA) antibodies were prepared as described (Gerhard et al., 1981 and Matlin et al., 1981, respectively). Polyclonal anti-caveolin 1 antibody was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA) or from Transduction Laboratories, Inc. (Lexington, KY). Polyclonal anti-annexin-V antibody was from Alexis Corp. (San Diego, CA). Monoclonal anti-TfR antibody was from Zymed (Bad Homburg, Germany). Monoclonal anti-FLAG M2 antibody was from Kodak (New Haven, CT). Goat anti-rabbit HRP-conjugated and goat anti–mouse HRP-conjugated antibodies were from BioRad (München, Germany). Goat anti–rabbit FITC-conjugated antibody was from Dianova (Hamburg, Germany). Protein A-coupled gold particles were purchased from the Department of Cell Biology, Faculty of Medicine, Utrecht, Netherlands.
Cell Culture and Virus Stocks
MDCK cells, strain II of low resistance, were cultured on Transwell™ filters (Pimplikar et al., 1994). For immunocytochemistry purposes, cells were seeded on 1.2-cm diameter filters with plating on 2.5 × 105 cells per filter. For biochemical experiments, cells were plated on 2.4- and 7.5-cm diameter Transwell™ filters at a cell density of 106 and 2.5 × 106 cells per filter, respectively, treated or not with mevalonate and lovastatin.
Stock of phenotypically mixed VSV (Indiana strain) grown in Chinese hamster ovary C15.CF1 cells which express HA on their plasma membrane and influenza stocks of N (A/chck/germany/49/Hav2Neq1) and PR8 (A/PR8/8/34) virus were prepared as described (Bennett et al., 1988; Matlin and Simons, 1983).
Immunofluorescence Staining and Confocal Analysis
Fixation, quenching, permeabilization, denaturation, blocking, and all the washing steps were performed at room temperature and with shaking of the filters as before (Fiedler et al., 1995). The affinity-purified rabbit anti– annexin XIIIb antibody was used diluted 1:10 (or 1:200 in experiments where unmyristoylated recombinant annexin XIIIb was added to permeabilized cells) for overnight incubation at 4°C. DNA was stained with propidium iodide (0.05 μg/ml) for 15 min at 37°C in PBS as described (Lafont et al., 1994). Cells were placed in mounting medium in PBS-glycerol (Merck, Darmstadt, Germany) 1:1 with 0.1% NaN3 and 100 mg/ml DABCO [1,4-diazabicyclo-2.2.2-octane]. Coverslips were perched on thin bridges cut from cellophane and sealed with nail polish.
Cells were observed using a LSM 410 Confocal equipped with an Axiovert 100 microscope (Carl Zeiss, Oberkochen, Germany). Images were processed using Adobe Photoshop™ Software (version 3.0.5; Mountain View, CA) and further with Canvas™ software (version 3.5.4; Deneba software, Miami, FL) on a Power MacIntosh™ computer (Apple, Cupertino, CA).
TGN-derived Carriers Isolation
Infected MDCK cells plated on 7.5-cm filters for 3.5 d were perforated by ripping off the apical plasma membrane with a nitrocellulose filter (Huber and Simons, 1994) with the following modifications. Proteins from the apical plasma membrane were biotinylated to allow, later on, their elimination by adsorbtion on immobilized streptavidin. To this aim, before perforation, cells were incubated 30 min at 4°C with 1 mg/ml Sulfo-NHS-LC-Biotin (Pierce, Rockford, IL) in PBS containing 0.9 mM CaCl2 and 0.5 mM MgCl2. Three 5-min washes with glycine 0.1 M and BSA 0.5% in PBS containing 0.9 mM CaCl2 and 0.5 mM MgCl2 were used to quench the cross-linking reaction. After perforation, cells were incubated for 15 min at 37°C in 500 μl of transport medium with a free calcium concentration fixed at 100 nM with Hepes/EGTA and Hepes/CaCO3 buffers according to concentrations calculated by the Max Chelator version 4.12 software (1990) from C. Patton (Stanford University, CA). The medium was then collected and adjusted to 30% Optiprep™ (Nycomed Pharma As, Oslo, Norway) in 600 μl before being overlaid with 1.1 ml of 25% Optiprep™ and 400 μl of 10% Optiprep™ prepared in transport medium. Exocytic carriers were floated for 2 h at 55,000 rpm in a TLS 55 Beckman rotor at 4°C. 200 μl of the interface between the 25% and 10% Optiprep™ steps was collected and incubated 30 min at 4°C with 50 μl of a slurry (1:1) of immunopure immobilized streptavidin (Pierce, Rockford, IL) in 25% Optiprep™ prepared in transport medium. The carriers were obtained in the supernatant after spinning the sample in a microfuge at 5,000 rpm 5 min at 4°C. Exocytic carriers were pelleted on a 2.0 M sucrose cushion at 30,000 rpm in a TLA 45 rotor and were processed for immunolabeling before electron microscopy analysis.
Electron Microscopy
For single immunolabeling, untransfected and non-infected polarized MDCK cells were used. For double immunolabeling, polarized MDCK cells expressing VSV G-tagged sialyltransferase were infected with influenza virus (Lafont et al., 1995). To this aim, human sialyltransferase cDNA tagged with the VSV G-epitope in pSRα vector (DNAX, Palo Alto, CA; Rabouille et al., 1995) was electroporated into MDCK cells that were grown under G418 selection for 2 weeks before selecting stably expressing clones. Single immunolabeling was done as described (Scheiffele et al., 1998) with affinity-purified anti-annexin XIIIb antibody diluted 1:5 to 1:25. For double labeling, the grids were incubated with affinity-purified rabbit anti-VSV G or monoclonal anti-HA antibodies, washed and incubated with protein A coupled 6- or 15-nm gold particles and further processed as for single labeling.
For immunolabeling of isolated TGN-derived carriers, see Scheiffele et al. (1998). Controls included the omission (or combinations) of primary antibodies after which the accompanying gold particles were not detected.
For double immunolabeling performed on basolaterally SLO-permeabilized cells expressing the VSV G epitope-tagged sialyltransferase, ultrathin cryosections were obtained and processed for immunostaining as described above using affinity-purified rabbit anti-VSV G and anti-annexin XIIIb antibodies.
Labeled ultrathin cryosections were examined under a Zeiss transmission 10 C electron microscope (Zeiss, Oberkochen, Germany).
Expression and Purification of Unmyristoylated and Myristoylated Recombinant Annexin XIIIb
The outer primers 5′-AATGGATCCAAAATGGGCAATCGTCATAGC-3′ and 5′-AAAATGCATTCAGTGCAAGAGGGCCAC-3′ were used for PCR amplification of the annexin XIIIb cDNA template in the Bluescript SK II plasmid (Fiedler et al., 1995). An insert fragment was obtained after BamHI and NsiI (blunt) digestions. The insert was cloned in the BamHI-SmaI digested pGAT2 expression vector. This vector contains a modified T7lac promoter and a fusion partner consisting of his6-glutathione-S-transferase (his6-GST) gene linked with a thrombin cleavage site in 5′ of the cDNA coding for annexin XIIIb (Peränen et al., 1996). JM109(DE3) cells (Promega, Madison, WI) harboring pGAT2-AnxXIIIB were cultured at 37°C in 2 liters of Luria broth medium with ampicillin (100 μg/ml). Expression was induced by addition of isopropylthio-β-D-galactoside to 0.5 mM at an OD600 of 0.5 for 4 h. The cells were harvested and pelleted at 4°C. Cells were resuspended in 30 ml of Buffer A (10 mM Tris-HCl, pH 7.5, 1% Triton X-100, 1 mM EDTA, 0.5 mM Pefabloc [Boehringer Mannheim, Mannheim, Germany], and 0.5 mg/ml lysozyme) and incubated for 30 min on ice. Cells were lysed with a French press (two times at 800 PSIG) in buffer A. After addition of 300 mM NaCl and 5 mM DTT, cell debris was removed by a 30-min spin at 18,000 rpm in a SW27 rotor at 4°C. The supernatant was filtered through a 0.45-μm pore Millex filter (Millipore, France) and loaded onto glutathione columns prepared as follows. Three columns were cast each with 2 ml slurry of glutathione Sepharose 4B (Pharmacia, Uppsala, Sweden) washed once with 10 ml PBS then equilibrated with 10 ml Buffer B (10 mM Tris, pH 7.5, 300 mM NaCl, and 1 mM EDTA). 8 ml of lysate was loaded per column and incubated in the cold room on a rotating wheel for overnight binding of the recombinant annexin XIIIb-GST to the glutathione. The columns were washed each with 50 ml Buffer B and the protein was eluted with 3 ml Buffer C (10 mM Tris, pH 7.5, 300 mM NaCl, 1 mM EDTA, 1 mM DTT, 0.5 mM Pefabloc, and 20 mM reduced glutathione). The eluted annexin XIIIb-GST was then dialyzed overnight in the cold room in Buffer D (10 mM Tris, pH 7.5, 50 mM NaCl, and 1 mM EDTA) and cleavage between annexin XIIIb and GST was achieved during dialysis upon incubation of the sample with 0.5 mg/ml of thrombin. Resolution of annexin XIIIb from the GST was subsequently obtained by loading the sample onto an anionic exchange Mono-Q HR 5/5 column (Pharmacia). 2 ml of the samples were loaded for each run and eluted with a salt gradient of 15 ml made with Buffer D and Buffer E (10 mM Tris, pH 7.5, and 300 mM NaCl). The separated cleaved products were collected in 0.3-ml fractions at a flow rate of 1 ml/min. Annexin XIIIb was eluted at 210 mM NaCl, and the uncleaved product, if any, was eluted at 240 mM NaCl. The purity of the fractions was tested after separation on 12% reducing SDS–polyacrylamide gel using either colloidal blue or silver staining kits from Novex (San Diego, CA). Fractions of pure protein were stored at −20°C with 10 mM DTT. The protein was dialyzed against KOAc buffer before being used in the in vitro transport or budding assays. The yield was routinely 5 mg of pure protein per liter of bacteria. We checked that the unmyristoylated recombinant protein was able to bind in a calcium-dependent manner liposomes made from lyophilized brain extract containing 80–85% phosphatidylserine.
To obtain regulated production at a high level of myristoylated recombinant annexin XIIIb, the protein was expressed in Drosophila Schneider (SL3) cells. For that purpose, the annexin XIIIb sequence was amplified by PCR to obtain an EcoRI-KpnI fragment containing 29 nucleotides before the ATG and all the sequence of the gene except the stop codon. The primers used for this amplification were: 5′-TCG GAA TTC TAC AGA ACA ACT GTC T-3′ and 5′-C GAC GGT ACC GTG CAA GAG GGC CAC-3′. The sequence has not been further modified because the Kozak sequence of annexin XIIIb is very close to the consensus Kozak sequence of Drosophila melanogaster. The annexin XIIIb fragment has been inserted between the EcoRI-KpnI sites in the polylinker of a modified pRmHa-3 vector provided by Dr. Jürgen Benting (EMBL, Heidelberg, Germany). In brief, this vector contains, like the parental one, the copper inducible Drosophila Metallothionein promoter in 5′ of the polylinker: EcoRI-SacI-NheI-KpnI-SmaI-BamHI-ClaI-Flag tag-EcoRV-10xHis-SalI, the KpnI and BamHI sites being in frame with the different tags. These tags can be cleaved off by numerous proteases after purification of the protein. Cotransfection of SL3 cells with pRmHa-3/AnxXIIIb and pUChsneo was done according to (Jackson et al., 1992; Wallny et al., 1995) with the modification that cells were cultured in presence of 1% FCS. The induction of the protein was achieved by incubating the cells for 72 h with 2 mM CuSO4. The routine volume was 600 ml which gave rise to 2 g of dry cells. After extensive washing, the cells were lysed (6 ml lysis buffer/g of dry cells) at 37°C for 30 min in PBS, 150 mM NaCl, 1.5% Triton X-100, 10 mM imidazole, and a cocktail of protease inhibitors (CLAP: chymostatin, leupeptin, antipain, pepstatin A; final concentration 25 mg/ml of each). The supernatant of a 20-min spin at 4,000 g was incubated overnight with equilibrated Ni-NTA Agarose beads (Qiagen, Hilden, Germany). The beads were then washed with increasing amount of imidazole in PBS, 150 mM NaCl, 1% Triton X-100. The last washes were performed without Triton X-100. The elution was obtained at 120 mM imidazole in PBS, 150 mM NaCl, 10 mM DTT, and CLAP. For the functional assays (see below), the protein was concentrated through using a 10 filter (Amicon, Beverly, MA) and dialyzed against KOAc buffer. We checked that the myristoylated recombinant protein was able to bind liposomes. To select the clones of Schneider cells expressing the highest amount of myristoylated annexin XIIIb, 1 ml of each clone was induced with 1 mM CuSO4 for 24 h and 20 μl [9.10(n)-3H] myristic acid (in ethanol solution at 1.89 TBq/mmol with 37 MBq/ml; Amersham, Braunschweig, Germany) was incorporated for the last 20 h of culture. Cells were then washed three times in ice-cold PBS and then lysed in PBS, 1% NP40, 1 mM DTT, and CLAP, for 30 min at room temperature with shaking. The solubilized material was analyzed by SDS-PAGE, gels were fixed and the signal was intensified (Intensify™; Du pont de Nemours, Brussels, Belgium), and exposed for 2 wk.
Lovastatin/Mevalonate Treatment, MeβCD, and Triton Extractions, Floatation
We followed a procedure based on the inhibition of de novo synthesis of cholesterol by lovastatin in the presence of mevalonate (used for the synthesis of nonsterol products) and extraction of cholesterol from the plasma membrane by MeβCD (Keller and Simons, 1998). When this procedure was used for MDCK cells grown on filters, cholesterol depletion <70% was accomplished (Keller and Simons, 1998). Control and MeβCD extracted cells were rinsed by dipping in ice-cold PBS containing 0.9 mM CaCl2 and 0.5 mM MgCl2 before they were transferred for 75 min to 20°C with 3 ml on both sides of infection medium containing 0.35 g/liter sodium bicarbonate. Then, filters were transferred to 37°C for 30 min. Cells were cooled down by dipping the filters in 10 mM ice-cold Hepes, pH 7.4 and 2 mM EGTA. Cells were scraped with a rubber policeman in this buffer containing 0.25 mM sucrose (United States Biochemical Corp., Cleveland, OH), 5 mM DTT, and CLAP. Cells were homogenized by 25 passages through a 25-G needle. For some floatation experiments cells were treated with 100 μM latrunculin B (Calbiochem, Bad Soden, Germany) and 33 μM nocodazole for 20 min at 37°C followed by 20 min on ice for complete disruption of the cytoskeleton. For Triton extraction, samples were incubated 30 min at 4°C with 1% (wt/vol) Triton X-100 (Serva, Heidelberg, Germany). Samples were then either centrifuged at 120,000 g for 30 min at 4°C on a 2 M sucrose cushion or submitted to floatation using OptiPrep™. In this latter case, samples were made in 900 μl 1.077 g/ml with OptiPrep™ and overlaid with a step gradient of 1.067, 1.043, and 1.037 g/ml (1,350, 900, and 900 μl, respectively) using a mix of 0.25 M sucrose and 30% OptiPrep™ both in 10 mM Hepes, pH 7.4, 2 mM EGTA, and 1% Triton X-100. Floatation was performed in SW 60 Beckman tubes at 40,000 rpm for 18 h at 4°C. Nine fractions were collected and proteins were methanol-chloroform precipitated for further analysis. Supernatants obtained after the 120,000 g centrifugation were methanol-chloroform precipitated.
The association of annexin XIIIb with rafts on TGN-derived vesicles was determined using exocytic carriers collected from control or mevalonate and lovastatin treated cells. These latter cells were further treated with MeβCD 10 mM for 30 min at 37°C before the 1% Triton X-100 extraction. Control and MeβCD-treated cells were Triton-extracted for 30 min at 4°C. Samples were made 1.159 g/ml using 200 μl of 10 mM ice-cold Hepes, pH 7.4, 60% OptiPrep™, 1% Triton X-100 buffer adjusted with Hepes/EGTA and Hepes/CaCO3 buffers such that the free calcium concentration was 100 nM. The sample was then overlaid with 1.130 g/ml (750 μl), 1.180 g/ml (750 μl), and 1.052 g/ml (250 μl) prepared using the same buffer. After 2 h spinning at 55,000 rpm in TLS 55 Beckman tubes at 4°C, nine fractions (250 μl) were collected from the top. Proteins were methanol-chloroform precipitated and analyzed by SDS-PAGE and Western blotting using 5% nonfat dried milk, 0.1% Tween 20 in PBS as blocking and incubation buffer.
In Vitro Cell Surface Transport Assay
This assay is performed on fully polarized SLO-permeabilized MDCK cells and is energy, cytosol, and temperature dependent (Lafont et al., 1995). The assay was carried out according to Pimplikar et al. (1994) with the following modifications. Both myristoylated and unmyristoylated recombinant annexin XIIIb and the annexin II–p11 heterotetramer complex were dialyzed against KOAc buffer (pH 7.4) before mixing with exogenous cytosol and CLAP. In all cases, the free calcium concentration was clamped to 100 nM with Hepes/EGTA and Hepes/CaCO3 buffers. After the transport, the cells were lysed, proteins were separated by SDS-PAGE (10% resolving gel), and band intensities were quantitated using PhosphorImager™ and ImageQuant software™ (Molecular Dynamics, Sunnyvale, CA). Calculations of transport efficiencies were done according to Pimplikar et al. (1994). Experiments were performed using duplicate filters and SEM of at least three independent experiments are reported. The values are expressed as control cytosol-dependent transport being 100% (transport in the presence of cytosol minus transport in the absence of added cytosol).
Exocytic Carriers Budding from Semi-intact Cells
Release of HA and VSV G transport carriers from the TGN of semi-intact cells grown on 10-cm dishes (Nunclon™; Nalge Nunc Int., Denmark) was studied according to published procedures (Müsch et al., 1996; Scheiffele et al., 1998). The budded fraction was obtained using a hypotonic swelling procedure (Xu and Shields, 1993) to selectively disrupt the plasma membrane (Beckers et al., 1987). The budding reaction was performed in prelubricated microcentrifuge tube from Costar (Corning Costar Corporation, Cambridge, MA) in order to avoid any binding of vesicles to the wall of the tube. The amount of HA or VSV G released in the budded vesicles from the pool associated with TGN were quantitated using PhosphorImager™ and ImageQuant software™ (Molecular Dynamics, Sunnyvale, CA). The values are expressed as control cytosol-dependent transport being 100% (transport in the presence of cytosol minus transport in the absence of added cytosol) and SEM of at least three independent experiments are reported.
Results
Annexin XIIIb Localization during Polarization
We have previously shown that in fully polarized MDCK cells, annexin XIIIb is located in the apical compartment (Fiedler et al., 1995; Fig. 1 A). In nonconfluent MDCK cells grown on coverslips, annexin XIIIb displays a punctate staining uniformly distributed over the cytoplasm (Fiedler et al., 1995). Here, we analyzed the distribution of annexin XIIIb during the development of polarity in filter-plated cells. We examined cells fixed after 24 h in culture (Fig. 1 B) before they are functionally polarized according to transepithelial resistance measurements and the localization of surface antigens (Balcarova-Ständer and Simons, 1984). At this stage, a mosaic of annexin XIIIb distribution was seen. In many cells annexin XIIIb was not polarized. Nevertheless, in some cells annexin XIIIb was already apically enriched (compare cells labeled with arrows in Fig. 1 B with those in Fig. 1 A). For comparison, we analyzed the distribution of annexin II, which was uniformly distributed over the cells (Fig. 1 C). Therefore, the polarization of annexin XIIIb is an early event.
Figure 1.
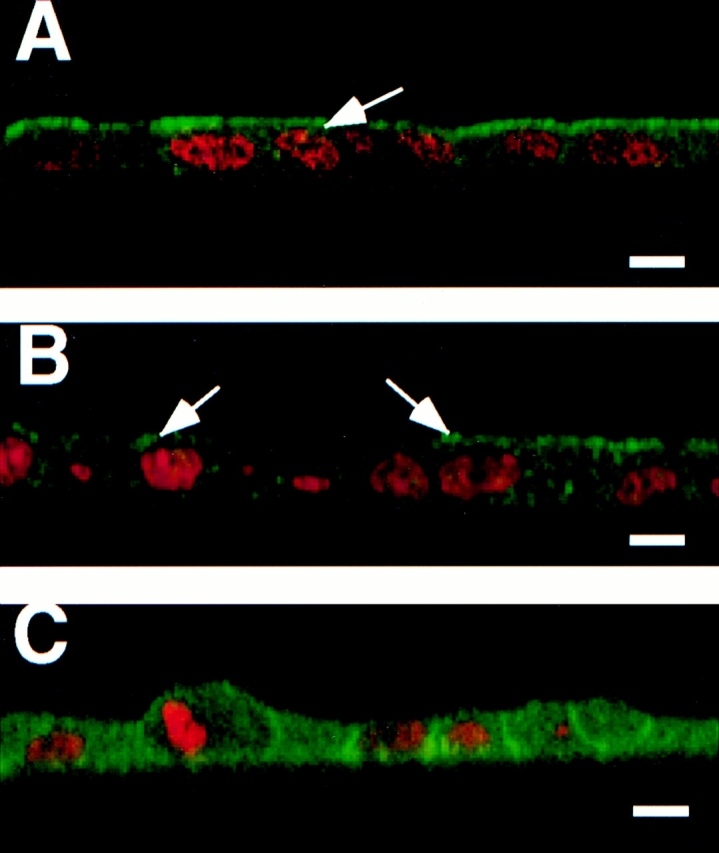
Survey of annexin XIIIb during MDCK cell polarization. Filter-grown MDCK cells were immunostained with anti-annexin XIIIb or anti-annexin II antibodies (green) and nuclei were labeled with propidium iodide (red). Samples were viewed by confocal microscopy. Confocal (x, z) scans are presented. (A) Fully polarized cell processed for annexin XIIIb immunostaining. (B and C) Cells fixed after 24 h in culture before immunolabeling with the anti-annexin XIIIb (B) or anti-annexin II (C) antibodies. Notice that some cells in B (arrows) display specific apical surface labeling like that observed for the entire monolayer when the cells are fully polarized (A, arrow). Bar, 10 μm.
Annexin XIIIb Distributes Along the Apical Route in Fully Polarized Cells
When analyzed in the confocal optical microscope, annexin XIIIb accumulated at the apex of fully polarized cells with a weak though significant intracellular staining (Fig. 1 A). To determine precisely the subcellular distribution of annexin XIIIb, we performed immunoelectron microscopy.
Annexin XIIIb was found associated with tubulovesicular structures nearby the Golgi stack, with vesicular structures (diameter ranging from 50 to 100 nm) and with the apical surface (Figs. 2 and 3 A–C). We quantified in cryosections the percentage of gold particles found in either the apical cytoplasmic region or the basolateral cytoplasmic region (Table I). Separately, we counted the gold particles within a region 50 nm from the apical, lateral or basolateral membrane. In 7 cells analyzed, we counted 178 ± 21 gold particles/cell cryosection located within the apical region, either membrane-associated or cytoplasmic (see Fig. 2), while 54 ± 13 gold particles/cell cryosection were counted in the basolateral region. This result indicated that ∼77% of annexin XIIIb is apically distributed. Moreover, in the apical region, we detected 42 ± 18 gold particles/cell cryosection directly associated with the plasma membrane which represents 24% of gold particles within the area above the cell nucleus. Only 6.5% of the gold particles in the apical plasma membrane were located between the microvilli (Fig. 3, C1 and C2). Surface biotinylation followed by agarose-coupled streptavidin binding or antibody binding at 4°C on living cells, demonstrated that annexin XIIIb was not accessible from the extracellular compartment suggesting that it was exclusively localized on the cytoplasmic side (data not shown). The Golgi stacks were often observed lateral to the nucleus as typically shown in Fig. 2. Therefore, of the 23% of annexin XIIIb not found in the apical region a significant part is due to labeling associated with membranes in the vicinity of the Golgi. On the other hand, annexin XIIIb was rarely observed along the lateral or basal membranes; only 11% of annexin XIIIb was found on the basolateral membrane (Table I).
Figure 2.
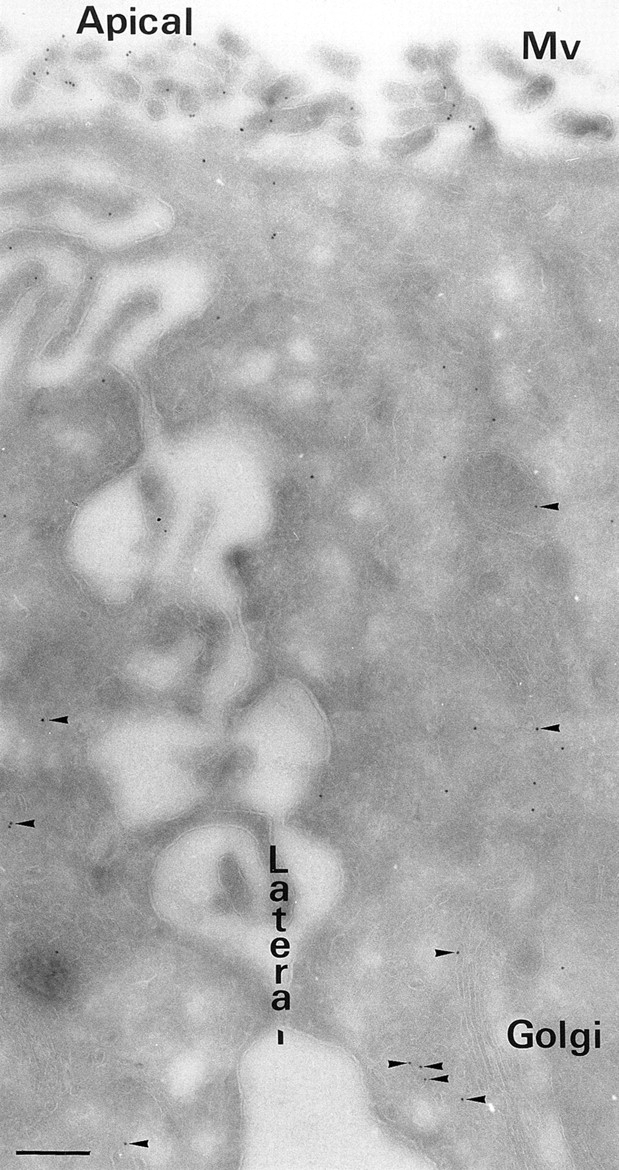
Overview of the ultrastructural distribution of annexin XIIIb in polarized MDCK cells. Polarized filter-grown MDCK cells were processed for immunoelectron microscopy with the anti-annexin XIIIb antibody. Arrowheads point out gold particles in the cytoplasm distributed in close vicinity of membranes. Bar, 400 nm.
Figure 3.
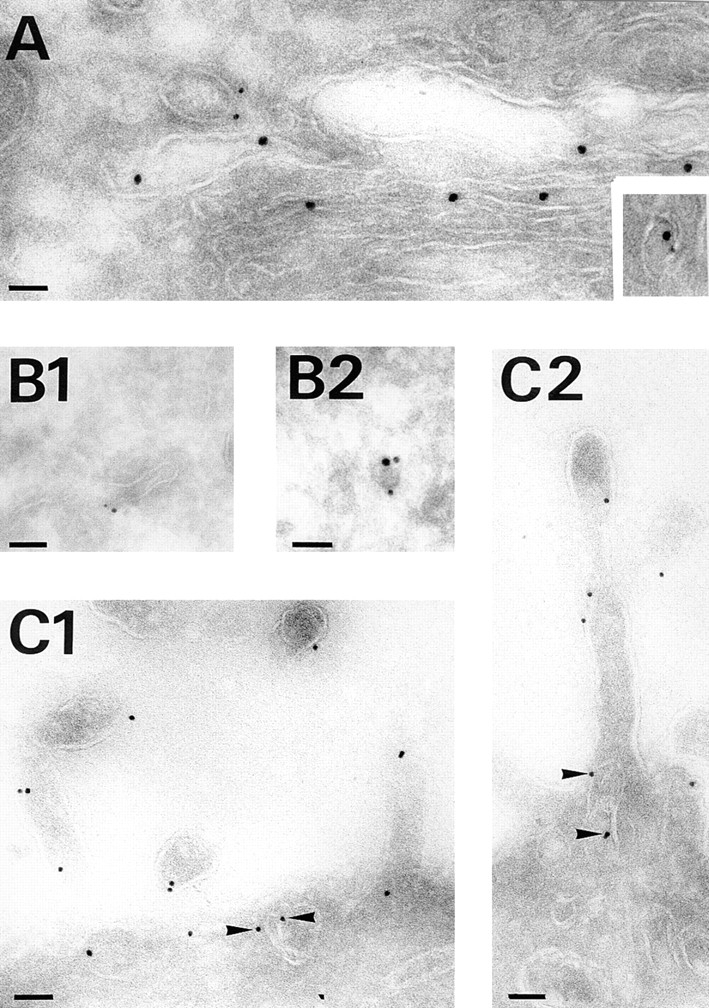
Annexin XIIIb distributes along the apical pathway. Double immunostaining was performed on influenza virus infected polarized filter-grown MDCK cells expressing the VSV G-tagged sialyltransferase before processing for electron microscopy. (A) Sialyltransferase detected with anti-VSV G antibody (15 nm gold) and anti-annexin XIIIb (6 nm gold) labeling at the Golgi level after a TGN block at 20°C for 75 min. The inset illustrates colocalization of both sialyltransferase and annexin XIIIb in the same vesicular profile found nearby the Golgi stacks. (B1 and B2) Examples of colocalization of annexin XIIIb (6 nm gold) and HA (15 nm gold) in tubulovesicular structures. (C1 and C2). Annexin XIIIb staining in tubulovesicular structures underneath the apical surface (arrowheads) and in microvilli. Bars: (B1) 120 nm; (B2) 100 nm; and (C1 and C2) 120 nm.
Table I.
Quantitative Distribution of Annexin XIIIb in Polarized MDCK Cells
| Cytoplasmic region | Number of gold particles/ cell cryosection | Plasma membrane | Number of gold particles/ cell cryosection | |||
|---|---|---|---|---|---|---|
| Apical | 178 ± 21 | Apical | 42 ± 18 | |||
| Basolateral | 54 ± 13 | Lateral | 16 ± 3 | |||
| Basal | 9 ± 4 |
The table shows quantitation done on one cryosection per cell processed for immunolabeling with the anti-annexin XIIIb antibody and protein A-coupled 10-nm gold particles. Samples were examined under transmission electron microscopy as described in Materials and Methods. Gold particles were counted in the cytoplasmic regions. For either apical or basolateral cytoplasmic regions the number of gold particles associated with the plasma membranes (apical, lateral, or basal) is also indicated. Seven cells were analyzed. Data are mean ± SEM.
To confirm that annexin XIIIb was in the TGN, we infected a cell line stably expressing an epitope-tagged sialyltransferase with influenza virus. To accumulate the viral glycoproteins in the TGN, we performed a 20°C TGN block for 75 min before fixing the cells. We found colocalization of the TGN-resident enzyme sialyltransferase (Fig. 3 A, large gold) with annexin XIIIb (Fig. 3 A, small gold), suggesting binding of annexin XIIIb to TGN membranes (Fig. 3 A, inset). We quantitated the gold particles associated with annexin XIIIb on membrane structures colabeled for the sialyltransferase. We found a ratio 1.4 ± 0.3: 8.3 ± 1.3 (small gold particles associated with annexin XIIIb/large gold particles associated with sialyltransferase, n = 25). For comparison, we found only 0.4 ± 0.1 small gold on mitochondria (unspecific labeling, n = 25). We also observed immunodecorated vesicular profiles likely to represent tubules or vesicles being released from the TGN (Fig. 3 A). However, we did not detect significant endoplasmic reticulum, cis or medial Golgi immunostaining (data not shown). When proteins were released from the 20°C block, tubular and vesicular-like structures (diameter ranging between 50 and 100 nm) displayed colocalization of annexin XIIIb with the apical marker HA (Fig. 3, B1 and B2, annexin XIIIb small gold and HA large gold). These structures could be localized rather distant from the Golgi stacks (Fig. 3 B2), suggesting that they were TGN-derived carriers. To strengthen this conclusion, we isolated TGN-derived carriers from infected and perforated cells (Fig. 4). Here, annexin XIIIb, HA and VIP21/caveolin-1 showed colocalization on the vesicles. Indeed, Table II shows the quantitation derived from the observation of 40 carriers positive for HA (Fig. 4 A, 15 nm gold) and VIP21/ caveolin-1 (Fig. 4 A, 5 nm gold) that were labeled for annexin XIIIb (Fig. 4 A, 10 nm gold). Using VSV infected cells, we similarly quantified in table II the average number of 10-nm gold particles associated with annexin XIIIb in 40 vesicles immunoreactive for both the basolateral marker vesicular stomatitis virus glycoprotein (Fig. 4 B, VSV G, 15 nm gold) and VIP21/caveolin-1 (Fig. 4 B, 5 nm gold). We also checked whether some of the VSV G positive structures would represent endocytic elements by colabeling with anti-Rab 5 antibody. Our results showed that Rab 5 positive structures contained very little annexin XIIIb (0.6 ± 0.1 gold particle per VSV G and Rab 5 labeled vesicles, n = 50). This result indicated that when the basolateral route is overstimulated upon VSV infection, only a minor pool of annexin XIIIb could be detected on the basolaterally routed carriers. Altogether these quantitations indicated that annexin XIIIb was clearly enriched in the apical HA-containing vesicles (more than threefold enrichment, compare Table I with Table II). The weak annexin XIIIb staining observed on these VSV G–containing carriers might be due to a partial missorting caused by the viral infection.
Figure 4.

Distribution of annexin XIIIb on exocytic TGN-derived carriers. (A) Immunostaining of HA (15 nm gold), annexin XIIIb (10 nm gold) and VIP21/caveolin-1 (5 nm gold) on TGN-derived carriers obtained from cells whose apical membrane have been ripped off (see Materials and Methods). (B) Immunostaining of VSV G (15 nm gold), annexin XIIIb (10 nm gold) and VIP21/ caveolin-1 (5 nm gold) on TGN-derived carriers. Bar, 100 nm. Note the enrichment of gold particles associated with annexin XIIIb on HA-containing carriers versus the amount observed on VSV G-containing carriers and see Table II. Bar, 100 nm.
Table II.
Quantitative Analysis of Annexin XIIIb Distribution in Exocytic Vesicles from MDCK Cells
| Annexin XIIIb | VIP21/caveolin-1 | |||
|---|---|---|---|---|
| HA | ||||
| 6.9 ± 0.7 | 7.1 ± 0.5 | 12 ± 0.9 | ||
| VSV G | ||||
| 4.4 ± 0.3 | 2.0 ± 0.4 | 8.7 ± 0.7 |
Numbers of gold particles per vesicle are indicated. Gold label on 40 HA and 40 VSV G positive vesicles from polarized cells was quantitated. Vesicles were considered positive for the viral markers when they contained at least two gold particles. Data are mean ± SEM.
We conclude that annexin XIIIb is enriched in the apical plasma membrane and in TGN-derived carriers en route to the apical plasma membrane where annexin XIIIb colocalizes with HA.
Annexin XIIIb Is Associated with Apical Rafts
Next, we analyzed whether annexin XIIIb could be specifically isolated in DIGs as has been shown for some other apically routed proteins (Scheiffele et al., 1997; Keller and Simons, 1998). Although annexins are known to bind phospholipids in the presence of calcium, we previously showed that annexin XIIIb had the unusual property of binding to membranes in the absence of calcium (Fiedler et al., 1995).
Indeed, in a typical experiment shown in Fig. 5 A, when cells were homogenized in EGTA-containing buffer, annexin XIIIb was clearly pelleted. After MeβCD treatment of lovastatin-mevalonate treated, annexin XIIIb also pelleted demonstrating that the MeβCD treatment alone did not lead to dramatic release of annexin XIIIb from membranes (Fig. 5 A). Moreover, we checked that the overall ultrastructure of the cells was not dramatically affected by such treatment (data not shown), only caveolae on the basolateral side were observed flattened in agreement with previous data (Hailstones et al., 1998). After Triton X-100 extraction at 4°C, a pool of annexin XIIIb remained insoluble although annexins are peripheral proteins that should be efficiently solubilized by detergent extraction (Fig. 5 A). To differentiate whether annexin XIIIb was associated with detergent-resistant membranes or with cytoskeletal elements, we performed floatation experiments of Triton X-100 extracts in sucrose-OptiPrep™ gradients in the absence of calcium (Fig. 5 B). We found that a pool of annexin XIIIb floated to low densities, indicating that annexin XIIIb was associated with detergent-resistant membranes and that this association was calcium independent. To prevent any association of annexin XIIIb with membranes via binding to the actin cytoskeleton, we treated the cell lysate with cytoskeleton-depolymerizing drugs but this treatment did not change the overall floatation behavior. This floatation experiment suggested that the association of annexin XIIIb with DIGs containing influenza HA did not require calcium.
Figure 5.
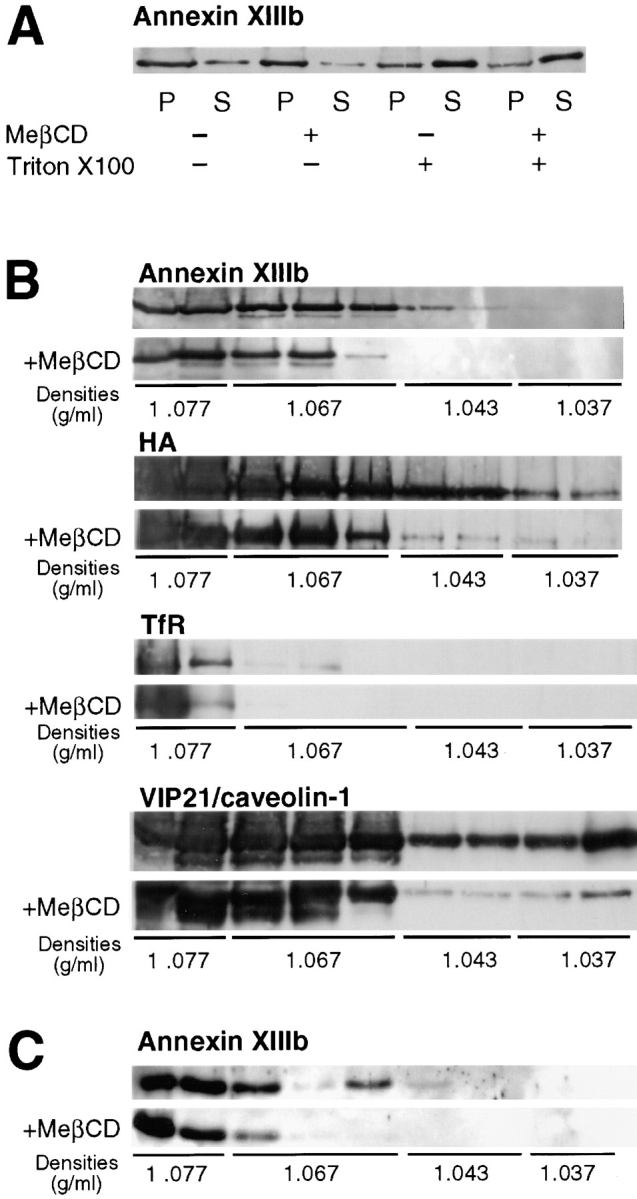
Annexin XIIIb is raft associated. Filter-grown MDCK cells were infected with influenza virus. Cells treated with mevalonate and lovastatin were extracted with MeβCD. Then, cells were submitted to a TGN block and chased at 37°C before lysis. (A) Lysed cells were either extracted with Triton X-100 or not before centrifugation. Percentages of insoluble (P) and soluble (S) annexin XIIIb were analyzed by Western blotting. Similar result were obtained in four independent experiments. (B) Lysed cells were extracted with Triton X-100 and membranes were floated in an OptiPrep™ step gradient. Western blotting was performed on methanol-chloroform precipitated fractions. The first two fractions correspond to the volume of the sample submitted to floatation. (C) TGN-derived carriers from influenza virus infected cells were extracted with Triton X-100 and membranes were floated in an Optiprep™ step gradient. Western blotting was performed on methanol-chloroform precipitated fractions.
As a more stringent test for raft association, we performed floatation after depleting cells of cholesterol by MeβCD treatment. Less annexin XIIIb was observed in the lightest density fractions after cholesterol depletion (Fig. 5 B, MeβCD) confirming that the floatation of annexin XIIIb was dependent on the integrity of rafts. After cholesterol depletion, floatation of HA was also reduced consistent with previous data (Scheiffele et al., 1997). The shift observed in the floatation pattern after cyclodextrin treatment was reproducible (n = 4). It is possible that some of the annexin XIIIb associated with internal membranes might not have been efficiently affected by the MeβCD treatment which was performed on intact cells (see Keller and Simons, 1998).
As a control for a nonraft protein, we used the transferrin receptor (TfR). This receptor is basolaterally targeted in MDCK cells (Fuller and Simons, 1986) and is not included in the DIGs (Kundu et al., 1996). This receptor was not found in the light gradient fractions where annexin XIIIb and HA could be observed. We excluded that annexin XIIIb associated in an unspecific manner with any detergent-resistant membranes because it was not found in the lightest buoyant fractions of the gradient that contained membranes as judged by the presence of VIP21/caveolin-1 (Fig. 5 B). The difference observed in the floatation patterns of annexin XIIIb and HA on one hand and VIP21/ caveolin-1 on the other hand could be explained by the presence of VIP21/caveolin-1 on both apical carriers and basolateral caveolae (Scheiffele et al., 1998) that would have different densities. However, this interpretation requires further studies.
Next, we determined whether annexin XIIIb was raft-associated in TGN-derived carriers. Isolated TGN-derived vesicles obtained from influenza virus-infected cells were Triton-extracted on ice and subsequently subjected to density gradient floatation. Annexin XIIIb was found associated with membranes floating in the gradient demonstrating its presence in DIGs on TGN-derived vesicles (Fig. 5 C). Furthermore, when cells were depleted of cholesterol (using MeβCD on mevalonate/lovastatin-treated cells) before Triton extraction, significantly less annexin XIIIb was associated with the floating membranes (Fig. 5 C).
Unmyristoylated and Myristoylated Recombinant Annexin XIIIb Affect Specifically the Apical Pathway
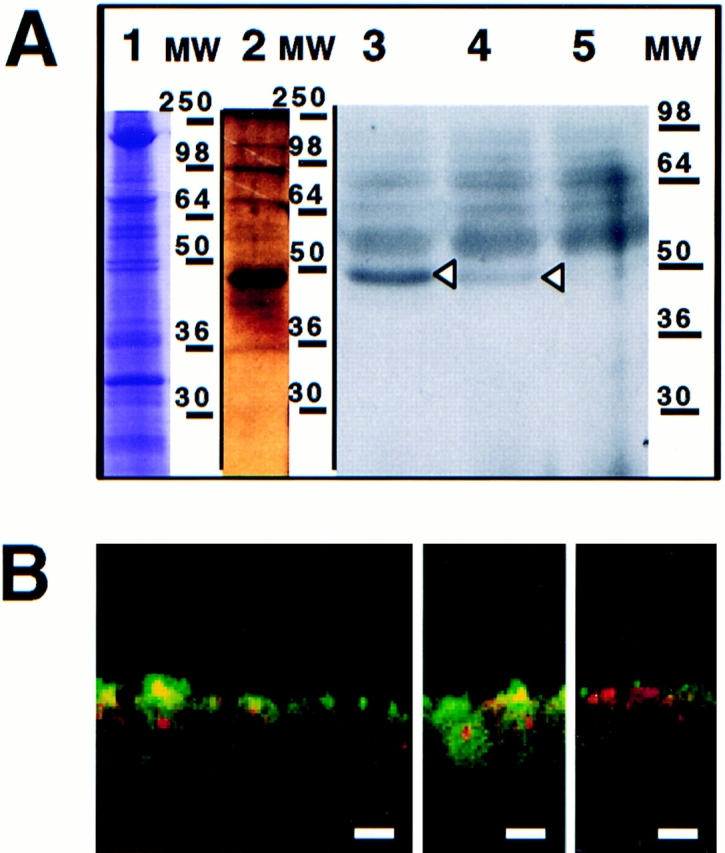
Previously we showed that a bivalent anti-annexin XIIIb antibody was able to inhibit apical delivery of HA while the VSV G delivery to the basolateral side was not affected (Fiedler et al., 1995). Importantly, no endogenous annexin XIIIb was leaking out of the cells after permeabilization and the ratio between membrane-bound and cytosolic pool was identical in both control and permeabilized cells (data not shown). Because of the localization of annexin XIIIb at the apical plasma membrane, it was not possible to exclude that apical carriers were blocked from reaching the surface due to steric hindrance by the bound antibody. Now, we checked whether addition of recombinant protein had an effect on transport. The GST-fused unmyristoylated recombinant annexin XIIIb expressed in Escherichia coli (Fig. 6 A, lane 1) was purified on a glutathione column (Fig. 6 A, lane 3) and after thrombin digestion (Fig. 6 A, lane 4), the cleaved products were separated by chromatography on a Mono-Q column. The purified unmyristoylated recombinant annexin XIIIb (Fig. 6 A, lane 5) was then used in further experiments.
Figure 6.


Purified unmyristoylated recombinant annexin XIIIb is efficiently reaching the intracellular compartment in permeabilized cells. (A) Purification of recombinant annexin XIIIb expressed in E. coli. Lane 1, E. coli lysate; lane 2, last wash, and lane 3, elution from glutathione column. Lane 4, Thrombin cleavage before run on Mono-Q column. Lane 5, Purified recombinant annexin XIIIb after Mono-Q column elution. Lanes 1–4, Coomassie blue staining. Lane 5, Silver staining. (B) Confocal (x,z) scans of cells either basolaterally (1, 2) or apically (3, 4) SLO-permeabilized. Recombinant annexin XIIIb (1.2 μM) was added to samples 1 and 3. Nuclei were labeled with propidium iodide (red) and annexin XIIIb and visualized using an anti-annexin XIIIb antibody (green). Note that the recombinant unmyristoylated protein is present in both the apical and basolateral compartments (B1 and B3). Bar, 10 μm.
We verified that exogenously added annexin XIIIb was located inside the permeabilized cells by confocal analysis on samples processed for immunocytochemistry with a low concentration of anti-annexin XIIIb antibody that shows only low signal for the endogenous protein. As shown in Fig. 6, B1 and B3, in SLO-permeabilized MDCK cells, the unmyristoylated recombinant annexin XIIIb was distributing in both the apical and basolateral compartments.
When the unmyristoylated recombinant annexin XIIIb was tested in the transport assay, we found a dose-dependent inhibition of apical delivery of HA (Fig. 7 A). Apical transport could be inhibited <80% with 1.2 μM of the unmyristoylated recombinant protein. On the other hand, transport of the VSV G protein was not impaired (Fig. 7 A). Importantly, even though the unmyristoylated recombinant annexin XIIIb was present in the entire cytoplasm including the basolateral compartment (Fig. 6, B1 and B3) it was only active in the apical pathway (Fig. 7 A). The lack of effect on the basolateral pathway suggested a specific role of annexin XIIIb in the apical route. To exclude a possible unspecific fusion event due to some functional property common to annexins (i.e., unspecific membrane aggregation), we analyzed whether the transport of HA would be sensitive to another annexin located in the apical compartment. We used the annexin II–p11 heterotetrameric complex. In fully polarized MDCK cells, we found annexin II localized in the apical but also in the basolateral compartment in agreement with previous result (Fig. 7 B; Harder and Gerke, 1993). When we tested this protein oligomer in the transport assay, we found no effect on apical transport (Fig. 7 A). This result suggests that the inhibitory effect observed with the recombinant annexin XIIIb was not due to a functional property shared by several annexins.
Figure 7.
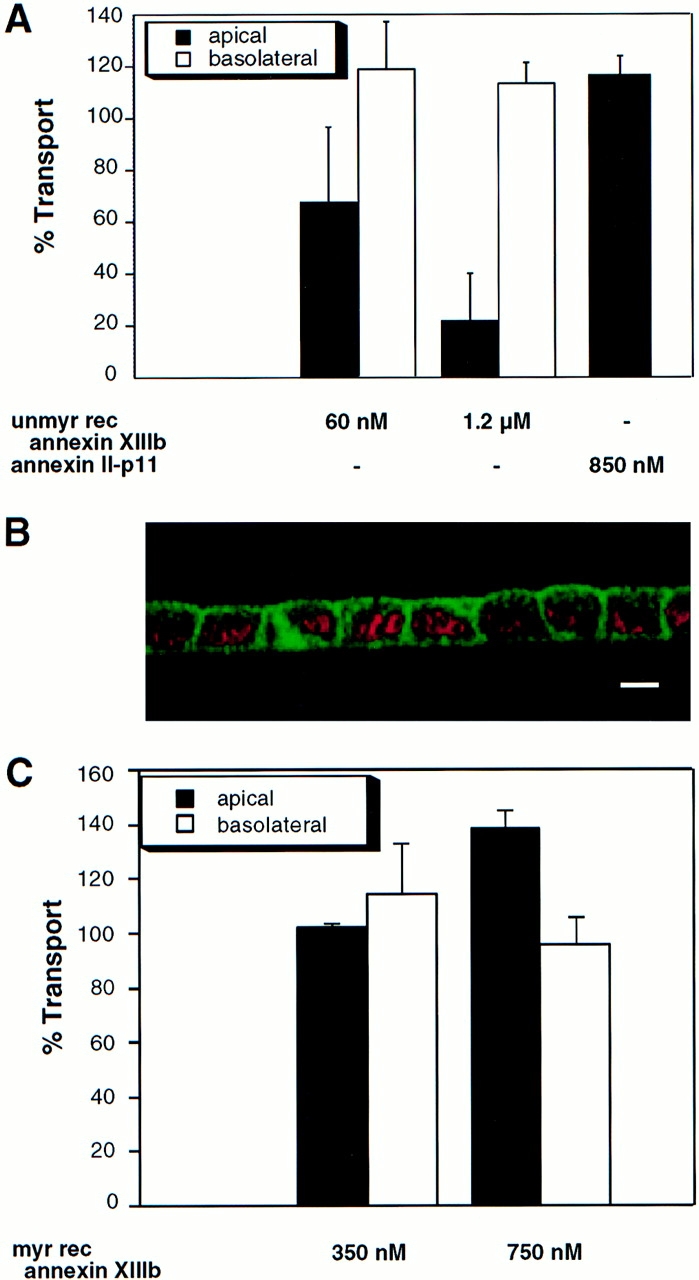
Exocytic apical delivery is specifically sensitive to unmyristoylated and myristoylated recombinant annexin XIIIb. (A) Dose-dependent effect of unmyristoylated recombinant annexin XIIIb (unmyr rec annexin XIIIb) expressed in E. coli in the in vitro transport assay with HA and VSV G as reporter proteins for the apical and basolateral exocytic pathway, respectively. Annexin II–p11 heterotetramer does not impair the apical transport of HA. (B) Confocal (x, z) scan of polarized MDCK cells double stained for nuclei (propidium iodide in red) and for annexin II (green). (C) Dose-dependent effect of myristoylated recombinant annexin XIIIb (myr rec annexin XIIIb) expressed in Drosophila Schneider SL3 cells in the in vitro transport assay with HA and VSV G as reporter proteins for the apical and basolateral exocytic pathway, respectively. Bar, 20 μm.
Since the endogenous annexin XIIIb is myristoylated, we also tested the effect of the myristoylated form of the recombinant protein. To obtain myristoylated recombinant annexin XIIIb, the protein was expressed in E. coli harboring a plasmid coding for the N-myristoylated transferase of S.cerevisiae (kindly provided by J. Gordon, St. Louis, MO). However, the yield of myristoylated annexin XIIIb was low but could be increased by expression in the Drosophila Schneider SL3 cells which in contrast to the E. coli possess endogenous N-myristoyl transferase activity. The histidine-tagged myristoylated recombinant annexin XIIIb (Fig. 8 A, lane 1) was purified on Ni-NTA beads (see Materials and Methods; Fig. 8 A, lane 2). We also checked that the selected clones were expressing the recombinant protein efficiently myristoylated (Fig. 8 A, lanes 3 and 4). The purified myristoylated recombinant annexin XIIIb was found to stimulate apical transport in a dose-dependent manner whereas the basolateral route was not affected by addition of the protein (Fig. 7 C). Strikingly, when added from the basolateral side to SLO-permeabilized cells, the myristoylated recombinant annexin XIIIb was visualized after immunostaining mainly in the apical compartment (Fig. 8 B) in contrast to the bacterial protein that was located in both compartments (Fig. 6 B1). To determine more precisely the internal distribution of the added myristoylated recombinant annexin XIIIb, we performed immunoelectron labeling using an anti-Flag antibody. As the endogenous protein, the myristoylated recombinant annexin XIIIb was located at the apical surface, on vesicular-like structures underneath the apical plasma membrane and on internal membrane located in the apical compartment (data not shown). It is important to note that the effects induced by the recombinant proteins in the transport assay were obtained with quantities that were not dramatically different from the physiological range. We estimated the amount of recombinant protein added being 2.5 (myristoylated) or 3 times (unmyristoylated) over the quantity of the endogenous annexin XIIIb found in control permeabilized cells (data not shown).
Figure 8.
Purified myristoylated recombinant annexin XIIIb is reaching the apical compartment of MDCK cells. (A) Purification of annexin XIIIb from Drosophila Schneider cells. Lane 1, Coomassie blue staining of the total lysate. Lane 2, Silver staining of eluted purified recombinant annexin XIIIb (see Materials and Methods). Arrowheads indicate the myristoylated annexin XIIIb. Note the appearance of dimers at 90 kD (each monomer is formed by the fusion of annexin XIIIb and histidine plus FLAG tags). Lanes 3–5, Autoradiography of annexin XIIIb expressing clones (lanes 3 and 4 clones BM17 and BM18, respectively) and of control cells (lane 5) after 3H-myristate labeling. (B) Distribution of the myristoylated recombinant annexin XIIIb in basolaterally SLO-permeabilized MDCK cells. The myristoylated recombinant protein was added from the basolateral side. Confocal (x, z) scans of cells double stained for nuclei (propidium iodide in red) and for the myristoylated recombinant annexin XIIIb with the anti-Flag antibody (green). Note that although in some cases the myristoylated recombinant protein was observed in both apical and basolateral compartments, the recombinant protein was mainly found in the apical compartment. Bar, 20 μm.
Anti-Annexin XIIIb Inhibits the Formation of HA-containing Carriers from the TGN
As myristoylated and unmyristoylated annexin XIIIb had different effects on apical surface delivery, we wanted to determine whether the added annexin XIIIb played a role in the budding of apical carriers from the TGN. We used an assay that allowed us to quantify the release in a cytosol- and energy-dependent manner of TGN-derived carriers containing either HA (apical carriers) or VSV G (basolateral carriers; Müsch et al., 1996). We tested the effect of the anti-annexin XIIIb antibody in this assay using another polyclonal antibody as a matched control. Since annexin V is located (though not exclusively) in the apical compartment of MDCK cells (Fig. 9 A), we tested an anti-annexin V in the budding assay and found that it did not interfere with the release of TGN-derived carriers; we obtained similar results with an anti-annexin VI antibody and an anti-annexin II antibody (MC2 affinity-purified rabbit and monoclonal HH7 antibodies, provided by S.E. Moss [University College, London, UK] and V. Gerke [University of Münster, Germany], respectively, data not shown). In contrast, the anti-annexin XIIIb antibody inhibited budding of HA-containing vesicles whereas the budding of the VSV G-containing vesicles was not impaired (Fig. 9 B). These results argue for a specific effect induced by the anti-annexin XIIIb antibody. We then analyzed the effect of the myristoylated recombinant annexin XIIIb on the budding assay using the annexin II–p11 heterotetramer as a control. While annexin II–p11 heterotetramer had no effect on the release of TGN-derived vesicles, myristoylated recombinant annexin XIIIb stimulated the release of apical carriers in a dose-dependent manner (Fig. 9 C). No effect of myristoylated recombinant annexin XIIIb was seen on basolateral carriers. Addition of unmyristoylated recombinant annexin XIIIb had no inhibitory action on the budding of apical carriers (Fig. 9 B). The unmyristoylated recombinant protein also did not affect the release of the basolateral carriers (Fig. 9 B). We also found an enrichment of the unmyristoylated recombinant protein on the TGN in permeabilized cells as determined by quantitating the extent of membrane structures colabeled with annexin XIIIb and the epitope-tagged sialyltransferase. We found that on the same membrane structures 8.2 ± 1.5 gold particles associated with sialyltransferase were colocalizing with 4.5 ± 0.7 gold particles associated with annexin XIIIb (n = 25). The average amount of annexin labeling associated with mitochondria was of 0.5 ± 0.2 (n = 25) excluding that the added recombinant protein was unspecifically binding any cell membrane. Thus, when compared with the quantitation performed for the endogenous annexin XIIIb in intact cells (see above), there was about three times more annexin XIIIb bound to the TGN while the unspecific labeling remained unchanged.
Figure 9.


Effects of anti-annexin XIIIb antibody, unmyristoylated and myristoylated recombinant annexin XIIIb on TGN-derived carriers release. (A) Confocal (x, z) scans of polarized MDCK cells double stained for nuclei (propidium iodide in red) and annexin V (green). (B) Anti-annexin XIIIb antibody can inhibit the budding of HA-containing carriers while it is not interfering with the budding of VSV G-containing carriers. Unmyristoylated recombinant annexin XIIIb (unmyr rec annexin XIIIb) is not affecting the release from the TGN of HA- or VSV G-containing carriers. The matched controls, i.e., anti-annexin V antibody and the annexin II–p11 complex have no effect on the budding of TGN-derived carriers (see text). (C) Myristoylated recombinant annexin XIIIb (myr rec annexin XIIIb) can stimulate in a dose- dependent manner the release of HA-containing carriers from the TGN while leaving the release of VSV G-containing carriers unaffected. Bar, 10 μm.
These findings lead us to suggest that annexin XIIIb regulates the formation and/or the budding of apical carriers and that its action might extend to the docking and fusion stage of apical delivery.
Discussion
In this study, we have demonstrated a specific role for annexin XIIIb in the delivery of influenza virus HA from the TGN to the apical surface. Several lines of evidence support this conclusion, namely the distribution of annexin XIIIb, its association with apical rafts and the results from functional assays.
Our subcellular localization data show that annexin XIIIb is uniquely placed to function in apical transport. In fully polarized MDCK cells, annexin XIIIb is localized along the route from the TGN to the apical plasma membrane. In the TGN, annexin XIIIb colocalized with the TGN resident enzyme sialyltransferase. In the apical cytoplasm, we could visualize annexin XIIIb on tubular or vesicular structures devoid of sialyltransferase. These structures could be viewed as carriers en route from the TGN. Supporting this interpretation, we observed that annexin XIIIb colocalized with HA in tubulovesicular and vesicular-like structures both in cryosections of MDCK cells and in isolated TGN-derived vesicles. This is in accordance with our previous biochemical results using immunoisolated apical transport vesicles (Wandinger-Ness et al., 1990; Fiedler et al., 1995). The results presented here thus extend these data by showing that annexin XIIIb colocalizes with HA in the same carrier. This vesicle fraction contains several other proteins that in subsequent studies were visualized in the apical compartment. However, these proteins, namely vesicular integral proteins (VIPs) of 17 kD, 21 kD (caveolin-1), and of 36 kD were also observed in the basolateral compartment although most likely associated in a different way as was recently shown for VIP 21/caveolin-1 (Scheiffele et al., 1998). Therefore, to date, among the proteins identified in immunoisolated apical carriers, annexin XIIIb is the only protein together with HA that has a predominantly apical distribution. Annexins are thought to mediate fusion events and several annexins have previously been shown to localize in the apical compartment of epithelial cells, i.e., annexins I, II, IV, and VI. However, these annexins are absent from immunoisolated apical vesicles (Wandinger-Ness et al., 1990). Thus, the localization of annexin XIIIb places it as a major candidate for a key role in the apical biosynthetic route of MDCK cells.
A role for annexin XIIIb in apical transport was further suggested by its association with apical rafts as shown by the inclusion of annexin XIIIb in DIGs. We found an association of annexin XIIIb with rafts that was not calcium dependent. This case is especially intriguing because annexin XIIIb is a peripheral membrane protein associated with the inner leaflet of the rafts. Our result raises the question of how a cytosolic peripheral protein can associate with a lipid microdomain with cholesterol and sphingolipids enriched in the exoplasmic leaflet.
One feature could be the myristoylation of annexin XIIIb (Wice and Gordon, 1992; Lafont, F., unpublished results). Myristoylation is a cotranslational and irreversible event that is found in many peripheral proteins partitioning between the cytosol and membranes (Johnson et al., 1994; Boutin, 1997). Doubly acylated proteins like the Gα subunit of heterotrimeric G proteins and the nonreceptor protein kinases also share the property of being associated with lipid rafts from the cytosolic side (Sargiacomo et al., 1993). Furthermore, for both Gα and the src-like kinases, double acylation by myristoylation and palmitoylation seems to be required for their binding to DIGs (Brown and London, 1997; Harder and Simons, 1997; Scheiffele and Simons, 1998). As for annexin XIIIb, the kinase fyn was shown to be increasingly solubilized after Triton extraction upon removal of cholesterol by MeβCD (Scheiffele et al., 1997).
However, the single acylation of annexin XIIIb is unlikely to be sufficient for DIG-association. Interestingly, other annexins (i.e., II, IV, V, and VI) have been shown to associate with DIGs and these proteins are neither myristoylated nor palmitoylated (Sargiacomo et al., 1993; Schnitzer et al., 1995; Parkin et al., 1996). A recent report analyzed annexin II behavior after treating a membrane fraction enriched in plasma membrane and endosomes with cholesterol-sequestering drugs or digitonin in the absence of calcium (Harder et al., 1997). Annexin II was extracted together with a subset of proteins all related to the actin cytoskeleton (ezrin, moesin, and α-actinin) that were immunoprecipitated with an anti-annexin II antibody. The interpretation of this result led the authors to conclude that annexin II could serve as an interface between actin and cholesterol-rich membrane domains. How annexin II binds to DIGs is not known. Either the protein binds to raft lipids or to raft proteins or to both. Altogether these and our studies suggest that annexins II, IV, V, VI, and XIII function in specific lipid environments and that they can be floated with DIGs. How far this can be generalized to the other members of the annexin superfamily and how important this property is for their physiological role remains to be elucidated.
Concerning annexin XIIIb, we combined cholesterol depletion and floatation analysis after detergent solubilization to demonstrate that annexin XIIIb binds to rafts. Although from a total cell extract a small pool of annexin XIIIb was observed floating after Triton extraction we think that this finding is highly significant because the soluble pool remains at the bottom of the gradient. We can therefore exclude a smearing effect since annexin XIIIb needs to bind membranes of light density to float. Annexin XIIIb was not found associated with all membranes floating since was not found in the very top fractions where a pool of VIP21/caveolin-1 was recovered. The shift observed after cholesterol extraction was reproducible and was also observed when the same experiment was performed with isolated apical carriers. The binding of annexin XIIIb to membranes of the same buoyant density as associated with HA did not depend on polymerized actin as treatment of the samples with latrunculin did not abolish DIG association. The binding to DIGs could be mediated both by myristoylation on the NH2 terminus and by the intrinsic lipid binding features of annexins such as those involved in the binding of annexin II to DIGs. What features of the inner lipid leaflet of sphingolipid-cholesterol rafts could determine how annexins associate with rafts? The main characteristic of annexins is their calcium-dependent binding to negatively charged phospholipids (Blackwood and Ernst, 1990). One interesting candidate is phosphatidylinositol (4,5) biphosphate (PIP2) which has been shown to be present in DIGs (Pike and Casey, 1996). Annexins have been described either not to be able to bind PIP2 when immobilized on hydrophobic support (Edwards and Crumpton, 1991) or to bind PIP2 when incorporated in liposomes although without preference over other charged phospholipids e.g., phosphatidic acid or phosphatidyl serine (Junker and Creutz, 1994). Clearly, analysis of the binding should be extended to lipid vesicles mimicking raft microdomains. Another important issue for future work is whether these interactions involve other proteins associated with the lipid rafts such as for instance VIP 21/caveolin-1. Annexin XIIIb and annexin II associate with DIGs that derive at least partially from different cellular compartments because these two annexins do not colocalize. This suggests that different annexins could be associated with DIGs present in different compartments and therefore raises the question of their function in different cellular locations. Annexins could regulate protein– protein and protein–lipid interaction in membrane microdomains involved in sorting events and signal transduction cascades. Different annexins could modulate raft function by binding to sphingolipid-cholesterol rafts in different organelles.
The role of annexin XIIIb in apical transport receive further support by our experiments with recombinant proteins in the SLO-permeabilized cells. Recombinant annexin XIIIb lacking its NH2-terminal myristoyl group was shown to inhibit apical delivery while addition of myristoylated annexin XIIIb to the permeabilized cells stimulated apical transport specifically. By analyzing the effects of these recombinant proteins in an assay that measures the release of apical and basolateral carriers from the TGN (Müsch et al., 1996) the stimulatory action of myristoylated annexin XIIIb could be pinpointed to the stage of the budding process. However, the unmyristoylated annexin XIIIb did not affect the release of apical carriers. Thus, the inhibition effect of this recombinant protein observed on the apical delivery of TGN-derived carriers must concern the delivery of the transport carriers or the docking/fusion stage. We exclude a general annexin effect of annexin XIIIb (i.e., general lipid-binding leading to aggregation of vesicles inside the cells) because the annexin II–p11 heterotetramer had no effect on apical transport although this annexin is present in MDCK cells and localized in the apical and basolateral compartments. We also did not observe the formation of giant vesicles upon addition of recombinant unmyristoylated annexin XIIIb (data not shown).
There are several mechanisms that could account for how annexin XIIIb exerts its effect on apical transport. Taking into account the dynamics of raft domains (Harder and Simons, 1997 a), one has to consider not only proteins recruited into these rafts but also proteins involved in clustering rafts into larger domains. In the TGN, annexin XIIIb could be involved in assembling raft lipids and proteins into the apical carriers. One important issue is the topology of the annexin XIIIb binding to the lipids. Annexin molecules have been shown to possess a slightly curved shape with the calcium and phospholipid binding domains on the convex face and with the NH2- and COOH-terminal domains on the concave face (Benz and Hofmann, 1997). If the NH2 terminus was associated with the lipid bilayer, would the calcium and phospholipid binding domains be exposed to the cytosol (see Fig. 10 A)? In that case, the membrane association of annexin XIIIb should be weak since the unitary Gibbs free energy for binding a myristoylated peptide to a phospholipid bilayer is not sufficient for membrane anchorage (Peitzsch and McLaughlin, 1993). Thus, other interactions would be required to stabilize the binding to the lipid microdomains.
Figure 10.
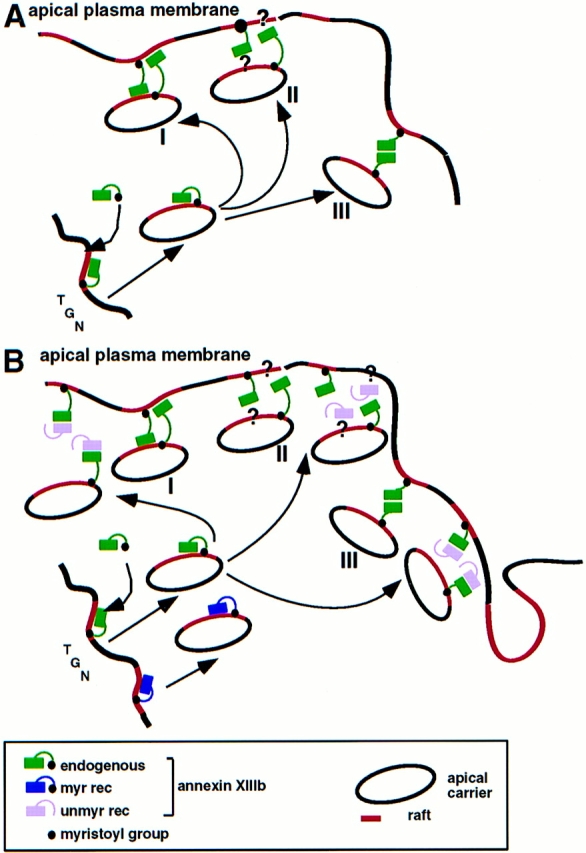
Model for the participation of annexin XIIIb and for the role played by the recombinant proteins in the apical exocytic pathway. (A) The raft-associated annexin XIIIb in the TGN level promotes assembly of apical carriers and participates in the docking and/or fusion steps at the plasma membrane. The raft depicted in red lines should be considered as membrane microdomains whose lipids are enriched in sphingolipid and cholesterol. In the last steps of exocytic transport annexin XIIIb could function either by binding to a membrane receptor (lipid domain I and/or proteins II), or by oligomerization (III). (B) Influence of unmyristoylated (unmy rec) and myristoylated (myr rec) recombinant annexin XIIIb on the apical exocytic pathway. The unmyristoylated protein inhibits the docking and/or fusion steps by interfering with the binding of the endogenous annexin XIIIb to its membrane ligand: lipid microdomains (I), protein complex, including putative cytosolic proteins (II), or itself, oligomerization (III). The myristoylated recombinant protein acts as the endogenous protein and increases the efficiency of the apical transport.
Another possibility is that annexin XIIIb is an adaptor that leads to the binding of other molecules to the apical transport carriers e.g., microtubule motors. Alternatively, annexin XIIIb could be involved in opening the way for the apical transport carriers through the cortical actin network underlying the apical plasma membrane. Indeed, annexin II has been proposed to mimic the effect on the cortical actin network by 14-3-3 proteins that might explain its effect on regulated exocytosis (Ali et al., 1989; Sarafian et al., 1991; Graham et al., 1997). We cannot exclude that annexin XIIIb would act in exocytosis by remodelling the actin cytoskeleton. However, we think that a direct role on the cytoskeleton is unlikely. We have not observed changes in the actin network when the unmyristoylated recombinant annexin XIIIb was introduced in permeabilized MDCK cells (Lecat, S., unpublished data). Moreover, we know that this recombinant protein does not bind actin (Lecat, S., unpublished data).
Although evidence is emerging for a role of SNAREs in the apical exocytic pathway, the sequence of events leading to the delivery is not understood. The relationship between SNAREs and annexins require further study. The fact that annexin XIIIb is present both on apical carriers and at the cytosolic surface of the apical plasma membrane suggests that annexin XIIIb could be involved in the docking/fusion stage of delivery of HA to the apical cell surface (Fig. 10 A). In this way, annexin XIIIb could also contribute to docking specificity. The flexibility of the molecule allowing one monomer to link donor and acceptor membranes would facilitate bilayer docking (Fig. 10 A, I) as has been proposed for other annexins e.g., (Ernst et al., 1991). Alternatively, annexin XIIIb on the carrier surface and on the cytoplasmic leaflet of the apical plasma membrane would allow the formation of homooligomers (Fig. 10 A, III), for instance by trimers on each side forming bridging hexamers as suggested for annexin XII (Luecke et al., 1995). Like many other annexins, recombinant annexin XIIIb can form dimers (Fig. 8 A), trimers, and hexamers (our unpublished data). Whether such oligomers are formed in MDCK cells remains to be seen. Identification of annexin XIIIb binding partners (see Fig. 10 A, II) will help to connect annexin XIIIb to the SNARE docking and fusion machinery (Weber et al., 1998) or possibly to define a novel mechanism for membrane delivery.
Acknowledgments
We are grateful to S. Bhakdi and V. Gerke for the generous supply of recombinant SLO and purified annexin II–p11 heterotetramer complex, respectively. We thank R. Luedecke from MSD Sharp & Dohme for the kind gift of lovastatin. We thank T. Nilson for providing the construct for the expression of the VSV G-tagged sialyl transferase. S. Eaton, T. Harder, P. Scheiffele, and M. Zerial are acknowledged for their critical reading of the manuscript. We thank S. Brendel and K. Ekroos for expert technical assistance and D. Zacchetti for help in calcium concentration determinations.
This work was supported by Commission of the European Communities and grant SFB 352.
Abbreviations used in this paper
- CLAP
chymostatin, leupeptin, antipain, pepstatin A
- DIG
detergent-insoluble glycosphingolipid complex
- GPI
glycosylphosphatidylinositol
- his6-GST
his6-glutathione-S-transferase
- HA
hemagglutinin
- MeβCD
methyl-β-cyclodextrin
- NSF
N-ethyl maleimide-sensitive factor
- PIP2
phosphatidylinositol (4,5) biphosphate
- SLO
streptolysin-O
- SNAP
soluble NSF-associated protein
- SNARE
SNAP receptor
- TfR
transferrin receptor
- VIP
vesicular integral protein
- VSV
vesicular stomatitis virus
Footnotes
Address all correspondence to Frank Lafont, European Molecular Biology Laboratory, Cell Biology and Biophysics Programme, Meyerhofstrasse 1, Postfach 10 2209, D-69012 Heidelberg, Germany. Tel: (49) 6221 387390. Fax: (49) 6221 387512. E-mail: lafont@embl-heidelberg.de
References
- Ahmed SN, Brown DA, London E. On the origin of sphingolipid/ cholesterol-rich detergent-insoluble cell membranes: physiological concentrations of cholesterol and sphingolipid induce formation of a detergent-insoluble, liquid-ordered lipid phase in model. Biochemistry. 1997;36:10944–10953. doi: 10.1021/bi971167g. [DOI] [PubMed] [Google Scholar]
- Ali SM, Geisow MJ, Burgoyne RD. A role for calpactin in calcium-dependent exocytosis in adrenal chromaffin cells. Nature. 1989;340:313–315. doi: 10.1038/340313a0. [DOI] [PubMed] [Google Scholar]
- Balcarova-Ständer J, Pfeiffer SE, Fuller SD, Simons K. Development of cell surface polarity in the epithelial Madin-Darby canine kidney (MDCK) cell line. EMBO (Eur Mol Biol Organ) J. 1984;3:2687–2694. doi: 10.1002/j.1460-2075.1984.tb02194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckers CJM, Keller DS, Balch WE. Semi-intact cells permeable to macromolecules: use in reconstitution of protein transport from the endoplasmic reticulum to the golgi complex. Cell. 1987;50:523–534. doi: 10.1016/0092-8674(87)90025-0. [DOI] [PubMed] [Google Scholar]
- Bennett M, Wandinger-Ness A, Simons K. Release of putative exocytic transport vesicles from perforated MDCK cells. EMBO (Eur Mol Biol Organ) J. 1988;7:4075–4085. doi: 10.1002/j.1460-2075.1988.tb03301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benz J, Hofmann A. Annexins: from structure to function. Biol Chem. 1997;378:177–183. [PubMed] [Google Scholar]
- Blackwood RA, Ernst JD. Characterization of Ca2+-dependent phospholipid binding, vesicle aggregation and membrane fusion by annexins. Biochem J. 1990;266:195–200. doi: 10.1042/bj2660195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutin JA. Myristoylation. Cell Signal. 1997;9:15–35. doi: 10.1016/s0898-6568(96)00100-3. [DOI] [PubMed] [Google Scholar]
- Brown RE. Sphingolipid organization in biomembranes: what physical studies of model membranes reveal. J Cell Sci. 1998;111:1–9. doi: 10.1242/jcs.111.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA, Rose JK. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell. 1992;68:533–544. doi: 10.1016/0092-8674(92)90189-j. [DOI] [PubMed] [Google Scholar]
- Brown DA, London E. Structure of detergent-resistant membrane domains: does phase separation occur in biological membranes? . Biochem Biophys Res Commun. 1997;240:1–7. doi: 10.1006/bbrc.1997.7575. [DOI] [PubMed] [Google Scholar]
- Burgoyne RD, Clague MJ. Annexins in the endocytic pathway. Trends Biochem Sci. 1994;19:231–232. doi: 10.1016/0968-0004(94)90143-0. [DOI] [PubMed] [Google Scholar]
- Cerneus DP, Ueffing E, Posthuma G, Strous GJ, van der Ende A. Detergent insolubility of alkaline phophatase during biosynthetic transport and endocytosis. Role of cholesterol. J Biol Chem. 1993;268:3150–3155. [PubMed] [Google Scholar]
- Creutz CE. The annexins and exocytosis. Science. 1992;258:924–931. doi: 10.1126/science.1439804. [DOI] [PubMed] [Google Scholar]
- Edwards HC, Crumpton MJ. Ca2+-dependent phospholipid and arachidonic acid binding by the placental annexins VI and IV. Eur J Biochem. 1991;198:121–129. doi: 10.1111/j.1432-1033.1991.tb15994.x. [DOI] [PubMed] [Google Scholar]
- Ernst JD, Hoye H, Blackwood RA, Mock TL. Identification of a domain that mediates vesicle aggregation reveals functional diversity of annexin repeats. J Biol Chem. 1991;266:6670–6673. [PubMed] [Google Scholar]
- Fath KR, Burgess DR. Golgi-derived vesicles from developing epithelial cells bind actin filaments and possess myosin-I as a cytoplasmically oriented peripheral membrane protein. J Cell Biol. 1993;120:117–127. doi: 10.1083/jcb.120.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiedler K, Lafont F, Parton RG, Simons K. Annexin XIIIb: a novel epithelial specific annexin is implicated in vesicular traffic to the apical plasma membrane. J Cell Biol. 1995;128:1043–1053. doi: 10.1083/jcb.128.6.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fra, A.M., E. Williamson, K. Simon, and R.G. Parton. 1995. De novo formation of caveolae in lymphocytes by expression of VIP21-caveolin. Proc. Natl. Acad. Sci. USA. 92:8655-–8659. [DOI] [PMC free article] [PubMed]
- Fuller SD, Simons K. Transferrin receptor polarity and recycling accuracy in “tight” and “leaky” strains of Madin-Darby canine kidney cells. J Cell Biol. 1986;103:1767–1779. doi: 10.1083/jcb.103.5.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhard W, Yewdell J, Frankel ME, Webster R. Antigenic structure of influenza virus haemagglutinin defined by hybridoma antibodies. Nature. 1981;290:713–717. doi: 10.1038/290713a0. [DOI] [PubMed] [Google Scholar]
- Graham ME, Gerke V, Burgoyne RD. Modification of annexin II expression in PC12 cell lines does not affect Ca2+-dependent exocytosis. Mol Biol Cell. 1997;8:431–442. doi: 10.1091/mbc.8.3.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenberg J, Emans N. Annexins in membrane traffic. Trends Cell Biol. 1993;3:224–227. doi: 10.1016/0962-8924(93)90116-i. [DOI] [PubMed] [Google Scholar]
- Gut A, Kappeler F, Hyka N, Balda MS, Hauri H-P, Matter K. Carbohydrate-mediated Golgi to cell surface transport and apical targeting of membrane proteins. EMBO (Eur Mol Biol Organ) J. 1998;17:1919–1929. doi: 10.1093/emboj/17.7.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hailstones D, Sleer LS, Parton RG, Stanley KK. regulation of caveolin and caveolae by cholesterol in MDCK cells. J Lipid Res. 1998;39:369–379. [PubMed] [Google Scholar]
- Hanada K, Nishijima M, Akamatsu Y, Pagano RE. Both sphingolipids and cholesterol participate in the detergent insolubility of alkaline phosphatase, a glycosylphosphatidylinositol-anchored protein, in mammalian membranes. J Biol Chem. 1995;270:6254–6260. doi: 10.1074/jbc.270.11.6254. [DOI] [PubMed] [Google Scholar]
- Hannan LA, Edidin M. Traffic, polarity, and detergent solubility of a glycosylphosphatidyl-anchored protein after LDL-deprivation of MDCK cells. J Cell Biol. 1996;133:1265–1276. doi: 10.1083/jcb.133.6.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder T, Gerke V. The subcellular distribution of early endosomes is affected by the annexin II2p11(2) complex. J Cell Biol. 1993;123:1119–1132. doi: 10.1083/jcb.123.5.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder T, Simons K. Caveolae, DIGs, and the dynamics of sphingolipid-cholesterol microdomains. Curr Opin Cell Biol. 1997;9:534–542. doi: 10.1016/s0955-0674(97)80030-0. [DOI] [PubMed] [Google Scholar]
- Harder T, Kellner R, Parton RG, Gruenberg J. Specific release of membrane-bound annexin II and cortical cytoskeleton elements by sequestration of membrane cholesterol. Mol Biol Cell. 1997;8:533–545. doi: 10.1091/mbc.8.3.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber, L.A., and K. Simons. 1994. Preparing and purification of post-Golgi transport vesicles from perforated Madin-Darby kidney cells. In Cell Biology: A Laboratory Handbook. Vol. 1. J.E. Celis, editor. Acad. Press, San Diego. 517–524.
- Ikonen E, Tagaya M, Ullrich O, Montecucco C, Simons K. Different requirements for NSF, SNAP, and Rab proteins in apical and basolateral transport in MDCK cells. Cell. 1995;81:571–580. doi: 10.1016/0092-8674(95)90078-0. [DOI] [PubMed] [Google Scholar]
- Jackson MR, Song ES, Yang Y, Peterson PA. Empty and peptide-containing conformers of class I major histocompatibility complex molecules expressed in Drosophila melanogastercells. Proc Natl Acad Sci USA. 1992;89:12117–12121. doi: 10.1073/pnas.89.24.12117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DR, Bhatnagar RS, Knoll LJ, Gordon JI. Genetic and biochemical studies of protein N-myristoylation. Annu Rev Biochem. 1994;63:869–914. doi: 10.1146/annurev.bi.63.070194.004253. [DOI] [PubMed] [Google Scholar]
- Junker M, Creutz CE. Ca2+-dependent binding of endonexin (annexin IV) to membranes: analysis of the effects of membrane lipid composition and development of a predictive model for the binding interaction. Biochemistry. 1994;33:8930–8940. doi: 10.1021/bi00196a010. [DOI] [PubMed] [Google Scholar]
- Keller P, Simons K. Cholesterol is required for surface transport of influenza virus hemagglutinin. J Cell Biol. 1998;140:1357–1367. doi: 10.1083/jcb.140.6.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein U, Gimpl G, Fahrenholz F. Alteration of the myometrial plasma membrane cholesterol content with b-cyclodextrin modulates the binding affinity of the oxytocin receptor. Biochemistry. 1995;34:13784–13793. doi: 10.1021/bi00042a009. [DOI] [PubMed] [Google Scholar]
- Kundu A, Avalos RT, Sanderson CM, Nayak DP. Transmembrane domain of influenza virus neuraminidase, a type II protein, possesses an apical sorting signal in polarized MDCK cells. J Virol. 1996;70:6508–6515. doi: 10.1128/jvi.70.9.6508-6515.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafont F, Simons K. The role of microtubule-based motors in the exocytic transport of polarized cells. Semin Cell Dev Biol. 1996;7:343–355. [Google Scholar]
- Lafont F, Burkhardt JK, Simons K. Involvement of microtubule motors in basolateral and apical transport in kidney cells. Nature. 1994;372:801–803. doi: 10.1038/372801a0. [DOI] [PubMed] [Google Scholar]
- Lafont, F., K. Simons, and E. Ikonen. 1995. Dissecting the molecular mechanisms of polarized membrane traffic: reconstitution of three transport steps in epithelial cells using Streptolysin-O permeabilization. Cold Spring Harb. Symp. Quant. Biol. LX:753–762. [DOI] [PubMed]
- Luecke H, Chang BT, Mailliard WS, Schlaepfer DD, Haigler HT. Crystal structure of the annexin XII hexamer and implications for bilayer insertion. Nature. 1995;378:512–515. doi: 10.1038/378512a0. [DOI] [PubMed] [Google Scholar]
- Low SH, Chapin SJ, Wimmer C, Whiteheart SW, Komuves LG, Mostov KE, Weimbs T. The SNARE machinery is involved in apical membrane trafficking in MDCK cells. J Cell Biol. 1998;141:1503–1513. doi: 10.1083/jcb.141.7.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matlin KS, Simons K. Reduced temperature prevents transfer of a membrane glycoprotein to the cell surface but does not prevent terminal glycosylation. Cell. 1983;34:233–243. doi: 10.1016/0092-8674(83)90154-x. [DOI] [PubMed] [Google Scholar]
- Matlin KS, Reggio H, Helenius A, Simons K. Infectious entry pathway of influenza virus in a canine kidney cell line. J Cell Biol. 1981;91:601–613. doi: 10.1083/jcb.91.3.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matter K, Mellman I. Mechanisms of cell polarity: sorting and transport in epithelial cells. Curr Opin Cell Biol. 1994;6:545–554. doi: 10.1016/0955-0674(94)90075-2. [DOI] [PubMed] [Google Scholar]
- Melkonian KA, Chu T, Tortorella LB, Brown DA. Characterization of proteins in detergent-resistant membrane complexes from Madin-Darby canine kidney epithelial cells. Biochemistry. 1995;34:16161–16170. doi: 10.1021/bi00049a031. [DOI] [PubMed] [Google Scholar]
- Moss, S.E. 1992. The Annexins. In The Annexins. S.E. Moss, editor. Portland Press, London. 1–9.
- Muallem S, Kwiatkowska K, Xu X, Yin HL. Actin filament disassembly is a sufficient final trigger for exocytosis in nonexcitable cells. J Cell Biol. 1995;128:589–598. doi: 10.1083/jcb.128.4.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata M, Peranen J, Schreiner R, Wieland F, Kurzchalia TV, Simons K. VIP21/caveolin is a cholesterol-binding protein. Proc Natl Acad Sci USA. 1995;92:10339–10343. doi: 10.1073/pnas.92.22.10339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müsch A, Xu H, Shields D, Rodriguez-Boulan E. Transport of vesicular stomatitis virus to the cell surface is signal mediated in polarized and nonpolarized cells. J Cell Biol. 1996;133:543–558. doi: 10.1083/jcb.133.3.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojakian GK, Schwimmer R. The polarized distribution of an apical cell surface glycoprotein is maintained by the interactions with the cytoskeleton of Madin-Darby canine kidney cells. J Cell Biol. 1988;107:2377–2387. doi: 10.1083/jcb.107.6.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkin ET, Turner AJ, Hooper NM. Isolation and characterization of two distinct low-density, triton-insoluble, complexes from porcine lung membranes. Biochem J. 1996;319:887–896. doi: 10.1042/bj3190887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peitzsch RM, McLaughlin S. Binding of acylated peptides and fatty acids to phospholipid vesicles: pertinence to myristoylated proteins. Biochemistry. 1993;32:10436–10443. doi: 10.1021/bi00090a020. [DOI] [PubMed] [Google Scholar]
- Peränen J, Auvinen P, Virta H, Wepf R, Simons K. Rab8 promotes polarized membrane transport through reorganization of actin and microtubules in fibroblasts. J Cell Biol. 1996;135:153–167. doi: 10.1083/jcb.135.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike LJ, Casey L. Localization and turnover of phosphatidylinositol 4,5-biphosphate in caveolae enriched membrane domains. J Biol Chem. 1996;271:26453–26456. doi: 10.1074/jbc.271.43.26453. [DOI] [PubMed] [Google Scholar]
- Pimplikar SW, Ikonen E, Simons K. Basolateral protein transport in streptolysin O-permeabilized MDCK cells. J Cell Biol. 1994;125:1025–1035. doi: 10.1083/jcb.125.5.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabouille C, Hui N, Hunte F, Kieckbush R, Berger EG, Warren G, Nilsson T. Mapping the distribution of Golgi enzymes involved in the construction of complex oligosaccharides. J Cell Sci. 1995;108:1617–1627. doi: 10.1242/jcs.108.4.1617. [DOI] [PubMed] [Google Scholar]
- Raynal P, Pollard HB. Annexins: the problem of assessing the biological role for a gene family of multifunctional calcium- and phospholipid-binding proteins. Biochim Biophys Acta. 1994;1197:63–93. doi: 10.1016/0304-4157(94)90019-1. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Boulan E, Nelson WJ. Morphogenesis of the polarized epithelial cell phenotype. Science. 1989;245:718–725. doi: 10.1126/science.2672330. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Boulan E, Powell SK. Polarity of epithelial and neuronal cells. Annu Rev Cell Biol. 1992;8:395–427. doi: 10.1146/annurev.cb.08.110192.002143. [DOI] [PubMed] [Google Scholar]
- Rothman JE. Mechanisms of intracellular protein transport. Nature. 1994;372:55–63. doi: 10.1038/372055a0. [DOI] [PubMed] [Google Scholar]
- Sarafian T, Pradel LA, Henry JP, Aunis D, Bader MF. The participation of annexin II (calpactin I) in calcium-evoked exocytosis requires protein kinase C. J Cell Biol. 1991;114:1135–1147. doi: 10.1083/jcb.114.6.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargiacomo M, Sudol M, Tang Z, Lisanti MP. Signal transducing molecules and GPI-linked proteins form a caveolin-rich insoluble complex in MDCK cells. J Cell Biol. 1993;122:789–807. doi: 10.1083/jcb.122.4.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiffele, P., and K. Simons. 1998. Epithelial polarity and sorting. In Epithelial morphogenesis in development and disease. W. Birchmeier, and C. Birchmeier, editors. Hardwood Publishers, In press.
- Scheiffele P, Peränen J, Simons K. N-glycans as apical sorting signals in epithelial cells. Nature. 1995;378:96–98. doi: 10.1038/378096a0. [DOI] [PubMed] [Google Scholar]
- Scheiffele P, Roth MG, Simons K. Interaction of influenza virus hemagglutinin with sphingolipid-cholesterol membrane domains via its transmembrane domain. EMBO (Eur Mol Biol Organ) J. 1997;16:5501–5508. doi: 10.1093/emboj/16.18.5501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiffele P, Verkade P, Fra AM, Virta H, Simons K, Ikonen E. Caveolin-1 and -2 in the exocytic pathway of MDCK cells. J Cell Biol. 1998;140:795–806. doi: 10.1083/jcb.140.4.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnitzer JE, Liu J, Oh P. Endothelial caveolae have the molecular transport machinery for vesicle budding, docking, and fusion including VAMP, NSF, SNAP, Annexins, and GTPases. J Biol Chem. 1995;270:14399–14404. doi: 10.1074/jbc.270.24.14399. [DOI] [PubMed] [Google Scholar]
- Schroeder R, London E, Brown D. Interactions between saturated acyl chains confer detergent resistance to lipids and glycosylphosphatidylinositol (GPI)-anchored proteins: GPI-anchored proteins in liposomes and cells show similar behavior. Proc Natl Acad Sci USA. 1994;91:12130–12134. doi: 10.1073/pnas.91.25.12130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K. Biogenesis of epithelial cell surface polarity. Harvey Lect. 1995;89:125–146. [PubMed] [Google Scholar]
- Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- van Meer G. Lipid traffic in animal cells. Annu Rev Cell Biol. 1989;5:247–275. doi: 10.1146/annurev.cb.05.110189.001335. [DOI] [PubMed] [Google Scholar]
- Wallny H-J, Sollami G, Karjalainen K. Soluble mouse major histocompatibility complex class II molecules produced in Drosophilacells. Eur J Immunol. 1995;25:1262–1266. doi: 10.1002/eji.1830250520. [DOI] [PubMed] [Google Scholar]
- Wandinger-Ness A, Bennett MK, Antony C, Simons K. Distinct transport vesicles mediate the delivery of plasma membrane proteins to the apical and basolateral domains of MDCK cells. J Cell Biol. 1990;111:987–1000. doi: 10.1083/jcb.111.3.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber T, Zemelman BV, McNew JA, Westerman B, Gmachl M, Parlati F, Söllner TH, Rothman JE. SNAREpins: Minimal machinery for membrane fusion. Cell. 1998;92:759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- Weimbs T, Low S-H, Chapin SJ, Mostov KE. Apical targeting in polarized epithelial cells: there's more afloat than rafts. Trends Cell Biol. 1997;7:393–399. doi: 10.1016/S0962-8924(97)01130-6. [DOI] [PubMed] [Google Scholar]
- Wice BM, Gordon JI. A strategy for isolation of cDNAs encoding proteins affecting human intestinal epithelial cell growth and differentiation: characterization of a novel gut-specific N-myristoylated annexin. JCell Biol. 1992;116:405–422. doi: 10.1083/jcb.116.2.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Shields D. Prohormone processing in the trans-Golgi network: endoproteolytic cleavage of prosomatostatin and formation of nascent secretory vesicles in permeabilized cells. J Cell Biol. 1993;122:1169–1184. doi: 10.1083/jcb.122.6.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yancey PG, Rodrigueza WV, Kilsdonk EP, Stoudt GW, Johnson WJ, Phillips MC, Rothblat GH. Cellular cholesterol efflux mediated by cyclodextrins: demonstration of kinetic pools and mechanism of efflux. J Biol Chem. 1996;271:16026–16034. doi: 10.1074/jbc.271.27.16026. [DOI] [PubMed] [Google Scholar]
- Yeaman C, Le Gall AH, Baldwin AN, Monlauzeur L, Le Bivic A, Rodriguez-Boulan E. The O-glycosylated stalk domain is required for apical sorting of neurotrophin receptors in polarized MDCK cells. J Cell Biol. 1997;139:929–940. doi: 10.1083/jcb.139.4.929. [DOI] [PMC free article] [PubMed] [Google Scholar]